

Abstract
Triple negative breast cancer (TNBC) typically exhibits rapid progression, high mortality and faster relapse rates relative to other breast cancer subtypes. In this report we examine the combination of taxanes (paclitaxel or docetaxel) with a breast cancer stem cell (CSC)-targeting agent sulforaphane for use against TNBC. We demonstrate that paclitaxel or docetaxel treatment induces IL-6 secretion and results in expansion of CSCs in TNBC cell lines. Conversely, sulforaphane is capable of preferentially eliminating CSCs, by inhibiting NF-κB p65 subunit translocation, downregulating p52 and consequent downstream transcriptional activity. Sulforaphane also reverses taxane-induced aldehyde dehydrogenase-positive (ALDH+) cell enrichment, and dramatically reduces the size and number of primary and secondary mammospheres formed. In vivo in an advanced treatment orthotopic mouse xenograft model together with extreme limiting dilution analysis (ELDA), the combination of docetaxel and sulforaphane exhibits a greater reduction in primary tumor volume and significantly reduces secondary tumor formation relative to either treatment alone. These results suggest that treatment of TNBCs with cytotoxic chemotherapy would be greatly benefited by the addition of sulforaphane to prevent expansion of and eliminate breast CSCs.
Keywords: Sulforaphane, Docetaxel, Paclitaxel, Triple negative breast cancer, Cancer stem cell
Introduction
Breast cancer is the most commonly diagnosed cancer in women in the United States, accounting for 29% of all new cases each year [1]. Breast cancer is a heterogeneous disease with high degree of diversity between and within tumors and among individual patients [2]. The heterogeneity of tumors leads to different incidence, disease progression, and various response to treatment [2]. Several different subtypes of breast cancers have been identified, among which the triple negative breast cancer (TNBC) is the most devastating disease with high morbidity and mortality rates [3]. TNBC is defined by the absence of three most commonly targeted receptors: estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor-2/neu receptors (HER2/neu) [4,5]. TNBC constitutes about 10–20% of newly diagnosed breast cancers and affect younger women more frequently [6]. At the time of diagnosis, patients with TNBC are generally present with larger tumor size and lymph node involvement, and the disease progression is biologically more aggressive [7]. Currently, treatment of patients with TNBC has been challenging due to the lack of receptors for targeting.
The use of neoadjuvant chemotherapy has increased in recent years with several clinical trials demonstrating more frequent pathologic complete response (pCR) in TNBC patients relative to other subtypes [8–11]. Therapies in the neoadjuvant setting typically consist of a taxane, paclitaxel or docetaxel, with other chemotherapeutic agents such as anthracyclines or cyclophosphamide [12]. However, with respect to patients who do not exhibit pCR overall survival is often worse in TNBC patients relative to non-TNBC patients due in part to higher rates of metastasis [9]. Strikingly, in advanced TNBC the duration of response to first line palliative chemotherapy has been reported to be less than 12 weeks and response to secondary and tertiary therapies even shorter (9 and 4 weeks respectively) [13].
Considerable effort has been given to identify new therapeutic targets or prognostic markers to improve the treatment efficacy against TNBC. Recently, the cancer stem cell (CSC) model has provided an attractive explanation for cancer relapse after primary chemotherapy [14–16]. The key properties of CSCs are self-renewal and differentiation potential, which allow for long term repopulation of tumors at the primary and metastatic sites [17,18]. This small population of CSCs have been previously reported to employ various strategies to resist drug treatment and escape cell death [19]. Breast cancer patients following neoadjuvant chemotherapy have exhibited an increase in phenotypic CSCs after therapy [20], which suggests the important role of CSCs in drug resistance, cancer metastasis and relapse. Thus, aiming to eliminate the CSCs is essential to improve the treatment outcomes against TNBC.
Sulforaphane, an isothiocyanate mainly derived from cruciferous vegetables, has been studied extensively as a cancer prevention agent [21,22]. Recently, sulforaphane has been shown to eliminate CSCs in a few cancer types [21,23,24]. In contrast, conventional cytotoxic chemotherapeutic agents may only inhibit the growth of differentiated cancer cells but fail to eliminate or may even expand CSC populations [25,26]. Taxanes, represented by paclitaxel and docetaxel, are a family of chemotherapeutic drugs widely used for breast cancer treatment [27]. In the clinical practice, the taxanes are standard therapy in both early-stage and metastatic breast cancer [27]. Similar to other chemotherapeuctic agents, the taxanes may only target differentiated cancer cell population and exert no effect against breast CSCs. Paclitaxel has been reported to induce IL-8 in human ovarian cancer cells [28] and docetaxel has been demonstrated to increase production of pro-inflammatory cytokines IL-6 and IL8 in patients [29]. In vitro studies have suggested that cytokine simulation is implicated in CSC survival [30–32]. Production of these cytokines is regulated by the activity of the transcription factor NF-κB, which has been demonstrated to be of critical importance in the regulation of CSC [33–36]. Therefore, we hypothesize that combination of taxanes (paclitaxel or docetaxel) with a breast CSC-targeting agent capable of NF-κB inhibition would be ideally suited therapy in TNBC patients.
In this report we demonstrate that taxane treatment increases the proportion of breast CSCs and enhances inflammatory cytokine production in TNBC cell lines. Conversely, the natural product sulforaphane reduces production of IL-6, IL-8, and NF-κB activity to inhibit CSCs. Combination of sulforaphane with paclitaxel or docetaxel can not only enhance the effect against bulk TNBC cells but sulforaphane suppresses docetaxel/paclitaxel mediated cytokine production and breast CSC expansion in vitro. Finally using an orthotopic mouse xenograft model with extreme limiting dilution analysis (ELDA) of serially reimplanted tumors we demonstrate that docetaxel increases breast CSC while the combination of sulforaphane and docetaxel demonstrates greater efficacy in primary tumors and reduces CSC frequency. These results provide a strong rationale for future studies aimed to utilize novel combination therapies for the treatment of TNBC patients.
Materials and methods
Cell lines and reagents
Human breast cancer cell lines SUM149 and SUM159 were gifts from Dr. Stephen Ethier (Karmanos Cancer Center, Detroit, Michigan) to the University of Michigan. The source of SUM159 cell line is primary breast anaplastic carcinoma. The SUM149 cell line is derived from a patient with primary inflammatory breast cancer (IBC). Both cell lines are estrogen receptor (ER) negative, progesterone receptor (PR) negative, and does not have Her2 overexpression. Both cell lines were tested and authenticated in their origin sources (http://www.asterand.com/asterand/BIOREPOSITORY/hbreastcancercelllines.aspx). Authentication of these cell lines included morphology analysis, growth curve analysis, isoenzyme analysis, short tandem repeat analysis, and mycoplasma detection. Both cell lines were passaged in our laboratory for fewer than 6 months after receipt. To maintain the integrity of collections, stocks of the earliest passage cells have been stored and cell lines have been carefully maintained in culture as described below. All cells were cultured at 37 °C under a 5% CO2 atmosphere. SUM149 and SUM159 cells were maintained in Ham’s F12 medium (Corning, NY) supplemented with 5% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA), 1% antibiotic-antimycotic (Thermo Fisher Scientific), and 5 μg/mL insulin (Thermo Fisher Scientific), 1 μg/ml hydrocortisone (Sigma–Aldrich, St Louis, MO), and 4 μg/mL gentamicin (Thermo Fisher Scientific). Sulforaphane, paclitaxel and docetaxel were obtained from LKT laboratories (St. Paul, MN).
MTS cell proliferation assay
SUM149 and SUM159 cells were plated in 96-well plates at a density of 3000–5000 cells per well. Cells were treated with various concentrations of sulforaphane, paclitaxel, docetaxel or a combination of sulforaphane with paclitaxel or docetaxel for 72 h. Cell viability was assessed by MTS assay (Promega, Madison, WI) according to the manufacturer’s instruction. The number of living cells in the sample is directly proportional to the quantity of a soluble formazan product estimated by its absorbance at 490 nm. The IC50s of cytotoxicity were calculated with WinNonlin software (Certara USA, Inc., Princeton, NJ).
Determination of secreted cytokines
Cells were seated at a density of 3000 cells per well in a 96-well plate and allowed to adhere overnight. Treatment with paclitaxel or docetaxel was performed in the presence or absence of sulforaphane for 8 h, afterward cells were washed with PBS and resuspended in cell culture media with and without sulforaphane where appropriate. After 72 h media was collected, centrifuged, and 200 μl was subjected to an ELISA to determine secretion of human IL-6 and IL-8. Assays were performed using paired antibody kits (Duosets, R&D Systems, Minneapolis, MN) according to manufacturer’s protocol with two exceptions: Samples were incubated overnight at 4 °C and the blocking/assay buffer was 0.2% casein in tris-buffered saline. Data was acquired with a BioTek Synergy HT plate reader and analyzed using Gen5 software (Winooski, VT).
Flow cytometry analysis
The Aldefluor assay (StemCell Technogies, Inc., Vancouver, BC, Canada) was used to identify the aldehyde dehydrogenase-positive (ALDH+) cell population. The assay was carried out according to manufacturer’s instructions. Following drug treatment adherent cells were collected, counted for determination of absolute cell numbers. The isolated cells were incubated with ALDH substrate for 45 min in a 37 °C water bath. For each replicate within a given treatment group, a fraction of cells from each sample was incubated under identical condition in the presence of the ALDH inhibitor diethylaminobenzaldehyde (DEAB) to serve as negative control. After staining, cells were washed with 2% FBS in HBSS followed by addition of DAPI immediately before analysis to determine viability. Flow cytometry was performed to measure the ALDH + population. For determination of CD44+/CD24−/EpCAM + cell population, after drug treatment, cells were stained with CD44-APC (BD Biosciences), CD24-PE (BD Biosciences), and EpCAM-PE-CY7 (Biolegend) in HBSS with 2% FBS on ice for 45 min before flow cytometry analysis.
Mammosphere formation assay
Mammosphere culture was done as previously described [23]. Cells were plated at a density of 2000 cells per well in an ultra-low attachment 6 well plate (Corning) in a serum-free medium. The serum-free medium consisted of MEBM base medium (Lonza, Inc.) supplemented with 2% B27 (Thermo Fisher Scientific), 1% antibiotic-antimycotic, 4 ug/ml gentamicin (Thermo Fisher Scientific), 5 ug/ml insulin, 20 ng/ml EGF (Sigma-aldrich), 20 ng/ml bFGF (Sigma-aldrich), 1 ug/ml hydrocortisone (Sigma-aldrich), and 1:25,000,000 β-ME (Sigma-aldrich). During primary culture, cells were treated with sulforaphane, paclitaxel, or the combination for seven days. Then formed spheres were counted manually and representative images were acquired using Olympus LSM 1000 confocal microscope with FluoView 10 software (Olympus Corp., Central Valley, PA, USA). The primary mammospheres were then collected using a nylon 40 μm mesh filter, followed by dissociation with trypsin in conjunction with passage through a 25 gauge needle. A fixed number of dissociated cells were then cultured for an additional seven days in the absence of drug. Secondary mammospheres were then counted as before.
