

Abstract
Understanding factors that contribute to the escalation of alcohol consumption are key to understanding how an individual transitions from non/social drinking to AUD and to providing better treatment. In this review, we discuss how the way ethanol is consumed as well as individual and environmental factors contribute to the escalation of ethanol consumption from intermittent low levels to consistently high levels. Moreover, we discuss how these factors are modelled in animals. It is clear a vast array of complex, interacting factors influence escalation of alcohol consumption. Some of these factors act early in the acquisition of ethanol consumption and initial escalation, while others contribute to escalation of ethanol consumption at a later stage and are involved in the development of alcohol dependence. It is apparent from our review that much of the literature examines factors contributing to the acquisition of ethanol consumption and on initial escalation from low levels to pharmacologically relevant levels of consumption. Some models capture escalation associated with the formation of dependence; however, neurobiological studies in these models usually focus on comparisons between the AUD model animals and alcohol naïve animals (or animals from other models), making it difficult to distinguish factors associated with the escalation of interest from those associated with consumption in the model per se. There is thus considerable need for more studies examining escalation associated with the formation of dependence as it is of considerable relevance to understanding and treating AUD.
Keywords: alcohol, ethanol, alcohol use disorder, alcohol dependence, alcoholic, alcoholism, incubation of craving, alcohol deprivation effect, drinking in the dark, sucrose fading, intermittent ethanol access, continuous ethanol access, social isolation, social anxiety
1. Introduction
People with AUD (and those without) do not begin voluntarily consuming alcohol at birth, but rather follow several different trajectories of consumption patterns over their lifespan. Ethanol consumption typically begins in adolescence or early adulthood, and for individuals who develop alcohol use problems there can be several different developmental trajectories. Some of these lead to AUD and alcohol dependence, while some do not. For instance, some include problem drinking during adolescence and early adulthood that does not lead to AUD or dependence in the long-term (Behrendt et al., 2008; Costanzo et al., 2007). Many factors are thought to contribute to the development of an escalating pattern of ethanol consumption, including: certain patterns of drinking, such as binge-drinking (Chassin et al., 2002); early life trauma (Davis et al., 2018; Meyers et al., 2019); early initiation of ethanol consumption (Pfefferbaum et al., 2018); parental alcohol and drug use/abuse patterns (Sternberg et al., 2018; Walden et al., 2007); permissive societal norms and/or regulations governing ethanol consumption, particularly in adolescence (Fairman et al., 2019); whether the individual lives in a rural or urban community (Donath et al., 2011); impulse control and other psychological features (Dick et al., 2009; Hardee et al., 2014); genetic factors (Hendershot et al., 2017); and other factors (Brunborg et al., 2018). Not surprisingly, many of these genetic, psychological and environmental factors interact to affect alcohol use/abuse trajectories (Kendler et al., 2011).
This review will discuss these factors alongside the many ways in which these factors have been modelled in animals. The purpose of this review is to identify and study the factors contributing to the escalation of ethanol consumption under controlled circumstances. We consider changes in consumption at different stages of the development of alcohol consumption. We refer to the change from no drinking, or low levels of consumption, to consumption of pharmacologically relevant amounts of alcohol as acquisition or initial escalation. We refer to increases in consumption that would be considered relevant to the formation of alcohol use disorder and dependence as escalation. As will be seen, the work conducted to date has provided valuable insights into factors that contribute to acquisition, initial escalation, and escalation of consumption, but also the biological mechanisms that change over the course of that escalation, especially in earlier stages. However, it is also clear that a detailed causal understanding of the underlying neurobiology of many of these factors contributing to the escalation of alcohol consumption remains lacking, especially those relating to escalation to consumption to levels associated with the formation of dependence and AUD. Discovering the essential mechanisms underlying escalation of ethanol consumption is of considerable important as it will identify points where intervention might be most effective, provide new targets for the development of interventions for AUD, and may also help provide biomarkers for identifying those most at risk. The discussion in this review will confirm and assert that understanding the process of escalation is critical to understanding the development of excessive alcohol consumption, alcohol dependence and AUD, and to better treating AUD.
Figure 1 presents a how different factors can contribute to escalation of ethanol consumption at different stages of drinking, aligned to the stages of life in which they usually occur, from initiation of drinking in first-time users to relapse in AUD, and lists some of the key animal models that facilitate exploration of these different factors.
Figure 1.

Factors contributing to escalation of alcohol consumption throughout the lifespan and at different stages of drinking, and animal models.
2. The impact of how ethanol is consumed on escalation of consumption
The way in which alcohol is consumed can have a significant impact on acquisition, initial escalation and escalation of alcohol consumption. This includes the solution in which alcohol is consumed, the time of day alcohol is consumed, and the frequency and quantity of alcohol consumed. The way alcohol is consumed can lead to changes in psychological functioning underpinned by alterations to neural systems subserving stress coping and reward, which not only appear to play a critical role in initial escalation of consumption, but also changes in craving that occur during abstinence that can reinvigorate consumption. These effects on both stress coping and reward, as well as other acute and chronic effects of alcohol, suggest the importance of both positive and negative reinforcement in increased ethanol consumption over the course of the development of alcohol dependence and AUD. These effects must also be considered in the context of not just ethanol withdrawal-induced impairments in affective and cognitive function, but also pre-existing psychopathology which could contribute to negative reinforcement upon symptom alleviation by alcohol.
In this section we will explore how sweetened alcohol consumption can facilitate acquisition and initial escalation of alcohol consumption and subsequent escalation to binge-drinking. Similarly, we will discuss circadian factors that appear to confer increased risk for escalation of alcohol consumption, which back-translates to animal models showing increased binge-like consumption when alcohol is provided during certain stages of the light-dark cycle. After acquisition of alcohol consumption and initial escalation, intermittent patterns of alcohol consumption appear to facilitate escalation to levels conferring significant risk of developing AUD. We will discuss studies exploring intermittent alcohol exposure and the neurobehavioural adaptations driving escalation. Finally, we will discuss how repeated cycles of withdrawal and abstinence promote changes that lead to an incubation of craving and a subsequent rebound in consumption that is escalated above baseline levels following a period of abstinence; a phenomenon known as the alcohol deprivation effect.
2.1. Sweetened alcoholic drinks
Most people start consuming alcohol during mid-to-late adolescence and it is common among young users to start consuming alcohol for the first time in sweetened beverages and to later transition to stronger and/or unsweetened alcoholic drinks (Roberts et al., 2015; Rossheim and Thombs, 2013). Some evidence suggests that sweetened alcoholic beverages promote acquisition and initial escalation of alcohol consumption among naïve and inexperienced drinkers (Roberts et al., 2015; Rossheim and Thombs, 2013). For instance, Roberts et al. (2015) found that sweetened alcoholic beverages (often referred to as “alcopops”) were among the most highly consumed alcoholic drinks in youth experiencing negative alcohol-related consequences. This study is also supported by epidemiological data showing that increasing popularity of alcopops in the 1990s and 2000s in a number of countries coincided with significant increases in the average amount of alcohol being consumed by adolescents (e.g. Romanus, 2000). This drove policy changes in many of these countries to reduce the accessibility of alcopops, with some evidence this has led to a reduction in alcohol consumption and alcohol-related harms among adolescents and young adults (Gale et al., 2015; Lensvelt et al., 2016; Mojica-Perez et al., 2020).
While the human data points to a possible connection between sweetened drinks, increased consumption in new drinkers, and transition to consumption of stronger alcoholic drinks, studies in animals provide causal evidence that sweetened alcoholic solutions promote acquisition and initial escalation of consumption. This evidence comes primarily from studies using the sucrose-fading initiation procedure in rats (Grant and Samson, 1985a, b; Samson, 1986; Tolliver et al., 1988). At its core, the procedure involves initiating consumption of ethanol by providing rats with access to ethanol in a palatable sucrose solution, then, once responding has stabilised, reducing the sucrose concentration until the rats are consuming a solution of ethanol in water. The procedure can successfully induce high levels of ethanol consumption (leading to BACs > 0.1 g/dL in some animals) consumed in solutions of up to 40% ethanol in water (Simms et al., 2010). Importantly, rats obtain these high levels of alcohol consumption without food or water deprivation. Interestingly, Tolliver et al. (1988) found that male rats with an initially low preference for ethanol show the greatest increase in ethanol preference following initiation using the sucrose-fading procedure. Thus, sweetened alcoholic drinks facilitate acquisition and initial escalation of ethanol consumption, perhaps especially in individuals who might not otherwise consume high levels of ethanol, consistent with conclusions drawn from the human data with alcopops, discussed above.
2.2. Circadian factors
In both humans and in animal models, circadian factors appear to play a key role in the acquisition and initial escalation of alcohol consumption, and may also contribute to escalation associated with AUD. In humans, circadian changes during late adolescence coincide with the initiation of alcohol consumption, and several sleep and circadian factors have been identified as potential contributors to initial escalation of alcohol consumption (for a recent review see Hasler and Pedersen, 2020). Later circadian timing, circadian misalignment, and a range of associated sleep disturbances have all been associated with increased alcohol consumption and in some studies with AUD (e.g. DeMartini and Fucito, 2014; Hasler et al., 2015). It has been proposed that both positive and negative reinforcement processes can play a role in circadian influences on alcohol consumption. Later circadian timing (associated with increased sensitivity to alcohol reward), perceived facilitation of sleep onset by alcohol, and perceived reductions in anxiety following alcohol consumption are among the most commonly reported reasons for drinking (Hasler and Pedersen, 2020).
In animal models, the critical role played by circadian factors in alcohol consumption is also borne out by the significant influence of when alcohol is provided during the light/dark cycle on acquisition of consumption. Rodents are nocturnal, so they show greater activity and ingestive behaviour during the dark cycle. Not surprisingly, alcohol consumption is higher during the dark phase. One model, in particular, warrants discussion here: the drinking-in-the-dark (DID) mouse model of binge-drinking ((Rhodes et al., 2005; Rhodes et al., 2007) and for review see Thiele et al. (2014)). The key goal of the model is to produce heavy drinking within a short period of time. However, the DID procedure might also be considered as accelerating the process of escalation, especially in susceptible animals. Numerous factors consistent with those discussed above affect DID, although considerable work has focussed upon genetic factors (Crabbe et al., 2009; Rhodes et al., 2007). One of those genetic approaches involved selection of high DID lines of mice. It is interesting to note, in the context of the previous discussion about circadian rhythms, that this selection resulted in alterations in circadian rhythms as well as binge-like alcohol consumption.
A common problem with rodent models of alcohol consumption is that subjects often will not voluntarily consume alcohol to levels of behavioural intoxication (BAC > 0.1 g/dl). The DID model overcomes this problem by capitalising on rodents increased ingestive behaviour about a quarter of the way into the dark cycle, although the limited periods of access during each day are also likely to contribute to these higher levels of consumption. In the most commonly utilised format, the DID model involves providing C57BL/6J mice with only 20% (v/v) ethanol in water for 2 hours, 3 hours into the dark cycle for 3 days in a row, with access provided for 4 hours on the fourth day. The model has high face validity as it produces binge-like drinking with short term access to the point of displaying behavioural intoxication – including impaired performance on the rotarod and balance beam test (Thiele and Navarro, 2014) – without requiring a long-history of alcohol consumption, sucrose-fading or long-term fluid or food deprivation.
As is the case with any model, the DID procedure is not without issues. The time-of-day dependency has led to criticisms it is simply capitalising on circadian factors that influence ingestive behaviour, although as discussed above, circadian factors also appear to play a role in increases in alcohol consumption in humans. The lack of choice, with only one bottle being provided, has also been criticised, as has the strain dependency, with C57BL/6J mice being the only strain reliably showing binge-like consumption in this model. That criticism is, arguably, misplaced, as it is clear in humans that genetic and environmental susceptibility contribute to the development of AUD. To put it another way, not all humans develop AUD after the same early alcohol drinking experiences, and the same should be expected of our models. In that sense, differential susceptibility based upon genetic or environmental predisposition, such as that seen with the DID paradigm, improve the validity of the model rather than diminish it. The predictive validity of the DID paradigm for pharmacotherapies further supports the model having translatability to humans with alcohol problems (Crabbe et al., 2017). Finally, given high, stable levels of consumption are usually established rapidly with the DID paradigm (providing a nice model of binge-like consumption), significant escalation is not commonly observed over time; this makes the model more useful for studying factors resulting in rapid acquisition of high levels of consumption, rather than escalation.
2.3. Intermittent access
Evidence from studies in humans suggests that intermittent exposure to ethanol, primarily binge-like consumption patterns, serves to escalate alcohol consumption over time and contributes to the risk of developing AUD. Binge drinking during adolescence and early adulthood is among the strongest predictors of developing AUD later in life; the earlier the pattern of binge-drinking commences, the greater the risk of later developing AUD (Chassin et al., 2002; Spear, 2015). For instance, a study of 21,137 individuals found binge drinking in high school was one of the strongest predictors of developing AUD by age 35 (Merline et al., 2008). Whilst studies in human populations have established a clear correlation between binge drinking and AUD, they are not able to provide evidence for binge drinking having a causal impact on propensity to develop AUD.
Animal studies help provide an understanding of the causal relationship between patterns of heavy intermittent alcohol consumption and establishment of AUD. Many studies have focused on comparing rodents provided with intermittent ethanol access (IEA) to those given continuous ethanol access (CEA). IEA more closely aligns with human patterns of alcohol consumption, in which ethanol usually completely clears from the drinker’s body between drinking sessions. A recent review (Spear, 2020), identified 14 studies that directly compared IEA to CEA in rodents; in all of these studies (Crabbe et al., 2012; Hopf et al., 2010b; Hwa et al., 2011; Hwa et al., 2016; Kimbrough et al., 2017b; Melendez, 2011; Osterndorff-Kahanek et al., 2013; Pinel and Huang, 1976; Rosenwasser et al., 2013; Simms et al., 2008; Sinclair, 1979; Spoelder et al., 2015; Tomie et al., 2006; Wise, 1973) rodents given IEA showed greater escalation of consumption than those given CEA (although note that Crabbe et al., 2012 reported escalation only in one of two strains of mice tested and only under certain conditions). Escalation of consumption with IEA relative to CEA has also been reported in male non-human primates (Lindell et al., 2017), providing further support for the validity of this paradigm and the translatability of findings across species.
Importantly, the escalation of consumption in IEA rodents is accompanied by other hallmarks of AUD which suggest it is an especially useful model for studying escalation relevant to the development of AUD and alcohol dependence. A major feature of AUD is the continuation of use despite negative consequences. One way this is modelled in animals is through assessment of the effect of adulteration of alcohol with quinine on consumption. In one study male IEA rats develop quinine resistance while CEA rats did not (Hopf et al., 2010a). Moreover, the development of the quinine resistance was linked to the escalation and duration of consumption with rats displaying resistance after 3 months, but not 1.5 months, of IEA. A study by one of the authors of this review provided particularly compelling evidence that binge-like drinking could play a causal role in development of AUD, demonstrating that adolescent male rats that had previously been given IEA showed faster escalation to compulsive alcohol consumption (as measured by progressive ratio responding and consumption of quinine-adultered alcohol) in adulthood using the chronic intermittent ethanol vapour exposure paradigm (Kimbrough et al., 2017b). Similarly, intermittent ethanol exposure in male mice during adolescence resulted in greater attenuation of the response to the aversive components of ethanol consumption in adulthood than those exposed to ethanol for the same duration continuously, as evidenced by greater attenuation of ethanol-induced conditioned taste-aversion in adulthood (Diaz-Granados and Graham, 2007). Another key indicator that escalation in IEA models is relevant to AUD comes from work demonstrating that male rats that received chronic IEA developed physical dependence and experienced withdrawal symptoms when ethanol was not available (Li et al., 2011).
2.4. Dependence, withdrawal, and negative reinforcement
It has been argued that negative reinforcement mechanisms play a key role in the escalation of alcohol consumption in dependent binge drinkers (Koob, 2013). This is supported by the observation that escalation of voluntary alcohol consumption is much greater in physically dependent rats (Buck et al., 2014; Vendruscolo and Roberts, 2014). Central to the link between physical dependence, intermittent access, and escalation of consumption, is a phenomenon referred to as “kindling”, which, in the context of alcohol withdrawal, refers to the increase in the severity of alcohol withdrawal symptoms that occurs over successive cycles of intoxication and withdrawal (Becker, 1998). Kindling has primarily been studied in the context of physical alcohol withdrawal symptoms, most notably seizures. However, kindling of physical withdrawal symptoms does not appear to greatly contribute to motivation to resume or increase alcohol consumption (Breese et al., 2005; Heilig et al., 2010). A growing body of evidence indicates kindling also occurs with the negative affective symptoms of withdrawal (for reviews see Becker, 1998; Breese et al., 2005; Heilig et al., 2010).
