

Abstract
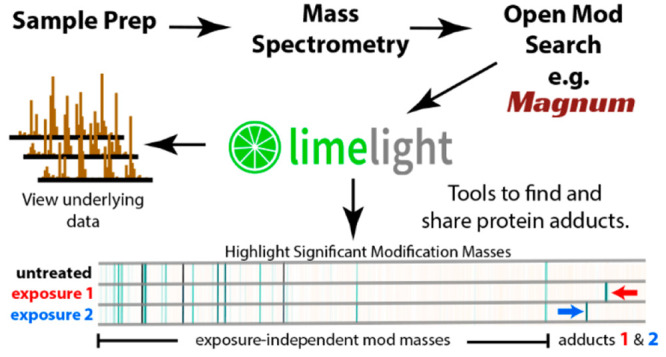
Drugs are often metabolized to reactive intermediates that form protein adducts. Adducts can inhibit protein activity, elicit immune responses, and cause life-threatening adverse drug reactions. The masses of reactive metabolites are frequently unknown, rendering traditional mass spectrometry-based proteomics approaches incapable of adduct identification. Here, we present Magnum, an open-mass search algorithm optimized for adduct identification, and Limelight, a web-based data processing package for analysis and visualization of data from all existing algorithms. Limelight incorporates tools for sample comparisons and xenobiotic-adduct discovery. We validate our tools with three drug/protein combinations and apply our label-free workflow to identify novel xenobiotic-protein adducts in CYP3A4. Our new methods and software enable accurate identification of xenobiotic-protein adducts with no prior knowledge of adduct masses or protein targets. Magnum outperforms existing label-free tools in xenobiotic-protein adduct discovery, while Limelight fulfills a major need in the rapidly developing field of open-mass searching, which until now lacked comprehensive data visualization tools.
Humans are constantly exposed to chemicals from their environment. Protein adducts result from covalent modification by xenobiotics, or their metabolites, and can also cause unintended toxicities and adverse drug reactions (ADRs).1−3 Adduct identification improves understanding of the mechanisms of ADRs and enables design of structural modifications to prevent them. Current identification methods, using radiolabeled compounds, trapping agents, immunological detection and mass spectrometry-based methods for detecting xenobiotic-protein adducts are labor-intensive and cannot sensitively measure the presence, abundance, and localization of protein adducts across a range of proteins without prior knowledge.1,4,5 Recent proteomics methods allow identification of unknown protein adducts but require isotopic labeling of test compounds6,7 or a complex combination of data-processing methods to reduce false discoveries and identify adducted peptides.8
Traditional database search algorithms9 search MS/MS spectra against protein sequences to identify the peptides and proteins that produced them. Known modifications can be identified if their masses are predefined. This is not possible for xenobiotic-protein adducts if their chemical composition is unknown. “Open-mass” search strategies10−18 solve this issue by allowing observed peptide masses to differ from identified peptide masses, returning mass differences as modifications of the identified peptides. This has allowed peptide spectrum matches (PSMs) to be made from a large proportion of previously unassigned spectra in shotgun proteomics data.10−18 However, few algorithms have been designed to identify xenobiotic-protein adducts that are typically low abundance and specific to the drug and treatment, and there are no graphical software tools to analyze and visualize data from open-mass searches.
Here we present Magnum, a purpose-built xenobiotic-protein adduct discovery algorithm, and Limelight, a web-based open modification analysis platform that rapidly highlights protein adducts that result from a specific treatment in a background of unrelated modifications. Limelight can combine and compare data from different pipelines empowering users to find the best tool or combination of tools for their specific application.
We validate our new tools using three drug/protein combinations and compare results from Magnum to other open-mass search tools. We apply our workflow to identify novel xenobiotic-protein adducts in the P450 enzyme CYP3A4 resulting from exposure to raloxifene. Our software and workflow enable rapid and accurate identification of novel xenobiotic–protein adducts with no prior knowledge of adduct masses or protein targets. These tools provide a highly accelerated and statistically rigorous label-free workflow for the discovery and characterization of xenobiotic-protein adducts and can be incorporated into drug discovery pipelines and environmental toxicology screening.
Materials and Methods
Reagents and Drug Incubations
The chemicals and reagents used in this study plus details of drug incubations are described in the Supplementary Methods section of the Supporting Information (SI).
Sample Preparation
β-lactam antibiotics and HSA: Aliquots (30 μL) of control or drug treated HSA (0.5 mg/mL) were reduced by adding 10 mM DTT, final concentration, and incubating at 37 °C for 30 min. Samples were alkylated with 16 mM iodoacetamide at room temp for 20 min in the dark. Tryptic digestion was done at 1:15 (enzyme:substrate) for 6 h at 37 °C in an Eppendorf Thermomixer with shaking (1000 rpm) prior to acidification with 250 mM HCl (final concentration). Samples were centrifuged at max speed in a benchtop microfuge for 10 min and supernatant transferred to autosampler vials and stored at −80 °C. Human plasma was prepared, treated, and digested as described in Supplementary Methods.
Raloxifene and CYP3A4: Aliquots (100 μL) of control or drug treated CYP3A4 incubation reaction mixture (63 μg total protein) were prepared as described but digested for 4 h. A second set of digests were performed as described in Supplementary Methods and labeled “extra digest” in results. After digestion, solid phase extraction was done on all CYP3A4 samples using Oasis MCX cartridges (see Supplementary Methods for details).
Mass Spectrometry
Sample digests (2 μL ∼ 1 μg) were loaded onto a 150 μm Kasil fritted trap packed with 2 cm of ReprosilPur C18AQ (3 μm bead diameter, Dr. Maisch) at a flow rate of 2 μL per min. Separation used a self-packed 75 μm i.d. 30 cm column. Peptides were eluted at 0.25 μL/min using a standard or higher concentration (labeled “highB” in results) acetonitrile gradient. A QExactive HF or Exploris 480 (Thermo Fisher Scientific) was used to perform MS in data dependent mode.
Data Processing
Acquired spectra were converted into mzML (for input to all algorithms except MODa) or mzXML (for input to MODa) using ProteoWizard’s msConvert.19 Proteins present in the samples were identified using Comet20 by standard closed searching against the entire human or E. coli proteomes, and smaller databases were made for subsequent open searching consisting only of proteins identified in initial comet searches by at least three peptides with a Percolator21 assigned q-value of ≤0.01. Decoy databases consisted of the corresponding set of reversed protein sequences and were provided to algorithms requiring pregenerated decoy sequences. All data are filtered at a false discovery rate (FDR) of 1% unless otherwise stated. Detailed procedures and parameters for each algorithm can be found in Supplementary Methods.
