

Abstract
Ex vivo programming of T cells can be efficacious but is complex and expensive; therefore, the development of methods to transfect T cells in situ is important. We developed and optimized anti-CD3-targeted lipid nanoparticles (aCD3-LNPs) to deliver tightly packed, reporter gene mRNA specifically to T cells. In vitro, targeted LNPs efficiently delivered mCherry mRNA to Jurkat T cells, and T-cell activation and depletion were associated with aCD3 antibody coating on the surface of LNPs. aCD3-LNPs, but not non-targeted LNPs, accumulated within the spleen following systemic injection, with mCherry and Fluc signals visible within 30 minutes after injection. At 24 h after aCD3-LNP injection, 2-4% of all splenic T cells and 2-7% of all circulating T cells expressed mCherry, and this was dependent on aCD3 coating density. Targeting and transfection were accompanied by systemic CD25+, OX40+, and CD69+ T-cell activation with temporary CD3e ligand loss and depletion of splenic and circulating subsets. Migration of splenic CD8a+ T cells from the white-pulp to red-pulp, and differentiation from naïve to memory and effector phenotypes, followed upon aCD3-LNP delivery. Additionally, aCD3-LNP injection stimulated the secretion of myeloid-derived chemokines and T-helper cytokines into plasma. Lastly, we administered aCD3-LNPs to tumor bearing mice and found that transfected T cells localized within tumors and tumor-draining lymph nodes following immunotherapy treatment. In summary, we show that CD3-targeted transfection is feasible, yet associated with complex immunological consequences that must be further studied for potential therapeutic applications.
Keywords: LNPs, lipid nanoparticles, immunotherapy, T cell, T cell receptor, CD3, T-cell transfection, T-cell activation, mRNA, Reporter gene, Lipid nanoparticle
INTRODUCTION
Cancer immunotherapy, including immune checkpoint therapy and adoptive cell transfer (ACT), has achieved impressive clinical outcomes in treatment of advanced melanoma and hematological malignancies [1–4]. T-cell recruitment and activation of CD8+ cytotoxic T cells are a major antitumor mechanism of the immune system response [5–7]. In many solid cancers, high levels of tumor-infiltrating T cells have been associated with good prognosis and enhanced survival [8, 9]. However, ex vivo programming of T cells is complex and expensive, and therefore, the development of methods to transfect T cells in situ remains an important endeavor. Direct T-cell targeting for effective gene therapies is accompanied by T-cell activation, depletion, and systemic cytokine production [10, 11]. Therefore, understanding the effect of T-cell targeting on T-cell activation, cytokine release, and toxicity is crucial to balance the efficacy and safety profiles for successful clinical development.
RNA delivery is an attractive approach for gene delivery and offers several advantages over DNA. RNA is readily translated in the cytoplasm, yielding high transfection efficiency. In addition, RNA does not need to enter the nucleus for gene expression and therefore can be expressed in both dividing and non-dividing cells [12–14]. The transient nature of gene expression by RNA allows for the pharmacological titration of T-cell dosing and RNA-based gene delivery and can bypass many safety concerns that arise from the stable genomic integration and prolonged expression of bioactive materials that are commonly associated with DNA-based transfection. For efficient systemic delivery, however, RNA requires encapsulation within vector systems that can protect it from degradation by nuclease activity. RNA delivery can be accomplished via viral and non-viral vector systems [15]. Viral-based gene therapies are difficult to implement due to the potential safety concerns and the complexity and cost of clinical vector production. Conversely, non-viral vectors can offer safe, versatile alternatives to viral vectors for in vivo gene delivery.
Among non-viral gene vectors, lipid nanoparticles (LNPs) represent the most advanced platform for systemic delivery of RNA [16]. Ease in scale up, titration, and particle surface modification to incorporate multifunctional ligands, as well as robust capability to condense and deliver gene constructs with low toxicity, are some of the advantages of gene delivery by LNPs. ionizable cationic lipids that effectively condense RNA without impacting the surface charge of the LNP can accommodate large RNA constructs or multiple genes in the interior core of LNPs [17–19]. When ionizable cationic lipids are mixed with polyanionic RNA at pH values lower than the pKa of the ionizable lipid head group, the protonated amino groups facilitate complexation of RNA [20, 21]. Upon internalization into the target cells, the ionizable lipids are protonated, which facilitates endosomal escape. Coronavirus disease 2019 (COVID-19) vaccines from Pfizer-BioNTech and Moderna deliver mRNA via LNPs and have driven the rapid exploration of the potential for such particles [22, 23]. However, these vaccine formulations are optimized for intramuscular injection, ideally clearing rapidly from the injection site [24].
In this study, we use targeted LNPs to transfect T-cells in situ with reporter genes and assess the resulting transfected cells. The mRNA is condensed and packaged inside LNPs with the help of the potent ionizable cationic lipid DLin-MC3-DMA (pKa of 6.44), also used in the formulation of the first FDA-approved RNA-based gene therapy, Onpattro® [25]. The resulting complex is coated with neutral and PEGylated lipids to enhance the stability of the assembled LNPs. DSPE-PEG2k is incorporated to improve LNP blood stability and to minimize hepatic uptake. Cy7 is conjugated to the lipid shell to facilitate tracking of the LNPs and provide imaging that is complementary to optical reporter genes.
Targeting T cells using CD3 specific antibodies offers an exciting approach for achieving T-cell specific transfection. The first monoclonal antibody specific against human CD3 (clone OKT3) was developed in 1979, and a murine monoclonal antibody (clone 145-2C11) was described soon thereafter in 1987 [26, 27]. Since then, anti-CD3 antibodies and their respective fragments have been used to study T-cell biology, prevent allograft rejection, treat cancer, and create bispecific antibodies (bsAbs) among many other applications [28, 29]. More relevant to our applications, aCD3-conjugated nanocarriers have recently been used to efficiently deliver chimeric antigen receptors (CARs) specifically to T cells in vitro for the treatment of various cancers in mice [30]. While anti-CD3 mAbs have been shown to deplete T cells while promoting anergy and broad cytokine release in vivo [31–34], F(ab’)2 CD3e fragments can in part avoid complement mediated opsonization and antibody-dependent cellular cytotoxicity due to their lack of an Fc antibody portion [28, 35]. Furthermore, CD3 bispecific antibodies have been widely explored in the clinic, and are the focus of intense antibody engineering efforts to optimize valency and affinity while suppressing activation and Fc-mediated effector functions [29]. we believe this approach has yet to be fully optimized for in vivo transfection and believe our findings will help inform ligand selection within the LNP literature.
Our approach contrasts with other recently reported work [10, 11, 36–39]. We evaluate the use of LNPs in direct, in situ transfection of T cells rather than administering CD3 antibodies or therapeutic cells previously engineered ex vivo [38]. Here, LNPs are targeted to mouse T cells using a CD3 F(ab’)2 or to human T cells using a CD3 antibody, each with varied surface coating densities. With a goal of intravenous injection and receptor-mediated targeting, we evaluate the circulation time, transfection efficiency, and resulting impact on T cells in the spleen and blood. We evaluate the time dependent impact of aCD3-LNPs on T-cell activation, depletion, and transfection, and elucidate the relative impact of the surface antibody and the mRNA concentration.
Finally, we evaluate the impact of aCD3-LNPs in a tumor model with and without immunotherapy in order to assess the impact on tumor-infiltrating T cell recruitment. In our previous work with local administration of CpG [40–42], we found that CpG-mediated local tumor cell death results in antigen-specific myeloid cells circulating within the blood and localizing within the spleen. Therefore, we hypothesized that not only selective T-cell transfection but also CpG-mediated T-cell activation could occur in the spleen, and activated T cells could localize in tumors. We incorporated this treatment to determine whether transfected T cells could be localized in tumors and whether immunotherapy would impact their density within tumors.
RESULTS:
aCD3-LNP encapsulation tightly packs mCherry mRNA and delivers mRNA to Jurkat T cells while resulting in activation and depletion
We first optimized mRNA encapsulation in aCD3-LNPs. mCherry mRNA was encapsulated with 90% efficiency in an optimized particle containing an ionizable cationic lipid (DLin-MC3-DMA) (Supplementary Figure S1). The particle size distribution by Dynamic Light Scattering (DLS) and zeta potential were narrow and unimodal with an average diameter of 71.2 nm (0.13 PDI) and surface charge of ~−9 mV at physiological pH (Supplementary Tables S1, S2). T-cell targeted and non-targeted LNPs were spherical on electron microscopy (EM) imaging with homogeneous structures enclosing a liquid crystal form of densely packaged nucleotides (Figure 1A) with an average LNP diameter of ~70 nm. Compared to non-targeted LNPs (Figure 1A–i), LNPs carrying anti-human CD3 mAbs were surrounded by a ~20 nm corona of increased EM density, as expected based on the grafting structure of the antibodies (Figures 1A–ii, A–iii). The assembled LNPs were coated by a lipid bilayer with a 6 nm distance between the outer and inner leaflet intensity peaks. The LNP interior was multilamellar with repeating units of lipid bilayer and nucleotides densely packed in the core and intercalated between adjacent lipid bilayers (Figure 1A–iv). We estimate that ~7 molecules of 1 kb mCherry mRNA are incorporated in each 70 nm lipid particle (see Supplemental Methods).
Figure 1. aCD3-LNPs containing mCherry mRNA transfect, deplete, and activate Jurkat T cells.
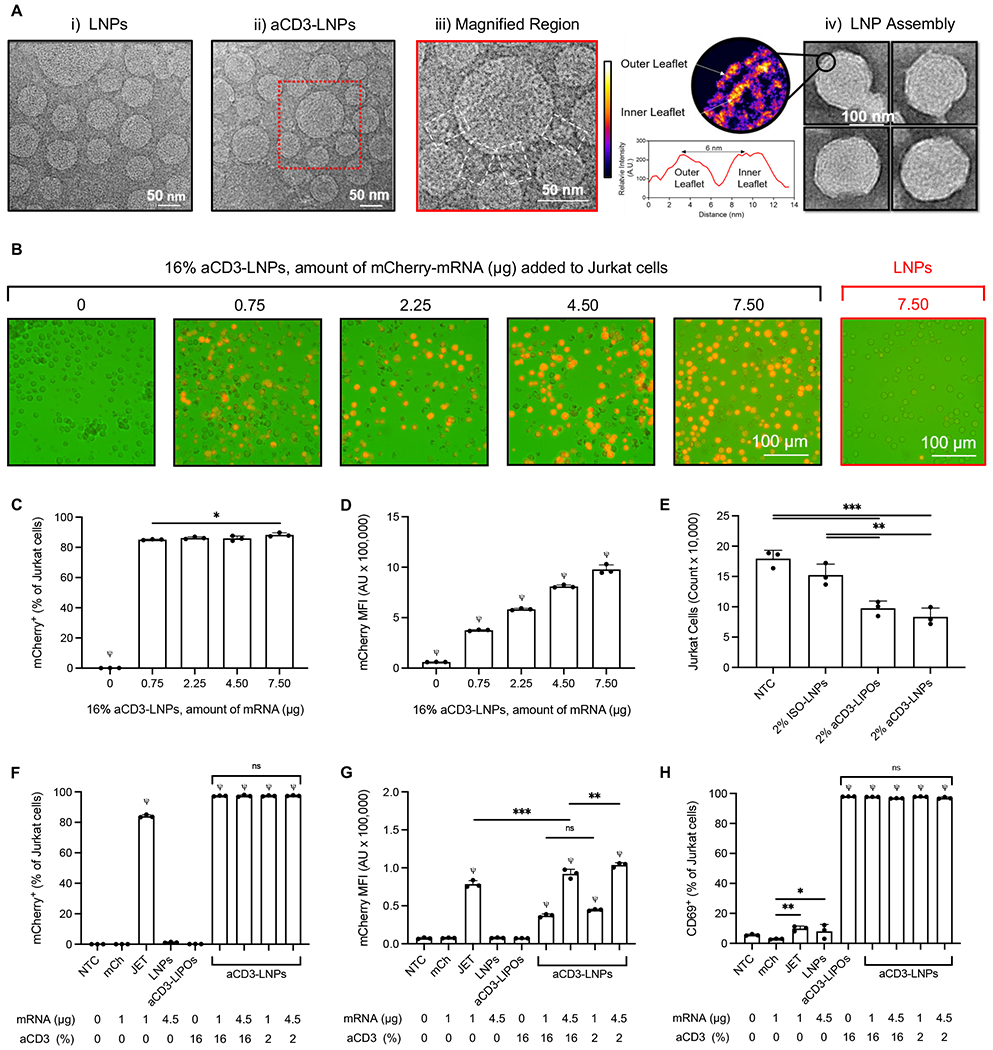
A) Transmission electron microscopy images of (i) non-targeted LNPs and (ii) T-cell targeted-LNPs, (iii) the magnified region of targeted LNP, and (iv) the structure of the lipid-nucleic acid assembly within the LNP. The red rectangle in (A-ii) defines the region magnified. The white rectangles in (A-iii) display the conjugated 16% anti-human CD3 on the surface of LNPs. The magnified region in (A-iv) demonstrates the lipid bilayer coating of the LNP with two leaflets at 6 nm apart from each other. The color scale details the electron scattering power (density) in each leaflet of the bilayer compared to the water in the background. Jurkat cells were treated with either non-targeted LNPs (LNPs) or 16% aCD3-LNPs for 24 h or remained untreated (NTC). B) mCherry fluorescence images of Jurkat cells treated with either non-targeted LNPs or 16% anti-human CD3-targeted LNPs including the corresponding amount of mCherry mRNA in the core of the particles. C, D) Percentage of mCherry+ cells (C), and the median mCherry fluorescence intensity (D) 24 h following incubation with various treatments or no treatment. E) Depletion of Jurkat cells 24 h after treatment with ISO-LNPs (with IgG on the surface), aCD3-LNPs, or aCD3-LIPOs (not containing mRNA), all with 2% antibody coating on the particle surface compared to no-treatment control (NTC) cells. Next, the transfection efficiency and activation of Jurkat cells treated with 16% aCD3-LNPs were compared with those of mCherry mRNA (mCh), mCherry mRNA complexed with jetMESSENGER (JET), non-targeted (0%) LNPs, 16% aCD3-liposomes (aCD3-LIPOs, not containing mRNA), and 2% aCD3-LNPs. F–H) Percentage of mCherry+ cells (F), the median mCherry fluorescence intensity (G), and percentage of CD69+ cells 24 h following incubation with various treatments or no treatment (H). Scale bars are 50 or 100 nm in A and 100 μm in B as displayed. All data are plotted as mean ± SD. Statistical analyses were performed using one-way ANOVA with Tukey’s multiple comparisons test. ns: not significant. *p < 0.05, **p < 0.01, ***p < 0.001. ψ represents p < 0.0001 of the below group across all other groups unless otherwise denoted with statistical analysis lines above. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
With 0.5% DSPE-PEG5000-Maleimide (DSPE-PEG5000-Mal) and an aCD3:Mal 1:1 molar ratio, we measured the amount of aCD3 on the surface of the LNPs. When 4 nmol aCD3 was added per mg lipid, we experimentally determined that 0.64 nmol (or ~16% of the added aCD3) was bound to LNPs (Supplementary Table S3). Assuming a surface area of 140 nm2 for anti-human CD3 with molecular weight of 150 kDa, we estimate a maximum of 72 molecules of aCD3 on the surface of a particle with an average diameter of ~80 nm and four repeated lamellar structures within the particle. The estimated 72 molecules of aCD3 per particle corresponds to 0.62 nmol of aCD3 per mg lipid of LNPs (Supplementary Table S3).
