

This case-control study assesses which genes contribute to the development of spontaneous coronary artery dissection.
Key Points
Question
Using genetic analyses of patients, what is the etiology of spontaneous coronary artery dissection (SCAD)?
Findings
In this case-control study of 130 patients with SCAD and 46 468 without who underwent whole-exome sequencing, those with SCAD were more likely to carry disruptive rare variants within fibrillar collagen genes. Mice deficient in fibrillar collagen genes demonstrated predilection to arterial dissection.
Meaning
Weakening of the extracellular matrix of the coronary arteries may contribute to the development of SCAD.
Abstract
Importance
Spontaneous coronary artery dissection (SCAD) is an increasingly recognized nonatherosclerotic cause of acute myocardial infarction enriched among individuals with early-onset myocardial infarction but is of unclear etiology.
Objective
To assess which genes contribute to the development of SCAD.
Design, Setting, and Participants
To prioritize genes influencing risk for SCAD, whole-exome sequencing was performed among individuals with SCAD in the discovery and replication cohorts from a tertiary care hospital outpatient specialty clinic, and gene set enrichment analyses were also performed for disruptive coding variants. All patients were sequentially enrolled beginning July 2013. Aggregate prevalence of rare disruptive variants for prioritized gene sets was compared between individuals with SCAD with population-based controls comprising 46 468 UK Biobank participants with whole-exome sequencing. Complementary mice models were used for in vivo validation. Analysis took place between June 2020 and January 2021.
Main Outcomes and Measures
The frequency and identity of rare genetic variants in individuals with SCAD.
Results
Of 130 patients, 109 (83.8%) were female (26 of 32 [81.2%] in the discovery cohort and 83 of 98 [84.7%] in the replication cohort) with mean (SD) age at first SCAD event of 48.41 (8.76) years in the discovery cohort and 47.74 (10.09) years in the replication cohort. Across all patients with SCAD, rare disruptive variants were found within 10 collagen genes (COL3A1, COL5A1, COL4A1, COL6A1, COL5A2, COL12A1, COL4A5, COL1A1, COL1A2, and COL27A1) were 17-fold (P = 1.5 × 10−9) enriched among individuals with SCAD compared with a background of 2506 constrained genes expressed in coronary artery. Furthermore, compared with individuals from the UK Biobank, individuals with SCAD were 1.75-fold (P = .04) more likely to carry disruptive rare variants within fibrillar collagen genes. Complementary mice models haploinsufficient for Col3a1 or Col5a1, the 2 most common collagen gene variants identified in SCAD cases, demonstrated increased risk of arterial dissection and increased size of arterial diameters especially in female mice, with resulting changes in collagen fibril organization and diameter.
Conclusions and Relevance
Unbiased gene discovery in patients with SCAD with independent human and murine validation highlights the role of the extracellular matrix dysfunction in SCAD.
Introduction
Spontaneous coronary artery dissection (SCAD) is a nonatherosclerotic cause of acute myocardial infarction (MI) frequently affecting young women.1,2,3,4,5,6 Up to 35% of women younger than 50 years presenting with MI may be due to SCAD.4,7,8 Other risk factors include fibromuscular dysplasia, pregnancy,9 and excessive emotional and/or physical stress.10 Imaging often identifies aneurysms, dissections, or luminal abnormalities in other arterial territories, suggesting a background of systemic arteriopathy. However, well-established genetically triggered arteriopathies are not the predominant cause of SCAD and familial cases are extremely rare,11,12,13,14,15,16 prompting further exploration to define the genetic architecture of SCAD. In this study, we perform prospective exome sequencing in a group of patients with SCAD, filtering variants to high confidence putative disruptive alleles in coronary artery–expressed genes and demonstrate enrichment within collagen genes of the extracellular matrix.
Methods
Discovery and Replication Populations
This study was approved by the Mass General Brigham institutional review board. Participants were identified by their treating physicians within the Division of Cardiology and Vascular Medicine at the Massachusetts General Hospital. Participants either presented to Massachusetts General Hospital with SCAD, were transferred after a SCAD event from another hospital, or came to our clinic at Massachusetts General Hospital for a second opinion after the SCAD occurrence. Data were collected prospectively thereafter. Inclusion criteria were patients 18 years or older with a compatible clinical history and imaging evidence of SCAD. Patients with known single-gene genetic diagnoses at the time of enrollment were excluded from analysis. All patients provided written informed consent and were sequentially enrolled in the Massachusetts General Hospital SCAD registry beginning July 2013. For the purpose of this analysis, the discovery cohort was collected from 2013 until December 2015, and the validation cohort was collected between 2016 and December 2019.
Each patient’s medical record was used to collect details of their SCAD hospitalization (clinician documentation of presentation, hospital course, and management), laboratory data, imaging results (electrocardiogram, echocardiogram, computed tomography, magnetic resonance imaging, stress testing), cardiac catheterization reports, and follow-up studies (extracoronary vascular imaging, genetic testing). Additionally, patients completed questionnaires asking about demographics (including race and ethnicity), symptoms at time of event, SCAD history, medical history, reproductive health, medications, allergies, social history, nutrition, and physical activity.
Whole-Exome Sequencing and Variant Annotation
Discovery whole-exome sequencing (WES) was performed on individuals with SCAD, with replication in additional individuals with SCAD. Details regarding the sequencing method used in WES are provided in the eMethods in Supplement 1. Reads were aligned to the GRCh38 reference genome and the discovery and replication genotypes were jointly called into 1 variant call set using the Genome Analysis Toolkit version 4 Haplotype Caller according to best practices.17 Only 1 sample was excluded from the SCAD analysis owing to a high degree of contamination (>10%). Variants were filtered to those that passed variant quality score recalibration filters, insertion/deletion variants with quality by depth of 3 or more, single-nucleotide variations with quality by depth of 2 or more, variants with call rate more than 95%, and Hardy-Weinberg equilibrium P ≥ 1 × 10−6. Variant annotation and filtration was performed using Hail version 0.218 and annotating using the Variant Effect Predictor version 95 plugin,19 dbNSFPv4.0 table,20 and allele frequencies from Genome Aggregation Database (gnomAD) release 3.021 across 123 136 whole-exome and 15 496 whole-genome sequences from diverse ancestries as well as 50 726 DiscovEHR whole-exome sequences from the Regeneron Genetics Center and Geisinger Health System.22
Filtration to Rare Disruptive Variants
We identified disruptive coding variants that are rare (frequency, <0.1%) or absent across each ethnic group characterized in the 15 496 gnomAD whole genomes and 123 136 gnomAD WES, as well as from the 50 726 DiscovEHR WES. We filtered to genes intolerant of disruption (probability of loss-of-function intolerance [pLI], >0.9)23 expressed in coronary artery (median >2 reads per kilobase of transcript per million in Genotype-Tissue Expression [GTEx]24). Predicted disruptive variants were defined as missense deleterious variants via MetaSVM25 predictor or a predicted high-confidence loss-of-function variants via LOFTEE.26
UK Biobank WES, Variant Annotation, and Variant Filtration
Individual-level genomic data and longitudinal phenotypic data from the UK Biobank, a large-scale population-based data set consisting of genotype and phenotype data in approximately 500 000 volunteer participants recruited from 2006 to 2010 was used.27 Baseline assessments were conducted at 22 assessment centers across the UK using touch screen questionnaire, computer-assisted verbal interview, physical tests, and sample collection including for DNA. Additional details regarding the study protocol are described online.28 Of 49 960 UK Biobank individuals who underwent WES,29 we used data across 46 559 consenting, unrelated individuals (removing one of each pair of first- or second-degree relatives at random). Variants were filtered to those with call rate of more than 95% and Hardy-Weinberg equilibrium P ≥ 1 × 10−6. The exact same methods as used in the SCAD WES was used here as well to annotate variants and filter to rare disruptive variants in genes expressed within coronary artery. Secondary data analysis of the UK Biobank was facilitated by UK Biobank application 7089 and approved by the Mass General Brigham institutional review board.