Native search results from all pipelines were converted to Limelight XML prior to uploading to the Limelight web application. We have written converters for many pipelines including Comet,20 Percolator,21 the Trans-Proteomic Pipeline (TPP),22 Crux,23 MSFragger,11 open-pFind,12 Comet-PTM,14 MetaMorpheus,17 MODa,13 TagGraph,18 and Magnum (this paper). A current list of converters is available on our documentation Web site: https://limelight-ms.readthedocs.io/. Further details can be found in Supplementary Note 3.
Quantification of CYP3A4 and P450-Reductase Adducts
Peptides were quantified using Skyline as previously described.24,25 Full details can be found in Supplementary Methods, processed data plus Limelight links can be found in Supplementary File 2, Sheet 3, and a complete interactive Skyline session is available on Panorama (https://panoramaweb.org/CYP3A4-raloxifene.url).
Software Availability
Magnum is written in C++ and is open source and freely available at http://magnum-ms.org. Full source code plus precompiled Magnum binaries are available for Windows and Linux. Docker images for the Magnum/Percolator/Limelight pipeline and associated documentation can be found at https://limelight-ms.readthedocs.io/en/latest/tutorials/magnum-pipeline.html. Magnum outputs results either as simple tab-delimited text or PepXML26 format for potential use with existing software supporting this format. For further details of Magnum see Supplementary Note 1.
Limelight is written in Java and TypeScript and is open source and freely available at https://limelight-ms.org/. Limelight source code plus preconfigured Docker containers are provided for running Limelight and Limelight XML converters. Extensive documentation is at https://limelight-ms.readthedocs.io/. Users not able to run their own Limelight installation may use a generally available installation at https://use.limelight-ms.org/. For full details of Limelight see Supplementary Note 3.
Data Availability
All raw and processed data discussed in this paper are available via Limelight at https://limelight.yeastrc.org/limelight/p/adduct-discovery. In addition, complete search algorithm configuration files, fasta search databases, raw search output, and raw MS data files were deposited to the ProteomeXchange Consortium via the PRIDE27 partner repository with the data set identifier PXD025019. Full Skyline quantification of CYP3A4/raloxifene was deposited to the ProteomeXchange Consortium via Panorama Public28 and is available at https://panoramaweb.org/CYP3A4-raloxifene.url with the data set identifier PXD024932.
Results
Development of Magnum
Magnum was developed to analyze MS/MS spectra to identify peptide sequences modified by adducts of unknown mass. The approach taken in Magnum is similar to cross-linked peptide identification in Kojak and makes use of the same spectral processing, analytical work-flow, and Xcorr scoring function.29 However, the algorithms are mutually exclusive. In Kojak search results contain two peptide sequences that sum together to produce the observed mass, whereas Magnum returns only one peptide sequence plus a modification mass equal to the mass unexplained by the predicted peptide. A detailed description the Magnum algorithm is presented in Supplementary Note 1.
Magnum incorporates several features that improve its ability to identify protein adducts. First, Magnum calculates and scores an adduct-modified MS/MS ion series. Some open search algorithms disregard adduct-modified fragment ions leading to a loss of sensitivity and preventing adduct localization.10,15−17 Magnum handles both MS-labile and stable modifications by including all possible scenarios in its search space for every spectrum. Adduct identification with or without adduct localization is therefore possible (see Supplemental Note 1 for details). Second, Magnum allows the restriction of adduct localization to specific amino acids during searching. This is useful if the reactivity of a xenobiotic is known or hypothesized based on its chemistry or through detection of thiol conjugates with reactive intermediates.1 Increased search sensitivity and statistical power can be gained by restricting the open modification search space to specific amino acids.30 Third, Magnum can flag MS/MS spectra containing diagnostic reporter ions. Some xenobiotics and endogenous post-translational modifications (PTMs) fragment predictably in the mass spectrometer producing reporter ions,31−33 the masses of which are constant regardless of the peptide to which the adduct was attached. If present, Magnum records this information in the PSM. Fourth, Magnum considers user defined peptide modifications and open modifications. This allows separation of masses due to defined versus open modifications and is important if both types of modification exist on a single peptide.
The product of a Magnum analysis is a set of PSMs for as many input spectra as possible, plus a set of metrics that can be used as input in a variety of PSM-validation21,34 algorithms.
Evaluation of Open Modification Search Tools for Xenobiotic-Protein Adduct Discovery
Open-mass searches have greater potential for false positive identifications than closed searches because discrepancies between theoretical and observed peptide masses are interpreted as open modifications. While enabling identification of undefined modifications, this can lead to incorrect identifications not otherwise possible. Additionally, large differences in unmodified and modified peptide search spaces and the small proportion of PSMs representing specific xenobiotic adducts of interest make accurate error estimation difficult35 (Supplemental Note 1). We therefore created a gold standard (true positive) data set to evaluate the accuracy and sensitivity of Magnum in identifying xenobiotic-protein adducts. We compared Magnum, which was designed specifically for xenobiotic-protein adduct detection, to several previously published open search algorithms.
For our gold standard data set, we acquired high resolution LC-MS/MS data of human serum albumin (HSA) exposed to the β-lactam antibiotics dicloxacillin and flucloxacillin. A significant portion of previous research identifying protein adducts from environmental exposures has focused on HSA36 as it is the most abundant protein in plasma and forms adducts with numerous xenobiotics.4 The β-lactam antibiotics dicloxacillin and flucloxacillin were chosen as there are published MS characterizations of their clinically significant adducts.31,37
Our gold standard data set consisted of 2979 MS/MS spectra extracted from 307 652 scans acquired from two dicloxacillin- and two flucloxacillin-treated HSA samples. Each of the 2979 spectra was definitively determined to result from a peptide containing one 469 Da (dicloxacillin) or 453 Da (flucloxacillin) adduct using the methods described in Supplementary Note 2. The four full data sets (307 652 MS/MS scans) were searched with 7 algorithms. Open modification masses associated with each PSM were extracted for the 2979 gold standard spectra. A precision/recall analysis was performed where precision was defined as the fraction of all answers that are correct (i.e., number of correct answers/total number of answers) and recall as the fraction of total possible correct answers identified (i.e., number of correct answers/number scans tested [2979]). An answer within ±3 Da of the known modification mass was considered correct, allowing for incorrect monoisotopic mass assignments,38 which in open searching are compensated for by changes in open modification masses. Both Magnum and MSFragger11 had excellent precision: >0.9 at 1% FDR (false discovery rate) (Figure 1). Recall of Magnum was better than all other algorithms. MSFragger, comet with a 500 Da wide precursor window, and MetaMorpheus17 performed next best. Recall of open-pFind12 was close to zero as it does not perform a truly unrestricted search, but rather a multinotch search,17 allowing only delta masses present in UniMod39 to be used as modification masses. As dicloxacillin and flucloxacillin adducts are not present in Unimod, open-pFind cannot identify them and returned results for only 9 of the 2979 scans (SI Table S6).