The size and zeta potential were compared by DLS and NanoSight with the aCD3 ligand (anti-mouse CD3 F(ab’)2 for mouse targeting or anti-human CD3 monoclonal Ab (mAb) for human cellular studies) covalently coupled to 0.5% maleimide (Mal) (DSPE-PEG5000-Mal) on the surface of LNPs. Surface conjugation with ~16% coverage increased the hydrodynamic particle size to ~160 nm but did not affect the particle surface charge as measured by DLS Zetasizer (Supplementary Tables S1, S2). Further measurements using NanoSight NS300 (Malvern, UK) estimated the particle size and particle number as 114 ± 17 nm and (7.75 ± 3) x 1012 particles/mL in 250 μL of the final volume of aCD3-LNPs when averaged among three serial dilutions. We then conjugated a range of antibody concentrations to the particles in order to evaluate the impact of the surface density by reducing the mass of antibody added to the particles. At the lowest concentration used within studies of the F(ab’)2 fragment (~1% of the sites were conjugated), the diameter increased to ~93 nm (PDI of 0.25), with the measured diameter increasing to 101.2 and 168 nm with 2 and 16% of the sites occupied.
The transfection efficiency of the assembled mCherry-containing LNPs was then evaluated in human Jurkat cells in suspension using formulations specified in Supplementary Table S4. At 24h after incubation with 16% aCD3-LNPs, mCherry fluorescence increased as a function of the number of LNPs incubated with Jurkat suspensions (Figure 1B). In contrast, mCherry fluorescence intensity was low in cells incubated with non-targeted LNPs (Figure 1B). More than 80% of 16% aCD3-LNP-treated Jurkat cells were mCherry+, and transfection efficiency was similar across a range aCD3-LNP concentrations (0.75 to 7.5 μg/mL mRNA delivered in 5 to 50 μL of aCD3-LNPs and added to 1 mL of 2.5 x 105 T cells in suspension) (Figure 1C). Further, mCherry fluorescence intensity increased in a dose-dependent fashion (Figure 1D).
In addition to transfection, we also found aCD3-LNPs depleted Jurkat cells and reduced CD3e/T-cell receptor (TCR) complex surface density. At 24 h after 2% aCD3-LNP administration (4.5 μg mRNA in 15 μL), Jurkat cell numbers were reduced ~2-fold as compared to no-treatment control (NTC) samples (Figure 1E). Depletion was dependent on aCD3-targeting and not on mRNA concentration, as depletion was similar for empty liposomes (aCD3-LIPOs) conjugated with the same amount of aCD3. Depletion was not observed for LNPs coated with IgG antibody (ISO-LNPs). Similar trends were observed with CD3 ligand internalization, as CD3 expression decreased in Jurkat cells incubated with aCD3-conjugated particles (Supplementary Figure S2A).
When incubated with Jurkat cells for 24 h, reducing the aCD3:Mal molar ratio on the LNP surface did not impact the binding and cellular uptake efficiency for a fixed mRNA dose (Supplementary Figure S2B). Increasing the dose from 0.075 to 7.5 μg increased the fraction of transfected cells (Supplementary Figure S2B). Similarly, reducing the molar percentage of DSPE-PEG5000-Mal on the LNP surface from 0.5 to 0.2 while maintaining the aCD3:Mal mole ratio at 1:1 and reducing aCD3 loading to 40% did not reduce the frequency of mCherry+ cells (Supplementary Figure S2C). However, when the reduced antibody coating was assessed in mice following systemic administration of LNPs, transfection was reduced. In particular, reducing DSPE-PEG5000-Mal on the surface of LNPs from 0.5 to 0.2 mole percent at the aCD3:Mal mole ratio of 1:1 reduced the mCherry transfection by ~80% and 50% in peripheral blood mononuclear cells (PBMCs) and splenocytes, respectively (Supplementary Figure S2D).
We labeled the lipid shell of aCD3-LNPs and aCD3-liposomes (each at 2% coating density) with DSPE-PEG2k-Cy7 to assess the relationship of the particle uptake and transfection. Confocal fluorescence images were acquired 24 h after incubation. The uptake of aCD3-LNPs-Cy7 (green) and mCherry transfection (red) was colocalized (yellow) in the cytoplasm of the cells treated with aCD3-LNP-Cy7 (Supplementary Figure S2E). Cy7 was visible in cells treated with aCD3-LIPO-Cy7, but not in cells treated with ISO-LNPs (Supplementary Figure S2E). mCherry signal was detected in cells transfected with aCD3-LNPs and the positive control jetMESSENGER at comparable fluorescence intensity levels. Cy7 and mCherry fluorescence were absent in cells treated with ISO-LNP-Cy7 containing mCherry mRNA or NTC (Supplementary Figure S2E).
To evaluate the relationship between T-cell transfection and activation, LNPs were loaded with mCherry mRNA and conjugated with 16% (saturated), 2% or 0% (non-targeted) aCD3 on the surface. Targeted vacant liposomes (aCD3-LIPOs) with matched concentration and lipid composition were coated with 16% aCD3. After 24 h of in vitro incubation, mCherry+ fluorescence was observed following treatment with aCD3-LNPs or jetMESSENGER particles each containing mCherry mRNA, but not with naked mCherry alone (Figure 1F, 1G). The median fluorescence intensity (MFI) of mCherry increased with a greater dose of mRNA LNPs (Figure 1G). Treatment with particles conjugated with aCD3 mAb upregulated CD69 expression in ~95% of Jurkat T cells regardless of the presence or absence of mRNA (Figure 1H).
Finally, we monitored the mCherry expression following treatment of Jurkat cells with 16% aCD3-LNPs at 5, 24, and 48 h. 68% of Jurkats were mCherry+ as early as 5 h, increasing to 79% and 86% at 24 and 48 h, respectively (Supplementary Figure S2F). mCherry+ fluorescence intensity also followed this trend and increased with time to achieve a maximal median fluorescence intensity at 48 h after 16% aCD3-LNP incubation (Supplementary Figure S2G).
T-cell targeted LNPs led to early luciferase expression in the spleen and liver with increased accumulation in lymph nodes
To track LNP and mRNA-mediated protein expression separately over time, we labeled LNPs by incorporating DSPE-PEG2000-Cy7 into the lipid shell and packaged Fluc mRNA or mCherry mRNA within LNPs, respectively. Fluorescence and luciferase bioluminescence signals (Supplementary Figure S3A) were imaged in vivo during circulation, and ex vivo after organ isolation. The blood half-life of Cy7-labeled LNPs with 16% aCD3 coating density on the surface carrying mCherry mRNA or Fluc mRNA was ~2h and particles had largely cleared by 24 h (Figure 2A, Supplementary Figure S3B). The bioluminescence radiance was ~5.5-fold and the fluorescence intensity was ~13.5-fold higher in blood at 5 h versus 24 h as compared to NTC with respective conditions (Fig. 2B).
Figure 2. Pharmacokinetics and transfection resulting from aCD3-LNPs reveal splenic and lymph node transfection.
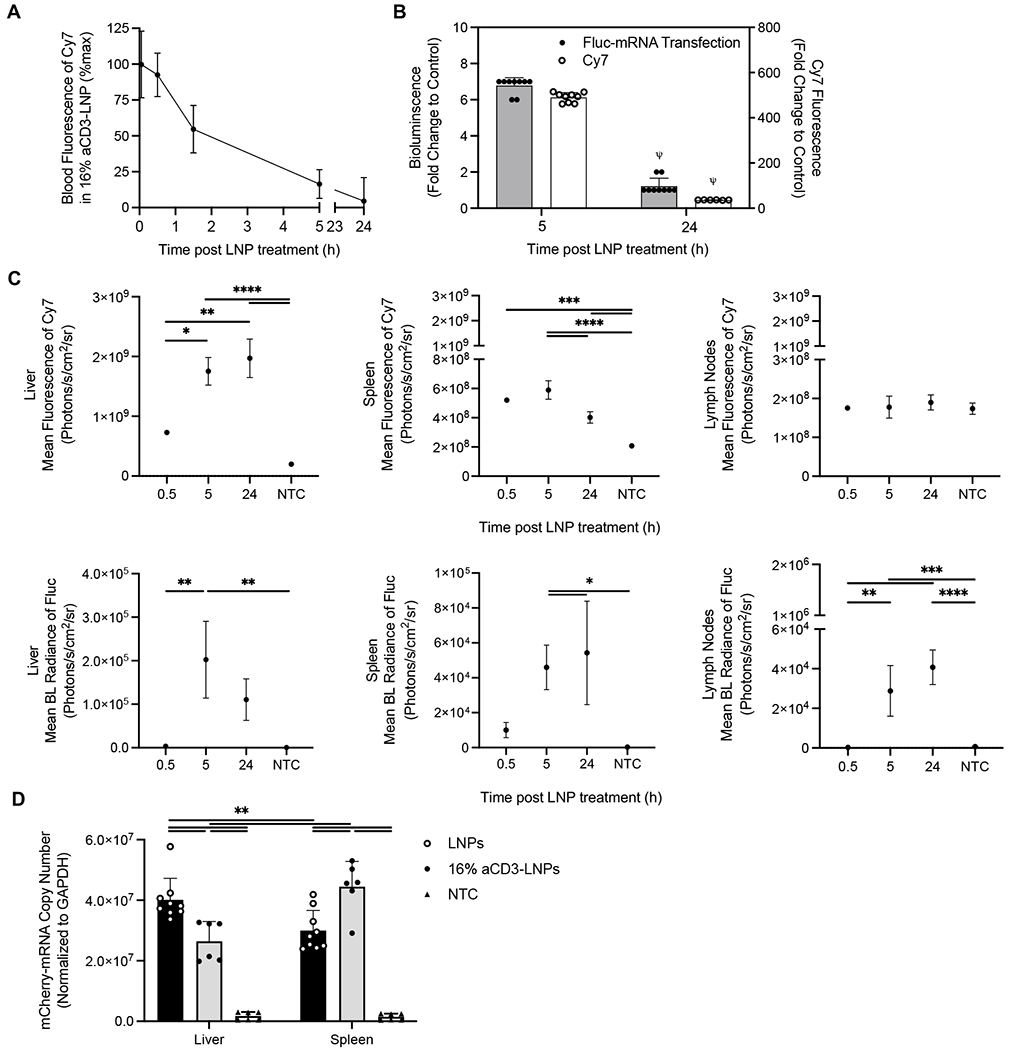
A-C) 16% aCD3-LNPs delivering Fluc mRNA with and without labeling the lipid shell with DSPE-PEG2k-Cy7 were injected in the tail vein in C57BL/6 mice (n = 18). For bioluminescence (BL) imaging of Fluc expression at each indicated time point, mice were injected with D-luciferin at 150 mg/kg body weight 10 min prior to collection of organ/tissue. Ex vivo images were acquired by a Lago optical imaging system, and fluorescence intensity of Cy7 and BL radiance of Fluc expression were quantified. A) Time-course of Cy7 fluorescence intensity of the labeled LNPs in blood over 24 h. B) Fold change of BL radiance of Fluc expression and Cy7 fluorescence intensity in blood at 5 h and 24 h following injection of 16% aCD3-Fluc-LNP-Cy7 to the corresponding control. C) Time-course of mean fluorescence intensity of Cy7 and mean BL radiance of Fluc expression in the liver, spleen, and lymph nodes (LNs). D) C57BL/6 mice (n = 24) were treated with systemic administration of either non-targeted LNPs (LNPs) or 16% aCD3-LNPs delivering mCherry mRNA. At 24 h post treatment, mice were perfused with cold PBS (−/−) and spleens and livers were harvested for RT-qPCR assay. Copy number of mCherry mRNA quantified by RT-qPCR and normalized to the housekeeping GAPDH gene in the liver and spleen 24 h after administration of targeted and non-targeted LNPs compared to the NTC. All data are presented as mean ± SD. Statistical analyses were performed using an unpaired T-test with Welch’s correction in B, one-way ANOVA and two-way ANOVA in C and D, respectively with Tukey’s or Dunnett’s multiple comparisons test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. ψ represents p < 0.0001 of the below group across all other groups unless otherwise denoted with statistical analysis lines above.
Cy7 fluorescence was detected in the spleen, liver, and kidneys as early as 0.5 h, reflecting the combination of particle circulation and accumulation, and continued to increase over 24 h in the liver and kidneys (Figure 2C, Supplementary Figures S3C and S3D). At 0.5 h, a low level of Fluc bioluminescence was detected in the spleen and liver and the normalized increase in bioluminescence was greater in the spleen than in the liver (Supplementary Figures S3E). The spleen and liver bioluminescence peaked at 5 h and the liver bioluminescence decreased by 24 h (Figure 2C). By contrast, while Cy7 fluorescence was not detected in lymph nodes at 0.5 h, Fluc expression was detected 5 h after treatment and increased over 24 h (Figure 2C, Supplementary Figure S3C–3D). The Fluc bioluminescence radiance in kidney and muscle peaked at 24 h (Supplementary Figure S3D). Following spleen targeting and transfection, spleen hypertrophy increased for 24 h, resulting in a 2-fold increase in the spleen weight (Supplementary Figure S3F).