Pathway Enrichment Analysis and Comparison With UK Biobank Controls and Patients With MI
Pathway enrichment analyses of resultant genes were performed using Gene Ontology’s Molecular Functions pathway through the online portal,30 which compares expected numbers of genes in each pathway expected to be found randomly with the actual number of genes present in a given list of genes to estimate an enrichment odds ratio and P value for each biological pathway. Enrichment analysis was performed across all identified unique genes with rare disruptive variants carried by individuals with SCAD (eTables 1 and 2 in Supplement 2). Significance was defined based on false discovery rate less than .05 across a background of constrained genes (pLI, >0.9)23 expressed in coronary artery in GTEx version 6.24 The top pathway (with the most significant enrichment P value) was carried forward for replication in the replication SCAD cohort. Finally, aggregate carrier status of disruptive variants in the top enriched pathway’s genes was associated with SCAD case status across the discovery and replication cohorts separately vs all UK Biobank individuals who underwent WES, UK Biobank MI-free controls, and, separately, UK Biobank individuals with MI using logistic regression under 3 adjustment models: (1) unadjusted; (2) sparsely adjusted for age, sex, and race; and (3) fully adjusted with the addition of hypertension, hypercholesterolemia, type 2 diabetes, and smoking status as covariates. For these analyses, UK Biobank individuals with MI were identified using the adjudicated MI phenotypes from the UK Biobank (field 42001), which combine self-report, hospital admission, and death codes for MI. UK Biobank baseline sample characteristics comparing individuals stratified by disruptive collagen variant carrier status are provided in eTable 4 in Supplement 2. Fixed-effects meta-analysis was used to meta-analyze across the SCAD discovery and replication enrichment associations. Statistical significance was assigned at α less than .05 for this analysis. Analyses were performed using R version 3.5 (R Foundation). Analysis took place between June 2020 and January 2021.
Clinical Annotation
We annotated the variants with Variant Effect Predictor version 10419 and obtained Human Genome Variation Society nomenclatures. Based on the obtained annotations, we queried American College of Medical Genetics and Genomics classification to VarSome framework,31 which provides automated variant classification following American College of Medical Genetics and Genomics guidelines32 using a series of evidence of variant characteristics and functional information as previously described.33
Mouse Models
All mice used in the study were cared for in accordance with the Partners Institutional Animal Care and Use Committee. Strain C.129S4(B6)-Col3a1tm1Jae/J (Col3a1+/−) mice were obtained from Jackson Laboratories (stock No. 002907). Col5a1+/− mice were generously provided by David E. Birk, PhD (University of South Florida, Tampa), and created by targeted deletion and described in detail previously.34 Col3a1+/−:Col5a1+/− mice were obtained by cross breeding. All experiments were performed on male and female animals at a 1:2 ratio, given additional analysis based on female mice parity.
All mice underwent echocardiogram evaluation at age 2 months and 4 months. Mice were euthanized at 6 months of age. Blood pressure was measured in mice prior to euthanasia using volume pressure recording sensor technology with the CODA mouse tail-cuff system (Kent Scientific), as previously described.35 Measurements of parasternal long-axis view were performed using Visualsonics Vevo 2100 and a 30-MHz probe on conscious mice after hair removal. Measurements were obtained by calculating the mean of 3 independent measurements at the aortic root and proximal ascending aorta.
Survival Curves
Survival curves (aortic dissection and euthanasia as end points) and statistical significance (χ2 and P values) between the survival functions for different strains were generated in GraphPad Prism8 by using the Kaplan-Meier estimator function for the log-rank, Breslow, and Tarone-Ware tests. Each gave similar results, and the log-rank test results are reported.
Histology
Removed tissues were then fixed in formalin, 10%, for 24 hours before transfer to ethanol, 70%, followed by paraffinization and sectioning (7μM) as previously described.36 Slides were produced for tissue with standard stains including hematoxylin-eosin or conjugated SM22α (Alexa Fluor 647; Santa Cruz Biotechnology). Slides were then visualized with the Leica TCS SP8 confocal microscopy station and the pictures were digitized with the Leica Application Suite X software.
Transmission Electron Microscopy
Fresh heart and ascending aortas were immersion fixed immediately on excision in a paraformaldehyde-lysine periodate solution for 24 hours at room temperature, then postfixed further in glutaraldehyde, 2.5%/paraformaldehyde, 2%, in 0.1 M sodium cacodylate buffer for several hours at room temperature and stored overnight at 4 °C. The next day, tissue was rinsed several times in cacodylate buffer, infiltrated for 1 hour in osmium tetroxide, 1%, then rinsed again several times. Specimens were then dehydrated through a graded series of ethanols to 100%, dehydrated briefly in propylene oxide, 100%, and allowed to preinfiltrate at least 2 hours in a 2:1 mix of propylene oxide and Eponate resin (Ted Pella). Samples were transferred into a 1:1 mix of propylene oxide and Eponate resin. The following day, specimens were allowed to infiltrate for at least 2 hours in a 2:1 mix of Eponate/propylene oxide, then several hours in fresh 100% Eponate resin. Tissue was transferred into flat molds with 100% Eponate resin and oriented such that aorta would be transversely sectioned; embeddings were placed in an oven at 60 °C for resin polymerization (24-48 hours). Thin (70 nm) sections were cut using a Leica EM UC7 ultramicrotome, collected onto formvar-coated grids, stained with uranyl acetate, 2%, and Reynold’s lead citrate and examined in a JEOL JEM 1011 transmission electron microscope at 80 kV. Images were collected using an AMT digital imaging system (Advanced Microscopy Techniques).
Results
To prioritize genes influencing risk for SCAD, we performed WES of 32 individuals with SCAD in the discovery cohort followed by 98 independent individuals with SCAD in the replication cohort, for a total of 130 individuals with SCAD from the Massachusetts General Hospital SCAD Registry (Table). Most of the patients evaluated were female (26 [81.2%] in the discovery cohort and 83 [84.7%] in the replication cohort) with mean (SD) age at first SCAD event of 48.41 (8.76) years in the discovery cohort and 47.74 (10.09) years in the replication cohort. The most common risk factors were hypertension, fibromuscular dysplasia, hypercholesterolemia, and smoking. Aneurysms and other dissections were infrequent among both the discovery (0% aneurysms; 1 of 32 dissections [3.1%]) and replication (7 of 98 aneurysms [7.1%]; 8 of 98 dissections [9.1%]) cohorts. The left anterior descending artery was the most commonly dissected coronary artery in both groups.