Figure 1.
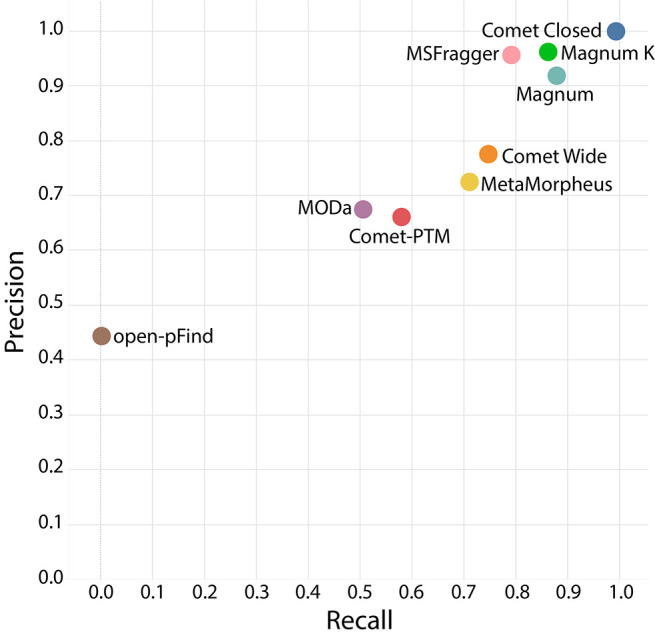
Evaluation of open modification search tools for xenobiotic-protein adduct discovery. Precision recall plot of adduct masses at 1% FDR by 7 open search algorithms. Closed comet searches using defined modifications were included as a positive control. Magnum was run allowing for open-masses on any amino acid (Magnum) or restricted to lysine only (Magnum K), the previously published residue modified by dicloxacillin and flucloxacillin adducts.31,37
Except for open-pFind, algorithms having a low recall did so mainly due to returning incorrect modification masses rather than by not returning a result for that scan. All algorithms except open-pFind returned over 2200 total answers at 1% FDR. These were distributed between correct and incorrect masses (SI Figure S6).
An analysis of all open-mass modifications returned by each algorithm within ±3 Da of the known correct modification masses shows Magnum had the most accurate (62–63%) monoisotopic mass assignment of the algorithms tested (SI Figure S7). MetaMorpheus and MSFragger were next best, and respectively assigned 53% and 49% of PSMs the correct monoisotopic mass. Like many pharmaceutical drugs, dicloxacillin and flucloxacillin contain chlorine, confounding monoisotopic mass assignment due to its unusual isotopic composition. To overcome these complexities, Magnum incorporates code that reduces peptide isotope distributions to a single monoisotopic mass (Supplementary Note 1).
Further validation of Magnum was done by comparing open modification masses returned by Magnum and MSFragger with those returned by closed comet searching of previously published phosphopeptide enriched data.40 Both Magnum and MSFragger results were almost exclusively (∼98%) a subset of closed search results indicating excellent accuracy by both algorithms (SI Figure S12). Magnum returned more PSMs (3101) than MSFragger (1760) at the same calculated FDR. Overall, our analyses showed Magnum accurately and sensitively identifies xenobiotic-protein adducts.
Development of Limelight
Mass spectrometry-based proteomics data is complex and open modification data even more so. Each search algorithm uses different scoring metrics and outputs results in different formats. PSM-validation tools21,34 are often used to assign statistical confidence to results, and add another layer of metrics to the data. If adduct data are to be assessable for answering specific questions, a simple graphical interface is needed. Limelight is a web application built to analyze, visualize, and share bottom-up MS proteomics data (Figure 2). It provides a generalized platform designed to support any MS database search pipeline. Data are displayed on a global level in protein, peptide, or modification centric views, which can be filtered on multiple criteria including any or all scores from the search algorithm and PSM-validation tools, and that provide access to all underlying raw data and associated metrics from every part of the analysis. Annotated spectra, including ions modified by open-masses where applicable, are viewable using a built in spectrum viewer.41 Data are unified and can be queried, viewed, and compared across multiple experiments and disparate software pipelines allowing the strengths of different algorithms to be leveraged.
Figure 2.
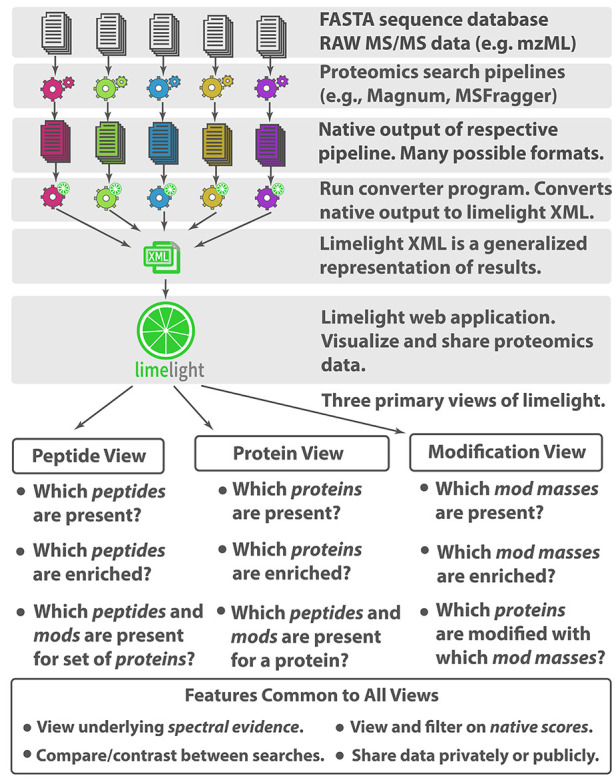
Limelight. A web-based application to interrogate, analyze, and visualize mass spectrometry proteomics results.
Limelight incorporates novel features designed for adduct and PTM analysis. Mass modifications identified in peptides may be viewed, analyzed, and visualized independently from the peptides or proteins in which they were identified and regardless of whether those modifications were localized or not. If desired, this allows interesting adduct masses to be identified and examined before segregation into protein, peptide, or residue level groups. To highlight exposure related adducts, we developed a suite of tools within Limelight for visualizing and statistically comparing modification masses between different experiments. These functions are used below and described in depth in Supplementary Note 3.