Immunocytochemistry
Cells were seated at a density of 3000 cells per well in black 96-well plates with optically clear bottoms and allowed to adhere overnight. Sulforaphane was incubated at the indicated concentration for 30 min, followed by the addition of 50 ng/ml TNF-α for 2 h. Treated cells were fixed using ice cold 1:1 methanol to acetone followed by blocking with 3% bovine serum albumin. Primary incubation with anti-p65 NF-κB (Cell Signaling Technology) was carried out at 4 °C overnight, followed by incubation with Alexa Fluor 488 conjugated secondary antibody (Invitrogen) for two hours at room temperature. Fluorescent imaging was carried out with an Olympus IX83 microscope and photos were acquired using cellSens software.
Luciferase reporter assay
Lentiviral particles containing luciferase reporter construct driven by NF-κB (system biosciences) were obtained and transfected into SUM159 cell line. Briefly, 4 copies of NF-κB TRE sequences “GGGACTTTCC” were inserted upstream of minimal essential CMV (mCMV) promoter which drives GFP-T2A-luciferase. Polyprotein is cleaved at the T2A site to give rise to GFP and Luciferase. Cell lines were plated at a density of 3000 cells per well in a clear bottom, white 96-well plate and treated with the indicated concentration with sulforaphane. Two hours following incubation with sulforaphane, TNF-α concentration in media was brought to 50 ng/ml. After 6 h Luciferase activity was measured according to manufacturer’s instructions using oneGlo assay (Promega) on Bio-Tek Synergy 2 plate reader.
Western blotting analysis
The cells were treated with sulforaphane, paclitaxel, docetaxel or the combination. Cells were then harvested, lysed in radioimmunoprecipitation assay (RIPA) buffer supplemented with Pierce™ protease inhibitors and phosphatase inhibitors (Thermo Fisher Scientific). Cell lysate was centrifuged at 14,000 rpm for 15 min and the supernatant was recovered. Protein concentration was determined with Pierce™ BCA Protein Assay Reagents (Thermo Fisher Scientific). Equal amounts of total protein were subjected to SDS-PAGE, transferred onto polyvinylidene difluoride (PVDF) membrane, and probed with appropriate antibodies. The first antibodies to cyclin D1, β-actin, NF-κB protein p105, p50, p100 and p52 were purchased from Cell Signaling Technology. The membranes were developed with an enhanced chemiluminescence (ECL) reagent kit (Thermo Fisher Scientific) and the pictures were taken with a UVP ChemiDoc-It™ Imager. The Western blots were quantitated using Image J program (http://rsb.info.nih.gov/ij/index.html).
NF-κB subunit knockdown
Pooled small interfering RNAs for RELB (cat: L-004767-00-0005) and NFKB2 (cat: L-003918-00-0005) were obtained from GE Healthcare. SUM159 cells were seeded at a density of 500,000 cells in 10 cm cell culture dishes and allowed to adhere overnight. Transfection of 50 nM targeted siRNA and negative siRNA control (Qiagen 1027281) was performed using Lipofectamine® RNAiMAX according to manufacturer’s instructions. Eight hours following incubation plates were washed to remove excess transfection reagent. Then 72 h after initial incubation cells were harvested for flow cytometry analysis. Residual cells were stained for aldehyde dehydrogenase activity using the Aldefluor assay (Stemcell technologies) with DAPI viability dye according to manufacturer’s instruction. DEAB controls were used to gate 0.1% background for each sample tested and analyzed using a MoFlo® Astrios (Beckman Coulter).
Advanced tumor model
All studies involving mice were conducted in accordance with a standard animal protocol approved by the University Committee on the Use and Care of Animals at the University of Michigan. Female 5 week old non-obese diabetic/severe combined immunodeficient (NOD/SCID) mice were obtained from Jackson Laboratory. Xenograft formation was generated by direct injection of 1.5 million SUM149 cells, suspended in 50 ul 25% F-12 in matrigel, into the exposed no.4 inguinal mammary pad. Tumor detection was assessed by palpation and once identified measurement of tumor volume was carried out using digital calipers every 5 days. Sulforaphane (50 mg/kg daily), docetaxel (10 mg/kg once every 7 days), or the combination of both were administered via I.P. injection beginning when tumor volume reached approximately 50 mm3. When the combined sulforaphane and docetaxel treatment began to cause significant tumor regression mice were euthanized by CO2 inhalation, tumors were isolated, and extreme limiting dilution analysis in secondary mice was performed.
Extreme limiting dilution analysis
Isolated primary tumors were mechanically dissociated using a gentleMACS octo tissue dissociator with C tubes (Miltenyi Biotec). In order to consistently obtain single cell suspensions a human tumor dissociation kit (Miltenyi Biotec) was used according to manufacturer’s instruction for “tough tumors”. Briefly, tumors were cut into 2–4 mm pieces and exposed to dissociation enzymes. Following incubation at 37 °C for 30 min on an orbital shaker mechanical dissociation was carried out with the genteMACS and this process was repeated 3 times. Dissociated cells were collected after passage through a 40 μm nylon mesh filter. Live human tumor cells from xenografts were obtained by FACS on a SY3200 (Sony Biotechnology) flow cytometer after selection of DAPI- and H2KD-cells. Secondary female, 5-week old, NOD/SCID mice were inoculated with 10,000, 1,000, or 100 cells from each treatment group as described above. Tumor formation rate in secondary mice was assessed 7 weeks following implanting cells by direct palpitation and used to assess BCSC frequency using the ELDA webtool (http://bioinf.wehi.edu.au/software/elda/) [37].
Statistical analysis
Statistical differences were determined using two-tailed Student’s t test. Data are presented as mean ± SD (n ≥ 3).
Results
Paclitaxel and docetaxel increase IL-6 and IL-8 secretion while suforaphane reduces their secretion
Recent analysis of inflammatory-related genes expressed in TNBCs has identified two highly expressed cytokines, IL-6 and IL-8, both of which are critical for TNBCs [38,39]. The coordinate expression of IL-6 and IL-8 are important for the growth, tumorigenicity, and resistance to apoptosis of TNBCs [38]. In clinical practice, higher expression of IL-6 and IL-8 is shown to be associated with poorer prognosis and decreased survival [29,38]. It has also been reported that IL-6 [31,32,40] and IL-8 [30,41] expression is implicated in expanding the breast CSC population and their resistance to chemotherapy in breast cancer.
Previous clinical trials have demonstrated that administration of taxanes (paclitaxel and docetaxel) to patients increases IL-6 in circulation [29]. Therefore, we first examined whether paclitaxel or docetaxel treatment could induce the secretion of IL-6 and IL-8 in TNBCs. SUM149 and SUM159 cells were treated with increasing concentrations of paclitaxel or docetaxel. As shown in Fig. 1A–D, both paclitaxel and docetaxel induce the expression of IL-6 in SUM149 and SUM159 cells relative to vehicle treatment control (e.g. first v.s. fourth column in A&B). Interestingly, the IL-8 secretion was only modestly increased upon docetaxel or paclitaxel treatment (Supplementary Fig. S1 A&B). These results suggest that the elevated release of IL-6 may be the primary response of the TNBC cell lines to taxane treatment. We next examined the effect of sulforaphane, the CSC-targeting agent, on the secretion of IL-6 and IL-8. The results show that sulforaphane (2.5 μM and 5 μM) reduces the secretion of both IL-6 and IL-8 by 40–90% in SUM149 and SUM159 cells in a concentration dependent manner (Fig. 1A–D, Supplementary Fig. S1 A&B). Further, the addition of 5 μM sulforaphane to either paclitaxel or docetaxel treatment can reduce the secretion of IL-6 and IL-8 to below control in the majority of combination treatments (Fig. 1A–D).
Fig. 1.
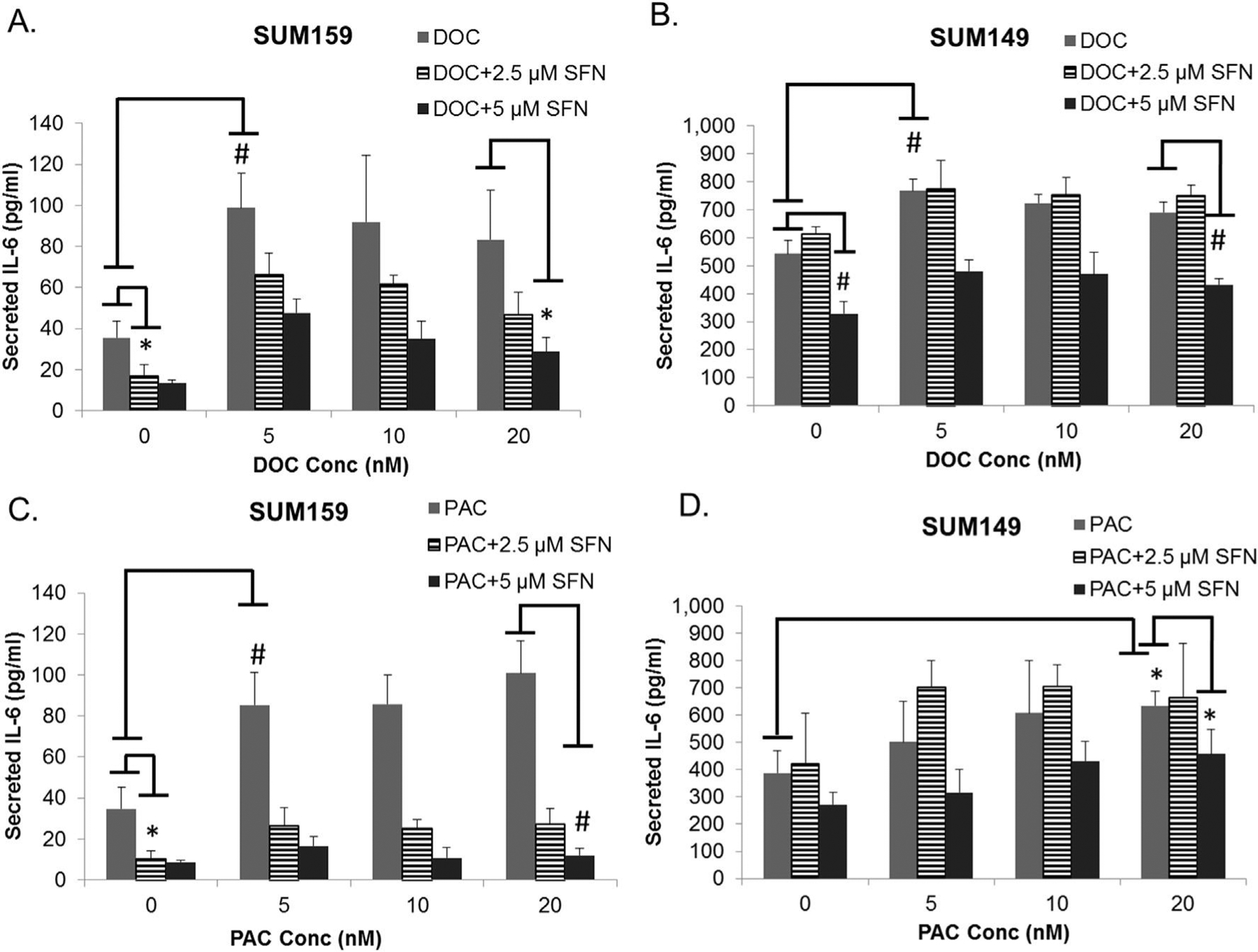
Docetaxel and paclitaxel increase cytokine expression, while sulforaphane decreases expression. A-D, concentration of secreted IL-6 protein in media from culture of SUM149 or SUM159 cells after being exposed to paclitaxel, docetaxel or the combination with suforaphane determined by enzyme-linked immunosorbent assay. N = 3. Columns represent mean ± standard deviation (SD). Representative statistics shown for significant changes in IL-6 expression relative to vehicle treated control for each drug, as well as, combination reduction relative to taxane alone. *p ≤ 0.05 and #p ≤ 0.01.