It appears that heightened anxiety, which is present during acute withdrawal and early abstinence, also becomes more severe over repeated cycles of intoxication and withdrawal and eventually leads to a more enduring state of heightened stress reactivity and impaired stress coping, a phenotype which is believed to play a major role in enduring relapse risk during protracted withdrawal (Koob and Le Moal, 2005). In this sense, repeated cycles of withdrawal can be thought of as a form of chronic stress, resulting in dysregulation of stress coping and socioemotional functioning. This greater intensity of negative emotional/motivational signs and symptoms during abstinence from substances of abuse has been coined hyperkatifeia (Shurman et al., 2010). Hyperkatifeia is associated with an increase in anxiety-like behaviours, stress-like behaviours, and pain-related behaviours during abstinence from alcohol and has been validated in both humans and rodents (Koob, 2021).
Negative reinforcement through alleviation of negative emotional/motivational symptoms, particularly anxiety, pain, and heightened stress reactivity, thus likely plays a crucial role in increasing motivation to consume alcohol when there is heavy intermittent exposure, especially in dependent animals. Consistent with this hypothesis, cues associated with negative affective states cause an escalation of alcohol consumption in male rats (Berger et al., 2013), and it is well established that stress, in particular social stress, can lead to escalation of alcohol consumption (which will be discussed in more detail later in this review). Hwa et al. (2016) specifically examined the link between stress, IEA and escalation of consumption in male mice. They found that a combination of social defeat stress and IEA led to the most pronounced escalation of ethanol intake. Newman et al. (2018a), extended on these findings, demonstrating that while socially stressed male mice with CEA reduced 24-hour alcohol consumption when administered a CRF-R1 antagonist, this effect was absent in stressed mice given IEA, suggesting that IEA leads to more complex neuroadaptations in stress pathways, conferring potential resistance to certain pharmacotherapies.
2.4.1. Biological systems implicated in negative reinforcement driven escalation
Negative reinforcement mechanisms underlying escalation of ethanol consumption may involve the κ-opioid receptor (KOR; OPRK) system. Chronic intermittent ethanol vapour exposure sensitizes KORs, and is associated with increased ethanol intake, ethanol preference and anxiety in male mice (Rose et al., 2016). These consequences were reduced by KOR antagonists and associated with a hypodopaminergic state, including reduced dopamine release and increased dopamine uptake. Conditions that induce affective impairments, and the mechanisms that underlie these changes, may be critical for escalation of ethanol consumption as the organism learns that the drug reverses these undesirable subjective states.
The biological changes underlying the development of negative reinforcement likely lie within a broad circuitry involving CRF, particularly those lying within the extended amygdala circuitry (Fig. 2). This circuitry is integrated into mesolimbic dopamine circuits underlying ethanol reinforcement as well as corticostriatal circuits underlying behavioural choice. The role of CRF in alcohol dependence is well-established (for review see Simpson et al. (2020) and Koob (2009)), and linked in part to the role that CRF plays in somatic and affective withdrawal symptoms (Kimbrough et al., 2017a; Schuckit, 2009). Chronic exposure to alcohol leads to down-regulation of periventricular nucleus CRF systems and the HPA axis, but increased CRF function in the extended amygdala (Logrip et al., 2013; Rivier et al., 1984), along with a network of other transcriptional changes (Contet et al., 2011). Moreover, over repeated binge drinking sessions in the DID paradigm, central amygdala CRF neurons become sensitised to ethanol effects in male and female mice (Aroni et al., 2021). At the same time that CRF function is elevated in the extended amygdala, neuroendocrine tolerance might contribute to sensitization of mesolimbic and mesocortical dopamine systems, and consequently to reduced prefrontal inhibitory control of mesolimbic reward and hypothalamic stress systems (for discussion of this theory see Blaine et al. (2016)). A further link in this cascade of events is CRF1A receptor dependent neuroimmune activation (Breese and Knapp, 2016).
Figure 2. Theories of brain regions that are involved in the neurobiology of alcohol use disorder.
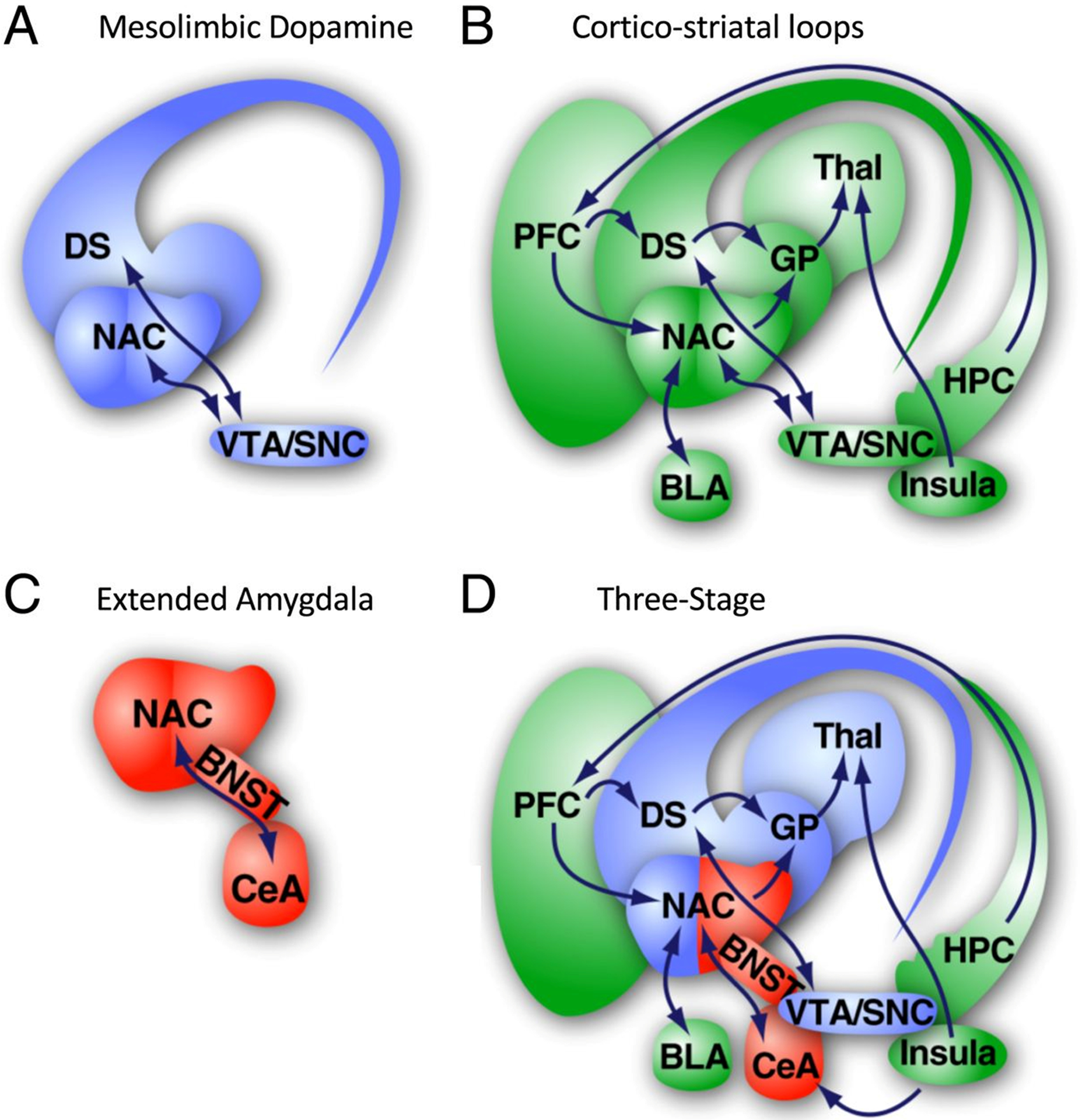
A. Brain regions of the mesolimbic dopamine system. B. Brain regions consisting of cortico-striatal loops. C. Extended amygdala brain regions. D. Three-stage theory. Reprinted from Kimbrough et al. (2020).
One of the important points to bear in mind when considering the state of CRF systems in AUD, is that this state is not constant, as reflected in animal models of ethanol dependence and withdrawal. Thus, CRF levels in the bed nucleus of the stria terminalis are elevated during withdrawal, but normalized by ethanol consumption in male rats (Olive et al., 2002). A 24-hr period of abstinence was associated with increased numbers of Fos-reactive cells in the medial prefrontal cortex (mPFC) and the central amygdala (CeA) in male rats that had been previously exposed using chronic alcohol administration (George et al., 2012). The majority of these Fos positive cells in the CeA are also CRF positive (De Guglielmo et al., 2019). Importantly, changes in the CeA were only observed after intermittent access, and not in animals that had been given continuous access (George et al., 2012). This elevated neuronal activity was normalized by ethanol consumption, so likely represents a counter-adaptation to the higher levels of ethanol consumption during intermittent access, the associated periods of forced abstinence, or the negative reinforcement involved in ethanol consumption after those periods of abstinence.
Importantly, these changes were found to affect the connectivity between important brain regions subserving alcohol seeking behaviour, as determined by correlations between Fos activity in corticolimbic brain regions. Abstinence after intermittent access was associated with reduced connectivity between the dorsal mPFC (dmPFC) and the CeA, the ventral mPFC (vmPFC) and the CeA, and also between the dmPFC and vmPFC. This contrasts with the strong correlation observed between these structures in rats that had been given continuous access to ethanol and did not show escalation of ethanol consumption. The activated neurons in the mPFC were shown to be GABA- and CRF-positive.
An important idea underlying this research is that coordinated activity within ensembles of neurons underlies responses to stimuli that drives drug-seeking behaviour (George and Hope, 2017). Inactivation of the neuronal ensemble comprised of Fos-positive cells in the CeA after 24h of abstinence using the Daun02 technique in non-dependent male rats with intermittent access to alcohol produced limited reductions in Fos-positive cells and only a transient (24 h) reduction in ethanol consumption (de Guglielmo et al., 2016). This result suggests that in non-dependent rats, the Fos-positive CeA neuronal ensemble activated during abstinence is labile and only partially controls alcohol drinking. To induce a greater degree of dependence, a vapour chamber approach was then used (de Guglielmo et al., 2016), along with intermittent access to operant ethanol self-administration. In this case, the reduction in the number of Fos-positive cells was greater, and the reduction in ethanol self-administration was long lasting (at least 2 weeks), suggesting that in dependent rats, the Fos-positive CeA neuronal ensemble activated during abstinence is stable and fully controls alcohol drinking. Optogenetic inhibition of CRF cells in the CeA that project to the bed nucleus of the stria terminalis (BNST) prevents recruitment of these neuronal ensembles, reduces escalation of ethanol consumption, and decreases somatic signs of withdrawal in male rats (De Guglielmo et al., 2019).
The importance of these CRF-dependent circuits are consistent with numerous studies demonstrating effects of CRF antagonists on ethanol consumption in dependent subjects, but not non-dependent subjects (Funk et al., 2007; Gilpin et al., 2008), including when injected only into the CeA (Funk and Koob, 2007). Moreover, these studies appear to dissociate the role of CRF between drinking after the establishment of dependence and in non-dependent binge-drinking (Ji et al., 2008). Of course, binge-drinking can contribute to the development of dependence and escalation of ethanol consumption due in part to its intermittent aspects, but CRF in the CeA circuit does not appear to be an important regulator of consumption until after dependence has developed. Many studies examining increases in ethanol consumption examine initial escalation in non-dependent individuals, rather than escalation to the point where the subject is physically dependent, although this is not always a line that is clearly drawn experimentally as physical dependence is not assessed in many studies. Withdrawal symptoms certainly can emerge early on in the establishment of addiction, and indeed may be an important part of the development of the dependent state. CRF sensitivity is reduced during withdrawal in BNST neurons projecting to the ventral tegmental area (VTA) (Silberman et al., 2013), while CRF-mediated potentiation of glutamatergic VTA afferents is enhanced after a binge ethanol procedure (DID) (Sparta et al., 2013). Intra-VTA CRF antagonists reduce ethanol consumption under these conditions, consistent with other observations (Hwa et al., 2013).
Collectively, the literature discussed above suggests that several CRF-modulated neural circuits are involved in alcohol dependence, and potentially have a role in escalation of ethanol consumption. Kimbrough et al. (2020) recently confirmed much of this model using immunolabeling-enabled three-dimensional imaging of solvent cleared organs (iDISCO) in male mice. Like the previous study from this group (De Guglielmo et al., 2019), single cell Fos activation was compared between brain regions, but on a much broader scale facilitated by the use of the iDISCO technique. The primary comparisons were between dependent and non-dependent ethanol exposed animals. This study confirmed many aspects of previous findings in alcohol dependent/abstinent animals, including the importance of changes in the connectivity of the extended amygdala and midbrain striatal modules discussed above, as well as the emergence of a novel cortico-hippocampo-thalamic module (the three-stage theory, Figure 2D). Overall, there was a broad functional reorganization, whereby dependence/abstinence was characterized by larger networks of co-activation within these regions, in contrast to the smaller, more diverse networks of co-activation that characterized non-dependent individuals. Many regions became incorporated into the cortico-hippocampo-thalamic module, becoming co-activated in dependent/abstinent individuals, while this activity was negatively correlated with activity within the extended amygdala module.
2.5. Incubation of craving and the alcohol deprivation effect
Intermittent patterns of ethanol consumption are thought to be at least partly driven by negative reinforcement from alleviation of acute withdrawal induced anxiety and dysphoria. However, whilst relapse risk is usually highest during acute withdrawal, relapse does occur in individuals who have remained abstinent beyond the acute withdrawal period (Kirshenbaum et al., 2009). The discussion above shows that initial escalation appears to occur in non-dependent individuals, but then further changes are necessary for the transition to alcohol dependence. As individuals who relapse beyond one week of entering sobriety are no longer experiencing acute withdrawal symptoms, other factors present during abstinence must play a key role in relapse in these individuals. These factors may include enduring heightened stress reactivity and alterations to reward and motivational systems (for review of the relationship of this protracted withdrawal syndrome to relapse, see Koob and Volkow (2016) and Beracochea et al. (2019)). An important factor that describes aspects of the change in drug seeking phenotypes over prolonged periods of abstinence is called incubation of craving.
Incubation of craving refers to the time-dependent increase in cue-induced craving or drug-seeking observed during abstinence. Numerous studies have shown that exposing individuals with AUD who are abstinent to alcohol-associated cues induces craving (Fox et al., 2007; Petrakis et al., 2001; Sinha and Li, 2007). Moreover, craving is a significant predictor of relapse (Bottlender and Soyka, 2004; Stohs et al., 2019). An important insight of these observations was that craving was often higher after an extended period of abstinence, beyond the period of initial withdrawal, than it was at earlier timepoints. This incubation of craving during abstinence thus may play an important role in relapse beyond the acute withdrawal period. This extended elevation of the likelihood of relapse suggests the involvement of processes other that just alleviation of acute withdrawal symptoms. Factors influencing this long-term increase in relapse rates have been linked to cue-induced or stress-induced relapse, and thus may involve learning that occurs early in the addictive process, during intermittent periods of consumption and abstinence, occurring before more consistent drug consumption develops. Of course, learning that occurs late in the addiction process is also likely a major contributor to persistent risk of relapse in abstinent individuals, with phenomenon such as conditioned withdrawal not expected in individuals with only limited exposure to alcohol.
Bienkowski et al. (2004) were the first to demonstrate incubation of craving for alcohol. In their study, male rats stably lever pressing for 8% alcohol for 30 days (following an ~20-day training and induction protocol) underwent either 24 h, 28 days, or 56 days of forced abstinence. Rats that underwent 28 days of abstinence showed the highest levels of lever pressing under extinction conditions, whereas those that underwent 56 days of abstinence showed the greatest cue-induced reinstatement of alcohol seeking. More recently, Li et al. (2015) provided the first experimental evidence for incubation of alcohol craving in humans. They assessed cue-induced alcohol craving in adult male inpatients with alcohol use disorder following 7, 14, 20 and 60 days of abstinence. Both between and within subjects, craving was at its highest after 60 days of abstinence, consistent with previous studies suggesting relapse risk remains high around this timepoint (Kirshenbaum et al., 2009). Importantly, this incubation of cue reactivity has been replicated in male alcohol dependent patients, and shown to be reduced by treatment with naltrexone (Bach et al., 2020b).