Development and Validation of Adduct Discovery Pipeline
Open search results contain many modification masses not related to exposures or treatment conditions. Numerous open-mass modifications are identified even in untreated purified HSA (SI Figure S14, Supplementary Note 4). We therefore designed an experimental workflow to allow discovery of protein adducts resulting specifically from exposure to xenobiotics (Figure 3). Our strategy was to produce, analyze, and compare both untreated and xenobiotic treated samples. This strategy is similar to that behind heavy/light isotope labeling: namely, that the difference between the two sets of samples allows adduct masses specific to the treatment of interest to be distinguished within a background of unrelated modification masses. We tested our label-free workflow using unexposed, dicloxacillin, and flucloxacillin exposed HSA.
Figure 3.

Adduct identification and visualization workflow using dicloxacillin or flucloxacillin exposed versus untreated HSA as an example. (a) Raw MS data was searched using Magnum and (b) resulting PSMs were analyzed by Limelight. Data visualization includes: (c) a heatmap of Global z-score for all modification masses across all samples highlighting modification masses enriched in the treated samples (red arrows); (d) results of statistical test of proportions on all modification masses automatically pinpoints dicloxacillin (469 Da) and (e) flucloxacillin (453 Da) adducts (red arrows). See Supplementary Note 3 for details. All results shown have a Percolator calculated PSM level q ≤ 0.01. Exact modification masses were binned into 1 Da bins by Limelight prior to analysis. Live view for (c) can be found on Limelight here: https://limelight.yeastrc.org/limelight/go/x1h2QRXqrE
Open-mass searching was performed using Magnum and PSMs were imported into Limelight for downstream analysis. Using Limelight, a two-tailed test of proportions comparing untreated with dicloxacillin or flucloxacillin treated HSA, easily picked out exposure specific modifications enriched in the treated samples (Figure 3d,e). Previously published studies determined that dicloxacillin and flucloxacillin produce 469 and 453 Da adduct modifications, respectively, on HSA lysine residues.31,37 These studies relied on MS analysis of unadducted antibiotics, combined with trapping experiments using N-acetyl-cysteine and N-acetyl-lysine plus extensive targeted MS experiments. Using our untargeted method, comparisons of dicloxacillin or flucloxacillin treated versus untreated samples resulted in 469 or 453 Da, being the most significantly enriched mass for their respective treatment with no prior knowledge or experimentation. These data show a two-tailed test of proportions comparing treated versus untreated samples, is effective in highlighting exposure specific adducts from PSMs generated by Magnum.
Limelight was designed to support output from any proteomics pipeline, and we performed the same analysis using PSMs from six additional open-mass search algorithms. In all cases Magnum identified the most treatment related PSMs as well as resulting in the largest z-score for the correct mass compared to other algorithms (SI Figure S17, Supplementary Note 4). Overall, Magnum identified 41 unique dicloxacillin- and 55 unique flucloxacillin-adducted HSA peptides of which 90% contained 1 or more reporter ions resulting from adduct fragmentation (SI Table S7). Manual validation using Limelight’s built-in spectrum viewer further confirmed Magnum’s automated results and example annotated spectra are shown in SI Figure S18 (dicloxacillin) and SI Figure S19 (flucloxacillin). These data demonstrate our software and workflow can correctly and automatically identify and highlight drug protein adducts and that Magnum is the most sensitive algorithm of those tested.
Identification of Penicillin Adducts in Human Plasma
To challenge our pipeline with a more complex sample we prepared unexposed and dicloxacillin exposed human plasma. Closed-mass comet searches identified 334 unique proteins in these samples. We performed open-mass searching using Magnum, and PSMs were imported into Limelight and analyzed as described above. As with purified HSA, Limelight’s two-tailed test of proportions, found 469 Da as the top scoring mass enriched in the treated sample (SI Figure S20a,b).
For comparison of modification masses observed across samples Limelight rounds all modifications to the nearest Da. We analyzed the distribution of observed open modification masses in PSMs within Limelight’s 469 Da bin. Observed masses in the treated sample peak at 469.01 Da, the exact mass of previously characterized dicloxacillin adducts. Observed masses in the unexposed sample do not result in any open-mass identifications in this region (SI Figure S20c). Overall, Magnum identified 15 unique peptides modified by 469 Da of which 80% were in HSA. 91% of identified PSMs contained all three reporter ions resulting from dicloxacillin fragmentation (SI Table S8). In addition to identifying dicloxacillin adducts in HSA our analysis found exposure specific adducts in transthyretin (TTHY), and Apolipoprotein A-II (APOA2). These data show our method can be successfully applied to complex protein samples. It should be noted that reduced depth of coverage is expected with increasing sample complexity. This is a function of individual peptides being a smaller fraction of total peptides as the number of different peptides in the sample increases. For highly complex samples, fractionation and enrichment techniques will improve sensitivity and depth of coverage.42
Magnum and Limelight Identify Novel Raloxifene Adducts in CYP3A4 and P450-Reductase
Most xenobiotics that ultimately form adducts undergo metabolic activation by cytochrome P450 (CYP) enzymes to form short-lived reactive intermediates that are difficult to predict and identify. Raloxifene, a drug commonly used to treat osteoporosis in postmenopausal women, is a mechanism-based inhibitor of CYP3A4.43 Raloxifene metabolism by CYP3A4 produces several electrophilic species (SI Figure S21), and 471.15 Da adducts on cysteine 239 and tyrosine 79 were previously identified.44,45 Past studies identifying adducts in CYP3A444−47 required extensive experimentation involving multiple techniques including (1) adduct trapping with GSH, N-acetyl cysteine, N-acetyl lysine, and potassium cyanide to determine the masses of likely adducts; (2) radiolabeled drugs combined with LC-radiochromatography in-line with MS; (3) whole-protein MS combined with heavy and light labeled drugs; (4) targeted MS experiments using predicted masses of adducts and/or (5) closed MS/MS database searching using predefined adduct masses previously determined by methods 1–4.
We incubated raloxifene in vitro with CYP3A4 plus P450-reductase and NADPH. LC-MS/MS data of exposed and untreated samples were acquired, searched using Magnum and analyzed using Limelight (Supplementary Note 6). A two-tailed test of proportions identified 471 Da as the most significantly (p = 0) enriched modification mass in raloxifene treated samples (SI Table S10). We searched these data using 6 additional open-mass search algorithms, however none proved as sensitive as Magnum (SI Figure S22).