Sulforaphane reduces NF-κB nuclear translocation and transcriptional activity
The expression of pro-inflammatory and inflammatory cytokines has been suggested to be mediated via the NF-κB signaling cascade in TNBCs [38,42]. Canonical activation of the NF-κB transcription factor is accomplished by a signal which results in translocation of the NF-κB subunits from the cytoplasm to the nucleus after dissociating from its endogenous inhibitor IκB [43]. In order to elucidate if sulforaphane is able to prevent this translocation, SUM159 and SUM149 cells were incubated with increasing concentrations of sulforaphane, followed by stimulation with TNF-α. In the absence of sulforaphane, TNF-α causes NF-κB p65 subunit to translocate into the nucleus of SUM159 and SUM149 cells (Fig. 2A). The addition of sulforaphane results in reduced nuclear NF-κB p-65 staining after TNF-α addition (Fig. 2A). In order to determine if blockade of translocation by sulforaphane translates to inhibition of transcriptional activity, cells were stably transfected with a reporter construct which produces luciferase mediated by a NF-κB response element in TNBC cell line SUM159. Following stimulation of transcription by TNF-α, in the presence of increasing concentrations of sulforaphane, luciferase activity was determined (Fig. 2B). Stimulation with TNF-α results in a 4.0 fold increase in luciferase activity for SUM159, which is reduced in a dose dependent manner by sulforaphane (Fig. 2B). In the case of 10 and 15 μM sulforaphane, the increase of NF-κB activity by TNF-α is completely repressed and luciferase activity is reduced to unstimulated levels in SUM 159 (10 μM p = 0.011, 15 μM p = 0.0002).
Fig. 2.
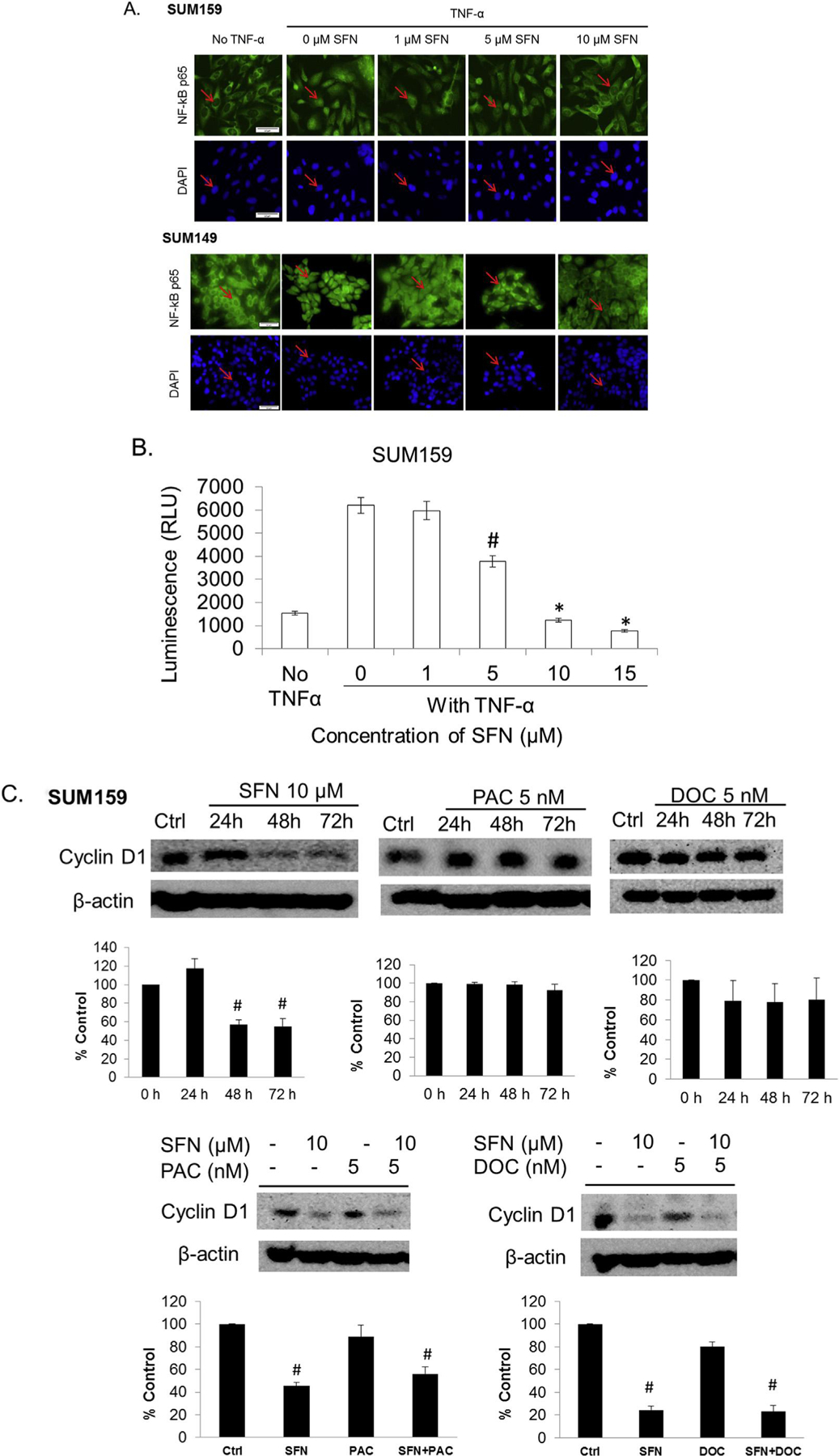
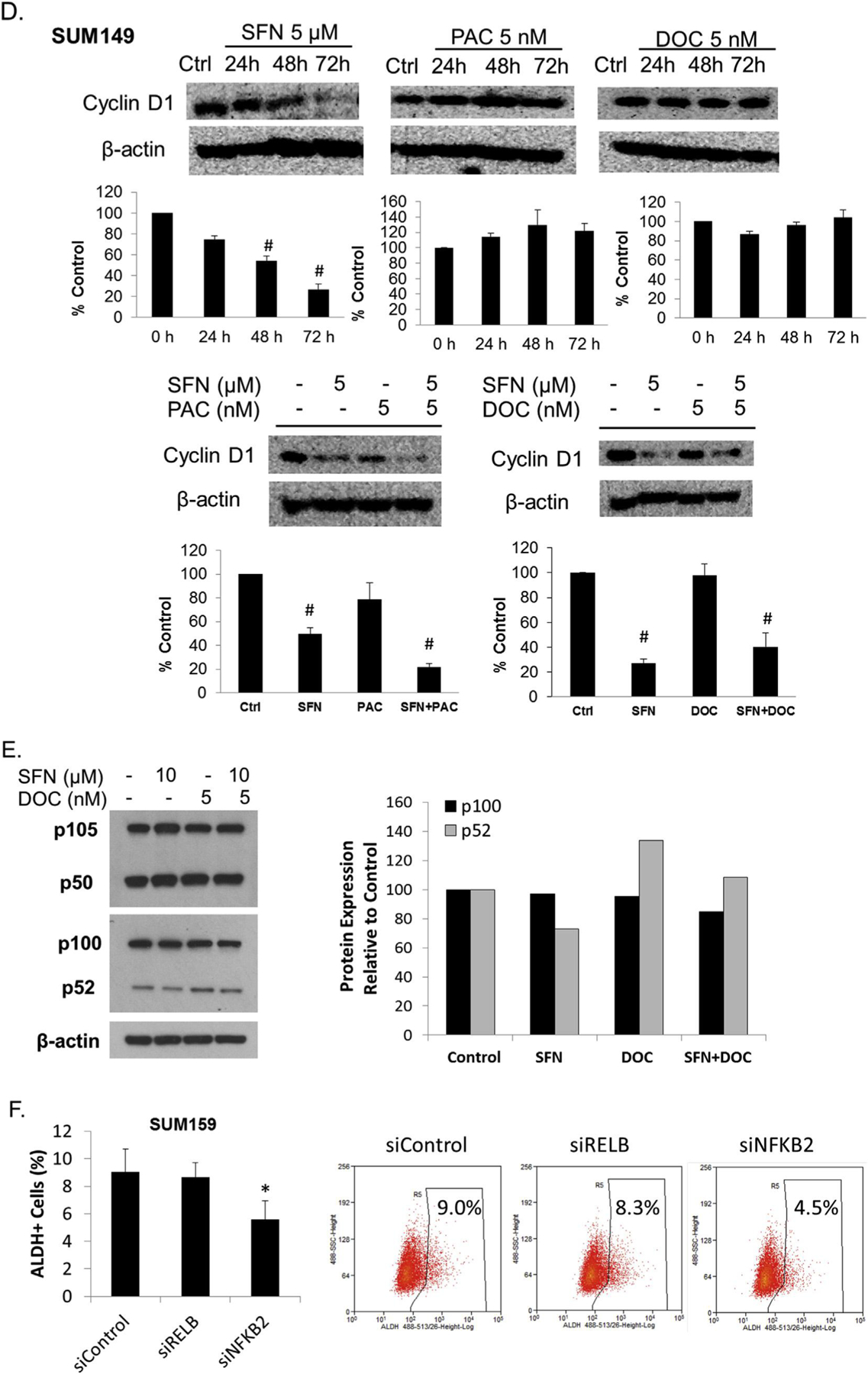
Sulforaphane inhibits NF-κB nuclear translocation, expression, transcriptional activity and downstream protein target cyclin D1 expression in triple negative breast cancer cells. A, representative immunocytochemical staining of the SUM159 or SUM149 cell lines for the p65 subunit of NF-kB after incubation without and with TNF-α, in the presence of increasing concentrations of sulforaphane. Counter staining of nucleus was carried out with DAPI immediately before imaging (bottom). Red arrow indicates the cytoplasmic-nuclear boarder. Images originally obtained using a 40× objective with scale bar = 50 μm. B. SUM159 cells were transfected with an NF-κB dependent luciferase reporter. Cell lines were treated with increasing concentrations of sulforaphane followed by the addition of TNF-α. N = 3. *p ≤ 0.05 and #p ≤ 0.01. C & D, SUM159 or SUM149 cells were treated with various concentrations of sulforaphane, paclitaxel, docetaxel or combination for 24–72 h and the levels of cyclin D1 were detected with Western blotting. The density of the bands were quantitated and normalize to control (beta-actin). E. SUM159 cells were treated with sulforaphane, docetaxel or combination for 72 h and the levels NF-kB signaling members were detected by Western blotting. F. The siRNA knockdown of RelB and NF-κB2 were performed in SUM159 cells, then Aldefluor assay and flow cytometry analysis were performed to measure the ALDH + cell population. N = 3. *p ≤ 0.05.