Given this incubation of craving, it is perhaps not surprising that relapse usually involves not just the rapid resumption of alcohol consumption, but escalation to levels of consumption that are initially above those consumed prior to entering abstinence (Burish et al., 1981; Ludwig and Wikler, 1974; Ludwig et al., 1974; O’Donnell, 1984). This transient escalation of consumption upon relapse is often referred to as the alcohol deprivation effect (ADE). ADE has been most closely studied in animals and has been shown in rats, mice and non-human primates (Sinclair, 1971). However, it should be noted that not all species display an alcohol deprivation effect and there are strain dependencies within species (for a review see Vengeliene et al., 2014). In general, the ADE is less consistently observed in mice than rats (Vengeliene et al., 2014). In at least some species and strains, the magnitude of the ADE increases with repeated phases of deprivation and access (Martin-Fardon and Weiss, 2013; Vengeliene et al., 2014). This suggests repeated cycles of abstinence and relapse can also serve to escalate consumption over time, which fits with the pattern of drinking observed in some humans with alcohol use disorder (Martin-Fardon and Weiss, 2013) and may be driven by strengthening of learning through repetition of negative reinforcement and changes in negative affect, along with underlying neuroadaptations.
Withdrawal-dependent plasticity underlies drug relapse behaviour studied in reinstatement and related procedures, including incubation of drug craving (for review see Dong et al. (2017)). These mechanisms include increased cell-surface expression of AMPA glutamate receptors in nucleus accumbens (NAc) medium spiny neurons (Christian et al., 2017; Werner et al., 2017), elevated glutamatergic synaptic activity (Conrad et al., 2008; Wolf and Tseng, 2012), increased GluA1 translation (Stefanik et al., 2018), and changes in subunit composition (Conrad et al., 2008). Similar changes occur under conditions that produce incubation of cocaine drug-seeking (Ma et al., 2014; McCutcheon et al., 2011). This process appears to be initiated by the early formation of silent synapses during withdrawal that are subsequently activated by increased cell-surface expression of AMPA receptors (Lee et al., 2013; Ma et al., 2014). Over the course of these adaptations in glutamatergic synapses there are also changes in receptor subunit expression, with increases in GluN2B expression in the first week of withdrawal followed by increased expression of GluN3 subunits 1 to 2 weeks later (Christian et al., 2017). Alterations in glutamatergic neurotransmission during abstinence/withdrawal also involve changes in expression of the glutamate transporter GLT1 (Kim et al., 2018). There are also changes in GABA receptors (Purgianto et al., 2016) that are a part of overall circuit-level changes that involve other brain regions, including the amygdala (Lu et al., 2007), ventromedial prefrontal cortex (Shin et al., 2016; Shin et al., 2018) and the strengthening of the prefrontal cortex–nucleus accumbens pathway (Luis et al., 2017). Incubation of cocaine seeking can be attenuated by mGluR2/3 agonist injection in the amygdala (Lu et al., 2007) and an mGluR1-dependent long term depression emerges in the nucleus accumbens after 35 days of withdrawal (Scheyer et al., 2018). The mechanisms of cellular change noted to occur during incubation, primarily in glutamatergic synapses also involve brain derived neurotrophic factor (Grimm et al., 2003; Schmidt et al., 2012), glial derived neurotrophic factor (Lu et al., 2009), and sensitized kinase signalling (Lu et al., 2006; Szumlinski and Shin, 2018).
The mechanisms that have been shown to be involved in incubation of cocaine-seeking behaviour are also thought to underlie incubation of drug-seeking for other drugs, although these mechanisms have not been studied as extensively for other drugs, particularly alcohol. ADE shows many similarities to incubation of drug seeking, as do other experimental procedures that utilize periods of abstinence to induce increases in ethanol consumption. Despite having been studied for much longer, only limited studies have addressed the underlying mechanisms of ADE. Glutamate has been shown to be important in ADE in ways that are very similar to incubation, but evidence is primarily based upon pharmacological approaches. NMDA receptor antagonist treatments reduce ADE-induced ethanol consumption (Holter et al., 1996, 2000; Vengeliene et al., 2005), as does an mGluR5 antagonist (Backstrom et al., 2004), and the AMPA antagonist GYKI 52466 (Sanchis-Segura et al., 2006). Other treatments that modify NMDA receptor activity also reduce ADE, including glycine transporter inhibition (Vengeliene et al., 2010), and inhibition of the kynurenine-3-monooxygenase (Vengeliene et al., 2016). Lamotrigine, a Na-channel inhibitor that reduces glutamate, dopamine and serotonin activity, also reduces ADE-induced ethanol consumption (Vengeliene et al., 2007). Acamprosate also reduces c-fos activation produced by ADE (Putzke et al., 1996). Like incubation, ADE also involves glutamate activity in the PFC as evidenced by the ability of injections of glutamate or acamprosate into the PFC to reduce ADE-induced ethanol consumption (Salimov and Salimova, 1993; Spanagel et al., 1996).
The long-term consequences of over a year of repeated ADE cycles in alcohol-preferring male rats was used to study the mechanisms underlying ADE (Vengeliene et al., 2006). Comparisons were made between rats that had undergone extensive and repeated ADE cycles, and those that had not been given alcohol; although to better identify factors specifically involved in escalation it would also have been useful to have additional control groups such as a single ADE cycle group with similar duration of alcohol exposure to the repeated cycle group, and a group given alcohol for a similar duration to the ADE groups but never deprived. Gene expression array analysis found 266 differentially expressed genes in the striatum of P rats and 140 differentially expressed genes in the striatum of HAD rats. The full gene list was not reported, unfortunately, but it is likely that there are substantial differences in the expression patterns between these strains as the majority of gene expression changes in P rats involved down-regulation, while the majority of changes in HAD rats involved upregulation. The authors focussed upon changes in dopaminergic system genes, which were substantially altered in P rats, but not HAD rats. Increased Drd3 receptor mRNA in P rats was confirmed by quantitative reverse transcription polymerase chain reaction (qRT-PCR) in the dorsal striatum, but not the nucleus accumbens. ADE-induced ethanol consumption was also reduced by a DRD3 receptor antagonist or partial agonist. Unfortunately, that study did not report changes in glutamatergic receptors. However, a subsequent study found that long-term ethanol consumption with repeated ADE cycles was associated with a reduction in the ratio of NR2C to NR2A subunit expression and increased NR1 subunit expression in male rats (Raeder et al., 2008). The importance of glutamatergic and dopaminergic systems in ADE was demonstrated by selective elimination of Grin1 (GluN1) or Gria1 (GluA1) gene expression in dopamine transporter (DAT) or DRD1 receptor expressing neurons in male mice (Eisenhardt et al., 2015). The genetic manipulations reduced ADE, which was duplicated by administration of NMDA receptor antagonists in the NAC or VTA, effects that could be reversed by potentiation of AMPA receptors.
ADE differs substantially between rats and mice, and among mouse strains (Vengeliene et al., 2014). These data implicate a genetic basis for the propensity to develop ADE. It might be thought that ethanol-preferring strains would be more sensitive to the development of ADE; or alternatively the ADE might exist in these strains prior to the ADE procedure, a type of pre-sensitization. Male ethanol-preferring AA rats did not show ADE in one study (Sinclair and Tiihonen, 1988), although a study from a previous generation of this line showed a longer-lasting ADE effect compared to ANA and outbred rats (Sinclair, 1979). The sP line develops only a short-lasting ADE (Agabio et al., 2000), and this effect does not change with repeated cycles of access and abstinence (Serra et al., 2003). There was also a shift towards a preference for higher ethanol concentrations beginning with the first ADE cycle in that study. Although the ethanol-preferring HAD line does not show an ADE effect after a single deprivation, it does show the effect after repeated deprivations in male rats (Rodd-Henricks et al., 2000). This presents an important link to intermittent access models, which produce escalation and show gradual increases in consumption over repeated periods of consumption and abstinence. Although several studies have not found ADE in C57BL/6J or C57BL/6N mice (Camp et al., 2011; Khisti et al., 2006; Tomie et al., 2013), the observation of ADE is dependent at least in part on experimental parameters (Melendez et al., 2006). A deprivation period of 1 week resulted in ADE in male C57BL/6 mice, but a longer period resulted in reduced ethanol consumption. An additional factor influencing the observation of ADE is the daily length of the ethanol exposure. An 18 hr availability (with 6 hr periods of no ethanol availability) resulted in ADE after a longer period of forced abstinence in male mice (Khisti et al., 2006), but ADE was not observed when initial ethanol consumption was continuous (Tomie et al., 2013).
One explanation for this lack of ADE effects in C57 strains in many studies is a ceiling effect. This is consistent with a study examining male and female disks large MAGUK scaffold protein 4 Dlg4 (PSD95) KO mice on a C57BL/6J background (Camp et al., 2011). No ADE was observed in wildtype (WT) mice that had very high levels of initial ethanol consumption, but ADE was observed in both male and female Dlg4 KO mice that had low initial ethanol consumption. C57BL/6NCrl mice had more moderate levels of ethanol consumption than C57BL/6J mice. C57BL/6J mice did not show ADE after single or repeated ADE cycles. However, C57BL/6NCrl mice did show an ADE effect after the initial deprivation, although the effect was modest, and tolerance developed over repeated ADE cycles rather than sensitization (e.g. escalation). However, the observation of ADE is clearly not dependent on low initial consumption levels alone. DBA/2J mice, which have low initial levels of ethanol consumption, show decreases in consumption after deprivation (Tomie et al., 2013). It may be that there must be a certain threshold level of initial ethanol consumption to see ADE or escalation, for instance consumption at least sufficient to achieve pharmacologically relevant brain levels of ethanol.
One form of the negative reinforcement hypothesis states that alcohol intake may be promoted by higher baseline pathology (such as anxiety) or greater withdrawal-induced anxiety. This hypothesis was explored in high anxiety-related behaviour (HAB) and low anxiety-related behaviour (LAB) rat lines (Henniger et al., 2002). Contrary to expectation, a substantial ADE was observed in both male and female LAB rats, but little or no ADE was observed in HAB rats. However, negative reinforcement may still play a role in ADE in other ways. It is clear that ethanol consumption in this situation is, to some extent, used to self-medicate since non-contingent administration of ethanol (IP) prior to oral access reduced ADE (Vengeliene et al., 2005). Moreover, it may be that contingency of the negative affective state is key as well as contrast to the normal state: i.e. HAB rats experience heightened anxiety all of the time, thus it may be less pronounced a change during withdrawal than for LAB rats.
Collectively, these studies are consistent with two important overall findings that come from animal models of alcohol consumption that are relevant to escalation: 1) paradigmatic differences (e.g. duration of exposure, number of exposures, length of abstinence periods, ethanol concentrations, etc.) have a great deal of influence over whether escalation of ethanol consumption occurs (see summary in Vengeliene et al. (2014)); and 2) genetic contributions to AUD, as seen in animal models, influence very specific phenotypic contributions to AUD. This last idea will be explored in more detail in a subsequent section, but it is made quite clear from a study that examined ADE and stress-induced ethanol consumption (Vengeliene et al., 2003). ADE was observed in Wistar and P rats, but not HAD or AA rats, whereas repeated swim stress increased ethanol intake in Wistar rats, but not in any of the ethanol-preferring strains. Thus, genetic factors can play a key role in determining whether or not environmental risk factors result in escalation of ethanol consumption. It is apparent, however, that the literature still relies heavily, especially in mechanistic studies, on comparisons between exposed vs not exposed animals or between preferring and non-preferring strains, with few a notable exceptions discussed above providing extremely valuable information on factors that might contribute specifically to escalation associated with the formation of dependence and AUD.
3. Individual and environmental factors that influence escalation of ethanol consumption
In the previous section of this review we focused on how the circumstances surrounding alcohol consumption can influence escalation of alcohol consumption. In this section, we will focus on genetic, biological and socioemotional factors that influence acquisition, initial escalation and escalation of alcohol consumption. The preceding discussion identified that increases in consumption relevant to the development of dependence and AUD can be modelled by repeated periods of consumption and abstinence in rodents. However, it is also clear that escalation of consumption in intermittent access, ADE, and incubation of craving models is not universally observed. Susceptibility to these effects is influenced by genetic and environmental factors, which will be further elaborated upon in this section. Moreover, it is also apparent that the underlying psychological and biological mechanisms mediating escalation are diverse, with multiple pathways facilitating escalation of consumption, and perhaps, requiring different treatment approaches in AUD patients depending upon the underlying mechanisms that are present.
3.1. Genetic factors and differences in response to alcohol
Variation in the function of neural and physiological systems associated with alcohol effects have been consistently shown to be of importance in conferring risk for the development of AUD. This involves alterations in dopaminergic, opioidergic, serotonergic, glutamatergic and GABAergic systems, among others. Current knowledge of the genetic basis of AUD rests on the findings of many genetic approaches that will be briefly considered here, including genetic studies in rodents and humans. Most human genetic studies have compared individuals with AUD (or other alcohol-related diagnoses, such as alcohol dependence) and non-affected individuals. Given the binary nature of many of these comparisons, not all of these findings will necessarily directly relate to escalation of ethanol consumption, but rather might be involved and thus should be considered and studied in the context of escalation in future studies. Similarly, mouse genetic studies have been used to confirm the role of particular genes in phenotypes relevant to AUD but have not always examined phenotypes that would necessarily relate to escalation of ethanol consumption. Nonetheless, these studies provide a starting point for understanding the potential genetic contributions to escalation.
Genetic alterations that are relevant to AUD likely involve additive and multiplicative interactions between multiple levels of regulation: genetic, epigenetic, transcriptional and translational. The advent of genomic studies of AUD clearly have shown that alcohol dependence is associated with many genetic changes, and that this underlying genetic causality is highly polygenic and heterogeneous (Salvatore et al., 2019; Tawa et al., 2016). Another way to look at the data from genome-wide association studies (GWAS) studies over the last 20 years is that they have been inconsistent; however, there are many reasons for this seeming inconsistency, not the least of which is that AUD is not a disorder with a singular underlying aetiology nor a singular phenotype. Not surprisingly, then, genetic effects in GWAS studies are stronger for more specific phenotypes, and different AUD phenotypes are associated with different underlying genetics. Research in the post-GWAS research era is beginning to overcome the shortcomings that have been recognized in GWAS studies of addiction for some time (for review see Hall et al. (2013) and Hall (2016)). Recent analytical advances allow the examination of multi-level omics datasets, integrating genetic, epigenetic, transcriptional and proteomic levels to identify complex networks of causality (Weighill et al., 2019), the ability to examine networks of related phenotypes (Chhetri et al., 2019), and the ability to examine complex genetic interactions (Joubert et al., 2018). These techniques will likely greatly advance our understanding of the genetics of AUD in coming years, but our present understanding is largely based upon other approaches that will be discussed here.
Efforts to characterize the genetic contribution to alcohol dependence and AUD liability began with comparisons between closely related individuals. Here it must be noted that most human genetic studies, even recent studies, have examined abuse or alcohol dependence, rather that the newer DSM-V diagnosis of AUD. Although these diagnoses are by no means synonymous, they are certainly overlapping, so here we will most often use the term AUD to broadly encompass alcohol use problems, unless otherwise noted. Estimates of the overall heritability of AUD have averaged about 50% (Verhulst et al., 2015). As summarized in an early review by Schuckit (1985), evidence of a genetic contribution to AUD includes 31% of individuals with AUD having a parent with AUD, monozygotic and dizygotic twin concordance rates of 55% and 28%, respectively, and adoption studies showing a 44% higher rate of AUD in the adopted offspring of parents with AUD.
This high (but by no means absolute) transmission of AUD traits among closely related individuals encouraged the use of genetic markers in linkage transmission studies among closely related individuals (for early reviews see Radouco-Thomas et al. (1979) and Jenkins and Thomas (1981)). However, these studies had trouble controlling for the shared environmental influences among closely related family members (Susser, 1985) and were only able to identify large areas of linkage. Candidate gene studies, primarily among unrelated individuals, thus became the dominant approach to studying the contribution of individual genes to AUD prior to the advent of whole genome approaches and continue to be commonly used.