Table. Demographics of SCAD Discovery and Replication Cohorts.
| Characteristic | No. (%) | P value | |
|---|---|---|---|
| Discovery (n = 32) | Replication (n = 98) | ||
| Age, mean (SD), y | 48.41 (8.76) | 47.74 (10.09) | .74 |
| Race | |||
| African American | 1 (3.1) | 3 (3.1) | .96 |
| Asian | 1 (3.1) | 5 (5.1) | |
| White | 29 (90.6) | 87 (88.8) | |
| Othera | 1 (3.1) | 3 (3.1) | |
| Female | 26 (81.2) | 83 (84.7) | .86 |
| Male | 6 (18.8) | 15 (15.3) | .86 |
| BMI, mean (SD) | 24.99 (4.26) | 27.55 (6.01) | .92 |
| Current or former smoker | 5 (16.7) | 26 (26.8) | .38 |
| Hypertension | 14 (46.7) | 39 (39.48) | .65 |
| Hypercholesterolemia | 13 (43.3) | 26 (26.5) | .13 |
| Diabetes | 0 | 1 (1.0) | >.99 |
| Cardiomyopathy | 3 (10.0) | 9 (9.1) | >.99 |
| Recurrent SCAD | 7 (23.3) | 9 (9.1) | .08 |
| Fibromuscular dysplasia | 10 (33.3) | 31 (32.3) | >.99 |
| Any aneurysm | |||
| None | 32 (100.0) | 91 (92.9) | .79 |
| Aortic | 0 | 2 (2.0) | |
| Cervical | 0 | 1 (1.0) | |
| Intracranial | 0 | 1 (1.0) | |
| Mesenteric | 0 | 1 (1.0) | |
| Splenic | 0 | 2 (2.0) | |
| Other dissection | |||
| None | 31 (96.9) | 90 (91.8) | .22 |
| Cervical | 0 | 6 (6.1) | |
| Intracranial | 1 (3.1) | 0 | |
| Mesenteric | 0 | 1 (1.0) | |
| Aortic | 0 | 1 (1.0) | |
| Therapy | |||
| Medical | 13 (40.6) | 69 (70.4) | .001 |
| PCI | 17 (53.1) | 25 (25.5) | |
| Surgical | 0 | 4 (4.1) | |
| None | 2 (6.2) | 0 | |
| STEMI or NSTEMI | |||
| NSTEMI | 11 (34.4) | 61 (62.2) | .02 |
| STEMI | 19 (59.4) | 34 (34.7) | |
| No MI | 2 (6.2) | 3 (3.0) | |
| Activity | |||
| Emotional stress | 4 (12.5) | 17 (17.3) | .498 |
| Exercise | 11 (34.4) | 32 (32.7) | |
| Postpartum | 1 (3.1) | 3 (3.0) | |
| Rest | 14 (43.8) | 30 (30.3) | |
| Location 1 | |||
| LAD | 23 (71.9) | 58 (58.6) | .31 |
| LCx | 0 | 2 (2.0) | |
| D1 | 0 | 3 (3.0) | |
| OM | 2 (6.2) | 18 (18.2) | |
| PDA | 1 (3.1) | 7 (7.1) | |
| RCA | 4 (12.5) | 8 (8.1) | |
| Location 2 | |||
| LAD | 2 (6.2) | 2 (2.0) | .73 |
| LCx | 0 | 2 (2.0) | |
| D1 | 3 (9.4) | 5 (5.1) | |
| D2 | 0 | 1 (1.0) | |
| D3 | 0 | 1 (1.0) | |
| OM | 2 (6.2) | 14 (14.1) | |
| PDA | 1 (3.1) | 6 (6.1) | |
| RCA | 2 (6.2) | 6 (6.1) | |
| No SCAD location 2 | 22 (68.8) | 61 (62.2) | |
| Tortuosity = 1 | 25 (86.2) | 44 (48.4) | .001 |
Abbreviations: BMI, body mass index (calculated as weight in kilograms divided by height in meters squared); D1, first diagonal artery; D2, second diagonal artery; D3, third diagonal artery; LAD, left anterior descending artery; LCx, left circumflex artery; MI, myocardial infarction; NSTEMI, non–ST-segment elevation myocardial infarction; OM, obtuse marginal artery; PCI, percutaneous coronary intervention; PDA, posterior descending artery; RCA, right coronary artery; SCAD, spontaneous coronary artery dissection; STEMI, ST-segment elevation myocardial infarction.
Other includes Middle Eastern/North African and Pacific Islander.
We implemented case-based filtration criteria (eFigure 1 in Supplement 1) to identify predicted disruptive coding variants that were rare (frequency, <0.1%) across 3 data sets (15 496 gnomAD whole-genome sequences, 123 136 gnomAD WES, and 50 726 DiscovEHR WES),21,22 within constrained genes intolerant of disruption (pLI, >0.9) and expressed in the coronary arteries (median, >2 reads per kilobase of transcript per million in GTEx version 624). Fifty-five rare disruptive coding variants in 53 constrained coronary artery-expressed genes were discovered in 32 individuals with SCAD in the discovery cohort (eFigure 2 in Supplement 1 and eTable 1 in Supplement 2). Unbiased enrichment analyses of these genes across all the Gene Ontology Molecular Function pathways found the strongest evidence of enrichment for 10 collagen genes among the extracellular matrix structural constituent conferring tensile strength (GO:0030020) pathway (COL3A1, COL5A1, COL4A1, COL6A1, COL5A2, COL12A1, COL4A5, COL1A1, COL1A2, and COL27A1), with 23.6-fold enrichment (P = 8.08 × 10−6, false discovery rate < .05) against a background of 2506 loss-of-function intolerant genes expressed in coronary artery (eTable 2 in Supplement 2). Replication of this finding was performed in 98 independent individuals with SCAD in the discovery cohort, identifying 120 unique genes with rare disruptive variants and also showed a consistent 13-fold enrichment (P = 4 × 10−5) of genes in the same Gene Ontology pathway (Figure 1A; eFigure 3 in Supplement 1 and eTable 3 in Supplement 2). Across all 130 samples, the most common genes with a rare disruptive variants were COL3A1, COL5A1, and COL4A1, which each had 3 carriers with SCAD. Other identified genes included COL6A1 (2 carriers) as well as COL5A2, COL12A1, COL4A5, and COL1A1 (1 carrier each) (Figure 1B and eTable 4 in Supplement 2). Clinical annotation of the 15 collagen variants demonstrated 2 pathogenic, 5 likely pathogenic, 7 variants of unknown significance, and 1 likely benign variant.32,33 Furthermore, subanalysis of the 7 variants of unknown significance revealed that 6 had features of pathogenicity.33 Because COL3A1 is the gene responsible for vascular Ehlers-Danlos syndrome, further enrichment analysis was performed excluding COL3A1, which showed consistent enrichment in this pathway (eFigure 4 in Supplement 1). Across all 130 individuals with SCAD, no significant (P < .05) differences were observed across baseline characteristics by collagen gene variant carrier status (eTable 1 in Supplement 1).
Figure 1. Enrichment of Disruptive Rare Variants Within Collagen Genes in Individuals With Spontaneous Coronary Artery Dissection (SCAD).