We next evaluated delivery of mCherry mRNA in the liver and spleen by qRT-PCR 24 h following systemic administration of T-cell targeted or non-targeted LNPs in mice. The mCherry mRNA copy number normalized to GAPDH was higher in the spleen than in the liver for mice treated with 16% aCD3-LNPs (4.0 x 107 ± 7.1 x 106 vs. 3.0 x 107 ± 6.6 x 106). For mice treated with non-targeted LNPs, the mCherry mRNA uptake was higher in the liver than in the spleen (4.5 x 107 ± 8.3 x 106 vs. 2.6 x 107 ± 6.5 x 106) (Figure 2D).
aCD3-LNPs transfect splenic and circulating T cells
At 24 h after 16% aCD3-LNP administration, mCherry was expressed in ~4% of splenic CD3e+ T cells (Supplementary Figure S4A). Of total T cells, ~4% of CD4+ and ~2.5% of CD8a+ T cells, respectively, expressed mCherry (Figures 3A, 3B). When we administered LNPs with a decreased aCD3 F(ab’)2 coating density, transfection efficiency declined and mCherry frequencies decreased in a linear fashion (Figures 3A, 3B, Supplementary Figure S4A). Transfection did not occur in other leukocyte populations (Supplementary Figure S4C), nor in control treatments that lacked a combination of aCD3 and mCherry mRNA, confirming that aCD3-LNPs specifically transfected T cells in vivo. By 48 h, mCherry+ CD3e+, CD4+, and CD8a+ T cells decreased to ~0.24%, ~0.41%, and ~0.14% of total T cells respectively (Figures 3A, 3B, Supplementary Figure S4A).
Figure 3. aCD3-LNPs transfect splenic and circulating CD4+/CD8a+ T cells.
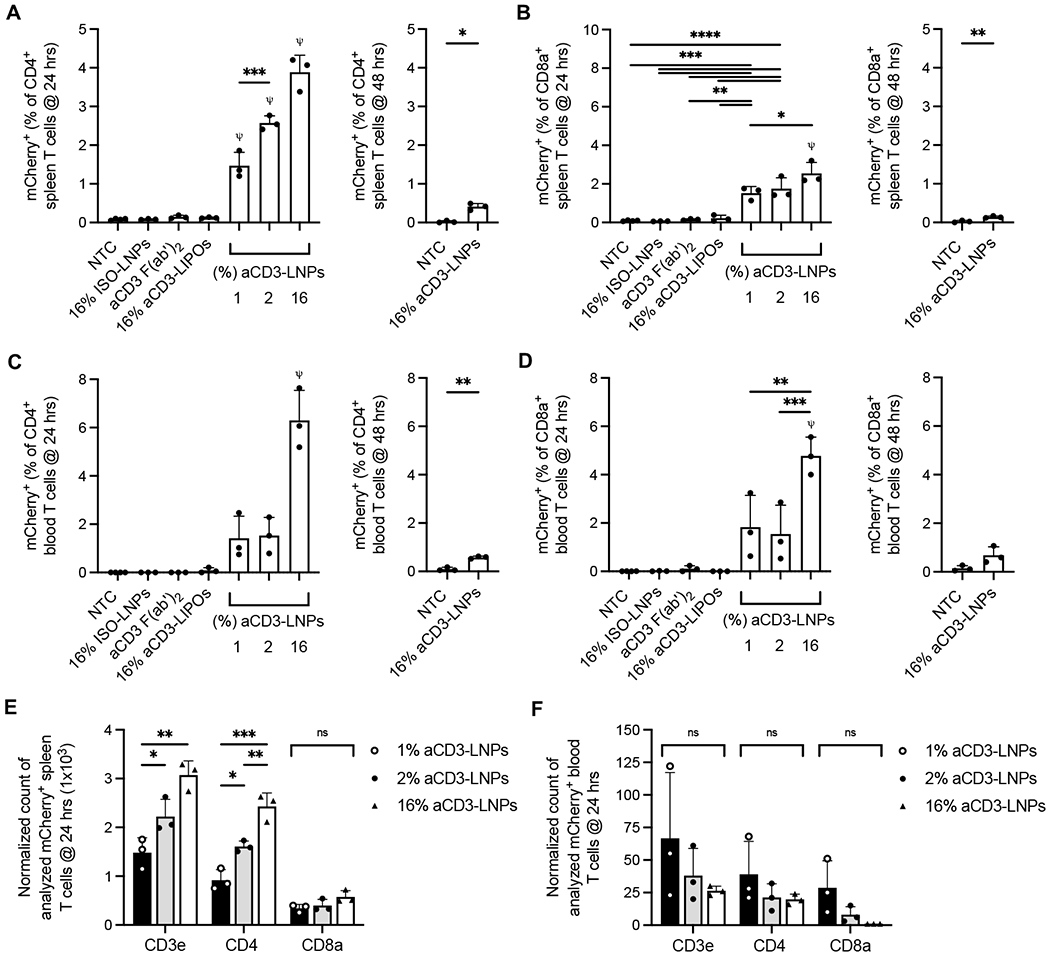
A-D) mCherry+ CD4+ (A,C) or mCherry+ CD8a+ (B, D) T cells as a percentage of CD4+ or CD8a+ T cells, respectively, at 24 and 48h after control (no treatment, isotype conjugated LNPs, or aCD3 F(ab’)2) or 1, 2, or 16% aCD3-LNP treatment in the spleen (A, B) or blood (C, D) (n=28). E-F) Normalized count of mCherry+ CD3e+, CD4+, and CD8a+ T cells at 24 h after 16% aCD3-LNP treatment from spleen (E) or blood (F). Each replicate was normalized in its respective 1, 2, or 16% group using the following: Normalized Count of Analyzed mCherry+ T cells = [(largest T-cell count among replicates in the group) / (T-cell count of replicate)] · mCherry+ count of replicate. All data are plotted as mean ± SD. For plots with more than 2 groups, statistical analyses were performed using ordinary one-way ANOVAs with Tukey’s multiple comparison test. Plots with 2 groups were statistically analyzed using an unpaired T test with Welch’s correction, ns: not significant. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001. ψ represents p<0.0001 of the below group across all other groups unless otherwise denoted with statistical analysis lines above.
Similar to the spleen, T-cell specific transfection was detected in blood at 24 and 48 h after aCD3-LNP administration (Fig. 3C and D and Supplementary Fig. S4B). Alternatively, mCherry expression was not detected in circulating T cells treated with mRNA-free liposomes, aCD3 antibody, or isotype control ISO-LNPs (Figures 3C, 3D, Supplementary Figure S4B). mCherry+ T cells as a percentage of CD4+, CD8a+, or CD3e+ T cells increased with increasing aCD3 coating. Importantly, the number of mCherry+ T cells increased with higher coating densities, confirming that splenic transfection is dependent on the aCD3 loading capacity of LNPs (Figure 3E). However, the number of circulating mCherry+ T cells was approximately equal between the three aCD3 LNP groups (Figure 3F). Taken together, these results indicate that circulating T cells were depleted as a result of aCD3-LNP treatment. Therefore, while transfection efficiency in the spleen depends on the amount of aCD3 ligand conjugated to LNPs, the same is not true for circulating T cells, as elevated levels of aCD3 coating have a greater impact on depletion rather than transfection.
aCD3-LNPs activate splenic and circulating T cells.
To measure in vivo activation over time, we again used flow cytometry to quantify the expression of a suite of activation markers in both the splenic and blood T cells. aCD3-LNPs activated >85% of splenic CD4+ and >90% CD8a+ T cells as early as 5 h after administration, and activation persisted at 24 h (Figures 4A, 4B). Lowering the aCD3 coating on LNPs decreased the frequency of CD69+ T cells slightly without altering the density of CD69 surface expression, suggesting that a low density of F(ab’)2 conjugation is sufficient for early and robust activation (Figures 4A, 4B, Supplementary Figures S5A). Injection of 16% aCD3-coated liposomes (aCD3-LIPOs) devoid of mCherry mRNA, or separate administration of free aCD3 antibody (aCD3), also stimulated robust CD69 expression. However, dramatically lower activation profiles were observed in LNPs conjugated with an isotype antibody (16% ISO-LNPs) (Figure 4A, 4B). Taken together, these results imply that in vivo activation is driven by aCD3 targeting and subsequent binding or crosslinking of T-cell receptors rather than inclusion of mRNA within LNPs. Circulating T cells displayed similar trends of CD69 activation after aCD3-LNP administration, however, activation was markedly reduced when compared to splenic populations (Figures 4C, 4D, Supplementary Figure S5B).
Figure 4. aCD3-LNPs activate splenic and circulating CD4+/CD8a+ T cells with distinct CD69, CD25, and OX40 kinetics (n=45 C57BL/6 mice).
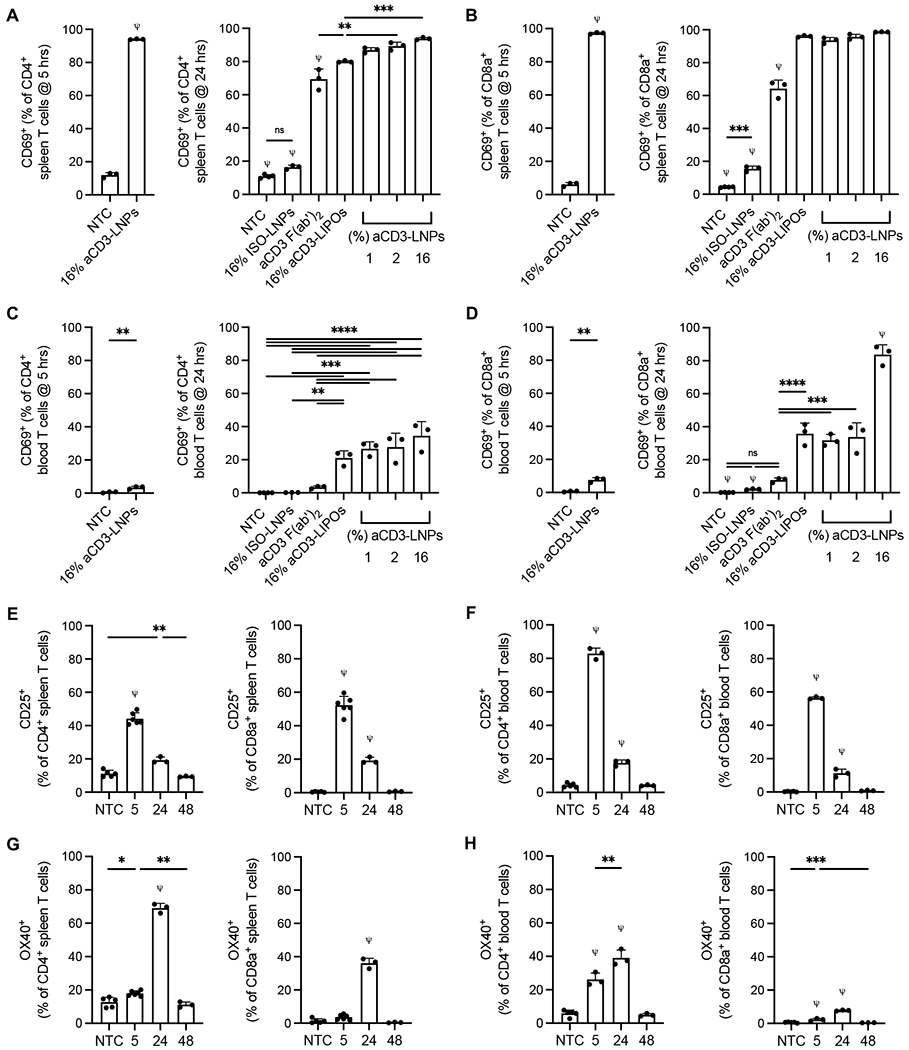
A-D) CD69+ CD4+ (A, C) or CD69+ CD8a+ (B, D) T cells as a percentage of CD4+ or CD8a+ T cells, respectively, at 5 and 24 h after control (no treatment, isotype conjugated LNPs, or aCD3 (Fab’)2) or 1, 2, or 16% aCD3-LNP treatment in the spleen (A, B) or blood (C, D). E-F) CD25+ CD4+or CD8a+ T cells as a percentage of respective total CD4+ or CD8a+ splenic (E) or circulating (F) T cells at 5, 24, and 48 h after 16% aCD3-LNP treatment. G-H) OX40+ CD4+ or CD8a+ T cells as a respective percentage of total CD4+ or CD8a+ splenic (G) or circulating (H) T cells at 5, 24, and 48 h after 16% aCD3-LNP treatment. All data are plotted as mean ± SD. For plots with more than 2 groups, statistical analyses were performed using ordinary one-way ANOVAs with Tukey’s multiple comparison test. Plots with 2 groups were statistically analyzed using an unpaired T-test with Welch’s correction, ns: not significant. *p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001. ψ represents p<0.0001 of the below group across all other groups unless otherwise denoted with statistical analysis lines above.
In addition to CD69 expression, CD25 and OX40 were differentially elevated by 48 h (Supplementary Figures S5C, S5D). In the spleen, 16% aCD3-LNPs induced peak CD25 expression on ~44% of CD4+ and ~52% of CD8a+ T cells at 5 h (Figure 4E). At 24 h, CD25+ populations dropped in frequency by more than 2-fold in both T-cell subsets and reached baseline NTC values by 48 h (Figure 4E). Circulating T cells mirrored this trend, as ~83% of CD4+ and ~56% of CD8a+ T cells expressed CD25 at 5 h followed by a return to NTC levels by 48 h (Figure 4F). While CD25+ populations dominated the splenic and circulating T-cell compartments at early timepoints, OX40 did not reach maximal expression until 24 h after 16% aCD3-LNP administration. In the spleen, ~69% of CD4+ and ~36% of CD8a+ T cells were OX40+ (Figure 4G) and (Figure 4H). OX40 expression was lower in the blood at ~39% of CD4+ and ~8% CD8a+ circulating T cells (Figure 4H). Similar to CD25, OX40 expression returned to NTC levels by 48 h, suggesting that aCD3-LNP mediated T-cell activation is a short-lived phenomenon.
aCD3-LNPs deplete splenic and circulating T cells while temporarily downregulating CD3 expression
Next, we analyzed the frequency of remaining T cells in both the spleen and blood to assess how aCD3-LNPs altered leukocyte architecture after treatment. At 5 h after 16% aCD3-LNP injection, splenic CD3e+ T cells were slightly, albeit non-significantly, reduced in frequency (Supplementary Figure S6A). A small, non-significant loss of CD8a+ T cells was responsible for this marginal reduction in CD3e+ T cells, as CD4+ T cells were unaffected after 16% aCD3-LNP treatment (Figures 5A, 5B). However, TCR-CD3 complex internalization was pronounced, with CD3e expression reduced in ~50% of both CD4+ and CD8a+ T cells (Figure 5C). By 24 h, CD3e− CD4+ and CD3e−CD8a+ populations were no longer present in any aCD3-LNP treatment group, suggesting targeting-dependent TCR internalization was temporary and only occurred at early time points in the spleen (Supplementary Figures S6B, S6C). At 24 h after aCD3-LNP administration, CD8a+, but not CD4+, T-cell numbers remained low, suggesting that LNPs have biased effects on the CD8a+ compartment (Figures 5A, 5B). This was further supported by the CD4+ to CD8a+ T-cell ratio, which was greater in mice treated with 1, 2, or 16% aCD3-LNPs compared to non-treated animals (Figure 5D). By 48 h, splenic T cells within the 16% aCD3-LNP group had fully recovered to match or exceed NTC levels (Figures 5A, 5B, Supplementary Figure S6A).