Candidate gene studies have identified a few large genetic contributions to AUD risk from individual gene variants, however, these are limited to particular circumstances and the majority of gene variants have much smaller effect sizes (for review see Edenberg et al. (2019) and (Sanchez-Roige et al., 2020)). The largest gene effects contributing to AUD involve variation within genes for the alcohol metabolizing enzymes alcohol dehydrogenase (ADH) and aldehyde dehydrogenase (ALDH). Although genetic variants are often considered in terms of predisposing individuals to AUD, in this case, based upon the “flushing” response associated with aldehyde accumulation, some alleles can be thought of as protective against AUD, particularly alleles in the ADH1B and ALDH2 genes ((Couzigou et al., 1994; Higuchi, 1994), and for review see Edenberg et al. (2019)).
Aside from these limited examples of large individual gene effects in AUD, the vast majority of genetic contributions to susceptibility to AUD, and other substance use disorders, are rather small and heterogeneous (for a discussion, see Hall et al. (2013) and Hall (2016)). This creates a problem for both candidate gene studies and GWAS as small effects, particularly if there are numerous different allelic variants within the same gene that may produce similar outcomes, are difficult to detect. Genes identified in candidate gene studies have also largely failed to be identified in GWAS (Olfson and Bierut, 2012) and the degree of replication of candidate genes is often less than is thought (Hall et al., 2013), especially when publication bias for positive associations is taken into account. Candidate gene studies necessitate a priori assumptions about the most important genes for AUD, whereas GWAS makes no such assumptions. GWAS findings have thus highlighted the importance of gene classes that have not been widely considered in the genetic variance that accounts for AUD liability.
We will not summarize all genes suggested to be associated with AUD, either based upon candidate gene studies or GWAS, and refer readers to recent reviews for a more thorough survey (Sanchez-Roige et al., 2020; Schuckit, 2018). However, a few points need to be made regarding these approaches. Clear associations in GWAS can only be seen with very large datasets (Visscher et al., 2017). However, numbers alone are insufficient. Even a study that included over 250,000 subjects only found 10 positive associations with AUD (Kranzler et al., 2019). Several of these were consistent with previous findings from candidate gene studies or GWAS, including several positive signals in ADH genes, as well as the dopamine DRD2 receptor, but many were novel. Moreover, findings in European Americans were not completely replicated in other ethnicities, who had even fewer positive associations identified. Techniques that allow examination of multiple “omic” levels simultaneously and interactive gene effects (Joubert et al., 2019; Joubert et al., 2018; Weighill et al., 2019) will help to unravel the genetics underlying AUD susceptibility.
These types of analyses have yet to be done for AUD, but they may also allow us to address another issue – the issue of which phenotypes should be used in association analyses. AUD is arguably a poor phenotype to examine in GWAS as it is a heterogeneous diagnosis. Associations may be much stronger if sub-phenotypes, or endophenotypes, are used. For example, Kranzler et al. (2019) found partially overlapping, but substantially different, genetic associations with AUD and alcohol consumption measures. This begs the question: should more specific symptoms or traits be examined in genetic studies, rather than the broad diagnoses that have primarily been used to date? In essence, this is part of the same question that underlies efforts to reconsider psychiatric diagnoses using Research Domain Criteria (Young et al., 2017) to better recognise their heterogeneity in both aetiology and presentation.
This heterogeneity is reflected in the wide range of neural and physiological systems that have been explored for their role in AUD. Candidate gene studies have chosen genes for study based upon a priori considerations about susceptibility to AUD (Hall et al., 2013) and this same logic has guided preclinical studies. However, human genetic findings present another issue for preclinical studies – if the contribution of any particular genetic variant is small, will this also mean preclinical manipulations of the same targets will have small effects? Luckily, this does not seem to be the case in many instances, but this may be because the genetic manipulations (most often homozygous gene knockouts) used in preclinical studies produce greater consequences than most genetic variants in humans, which may alter protein functioning or gene expression, but not entirely eliminate expression. This does raise a question of translational validity, but at least makes the experiments tractable.
Bearing these general caveats in mind, the following sections consider many of the systems that have been associated with AUD or alcohol-related phenotypes in human and preclinical genetic studies. Because of the nature of escalation, human studies cannot directly assess the phenomenon as they study AUD after this process occurs, but without doubt at least some of the systems identified contribute to escalation. It is important to note that although positive findings are referenced for many genes/gene variants, there are also many examples of negative findings for the same genes/gene variants. Most preclinical studies in this area also have not directly examined the factors that may contribute to escalation. Thus, these human and preclinical studies may provide some insight into mechanisms that should be examined specifically for their role in the escalation of ethanol consumption in future work.
3.1.1. Metabolic Enzymes
Polymorphisms in the alcohol metabolizing enzymes ADH and ALDH have been consistently associated with AUD ((Chen et al., 1999; Couzigou et al., 1994; Higuchi, 1994), and for review see Edenberg et al. (2019)). Moreover, variation in multiple alcohol metabolism genes combine to contribute to overall risk/protection phenotypes (Chen et al., 1999). These genes not only affect ethanol metabolism and ethanol blood levels, but also subjective effects of ethanol and long-term outcomes of ethanol intake (Agarwal and Goedde, 1992). Much of the focus in this research has been on the negative effects of ethanol, in particular the “flushing” responses resulting from accumulation of acetaldehyde (Harada et al., 1981). The presence of these negative effects is protective, with homozygosity of an ADH1B variant, most often identified in East Asian populations, reducing the risk of alcohol dependence by 8-fold and homozygosity of an ALDH2 variant further enhancing this protective effect (Peng and Yin, 2009). More recent evidence has also identified this protective effect in people of European and African ancestry (Gelernter et al., 2014).
Some animal models have supported the idea that genetic variation in alcohol metabolism genes contributes to alcohol consumption phenotypes. An example of the evidence comes from studies in males and females from the selectively-bred UChA (low-consuming) and UChB (high-consuming) rat lines (Quintanilla et al., 2006). Of particular interest is the description in that paper that high-consuming lines “learn” to drink higher amounts, progressing from low levels of ethanol consumption to higher levels of consumption (i.e. escalation). This escalation may at least partly result from faster acquisition of tolerance in UChB rats and greater ability to metabolize acetaldehyde. Some other selectively-bred ethanol-preferring rat lines also show differences in ethanol metabolism that involve ADH and ALDH activity (Koivisto and Eriksson, 1994; Lodge and Lawrence, 2003a), although such changes in metabolism are accompanied by other wide ranging changes in gene expression (Ciccocioppo et al., 2006). Among the mutations in the human ADH1B gene is a point mutation resulting in an amino acid substitution (Arg47His) that greatly increases enzyme activity and is protective against AUD (Whitfield, 1997, 2002). The rat homologue of this mutation was introduced into UChB rats and produced a similar pattern of phenotypes, including increased liver ADH activity and reduced ethanol consumption in female rats (Rivera-Meza et al., 2010).
Like UChA rats, Aldh2 KO mice show reduced ethanol consumption and increased behavioural responses to ethanol associated with their increased relative acetaldehyde levels (Fernandez et al., 2006; Isse et al., 2005a; Isse et al., 2002; Isse et al., 2005b). Reductions in ethanol consumption in global Aldh2 KO mice were only partially recapitulated by hepatic specific Aldh2 KO or hepatic specific shRNA knockdown (Guillot et al., 2019). This clearly indicates that Aldh2 expression outside of the liver contributes to the overall effects of global Aldh2 reduction.
Adh gene mutants should also greatly affect ethanol metabolism and blood ethanol levels in mice. Notably, although the human ADH1 gene family consists of 3 genes, ADH1A, ADH1B, and ADH1C, the mouse has only one Adh1 gene. Male Adh1 KO mice have been shown to have greatly increased blood ethanol levels and reduced metabolism after a bolus ethanol injection (Okuda et al., 2018). A follow-up study examined chronic continuous access to 10% ethanol over a one-month period (Haseba et al., 2020). Male Adh1 KO mice had much higher blood ethanol levels despite reduced consumption. Moreover, after two weeks of ethanol exposure these mice began dying, with 100% mortality observed by the end of the 8-week period.
As previous discussion in this review has shown, it is likely that escalation requires the attainment of sufficient blood ethanol levels, and consequent behavioural effects, in order to produce counter-adaptations, tolerance, withdrawal and negative reinforcement. It seems likely that differences in the activity of alcohol metabolism genes would contribute to this process, but their specific contribution to escalation of ethanol consumption remains poorly defined.
3.1.2. Dopaminergic systems
Dopamine (DA) is involved in the rewarding and reinforcing effects of ethanol and variation in dopaminergic genes have been thought to modulate these effects, thereby contributing to AUD. Polymorphisms in genes such as SLC6A3, which encodes the dopamine transporter (DAT) protein, have been implicated in the propensity to develop alcohol dependence. A9 carriers of the 40-basepair variable number of tandem repeats (VNTR) polymorphism (rs28363170) were found to have higher synaptic dopamine levels than A10 homozygotes (Fuke et al., 2001), and this polymorphism has been associated with severe alcohol dependence (Du et al., 2011; Köhnke et al., 2005). Consistent with the polygenic nature of the disease, SLC6A3 A10 homozygotes who are also μ-opioid receptor gene (OPRM1) G-allele carriers report steeper increases in the effect of alcohol dosage on stimulation and positive mood (Ray et al., 2014). Therefore, variation in this dopamine transporter gene, in combination with others, influences responses to alcohol and AUD liability.
In addition to SLC6A3 variants, polymorphisms of DRD2 have also been associated with alcohol abuse liability. For instance, an E8 SNP in the 3’ untranslated region of DRD2 and the A/A genotype at this locus have been associated with increased daily alcohol intake and reduced DRD2 function (Finckh et al.; Lucht et al., 2001). Again exemplifying the polygenic nature of AUD, haplotypes composed of both DRD2 and ANKK1 polymorphisms may pre-dispose alcohol dependent individuals to greater incidence of delirium and seizures during withdrawal (Kucharska-Mazur et al., 2012). Collectively, these polymorphisms may affect abuse liability and escalation, especially considering the role that withdrawal symptom severity can play in the escalation of ethanol consumption through negative reinforcement, discussed earlier in this review.
Studies using DAT (Slc6a3) KO mice have provided some information about the potential role of the DAT gene in alcohol dependence, but the results have been somewhat contradictory. A two-bottle choice paradigm that presented increasing concentrations of ethanol (0%, 3%, 6%, 10%, and 15%) showed no difference in ethanol preference or consumption between female heterozygous DAT KO mice and wildtype (WT) mice, but female homozygous DAT KO mice had reduced consumption and preference (Savelieva et al., 2002). In contrast, Hall et al. (2003) found that heterozygous and male homozygous DAT KO mice had greater preference and consumption of ethanol at higher concentrations. Morice et al. (2010) found that DAT KO mice show increased behavioural sensitization to the locomotor stimulant effects of ethanol. Together, these studies indicate that altered DAT expression may affect consumption and other behavioural effects of ethanol, although the results are not entirely consistent. This may indicate that there are additional mediating factors affecting the role of DAT in ethanol consumption and AUD-related phenotypes. The different methods of assessing ethanol consumption, including duration of exposure and ethanol concentration, could explain some differences between these studies of ethanol consumption in DAT KO mice. It is interesting to note that these are also key factors regulating escalation of ethanol consumption.
Dopamine D2 receptors (DRD2) have long (DRD2L) and short (DRD2S) isoforms that are thought to influence motivation and reinforcement for many drugs of abuse. Both male and female DRD2L KO mice have been shown to drink significantly more ethanol in a 4-day DID paradigm, but were less active, leading to the conclusion that the overrepresentation of DRD2S, relative to DRD2L, in DRD2L KO mice contributes to increases in ethanol intake (Bulwa et al., 2011). Stress, specifically chronic mild stress (CMS), significantly increases ethanol intake and preference in male DRD2 −/− and DRD2 +/− mice (Delis et al., 2013). Moreover, ethanol was shown to reverse CMS-induced immobility during a forced swim test in DRD2 +/− mice, but not DRD2 −/− or WT mice. Furthermore, a study measuring receptor levels in the basal forebrain of DRD2 KO mice found that D2 receptor levels were higher in the lateral and medial striatum of WT mice after CMS in mice previously exposed to ethanol than in non-stressed controls that had also been exposed to ethanol. This indicates that chronic exposure to ethanol can prime individuals to changes triggered by other events. Importantly, DRD2 levels were negatively correlated with ethanol intake in male WT mice (Delis et al., 2015), suggesting that the ability to up-regulate DRD2 expression may be adaptive.
3.1.3. Opioidergic systems
Additional potential genetic modulators of the response to alcohol include opioidergic genes such as OPRM1, which encodes the μ opioid receptor. For instance, some studies have associated a SNP (A118G, rs1799971) in OPRM1 with increased susceptibility to alcohol dependence (Bart et al., 2005; Town et al., 1999), although a meta-analysis found no effect of this SNP on risk of alcohol or other substance dependence (Arias et al., 2006). Nonetheless, G-allele carriers were reported to have significantly greater alcohol-induced stimulation, vigour, and positive mood than A-allele homozygotes, supporting the idea that these individuals display greater sensitivity to the hedonic effects of alcohol (Ray et al., 2014; Ray and Hutchison, 2004; Ray et al., 2010), which could be especially relevant for acquisition and initial escalation of consumption. More recent evidence has not shown a link between the A118G SNP and the subjective response to intravenous alcohol, although G-allele carriers made significantly more alcohol requests than A-allele homozygotes when allowed to self-administer alcohol (Hendershot et al., 2016).
Several mouse genetic models have been used to study the role of opioidergic systems in AUD. Oprm KO mice on a 129/Sv x C57BL/6J background have been consistently shown to have decreased ethanol consumption using a number of ethanol consumption procedures (Becker et al., 2002; Hall et al., 2001; Roberts et al., 2000), although effects on two-bottle continuous access ethanol preference are also dependent on genetic background and sex (Hall et al., 2001). Oprm KO also affects ethanol induced dopamine release, which is also dependent on the genetic background of the mice (Job et al., 2007; Ramachandra et al., 2011), suggesting that genetic differences, whether resulting from direct manipulation or genetic background, are highly interactive. Another study found that ethanol consumption was altered in Oprm KO mice, but in a manner that was dependent on early rearing experience and sex (Moriya et al., 2015). Ethanol consumption was increased in male isolation-reared Oprm KO mice, but this effect was the opposite in female mice, in which socially-reared KO mice had increased ethanol consumption. It is clear from these studies that effects of Oprm deletion on ethanol consumption are not always observed, and tend to interact substantially with other factors, either characteristics of the subjects, characteristics of the ethanol exposure, or other environmental factors.
Oprm KO mice have not been explicitly studied under conditions that are likely to show escalation of ethanol consumption. The Moriya et al. (2015) study, like others that have utilized an ascending presentation of ethanol concentrations under continuous access conditions, do not generally show any type of escalation, although this might also be obscured by the procedure. Increases in consumption are observed from low to high doses, which is assumed to result from changing concentrations, but this is also confounded with the length of overall access to ethanol. It will be interesting to determine if escalation is affected in these mice. Some observations suggest that this may be the case. LaBuda et al. (2000) found that Oprm KO mice have blunted anxiolytic responses to ethanol and exhibit withdrawal symptoms earlier than WT mice after deprivation, while another study found greater anxiety-like responses during ethanol withdrawal (Ghozland et al., 2005). During an intermittent access procedure, such effects might promote increases in consumption over time.
Based on the findings discussed above, genetic alterations affecting levels of the primary endogenous ligand for Oprm, β-endorphin, should also be expected to affect ethanol consumption. Homozygous male and female β-endorphin KO mice on a C57BL/6J background showed increased consumption at a low ethanol concentration of 7% (Grisel et al., 1999), with greater intake occurring during a daily 2-hour test and after 2 days of alcohol deprivation in comparison to controls, but there was no difference during 28 days of two-bottle continuous access (Grahame et al., 2000). Although escalation of ethanol consumption was not explicitly studied, this modest ADE suggests that greater effects might be seen with longer or repeated periods of deprivation. Curiously, heterozygous β-endorphin KO mice showed increased drinking at all concentrations in a two-bottle continuous access preference test. This clearly shows that not only are there genetic contributions to ethanol drinking under different circumstances, but also that different degrees of alteration of the same genes can have different effects. Whether this is a result of differential receptor reserve, as has been suggested for some other effects of opioids in Oprm KO mice (Sora et al., 2001), or a matter of different degrees of compensatory changes, is uncertain. However, these types of functional and adaptive complexities are likely to play a role in the consequences of allelic variation on responses to alcohol and the development of AUD in humans.