A fully unrestricted Magnum search (modifications allowed on any amino acid) found 146 PSMs containing a 471 Da modification mass in treated samples versus 14 in untreated samples (10:1 ratio) indicating that >90% of 471 Da identifications made by Magnum in raloxifene treated samples are exposure-specific adducts. Manual evaluation, using Limelight’s built-in spectrum viewer, of all 146 PSMs revealed that close to 90% of 471 Da adducts were on cysteine, tryptophan or tyrosine (Supplementary File 2, Sheet 2). The only two previously identified adducts were on a cysteine and a tyrosine, and raloxifene adducts have previously been thought to occur primarily on cysteine. Cysteine, tryptophan and tyrosine contain nucleophilic moieties (the thiol group of cysteine, the aromatic nitrogen of tryptophan and the phenolic group of tyrosine) which can react with electrophilic sites of the reactive raloxifene metabolite to form adducts. We further improved search sensitivity by using Magnum’s ability to restrict open-mass modifications to specific amino acids: searches were performed restricting open-mass modifications to cysteine only, or, to cysteine (C), tryptophan (W), and tyrosine (Y) collectively. Cysteine-restricted searches yielded fewer (67) PSMs containing 471 Da adduct masses confirming the presence of adducts on residues other than cysteine. CWY-restricted searches resulted in more 471 Da PSMs in treated samples (157), and less in untreated samples (3), than any other search (a 52:1 ratio, SI Figure S22).
Extracted ion chromatograms (XICs) were produced and quantified in Skyline24,25 and showed treatment specific signal for all 471 Da modified peptides identified by Magnum as exclusively in treated samples (SI Figure S23). We searched an additional 6 untreated and 6 raloxifene treated CYP3A4 samples using CWY-restricted Magnum resulting in a total of 443 PSMs containing a 471 Da modification in CYP3A4 and 91 PSMs in P450-reductase, with just 7 total PSMs showing this mass in the untreated samples (a 76:1 ratio, Supplementary File 2, Sheet 5). These PSMs mapped to 12 distinct amino acids in CYP3A4 and 11 in P450-reductase (Figure 4 and SI Figure S30) and 22/23 represent novel raloxifene-protein adducts.
Figure 4.

Identification of novel raloxifene adducts in CYP3A4. (a) Magnum identifies multiple 471 Da protein adducts in CYP3A4 after exposure to raloxifene. Adducted residues are mapped to defined regions of CYP3A4.50 (b) Observed 471 Da modifications are shown on the structure of CYP3A4 as magenta spheres. Results were identified by ≥3 PSMs, 1% FDR. Limelight view: https://limelight.yeastrc.org/limelight/go/0UjwIJNz45 (c) Extracted ion chromatograms (XICs) of 2+, 3+, and 4+ precursor ions corresponding to CYP3A4 residue W126 elute as four distinct chromatographic peaks likely representing regioisomers resulting from the different positions43,48 in the raloxifene metabolite, diquinone methide, that are subject to nucleophilic attack (inset structure, red arrows). Unexposed control XICs show no signal (inset box, left).
We analyzed the distribution of observed open modification masses in PSMs within Limelight’s 471 Da bin. Observed masses (536 PSMs) in the treated sample peak at 471.15 Da, the exact mass of previously characterized raloxifene diquinone methide adduct. Only seven PSMs were observed in the 471 Da bin in unexposed samples (SI Figure S31).
Interestingly, W126 was associated with four distinct chromatographically separated peaks (Figure 4c) that might represent regioisomers resulting from the different positions in the raloxifene metabolite subject to nucleophilic attack (Figure 4c, red arrows).43,48 Several other 471 Da modified peptides showed multiple distinguishable chromatographic peaks (e.g., SI Figures S27 and S28) and represent the first evidence for regioisomers in native P450 enzymes.
The identification of multiple novel raloxifene-protein adducts within both CYP3A4 and P450-reductase demonstrates our methodology and software provide greatly expanded power to better understand the structure/function and ligand interactions of catalytic membrane proteins such as cytochrome P450s. Past biochemical studies have typically identified single residues in CYP3A4 that can be modified by xenobiotics despite the expected exposure of multiple nucleophilic residues to reactive metabolites formed by CYP3A4.43,46,47,49 Additionally, previous studies required extensive experimentation to define adducts while the methods and tools presented here required no prior knowledge or experimentation.
Conclusions
Open-mass search algorithms have become increasingly important in the past decade and can now identify over 50% of spectra invisible to traditional search methods.10 Previous open-mass search analyses of proteomics data sets have found hundreds of yet-to-be-identified modifications in untreated protein samples,10−13,17,18 but until now there were few pipelines dedicated to discovering exposure related xenobiotic-protein adducts in the background of modifications identified by open searching. The pipeline presented here allows the use of standard shotgun proteomics to identify xenobiotic-protein adducts in an automated manner with no prior knowledge of the adduct mass or the adducted peptides.
Magnum is optimized to detect adduct modified peptides and outperforms existing label-free open-search based methods in xenobiotic-protein adduct discovery. Limelight provides specific tools to automatically highlight exposure specific modifications while also fulfilling a major need in the rapidly developing field of open-mass searching, which until now has had no comprehensive data visualization tools. Users are thus empowered to utilize the best algorithm, or combination of algorithms to fulfill their needs.
Acknowledgments
We thank Jimmy Eng and David D. Shteynberg for help with comet-PTM and PTMProphet, respectively. Purified recombinant human CYP3A4 and the liposome stock were gifts from Dr. William Atkins. This research was supported by the National Institutes of Health, National Institute of General Medical Sciences under Award P41GM103533 (to M.J.M.), and National Institute On Aging under Award Number U19AG023122 (to R.L.M.). This work was supported in part by the University of Washington’s Proteomics Resource (UWPR95794).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.1c04101.
File 1: Additional experimental details, in depth data analysis, supplementary figures and tables, plus a full description of Magnum and Limelight (PDF)
File 2: Annotated spectra of all results from an unrestricted Magnum search of raloxifene treated CYP3A4. Raw data and quantification of raloxifene treated and untreated CYP3A4 samples searched with Magnum allowing for adducts on C, W, or Y residues only. Results from two-tailed test of proportions on untreated versus penicillin treated HSA using PSMs from seven open search algorithms. Results from two-tailed test of proportions on untreated versus raloxifene treated CYP3A4 using PSMs from seven open search algorithms. PSMs identified by Magnum from untreated and dicloxacillin treated human plasma samples (XLSX)
File 3: PyMOL sessions showing observed 471 Da modifications on the structures of CYP3A4 and P450-reductase (ZIP)
Author Contributions
A.Z., G.Z., M.R., M.R.H., and N.I. conceived the experiments. G.Z. performed drug–protein incubations. A.Z. carried out the MS experiments and MS data analysis. M.R.H. developed Magnum. M.R. and D.J. developed Limelight. The manuscript was written by A.Z. with contributions from M.R., M.R.H., N.I., and G.Z. All authors discussed the results and commented on the manuscript. All authors have given approval to the final version of the manuscript.