With this observation we sought to determine if sulforaphane would reduce endogenous NF-κB targets in the absence of any stimulator factor. As a central mediator of immune and inflammatory responses, NF-κB regulates many signaling cascades involved in stress responses and proliferation and apoptosis [43]. As previously shown, suforaphane can inhibit NF-κB mediated inflammatory processes by downregulating cytokine secretion (Fig. 1). In order to evaluate if inhibition of NF-κB signaling by sulforphane would repress proteins involved in cell proliferation and survival, cyclin D1 expression was evaluated. The human cyclin D1 promoter contains two putative NF-κB binding sites and the nuclear translocation of NF-κB p65 subunit directly regulates its expression [44]. Upon sulforaphane treatment, there is a remarkable decrease of cyclin D1 in both SUM149 and SUM159 cells (Fig. 2C&D). In contrast, neither paclitaxel nor docetaxel reduces the cyclin D1 expression (Fig. 2C&D). The combination of sulforaphane with paclitaxel or docetaxel reduces the cyclin D1 levels comparable to suforaphane treatment alone (Fig. 2C&D).
In order to determine if sulforphane’s action is limited to primarily NF-κB activity or downregulation of individual protein subunits further protein expression studies are warranted. The NF-κB family of transcription factors consists of five members: p65 (RelA), RelB, c-Rel, p105/p50, and p100/p52 [45]. The p65 (RelA), c-Rel and the P105/p50 dimer are members of the canonical NF-κB pathway, while RelB and p100/p52 dimer are members of the noncanonical NF-κB signaling [46]. In Fig. 2A we have shown that sulforaphane inhibits the canonical NF-κB p65 subunit from translocating into the nucleus. We next investigated the protein expression of both canonical and noncanonical members upon sulforaphane treatment at a longer 72 h time point. As shown in Fig. 2E, p52 protein level is weakly decreased upon sulforaphane treatment, increased upon docetaxel treatment, and the increase is reversed by the combination treatment. However, those changes are subtle. The expression levels of p100, p105 and p50 protein do not seem to be affected upon either sulforaphane or docetaxel treatment (Fig. 2E). The p52 subunit belongs to the noncanonical NF-κB pathway and is generated from cotranslational processing of p100, encoded by the NFKB2 gene. To determine if noncanonical NF-κB member expression exerts an effect on CSCs siRNA knockdown was performed in conjuction with the Aldefluor assay. Knockdown of the NF-κB2 gene was uniquely capable of decreasing the ALDH + cell population (Fig. 2F), similar to the results upon sulforaphane treatment. This suggests that sulforaphane may also exert its effect via regulation of the p52 protein during noncanonical signaling in addition to inhibiting p65 translocation in the canonical pathway. Taken together with the result from overall reporter inhibition these results support that sulforaphane is capable of reducing NF-κB function thus suppressing the expression cytokines and promoters for CSC growth.
Sulforaphane potentiates antiproliferation effects of taxanes in bulk tumor cells
The suppression of taxane-induced cytokine expression by sulforaphane provides a rationale to evaluate the therapeutic efficacy of these drug combinations. As a results of these unique interactions the combination of sulforphane and taxanes may allow for a reduction in individual drug doses while achieving the same therapeutic effect. To investigate the anti-proliferative effects of combination therapy against the bulk tumor cells the MTS assay was performed. SUM149 and SUM159 cells were treated with increasing concentrations of each therapeutic agent and the percentage of viable cells relative to control was plotted in Fig. 3.
Fig. 3.
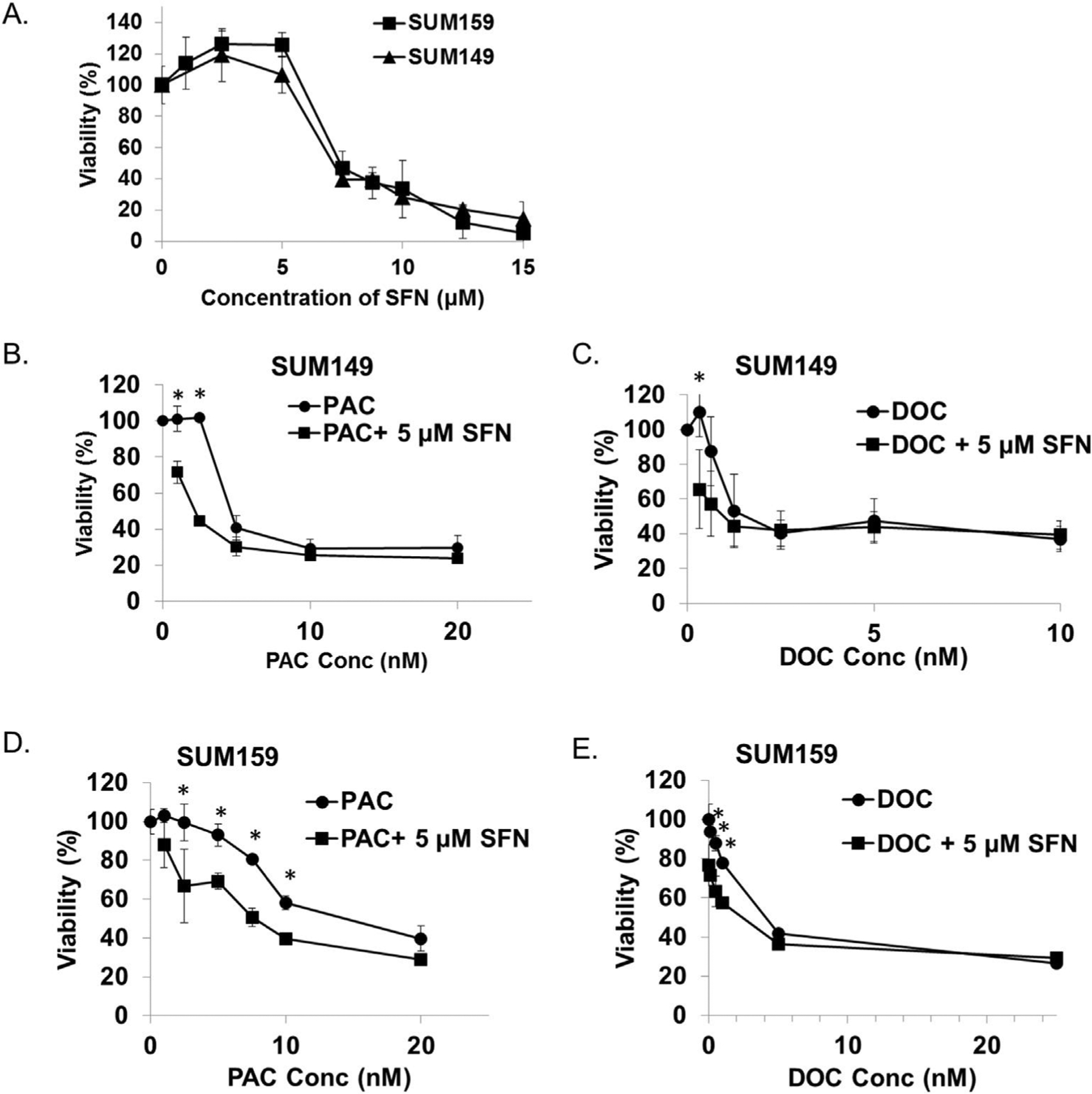
Sulforaphane enhances the anti-proliferative activity of paclitaxel and docetaxel. The TNBC cells SUM149 and SUM159 were treated with sulforaphane, paclitaxel, docetaxel or the combination at indicated concentrations, and cell viability was determined by MTS assay. A, cells were treated with increasing concentrations sulforaphane for 72 h. B-E, cells were treated with increasing concentrations of paclitaxel or docetaxel alone or in combination with 5 μM sulforaphane for 72 h. Points represent mean ± standard deviation (SD). SFN, sulforaphane; DOC, docetaxel; PAC, paclitaxel. *p ≤ 0.05.
In SUM149 cells the IC50s of sulforaphane, Paclitaxel, and docetaxel were determined to be 7.5 μM, 5.6 nM and 2.6 nM, respectively (Fig. 3A, B & C). Similarly, SUM159 which are relatively more resistant to treatment exhibit IC50s of 7.8 μM, 14 nM and 5.0 nM (Fig. 3A, D & E). After combination with 5 μM sulforaphane, a concentration at which alone exhibits minimal anti-proliferative effect in both cell lines, the IC50 of paclitaxel and docetaxel was reduced to 2.2 and 1.4 nM in SUM 149 (Fig. 3B &C). Similarly, the IC50s of paclitaxel and docetaxel were reduced to 7.5 nM and 1.9 nM in SUM159 (Fig. 3D and E). These results demonstrate that the combination of sulforaphane with taxanes can inhibit bulk TNBC cells better than using any single agent alone.
Sulforaphane reverses taxane-induced breast cancer stem/progenitor cells enrichment
It has been suggested that the resistance and eventual relapse of cancer are attributed to the presence of the CSC population [47]. Since IL-6 and IL-8 are suggested to increase breast CSC population and enhance their resistance to chemotherapy the most critical advantage of the identified combinations is the elimination of multiple populations of the heterogeneous tumors [31,32,40,30,41]. The ALDH-positive cell population is enriched for tumorigenic stem/progenitor cells, capable of self-renewal and generating tumors resembling the parental tumor in breast cancer [48]. Therefore, we first examined the effect of individual compound on ALDH-positive population in SUM149 and SUM159 cells. Cells were incubated with sulforaphane, paclitaxel or docetaxel for 72 h before Aldefluor assay was performed. The results show that either paclitaxel or docetaxel treatment increases the ALDH-positive CSC by 2–3 fold (Fig. 4A and B), suggesting that taxane treatment can enrich the CSC population. On the contrary, sulforaphane (2.5 and 5 μM) decreases the ALDH-positive cells by 40–50% (Fig. 4C). Strikingly, the combination of sulforaphane with paclitaxel (Fig. 4D) or docetaxel (Fig. 4E) not only prevents ALDH-positive cell expansion caused by paclitaxel or docetaxel but actually reduces the population to the similar extent as sulforaphane treatment alone. Similar results have been observed with another set of breast CSC marker CD44+/CD24−/Epcam+. As shown in Supplementary Fig. 2, docetaxel treatment increases the CD44+/CD24−/Epcam + cell population by 2-fold (Suppl. Fig. 2A&B), and sulforaphane (5 μM) decreases it by 50% (Suppl. Fig. 2B). The combination of sulforaphane with or docetaxel reduces the CD44+/CD24−/Epcam + cell population to the similar extent as sulforaphane treatment alone (Suppl. Fig. 2C). To further support this point, we evaluated the ALDH+ and CD44+/CD24−/Epcam + overlapping cell population, which is the most tumorigenic cell population [49,50]. Not surprisingly, docetaxel treatment increases the CD44+/CD24−/Epcam+/ALDH + cell population and sulforaphane decreases this population (Suppl. Fig. 2D&E).