Thus far, manipulations of dynorphin signalling appear to have inconsistent effects on ethanol consumption. Male and female mice lacking the dynorphin receptor (κ-opioid, Oprk) had reduced ethanol intake in a two-bottle continuous access procedure (Kovacs et al., 2005). In contrast, one study reported increased ethanol consumption in male prodynorphin (Pdyn) KO mice (Femenia and Manzanares, 2012), but the increase was accompanied by compensatory changes in both dopaminergic and opioidergic systems that might account for these effects. The problem of compensatory alterations has been a significant issue for studies in genetically modified mice, particularly homozygous gene KO mice. In any case, another study found that ethanol consumption and preference were reduced in female, but not male, Pdyn KO mice (Blednov et al., 2006), adding further to this inconsistent picture. An additional factor influencing the observation of effects in genetically modified mice is genetic background (the collective genetic variants against which the gene of interest is studied), which differed in these studies. Genetic background is certainly also an issue in human genetic studies, and gene-gene interactions based on differences in genetic background likely account for some of the apparent inconsistencies of genetic effects in humans as well as in mouse models.
3.1.4. Serotonergic systems
Genetic variation in serotonergic system genes have been widely implicated in AUD, either through their effects on responses to ethanol (Matsushita and Higuchi, 2014) or through their effects on traits that may contribute to AUD (Oreland et al., 2018). However, although candidate gene studies have suggested a role for serotonergic genes in AUD, GWAS have generally failed to confirm these findings. Well-established genetic polymorphisms are related to the effects of tryptophan hydroxylase 1 (TPH1) gene variants on suicidality, impulsivity and AUD (Nielsen et al., 1994; Nielsen et al., 1998). The relationship of these gene polymorphisms to central serotonin function is uncertain since the more brain specific TPH gene, TPH2, does not appear to be associated with AUD (Plemenitas et al., 2015). Nonetheless, alterations in serotonin homeostasis, often measured in terms of reduced cerebrospinal fluid levels of 5-hydroxyindoleacetic acid (5-HIAA), are related to AUD. Perhaps not surprisingly, the serotonin transporter (SERT; SLC6A4) has also been widely associated with AUD, although a recent meta-analysis did not support this assertion (Villalba et al., 2015). As for other genetic findings, there may be a variety of reasons for this inconsistency, including that differences in SLC6A4 function may occur only in certain individuals with AUD, or only affect certain AUD- or alcohol-related phenotypes. For example, the 5-HTTLPR polymorphism of SLC6A4 yields two functionally different alleles, a long (L) and a short (S) allele, the latter of which has been associated with increased risk of alcohol-related delirium and seizures (Sander et al., 1997). In the context of escalation of ethanol consumption, greater severity of withdrawal symptoms might promote self-medication and escalation through greater negative reinforcement. Moreover, consistent with the complex nature of genetic causality in AUD, interactions between 5-HTTLPR and DRD2 exon 8 SNP (DRD2 E8, rs6276) influence the likelihood of delirium tremens during alcohol withdrawal (Karpyak et al., 2010). This is just a small number of examples of serotonergic genes associated with AUD, and it remains to be seen which of these may contribute to the escalation process during the development of AUD. Given the nature of genetic studies in humans the role of serotonergic gene polymorphisms specifically in escalation of ethanol consumption is difficult to study, although utilisation of some of the approaches for defining trajectories of addiction would allow useful insights to be gained.
Perhaps not surprisingly given the discussion above, male SERT KO mice have reduced ethanol consumption (Kelai et al., 2003), although only modestly, and this may actually result from increased ethanol sensitivity (Boyce-Rustay et al., 2006). In an operant ethanol self-administration study male SERT KO mice had lower breakpoints when tested on a progressive ratio (Lamb and Daws, 2013). Although much remains to be explored here, this suggests that greater differences may emerge in these mice under certain conditions, such as intermittent access, which remain to be examined. Deletion of the MOA A gene (MAOA) in male mice does not affect free-choice ethanol consumption, but does reduce ethanol-induced sleep and hypothermia (Popova et al., 2000). Such effects are sometimes referred to as “innate tolerance” and might lead to greater or more rapid escalation of ethanol consumption, although this remains to be explored in these mice.
One problem with these sorts of studies is that the extreme nature of a homozygous KO may not closely replicate the sort of variation in gene function that results from the human gene variants associated with AUD, which are generally more subtle. Thus, one approach would be to “knock-in” a human gene variant, or modification similar to such a variant. Even just examining heterozygous KO mice might provide some insight into the effects of more subtle modifications, but most studies tend to ignore the heterozygous condition. One example of this type of model is the insertion of a hypofunctioning R439H Tph2 mutation in a knock-in mouse line, analogous to the R441H polymorphism in humans (Zhang et al., 2005) that reduces brain serotonin levels in mice by 60–80% (Sachs et al., 2014). These mice showed reduced initial ethanol-induced ataxia and tolerance, as well as increased ethanol consumption and preference. Although longer-term ethanol consumption was not examined, differences in initial responses and tolerance suggest that this polymorphism might affect at least initial escalation of ethanol consumption.
Another important regulator of 5-HT homeostasis is the serotonin 5-HT1B receptor (Htr1b). An initial study reported that male and female Htr1b KO mice had increased ethanol consumption compared to WT mice, which was associated with reduced ethanol-induced ataxia and tolerance (Crabbe et al., 1996). This is slightly surprising since Htr1b KO was shown to reduce the rewarding effects of ethanol (Risinger et al., 1996), although increased consumption could also be interpreted as a compensatory response for reduced rewarding effects of ethanol. In any case, a subsequent operant self-administration study, which largely maintained continuous ethanol access (e.g. 23 h/day)), did not find effects of Htr1b KO across most test conditions (Risinger et al., 1999). Studies also failed to find differences in ethanol consumption under continuous access conditions (Bouwknecht et al., 2000; Gorwood et al., 2002).
Although these findings would seem to suggest that Htr1b does not have a role in ethanol-mediated effects or phenotypes relevant to AUD, these few studies are not a sufficient test of this relationship – something that plagues this field in which the simplest situations are generally examined first, and then subsequent studies looking at more complex situations are not conducted because of the initial negative findings. In particular, no studies examining Htr1b KO have examined more chronic ethanol exposure, particularly under circumstances that might produce escalation of ethanol consumption in a manner that is more relevant to the development of AUD. This is true of most other studies of 5-HT receptor gene modifications in mice, including the 5-HT6 (Htr6) receptor KO mouse (Bonasera et al., 2006), and mice over-expressing the 5-HT3A (Htr3a) receptor overall (Metz et al., 2006), or selectively in the forebrain (Engel et al., 1998). Although the studies by Bonasera et al. (2006) and Metz et al. (2006) did find reduced ethanol consumption.
Genetic studies of the 5-HT1A receptor (Htr1a) are lacking, although a post-mortem human brain study found altered 5-HT1A receptor binding was associated with AUD in those who had committed suicide (Underwood et al., 2018). Consistent with this, and suggestive of a potential role in escalation, changes in 5-HT1A receptor expression, especially downregulation in the hippocampus, have been associated with the emergence of alcohol withdrawal-induced anxiety and anxiety during protracted withdrawal following chronic forced alcohol exposure in mice (Breese et al., 2004; Lowery-Gionta et al., 2015; Overstreet et al., 2006; Wills et al., 2009). Another study found a 5-HT1A receptor partial agonist prevented alcohol withdrawal-induced anxiety in mice undergoing chronic binge-like alcohol consumption in the DID paradigm and reversed deficits in hippocampal neurogenesis following chronic DID in male C57BL/6 mice (Belmer et al., 2018). Taken together, these findings suggest changes in 5-HT1A signalling may contribute to escalation-relevant alterations in emotional regulation linked to chronic binge-drinking and withdrawal.
3.1.5. GABAergic systems
Several behavioural effects of alcohol involve gamma-aminobutyric acid, type A receptors (GABAA) (Davies, 2003; Lobo and Harris, 2008) and genetic variance in this receptor has been investigated for its effects on alcohol responses and its role in AUD. A SNP in the gene encoding the GABA α2 subunit (GABRA2) has been associated with alcohol dependence (Edenberg et al., 2004; Vengeliene et al., 2014). Moreover, the synonymous A-to-G substitution in exon 4 of GABRA2 (rs279858) has been associated with differences in the subjective effects of alcohol, including greater stimulatory and euphoric effects in carriers of the C allele (Arias et al., 2014). However, another study reported that homozygotes for the major A allele of rs279858 had greater subjective effects of ethanol (Pierucci-Lagha et al., 2005). Carriers of the minor allele of GABRA2 SNPs rs279858, rs279844, rs279845, rs279826, rs279828, and rs279836 have also been reported to have reduced aversive effects of alcohol (Uhart et al., 2013).
Some of these gene variants may ultimately affect levels of mRNA expression, as post mortem hippocampal samples from people who had AUD also show lower expression of multiple GABAA and GABAB subunit genes including GABBR1, GABRA2, GABRG1, and GABRG2 (Enoch et al., 2012; Zhou et al., 2011). Differential expression of GABA receptor subunits has also been reported in post mortem prefrontal cortex samples (Farris and Mayfield, 2014). Genes implicated in GABA synthesis and transport, including GAD1, SLC6A1, PRAF2, and GPHN, are also down-regulated in hippocampal tissue of humans with AUD (Enoch et al., 2013; Enoch et al., 2012; Zhou et al., 2011). Whether these post mortem observations reflect pre-existing differences, or differences that develop over the course of the disease cannot be determined from these studies. Nonetheless, although this story is certainly complex, it is clear that genetic variation in GABAergic genes has some role in AUD and responses to alcohol.
The involvement of the GABAA receptor in responses to ethanol is also seen in rodent genetic models. Increased ethanol intoxication is observed in male and female long-sleep mice and also in mice with reduced levels of protein kinase C (Harris et al., 1995). Long-sleep mice have an increased latency to regain the righting reflex following acute ethanol injections, and elevated ethanol induced Cl- uptake (Allan and Harris, 1986). Indeed, many of the top candidate genes implicated in AUD code for particular GABAA receptor subunits, including the α2, α6 and γ2 subunits ((Li et al., 2014); and see review by Trudell et al. (2014)).
There are mutant mouse lines available for several GABAA receptor subunits (Boehm II et al., 2004), including null and overexpressing transgenic lines. Deletion of the α1 subunit in male and female mice has been shown to decrease alcohol consumption in an operant self-administration task and to reduce ethanol consumption under limited access conditions (June et al., 2007). However, as noted previously, operant procedures generally involve limited access, and the sucrose-fading procedure used to initiate consumption might have obscured any differences in acquisition or initial escalation (see section 2.1 for a review of sucrose fading procedures and effects on acquisition and initial consumption). Indeed, this approach is somewhat typical of these types of procedures, particularly for operant approaches, in which observations are made only after “stabilization” of responding or intake. In conducting the experiments in this way one of the most important portions of the data may be lost, that which occurs over the course of acquisition and initial escalation, showing the transition from low levels of consumption to pharmacologically relevant levels of consumption.
Other genetically modified mouse lines have also been studied, and the results suggest potential roles for many GABAA receptor subunits in responses to ethanol. However, effects are often complex and interactive. Knockdown of α5 GABAA receptor subunits reduces consumption in male (Boehm II et al., 2004), but not female (Stephens et al., 2005) mice. Deletion of δ or ρ1 subunits also decreases ethanol drinking in a two-bottle choice continuous access procedure (Blednov et al., 2014; Mihalek et al., 2001). Deletion of glutamic acid decarboxylase (GAD) increases consumption in mice on a mixed C57BL/6J x 129/SvJ (N2) background, but not in mice on a congenic C57BL/6J background (Blednov et al., 2010), as assessed in a two-bottle IEA procedure. Ethanol-insensitive α1 knock-in mice did not differ from WT mice in ethanol consumption tested through a continuous two-bottle access procedure (Werner et al., 2006), but mice with α2 subunit knock-in drank more in a two-bottle IEA procedure (Blednov et al., 2011). All of these effects might impact escalation of ethanol consumption, although this has not been explicitly examined for any of these genetically modified mouse lines.
3.1.6. Glutamatergic systems
Ionotropic and metabotropic glutamate receptors are important mediators of the actions of alcohol (D’Souza, 2015) and likely contribute to a broad network of genes involved in AUD (Spanagel et al., 2010). Hypotheses suggesting the fundamental importance of glutamatergic dysfunctions in AUD have existed for some time (see Dodd et al. (2000)). Evidence from many types of studies supports this view, including post-mortem transcriptomic analyses. For instance, a transcriptomic analysis of prefrontal cortex samples from people who had AUD found that the ionotropic receptor NMDA Type Subunit 2B (GRIN2B) and AMPA Type Subunit 1 (GRIA1) were hub genes in a network of transcriptomic changes (Farris and Mayfield, 2014). Moreover, GRIN2B expression is specifically dysregulated in the hippocampus of AUD patients (Zhou et al., 2011). Analysis of frontal cortex modules of AUD patients revealed an upregulation of genes involved in synaptic transmission at glutamatergic synapses, including GRIN1, dynamin (DNM1), syntaxin 1A (STX1A), synapsin 1 (SYN1), synaptophysin (SYP), and the vesicular glutamate transporter 1 (VGLUT1, SLC7A7) (Ponomarev et al., 2012). In another study of post mortem hippocampal tissue samples from AUD patients expression of GRIN2B (encoding GluN2B), GRIA4 (encoding GluA4), GRIK3 (GluR7), GRM3 (mGluR3), and GRIN2D (encoding GluN2D) were upregulated (Enoch et al., 2014). These differences in gene expression no doubt reflect mechanisms that are relevant to AUD, but they do not indicate how this came to pass; whether these effects are the result of genetic differences in these individuals to begin with, the effects of some experience on gene expression, or the effect of the long-term exposure to alcohol. In the context of the present discussion, it is impossible to know how the expression of these genes changed over the course of the development of AUD.
Although changes may be occurring at multiple levels, there is some evidence for genetic differences in some of the genes mentioned above. For instance, a functional polymorphism (Ser310A1a) of the GRIK3 gene, which encodes glutamatergic kainate receptor subunit GlurR7, is associated with delirium tremens during alcohol withdrawal (Preuss et al., 2006). A GRIN2B polymorphism is also associated with earlier onset of alcohol withdrawal symptoms, which may reflect an accelerated trajectory of the development of alcohol dependence (Paul et al., 2017), i.e. faster escalation. Other gene differences, such as the rs6465084 and rs1468412 polymorphisms of the GRM3 gene may result in AUD-associated prefrontal cortical functional changes that result in executive function deficits, which might contribute to escalation of alcohol consumption (Xia et al., 2012). Some polymorphisms in glutamatergic genes associated with AUD also interact with stressful life events (Vrettou et al., 2019), suggesting that gene-environment interactions may underlie some contributions of changes in glutamatergic systems to the development of AUD.
Several studies have used genetically modified lines of mice to examine the role of glutamate in alcohol consumption. Male mice with a genetic deletion of the metabotropic glutamate receptor 2 gene (Grm2) had increased consumption and preference at high concentrations of ethanol in a 2-bottle choice procedure that gradually increased ethanol concentration from 3 to 17% over 80 days (Zhou et al., 2013). As has been mentioned before, because this type of approach uses an ascending presentation of concentrations it confounds consumption at higher concentrations with the potential escalation of consumption occurring over time. The increased consumption seen in Grm2KO mice was seen both at higher concentrations and after a long period of ethanol consumption. This was a long version of this protocol (80 days). Another study using a similar procedure, but with fewer ethanol concentrations (3–9%), did not find differences in consumption or preference between Grm5KO mice and WT mice (Blednov and Harris, 2008). The length of testing was not clearly stated in that study, so it is difficult to compare these two studies. Additionally, these studies showed quite low levels of ethanol consumption overall, which may have influenced the nature of the increase, drawing into question the relevance to understanding escalation pertaining to AUD as well as the interpretation of the observed genotypic effects. This low level of consumption is likely due to the background strain of the mice being used in each case: CD1 (Zhou et al., 2013) and a mixed 129/SvJ-C57Bl/6J background (Blednov and Harris, 2008). This is an important question in evaluating the consequences of genetic manipulations on ethanol consumption overall: whether the procedure used produces escalation in control mice. Genetic background clearly affects ethanol consumption (Belknap et al., 1993), although it is less clear how it affects escalation. If escalation is dependent on initial levels of consumption, it would be clear that consequences of genetic manipulations will only be seen when they exist on particular genetic backgrounds. It remains to be seen how genetic background might interact with genetic manipulations to affect ethanol consumption or escalation, although it is clear that genetic background not only influences the observation of phenotypes, but at times can reverse the direction of effects, as shown in the seminal study by Sittig et al. (2016).