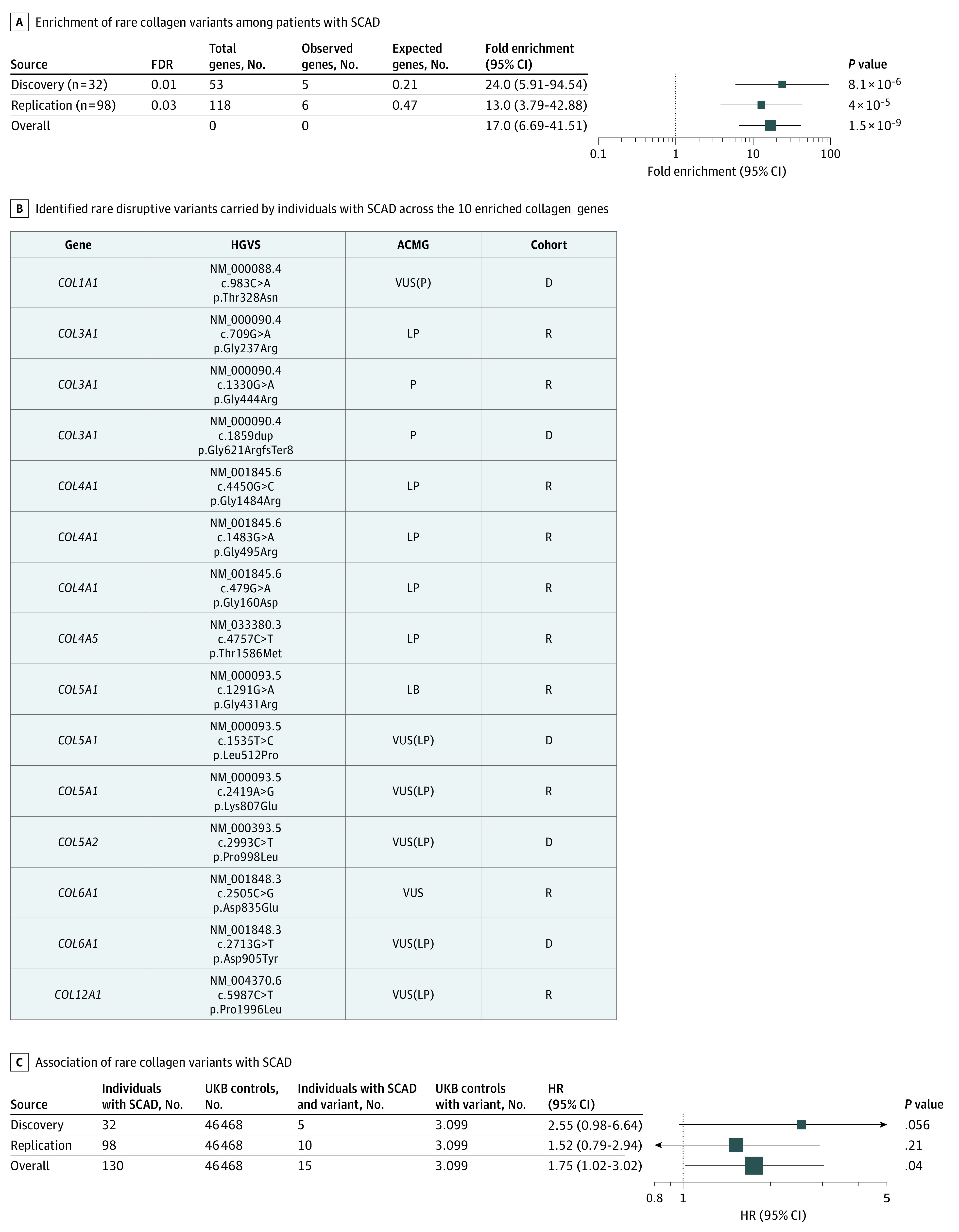
A, Enrichment of genes with rare disruptive variants identified the 10 genes within the extracellular matrix structural constituent conferring tensile strength pathway (GO:0030020). B, Identified rare disruptive variants carried by individuals with SCAD across the 10 enriched collagen genes annotated by Human Genome Variation Society (HGVS) nomenclature and American College of Medical Genetics (ACMG) pathogenicity designation. Variants of unknown significance (VUS) were further designated as either VUS(P) to indicate some pathologic evidence or VUS(LP) to indicate minor pathologic evidence. Discovery (D) or replication (R) cohorts are indicated. C, Association of rare disruptive variants across the 10 collagen genes in individuals with SCAD vs UK Biobank (UKBB) controls. FDR indicates false discovery rate; HR, hazard ratio, LB, likely benign.
Aggregate carrier status of disruptive variants in the top enriched pathway’s genes was associated with SCAD case status vs WES UK Biobank participants (eTable 2 in Supplement 1). Comparisons were made across (1) unadjusted, (2) sparsely adjusted models adjusted for age, race, and sex, and (3) fully adjusted models additionally adjusting for prevalent hypertension, hyperlipidemia, type 2 diabetes, and smoking status (eFigure 5 in Supplement 1). Across the discovery and replication SCAD cohorts together, rare disruptive variants in the 10 collagen genes were found to be 1.75-fold enriched among individuals with SCAD (15 of 130 [11.5%]) compared with all included UK Biobank individuals (3099 of 46 468 [6.7%]) (95% CI, 1.02-3.02; P = .04) (Figure 1C) in models adjusted for age, sex, and race, with similar effect estimates across all 3 adjustment models and across UK Biobank MI-free controls and individuals with MI (eFigure 5 in Supplement 1). Analyses were performed to compare the distributions of rare predicted disruptive variants among individuals with SCAD vs UK Biobank participants across all pLI more than 0.9 genes expressed in the coronary artery, suggesting that results at baseline are biased in the opposite direction (UK Biobank participants have higher mean counts of rare disruptive variants) owing to technical differences (eFigure 6 in Supplement 1).
These data implicate fibrillar collagen deficiency; thus, presumably, SCAD may also be seen in patients with disorders of collagen fibrillar assembly. This is consistent with observations in a patient evaluated at our center (not included in the sequencing cohort) who experienced a SCAD event in the left anterior descending artery at in her early 30s (eFigure 7 in Supplement 1) and was known to have a biallelic duplication of exons 10 to 16 of the PLOD1 gene encoding a hydroxylase necessary for cross-linking and stabilization of collagen fibrils.37
Focusing on the 2 most common collagen gene isoforms disrupted in patients with SCAD, we obtained mice deficient in Col3a1 and Col5a1 and generated Col3a1+/−, Col5a1+/−, and Col3a1+/−:Col5a1+/− genotypes to gain insights into the observed collagen perturbations in patients with SCAD. Targeted disruption of Col3a1 and Col5a1 had been previously described to result in 50% reduction in messenger RNA and protein expression in mice.34,38 Under observation, Col5a1+/− mice died spontaneously of aortic dissection, and postmortem examination showed hemothorax without any other gross pathology (Figure 2A and B). Col3a1+/−:Col5a1+/− mice had a more severe dissection phenotype than either Col3a1+/− or Col5a1+/− alone (log-rank P < .005). However, no Col3a1+/− mouse died from aortic dissection or rupture, a common occurrence in mice modeling glycine substitutions in Col3a1.39
Figure 2. Combinatorial Murine Models of Collagen Deficiency Demonstrate Vascular Phenotypes.