Fig. 4.
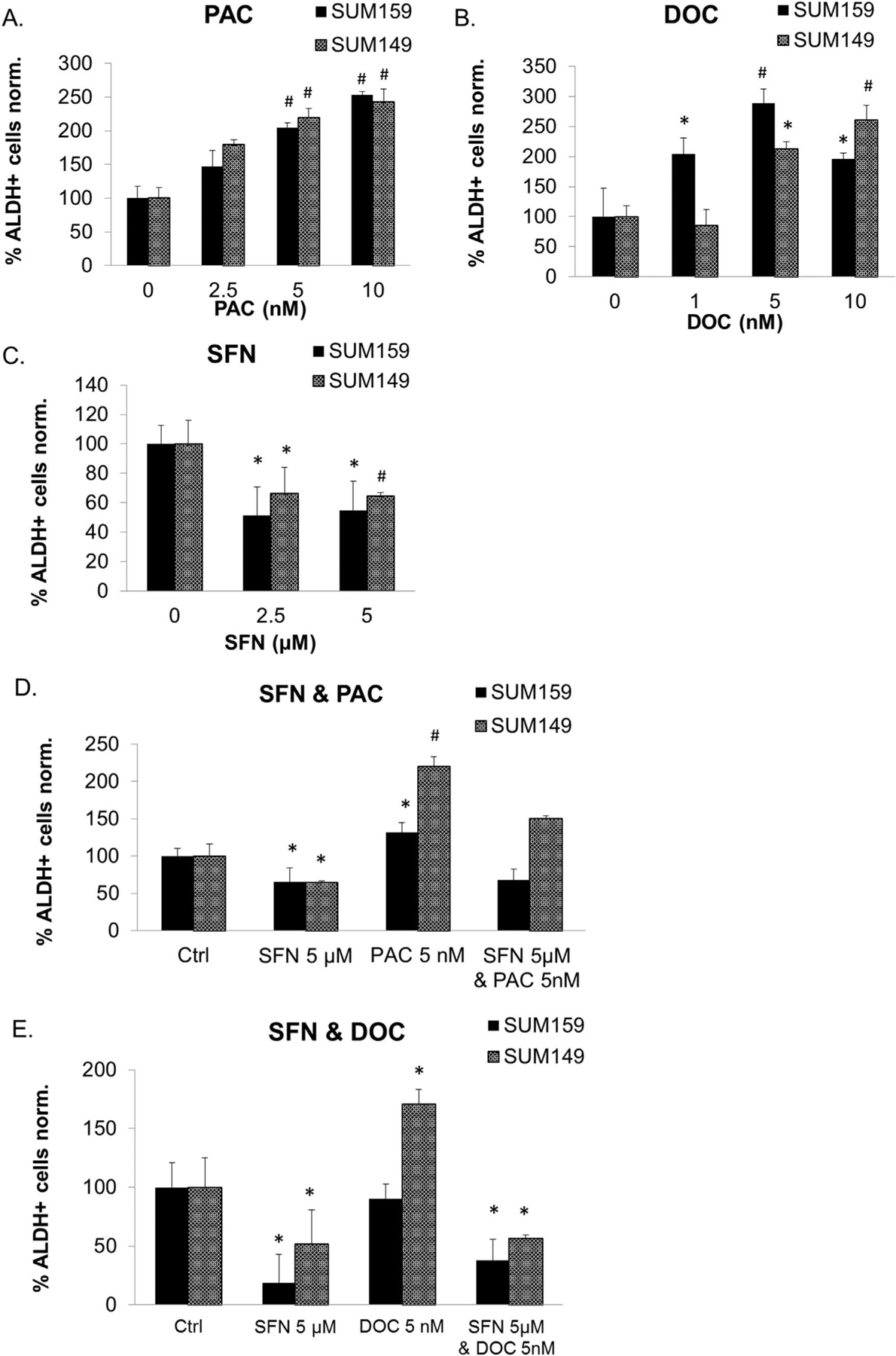
Sulforaphane reverses paclitaxel- or docetaxel-induced ALDH-positive cell population increase. Cells were treated with DMSO, sulforaphane (2.5 and 5 μM), paclitaxel (2.5–10 nM), or docetaxel (1–10 nM) alone or in combination for 72 h and then subject to Aldefluor assay and flow cytometry analysis. A, sulforaphane decreases the percentage of ALDH-positive cells. B, paclitaxel increases the percentage of ALDH-positive cells. C, docetaxel increases the percentage of ALDH-positive cells. D & E, Combination of sulforaphane with paclitaxel or docetaxel decreases ALDH-positive cell population compared with treatment with either paclitaxel or docetaxel alone. SFN, sulforaphane; DOC, docetaxel; PAC, paclitaxel. *p ≤ 0.05 and #p ≤ 0.01.
While the Aldefluor assay is an established method to determine the regulation of breast CSCs in vitro, to further support this conclusion it is necessary to perform additional functional assays. Using the in vitro mammosphere formation assay our results show that in SUM149 cells, primary sphere formation rate is unaltered in the presence of 1 nM docetaxel relative to the vehicle treated cells (Fig. 5A, Left). However, upon passaging in the absence of further drugs more secondary spheres form. Conversely, 2.5 μM sulforaphane alone or in combination with 1 nM docetaxel both inhibit primary sphere formation and alter the long term self-renewal of breast CSCs and progenitors, suggested by decrease in secondary mammosphere formation. The size of primary spheres formed in all cases is reduced relative to control in the case of any drug treatment (Fig. 5A, Right). In SUM159 cells, primary sphere formation rate decline by ~50% and ~60% upon sulforaphane (5 μM) and paclitaxel (5 nM) treatment, respectively (Fig. 5B, Left). Cells dissociated from primary mammospheres under sulforaphane treatment show reduced capacity of secondary mammosphere formation in the absence of further drug treatment, while those from paclitaxel treatment group returned to control levels. The combination of both sulforaphane (5 μM) and paclitaxel (5 nM) achieve the maximum inhibition of both primary and secondary mammosphere formation (Fig. 5B, Right). These results suggest that sulforaphane in combination with paclitaxel or docetaxel can effectively eliminate breast CSCs and attenuate the self-renewal ability in TNBCs.
Fig. 5.
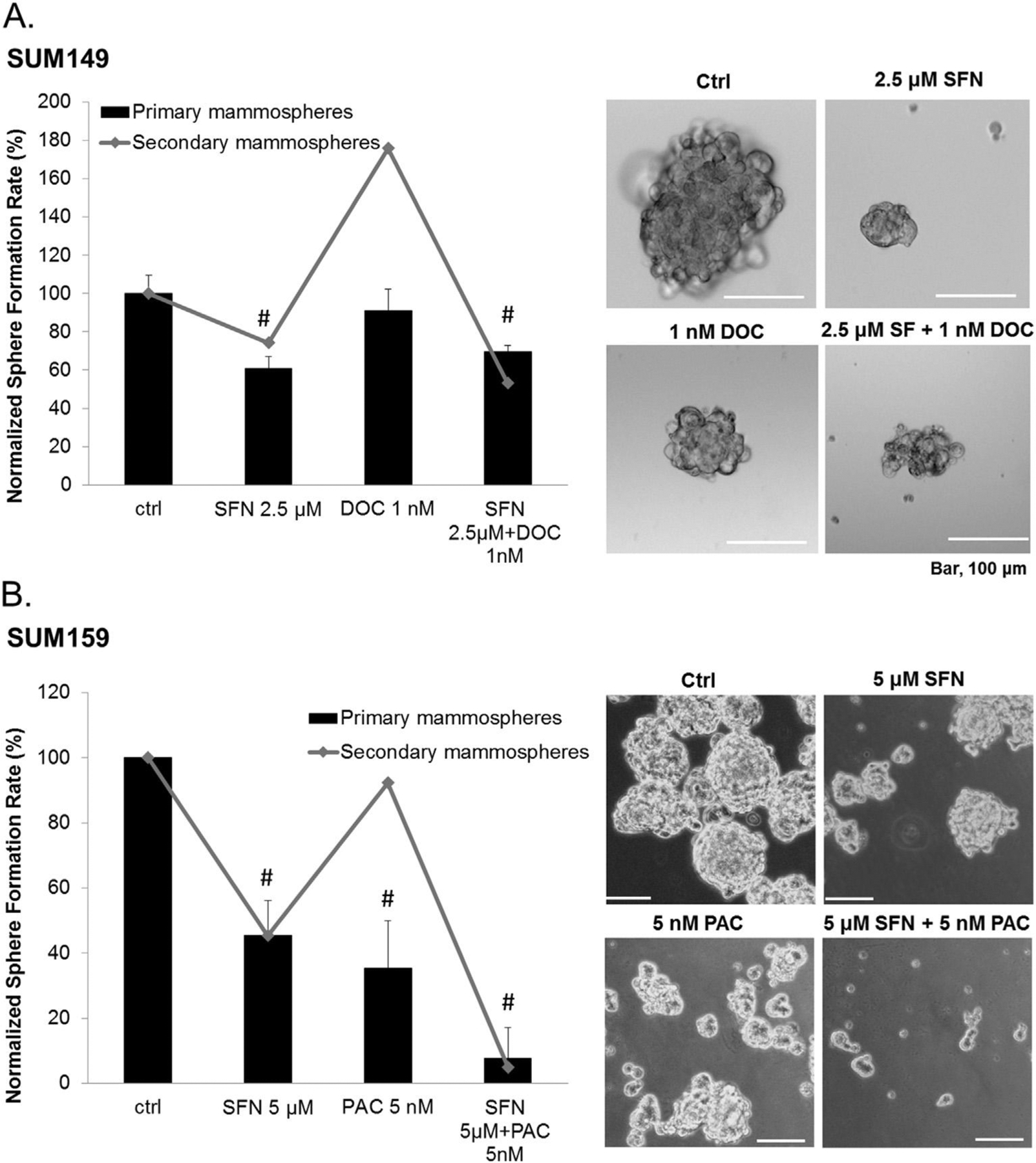
Inhibitory effects of suforaphane, paclitaxel, docetaxel or combination on mammosphere formation. SUM149 and SUM159 cells were cultured in mammosphere-forming media and treated with the indicated compounds for seven days to form the primary mammospheres. After that, the primary mammsospheres were dissociated into single cells and cultured without drug treatment to form the secondary mammospheres. A, Left, normalized number of formed primary and secondary mammospheres upon sulforaphane (2.5 μM), docetaxel (1 nM), or combination treatments. Right, representative images of primary mammospheres formed after seven days. Bar, 100 μm. B, normalized number of formed primary and secondary mammospheres upon sulforaphane (5 μM), paclitaxel (5 nM), or combination treatments. Right, representative images of primary mammospheres formed in the absence or presence of sulforaphane, paclitaxel or combination treatment for seven days. Bar, 100 μm *p ≤ 0.05 and #p ≤ 0.01. SFN, sulforaphane; DOC, docetaxel; PAC, paclitaxel.