Figure 5. aCD3-LNPs temporarily deplete splenic and circulating CD4+/CD8a+ T cells while reducing CD3 surface expression (n=31 C57BL/6 mice).
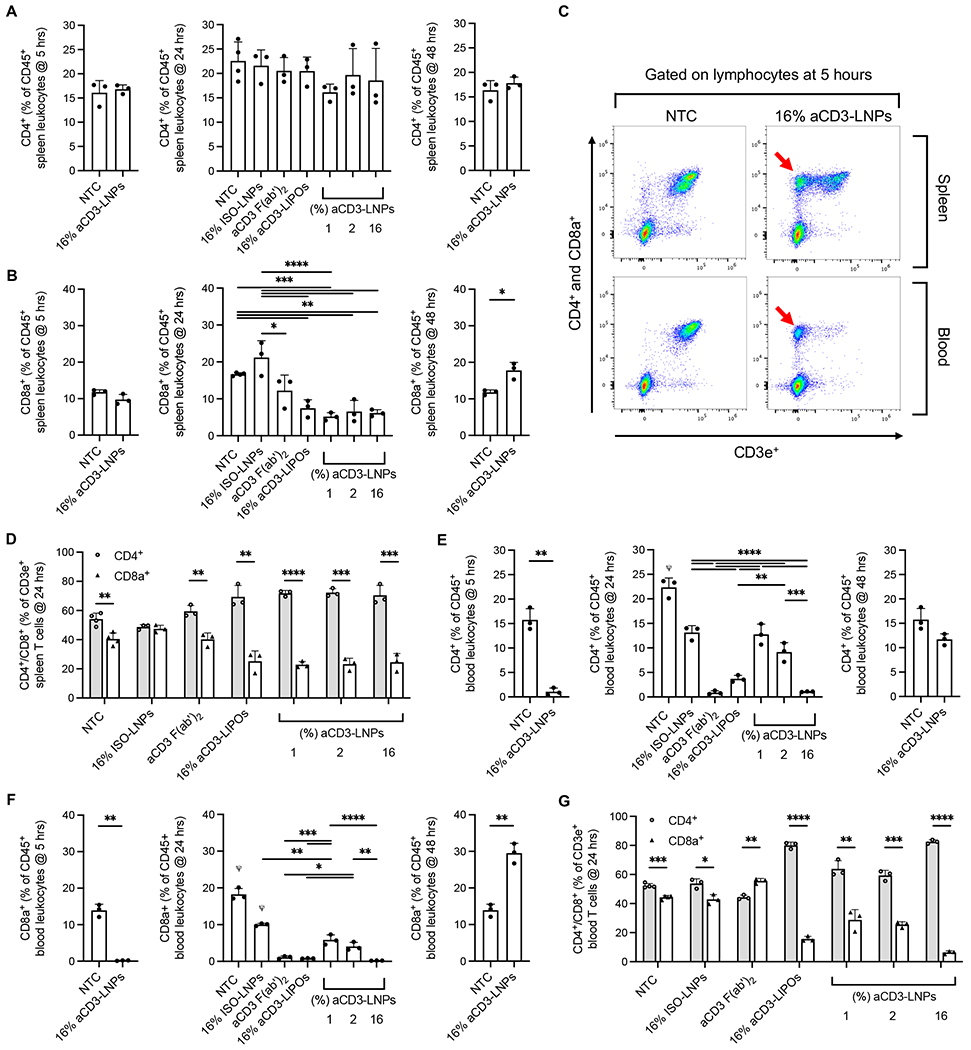
A-B) CD4+ (A) or CD8a+ (B) T cells as percentage of total CD45+ splenic leukocytes at 5, 24, and 48 h after control or 1, 2, or 16% aCD3-LNP treatment. C) Representative flow cytometry dot plots (down sampled to 15,000 events each) of splenic and circulating lymphocytes at 5 h after 16% aCD3-LNP or NTC treatment showing CD3e ligand internalization as denoted by red arrows. D) CD4+ and CD8a+ as a percentage of CD3e+ splenic T cells at 24 h after control or 1, 2, or 16% aCD3-LNP treatment. E-F) CD4+ (E) or CD8a+ (F) T cells as a percentage of total CD45+ circulating leukocytes at 5, 24, and 48 h after control or 1, 2 or 16% aCD3-LNP treatment. G) CD4+ and CD8a+ as a percentage of CD3e+ circulating T cells at 24 h after control or 1, 2, or 16% aCD3-LNP treatment. All data are plotted as mean ± SD. For plots with more than 2 groups, statistical analyses were performed using ordinary one-way ANOVAs with Tukey’s multiple comparison test. Plots with 2 groups were statistically analyzed using an unpaired T-test with Welch’s correction. CD4+ and CD8a+ were compared in D and E with an unpaired T-test with Welch’s correction. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001. ψ represents p<0.0001 across all groups unless otherwise denoted with statistical analysis lines above.
In the blood, T-cell loss was highly pronounced. Depletion was directly correlated with LNP aCD3 coating density, as CD3e+ T-cell fractions of CD45+ leukocytes were approximately 19.6, 15 and 1.4% with 1, 2, and 16% aCD3-LNPs, respectively (Supplementary Figure S6D). Liposomes coated with 16% aCD3 displayed similar depletion profiles to the 16% aCD3-LNP, while isotype-conjugated LNPs exhibited the lowest levels of depletion (Supplementary Figure S6D). Furthermore, high levels of circulating T-cell depletion and TCR internalization at 24 h after treatment occurred in animals treated with an aCD3 antibody alone, cementing the relationship between aCD3-TCR targeting and T-cell loss (Supplementary Figure S6B, S6C, S6D). We then dissected CD4+ and CD8a+ T-cell populations. At 5 h after 16% aCD3-LNP administration, CD4+ and CD8a+ T cells were heavily depleted, decreasing by ~15-fold and ~77-fold respectively when compared to NTC (Figures 5E, 5F). By 24 h, circulating T cells had not recovered in frequency (Figures 5E, 5F) but now fully expressed CD3e (Supplementary Figures S6B, S6C), suggesting that similar to the spleen, CD3e internalization was short lived in the blood. Both CD4+ and CD8a+ subsets made up a smaller percentage of total leukocytes, with CD4+ T cells increasingly dominating the CD3e+ T cell compartment when higher densities of aCD3 F(ab’)2 were conjugated to the LNPs (Figures 5E, 5F, 5G). By 48 h, circulating T-cell frequencies in 16% aCD3-LNP treated animals equaled or exceeded baseline level (Figures 5E, 5F). Taken together, our results indicate T-cell depletion is a temporary effect of aCD3-LNP administration.
aCD3-LNPs Induce Splenic T-cell Migration
Following effective T-cell transfection and activation in the spleen, we performed splenectomies to measure the physical characteristics of whole spleens. At 24 h after 16% aCD3-LNP treatment, the size and weight of spleens increased ~2-fold as compared to mice treated with non-targeted LNPs or NTC (Figures 6A, 6B), which confirmed that spleen enlargement was due to aCD3 surface coating on LNPs. Non-targeted LNPs did not induce liver or splenic hypertrophy as compared with NTC (Figures 6A, 6B, Supplementary Figure S7A). After one week, aCD3-LNP-treated spleens had returned to a normal size and weight, suggesting that splenic hypertrophy was a relatively short-lived process (Figures 6C, 6D).
Figure 6. aCD3-LNPs mobilize and activate splenic T cells (n=21 C57BL/6 mice).
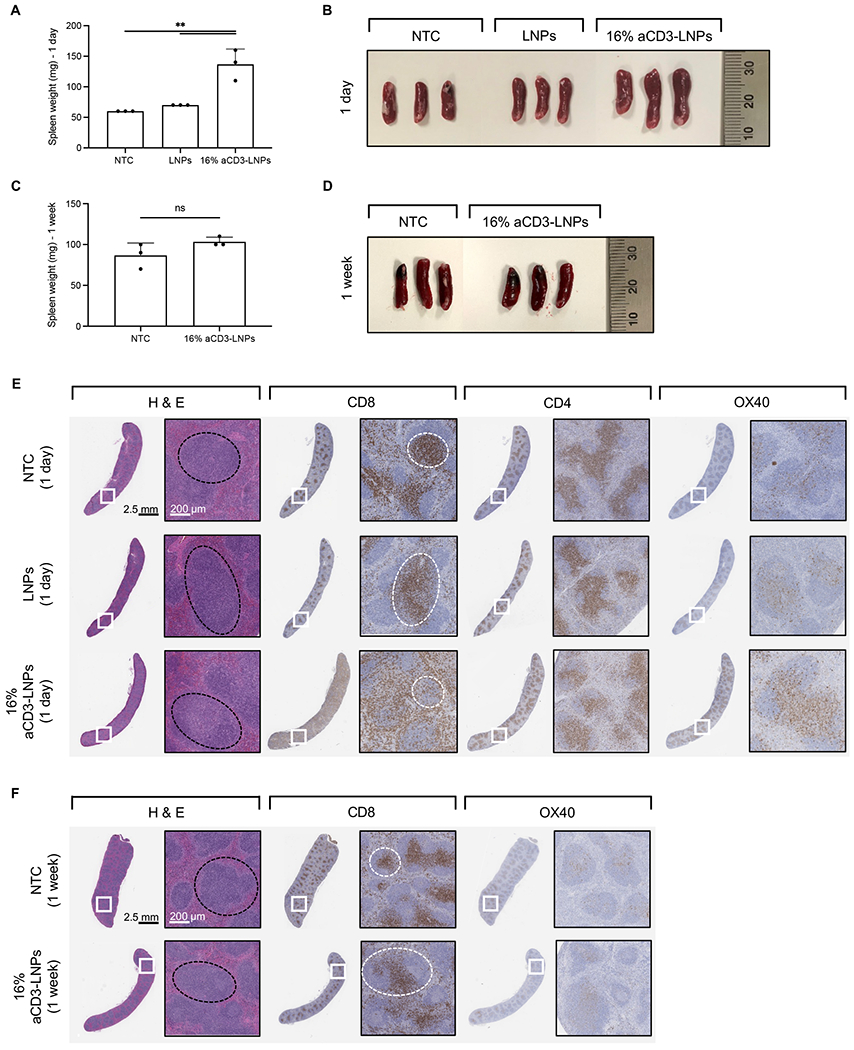
Mice were treated with systemic administration of either 16% aCD3-LNPs or non-targeted LNPs, each carrying mCherry, and compared to no-treatment control (NTC). A, B) Spleen weight (A) and size (B) one day after treatment. C, D) Spleen weight (C) and size (D) one-week after treatment. E, F) Histological sections of the spleens harvested one day (E) and one week (F) following each treatment and stained with H&E, aCD8, aCD4, and aOX40. Black and white dashed circles represent the white pulp and the periarterial lymphatic sheaths (PALS) in the spleen sections, respectively. Scale bars are 2.5 mm for the whole section and 200 μm for the magnified section. In A and C, all data are presented as mean ± SD. Statistical analyses were performed using an ordinary one-way ANOVA with Tukey’s multiple comparisons test in A and unpaired T-tests with Welch’s correction in C. ns: not significant. **p<0.01
Next, we evaluated the spatial distribution of CD4, CD8, and OX40 in histological sections of these spleens. At 24 h after 16% aCD3-LNP treatment, LNPs mobilized CD8+ cells from the white pulp (black dashed circle) and periarterial lymphatic sheath (PALS) to the marginal zone (MZ) and red pulp, while CD4+ cells were largely localized in the PALS (white dashed circle) (Fig. 6E). Expression of OX40 increased with 16% aCD3-LNP treatment compared to non-targeted LNP treatment and NTC (Fig. 6E). At 24 h after LNP injection, OX40− expressing cells were found largely in the PALS after 16% aCD3-LNP treatment (Fig. 6E). Histological sections of spleens one week after 16% aCD3-LNP treatment showed that CD8+ cells were localized in the PALS with OX40 activation equal to that of control spleen (Fig. 6F). These results suggest that T cells encountered aCD3-LNPs in the spleen and altered their spatial distribution.
In the liver, no noticeable difference was observed in the H&E-stained sections across treatments, however, a small increase in the frequency of CD8+, CD4+, and OX40+ T cells was noted in the livers of mice treated with 16% aCD3-LNPs (Supplementary Figure S7B).
Cytokine concentrations were elevated in blood plasma 5 and 24h after aCD3-LNP injection
In addition to transfection, activation, depletion, and migration of T-cell subsets, aCD3-LNPs stimulated dramatic levels of cytokine release. Compared to NTC animals, 16% aCD3-LNP treated animals exhibited large fold-changes in the systemic secretion of pro-inflammatory factors IFN-γ, IL-2, IL-3, TNF-α, indicative of aTh1 response (Figure 7A). Complimentary Th2 responses were also upregulated, including anti-inflammatory interleukins IL-4, IL-5, IL-9, and IL-13 (Figure 7B). Furthermore, 16% aCD3-LNPs induced early and high secretion of IL-2Rα (CD25) that decreased with time, matching the kinetic activation profiles of CD25+ splenic and circulating T cells (Figure 7C). This finding suggests that CD25 is shed from both CD4+ and CD8a+ activated T cells within hours of aCD3-LNP administration. Factors associated with T-regulatory cells, Th17, Th22, and Th25 phenotypes, such as IL-10, IL-17α, IL-22, and IL-25, respectively, were also upregulated with time (Figure 7C). Interestingly, secreted factors likely originating from myeloid cells, such as G-CSF, M-CSF, and IL-1β were elevated shortly after LNP injection, to suggesting aCD3-LNPs have downstream effects on leukocytes beyond T cells (Supplementary Figure S8A). Additional factors of interest can be found in Supplementary Figure S8B.
Figure 7. 16% aCD3-LNPs promote secretion of T-helper cytokines into murine plasma (n=9 C57BL/6 mice).