A, Survival curve demonstrates Col5a1+/− and Col3a1+/−:Col5a1+/− mice exhibit sudden death due to aortic rupture (log-rank P < .005). B, Postmortem Col5a1+/− and Col3a1+/−:Col5a1+/− mice demonstrate hemothorax indicative of aortic failure. Latex-injected murine aortas (C) and parasternal long-axis echocardiographic still images of genotypes (D; white arrowheads indicate aortic root). E, Aortic root dimensions by genotype.
Cardiac ultrasonography demonstrated enlargement of the aortic root in Col3a1+/−, but not in Col5a1+/−, mice although the magnitude of this difference was very small (mean [SD] at 2 months: wild type, 1.6 [0.01] mm vs Col3a1+/−, 1.7 [0.1] mm; P < .05) (Figure 2C-E). Although Col3a1+/− mice had larger aortic roots than wild-type, Col5a1+/−, or Col3a1+/−:Col5a1+/− mice, no Col3a1+/− mouse died from aortic dissection or rupture, clinically similar to coronary artery dissection where aneurysm does not typically precede dissection. Unlike most models of murine arterial disease, female Col3a1+/− (and Col3a1+/−:Col5a1+/−) mice exhibited worsened aortic dimensions than male mice, consistent with female enrichment in SCAD cases in humans (eFigure 8 in Supplement 1). Subgroup analysis also demonstrated this enlargement was exacerbated by pregnancy, a known risk factor for SCAD (eFigure 9 in Supplement 1).9
Collagen ultrastructure of Col3a1+/− and Col5a1+/− murine aortas by electron microscopy showed decreased adventitial collagen content, most severely in Col3a1+/−:Col5a1+/− animals (Figure 3A and B). Individual collagen fibrils were disorganized, with uneven fiber sizes and diameters (Figure 3C). We did not observe split or cauliflower forms but did observe increased fibril diameters among Col5a1+/− and Col3a1+/−:Col5a1+/− mice and decreased fibril diameter among Col3a1+/− mice when compared with wild-type mice, as described previously.34,40 Vascular smooth muscle cells between elastic fibers within collagen deficient aortas demonstrated morphologic changes with a more rounded appearance, sparser interaction with elastin fibers, increased intracellular vesicles, and nuclear reorganization (Figure 3 and eFigure 10 in Supplement 1). These data suggest that changes in expression levels between different forms of fibrillar collagens may have profound effects on vascular cell structure and function.
Figure 3. Vascular Tissue Ultrastructure in Col3a1+/− and Col5a1+/− Mice.

A, Transmission electron microscopic examination of mouse vascular tissue shows smooth muscle cell nuclear reorganization, sparse attachments to elastin, and widening between elastin layers. B, Enlarged view of boxed region in A. C, Collagen fibril diameters by mouse genotype indicates smaller fibers in Col3a1+/− and larger fibers in Col5a1+/− haploinsufficient mice.
aP < .001.
Discussion
In humans, genetic fibrillar collagen deficiency results in varied conditions including subtypes of Ehlers-Danlos syndrome,41 osteogenesis imperfecta,42 kidney disease,43 and forms of muscular dystrophy.44 Glycine substitutions in COL1A1 can cause arterial dissections without bone fragility.45,46 Variants affecting the N-terminal procollagen region of the COL1A1 protein have also been associated with arterial dissections,47 and Col1a1-deficient mice exhibit arterial dissection.48 Variants in COL5A1 and COL5A249 most frequently cause joint and skin abnormalities; however, arterial dissection has been reported.50,51,52 COL5A1 variants have also been associated with fibromuscular dysplasia and arterial dissections,53 and Col5a2-deficient mice have been shown to exhibit arterial dissection.54 We have now demonstrated spontaneous dissections in Col5a1 haploinsufficient mice (Figure 2). Arterial dissections have been reported in individuals with variants in COL4A5,55 including SCAD.56,57 Finally, COL3A1 has the most well-known association with arterial dissections and/or ruptures, with variable severity associated with variant subtype.58 Our group and others have reported COL3A1 variants discovered in patients with SCAD with syndromic findings of vascular Ehlers-Danlos syndrome,59,60 and the 3 patients identified in this study with COL3A1 variants demonstrate the range of clinical findings in vascular Ehlers-Danlos syndrome.
Recently, 2 genome-wide association studies of SCAD have identified 5 associated loci (near PHACTR1, LRP1, LINC00310, and FBN1) with opposing effects at corresponding atherosclerotic cardiovascular disease risk loci.61,62 These observations suggest that risk for atherogenic disease is balanced against a risk for arterial dissection for some aspects of mural wall biology. While the role of collagens in post-MI recovery is well documented,63 there is a lack of evidence that collagen isoforms play a role in primary atherothrombotic MI risk. Using a different filtering strategy, a previous study examining genome sequencing in patients with SCAD found enrichment of rare variation in the gene PKD1 and variant enrichment in genes impacting kidney function.64 In this study, we concentrated on rare variation imposing a risk for SCAD expressed in the coronary arteries and identified defects within genes encoding fibrillary collagens. In support of these findings, we also observed a patient with a homozygous deletion variant of the gene PLOD1, encoding a hydroxylase necessary for cross-linking of collagen fibrils (eFigure 7 in Supplement 1), consistent with a recent report of LOX variants in a SCAD cohort.60 LOX encodes lysyl oxidase, also a protein with collagen cross-linking activity. These results may inform clinical exome or genome sequencing in individuals and families with disorders of arterial dissections including SCAD.
Limitations
Our study has several limitations. First, exome sequencing may not identify rare variation found outside of coding regions or may have incomplete sequence depth. Second, technical differences between individuals with SCAD and external controls limits precision of association analyses. Since our sensitivity analyses indicate a higher number of observed rare predicted disruptive variants across the genome in the UK Biobank compared with individuals with SCAD (eFigure 6 in Supplement 1), the true effect estimate may truly be larger than what is reported here. Third, although we filtered for deleterious coding variation, we did not model the effect of the individually identified variants directly on collagen production or function. Fourth, because mice do not exhibit SCAD, animal modeling of coronary dissection is examined by proxy outcomes such as aortic dysfunction. Finally, the lack of ethnic diversity in out cohort precludes applicability of these findings across diverse populations.
Conclusions
In this study, we prospectively performed exome sequencing on a group of individuals with SCAD, filtering variants to high confidence putative disruptive alleles in coronary artery expressed genes and demonstrated enrichment within collagen genes. Compared with external controls, individuals with SCAD were enriched for variants in this pathway. Small animal modeling of partial collagen deficiency revealed cardiovascular phenotypes including mild aortic dilation and spontaneous arterial dissections, accentuated in female mice. Transmission electron microscopy analysis of collagen fibrils ultrastructure showed abnormalities of structure and fibril quality. Interestingly, vascular smooth muscle cells phenotypic derangement was profound even with these mild changes in collagen expression, likely representing abnormalities of extracellular matrix sensing imposing cellular behavioral changes. Taken together, these data indicate that deficits in the fibrillar collagen system impose arterial fragility and a susceptibility to SCAD.
eMethods.