Combination of sulforaphane and docetaxel significantly inhibits tumor growth and breast CSCs in vivo
Elimination of breast CSCs is critical to ultimately curing patients. However it is also necessary to eliminate more differentiated cells, which are responsible for the majority of a tumors volume, in order to reduce symptoms of disease progression. Further, due to the genetically unstable nature of cancer cells it is possible that overtime a more differentiated cell could acquire mutations or experience enough environmental or stochastic influence to reactive genes responsible for self-renewal. These potential problems could then lead to the acquisition of breast CSC characteristics and little would be done to eliminate the disease. In order to evaluate the ability of sulforaphane and taxanes to inhibit both bulk tumor volume and breast CSCs in vivo we utilized an advanced treatment orthotopic mouse xenograft model and performed extreme limiting dilution analysis in secondary mice (ELDA) with residual primary tumors [37].
After implantation of 1.5 million SUM149 cells into the 4th mammary pad of NOD/SCID mice, tumors were allowed to reach an average volume of 50 mm3 and randomized into 4 separate groups before treatment began. Treatment groups included control mice which received 0.9% saline, mice administered 50 mg/kg sulforaphane daily, 10 mg/kg docetaxel administered weekly, and 50 mg/kg sulforaphane daily with 10 mg/kg docetaxel weekly. All treatments were administered via intraperitoneal (I.P.) administration. When control tumors reached the protocol specific endpoint sulforphane treatment reduced bulk tumor volume by 37.4% (p = 0.011), whereas docetaxel reduced tumor volume by 83.2% and the combination by 92.5% (Fig. 6A). Interestingly, only the combination of sulforaphane and docetaxel causes significant (p = 0.039) tumor regression relative to the maximum tumor volume for that treatment group (day 27 vs 51). Mouse body weight was consistent throughout the course of study, demonstrating no dramatic toxicity at the indicated dose regimens (Fig. 6B).
Fig. 6.
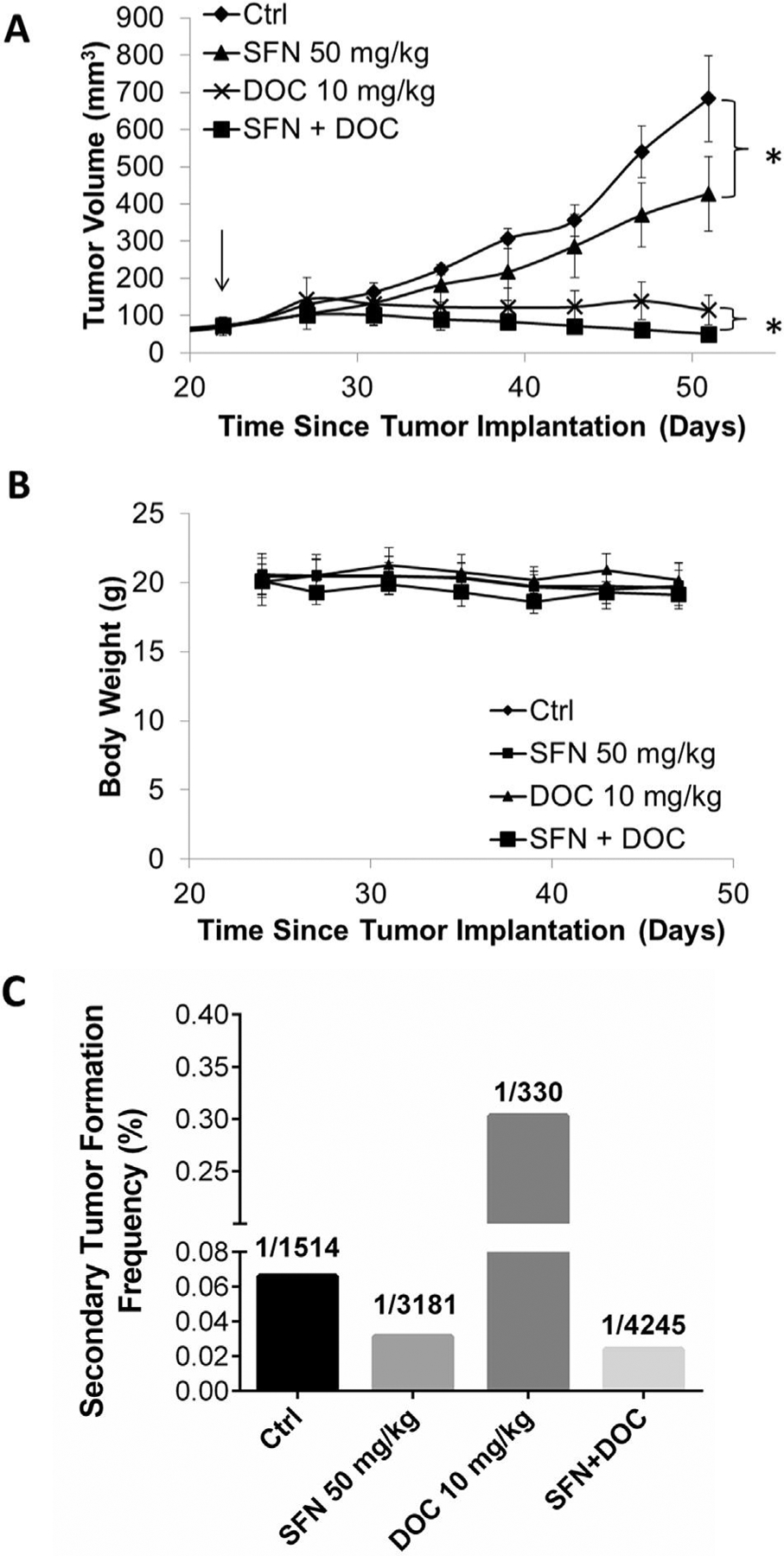
Docetaxel and sulforaphane cooperate to eliminate both bulk tumor volume and breast CSCs in vivo. A, NOD/SCID mice bearing an average tumor volume of 50 mm3 SUM149 xenografts were randomized into treatment groups which received daily 0.9% saline (Control), daily 50 mg/kg sulforaphane, weekly 10 mg/kg docetaxel, or daily 50 mg/kg sulforaphane in combination with weekly 10 mg/kg docetaxel as a cassette dose. Each drug was administered via I.P. injection. Arrow denotes the beginning of treatment in primary mice. N = 5. *p ≤ 0.05 in final tumor volume comparisons. B, body weight of mice receiving each treatment regimen over the course of administration. N = 5. C. The frequency of secondary tumor formation after inoculating of the isolated tumor cells from primary mice xenograft. Equal number of tumor cells isolated from mice treated above (panel A) were implanted in to new set of recipient mice, which did not received any therapies and were monitored for tumor initiation. The tumor initiating CSC rate was determined and plotted.
In order to evaluate the therapeutic efficacy of the given treatment groups with respect to breast CSCs. Primary tumors were harvest, dissociated into a single cell suspension, and residual live human cells collect with FACS (DAPI-, H2KD-). Three separate dilutions of cells from each treatment group were implanted in to recipient mice, which had not received any therapies, to quantify the frequency of tumor initiating breast CSCs by ELDA (Fig. 6C & Table 1). As shown in Table 1, the tumor cells isolated from control treatment group have high tumor formation capability (5/6, 4/4, and 4/6), which means 5 out of 6 mice, 4 out of 4 mice, and 4 out of 6 mice have tumor formed when 10,000, 1,000, and 100 cells were inoculated into the mice and observed for 7 weeks (Table 1). Even higher tumor formation rate is shown in cells isolated from docetaxel treatment groups, with rate of 6/6, 5/6, and 3/4 when 10,000, 1,000, and 100 cells were inoculated (Table 1). In contrast, the tumor formation frequency of the cells isolated from sulforaphane and combination group are lower. When only 100 and 1000 cells were inoculated, 1 out of 6 mice and 3 out of 6 mice have tumor formed after inoculating tumor cells from sulforaphane-treated primary xenograft; 2 out of 6 mice, and 3 out of 6 mice have tumor formed after inoculating tumor cells from combination treated-primary xenograft. Further analysis of all the limiting dilution data demonstrates that tumor formation rates is 1 in 1514 in cells from control primary mice (Fig. 6C & Table 1). Docetaxel significantly increases this frequency to 1 in 330 cells whereas both sulforaphane and combination therapy reduce the rate of tumor formation to 1 in 3181 and 1 in 4245 cells, respectively (Fig. 6C & Table 1). Taken together, these results suggest that combination of sulforaphane and docetaxel are effective in vivo at not only reducing bulk tumor volume, but more importantly, inhibiting tumor initiation ability.
Table 1.
The extreme limiting dilution analysis (ELDA) for calculation of tumor initiating CSC frequency after reimplantation of primary xenograft tumor cells into secondary mice.
| Group | Limiting dilutions (Tumors/Implantations) | CSC frequency (1 in/…) | ||
|---|---|---|---|---|
| 10,000 | 1000 | 100 | ||
| Control | 5/6 | 4/4 | 4/6 | 1514 |
| SFN | 5/6 | 3/6 | 1/6 | 3181 |
| DOC | 6/6 | 5/6 | 3/4 | 330 |
| SFN + DOC | 4/6 | 2/6 | 3/6 | 4245 |
Discussion
Treatment options for breast cancer vary depending on the different subtypes, which are primarily classified based on the receptors present [4]. Luminal A (ER + or PR+, Her2−) and luminal B (ER + or PR+, Her2−, Ki-67 high or ER-PR+, Her2+) breast cancers have multiple treatment options, including endocrine, anti-HER2, and conventional cytotoxic chemotherapy [4]. Her2+ (ER and PR−, Her2+) breast cancers may respond well to anti-HER2 therapy in combination with cytotoxic chemo-therapy [51]. However, TNBC which represent the vast majority of basal and claudin-low subtypes, are resistant to most current treatment options and are largely restricted to treatment with conventional cytotoxic chemotherapy [52]. The efficacy of chemotherapeutic agents is generally evaluated by the preclinical methods and clinical trial endpoints focusing on reducing tumor volume to delay disease progression. Determined with these endpoints, it is possible that conventional therapies are very effective in reducing bulk tumor volume but fail to eliminate the breast CSCs which may make up only a small portion of the heterogeneous tumor.
The occurrence of cancer metastasis and relapse is common and has recently been attributed to the existence of CSCs after chemotherapy [47]. In order to cure TNBCs there is a critical need to develop effective strategies that eliminate both breast CSCs, responsible for metastasis and relapse after therapy, as well as, more differentiated cancer cells which may cause unwanted symptoms at metastatic sites. However, accumulating preclinical evidence suggest that many anticancer agents such as sunitinib, doxorubicin and gemcitabine only inhibit the growth of differentiated cancer cells but fail to eliminate or may even expand CSC populations [25,26]. In this report we demonstrate that the conventional cytotoxic chemotherapeutic agents of the taxane family, docetaxel and paclitaxel, which inhibit microtubule polymerization to shrink bulk tumor volume, actually enhance IL-6 and IL-8 production thereby increasing breast CSCs. This data is consistent with previous in vivo findings demonstrating that docetaxel increased IL-6 and IL8 production in patients, and that direct stimulation with IL-6 and IL-8 can increase breast CSCs [29,30,32,53]. Production of both these cytokines is regulated by the activity of the transcription factor NF-κB, which has been demonstrated to be of critical importance in the regulation of CSC [33–35,54,55]. Further, IL6 is known to regulate NF-κB through STAT3, an interaction capable of establishing a positive feedback loop [26,32].