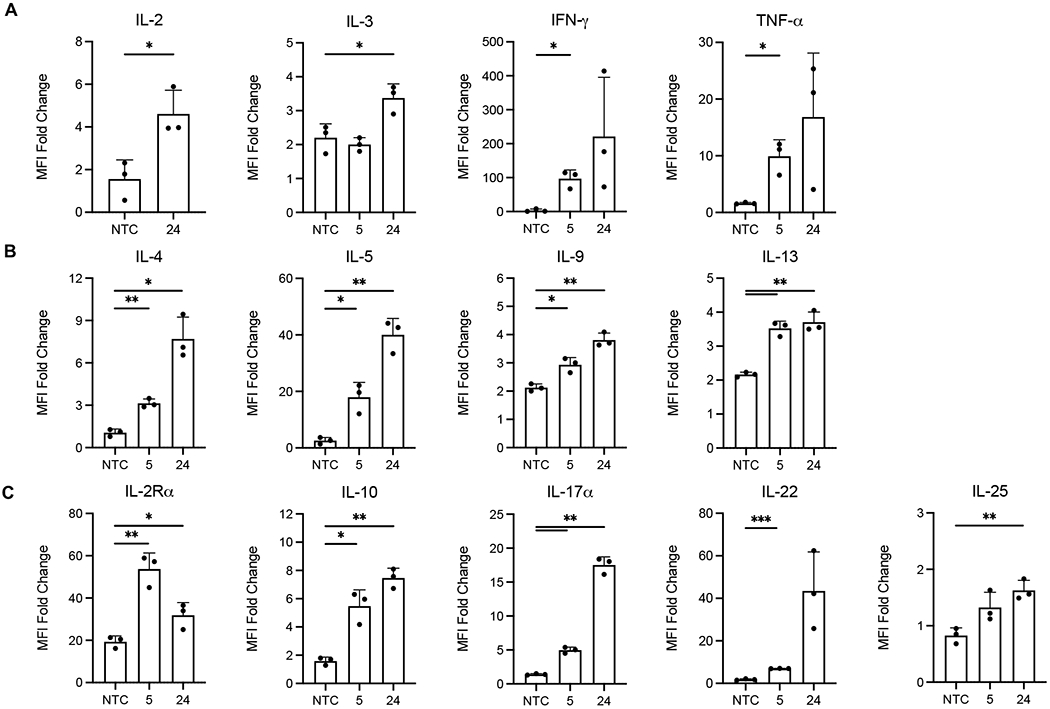
A-C) Fold change of Th1 (A), Th2 (B), or T-regulatory, Th17, Th22, and Th25 (C) associated cytokines secreted in murine plasma at 5 h and 24 h after 16% aCD3-LNP administration. Median Fluorescence Intensity (MFI) Fold Change = average MFI of sample / average MFI of PBS buffer associated with that sample. Raw data passed internal quality control such that each replicate’s MFI was plotted only if at least 20 Luminex beads were recovered during acquisition. All data are plotted as mean ± SD. For Luminex plots, statistical analyses were performed with NTC against 5 or 24 h using an unpaired T-test with Welch’s correction. * p<0.05, ** p<0.01, *** p<0.001.
aCD3-LNP injection shifts the phenotypic composition of splenic and circulating T cells
At 5 h after administration of 16% aCD3-LNPs, both CD4+ and CD8a+ CD44−CD62L+ and CD44+CD62L+ T cells in the spleen decreased as a percentage of total CD3e+ T cells, likely due to early activation-dependent CD62L shedding (Figure 8A). Conversely, CD4+ CD44+CD62L− and CD44−CD62L− T cells expanded while CD44−CD62L− cells filled the majority of CD8a compartment (Figure 8A and Supplementary Figure S9). By 24 h, CD44+CD62L− CD4+ T cells returned to NTC levels, while CD44+CD62L+ CD4+ T cells increased by ~20% and CD44−CD62L+ T cells remained low (Figure 8A). Similar trends were observed in CD8a+ T cells except for CD44+CD62L− cells, which remained elevated as compared to non-treated animals (Figure 8A). These 24 h trends were consistent at the 48 h timepoint, except for CD44+CD62L− CD8a+ T cells which had returned to NTC baseline (Figure 8A). In summary, CD44−CD62L+ CD4+ and CD8a+ T cells, consistent with a naïve phenotype, declined up to ~3-fold with time and reached their lowest frequency within the splenic T-cell compartment by 24 h. In parallel, CD44+CD62L+ T cells were consistent with an activated phenotype, which increased with time and comprised the dominant T-cell phenotype at 24 h. While central memory T cells also express CD44 and CD62L, we hypothesize that the observed increase in frequency was due to the activation of naïve T cells rather than the expansion of memory cells, as aCD3-LNPs stimulate robust activation.
Figure 8. 16% aCD3-LNPs shift the phenotype of splenic and circulating CD4+/CD8a+ T cells (n=12 C57BL/6 mice).
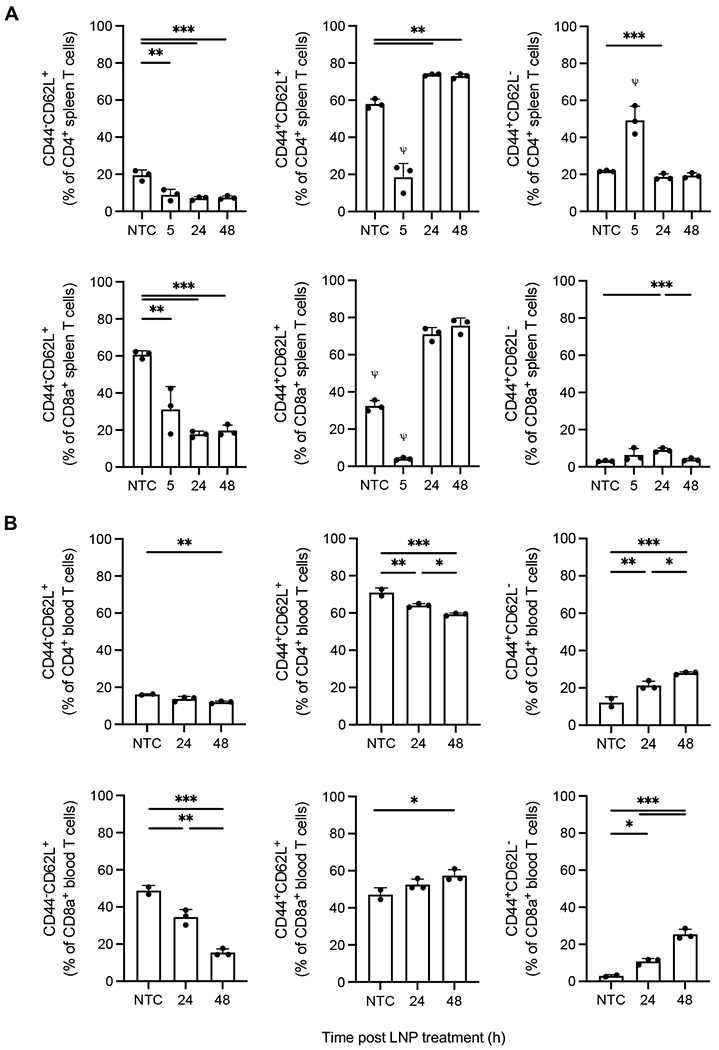
A-B) Splenic (A) and circulating (B) CD4+ and CD8a+ CD44+/−CD62L+/− T-cell frequencies at 5, 24, and 48 h after no-treatment control or 16% aCD3-LNP administration. All data are plotted as mean ± SD. Statistical analyses were performed using ordinary one-way ANOVAs with Tukey’s multiple comparison test. * p<0.05, ** p<0.01, *** p<0.001. ψ represents p<0.0001 across all groups unless otherwise denoted with statistical analysis lines above.
Similar to splenic T cells, the frequency of CD44−CD62L+ naïve circulating T cells decreased with time (Figure 8B). CD4+ CD62L+CD44+ cells also decreased temporally, but the CD8a+ equivalent followed splenic trends and gradually increased in frequency (Figure 8B). These results suggest that circulating CD4+ and CD8a+ T cells may have specialized responses after being targeted by aCD3-LNPs. Interestingly, CD62L−CD44+ subsets, consistent with an effector phenotype, dramatically increased with time (~2.3-fold and ~8.6-fold in frequency for CD4+ and CD8a+ T cells respectively by 48 h) (Figure 8B). This trend was not observed in the spleen, as splenic effector T cells had returned to NTC levels by 48 h, revealing that discrete phenotypes persist across tissues after aCD3-LNP administration. Altogether, our results show both splenic and circulating CD62L+CD44− naïve T cells decrease with time after 16% aCD3-LNP administration while CD44+CD62L+ activated phenotypes generally expand and dominate both CD4+ and CD8a+ compartments.
Tumor and tumor draining lymph node accumulation of T cells
We then set out to determine whether the signal from transfected T cells would be detected in tumors and tumor draining lymph nodes (TDLNs) in mice treated with immunotherapy. In previous reports [40–42], we demonstrated that a combination of synergistic CpG and aPD-1 is highly efficacious in models of melanoma and breast cancer via the accumulation of tumor infiltrating leukocytes and systemic antigen-specific myeloid and T cells, which produce tumor cell death over a period of days. Therefore, we first evaluated whether the injection of LNPs 24 h after immunotherapy treatment would result in the accumulation of transfected T cells within the tumors and TDLNs (Supplementary Figure S10A). In fact, we observed significant increases in mCherry fluorescence in both the CpG-treated tumor and the nearby TDLN 24 h after aCD3-LNP injection, with similar trends observed in the distant tumor (Supplementary Figure S10B). Further, mCherry fluorescence was greater in the tumor and TDLNs following immunotherapy and aCD3-LNP injection (CpG + aPD-1 + aCD3-LNPs) than a separate cohort without immunotherapy (aCD3-LNPs only). Fluorescence was not enhanced in mice injected with LNPs only (CD3-LNPs only) when compared to NTC mice (Supplementary Figures S10C–D).
Immunohistochemistry of tumors excised at 24 h after aCD3-LNP treatment (48 h after immunotherapy) further confirmed that CpG treatment enhanced tumor cell death and increased the infiltration and activation of T cells in a manner that corresponded with observed mCherry fluorescence (Supplementary Figure S11A). CD8+ T-cell numbers increased 3.2-fold and 2.1-fold in the CpG-treated and distant tumors compared to the no-treatment control tumors, respectively (Supplementary Figure S11B). Activated T cells increased 15-fold and 11-fold in the CpG-treated and distant tumors, respectively, compared to non-treated tumors (Supplementary Figure S11C).
Finally, we confirmed transfected T-cell infiltration in a second tumor model (E0771) with a modified study in which aCD3-LNPs were injected 5 h before immunotherapy was administered (Supplementary Figure S12A). Both mCherry and Fluc mRNA were evaluated in this model. Here, we also found that mCherry fluorescence increased in the tumor or TDLNs (in this study the aggressive tumor and TDLN merge) following immunotherapy treatment (Supplementary Figures S12 B–C). Finally, we conjugated Cy7 to aCD3-LNPs and confirmed that they accumulated in tumors but rarely in TDLNs (Supplementary Figure S12D). LNP accumulation was not impacted by the immunotherapy treatment applied in this study, as Cy7 fluorescence intensity in tumors was identical between aCD3-LNPs alone and aCD3-LNPs supplemented with immunotherapy (Supplementary Figure S12D). Taken together, these preliminary data suggest that the observed transfected T-cell signal was due to T cells entering the tumors and TDLNs rather than aCD3-LNPs directly transfecting tumor-resident T cells. However, further studies are required to evaluate the precise sequence of events that produce mCherry fluorescence in tumors and TDLNs.
DISCUSSION
We set out to use targeted LNPs to transfect T cells in situ. Systemic silencing of CD45 in CD4+ T cells was previously reported using CD4-targeted LNPs to specifically delivery siRNA [36]. In another report, mRNA-based polymeric nanocarriers were fabricated for transient and targeted transfection of chimeric antigen receptor (CAR)-T cells ex vivo to knockout the T-cell receptor and enhance memory T-cell functionality [38]. Here, we fabricated T-cell targeted LNPs by incorporating DLin-MC3-DMA into particles containing mCherry mRNA or Fluc mRNA and conjugating them with an aCD3 F(ab’)2.
With 90% mCherry mRNA encapsulation efficiency, the purified LNPs were negatively charged with an average hydrodynamic diameter of 70 nm. Electron microscopy confirmed the highly packed mCherry-mRNA within the LNPs and 20 nm targeting moieties on the surface of CD3-targeted LNPs (the typical size of an IgG molecule such as an anti-CD3 monoclonal antibody [43]). Lipid lamella intercalating with mRNA were visible in the magnified EM images. Similar structures have been visualized with EM previously [44]. Alternatively, bleb-like structures were reported for LNPs encapsulating multiple mRNA or with the replacement of cholesterol by alternative cholesterol analogues [44–46]. With the 1 kb mCherry mRNA, we estimated 7-8 molecules of RNA to be compacted within 80-nm LNPs.
LNPs were targeted with aCD3 using the thiol-Mal conjugation method by incorporating 0.5 molar of DSPE-PEG5k-Mal in the lipid formulation of LNPs. The resulting aCD3-targeting LNPs circulated in blood with a half-life of 2 h and expressed Fluc 5 h after injection. At 0.5 h after injection, Fluc expression was first detected at a low level in the spleen. Bioluminescence increased with time in the spleen, liver, and kidney, peaking at 5 h in most organs while following a similar trend to Cy7-labeled LNPs. In the lymph nodes, bioluminescence increased over 24 h without an increase in Cy7, suggesting that transfected T cells were temporarily migrating to these tissues.
Transfection
Labeling the lipid shell with Cy7 and loading the LNP with mCherry mRNA demonstrated that cellular uptake, internalization, and transfection are specific to CD3 targeting in cell culture. LNPs surface-conjugated with control IgG did not internalize, whereas CD3-targeted LNPs and liposomes were internalized. The CD3-mediated LNP internalization was not sensitive to aCD3 mAb coating density in cell culture, but mCherry fluorescence intensity was dose dependent. In vivo, 16% aCD3-LNPs transfected ~2-7% of circulating T cells and ~2-4% of splenic T cells 24 h after treatment. Unlike the in vitro results, in vivo transfection efficiency was dependent on the density of aCD3 F(ab’)2 coating such that 16% aCD3-LNPs yielded the highest frequencies of mCherry+ T cells as a percentage of total T cells remaining in the tissue. While 1% and 2% aCD3-LNPs transfected a comparable number of circulating T cells as 16% aCD3-LNPs due to aCD3 targeting-mediated depletion, they transfected fewer splenic T cells. We focused here on the 24 h transfection results as they are likely to be the most meaningful for future assays. Other groups have also achieved broad in vivo leukocyte transfection using non-targeted lipid delivery vehicles although a smaller fraction of T cells were transfected [47, 48].