eFigure 1. Variant filtration schematic
eFigure 2. STRING Protein-protein interactions from genes carrying rare disruptive variants in the SCAD Discovery cohort (32 cases)
eFigure 3. STRING protein-protein interactions from genes carrying rare disruptive variants in the SCAD replication cohort (99 cases)
eFigure 4. Enrichment of disruptive rare variants within collagen genes in SCAD cases, excluding COL3A1
eFigure 5. Association of rare disruptive variants within the 10 collagen genes in SCAD discovery and replication cases versus the UK Biobank
eFigure 6. Sensitivity analysis comparing the distribution of rare disruptive variants per person among constrained genes expressed in coronary artery in the SCAD WES cases versus UK Biobank WESed individuals
eFigure 7. Contrast angiogram demonstrating spontaneous coronary artery dissection in a woman in her early 30s with homozygous PLOD1 mutation
eFigure 8. Murine echocardiography in male versus female mice demonstrates increased aortic dimensions in female collagen mice
eFigure 9. Murine echocardiography in nulliparous versus multiparous mice demonstrates increased aortic dimensions in mice after pregnancy in all genotypes, accentuated in Col5a1-/+ mice
eFigure 10. Coronary and aortic histology sections of Col3a1+/- and Col5a1+/- haploinsufficient mice demonstrate deficiency of contractile Sm22a protein
eTable 1. Comparing SCAD patient characteristics across collagen gene carriers vs controls in both discovery and replication datasets combined
eTable 2. UK Biobank sample characteristics
eTable 1. Rare disruptive variants among genes expressed in Coronary Artery in the SCAD Discovery cohort (N=32)
eTable 2. Enrichment analysis among the 53 rare predicted disruptive genes identified in the Discovery SCAD cohort, compared to a list of 2507 genes with pLI>0.9 and expressed in coronary artery among GTEx
eTable 3. Rare disruptive variants among genes expressed in Coronary Artery in the SCAD Replication cohort (N=98)
eTable 4. Collagen variants with HGVS assignment and chromosomal position
References
- 1.Krittanawong C, Kumar A, Wang Z, et al. Clinical features and prognosis of patients with spontaneous coronary artery dissection. Int J Cardiol. 2020;312:33-36. doi: 10.1016/j.ijcard.2020.03.044 [DOI] [PubMed] [Google Scholar]
- 2.Luong C, Starovoytov A, Heydari M, Sedlak T, Aymong E, Saw J. Clinical presentation of patients with spontaneous coronary artery dissection. Catheter Cardiovasc Interv. 2017;89(7):1149-1154. doi: 10.1002/ccd.26977 [DOI] [PubMed] [Google Scholar]
- 3.Mortensen KH, Thuesen L, Kristensen IB, Christiansen EH. Spontaneous coronary artery dissection: a Western Denmark Heart Registry study. Catheter Cardiovasc Interv. 2009;74(5):710-717. doi: 10.1002/ccd.22115 [DOI] [PubMed] [Google Scholar]
- 4.Nishiguchi T, Tanaka A, Ozaki Y, et al. Prevalence of spontaneous coronary artery dissection in patients with acute coronary syndrome. Eur Heart J Acute Cardiovasc Care. 2016;5(3):263-270. doi: 10.1177/2048872613504310 [DOI] [PubMed] [Google Scholar]
- 5.Saw J, Humphries K, Aymong E, et al. Spontaneous coronary artery dissection: clinical outcomes and risk of recurrence. J Am Coll Cardiol. 2017;70(9):1148-1158. doi: 10.1016/j.jacc.2017.06.053 [DOI] [PubMed] [Google Scholar]
- 6.Vanzetto G, Berger-Coz E, Barone-Rochette G; et al. Prevalence, therapeutic management and medium-term prognosis of spontaneous coronary artery dissection: results from a database of 11,605 patients. Eur J Cardiothorac Surg. 2009;35(2):250-254. doi: 10.1016/j.ejcts.2008.10.023 [DOI] [PubMed] [Google Scholar]
- 7.Nakashima T, Noguchi T, Haruta S, et al. Prognostic impact of spontaneous coronary artery dissection in young female patients with acute myocardial infarction: a report from the Angina Pectoris-Myocardial Infarction Multicenter Investigators in Japan. Int J Cardiol. 2016;207:341-348. doi: 10.1016/j.ijcard.2016.01.188 [DOI] [PubMed] [Google Scholar]
- 8.Rashid HN, Wong DT, Wijesekera H, et al. Incidence and characterisation of spontaneous coronary artery dissection as a cause of acute coronary syndrome: a single-centre Australian experience. Int J Cardiol. 2016;202:336-338. doi: 10.1016/j.ijcard.2015.09.072 [DOI] [PubMed] [Google Scholar]
- 9.Hayes SN, Kim ESH, Saw J, et al. ; American Heart Association Council on Peripheral Vascular Disease; Council on Clinical Cardiology; Council on Cardiovascular and Stroke Nursing; Council on Genomic and Precision Medicine; and Stroke Council . Spontaneous coronary artery dissection: current state of the science: a scientific statement from the American Heart Association. Circulation. 2018;137(19):e523-e557. doi: 10.1161/CIR.0000000000000564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saw J, Aymong E, Sedlak T, et al. Spontaneous coronary artery dissection: association with predisposing arteriopathies and precipitating stressors and cardiovascular outcomes. Circ Cardiovasc Interv. 2014;7(5):645-655. doi: 10.1161/CIRCINTERVENTIONS.114.001760 [DOI] [PubMed] [Google Scholar]
- 11.Frank M, Albuisson J, Ranque B, et al. The type of variants at the COL3A1 gene associates with the phenotype and severity of vascular Ehlers-Danlos syndrome. Eur J Hum Genet. 2015;23(12):1657-1664. doi: 10.1038/ejhg.2015.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garcia-Bermudez M, Moustafa AH, Barros-Membrilla A, Tizon-Marcos H. Repeated loss of consciousness in a young woman: a suspicious SMAD3 mutation underlying spontaneous coronary artery dissection. Can J Cardiol. 2017;33(2):292.e1-292.e3. doi: 10.1016/j.cjca.2016.09.004 [DOI] [PubMed] [Google Scholar]
- 13.Henkin S, Negrotto SM, Tweet MS, et al. Spontaneous coronary artery dissection and its association with heritable connective tissue disorders. Heart. 2016;102(11):876-881. doi: 10.1136/heartjnl-2015-308645 [DOI] [PubMed] [Google Scholar]
- 14.Maas AHEM, Bouatia-Naji N, Persu A, Adlam D. Spontaneous coronary artery dissections and fibromuscular dysplasia: current insights on pathophysiology, sex and gender. Int J Cardiol. 2019;286:220-225. doi: 10.1016/j.ijcard.2018.11.023 [DOI] [PubMed] [Google Scholar]
- 15.Nakamura M, Yajima J, Oikawa Y, et al. Vascular Ehlers-Danlos syndrome: all three coronary artery spontaneous dissections. J Cardiol. 2009;53(3):458-462. doi: 10.1016/j.jjcc.2008.09.007 [DOI] [PubMed] [Google Scholar]
- 16.Pepin MG, Schwarze U, Rice KM, Liu M, Leistritz D, Byers PH. Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type IV). Genet Med. 2014;16(12):881-888. doi: 10.1038/gim.2014.72 [DOI] [PubMed] [Google Scholar]
- 17.Van der Auwera GA, Carneiro MO, Hartl C, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43(1110):11.10.1-11.10.33. doi: 10.1002/0471250953.bi1110s43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hail. Index page. Accessed January 25, 2022. https://hail.is/index.html