Author Contributions
⊥ M.R. and M.H. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Gan J.; Zhang H.; Humphreys W. G. Drug-Protein Adducts: Chemistry, Mechanisms of Toxicity, and Methods of Characterization. Chem. Res. Toxicol. 2016, 29 (12), 2040–2057. 10.1021/acs.chemrestox.6b00274. [DOI] [PubMed] [Google Scholar]
- Whitby L. R.; Obach R. S.; Simon G. M.; Hayward M. M.; Cravatt B. F. Quantitative Chemical Proteomic Profiling of the in Vivo Targets of Reactive Drug Metabolites. ACS Chem. Biol. 2017, 12 (8), 2040–2050. 10.1021/acschembio.7b00346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans D. C.; Watt A. P.; Nicoll-Griffith D. A.; Baillie T. A. Drug–Protein Adducts: An Industry Perspective on Minimizing the Potential for Drug Bioactivation in Drug Discovery and Development. Chem. Res. Toxicol. 2004, 17 (1), 3–16. 10.1021/tx034170b. [DOI] [PubMed] [Google Scholar]
- Sabbioni G.; Turesky R. J. Biomonitoring Human Albumin Adducts: The Past, the Present, and the Future. Chem. Res. Toxicol. 2017, 30 (1), 332–366. 10.1021/acs.chemrestox.6b00366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tailor A.; Waddington J. C.; Meng X.; Park B. K. Mass Spectrometric and Functional Aspects of Drug-Protein Conjugation. Chem. Res. Toxicol. 2016, 29 (12), 1912–1935. 10.1021/acs.chemrestox.6b00147. [DOI] [PubMed] [Google Scholar]
- Leeming M. G.; Donald W. A.; O’Hair R. A. J. Nontargeted Identification of Reactive Metabolite Protein Adducts. Anal. Chem. 2017, 89 (11), 5748–5756. 10.1021/acs.analchem.6b04604. [DOI] [PubMed] [Google Scholar]
- Wang H.; Leeming M. G.; Cochran B. J.; Hook J. M.; Ho J.; Nguyen G. T. H.; Zhong L.; Supuran C. T.; Donald W. A. Nontargeted Identification of Plasma Proteins O-, N-, and S-Transmethylated by O-Methyl Organophosphates. Anal. Chem. 2020, 92 (23), 15420–15428. 10.1021/acs.analchem.0c03077. [DOI] [PubMed] [Google Scholar]
- Golizeh M.; Leblanc A.; Sleno L. Identification of Acetaminophen Adducts of Rat Liver Microsomal Proteins Using 2D-LC-MS/MS. Chem. Res. Toxicol. 2015, 28 (11), 2142–2150. 10.1021/acs.chemrestox.5b00317. [DOI] [PubMed] [Google Scholar]
- Eng J. K.; Searle B. C.; Clauser K. R.; Tabb D. L. A Face in the Crowd: Recognizing Peptides Through Database Search. Mol. Cell. Proteomics 2011, 10, 1–9. 10.1074/mcp.R111.009522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chick J. M.; Kolippakkam D.; Nusinow D. P.; Zhai B.; Rad R.; Huttlin E. L.; Gygi S. P. A Mass-Tolerant Database Search Identifies a Large Proportion of Unassigned Spectra in Shotgun Proteomics as Modified Peptides. Nat. Biotechnol. 2015, 33 (7), 743–749. 10.1038/nbt.3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A. T.; Leprevost F. V.; Avtonomov D. M.; Mellacheruvu D.; Nesvizhskii A. I. MSFragger: Ultrafast and Comprehensive Peptide Identification in Mass Spectrometry-Based Proteomics. Nat. Methods 2017, 14 (5), 513–520. 10.1038/nmeth.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi H.; Liu C.; Yang H.; Zeng W. F.; Wu L.; Zhou W. J.; Wang R. M.; Niu X. N.; Ding Y. H.; Zhang Y.; Wang Z. W.; Chen Z. L.; Sun R. X.; Liu T.; Tan G. M.; Dong M. Q.; Xu P.; Zhang P. H.; He S. M. Comprehensive Identification of Peptides in Tandem Mass Spectra Using an Efficient Open Search Engine. Nat. Biotechnol. 2018, 36 (11), 1059–1066. 10.1038/nbt.4236. [DOI] [PubMed] [Google Scholar]
- Na S.; Bandeira N.; Paek E. Fast Multi-Blind Modification Search through Tandem Mass Spectrometry. Mol. Cell. Proteomics 2012, 11 (4), M111.010199. 10.1074/mcp.M111.010199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagwan N.; Bonzon-kulichenko E.; Calvo E.; Lechuga-vieco A. V.; Michalakopoulos S.; Trevisan-Herraz M.; Ezkurdia I.; Rodrıiguez J.; Magni R.; Latorre-pellicer A.; Enrı A.; Enriquez J.; Vazquez J. Comprehensive Quantification of the Modified Proteome Reveals Oxidative Heart Damage in Mitochondrial Heteroplasmy. Cell Rep. 2018, 23, 3685–3697. 10.1016/j.celrep.2018.05.080. [DOI] [PubMed] [Google Scholar]
- Li Q.; Shortreed M. R.; Wenger C. D.; Frey B. L.; Schaffer L. V.; Scalf M.; Smith L. M. Global Post-Translational Modification Discovery. J. Proteome Res. 2017, 16 (4), 1383–1390. 10.1021/acs.jproteome.6b00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David M.; Fertin G.; Rogniaux H.; Tessier D. SpecOMS: A Full Open Modification Search Method Performing All-to-All Spectra Comparisons within Minutes. J. Proteome Res. 2017, 16, 3030–3038. 10.1021/acs.jproteome.7b00308. [DOI] [PubMed] [Google Scholar]
- Solntsev S. K.; Shortreed M. R.; Frey B. L.; Smith L. M. Enhanced Global Post-Translational Modification Discovery with MetaMorpheus. J. Proteome Res. 2018, 17, 1844–1851. 10.1021/acs.jproteome.7b00873. [DOI] [PubMed] [Google Scholar]