Activation and migration
In addition to successful transfection in vitro and in vivo, we found aCD3-LNPs to be potent T-cell activators. In vitro, ~97% of all Jurkat cells were CD69+ regardless of the amount of mCherry mRNA packed into LNPs or surface density of aCD3 mAb coating. In vivo, aCD3-targeting dependent activation yielded high, early spikes of CD69+ and CD25+ activation followed by OX40+ expression in both circulating and splenic T cells. By 48 h, CD25+ and OX40+ expression had returned to baseline levels in both tissues, showing that activation was a transient effect. LNPs coated with isotype IgG did not cause increased activation as compared to non-treated controls in the blood, and only slightly elevated splenic T-cell activation, suggesting that the aCD3 F(ab’)2, rather than LNP lipid composition or mCherry mRNA, was the main component influencing T-cell activation. While aCD3-LNPs enable short lived and reversible activation, additional studies are required to understand the long-term effects of this phenomenon or if alternative targeting ligands, such as nanobodies or monovalent antibody fragments, can bypass T-cell activation pathways altogether.
Within 24 h after aCD3-LNP injection, CD8+ T cells had relocated from the PALS to the MZ and then to the red pulp in the spleen. OX40 expression in the PALS, the red pulp, and in blood were enhanced. In wild type mice, RT-qPCR confirmed that CD3-targeted LNPs delivered significantly greater copy numbers of mCherry mRNA to the spleen than the liver at the 24 h timepoint, whereas non-targeted LNPs deposited higher mCherry mRNA in the liver than the spleen.
Depletion
In vitro, Jurkat cell depletion resulted from 2% aCD3-LNP incubation and TCR-CD3 complexes were absent in ~99% of all T cells. These findings were replicated with aCD3-conjugated liposomes, confirming depletion and antigenic modulation were dependent on aCD3 mAb coating rather than lipid components or mCherry mRNA. In vivo, early circulating CD3e+ T-cell depletion was dependent on the amount of aCD3 coating on LNPs such that a lower amount of F(ab’)2 preserved more T cells. However, this trend was not observed in the spleen, where we observed biased CD8a+ T-cell depletion that was consistent across all aCD3 coating concentrations. Across both tissues, depletion was short-lived, as T cells either equaled or exceeded non-treatment control levels by 48 h after aCD3-LNP administration.
Other reports in the literature have described a similar pattern of T-cell depletion in the blood after anti-CD3 antibody injection, and that this phenomenon was reversible upon antibody clearance from systemic circulation [32]. In fact, we found that once LNPs had completely cleared circulation by ~24 h, circulating T cells had recovered in frequency shortly thereafter. There are many proposed mechanisms for T-cell depletion, including activation-induced cell death [28, 32, 33] and possible toxicity due to an abundant release of myeloid-derived cytokines. Further functional studies will be needed to fully understand the long-term effects of aCD3-LNP mediated depletion, including immune tolerance and T-cell memory. At early timepoints, we also found IL-10 to be ~3 fold higher in 16% aCD3-LNP treated animals when compared to non-treated animals (Fig. 7C). Taken together with the literature, our results suggest that aCD3-LNPs have discrete influences on T cells depending on the tissue type [31]. While outside the scope of this paper’s objectives, it would be important to examine how splenic T cells respond to antigen after aCD3-LNP exposure, as this would inform the types of therapeutic mRNAs that could be viable for T-cell transfection.
Cytokine Secretion
Along with the changes in activation and depletion after administration of aCD3-LNPs in vivo, Th1 and Th2 cytokines increased in plasma. We found that treated mice had equal Th1 and Th2 profiles with identical kinetics that increased with time. Similar results have been observed after use of an in vivo anti-CD3 antibody (clone 1452C11), in which IL-2, IL-3, IL-4, TNF-α, and IFN-γ were detected via northern blot or in the serum of treated mice [34, 49]. We also found evidence of Th17, Th22, Th25, and T-regulatory T-cell phenotypes due to the secretion of associated interleukins. These results suggest that naïve Th0 T cells non-discriminately pursue a spectrum of helper phenotypes after CD3 receptor binding to aCD3-LNPs. Additionally, aCD3-LNPs stimulated the secretion of several myeloid-derived cytokines, suggesting immune cells other than lymphocytes could be affected downstream of T-cell targeting. Further studies utilizing scRNA-seq will be necessary to validate the precise cell types responsible for these secreted chemokines. Previous reports have shown that targeting CD4+ or CD8a+ T cells with monoclonal antibodies yielded a reduced cytokine profile and less circulating T-cell depletion [32]. aCD4 or aCD8a targeted LNPs could offer alternative routes for avoiding activation and subsequent cytokine release while still serving as candidates for T-cell specific transfection.
Phenotype
In both the blood and spleen, the T cells that existed after aCD3-LNP treatment had shifted expression profiles of CD44 and CD62L as a percentage of their respective CD4+ and CD8a+ compartments. In the spleen, a CD44+CD62L+ phenotype had increased from ~58% to ~73% in CD4+ T cells and from ~32% to ~76% in CD8a+ T cells at 48 h after 16% aCD3-LNP treatment. Elevated levels of CD44 in both tissues coincided with increased T-cell activation and we indeed observed a higher intensity of CD44 expression at this timepoint as compared to NTC animals. While additional markers are needed to separate true central memory cells from activated naïve cells, we believe the increase in this phenotype is primarily due to naïve T cells expressing higher levels of CD44. In the blood, while CD4+ CD44+CD62L+ subsets slightly declined and CD8a+ CD44+CD62L+ subsets marginally increased, the greatest phenotypic change was observed in CD44+CD62L− cells, in which CD4+ subsets increased from ~12% to ~28% and CD8a+ subsets, from ~3% to ~25%, at 48 h after 16% aCD3-LNP treatment. While these cells express the markers consistent with an effector memory phenotype, we found heterogeneous dot plot densities in the CD8a+ compartment, suggesting this profile may not be the sole population of cells present. Additional studies are necessary to not only separate these cells, but to determine how long this altered T-cell compartment persists and its tolerance to antigen.
Tumor accumulation of in situ transfected T cells
We observed transfected T cells in tumors only after immunotherapy was added. We evaluated 2 protocols. When CpG and aPD-1 were injected first, tumor cell death was initiated, leukocytes increased in the tumor, and T cells were activated. In fact, CD8+ T cells increased 3.2-fold and 2.1-fold in the CpG-treated and distant tumors compared to the no-treatment control tumors, respectively. Also, activated T cells increased 15-fold and 11-fold in the CpG-treated and distant tumors, respectively, compared to non-treated tumors. We have previously shown that this protocol in this tumor model also results in antigen specific T cells in the spleen [40, 42]. Twenty-four hours later, LNPs were injected. The accumulated transfected T cells in the tumor and TDLN could result from migration from the spleen or from the local transfection of resident T cells. We also reversed the order of treatment. When LNPs were injected first, circulating T cells were depleted within 5 hours (before immunotherapy could enhance their tumor accumulation), and therefore they would not be localized in the tumor when immunotherapy was administered. Yet, transfected T cells were observed in tumors 24 or 48 hrs later, likely migrating from the spleen or lymph nodes.
It was encouraging to observe transfected T cells within tumors after immunotherapy but premature to conclude that transfected T cells from the spleen reach tumors. Alternative hypotheses include transfected T cells entering from local lymph nodes or T cell directly transfected in the tumor. Evidence supporting the first hypothesis includes the fact that Cy7 imaging revealed relatively few LNPs within local lymph nodes. Further, the relative paucity of T cells within the tumor as compared with the spleen and lymph nodes suggests that transfection in the spleen would be more common. Additionally, colocalization of the T cells and LNPs could be impeded within the tumor, due to fibrosis or extracellular matrix stratification as compared to the lymph nodes or spleen.
Potential limitations and future directions
In this study, we explored an aCD3 mAb targeting human Jurkat cells and a F(ab’)2 antibody fragment targeting murine T cells to specifically transfect T cells both in vitro and in vivo. While the murine antibody fragment lacked an Fc region, such that complement-mediated depletion and antibody-dependent cellular cytotoxicity were avoided, T cells were depleted in vivo, likely due to activation dependent apoptosis. Although T-cell specific transfection was accomplished in vitro and in vivo, future studies exploring monovalent aCD3 ligands, such as nanobodies, Fab, or scFv fragments, will be crucial for minimizing TCR crosslinking and reducing activation associated with bivalent aCD3 targeting. Furthermore, whether by modifying the lipid composition, targeting ligands, or size of targeted LNPs, it is essential to engineer this delivery system to optimize for a specific response, ideally sterile T-cell transfection, without rampant cytokine release.
More studies are required to understand the long-term effects of aCD3-mediated T-cell activation and phenotypic shifting with regards to antigen sensing. Given that aCD3-LNP activation is independent of MHC Class I/II and CD28 signaling, short term CD69, CD25, OX40, and CD44 expression likely resulted in an anergic immune microenvironment. Antigen challenge experiments accompanied by deep immunophenotyping of splenic and circulating T-cell compartments would be necessary to test this hypothesis and dissect the time scales at which these cells persist.
For extended observation, an attractive alternative to synthetic mRNA is self-amplifying RNA (sa-RNA) currently applied in vaccine production. Sa-RNA is attractive for its longer and greater profile of transfection [50, 51], and LNPs would provide an attractive platform for delivery of the relatively large and highly negative nucleic acid construct [52–54].
CONCLUSIONS
We successfully packaged an mRNA reporter gene within lipid-based nanoparticles and targeted the resulting particles to T cells using aCD3. In vitro, aCD3-LNPs transfected and activated ~97% of Jurkat cells. In vivo, aCD3-LNPs transfected 2-7% of circulating T cells and 2-4% of splenic T cells respectively, while causing temporary activation, depletion, migration, cytokine release, and phenotypic shifts. The use of a single chain aCD3 antibody or affibody without baseline activation can improve the clinical translational of this strategy for cancer immunotherapy.
EXPERIMENTAL SECTIONS
Materials.
The ionizable cationic lipid DLin-MC3-DMA was synthesized by Biofine International Inc. (Vancouver, BC, Canada). All other lipids including 1,2-stearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2- distearoyl-sn-glycero-3-phosphoethanolamine-N-methoxypolyethyleneglycol-2000 (DSPE-PEG2k),1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-5000] (DSPE-PEG5k-Mal), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000]-N-(Cyanine 7) (DSPE-PEG2k-Cy7) and cholesterol (Chol) were from Avanti Polar Lipids Inc. (Alabaster, AL, USA). CleanCap mCherry mRNA (996 bp, 319377 Da) and Fluc mRNA (1929 bp, 618522 Da) were purchased from TriLink Biotechnology (San Diego, CA, USA). CpG-ODN 1826 (5’-tccatgacgttcctgacgtt-3’; total backbone phosphorothioated) was obtained from InvivoGen (San Diego, CA, USA). The checkpoint inhibitor, rat anti-mouse PD-1 antibody (clone RMP1-14) was from Bio X Cell (West Lebanon, NH, USA). The neu deletion (NDL) murine metastatic mammary carcinoma cell line was a generous gift from the Alexander Borowsky Laboratory (UC Davis)[7, 55]. The E0771 murine medullary breast adenocarcinoma cell line was purchased from CH3 BioSystems (Amherst, NY, USA). NDL cells were cultured in high-glucose DMEM (Gibco #11995) supplemented with 10% FBS (Omega Scientific #FB-11) and 1% penicillin-streptomycin (Gibco #15140) in a 37 °C humidified CO2 incubator. E0771 cells were cultured in a 37 °C humidified incubator with 5% carbon dioxide with RPMI 1640 (Gibco #11835), supplemented with 10% FBS, 1% Penicillin/Streptomycin and grown to 80-90% confluency. For E0771 tumor generation in mice, media was removed, and cells were trypsinized with TrypLE™ Express Enzyme (1X) without phenol red (Gibco #12604). The cells were then centrifuged at 100 x g for 5 minutes at room temperature, TrypLE™ was removed and replaced with sterile PBS without calcium and magnesium (PBS−/−) for subcutaneous injection.
Preparation of mRNA encapsulating lipid nanoparticles (LNPs).
Lipids composed of DLin-MC3-DMA:DSPC:DSPE-PEG2k:DSPE-PEG5k-Mal:Chol (50:10:1.5:0.5:38, molar ratio) or DLin-MC3-DMA:DSPC:DSPE-PEG2k:DSPE-PEG5k-Mal:DSPE-PEG2k-Cy7:Chol (50:10:1.32:0.5:0.18:38), molar ratio) were dissolved in chloroform and dried under a stream of nitrogen gas and then under vacuum overnight. The dried lipid was dissolved in ethanol at 10 mg/mL prior to assembly in LNPs. mCherry or Fluc mRNA was dissolved in 25 mM sodium acetate buffer, pH 4.0 at 116 μg/mL. mCherry mRNA was used in studies involving flow cytometry, and the notation aCD3-LNPs is used for these particles. Fluc mRNA was selected for studies involving whole body imaging, and the notation aCD3-Fluc-LNPs is used unless otherwise noted. LNPs were assembled by mixing one volume of the lipid solution in ethanol with three volumes of mRNA in buffer through a microfluidic micromixer performed on a benchtop NanoAssemblr™ (Precision Nanosystems, Vancouver, BC, Canada) at a combined flow rate of 2 mL/min (Supplementary Fig. S1). To remove ethanol, the resulting LNP solution in 25% ethanol was then dialyzed overnight at 4 °C against PBS (−/−) using Spectro/Por dialysis membranes (molecular weight cutoff 1000 kDa, Spectrum Laboratories, Rancho Dominguez, CA, USA). The LNP solution was concentrated using Amicon Ultra centrifugal tubes (molecular weight cutoff 100 kDa, Merck Millipore, Billerica, MA, USA) at 2000×g, 10 °C and purified through a spin column of Sepharose CL-4B (5 × 1 cm, Sigma, St. Louis, MO, USA) equilibrated with PBS (−/−). The average hydrodynamic size, the polydispersity index (PDI), and the zeta potential values of the LNPs after dialysis were determined as 71 nm, 0.13, and −9.8 mV, respectively, using dynamic light scattering (intensity mode; Zetasizer Nano ZS, Malvern Instruments Ltd., Malvern, UK). Lipid concentrations were determined using the Phospholipids C assay kit and Cholesterol E Assay Kit (Wako Chemicals USA, Richmond, VA, USA) according to manufacturer’s instructions. Encapsulation efficiency was measured by determining unencapsulated mRNA using fluorescence measurement upon the addition of RiboGreen (Molecular Probes, Eugene, OR, USA) to LNPs (Fi) and comparing this value to the total mRNA content that is obtained upon lysis of LNP by 0.2% Triton X-100 (Ft): % encapsulation = (Ft – Fi)/Ft × 100. The fluorescence intensity was determined using a Tecan (San Jose, CA, USA) Infinite® M1000 microplate reader. Total particle count of the purified LNPs was determined using a NanoSight NS300 System (Malvern, UK). For LNPs labeled with Cy7, the fluorescence intensity of Cy7 was detected at the excitation and emission wavelengths of 750 nm, 780 nm, respectively.