- 19.McLaren W, Gil L, Hunt SE, et al. The Ensembl variant effect predictor. Genome Biol. 2016;17(1):122. doi: 10.1186/s13059-016-0974-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu X, Li C, Mou C, Dong Y, Tu Y. dbNSFP v4: a comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med. 2020;12(1):103. doi: 10.1186/s13073-020-00803-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karczewski KJ, Francioli LC, Tiao G, et al. ; Genome Aggregation Database Consortium . The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434-443. doi: 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dewey FE, Murray MF, Overton JD, et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science. 2016;354(6319):354. doi: 10.1126/science.aaf6814 [DOI] [PubMed] [Google Scholar]
- 23.Lek M, Karczewski KJ, Minikel EV, et al. ; Exome Aggregation Consortium . Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285-291. doi: 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Battle A, Brown CD, Engelhardt BE, Montgomery SB; GTEx Consortium; Laboratory, Data Analysis &Coordinating Center (LDACC)—Analysis Working Group; Sta tistical Methods groups—Analysis Working Group; Enhancing GTEx (eGTEx) groups; NIH Common Fund; NIH/NCI; NIH/NHGRI; NIH/NIMH; NIH/NIDA; Biospecimen Collection Source Site—NDRI; Biospecimen Collection Source Site—RPCI; Biospecimen Core Resource—VARI; Brain Bank Repository—University of Miami Brain Endowment Bank; Leidos Biomedical—Project Management; ELSI Study; Genome Browser Data Integration & Visualization—EBI; Genome Browser Data Integration & Visualization—UCSC Genomics Institute, University of California Santa Cruz; Lead analysts; Laboratory, Data Analysis & Coordinating Center (LDACC); NIH program management; Biospecimen collection; Pathology; eQTL manuscript working group . Genetic effects on gene expression across human tissues. Nature. 2017;550(7675):204-213. doi: 10.1038/nature24277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dong C, Wei P, Jian X, et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet. 2015;24(8):2125-2137. doi: 10.1093/hmg/ddu733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.MacArthur DG, Balasubramanian S, Frankish A, et al. ; 1000 Genomes Project Consortium . A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335(6070):823-828. doi: 10.1126/science.1215040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bycroft C, Freeman C, Petkova D, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562(7726):203-209. doi: 10.1038/s41586-018-0579-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.UK Biobank. Accessed January 25, 2022. https://www.ukbiobank.ac.uk/
- 29.Van Hout CV, Tachmazidou I, Backman JD, et al. Whole exome sequencing and characterization of coding variation in 49,960 individuals in the UK Biobank. bioRxiv. Preprint posted March 9, 2019. doi: 10.1101/572347 [DOI]
- 30.Gene Ontology Resource. Accessed January 25, 2022. http://geneontology.org/
- 31.Varsome. Accessed January 25, 2022. https://varsome.com/
- 32.Richards S, Aziz N, Bale S, et al. ; ACMG Laboratory Quality Assurance Committee . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kopanos C, Tsiolkas V, Kouris A, et al. VarSome: the human genomic variant search engine. Bioinformatics. 2019;35(11):1978-1980. doi: 10.1093/bioinformatics/bty897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wenstrup RJ, Florer JB, Davidson JM, et al. Murine model of the Ehlers-Danlos syndrome: col5a1 haploinsufficiency disrupts collagen fibril assembly at multiple stages. J Biol Chem. 2006;281(18):12888-12895. doi: 10.1074/jbc.M511528200 [DOI] [PubMed] [Google Scholar]
- 35.Xiao L, Salem JE, Clauss S, et al. Ibrutinib-mediated atrial fibrillation attributable to inhibition of C-terminal Src kinase. Circulation. 2020;142(25):2443-2455. doi: 10.1161/CIRCULATIONAHA.120.049210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lino Cardenas CL, Kessinger CW, Chou EL, et al. HDAC9 complex inhibition improves smooth muscle-dependent stenotic vascular disease. JCI Insight. 2019;4(2):4. doi: 10.1172/jci.insight.124706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yeowell HN, Walker LC. Mutations in the lysyl hydroxylase 1 gene that result in enzyme deficiency and the clinical phenotype of Ehlers-Danlos syndrome type VI. Mol Genet Metab. 2000;71(1-2):212-224. doi: 10.1006/mgme.2000.3076 [DOI] [PubMed] [Google Scholar]
- 38.Smith LB, Hadoke PW, Dyer E, et al. Haploinsufficiency of the murine Col3a1 locus causes aortic dissection: a novel model of the vascular type of Ehlers-Danlos syndrome. Cardiovasc Res. 2011;90(1):182-190. doi: 10.1093/cvr/cvq356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bowen CJ, Calderón Giadrosic JF, Burger Z, et al. Targetable cellular signaling events mediate vascular pathology in vascular Ehlers-Danlos syndrome. J Clin Invest. 2020;130(2):686-698. doi: 10.1172/JCI130730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hausser I, Anton-Lamprecht I. Differential ultrastructural aberrations of collagen fibrils in Ehlers-Danlos syndrome types I-IV as a means of diagnostics and classification. Hum Genet. 1994;93(4):394-407. doi: 10.1007/BF00201664 [DOI] [PubMed] [Google Scholar]
- 41.Malfait F, Francomano C, Byers P, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175(1):8-26. doi: 10.1002/ajmg.c.31552 [DOI] [PubMed] [Google Scholar]
- 42.Byers PH, Wallis GA, Willing MC. Osteogenesis imperfecta: translation of mutation to phenotype. J Med Genet. 1991;28(7):433-442. doi: 10.1136/jmg.28.7.433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Plaisier E, Gribouval O, Alamowitch S, et al. COL4A1 mutations and hereditary angiopathy, nephropathy, aneurysms, and muscle cramps. N Engl J Med. 2007;357(26):2687-2695. doi: 10.1056/NEJMoa071906 [DOI] [PubMed] [Google Scholar]
- 44.Jobsis GJ, Bolhuis PA, Boers JM, et al. Genetic localization of Bethlem myopathy. Neurology. 1996;46(3):779-782. doi: 10.1212/WNL.46.3.779 [DOI] [PubMed] [Google Scholar]
- 45.Mayer SA, Rubin BS, Starman BJ, Byers PH. Spontaneous multivessel cervical artery dissection in a patient with a substitution of alanine for glycine (G13A) in the alpha 1 (I) chain of type I collagen. Neurology. 1996;47(2):552-556. doi: 10.1212/WNL.47.2.552 [DOI] [PubMed] [Google Scholar]
- 46.Adham S, Dupuis-Girod S, Charpentier E, Mazzella JM, Jeunemaitre X, Legrand A. Classical Ehlers-Danlos syndrome with a propensity to arterial events: a new report on a French family with a COL1A1 p.(Arg312Cys) variant. Clin Genet. 2020;97(2):357-361. doi: 10.1111/cge.13643 [DOI] [PubMed] [Google Scholar]
- 47.Malfait F, Symoens S, De Backer J, et al. Three arginine to cysteine substitutions in the pro-alpha (I)-collagen chain cause Ehlers-Danlos syndrome with a propensity to arterial rupture in early adulthood. Hum Mutat. 2007;28(4):387-395. doi: 10.1002/humu.20455 [DOI] [PubMed] [Google Scholar]