In addition to the influence of genetic background on the observation of effects after genetic manipulations, factors involved in different ethanol consumption procedures clearly are important. The Blednov and Harris (2008) study is particularly interesting in this regard, as it is one of the few genetic studies to extensively examine ethanol consumption using multiple procedures, including a 2-bottle choice continuous access procedure using ascending ethanol concentrations (3–12% EtOH), a 4-bottle continuous access procedure (0, 4, 8 and 12% EtOH), a two-bottle DID procedure (15%), a one bottle DID (15% EtOH), and a limited access procedure with fluid deprivation (5% EtOH). No differences were observed in the 2-bottle choice experiment, the one-bottle DID or the limited access procedure with fluid deprivation, and none of these procedures showed clear escalation in WT mice. In the 4-bottle continuous access procedure there was a slight reduction in ethanol consumption and preference in Grm5 KO mice, which contrasted to the escalation observed in WT mice. More robust escalation was observed in WT mice in the 2-bottle DID, and again this escalation was eliminated in Grm5 KO mice. Another study found reduced consumption and preference for 5 or 10% ethanol in a 2-bottle continuous access consumption model (Bird et al., 2008) in male Grm5 KO mice compared to WT mice. Reduced consumption coincided with observations of increased sensitivity to ethanol in conditioned place preference and loss of righting assays. Reduced initial consumption might prevent escalation, or perhaps itself reflect reduced escalation, although the conditions used here are not generally conducive to escalation.
Other glutamate receptor subunit mutants have also been studied, although less extensively than Grm5 mutants. Male Gria1 KO mice did not differ from controls in voluntary ethanol consumption, stress induced drinking, or ADE (Cowen et al., 2003). Similarly, male Gria3 KO mice did not differ from WT mice in voluntary ethanol consumption or ethanol preference (2-bottle choice, continuous access), or in baseline responding in an operant self-administration task (Sanchis-Segura et al., 2006). However, while having no effect under baseline conditions, Gria3 KO blunted consumption after ethanol deprivation (ADE) and reduced cue-induced reinstatement in the operant task. Projected over repeated experiences, these findings might lead to reduced escalation.
Taken together, this research in genetically modified mice supports the extensive evidence of an important role for glutamate in responses to ethanol and the propensity to develop AUD. Moreover, some of the findings point to differences in the expression of particular glutamate receptor subunits potentially being involved in individual differences in the escalation of ethanol consumption.
3.1.7. Stress-related genes
Many polymorphisms in stress-related genes have been associated with AUD or related phenotypes, including many in the noradrenergic system (Clarke et al., 2012; Preuss et al., 2013), particularly the high-activity COMT allele (Kauhanen et al., 2000; Nakamura et al., 2001; Sery et al., 2006; Tiihonen et al., 1999). However, the most consistently observed stress-related genes associated with AUD are in the CRF system. The role of CRF in responses to alcohol and changes in CRF in response to chronic alcohol exposure have already been discussed 2.4.1.. Given these findings, it is not surprising that variation in CRF system genes, or other stress-related systems, are associated with AUD. Variations in the CRF receptor 1 gene (CRHR1) are associated with several AUD-related phenotypes, including binge drinking in adolescents (Chen et al., 2010; Treutlein et al., 2006). The binge-drinking phenotype is particularly relevant to much of the literature on the effects of intermittent alcohol access discussed in this review. Moreover, given the role of CRF systems in both neural and endocrine responses to stress, and the inter-relationships between chronic stress and AUD, it is not at all surprising that there are genotype x environment interactions for CRF system genes, notably between CRHR1 polymorphisms and trauma (Ray et al., 2013). The association of CRHR1 polymorphisms and AUD were also confirmed by GWAS (Gelernter et al., 2019).
Genetic manipulations in mice have confirmed a functional role for many of the genes identified in human studies of AUD-related phenotypes. CRF-deficient male mice consume much more ethanol than control mice, but this is associated with reduced locomotor stimulant and reinforcing effects of ethanol (Olive et al., 2003). As would be expected based on that result, over-expression of CRF in male mice decreases ethanol consumption (Palmer et al., 2004). Alcohol self-administration is not increased after a period of abstinence in Crhr1 KO male mice, as it is in WT mice (Chu et al., 2007). These mice also fail to show sensitization of locomotor responses to ethanol (Pastor et al., 2008). An interesting manipulation involved selective elimination of Crhr1 from the brain of male mice (Molander et al., 2012). Reduction in brain Crhr1 did not affect baseline ethanol consumption, and only slightly reduced stress-induced ethanol consumption. A subsequent period of alcohol deprivation did produce an ADE, but this was not affected by the genetic manipulation. This study also examined complete Crhr1 KO, which similarly did not affect basal consumption, nor did it affect ADE or swim stress-induced increases in ethanol consumption, but it greatly reduced consumption of ethanol after a social stressor. In a final experiment in that study, mice were allowed free access to 8% ethanol for 3 months and then exposed to 4 repeated cycles of ethanol vapour exposure (16 hrs) and withdrawal (8 hrs), followed by a return to free-access 24 hrs later. The vapour exposure was associated with a large increase in ethanol consumption, but this increase was eliminated in Crhr1 KO mice. These studies demonstrate a clear role for Crhr1 in stress- and repeated-withdrawal-induced escalation of ethanol consumption in mice. Moreover, they clearly demonstrate the need for detailed assessments of gene effects in ethanol consumption procedures that produce escalation and dependence.
3.1.8. Circadian clock genes
An interesting class of genes involved in AUD are circadian clock genes (Takahashi et al., 2008). From the point of view of escalation this class is interesting because part of the underlying changes that lead to escalation may involve alterations in circadian rhythms, either as a pre-existing risk factor or as something that develops over the course of disease progression. Supporting this idea, alterations in circadian period have been associated with AUD symptom severity (McCarthy et al., 2013) and several circadian clock genes have been associated with AUD or associated phenotypes (Kovanen et al., 2010; Spanagel et al., 2005).
Mice with mutations of the period genes that prevent normal circadian function (Per1BRDM1, Per2BRDM1 and Per3BRDM1 mice) have been studied in multiple alcohol consumption procedures. Per1BRDM1 mice showed no differences in baseline consumption, self-administration, reinstatement or ADE-induced increases in ethanol consumption in an initial study (Zghoul et al., 2007), but showed increased consumption after social stress (Dong et al., 2011b). Moreover, an association between heavy drinking and hPer1 SNP rs3027172 was identified in human adolescents who had suffered psychosocial adversity (Dong et al., 2011a). It is also important to note that alcohol consumption can have significant impacts on circadian rhythms, potentially creating a vicious cycle (Ruby et al., 2009). However, a subsequent study showed that both Per1BRDM1, Per2BRDM1, and double mutant male and female mice have increases in ethanol consumption in a standard two-bottle test assessing consumption of ascending concentrations of ethanol, as well as slightly increased ethanol conditioned place preference (Gamsby et al., 2013). These changes were also associated with alterations in ethanol metabolism, primarily in females.
3.2. Sex Differences
There are clear sex differences in the presentation of AUD (Flores-Bonilla and Richardson, 2020), which are also observed in animal models of alcohol consumption and alcohol dependence (Bell et al., 2017). Moreover, factors related to sex influence different stages of the addiction process, including acquisition, initial escalation and escalation associated with dependence (Carroll et al., 2004). In many sections throughout this review we have highlighted sex differences, or lack of sex differences, when both males and females have been studied, but in this section we specifically focus on sex as a biological variable in relation to changes in ethanol consumption.
Female rodents are very well known to consume more ethanol than males (see summary table of this literature in Priddy et al. (2017)), but generally the ethanol consumption models that have shown this relate to early stages of the addiction process, and may primarily involve initial differences in the pharmacological responses to ethanol (Blanchard et al., 1993). It has been proposed that males are more driven by positive reinforcement from ethanol and females more by negative reinforcement from ethanol, and that these differences in motivation might contribute to differences in consumption at different stages of the alcohol addiction cycle and in different models (Varlinskaya et al., 2015b). For instance, this difference would impact observations of ethanol consumption under “basal” conditions in males and females since almost all methods involve social isolation, which may tend to potentiate consumption in females but not males. With regard to changes over time, some studies report accelerated or greater escalation of ethanol consumption in females under some intermittent access conditions (Varlinskaya et al., 2015a). However, another study found that males showed escalation of ethanol consumption during an intermittent access procedure, while females did not, and that consumption was increased further in males, but not females, after a period of vapour ethanol exposure (Morales et al., 2015). It is uncertain to what extent this lack of escalation in females was a result of their initially higher levels of ethanol consumption.
This type of pattern, in which males initially consume lower amounts of ethanol, but when tested for an extended period reach levels equal to females, is often observed. For example, Moore and Lynch (2015) showed escalation in males under continuous access 3-bottle choice conditions. Moreover, ethanol preference increased even more than consumption over this period, eventually reaching levels higher than females. A two-bottle choice procedure with 8% ethanol showed much more continuous escalation over the entire period (90 days); again, only in males. When tested in an operant (limited access) procedure, both males and females showed initial escalation over the first two weeks, and then consumption was maintained at the same level for the rest of the experiment. These effects during and soon after acquisition are clearly relevant to acquisition and initial escalation, but it is not clear whether they are relevant to escalation associated with the formation of dependence and AUD. Moreover, they are dependent on initial levels of consumption, and as such the sex differences that are seen may have more to do with those initial levels of consumption than in differences in the process of escalation itself.
Initial levels of consumption are not the only determinant of escalation. Hwa et al. (2011) used a method in which the ethanol concentration was gradually increased over the first week of exposure from 3 to 20%, and thereafter kept at 20%. These authors compared continuous access and intermittent access, with intermittent access achieving much higher levels of ethanol consumption, although consumption stabilized once the concentration was maintained at 20% and did not increase further. Of relevance to the topic of sex differences, although males and females consumed similar amounts initially during IA, females had faster initial escalation and stabilized at a higher level of consumption. Given that the study confounded time and concentration preference it is difficult to say whether this just represents greater consumption at the higher concentrations in females or greater initial escalation.
Much work remains before it can be determined whether there are sex differences specifically in the escalation of ethanol consumption, especially to levels most relevant to AUD, and what factors determine such differences.
3.3. Socioemotional factors
3.3.1. Social isolation and social support
Studies in human populations have long pointed to social isolation and subsequent loneliness being important factors contributing to the establishment and maintenance of problematic alcohol use (for a review of early work in this area, see Åkerlind and Hörnquist, 1992). A study of elderly Germans found that living alone was associated with increased alcohol use (Du et al., 2008) and another study found that loneliness was a significant predictor of problem drinking in middle to late life (Kuerbis et al., 2018). This association does not appear to be restricted to older individuals, with a systematic review exploring alcohol use among adolescents in Brazil finding loneliness was associated with increased risk for heavy alcohol use (Barbosa Filho et al., 2012). Moreover, systematic reviews examining the impact of social isolation and loneliness on health also identified excessive alcohol consumption as a potential consequence (Leigh-Hunt et al., 2017). These findings linking loneliness to increased alcohol consumption have been further supported by emerging data from the COVID-19 pandemic. One study found that adults in the USA who felt, on average, lonelier during the COVID-19 restrictions of the 2020 northern hemisphere summer consumed significantly more alcohol each day (Bragard et al., 2021). Another study found 65% of participants reported increased loneliness during the pandemic and 58% of these participants reported increased drinking, with change in loneliness and change in consumption significantly correlated (Horigian et al., 2021). However, analysis of data collected in 2004–05 from over 30,000 subjects in the National Epidemiologic Survey on Alcohol and Related Conditions highlights the complex nature of the interaction between social isolation and alcohol consumption. Unexpectedly, the study found that less frequent contact with close friends was associated with reduced risk of AUD. In contrast, less frequent contact with members of their religious group within their social network was associated with increased risk of AUD. The authors argue that the nature of the relationship is likely a key determinant of the outcome, as individuals with already established AUD tend to have fairly large social networks, with over half of those also having AUD (Chou et al., 2011). Therefore, increased contact with those friends may increase opportunities for drinking. A critical emergent factor across these studies is perhaps the lack of a positive social support network increases risk of developing an AUD.
These human studies have emphasized the relationship between current social circumstances and problem drinking, yet problem drinking is most likely to develop as a result of longer-term issues, often beginning before adulthood. Problem drinking most often has its roots in adolescence, and adolescent social isolation has most often been associated with increased risk of AUD (for review see (Butler et al., 2016; Spear, 2015)). Social isolation at different ages has been extensively studied in rodents.
Social isolation induces many phenotypes that are relevant to AUD, and the specific phenotype is highly dependent on age and the nature and duration of the experience (Hall, 1998). Although often discussed in terms of “social stress”, social isolation early in life is best considered as the interruption of a developmental epigenetic program that guides neural and physiological changes in response to social experience, with the “isolation phenotype” being a pre-programmed alternative to the “social phenotype” (for a discussion of this view of isolation see Hall and Perona (2012)).
In contrast to isolation in adulthood, social isolation beginning in adolescence in rodents (often termed isolation rearing) has been consistently found to increase ethanol consumption (Deehan et al., 2007; Deehan et al., 2011; Hall et al., 1998b; Lopez et al., 2011; McCool and Chappell, 2009; Schenk et al., 1990; Wolffgramm, 1990), and persists even if the isolation is limited to adolescence (Lesscher et al., 2015). In adult rodents, isolation usually has little effect on alcohol consumption (Andreas et al., 1985; Schenk et al., 1990), and under some circumstances actually reduces consumption (Doremus et al., 2005). Increased ethanol consumption after chronic adolescent social isolation, usually assessed in adulthood, is associated with increased reward sensitivity and anxiety, which are potential mediators. For example, isolation-rearing increased ethanol preference in ethanol preferring male fawn hooded rats (Hall et al., 1998b; Lodge and Lawrence, 2003c), which was associated with anxiety (Djouma et al., 2006; Hall et al., 1998a). Consistent with this role for isolation-induced increases in anxiety driving ethanol consumption in these rats, both diazepam and the CRFR1 Antagonist CP-154,526 reduced ethanol consumption in isolation-reared fawn hooded rats (Lodge and Lawrence, 2003b).
Several factors may affect how rodents respond to social isolation, and these overlap with factors influencing susceptibility and resilience to the development of AUD in humans. As previously noted, factors may contribute differently to alcohol consumption and dependence in males and females, which may interact with aspects of experimental procedures used to examine ethanol consumption in animal models. Isolation-rearing increased ethanol consumption in males but not females in one study (Lopez et al., 2011). Interestingly, in the same study, isolation of adult animals increased ethanol consumption in females but not males. In this study, as in most of the studies mentioned previously, ethanol consumption was measured after a period of chronic social or isolation housing by subsequently isolating both groups. It is important to note that allowing animals to consume alcohol in social circumstances affects ethanol consumption, particularly in adolescent mice. Thus, adolescent male mice, but not females or adult male mice, were found to drink more when consumption occurred in the presence of conspecifics (Logue et al., 2014). As is clear from some of the studies discussed in a previous section, genetic predisposition also clearly affects the outcome of social deprivation. For instance, one study found increased ethanol consumption after isolation rearing only in alcohol-preferring P rats (Ehlers et al., 2007). Another study found that ethanol consumption was altered in Oprm KO mice in a manner dependent on isolation rearing and sex (Moriya et al., 2015). Ethanol consumption was increased in isolation reared male Oprm KO mice, but decreased in isolation reared female KO mice.