- Devabhaktuni A.; Lin S.; Zhang L.; Swaminathan K.; Gonzalez C. G.; Olsson N.; Pearlman S. M.; Rawson K.; Elias J. E. TagGraph Reveals Vast Protein Modification Landscapes from Large Tandem Mass Spectrometry Datasets. Nat. Biotechnol. 2019, 37 (4), 469–479. 10.1038/s41587-019-0067-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers M. C.; Maclean B.; Burke R.; Amodei D.; Ruderman D. L.; Neumann S.; Gatto L.; Fischer B.; Pratt B.; Egertson J.; Hoff K.; Kessner D.; Tasman N.; Shulman N.; Frewen B.; Baker T. A.; Brusniak M. Y.; Paulse C.; Creasy D.; Flashner L.; Kani K.; Moulding C.; Seymour S. L.; Nuwaysir L. M.; Lefebvre B.; Kuhlmann F.; Roark J.; Rainer P.; Detlev S.; Hemenway T.; Huhmer A.; Langridge J.; Connolly B.; Chadick T.; Holly K.; Eckels J.; Deutsch E. W.; Moritz R. L.; Katz J. E.; Agus D. B.; MacCoss M.; Tabb D. L.; Mallick P. A Cross-Platform Toolkit for Mass Spectrometry and Proteomics. Nat. Biotechnol. 2012, 30 (10), 918–920. 10.1038/nbt.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng J. K.; Jahan T. A.; Hoopmann M. R. Comet: An Open-Source MS/MS Sequence Database Search Tool. Proteomics 2013, 13 (1), 22–24. 10.1002/pmic.201200439. [DOI] [PubMed] [Google Scholar]
- Kall L.; Canterbury J. D.; Weston J.; Noble W. S.; MacCoss M. J. Semi-Supervised Learning for Peptide Identification from Shotgun Proteomics Datasets. Nat. Methods 2007, 4 (11), 923–925. 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- Deutsch E. W.; Mendoza L.; Shteynberg D.; Farrah T.; Lam H.; Tasman N.; Sun Z.; Nilsson E.; Pratt B.; Prazen B.; Eng J. K.; Martin D. B.; Nesvizhskii A. I.; Aebersold R.. A Guided Tour of the Trans-Proteomic Pipeline. Proteomics. NIH Public Access March 2010; pp 1150–1159. 10.1002/pmic.200900375. [DOI] [PMC free article] [PubMed]
- McIlwain S.; Tamura K.; Kertesz-Farkas A.; Grant C. E.; Diament B.; Frewen B.; Howbert J. J.; Hoopmann M. R.; Käll L.; Eng J. K.; MacCoss M. J.; Noble W. S. Crux: Rapid Open Source Protein Tandem Mass Spectrometry Analysis. J. Proteome Res. 2014, 13 (10), 4488–4491. 10.1021/pr500741y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B.; Tomazela D. M.; Shulman N.; Chambers M.; Finney G. L.; Frewen B.; Kern R.; Tabb D. L.; Liebler D. C.; MacCoss M. J. Skyline: An Open Source Document Editor for Creating and Analyzing Targeted Proteomics Experiments. Bioinformatics 2010, 26 (7), 966–968. 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling B.; Rardin M. J.; MacLean B. X.; Zawadzka A. M.; Frewen B. E.; Cusack M. P.; Sorensen D. J.; Bereman M. S.; Jing E.; Wu C. C.; Verdin E.; Kahn C. R.; MacCoss M. J.; Gibson B. W. Platform-Independent and Label-Free Quantitation of Proteomic Data Using MS1 Extracted Ion Chromatograms in Skyline: Application to Protein Acetylation and Phosphorylation. Mol. Cell. Proteomics 2012, 11 (5), 202–214. 10.1074/mcp.M112.017707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A.; Eng J.; Zhang N.; Li X.-j.; Aebersold R.. A Uniform Proteomics MS/MS Analysis Platform Utilizing Open XML File Formats. Mol. Syst. Biol. 2005, 1. 10.1038/msb4100024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaíno J. A.; Côté R.; Reisinger F.; Foster J. M.; Mueller M.; Rameseder J.; Hermjakob H.; Martens L.. A Guide to the Proteomics Identifications Database Proteomics Data Repository. Proteomics. Wiley-Blackwell, September 2009; 4276–4283. 10.1002/pmic.200900402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V.; Eckels J.; Schilling B.; Ludwig C.; Jaffe J. D.; MacCoss M. J.; MacLean B. Panorama Public: A Public Repository for Quantitative Data Sets Processed in Skyline. Mol. Cell. Proteomics 2018, 17 (6), 1239–1244. 10.1074/mcp.RA117.000543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoopmann M. R.; Zelter A.; Johnson R. S.; Riffle M.; MacCoss M. J.; Davis T. N.; Moritz R. L. Kojak: Efficient Analysis of Chemically Cross-Linked Protein Complexes. J. Proteome Res. 2015, 14 (5), 2190–2198. 10.1021/pr501321h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble W. S. Mass Spectrometrists Should Search Only for Peptides They Care About. Nat. Methods 2015, 12 (7), 605–608. 10.1038/nmeth.3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins R. E.; Meng X.; Elliott V. L.; Kitteringham N. R.; Pirmohamed M.; Park B. K. Characterisation of Flucloxacillin and 5-Hydroxymethyl Flucloxacillin Haptenated HSA in Vitro and in Vivo.. Proteomics - Clin. Appl. 2009, 3 (6), 720–729. 10.1002/prca.200800222. [DOI] [PubMed] [Google Scholar]
- Ariza A.; Garzon D.; Abánades D. R.; de los Ríos V.; Vistoli G.; Torres M. J.; Carini M.; Aldini G.; Pérez-Sala D. Protein Haptenation by Amoxicillin: High Resolution Mass Spectrometry Analysis and Identification of Target Proteins in Serum. J. Proteomics 2012, 77, 504–520. 10.1016/j.jprot.2012.09.030. [DOI] [PubMed] [Google Scholar]