Preparation of empty liposomes.
Liposomes were prepared with the lipid composition mimicking the LNPs as described above. DLin-MC3-DMA:DSPC:DSPE-PEG2k:DSPE-PEG5k-Mal:Chol (50:10:1.5:0.5:38, molar ratio) were dissolved in chloroform and the dried lipid was dissolved in ethanol at 10 mg/mL. Vacant liposomes were prepared by mixing one volume of the lipid solution in ethanol against three volumes of PBS (−/−) in the absence of mRNA using the benchtop NanoAssemblr at a flow rate of 10 mL/min. The resulting liposomes were dialyzed overnight at 4 °C against PBS (−/−) using Spectro/Por dialysis membranes (molecular weight cutoff 1000 kDa) and concentrated using Amicon Ultra centrifugal tubes (molecular weight cutoff 100 kDa) at 2000×g, 10 °C. The empty liposomes had an average size and PDI of 76 nm and 0.3, respectively, and the zeta potential value of −7.7 ± 4 mV prior to conjugation with anti-CD3.
T-cell targeting of mCherry-LNPs.
LNPs or liposomes carrying DSPE-PEG5k-Mal (0.5 mol%) on the surface were either targeted to human Jurkat cells for in vitro studies or to mouse T cells for in vivo studies using InVivoMAb anti-human CD3 or InVivoMAb anti-mouse-CD3ε F(ab′)2 fragment (Bio X Cell, West Lebanon, NH, USA), respectively. In subsets of experiments, LNPs were conjugated with InVivoMAb hamster IgG F(ab’)2 fragment (Bio X Cell) as the isotype control (ISO-LNPs). The cysteine residues of the employed antibodies were reduced using TCEP (Tris[2-carboxyethyl] phosphine-HCl) Bond Breaker solution (0.5 M, Pierce, Rockford, IL, USA) at 10-fold higher molar ratio of the antibodies at pH 7–7.4 at room temperature for 30 min. The antibodies with the available SH groups were then added to the purified LNPs or liposomes containing 0.5 mol% Mal at a molar ratio of 1:1 of Mal:antibody in PBS (−/−, pH 7.4) at room temperature for 1 h (Supplementary Fig. S1). LNPs or liposomes conjugated with antibodies at 1:1 of antibody:Mal were labeled as 16% aCD3-LNPs, 16% ISO-LNPs, and aCD3-LIPOs. In subsets of experiments, reduced molar ratios of antibody:Mal were studied. The resulting aCD3-LNPs or aCD3-LIPOs or ISO-LNPs were dialyzed overnight at 4 °C against PBS (−/−) using Spectro/Por dialysis membranes (molecular weight cutoff 1000 kDa) and concentrated using Amicon Ultra centrifugal tubes (molecular weight cutoff 100 kDa) at 2000×g, 10 °C. The purified particles were eluted from a spin column of Sepharose CL-4B (5 × 1 cm, Sigma, St. Louis, MO, USA) equilibrated with PBS (−/−). Supplementary Table S1 and 2 summarizes the average diameter, the PDI, and the surface charge of the antibody-conjugated particles used in this study.
T-cell culture and activation.
In vitro T-cell transfection using aCD3-LNPs carrying mCherry was evaluated in Jurkat cells, an immortalized line of human T lymphocyte cells. Jurkat (clone E6-1) T cells were obtained from the Stanley Qi Laboratory (Stanford University) and cultured in a 37 °C humidified incubator with 5% carbon dioxide using RPMI 1640 (Gibco #11835), supplemented with 10% FBS (#FB-11, Omega Scientific), 1% Penicillin/Streptomycin (#15140, Gibco), and 10 mM of HEPES (#15630080, Gibco). This media formulation will be referred to as T-cell media (TCM). Jurkat cells natively are activated, and therefore, T-cell transfection was performed without a prerequisite T-cell activation process.
In vitro T-cell transfection study.
T-cell transfection was performed by addition of aCD3-targeted and non-targeted LNPs delivering mCherry mRNA at various concentrations to 5 × 105 T cells in 500 μL of Opti-MEM Reduced Serum Medium (#31985070, Gibco) in a 12-well plate and incubated for 2 h in a 37 °C humidified incubator with 5% carbon dioxide. Transfection efficiency and T-cell activation were compared with those of jetMESSENGER® (Polyplus-transfection®, NY, USA) as the positive control and aCD3-liposomes as the particles not containing mRNA.
Following the incubation, 600 μL of TCM was added, and the incubation was continued for 24 h. Expression of mCherry protein was visualized by imaging of the cell plates with a fluorescence microscope (Mikron IV 600 L, San Diego, CA, USA) using a 20x water-immersion objective and TRITC filter set or using a Leica DMi8 Inverted Microscope (Leica Microsystems, Wetzlar, Germany). Following fluorescence imaging, flow cytometry was also performed to quantify the transfection efficiency after 24 h incubation. T-cells were collected, and viable cell number was assessed using a hemocytometer with Trypan Blue dead cell exclusion. mCherry expression in cells and T cell activation was measured on a CytoFLEX flow cytometer (Beckman Coulter, Indianapolis, IN, USA) or Cytek Aurora Cytometer (Cytek Biosciences, Fremont, CA, USA) at the Stanford Shared FACS Facility (Stanford University, Palo Alto, CA, USA), and data were analyzed using FlowJo v10.7.1 software (Tree Star Inc, Ashland OR, USA).
Animals and in vivo procedures.
All animal studies were conducted according to guidelines for the Care and Use of Laboratory Animals provided by the National Institutes of Health and all animal work was performed under the protocols approved by the Institutional Animal Care and Use Committee (IACUC) of University of California, Davis, and Stanford University. Female FVB/n mice and female C57BL/6 mice (6–8 weeks old, 20–25 g) were purchased from (Charles River, Wilmington, MA, USA). To generate the bilateral syngeneic NDL tumor model, FVB/n mice were orthotopically transplanted with NDL tumor biopsies into the #4 and #9 inguinal mammary fat pads [56]. For the bilateral E077-1 tumor model, C57BL/6 mice were subcutaneously (s.c.) injected with E0771 cells (5 × 105 in 25 μL PBS−/−) into the #4 and #9 inguinal mammary fat pads under dissecting microscope.
Among a total of 186 mice (Supplementary Table S5), 53 FVB mice were used for immune profile analysis of blood and spleen after intravenous administration of therapies that included mCherry mRNA or a control. These groups included 16% aCD3-LNPs (n = 20), non-targeted LNPs (n = 4) and saline or PBS-injected control (n =17). Twelve FVB mice were transplanted with NDL tumors bilaterally and distributed among four groups: CpG+αPD-1 and 16% aCD3-LNPs (24 h, n = 3), CpG+αPD-1 and 16% aCD3-LNPs (48 h, n = 3), 16% aCD3-LNPs (24 h, n = 3) and NTC (n = 3).
Among a total of 133 C57BL/6 mice, 18 mice were used to study pharmacokinetics and biodistribution of 16% aCD3-LNP-Cy7 containing Fluc mRNA. C57BL/6 mice (n = 85) were used for RT-qPCR quantification of mCherry mRNA, immunohistochemistry staining and flow analysis. The mice were distributed among cohorts including 16% aCD3-LNPs containing mCherry mRNA (n = 28), non-targeted LNPs containing mCherry mRNA (n = 11), 16% aCD3-Liposomes (n = 3), 16% (IgG) ISO-LNPs containing mCherry mRNA (n = 3), aCD3 (n = 3), 1% aCD3-Fluc-LNPs (n = 3), 2% aCD3-Fluc-LNPs (n = 3), 2% aCD3-LNPs containing mCherry mRNA (n = 3), and NTC (n = 28).
To assess the kinetics of immune response following treatment, 12 E0771 tumor-bearing C57BL/6 mice were studied in treatment cohorts of LNPs containing mCherry mRNA including: 16% aCD3-LNPs (5 h, n = 3), CpG+aPD-1 and 16% aCD3-LNPs (29 h, n = 3), CpG+aPD-1 and 16% aCD3-LNPs (53 h, n = 3), NTC (n = 3). For ex vivo optical imaging, 18 E0771 tumor-bearing C57BL/6 mice were studied in the treatment cohorts of CpG+aPD-1 and 16% aCD3-Fluc-LNPs-Cy7 (5 h, n = 3), CpG+aPD-1 and 16% aCD3-Fluc-LNPs-Cy7 (24 h, n = 3), CpG+aPD-1 and 16% aCD3-Fluc-LNPs-Cy7 (48 h, n = 3). Three groups were also studied containing mCherry mRNA: 16% aCD3-LNPs-Cy7 (5h, n = 2), 16% aCD3-LNPs-Cy7 (24h, n = 2), 16% aCD3-LNPs-Cy7 (48h, n = 2), and NTC (n = 3).
Tumor diameters were measured with ultrasound and treatment started when tumors reached ~4–5 mm (~ 35–65 mm3) in longitudinal diameter. For treatments involving CpG + aPD-1, a dose of 25 μg or 100 μg CpG in 50 μL of endotoxin-free water was administered intratumorally in a single injection to the treated tumor and aPD-1 (200 μg) was delivered intraperitoneal (i.p.) in 50 μL volume of PBS−/−. In the treatment cohorts involving LNPs or liposomes, mice were injected via the tail vein with ~4 × 1013 particles/kg body weight carrying ~0.6 mg mCherry mRNA (2 nmol) or Fluc mRNA (1 nmol)/kg body weight, and ~25 mg lipid/kg body weight. No treatment control mice received ~90 μL saline or PBS injection.
Flow cytometry antibodies.
Pacific Blue rat anti-mouse CD45 (30-F11), PE-Cy7 Armenian hamster anti-mouse CD3e (145-2C11), Alexa Fluor 700 rat anti-mouse CD8a (53-6.7), APC Armenian hamster anti-mouse CD69 (H1.2F3), FITC mouse anti-human CD3 (HIT3a), and APC mouse anti-human CD69 (FN50) were purchased from BioLegend (San Diego, CA, USA). Alexa Fluor 532 rat anti-mouse CD45 (30-F11), Alexa Fluor 700 rat anti-mouse CD8a (53-6.7), and PE-Cy7 rat anti-mouse OX40 (OX-86) were purchased from Thermo Fisher Scientific. Rat anti-mouse CD16/CD32 antibody (2.4G2), Brilliant™ violet 480 rat anti-mouse CD25 (PC61), Brilliant™ Violet 786 rat anti-mouse CD19 (1D3), Brilliant™ ultraviolet 496 hamster anti-mouse CD3e (145-2C11), Brilliant™ ultraviolet 805 rat anti-mouse CD4 (GK1.5), FITC rat anti-mouse CD4 (GK1.5), Brilliant™ violet 510 rat anti-mouse CD44 (IM7), and Brilliant™ violet 650 rat anti-mouse CD62L (MEL-14) were purchased from BD Biosciences. Human Fcγ receptors were blocked using Human BD Fc Block™ (BD Biosciences #564220).
In vivo flow cytometry.
The spleen was collected in 2-mL Eppendorf tubes containing 500 μL of sterile PBS (−/−). The spleen was poured into a 35-millimeter petri dish containing a 0.7-micron mesh filter (Corning) and 1.5 mL of PBS (−/−) where it was mechanically disrupted to a single cell suspension using the plunger end of a 3-mL disposable syringe. Cell suspensions were spun down at 300 x g for 5 min at 4°C, after which the supernatant was removed, and the pellet mixed/incubated with 1mL of ACK Lysis Buffer (Gibco) for 5 min at room temperature. Lysis buffer was diluted with 9 mL of PBS (−/−), and the solution was spun down at 300 x g for 5 min at 4°C, after which the supernatant was removed, and the pellet resuspended in 1mL of FACS buffer for hemocytometer counting and antibody staining.
Blood was collected in EDTA-treated tubes (BD Biosciences) and 10 mL of ACK lysing buffer (Invitrogen) was added to 400-600 μL of blood for an incubation period for 5 min at room temperature. Lysis buffer was diluted with 20 mL of PBS (−/−), and the solution was spun down at 300 x g for 5 min at 4°C. The supernatant was removed, and the pellet was again incubated with 5 mL of ACK lysis buffer for 5 min at room temperature. The solution was diluted with 10 mL of PBS (−/−) and spun down at 300 x g for 5 min at 4°C, after which the supernatant was removed, and the pellet resuspended in 25 uL of FACS buffer for antibody staining.
Cell suspensions were stained using the LIVE/DEAD® Fixable Aqua Dead Cell Stain Kit (Invitrogen) according to the manufacturer’s instructions for dead cell exclusion from analysis. After, cells were washed, pelleted, and incubated with 2.4G2 mAb for 30 min in the dark at 4°C to block nonspecific antibody binding. Finally, blocked cells were stained with combinations of fluorochrome-conjugated anti-mouse antibodies for 30 min in the dark at 4°C. Antibody combinations used to distinguish immune cell populations were: CD45+ (all leukocytes), CD19 (B cells), CD3e (all T cells), CD4 (CD4+ T cells), CD8a (CD8a+ T cells), CD25/OX40/CD69 (activation suite), CD44/CD62L (phenotype suite). Cells were fixed in 4% paraformaldehyde in PBS (−/−) for 30 min at room temperature (PFA) and resuspended in fresh FACS buffer prior to acquisition. Stained cells were analyzed within 24 h on a Cytek Aurora spectral flow cytometer. All datasets were analyzed using FlowJo software v10.7.1 (TreeStar).
Luminex Assay.
Mouse blood was collected in EDTA-treated tubes and spun down at 1000 x g for 10 min at RT. The isolated plasma samples were stored at −80°C for Luminex analysis. Concentrations of cytokines and chemokines were analyzed by the Human Immune Monitoring Center (HIMC) at Stanford University using a 48-plex Luminex assay.