The ethanol consumption studies mentioned above primarily used continuous access paradigms, assessed initial phases of the acquisition of ethanol consumption, and did not examine conditions that tend to produce escalation of ethanol consumption relevant to the formation of dependence. One study did examine a 2-bottle choice DID procedure over an extended period of time with alternating 2-day periods of forced abstinence and 5-day periods of DID access (Holgate et al., 2017). This study utilized the IntelliCage apparatus which uses subcutaneous transponders to individually identify socially housed mice so that consumption can be assessed without short-term isolation for measurement of ethanol consumption. Socially isolated male mice had much greater initial ethanol preference than social housed mice, with testing beginning in late adolescence (6 weeks of age). Moreover, physical environmental enrichment, in addition to social enrichment, further reduced initial ethanol preference. Isolated mice showed a slight increase in ethanol preference over the four weeks, whereas preference mice in the enriched environment decreased their preference. Of most relevance to escalation, when mice consuming ethanol in social housing were moved to isolated housing they showed a rapid and drastic increase in ethanol preference, consistent with the human literature suggesting that social isolation can be a trigger for substantial increases in consumption. In contrast, mice moved from isolated to enriched housing rapidly and drastically reduced their ethanol preference. These findings clearly highlight a potentially significant interaction between social experience/circumstances and alcohol consumption, and future work would might examine whether the switch from social to isolated housing is sufficient to escalate established consumption to levels that induce dependence and other features relevant to AUD.
As some of the studies discussed above suggest, the impact of social isolation on alcohol consumption extends beyond possibly facilitating escalation of consumption to hindering cessation of problematic use. Here, social support, or more precisely, a lack thereof, appears critically important. Greater social support is consistently found to be a predictor of recovery from excessive alcohol consumption and protective against escalation of consumption (Fuehrlein et al., 2018; Weitzman and Chen, 2005). Moreover, numerous studies point to an individual’s level of engagement in the positive social support network provided by programs like Alcoholics Anonymous as the most important predictor of a successful treatment outcome (Bond et al., 2003; Groh et al., 2008; Kaskutas et al., 2002; Longabaugh et al., 1998; Nealon-Woods et al., 1995; Timko et al., 2015). It is important to note here that heavy alcohol consumption can alienate positive social supports and mounting evidence points to disruption of social motivation and capacity for normal social interactions with chronic heavy alcohol use (Moos et al., 2010; Trezza et al., 2014; Zou et al., 2009).
In light of this observation, there is growing interest in how drugs that act to enhance social motivation and facilitate functional social interactions might play an important role in treating AUD (Baskerville and Douglas, 2010; Bowen et al., 2016; Bowen and Neumann, 2017, 2018; McGregor and Bowen, 2012; McGregor and Bowen, 2013). The brain oxytocin system has received considerable interest in this regard. Among other things, oxytocin plays a critical role in the regulation of social behaviours, anxiety, and stress and fear responses (Jurek and Neumann, 2018). Of specific relevance here, OXT genotype appears to moderate the effect of social support on psychological distress in patients with AUD (Love et al., 2018) and chemogenetic activation of hypothalamic OXT neurons was sufficient to inhibit binge-like alcohol consumption in male mice in the DID model (King et al., 2021). Importantly, both clinical and preclinical studies highlight that targeting the brain oxytocin system pharmacologically has potential to prevent the escalation of alcohol consumption, reduce established alcohol consumption, inhibit relapse and interfere with effects of ethanol on neurotransmitter systems involved in ethanol reward and intoxication (Bach et al., 2020a; Bach et al., 2019; Betka et al., 2018; Bowen et al., 2011; Bowen et al., 2015; Dannenhoffer et al., 2018; Hansson et al., 2018; King and Becker, 2019; King et al., 2017; MacFadyen et al., 2016; McGregor and Bowen, 2012; Mitchell et al., 2016; Pedersen et al., 2012; Peters et al., 2013; Peters et al., 2017; Stevenson et al., 2017; Tunstall et al., 2019; Walcott and Ryabinin, 2020). One study that is particularly relevant to the effects of OXT on escalation (Bowen et al., 2011) found that male rats sub-chronically pre-treated with OXT showed reduced initial escalation of ethanol consumption over the course of a 25 day continuous access two bottle paradigm. Moreover, the pre-treatment with OXT was associated with a phenotype characterised by reduced anxiety-like behaviour and increased social interaction.
3.3.2. Social anxiety
There is a clear association between alcohol consumption and social anxiety. As many as 10% of those with AUD have comorbid social anxiety disorder (Gabriels et al., 2019) and those with social anxiety disorder are up to 4.5 times more likely to have AUD (Buckner et al., 2008). Some studies suggest that use of alcohol to cope with social anxiety may contribute to the establishment of problematic alcohol use. For instance, coping with social anxiety symptoms has been identified as a major motivation for alcohol consumption, especially among adolescents and young adults (Caruso et al., 2018; Simons et al., 2017). Further supporting a role for social anxiety in escalation of alcohol consumption, social anxiety disorder occurs first in 80% of comorbid cases of social anxiety disorder and AUD (Schneier et al 2010). A recent human laboratory study found that participants without AUD reported increased craving for alcohol after performing the Trier Social Stress Test (Clay et al., 2018). However, another study found this effect was absent in participants with AUD (Bacon and Thomas, 2013). Together, these studies suggest that acute social anxiety might play a role in driving initial escalation of alcohol consumption, and perhaps establishment of AUD, but that once an individual has AUD it has less influence on consumption.
Due to the challenges in identifying appropriate placebos for alcohol studies in humans, it is difficult to ascertain whether alcohol has any real impact on social anxiety through its pharmacological actions, or whether people merely have an expectancy bias due to commonly held beliefs about alcohol’s social lubricant effects (Raymond et al., 2019). In animal studies, the effects of alcohol on social behaviour vary, with the observation of social inhibition or social facilitation depending on a variety of factors, including sex, age and context (Raymond et al., 2019; Varlinskaya and Spear, 2004, 2006). Although these studies provide insight into ethanol interactions with social anxiety-like behaviour, the stress-paradigms used to induce social fear can also induce non-social-specific changes in behaviour, confounding interpretations.
To address this, Raymond et al. (2019) used a social fear conditioning paradigm that models the acute social avoidance and social fear aspects of social anxiety disorder, while addressing the issue of non-specific effects found in other models (see Toth et al., 2012 for more information on the social fear conditioning paradigm). Raymond et al. (2019) found that alcohol reduced social avoidance in socially fear conditioned adolescent male mice (Figure 3), but only at a low dose. In contrast, the low dose of ethanol had no effect in adult mice and a high dose inhibited social interaction irrespective of social fear status. The results were social specific as they were not observed when mice were conditioned to a non-social stimulus using the same procedure. The reduction of social avoidance only in the adolescent mice aligns with the human literature discussed above, suggesting adolescence is a period during which alleviation of social anxiety symptoms is an important driver of alcohol consumption.
Figure 3. Exploring the interactions between alcohol and social anxiety using murine models.
A. When adult male C57BL/6 mice were administered 0.25 g/kg ethanol i.p. prior to an extinction session after undergoing social fear conditioning (SFC+) or no fear conditioning (SFC-), it had no impact on social investigation time in either the conditioned or unconditioned mice. B. In contrast, in adolescent mice, the same dose of ethanol significantly inhibited social avoidance in the conditioned mice, while having no impact on social investigation time in unconditioned mice. A and B are adapted from Raymond et al. (2016). C. N = 10 male C57BL/6 mice underwent daily (Monday – Friday) 2 h DID sessions two hours into the dark cycle with 20% ethanol available and an empty wire mesh stimulus cage (7 × 7 × 6 cm) in the back corner of their home cage. After two weeks, mice underwent the social fear conditioning procedure, with half receiving conditioning (SFC+) and half not (SFC-). DID resumed as normal the following day. The next day (~42 h after social fear conditioning), mice underwent DID with a social stimulus (novel mouse inside a cage) for the duration of the drinking session. Cage in the graph is the average consumption on the three days when only the cage was present in the home cage during the drinking session. Consumption was measured 30 min into each session and 2 h into each session. Only data for the first 30 min is shown as the manipulation only impacted consumption in the first 30 min. Conditioned mice showed no difference in ethanol consumption during the first 30 min of their drinking session whether a social stimulus or stimulus cage was present. In contrast, unconditioned mice reduced their consumption of ethanol in the first 30 min of the drinking session when a social stimulus was present (SFC- cage vs social p < 0.05). This indicates that induction of social anxiety-like behaviour in mice removes the social buffering effect of social interactions on alcohol consumption in the first 30 min of DID.
In subsequent work, the same group showed that socially fear conditioned male mice maintain binge-level alcohol consumption in the DID paradigm when a social stimulus is placed into their cage during a drinking session, whereas unconditioned mice reduce their consumption to non-intoxicating levels (Figure 3). Conditioned social fear thus appears to eliminate social buffering of alcohol consumption in this model.
Taken together, these findings exploring the relationship between social anxiety and alcohol consumption in both humans and animal models suggest alcohol might initially serve to alleviate social anxiety symptoms, which might facilitate the establishment of problematic patterns of alcohol use through negative reinforcement. Moreover, under normal conditions, social interactions may serve to inhibit excessive alcohol consumption, which is consistent with some of the literature exploring the effects of social isolation on escalation of alcohol consumption, discussed above. However, more work needs to be conducted in models of consumption that capture escalation associated with the formation of dependence and AUD.
3.3.3. Trauma
Trauma is consistently linked with increased risk of developing AUD. A comorbidity study of 5,877 individuals found that those with post-traumatic stress disorder (PTSD) were twice as likely to have AUD as those without (Kessler, 1995). A systematic review examining comorbidity between AUD and PTSD found 9.8 to 61.3% of those with PTSD misused alcohol, with odds ratios as high as 4.87 (Debell et al., 2014). A recent review of the epidemiological data noted the consistency of the comorbidity between AUD and PTSD across different populations and over time (Smith and Cottler, 2018). Of interest, not only do those with PTSD have an increased likelihood of developing AUD, possibly to self-medicate, those with AUD also appear to have an increased likelihood of developing PSTD, perhaps through increased likelihood of being exposed to traumatic situations (Debell et al., 2014; Smith and Cottler, 2018).
Social defeat stress is commonly used in animal models to explore the impact of social trauma and stress on alcohol consumption. Studies in several rodent species and non-human primates report long-lasting elevations in voluntary alcohol consumption following exposure to chronic, continuous social defeat stress (for a review see Newman et al., 2018b). In these models, the impact of social defeat stress on alcohol consumption appears independent of baseline levels of consumption prior to stress exposure. Other studies use models which involve exposing subjects to short periods of social defeat stress intermittently over a period of days or weeks. The effect of defeat stress in these models is more subtle than in models involving continuous chronic exposure, with stressed subjects initially consuming less alcohol, possibly due to suppression of ingestive behaviour by autonomic nervous system arousal, but then developing escalated consumption from days up to weeks after the cessation of stressor exposure (Newman et al., 2018b). It is also important to note that the relationship between alcohol and social defeat stress is bidirectional, with alcohol exposure increasing sensitivity to social defeat stress (Nelson et al., 2018; Nennig et al., 2020), which is consistent with the human literature on PTSD and AUD discussed above. Finally, recent work by Newman et al. (2021) suggests changes in CRF may be involved in effects of social stress on escalation of alcohol consumption. The study identified a population of CRF positive neurons in the anterior central medial thalamus that are active during social interactions in non-stressed but not socially stressed female mice. Optogenetic activation of these cells in stressed and non-stressed female mice inhibited abstinence-escalated drinking.
Whereas social defeat stress is used to model social trauma, non-social stressors are also used in models of trauma. Two commonly used non-social stressors are restraint stress and predator odour. Evidence for escalation of alcohol consumption in rodents following exposure to restraint stress is inconsistent (for a review see Suh and Ressler, 2018). In contrast, escalation of alcohol consumption has been more consistently demonstrated in rodents following exposure to predator odour stress, although variables that appear to mediate escalation of consumption in response to predator odour are not always consistent. Edwards et al. (2013) found increased escalation of alcohol consumption from baseline levels in passive, but not active, stress coping male Wistar rats following exposure to predator odour (bobcat urine), an effect which the group has replicated (Weera et al., 2020). It should be noted that these individual differences should be expected from a valid animal model for exploring the effect of trauma on alcohol consumption – just as not all humans who experience trauma develop AUD, we do not expect all outbred rats to escalate alcohol consumption in response to a stressor – and stress coping style is a mediator of this relationship consistent with the human literature. In contrast to Edwards et al. (2013), Ornelas et al. (2021) reported increased escalation of ethanol consumption in active, but not passive, female, but not male, Long-Evans rats following exposure to the predator odour TMT. Edwards et al. (2013) did not test females so it is possible they also would have observed this pattern in female rats. Moreover, the two studies used different protocols, different strains of rats, and different predator odours. Some research suggests that TMT is not a true predator odour, but rather an aversive smell (McGregor et al., 2002). However, another recent study did report increased alcohol consumption in male rats following exposure to TMT (Makhijani et al., 2021). In mice, predator odour exposure (dirty rat bedding) resulted in escalation of alcohol consumption in both males and females; however the stressor-induced escalation was only observed in males with prior binge-like drinking experience and only in females with low baseline levels of consumption (Finn et al., 2018).
The involvement of hyperactivity and hyperreactivity of the amygdala in the neurobiology of PTSD is well-established. However, few human studies have examined the amygdala in the context of comorbid PTSD and AUD, and well-controlled studies are even scarcer. One neuroimaging study found a combination of low reward-related activity in the ventral striatum and high reactivity to threat in the amygdala was associated with problem drinking (Nikolova et al., 2016). Supporting a causal role of the amygdala in escalation of alcohol consumption, a recent preclinical study found that escalation of alcohol consumption in passive stress-coping male rats following predator odour exposure was associated with increased fos immunoreactivity in CRF-positive cells in the central amygdala and that infusion of a CRFR1 antagonist into this region reversed escalation of alcohol consumption (Weera et al., 2020). In another study, infusion of a CRFR1 antagonist into the ventral tegmental area was able to reverse social-defeat stress-escalated alcohol consumption in male mice (Newman et al., 2018a). Several other regions and pathways have been identified as potential mediators of the link between PTSD and AUD: the medial PFC, and specifically lack of inhibition of the amygdala by the mPFC; complex changes in HPA axis function; elevated noradrenergic signalling; hippocampal dysfunction; and mesolimbic reward pathway hypoactivity. However, causal evidence for their role specifically in escalation of alcohol consumption in response to trauma is currently lacking and thus they are not discussed in detail here (for an excellent review see Gilpin and Weiner, 2017).
There is a strong link between childhood adversity, specifically, and a range of psychiatric disorders in adulthood, including AUD (for reviews of the human literature see Brady and Back, 2012; Keyes et al., 2011). Maternal separation is a commonly used technique to model early-life adversity in rodents. Maternal separation causes escalation of alcohol consumption in rodents and, not surprisingly, longer periods of maternal separation more reliably cause increases in consumption. Interestingly, several studies report that heightened alcohol consumption in maternally separated rodents only emerges when they reach adulthood. Male mice who were maternally separated showed more pronounced escalation of alcohol consumption in response to stress and a greater ADE (Portero-Tresserra et al., 2018). For an in-depth review on the impact of early life stress on susceptibility for substance use disorders with a particular focus on animal models, see Baracz et al. (2020).
One study found maternally separated male and female mice had altered gene expression in the serotonin, reward and HPA axis systems that was associated with increased alcohol consumption (De Almeida Magalhães et al., 2018). Ethanol consumption reversed heightened stress behaviour and altered gene expression in some parts of the aforementioned systems, suggesting that escalation of consumption may be driven by self-medication under some circumstances. Another study reported that ethanol exposure resulted in greater elevations of plasma corticosterone and brain monoamines in mice that had been maternally separated, with the latter effect being sex-dependent (Kawakami et al., 2013). Elevated CRF and GABA-A receptor α2 subunit expression have been reported in stress and reward nuclei following maternal separation (Gondré-Lewis et al., 2016). Infusion of either a CRF1 receptor antagonist or negative allosteric modulator of ethanol effects at α2 subunit containing GABA-A receptors prevented maternal separation-induced binge-like ethanol consumption, suggesting mechanisms which drive escalation in other models may also be involved in heightened consumption in maternal separation models.