- Paul Zolg D.; Wilhelm M.; Schmidt T.; Médard G.; Zerweck J.; Knaute T.; Wenschuh H.; Reimer U.; Schnatbaum K.; Kuster B. Proteometools: Systematic Characterization of 21 Post-Translational Protein Modifications by Liquid Chromatography Tandem Mass Spectrometry (Lc-Ms/Ms) Using Synthetic Peptides. Mol. Cell. Proteomics 2018, 17 (9), 1850–1863. 10.1074/mcp.TIR118.000783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shteynberg D.; Deutsch E. W.; Lam H.; Eng J. K.; Sun Z.; Tasman N.; Mendoza L.; Moritz R. L.; Aebersold R.; Nesvizhskii A. I. IProphet: Multi-Level Integrative Analysis of Shotgun Proteomic Data Improves Peptide and Protein Identification Rates and Error Estimates. Mol. Cell. Proteomics 2011, 10 (12), 1–16. 10.1074/mcp.M111.007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi X.; Gong F.; Fu Y. Transfer Posterior Error Probability Estimation for Peptide Identification. BMC Bioinformatics 2020, 21 (1), 1–17. 10.1186/s12859-020-3485-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappaport S. M.; Li H.; Grigoryan H.; Funk W. E.; Williams E. R. Adductomics: Characterizing Exposures to Reactive Electrophiles. Toxicol. Lett. 2012, 213 (1), 83–90. 10.1016/j.toxlet.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker C. E.; Perkins J. R.; Tomer K. B.. Nanoscale Packed Capillary Liquid Chromatography- Electrospray Ionization Mass Spectrometry: Analysis of Penicillins and Cephems. J. Chromatogr. 1993, 616, 45–51 10.1016/0378-4347(93)80470-o. [DOI] [PubMed] [Google Scholar]
- Rad R.; Li J.; Mintseris J.; O’Connell J.; Gygi S. P.; Schweppe D. K.. Improved Monoisotopic Mass Estimation for Deeper Proteome Coverage. J. Proteome Res. 2021. 20591. 10.1021/acs.jproteome.0c00563. [DOI] [PubMed] [Google Scholar]
- Creasy D. M.; Cottrell J. S.. Unimod: Protein Modifications for Mass Spectrometry. Proteomics.; John Wiley & Sons, Ltd, June 1, 2004; pp 1534–1536. 10.1002/pmic.200300744. [DOI] [PubMed] [Google Scholar]
- Lawrence R. T.; Searle B. C.; Llovet A.; Villén J. Plug-and-Play Analysis of the Human Phosphoproteome by Targeted High-Resolution Mass Spectrometry. Nat. Methods 2016, 13 (5), 431–434. 10.1038/nmeth.3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V.; Eng J. K.; Maccoss M. J.; Riffle M. A Mass Spectrometry Proteomics Data Management Platform. Mol. Cell Proteomics 2012, 11 (9), 824–831. 10.1074/mcp.O111.015149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faca V.; Pitteri S. J.; Newcomb L.; Glukhova V.; Phanstiel D.; Krasnoselsky A.; Zhang Q.; Struthers J.; Wang H.; Eng J.; Fitzgibbon M.; McIntosh M.; Hanash S. Contribution of Protein Fractionation to Depth of Analysis of the Serum and Plasma Proteomes. J. Proteome Res. 2007, 6 (9), 3558–3565. 10.1021/pr070233q. [DOI] [PubMed] [Google Scholar]
- Chen Q.; Ngui J. S.; Doss G. A.; Cai X.; Wang R. W.; DiNinno F. P.; Blizzard T. A.; Hammond M. L.; Stearns R. A.; Evans D. C.; Baillie T. A.; Tang W. Cytochrome P450 3A4-Mediated Bioactivation of Raloxifene: Irreversible Enzyme Inhibition and Thiol Adduct Formation. Chem. Res. Toxicol. 2002, 15 (7), 907–914. 10.1021/tx0200109. [DOI] [PubMed] [Google Scholar]
- Baer B. R.; Wienkers L. C.; Rock D. A. Time-Dependent Inactivation of P450 3A4 by Raloxifene: Identification of Cys239 as the Site of Apoprotein Alkylation. Chem. Res. Toxicol. 2007, 20 (6), 954–964. 10.1021/tx700037e. [DOI] [PubMed] [Google Scholar]
- Yukinaga H.; Takami T.; Shioyama S. H.; Tozuka Z.; Masumoto H.; Okazaki O.; Sudo K. I. Identification of Cytochrome P450 3A4Modification Site with Reactive Metabolite Using Linear Ion Trap-Fourier Transform Mass Spectrometry. Chem. Res. Toxicol. 2007, 20 (10), 1373–1378. 10.1021/tx700165q. [DOI] [PubMed] [Google Scholar]
- Lin H. L.; Kenaan C.; Hollenberg P. F. Identification of the Residue in Human CYP3A4 That Is Covalently Modified by Bergamottin and the Reactive Intermediate That Contributes to the Grapefruit Juice Effect. Drug Metab. Dispos. 2012, 40 (5), 998–1006. 10.1124/dmd.112.044560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock B. M.; Hengel S. M.; Rock D. A.; Wienkers L. C.; Kunze K. L. Characterization of Ritonavir-Mediated Inactivation of Cytochrome P450 3A4. Mol. Pharmacol. 2014, 86 (6), 665–674. 10.1124/mol.114.094862. [DOI] [PubMed] [Google Scholar]
- Yu L.; Liu H.; Li W.; Zhang F.; Luckie C.; Van Breemen R. B.; Thatcher G. R. J.; Bolton J. L. Oxidation of Raloxifene to Quinoids: Potential Toxic Pathways via a Diquinone Methide and o-Quinones. Chem. Res. Toxicol. 2004, 17 (7), 879–888. 10.1021/tx0342722. [DOI] [PubMed] [Google Scholar]
- Wen B.; Lampe J. N.; Roberts A. G.; Atkins W. M.; David Rodrigues A.; Nelson S. D. Cysteine 98 in CYP3A4 Contributes to Conformational Integrity Required for P450 Interaction with CYP Reductase. Arch. Biochem. Biophys. 2006, 454 (1), 42–54. 10.1016/j.abb.2006.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkaidali L.; André F.; Moroy G.; Tangour B.; Maurel F.; Petitjean M. Four Major Channels Detected in the Cytochrome P450 3A4: A Step toward Understanding Its Multispecificity. Int. J. Mol. Sci. 2019, 20 (4), 987. 10.3390/ijms20040987. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw and processed data discussed in this paper are available via Limelight at https://limelight.yeastrc.org/limelight/p/adduct-discovery. In addition, complete search algorithm configuration files, fasta search databases, raw search output, and raw MS data files were deposited to the ProteomeXchange Consortium via the PRIDE27 partner repository with the data set identifier PXD025019. Full Skyline quantification of CYP3A4/raloxifene was deposited to the ProteomeXchange Consortium via Panorama Public28 and is available at https://panoramaweb.org/CYP3A4-raloxifene.url with the data set identifier PXD024932.