- 48.Rahkonen O, Su M, Hakovirta H, et al. Mice with a deletion in the first intron of the Col1a1 gene develop age-dependent aortic dissection and rupture. Circ Res. 2004;94(1):83-90. doi: 10.1161/01.RES.0000108263.74520.15 [DOI] [PubMed] [Google Scholar]
- 49.Symoens S, Syx D, Malfait F, et al. Comprehensive molecular analysis demonstrates type V collagen mutations in over 90% of patients with classic EDS and allows to refine diagnostic criteria. Hum Mutat. 2012;33(10):1485-1493. doi: 10.1002/humu.22137 [DOI] [PubMed] [Google Scholar]
- 50.Mehta S, Dhar SU, Birnbaum Y. Common iliac artery aneurysm and spontaneous dissection with contralateral iatrogenic common iliac artery dissection in classic Ehlers-Danlos syndrome. Int J Angiol. 2012;21(3):167-170. doi: 10.1055/s-0032-1325118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Borck G, Beighton P, Wilhelm C, Kohlhase J, Kubisch C. Arterial rupture in classic Ehlers-Danlos syndrome with COL5A1 mutation. Am J Med Genet A. 2010;152A(8):2090-2093. doi: 10.1002/ajmg.a.33541 [DOI] [PubMed] [Google Scholar]
- 52.Monroe GR, Harakalova M, van der Crabben SN, et al. Familial Ehlers-Danlos syndrome with lethal arterial events caused by a mutation in COL5A1. Am J Med Genet A. 2015;167(6):1196-1203. doi: 10.1002/ajmg.a.36997 [DOI] [PubMed] [Google Scholar]
- 53.Richer J, Hill HL, Wang Y, et al. A novel recurrent COL5A1 genetic variant is associated with a dysplasia-associated arterial disease exhibiting dissections and fibromuscular dysplasia. Arterioscler Thromb Vasc Biol. 2020;40(11):2686-2699. doi: 10.1161/ATVBAHA.119.313885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Park AC, Phan N, Massoudi D, et al. Deficits in Col5a2 expression result in novel skin and adipose abnormalities and predisposition to aortic aneurysms and dissections. Am J Pathol. 2017;187(10):2300-2311. doi: 10.1016/j.ajpath.2017.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kashtan CE, Segal Y, Flinter F, Makanjuola D, Gan JS, Watnick T. Aortic abnormalities in males with Alport syndrome. Nephrol Dial Transplant. 2010;25(11):3554-3560. doi: 10.1093/ndt/gfq271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Díez-del Hoyo F, Sanz-Ruiz R, Díez-Villanueva P, et al. A novel cardiovascular presentation of Alport syndrome: spontaneous coronary artery dissection. Int J Cardiol. 2014;177(3):e133-e134. doi: 10.1016/j.ijcard.2014.09.065 [DOI] [PubMed] [Google Scholar]
- 57.Anuwatworn A, Sethi P, Steffen K, Jonsson O, Petrasko M. Spontaneous coronary artery dissection: a rare manifestation of Alport syndrome. Case Rep Cardiol. 2017;2017:1705927. doi: 10.1155/2017/1705927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Byers PH, Belmont J, Black J, et al. Diagnosis, natural history, and management in vascular Ehlers-Danlos syndrome. Am J Med Genet C Semin Med Genet. 2017;175(1):40-47. doi: 10.1002/ajmg.c.31553 [DOI] [PubMed] [Google Scholar]
- 59.Kaadan MI, MacDonald C, Ponzini F, et al. Prospective cardiovascular genetics evaluation in spontaneous coronary artery dissection. Circ Genom Precis Med. 2018;11(4):e001933. doi: 10.1161/CIRCGENETICS.117.001933 [DOI] [PubMed] [Google Scholar]
- 60.Verstraeten A, Perik MHAM, Baranowska AA, et al. ; European/International Fibromuscular Dysplasia Registry and Initiative (FEIRI); Collaborators of the European/International Fibromuscular Dysplasia Registry and Initiative (FEIRI) . Enrichment of rare variants in Loeys-Dietz Syndrome genes in spontaneous coronary artery dissection but not in severe fibromuscular dysplasia. Circulation. 2020;142(10):1021-1024. doi: 10.1161/CIRCULATIONAHA.120.045946 [DOI] [PubMed] [Google Scholar]
- 61.Turley TN, O’Byrne MM, Kosel ML, et al. Identification of susceptibility loci for spontaneous coronary artery dissection. JAMA Cardiol. 2020;5(8):929-938. doi: 10.1001/jamacardio.2020.0872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saw J, Yang ML, Trinder M, et al. ; Million Veteran Program . Chromosome 1q21.2 and additional loci influence risk of spontaneous coronary artery dissection and myocardial infarction. Nat Commun. 2020;11(1):4432. doi: 10.1038/s41467-020-17558-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zamilpa R, Lindsey ML. Extracellular matrix turnover and signaling during cardiac remodeling following MI: causes and consequences. J Mol Cell Cardiol. 2010;48(3):558-563. doi: 10.1016/j.yjmcc.2009.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carss KJ, Baranowska AA, Armisen J, et al. Spontaneous coronary artery dissection: insights on rare genetic variation from genome sequencing. Circ Genom Precis Med. 2020;13(6):e003030. doi: 10.1161/CIRCGEN.120.003030 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eMethods.
eFigure 1. Variant filtration schematic
eFigure 2. STRING Protein-protein interactions from genes carrying rare disruptive variants in the SCAD Discovery cohort (32 cases)
eFigure 3. STRING protein-protein interactions from genes carrying rare disruptive variants in the SCAD replication cohort (99 cases)
eFigure 4. Enrichment of disruptive rare variants within collagen genes in SCAD cases, excluding COL3A1
eFigure 5. Association of rare disruptive variants within the 10 collagen genes in SCAD discovery and replication cases versus the UK Biobank
eFigure 6. Sensitivity analysis comparing the distribution of rare disruptive variants per person among constrained genes expressed in coronary artery in the SCAD WES cases versus UK Biobank WESed individuals
eFigure 7. Contrast angiogram demonstrating spontaneous coronary artery dissection in a woman in her early 30s with homozygous PLOD1 mutation
eFigure 8. Murine echocardiography in male versus female mice demonstrates increased aortic dimensions in female collagen mice
eFigure 9. Murine echocardiography in nulliparous versus multiparous mice demonstrates increased aortic dimensions in mice after pregnancy in all genotypes, accentuated in Col5a1-/+ mice
eFigure 10. Coronary and aortic histology sections of Col3a1+/- and Col5a1+/- haploinsufficient mice demonstrate deficiency of contractile Sm22a protein
eTable 1. Comparing SCAD patient characteristics across collagen gene carriers vs controls in both discovery and replication datasets combined
eTable 2. UK Biobank sample characteristics
eTable 1. Rare disruptive variants among genes expressed in Coronary Artery in the SCAD Discovery cohort (N=32)
eTable 2. Enrichment analysis among the 53 rare predicted disruptive genes identified in the Discovery SCAD cohort, compared to a list of 2507 genes with pLI>0.9 and expressed in coronary artery among GTEx
eTable 3. Rare disruptive variants among genes expressed in Coronary Artery in the SCAD Replication cohort (N=98)
eTable 4. Collagen variants with HGVS assignment and chromosomal position