Quantitative reverse transcription PCR (RT-qPCR).
Following perfusion of mice with cold PBS (−/−), organs were dissected, weighed, snap frozen in liquid nitrogen, and stored at −80 °C until used for RNA extraction. Organs were homogenized in lysis buffer (100 mg tissue: 300 μL lysis buffer) and total RNA was extracted using the mirVANA™miRNA isolation kit (Life Technologies, AM1560) according to the manufacturer’s protocol. The extracted RNA was quantified using a NanoDrop spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA), and 1 ug of RNA was used for cDNA synthesis using qScript cDNA Supermix (QuantaBio, #95048-100) in a total reaction volume of 20 μL. The reaction was carried out for 5 min at 25 °C and 1 h at 40 °C followed by 5 min at 80 °C to inactivate the reverse transcriptase in a DNA Thermal Cycler 480 (PerkinElmer, Waltham, MA, USA). The quantitative RT-PCR assay was then performed in triplicate using 100 ng RNA equivalent of cDNA in a total reaction volume of 20 μL containing TaqMan Master Mix (Thermofisher Scientific, #4444557). GAPDH was used as a house-keeping gene (Life Technology, Assay ID Mm99999915-g1, FAM) for normalizing results. We used custom designed forward and reverse primers and FAM™ Dye-labeled TaqMan® MGB probe (Custom Plus TaqMan™ RNA Assay, FAM Assay ID: ARH6CMC Assay) for mCherry mRNA. The q-RT-PCR forward primer (5′ GAGCGGGTGATGAACTTCGA), the reverse primer (5′ CCGCAGCTTCACCTTGTAGAT), and the probe sequence (5′ CAGGACGGCGAGTTC) were custom designed based on the mCherry mRNA open reading frame sequence. The reactions were performed using a CFX96 real-time PCR system (Bio-Rad, Hercules, CA, USA) with the following program: 2 min at 50 °C (UNG activity), 10 min at 95 °C (polymerase activation), followed by 50 cycles of denaturation at 95°C for 15 s and extension at 60 °C for 60 s. The mCherry copy number was calculated from the obtained cycle quantification (Cq) values using the mCherry mRNA Standard plot (Supplementary Figure S1B).
Statistical analyses.
Statistical analyses were performed using Prism 9 software (GraphPad Software Inc.). Statistical tests are reported in the relevant captions. Values of P <0.05 were considered significant and were adjusted for multiple comparisons as indicated in the legends. Analysis of differences between two normally distributed test groups was performed using an unpaired t test assuming unequal variance. For t tests comparing only two groups within a larger data set, the mean of each treatment group was compared only to the mean of the control group and the Welch’s t test with multiple comparisons correction was applied. Data are expressed as mean ± SD, unless otherwise indicated.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by funding from National Institute of Health grants R01CA253316, R01CA250557, R01CA210553, R01CA112356, NIH T32 CA118681, and T32GM007276. Illustrations were created in part using Biorender.com. The content is solely the responsibility of the authors and does not necessarily represent the official view of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
The authors declare no competing financial of interest.
Supplementary Information Available: Additional information for Methods, Supplementary information (SI)
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
DATA AVAILABILITY
The raw data required to reproduce these findings are available to download from Figshare at https://doi.org/10.6084/m9.figshare.17869010.v1.
REFERENCES
- 1.Blattman JN and Greenberg PD, Cancer immunotherapy: a treatment for the masses. Science, 2004. 305(5681): p. 200–5. [DOI] [PubMed] [Google Scholar]
- 2.Marin-Acevedo JA, et al. Cancer immunotherapy beyond immune checkpoint inhibitors. J Hematol Oncol, 2018. 11(1): p. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Castellarin M, et al. Driving cars to the clinic for solid tumors. Gene Ther, 2018. 25(3): p. 165–175. [DOI] [PubMed] [Google Scholar]
- 4.Khalil DN, et al. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol, 2016. 13(5): p. 273–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Appay V and Sauce D, Immune activation and inflammation in HIV-1 infection: causes and consequences. J Pathol, 2008. 214(2): p. 231–41. [DOI] [PubMed] [Google Scholar]
- 6.Marelli-Berg FM and Jarmin SJ, Antigen presentation by the endothelium: a green light for antigen-specific T cell trafficking? Immunol Lett, 2004. 93(2-3): p. 109–13. [DOI] [PubMed] [Google Scholar]
- 7.Cardiff RD, et al. Quantitation of fixative-induced morphologic and antigenic variation in mouse and human breast cancers. Lab Invest, 2013. 93(4): p. 480–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chew V, et al. Chemokine-driven lymphocyte infiltration: an early intratumoural event determining long-term survival in resectable hepatocellular carcinoma. Gut, 2012. 61(3): p. 427–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dahlin AM, et al. Colorectal cancer prognosis depends on T-cell infiltration and molecular characteristics of the tumor. Mod Pathol, 2011. 24(5): p. 671–82. [DOI] [PubMed] [Google Scholar]
- 10.Haber L, et al. Generation of T-cell-redirecting bispecific antibodies with differentiated profiles of cytokine release and biodistribution by CD3 affinity tuning. Sci Rep, 2021. 11(1): p. 14397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li J, et al. CD3 bispecific antibody-induced cytokine release is dispensable for cytotoxic T cell activity. Sci Transl Med, 2019. 11(508). [DOI] [PubMed] [Google Scholar]
- 12.Trepotec Z, et al. Delivery of mRNA Therapeutics for the Treatment of Hepatic Diseases. Mol Ther, 2019. 27(4): p. 794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kowalski PS, et al. Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Mol Ther, 2019. 27(4): p. 710–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Orlandini von Niessen AG, et al. Improving mRNA-Based Therapeutic Gene Delivery by Expression-Augmenting 3′ UTRs Identified by Cellular Library Screening. Mol Ther, 2019. 27(4): p. 824–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou Z, et al. Nonviral cancer gene therapy: Delivery cascade and vector nanoproperty integration. Adv Drug Deliv Rev, 2017. 115: p. 115–154. [DOI] [PubMed] [Google Scholar]
- 16.Buck J, et al. Lipid-Based DNA Therapeutics: Hallmarks of Non-Viral Gene Delivery. ACS Nano, 2019. 13(4): p. 3754–3782. [DOI] [PubMed] [Google Scholar]
- 17.Habrant D, et al. Design of Ionizable Lipids To Overcome the Limiting Step of Endosomal Escape: Application in the Intracellular Delivery of mRNA, DNA, and siRNA. J Med Chem, 2016. 59(7): p. 3046–62. [DOI] [PubMed] [Google Scholar]
- 18.Zhang J, et al. Interaction of cholesterol-conjugated ionizable amino lipids with biomembranes: lipid polymorphism, structure-activity relationship, and implications for siRNA delivery. Langmuir, 2011. 27(15): p. 9473–83. [DOI] [PubMed] [Google Scholar]
- 19.Swaminathan G, et al. A Tetravalent Sub-unit Dengue Vaccine Formulated with Ionizable Cationic Lipid Nanoparticle induces Significant Immune Responses in Rodents and Non-Human Primates. Sci Rep, 2016. 6: p. 34215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leung AK, et al. Microfluidic Mixing: A General Method for Encapsulating Macromolecules in Lipid Nanoparticle Systems. J Phys Chem B, 2015. 119(28): p. 8698–706. [DOI] [PubMed] [Google Scholar]
- 21.Kulkarni JA, et al. On the Formation and Morphology of Lipid Nanoparticles Containing Ionizable Cationic Lipids and siRNA. ACS Nano, 2018. 12(5): p. 4787–4795. [DOI] [PubMed] [Google Scholar]
- 22.Hou X, et al. Lipid nanoparticles for mRNA delivery. Nature Reviews Materials, 2021. 6, 1078–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schoenmaker L, et al. mRNA-lipid nanoparticle COVID-19 vaccines: Structure and stability. Int J Pharm, 2021. 601: p. 120586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hassett KJ, et al. Optimization of Lipid Nanoparticles for Intramuscular Administration of mRNA Vaccines. Mol Ther Nucleic Acids, 2019. 15: p. 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wood H, FDA approves patisiran to treat hereditary transthyretin amyloidosis. Nat Rev Neurol, 2018. 14(10): p. 570. [DOI] [PubMed] [Google Scholar]
- 26.Leo O, et al. Identification of a monoclonal antibody specific for a murine T3 polypeptide. Proc Natl Acad Sci U S A, 1987. 84(5): p. 1374–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kung P, et al. Monoclonal antibodies defining distinctive human T cell surface antigens. Science, 1979. 206(4416): p. 347–9. [DOI] [PubMed] [Google Scholar]
- 28.Chatenoud L, CD3-specific antibody-induced active tolerance: from bench to bedside. Nat Rev Immunol, 2003. 3(2): p. 123–32. [DOI] [PubMed] [Google Scholar]
- 29.Labrijn AF, et al. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov, 2019. 18(8): p. 585–608. [DOI] [PubMed] [Google Scholar]
- 30.Parayath NN, et al. In vitro-transcribed antigen receptor mRNA nanocarriers for transient expression in circulating T cells in vivo. Nature Communications, 2020. 11(1): p. 6080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wong JT and Colvin RB, Selective reduction and proliferation of the CD4+ and CD8+ T cell subsets with bispecific monoclonal antibodies: evidence for inter-T cell-mediated cytolysis. Clin Immunol Immunopathol, 1991. 58(2): p. 236–50. [DOI] [PubMed] [Google Scholar]
- 32.Loubaki L, Tremblay T, and Bazin R, In vivo depletion of leukocytes and platelets following injection of T cell-specific antibodies into mice. J Immunol Methods, 2013. 393(1-2): p. 38–44. [DOI] [PubMed] [Google Scholar]
- 33.Wesselborg S, Janssen O, and Kabelitz D, Induction of activation-driven death (apoptosis) in activated but not resting peripheral blood T cells. J Immunol, 1993. 150(10): p. 4338–45. [PubMed] [Google Scholar]
- 34.Ferran C, et al. Cytokine-related syndrome following injection of anti-CD3 monoclonal antibody: further evidence for transient in vivo T cell activation. Eur J Immunol, 1990. 20(3): p. 509–15. [DOI] [PubMed] [Google Scholar]
- 35.Perruche S, et al. CD3-specific antibody-induced immune tolerance involves transforming growth factor-beta from phagocytes digesting apoptotic T cells. Nat Med, 2008. 14(5): p. 528–35. [DOI] [PubMed] [Google Scholar]
- 36.Ramishetti S, et al. Systemic Gene Silencing in Primary T Lymphocytes Using Targeted Lipid Nanoparticles. ACS Nano, 2015. 9(7): p. 6706–16. [DOI] [PubMed] [Google Scholar]
- 37.Lee SH, et al. Feasibility of real-time in vivo 89Zr-DFO-labeled CAR T-cell trafficking using PET imaging. PLoS One, 2020. 15(1): p. e0223814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moffett HF, et al. Hit-and-run programming of therapeutic cytoreagents using mRNA nanocarriers. Nat Commun, 2017. 8(1): p. 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith TT, et al. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat Nanotechnol, 2017. 12(8): p. 813–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chavez M, et al. Distinct immune signatures in directly treated and distant tumors result from TLR adjuvants and focal ablation. Theranostics, 2018. 8(13): p. 3611–3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seo JW, et al. CD8(+) T-Cell Density Imaging with (64)Cu-Labeled Cys-Diabody Informs Immunotherapy Protocols. Clin Cancer Res, 2018. 24(20): p. 4976–4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Silvestrini MT, et al. Priming is key to effective incorporation of image-guided thermal ablation into immunotherapy protocols. JCI Insight, 2017. 2(6): p. e90521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tan YH, et al. A nanoengineering approach for investigation and regulation of protein immobilization. ACS nano, 2008. 2(11): p. 2374–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brader ML, et al. Encapsulation state of messenger RNA inside lipid nanoparticles. Biophys J, 2021. 120(14): p. 2766–2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Patel S, et al. Naturally-occurring cholesterol analogues in lipid nanoparticles induce polymorphic shape and enhance intracellular delivery of mRNA. Nat Commun, 2020. 11(1): p. 983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eygeris Y, et al. Deconvoluting Lipid Nanoparticle Structure for Messenger RNA Delivery. Nano Letters, 2020. 20(6): p. 4543–4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McKinlay CJ, et al. Enhanced mRNA delivery into lymphocytes enabled by lipid-varied libraries of charge-altering releasable transporters. Proc Natl Acad Sci U S A, 2018. 115(26): p. E5859–e5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fenton OS, et al. Synthesis and Biological Evaluation of Ionizable Lipid Materials for the In Vivo Delivery of Messenger RNA to B Lymphocytes. Adv Mater, 2017. 29(33). [DOI] [PubMed] [Google Scholar]
- 49.Scott DE, et al. Anti-CD3 antibody induces rapid expression of cytokine genes in vivo. J Immunol, 1990. 145(7): p. 2183–8. [PubMed] [Google Scholar]
- 50.Pepini T, et al. Induction of an IFN-Mediated Antiviral Response by a Self-Amplifying RNA Vaccine: Implications for Vaccine Design. J Immunol, 2017. 198(10): p. 4012–4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vogel AB, et al. Self-Amplifying RNA Vaccines Give Equivalent Protection against Influenza to mRNA Vaccines but at Much Lower Doses. Mol Ther, 2018. 26(2): p. 446–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reinhard K, et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science, 2020. 367(6476): p. 446–453. [DOI] [PubMed] [Google Scholar]
- 53.Blakney AK, et al. Big Is Beautiful: Enhanced saRNA Delivery and Immunogenicity by a Higher Molecular Weight, Bioreducible, Cationic Polymer. ACS Nano, 2020. 14(5): p. 5711–5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huysmans H, et al. Expression Kinetics and Innate Immune Response after Electroporation and LNP-Mediated Delivery of a Self-Amplifying mRNA in the Skin. Mol Ther Nucleic Acids, 2019. 17: p. 867–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miller JK, et al. Suppression of the negative regulator LRIG1 contributes to ErbB2 overexpression in breast cancer. Cancer Res, 2008. 68(20): p. 8286–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Borowsky AD, et al. Syngeneic mouse mammary carcinoma cell lines: two closely related cell lines with divergent metastatic behavior. Clin Exp Metastasis, 2005. 22(1): p. 47–59. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data required to reproduce these findings are available to download from Figshare at https://doi.org/10.6084/m9.figshare.17869010.v1.