The anticancer efficacy and mechanism of sulforaphane have been studied in a variety of cancers including breast, colon, leukemia, prostate, and pancreatic cancer [56]. Most of these studies have been focused on its effect to induce apoptosis in the bulk tumor population, which usually requires higher concentrations of sulforaphane as it is not a very potent cytotoxic compound. We first reported that sulforaphane may selectively inhibit self-renewal of breast CSCs at relatively low concentrations [23]. These results were subsequently confirmed by other researchers demonstrating that sulforaphane can eliminate cancer stem-like cells in other cancer types such as pancreatic and prostate cancer [57,58]. The current results are consistent with these findings by showing that sulforaphane can decrease CSC population in TNBC cell lines. A staggering number of potential mechanisms of action have reported to explain sulforaphane’s efficacy; regulation of Nrf2, HDAC, Chk2, p21, MAPK, death receptor, NF-κB, Stat3, and Hsp90 in various cancer cells [59–61]. While sulforaphane may indeed regulate these molecules it is unclear what their relative contributions are to efficacy and in what contexts each are relevant. In this report we identify that sulforaphane inhibits NF-κB function by preventing intracellular translocation of p65 subunit and its transcriptional activity. This is consistent with the finding that in human umbilical vein endothelial cells sulforaphane inhibits tumor necrosis factor-α (TNF-α) induced NF-κB signaling [62]. In glioblastoma, it has been reported that CSCs exhibited increased nuclear localization of p65, suggesting the important role of p65 subunit in CSC self-renewal [63]. Song et al. also reported that inhibition of NF-B reduced self-renewal and blocked xenograft tumor growth [64]. In addition to the canonical NF-κB pathway, we also find that sulforaphane can inhibit p52 in the noncanonical NF-κB signaling pathway as well. Knockdown of the NF-κB 2 gene, encoding p52, can decrease CSC populations similar to that upon sulforaphane treatment. Noting that both breast CSCs and TNBCs in general are more dependent on cytokine-NF-κB signaling and that we have shown sulforaphane preferentially inhibits these factors, we hypothesize that in breast cancers NF-κB inhibition is the major mechanism of action for efficacy. This is consistent with the report by Kallifatidis et al. who demonstrate pancreatic cancer cell lines and their respective CSC population are sensitive to sulforaphane mediated NF-κB inhibition [65].
The combination of anti-CSC agents with chemotherapeutic agents thus should provide much more benefits to the patients. As demonstrated by the in vitro results shown here, sulforaphane, at subcytotoxic concentrations, has decreased the CSC population significantly, and reversed taxane-induced CSC increase. Meanwhile, paclitaxel or docetaxel, at therapeutically relevant concentrations, is efficient to kill the bulk of differentiated cells. In vivo using an orthotopic mouse xenograft tumor model I.P. administration of daily sulforaphane in combination with weekly docetaxel leads to a dramatic reduction in primary tumor growth. Further, ELDA with residual cells from the primary tumors illustrates that docetaxel indeed increases breast CSC frequency while the combination dramatically reduces them. The combination of both compounds exhibits dramatic enhancement in efficacy to inhibit proliferation and lead to death of all the cancer cells, which provides strong evidence for further investigation in the clinic. The concurrent use of two or more therapeutic agents with unrelated mechanisms of action has been an effective strategy in breast cancer management [66]. The combinations can usually facilitate the attack on multiple intercellular processes and result in more efficient tumor responses [66]. Considering that the CSC population is root of cancer resistance and relapse, combination of an anti-CSC agent with a chemotherapeutic agent may achieve much better clinical benefit with respect to long term survival.
In this report we demonstrated that paclitaxel or docetaxel treatment enriches the breast CSC population while increasing IL-6 in TNBC cell lines. Conversely, the natural compound sulforaphane is capable of preferentially eliminating CSCs and inhibiting NF-κB. Combination of sulforaphane with either paclitaxel or docetaxel not only exerts a dramatic enhancement of cytotoxic potency against the bulk tumor cells, but also greatly suppresses the CSC population compared with paclitaxel or docetaxel alone. Taken together, these results demonstrate that treatment of TNBCs with cytotoxic chemotherapy would be greatly benefited by the addition of sulforaphane to prevent expansion of and eliminate breast CSCs.
Supplementary Material
Funding
This work was partially supported by American Association of Colleges of Pharmacy (AACP) New Investigator Award (NIA) (to T. Zhang), Husson University research fund and Husson University School of Pharmacy research fund (to T. Zhang).
Abbreviations:
- ALDH+
Aldehyde dehydrogenase-positive
- CSC
Cancer stem cell
- DOC
Docetaxel
- ELDA
Extreme limiting dilution analysis
- IL
Interleukin
- PAC
Paclitaxel
- SFN
Sulforaphane
- TNBC
Triple negative breast cancer
Footnotes
Conflicts of interest
The authors declare no potential conflicts of interest.
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.canlet.2017.02.023.
References
- [1].Cancer facts & figures, American Cancer Society; 2015, 2015. [Google Scholar]
- [2].Polyak K, Heterogeneity in breast cancer, J. Clin. Investig 121 (10) (2011) 3786–3788, 10.1172/ja60534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hudis CA, Gianni L, Triple-negative breast cancer: an unmet medical need, Oncol. 16 (Suppl 1) (2011) 1–11, 10.1634/theoncologist.2011-Sl-01. [DOI] [PubMed] [Google Scholar]
- [4].Parise CA, Caggiano V, Breast cancer survival defined by the ER/PR/HER2 subtypes and a surrogate classification according to tumor grade and immunohistochemical biomarkers, J. Cancer Epidemiol 2014 (2014) 469251, 10.1155/2014/469251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Onitilo AA, Engel JM, Greenlee RT, Mukesh BN, Breast cancer subtypes based on ER/PR and Her2 expression: comparison of clinicopathologic features and survival, Clin. Med. Res 7 (1–2) (2009) 4–13, 10.3121/cmr.2009.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Boyle P, Triple-negative breast cancer: epidemiological considerations and recommendations, Ann. Oncol. Off. J. Eur. Soc. Med. Oncol./ESMO 23 (Suppl 6) (2012), 10.1093/annonc/mdsl87 vi7–12. [DOI] [PubMed] [Google Scholar]
- [7].Podo F, Buydens LM, Degani H, Hilhorst R, Klipp E, Gribbestad IS, et al. , Triple-negative breast cancer: present challenges and new perspectives, Mol. Oncol 4 (3) (2010) 209–229, 10.1016/j.molonc.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Carey LA, Dees EC, Sawyer L, Gatti L, Moore DT, Collichio F, et al. , The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes, Clin. Cancer Res 13 (8) (2007) 2329–2334, 10.1158/1078-0432.ccr-06-1109. [DOI] [PubMed] [Google Scholar]
- [9].Liedtke C, Mazouni C, Hess KR, André F, Tordai A, Mejia JA, et al. , Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer, J. Clin. Oncol 26 (8) (2008) 1275–1281, 10.1200/jco.2007.14.4147. [DOI] [PubMed] [Google Scholar]
- [10].Huober J, von Minckwitz G, Denkert C, Tesch H, Weiss E, Zahm D, et al. , Effect of neoadjuvant anthracycline–taxane-based chemotherapy in different biological breast cancer phenotypes: overall results from the GeparTrio study, Breast Cancer Res. Treat 124 (1) (2010) 133–140, 10.1007/s10549-010-1103-9. [DOI] [PubMed] [Google Scholar]
- [11].von Minckwitz G, Martin M, Neoadjuvant treatments for triple-negative breast cancer (TNBC), Ann. Oncol 23 (suppl 6) (2012), 10.1093/annonc/mds193 vi35–vi9. [DOI] [PubMed] [Google Scholar]
- [12].von Minckwitz G, Martin M, Neoadjuvant treatments for triple-negative breast cancer (TNBC), Ann. Oncol. Off. J. Eur. Soc. Med. Oncol./ESMO 23 (Suppl 6) (2012), 10.1093/annonc/mds193 vi35–9. [DOI] [PubMed] [Google Scholar]
- [13].Kassam F, Enright K, Dent R, Dranitsaris G, Myers J, Flynn C, et al. , Survival outcomes for patients with metastatic triple-negative breast cancer: implications for clinical practice and trial design, Clin. Breast Cancer 9 (1) (2009) 29–33, 10.3816/CBC.2009.n.005. [DOI] [PubMed] [Google Scholar]
- [14].Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, et al. , Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy, J. Natl. Cancer Inst 100 (9) (2008) 672–679, 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- [15].Tanei T, Morimoto K, Shimazu K, Kim SJ, Tanji Y, Taguchi T, et al. , Association of breast cancer stem cells identified by aldehyde dehydrogenase 1 expression with resistance to sequential Paclitaxel and epirubicin-based chemotherapy for breast cancers, Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res 15(12) (2009) 4234–4241, 10.1158/1078-0432.CCR-08-1479. [DOI] [PubMed] [Google Scholar]
- [16].Shafee N, Smith CR, Wei S, Kim Y, Mills GB, Hortobagyi GN, et al. , Cancer stem cells contribute to cisplatin resistance in Brca1/p53-mediated mouse mammary tumors, Cancer Res. 68 (9) (2008) 3243–3250, 10.1158/0008-5472.CAN-07-5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bonnet D, Dick JE, Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell, Nat. Med 3 (7) (1997) 730–737. [DOI] [PubMed] [Google Scholar]
- [18].Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF, Prospective identification of tumorigenic breast cancer cells, Proc. Natl. Acad. Sci. U. S. A 100 (7) (2003) 3983–3988, 10.1073/pnas.0530291100 0530291100 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Reya T, Morrison SJ, Clarke MF, Weissman IL, Stem cells, cancer, and cancer stem cells, Nature 414 (6859) (2001) 105–111, 10.1038/3510216735102167 [pii]. [DOI] [PubMed] [Google Scholar]
- [20].Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, et al. , Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features, Proc. Natl. Acad. Sci. U. S. A 106 (33) (2009) 13820–13825, 10.1073/pnas.0905718106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Li Y, Zhang T, Targeting cancer stem cells with sulforaphane, a dietary component from broccoli and broccoli sprouts, Future Oncol 9 (8) (2013) 1097–1103, 10.2217/fon.13.108. [DOI] [PubMed] [Google Scholar]
- [22].Cheung KL, Kong AN, Molecular targets of dietary phenethyl isothiocyanate and sulforaphane for cancer chemoprevention, AAPS J 12 (1) (2010) 87–97, 10.1208/s12248-009-9162-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Li Y, Zhang T, Korkaya H, Liu S, Lee HF, Newman B, et al. , Sulforaphane, a dietary component of broccoli/broccoli sprouts, inhibits breast cancer stem cells, Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res 16 (9) (2010) 2580–2590, 10.1158/1078-0432.CCR-09-2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sun L, Burnett J, Gasparyan M, Xu F, Jiang H, Lin C-C, et al. , Novel Cancer Stem Cell Targets during Epithelial to Mesenchymal Transition in PTEN-deficient Trastuzumab-resistant Breast Cancer, 2016. [DOI] [PMC free article] [PubMed]
- [25].Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, et al. , Identification of selective inhibitors of cancer stem cells by high-throughput screening, Cell 138 (4) (2009) 645–659, 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K, Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission, Cancer Res. 69 (19) (2009) 7507–7511 doi: 0008-5472.CAN-09-2994 [pii] 10.1158/0008-5472.CAN-09-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].King KM, Lupichuk S, Baig L, Webster M, Basi S, Whyte D, et al. , Optimal use of taxanes in metastatic breast cancer, Curr. Oncol 16 (3) (2009) 8–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lee LF, Hellendall RP, Wang Y, Haskill JS, Mukaida N, Matsushima K, et al. , IL-8 reduced tumorigenicity of human ovarian cancer in vivo due to neutrophil infiltration, J. Immunol 164 (5) (2000) 2769–2775. [DOI] [PubMed] [Google Scholar]
- [29].Tsavaris N, Kosmas C, Vadiaka M, Kanelopoulos P, Boulamatsis D, Immune changes in patients with advanced breast cancer undergoing chemotherapy with taxanes, Br. J. Cancer 87 (1) (2002) 21–27, 10.1038/sj.bjc.6600347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ginestier C, Liu S, Diebel ME, Korkaya H, Luo M, Brown M, et al. , CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts, J. Clin. Investig 120 (2) (2010) 485–497, 10.1172/jci39397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sansone P, Storci G, Tavolari S, Guarnieri T, Giovannini C, Taffurelli M, et al. , IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland, J. Clin. Investig 117 (12) (2007) 3988–4002, 10.1172/jci32533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Korkaya H, Kim GI, Davis A, Malik F, Henry NL, Ithimakin S, et al. , Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population, Mol. Cell 47 (4) (2012) 570–584, 10.1016/j.molcel.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Libermann TA, Baltimore D, Activation of interleukin-6 gene expression through the NF-kappa B transcription factor, Mol. Cell. Biol 10 (5) (1990) 2327–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Elliott CL, C Allport V, Loudon JAZ, Wu GD, Bennett PR, Nuclear factor-kappa B is essential for up-regulation of interleukin-8 expression in human amnion and cervical epithelial cells, Mol. Hum. Reprod 7 (8) (2001) 787–790, 10.1093/molehr/7.8.787. [DOI] [PubMed] [Google Scholar]
- [35].Yamamoto M, Taguchi Y, Ito-Kureha T, Semba K, Yamaguchi N, Inoue J-i. NF-κB non-cell-autonomously regulates cancer stem cell populations in the basal-like breast cancer subtype, Nat. Commun 4 (2013), 10.1038/ncomms3299. [DOI] [PubMed] [Google Scholar]
- [36].Burnett JP, Korkaya H, Ouzounova MD, Jiang H, Conley SJ, Newman BW, et al. , Trastuzumab resistance induces EMT to transform HER2+ PTEN− to a triple negative breast cancer that requires unique treatment options, Sci. Rep 5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hu Y, Smyth GK, ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays, J. Immunol. Methods 347 (1–2) (2009) 70–78, 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- [38].Hartman ZC, Poage GM, den Hollander P, Tsimelzon A, Hill J, Panupinthu N, et al. , Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8, Cancer Res. 73 (11) (2013) 3470–3480, 10.1158/0008-5472.CAN-12-4524-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chavey C, Bibeau F, Gourgou-Bourgade S, Burlinchon S, Boissiere F, Laune D, et al. , Oestrogen receptor negative breast cancers exhibit high cytokine content, Breast Cancer Res. BCR 9 (1) (2007) R15, 10.1186/bcr1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Marotta LL, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR, et al. , The JAK2/STAT3 signaling pathway is required for growth of CD44(+) CD24(−) stem cell-like breast cancer cells in human tumors, J. Clin. Investig 121 (7) (2011) 2723–2735, 10.1172/ja44745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bhola NE, Balko JM, Dugger TC, Kuba MG, Sanchez V, Sanders M, et al. , TGF-beta inhibition enhances chemotherapy action against triple-negative breast cancer, J. Clin. Investig 123 (3) (2013) 1348–1358, 10.1172/ja65416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Vyas D, Laput G, Vyas AK, Chemotherapy-enhanced inflammation may lead to the failure of therapy and metastasis, Onco Targets Ther. 7 (2014) 1015–1023, 10.2147/OTT.S60114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Oeckinghaus A, Ghosh S, The NF-kappaB family of transcription factors and its regulation, Cold Spring Harb. Perspect. Biol 1 (4) (2009), 10.1101/cshperspect.a000034 a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hinz M, Krappmann D, Eichten A, Heder A, Scheidereit C, Strauss M, NF-kappaB function in growth control: regulation of cyclin Dl expression and G0/G1-to-S-phase transition, Mol. Cell. Biol 19 (4) (1999) 2690–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Shostak K, Chariot A, NF-kappaB, stem cells and breast cancer: the links get stronger, Breast Cancer Res. BCR 13 (4) (2011) 214, 10.1186/bcr2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Rinkenbaugh AL, Baldwin AS, The NF-kappaB pathway and cancer stem cells, Cells 5 (2) (2016), 10.3390/cells5020016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Yu Y, Ramena G, Elble RC, The role of cancer stem cells in relapse of solid tumors, Front. Biosci 4 (2012) 1528–1541. [DOI] [PubMed] [Google Scholar]
- [48].Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. , ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome, Cell Stem Cell 1 (5) (2007) 555–567 doi: S1934–5909(07)00133–6 [pii] 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Nie S, McDermott SP, Deol Y, Tan Z, Wicha MS, Lubman DM, A quantitative proteomics analysis of MCF7 breast cancer stem and progenitor cell populations, Proteomics 15 (22) (2015) 3772–3783, 10.1002/pmic.201500002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, et al. , Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts, Stem Cell Rep. 2 (1) (2014) 78–91, 10.1016/j.stemcr.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Cardoso F, Harbeck N, Fallowfield L, Kyriakides S, Senkus E, Group EGW, Locally recurrent or metastatic breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up, Ann. Oncol. Off. J. Eur. Soc. Med. Oncol./ESMO 23 (Suppl 7) (2012), 10.1093/annonc/mds232 viill–9. [DOI] [PubMed] [Google Scholar]
- [52].Andre F, Zielinski CC, Optimal strategies for the treatment of metastatic triple-negative breast cancer with currently approved agents, Ann. Oncol. Off. J. Eur. Soc Med. Oncol./ESMO 23 (Suppl 6) (2012), 10.1093/annonc/mds195 vi46–51. [DOI] [PubMed] [Google Scholar]
- [53].Sansone P, Storci G, Tavolari S, Guarnieri T, Giovannini C, Taffurelli M, et al. , IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland, J. Clin. Investig 117 (12) (2007) 3988–4002, 10.1172/JCI32533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Guzman ML, Neering SJ, Upchurch D, Grimes B, Howard DS, Rizzieri DA, et al. , Nuclear Factor-κB is Constitutively Activated in Primitive Human Acute Myelogenous Leukemia Cells, 2001, pp. 2301–2307. [DOI] [PubMed]
- [55].Hinohara K, Kobayashi S, Kanauchi H, Shimizu S, Nishioka K, Tsuji E, et al. , ErbB receptor tyrosine kinase/NF-kappaB signaling controls mammosphere formation in human breast cancer, Proc. Natl. Acad. Sci. U. S. A 109 (17) (2012) 6584–6589, 10.1073/pnas.1113271109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Pawlik A, Wiczk A, Kaczynska A, Antosiewicz J, Herman-Antosiewicz A, Sulforaphane inhibits growth of phenotypically different breast cancer cells, Eur.J. Nutr 52 (8) (2013) 1949–1958, 10.1007/s00394-013-0499-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kallifatidis G, Labsch S, Rausch V, Mattern J, Gladkich J, Moldenhauer G, et al. , Sulforaphane increases drug-mediated cytotoxicity toward cancer stem-like cells of pancreas and prostate. Molecular therapy, J. Am. Soc. Gene Ther 19 (1) (2011) 188–195, 10.1038/mt.2010.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Li SH, Fu J, Watkins DN, Srivastava RK, Shankar S, Sulforaphane regulates self-renewal of pancreatic cancer stem cells through the modulation of Sonic hedgehog-GLI pathway, Mol. Cell. Biochem 373 (1–2) (2013) 217–227, 10.1007/s11010-012-1493-6. [DOI] [PubMed] [Google Scholar]
- [59].Juge N, Mithen RF, Traka M, Molecular basis for chemoprevention by sulforaphane: a comprehensive review, Cell. Mol. Life Sci 64 (9) (2007) 1105–1127, 10.1007/s00018-007-6484-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Hahm E-R, Singh SV, Sulforaphane inhibits constitutive and interleukin-6–induced activation of signal transducer and activator of transcription 3 in prostate cancer cells, Cancer Prev. Res 3 (4) (2010) 484–494, 10.1158/1940-6207.capr-09-0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Li Y, Karagoz GE, Seo YH, Zhang T, Jiang Y, Yu Y, et al. , Sulforaphane inhibits pancreatic cancer through disrupting Hsp90-p50(Cdc37) complex and direct interactions with amino acids residues of Hsp90, J. Nutr. Biochem 23 (12) (2012) 1617–1626, 10.1016/j.jnutbio.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Hung CN, Huang HP, Wang CJ, Liu KL, Lii CK, Sulforaphane inhibits TNF-alpha-induced adhesion molecule expression through the Rho A/ROCK/NF-kappaB signaling pathway, J. Med. Food 17 (10) (2014) 1095–1102, 10.1089/jmf.2013.2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Garner JM, Fan M, Yang CH, Du Z, Sims M, Davidoff AM, et al. , Constitutive activation of signal transducer and activator of transcription 3 (STAT3) and nuclear factor kappaB signaling in glioblastoma cancer stem cells regulates the Notch pathway, J. Biol. Chem 288 (36) (2013) 26167–26176, 10.1074/jbc.M113.477950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Song L, Liu L, Wu Z, Li Y, Ying Z, Lin C, et al. , TGF-beta induces miR-182 to sustain NF-kappaB activation in glioma subsets, J. Clin. Investig 122 (10) (2012) 3563–3578, 10.1172/JCI62339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Kallifatidis G, Rausch V, Baumann B, Apel A, Beckermann BM, Groth A, et al. , Sulforaphane targets pancreatic tumour-initiating cells by NF-kappaB-induced antiapoptotic signalling, Gut 58 (7) (2009) 949–963 doi: gut.2008.149039 [pii] 10.1136/gut.2008.149039. [DOI] [PubMed] [Google Scholar]
- [66].Yardley DA, Drug resistance and the role of combination chemotherapy in improving patient outcomes, Int. J. Breast Cancer 2013 (2013) 137414, 10.1155/2013/137414. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.