Several studies in rodents point to alterations in the endocannabinoid system being involved in escalated alcohol consumption following maternal separation. However, studies demonstrating a mechanistic link are lacking. Male mice that underwent maternal separation and early weening showed increased escalation of alcohol consumption in at two week DID procedure, which was associated with reduced endocannabinoid levels in the PFC, reduced endocannabinoid and monoamine levels in the striatum, and reduced sensitivity to the rewarding effects of ethanol (Portero-Tresserra et al., 2018). Another study reported an association between increased cannabinoid type 1 receptor expression in the nucleus accumbens and elevated baseline ethanol consumption as well as greater escalation of ethanol intake in male rats that underwent maternal separation relative to controls (an effect further exacerbated by isolation housing during adolescence) (Amancio-Belmont et al., 2020). More studies examining the biological basis of the link between early life adversity and alcohol consumption in clinical populations are required, especially those with a specific focus on the link between early life adversity and escalation of alcohol consumption. The preclinical literature provides some guidance, although studies establishing causal links between early life trauma and escalation of alcohol consumption are few.
4. Summary and Future Directions
Many studies, especially those examining mechanisms, focus on examining factors that influence ethanol consumption in limited access paradigms in animals that likely do not form dependence or other hallmark features of AUD. These studies, which often use sucrose fading, and/or short-term operant or free consumption procedures, have provided valuable insights into acquisition of ethanol consumption and initial escalation to pharmacologically relevant levels of consumption. However, these studies do not provide clear insight into factors that specifically contribute to escalation in latter stages, which are arguably most relevant to better understanding and treating dependence and AUD.
Dependence was identified as a particularly important factor from our review. Studies which involve IEA combined with vapour chamber ethanol exposure have been especially useful in identifying repeated cycles of withdrawal and abstinence as a likely key driver of escalation in latter stages of AUD as well as highlighting the potential involvement of numerous biological mechanisms in this process. More studies are needed that focus on comparing changes at different stages of the cycles, and how the repeated cycling itself is driving changes that differ from change resulting simply from cumulative ethanol exposure over time. Yoked control groups that receive similar ethanol exposure without the repeated cycling would be particularly useful, especially in mechanistic studies. This will facilitate identification of mechanisms involved specifically in the repeated cycling that seems to drive escalation and formation of an AUD-like phenotype.
Studies on the involvement of specific genes in escalation are challenging as they often rely on a comparison between, for example, subjects with a particular gene or gene variant, and subjects without. These studies have provided a wealth of insights, including identifying particular genes associated with differences in alcohol consumption, dependence and withdrawal. However, the constitutive gene expression in these studies makes it challenging to assess the contribution of these genes specifically to escalation and factors that contribute to escalation. Here, preclinical studies in inducible lines, or using other techniques with appropriate temporal precision, will allow manipulation at key periods involved in escalation (e.g. during repeated cycles of withdrawal and abstinence) which could help provide a more precise exploration of the involvement of genes in escalation.
Across models, chronic IEA is consistently the most reliable means to induce escalation that is relevant to AUD. Given the importance of dependence, and the subsequent cycles of withdrawal and abstinence, in escalation during latter stages, it of considerable interest to identify what factors result in animals escalating voluntary consumption to levels where they form dependence and experience withdrawal. In this regard, there should be more emphasis in future studies on assessing physical dependence alongside escalation of consumption. This should be done in chronic IEA paradigms, and other models of interest, that do not involve forced induction of dependence.
Finally, studies examining sex as a biological variable influencing escalation are lacking. The majority of preclinical studies reviewed used male subjects only, some used both, a small number used female subjects only, and a small number did not specify the sex of the subjects used. The bias toward male subjects was far less prevalent amongst the human studies, although a small number of studies only included male subjects.
5. Conclusions
As is clear from the literature explored in this review, a vast array of complex, interacting factors influence alcohol consumption and the development of AUD. Some of these factors act early in the acquisition and initial escalation of ethanol consumption, while others contribute to escalation of ethanol consumption at a later stage and are involved in the development of alcohol dependence and AUD. Many of these processes can now be modelled in animals, which provides a pathway for not only probing genetic and neurobiological substrates involved in these factors, but also a platform for screening novel compounds to treat AUD. From a clinical perspective, appreciation of the different factors that contribute to escalation of alcohol consumption, and maintenance of escalated consumption, could provide important insights into the most appropriate treatment approach to use for a particular individual. Unfortunately, as is clear from the discussion of biological and environmental factors that may contribute to escalation of alcohol consumption, much of this research has yet to be done. Preclinical research investigating genetic mechanisms has tended to use primarily the simplest models of ethanol consumption that produce neither escalation of ethanol consumption nor alcohol dependence. These studies have managed to capture certain factors, including the contribution of underlying co-morbidities, like anxiety, to the development of high levels of alcohol consumption. Nonetheless, they have largely failed to capture more meaningful aspects of negative reinforcement or self-treatment.
Different models capture different aspects of acquisition and escalation of alcohol consumption that can be relevant to different stages of the dependence process, as well as different demographics. For instance, sucrose fading is a relevant approach for exploring factors relevant to increases in consumption from non-pharmacologically relevant levels to pharmacologically relevant levels in those who might otherwise not consume high levels of alcohol. In contrast, intermittent access and binge paradigms are particularly relevant for exploring the transition from social drinking to excessive alcohol consumption. Studying the impact of dependence on consumption, especially repeated cycles of withdrawal and abstinence, as well as incubation of craving and ADE, has provided insights into the transition to AUD and to more severe AUD-like phenotypes. These approaches can now be used to better understand the biological changes that occur during the process of escalation, paving the way for identifying biomarkers for escalation risk and providing the opportunity to develop more targeted therapeutics for AUD.
We have learnt much about the factors that lead to the initiation of consumption and escalation from low, non-intoxicating to intoxicating levels. In comparison, far fewer studies have been able to capture the escalation of consumption to levels that result in physical dependence and that truly model latter stage AUD. However, suitable models have now been identified and breakthroughs, especially in our understanding of genetic and biological mechanisms driving escalation, will depend on utilising these models with appropriate controls and techniques that allow factors specifically involved in escalation relevant to dependence and AUD to be explored.
8. Acknowledgements
We would like to acknowledge Bianca Wilson from MTB’s team, who assisted in the collection of the data presented in Figure 3C.
6. Funding
This work was supported by NHMRC funding to MTB: GNT1092046, GNT1108546.
Footnotes
Declaration of Competing Interest
MTB is listed as an inventor on patents for novel oxytocin-based therapeutic candidates and for therapeutic candidates for alcohol use disorder and is co-founder and chief scientific officer of a company, Kinoxis Therapeutics Pty Ltd, commercialising some of this technology. The other authors declare they have no conflicts of interest.
9. References
- Agabio R, Carai MA, Lobina C, Pani M, Reali R, Vacca G, Gessa GL, Colombo G, 2000. Development of short-lasting alcohol deprivation effect in sardinian alcohol-preferring rats. Alcohol 21, 59–62. [DOI] [PubMed] [Google Scholar]
- Agarwal DP, Goedde HW, 1992. Pharmacogenetics of alcohol metabolism and alcoholism. Pharmacogenetics 2, 48–62. [DOI] [PubMed] [Google Scholar]
- Åkerlind I, Hörnquist JO, 1992. Loneliness and alcohol abuse: A review of evidences of an interplay. Social Science & Medicine 34, 405–414. [DOI] [PubMed] [Google Scholar]
- Allan AM, Harris RA, 1986. Gamma-aminobutyric acid and alcohol actions: neurochemical studies of long sleep and short sleep mice. Life sciences 39, 2005–2015. [DOI] [PubMed] [Google Scholar]
- Amancio-Belmont O, Becerril Meléndez AL, Ruiz-Contreras AE, Méndez-Díaz M, Prospéro-García O, 2020. Maternal separation plus social isolation during adolescence reprogram brain dopamine and endocannabinoid systems and facilitate alcohol intake in rats. Brain Research Bulletin 164, 21–28. [DOI] [PubMed] [Google Scholar]
- Andreas K, Dienel A, Fischer HD, Oehler J, Wustmann C, Schmidt J, 1985. The influence of social isolation on ethanol preference behavior and dopamine release in telencephalon slices in mice. Pol J Pharmacol Pharm 37, 851–854. [PubMed] [Google Scholar]
- Arias A, Feinn R, Kranzler HR, 2006. Association of an Asn40Asp (A118G) polymorphism in the μ-opioid receptor gene with substance dependence: a meta-analysis. Drug and alcohol dependence 83, 262–268. [DOI] [PubMed] [Google Scholar]
- Arias AJ, Covault J, Feinn R, Pond T, Yang B-Z, Ge W, Oncken C, Kranzler HR, 2014. A GABRA2 variant is associated with increased stimulation and ‘high’following alcohol administration. Alcohol and Alcoholism 49, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aroni S, Marino RAM, Girven KS, Irving JM, Cheer JF, Sparta DR, 2021. Repeated binge ethanol drinking enhances electrical activity of central amygdala corticotropin releasing factor neurons in vivo. Neuropharmacology 189, 108527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach P, Koopmann A, Bumb JM, Zimmermann S, Bühler S, Reinhard I, Witt SH, Rietschel M, Vollstädt-Klein S, Kiefer F, 2020a. Oxytocin attenuates neural response to emotional faces in social drinkers: an fMRI study. European Archives of Psychiatry and Clinical Neuroscience. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach P, Reinhard I, Bühler S, Vollstädt-Klein S, Kiefer F, Koopmann A, 2019. Oxytocin modulates alcohol-cue induced functional connectivity in the nucleus accumbens of social drinkers. Psychoneuroendocrinology 109, 104385. [DOI] [PubMed] [Google Scholar]
- Bach P, Weil G, Pompili E, Hoffmann S, Hermann D, Vollstadt-Klein S, Mann K, Perez-Ramirez U, Moratal D, Canals S, Dursun SM, Greenshaw AJ, Kirsch P, Kiefer F, Sommer WH, 2020b. Incubation of neural alcohol cue reactivity after withdrawal and its blockade by naltrexone. Addict Biol 25, e12717. [DOI] [PubMed] [Google Scholar]
- Backstrom P, Bachteler D, Koch S, Hyytia P, Spanagel R, 2004. mGluR5 antagonist MPEP reduces ethanol-seeking and relapse behavior. Neuropsychopharmacology 29, 921–928. [DOI] [PubMed] [Google Scholar]
- Bacon AK, Thomas SE, 2013. Stress Reactivity, Social Anxiety, and Alcohol Consumption in People With Alcoholism: A Laboratory Study. Journal of Dual Diagnosis 9, 107–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baracz SJ, Everett NA, Cornish JL, 2020. The impact of early life stress on the central oxytocin system and susceptibility for drug addiction: Applicability of oxytocin as a pharmacotherapy. Neuroscience & Biobehavioral Reviews 110, 114–132. [DOI] [PubMed] [Google Scholar]
- Barbosa Filho VC, Campos W.d., Lopes A.d.S., 2012. Prevalence of alcohol and tobacco use among Brazilian adolescents: a systematic review. Revista de Saúde Pública 46, 901–917. [DOI] [PubMed] [Google Scholar]
- Bart G, Kreek MJ, Ott J, LaForge KS, Proudnikov D, Pollak L, Heilig M, 2005. Increased attributable risk related to a functional μ-opioid receptor gene polymorphism in association with alcohol dependence in central Sweden. Neuropsychopharmacology 30, 417–422. [DOI] [PubMed] [Google Scholar]
- Baskerville TA, Douglas AJ, 2010. Dopamine and oxytocin interactions underlying behaviors: potential contributions to behavioral disorders. CNS Neurosci. Ther 16, e92–e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker A, Grecksch G, Kraus J, Loh HH, Schroeder H, Höllt V, 2002. Rewarding effects of ethanol and cocaine in μ opioid receptor-deficient mice. Naunyn-Schmiedeberg’s archives of pharmacology 365, 296–302. [DOI] [PubMed] [Google Scholar]
- Becker HC, 1998. Kindling in alcohol withdrawal. Alcohol Health Res World 22, 25–33. [PMC free article] [PubMed] [Google Scholar]
- Behrendt S, Wittchen HU, Hofler M, Lieb R, Low NC, Rehm J, Beesdo K, 2008. Risk and speed of transitions to first alcohol dependence symptoms in adolescents: a 10-year longitudinal community study in Germany. Addiction 103, 1638–1647. [DOI] [PubMed] [Google Scholar]
- Belknap JK, Crabbe JC, Young ER, 1993. Voluntary consumption of ethanol in 15 inbred mouse strains. Psychopharmacology (Berl) 112, 503–510. [DOI] [PubMed] [Google Scholar]
- Bell RL, Hauser SR, Liang T, Sari Y, Maldonado-Devincci A, Rodd ZA, 2017. Rat animal models for screening medications to treat alcohol use disorders. Neuropharmacology 122, 201–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmer A, Patkar OL, Lanoue V, Bartlett SE, 2018. 5-HT1A receptor-dependent modulation of emotional and neurogenic deficits elicited by prolonged consumption of alcohol. Scientific Reports 8, 2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beracochea D, Mons N, David V, 2019. Targeting the Glucocorticoid Receptors During Alcohol Withdrawal to Reduce Protracted Neurocognitive Disorders. Front Psychiatry 10, 580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger AL, Williams AM, McGinnis MM, Walker BM, 2013. Affective Cue-Induced Escalation of Alcohol Self-Administration and Increased 22-kHz Ultrasonic Vocalizations during Alcohol Withdrawal: Role of Kappa-Opioid Receptors. Neuropsychopharmacology 38, 647–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betka S, Gould Van Praag C, Paloyelis Y, Bond R, Pfeifer G, Sequeira H, Duka T, Critchley H, 2018. Impact of intranasal oxytocin on interoceptive accuracy in alcohol users: an attentional mechanism? Social Cognitive and Affective Neuroscience 13, 440–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienkowski P, Rogowski A, Korkosz A, Mierzejewski P, Radwanska K, Kaczmarek L, Boguckia-Bonikowska A, Kostowski W, 2004. Time-dependent changes in alcohol-seeking behaviour during abstinence. European Neuropsychopharmacology 14, 355–360. [DOI] [PubMed] [Google Scholar]
- Bird MK, Kirchhoff J, Djouma E, Lawrence AJ, 2008. Metabotropic glutamate 5 receptors regulate sensitivity to ethanol in mice. Int J Neuropsychopharmacol 11, 765–774. [DOI] [PubMed] [Google Scholar]
- Blaine SK, Milivojevic V, Fox H, Sinha R, 2016. Alcohol Effects on Stress Pathways: Impact on Craving and Relapse Risk. Can J Psychiatry 61, 145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard BA, Steindorf S, Wang S, Glick SD, 1993. Sex differences in ethanol-induced dopamine release in nucleus accumbens and in ethanol consumption in rats. Alcohol Clin Exp Res 17, 968–973. [DOI] [PubMed] [Google Scholar]
- Blednov Y, Borghese C, McCracken M, Benavidez J, Geil C, Osterndorff-Kahanek E, Werner D, Iyer S, Swihart A, Harrison N, 2011. Loss of ethanol conditioned taste aversion and motor stimulation in knockin mice with ethanol-insensitive α2-containing GABAA receptors. Journal of Pharmacology and Experimental Therapeutics 336, 145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Benavidez JM, Black M, Leiter CR, Osterndorff-Kahanek E, Johnson D, Borghese CM, Hanrahan JR, Johnston GA, Chebib M, 2014. GABAA receptors containing ρ1 subunits contribute to in vivo effects of ethanol in mice. PLoS One 9, e85525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Harris RA, 2008. Metabotropic glutamate receptor 5 (mGluR5) regulation of ethanol sedation, dependence and consumption: relationship to acamprosate actions. Int J Neuropsychopharmacol 11, 775–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Walker D, Martinez M, Harris RA, 2006. Reduced alcohol consumption in mice lacking preprodynorphin. Alcohol 40, 73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Walker DL, Iyer SV, Homanics G, Harris AR, 2010. PRECLINICAL STUDY: Mice lacking Gad2 show altered behavioral effects of ethanol, flurazepam and gabaxadol. Addict Biol 15, 45–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm II SL, Ponomarev I, Jennings AW, Whiting PJ, Rosahl TW, Garrett EM, Blednov YA, Harris RA, 2004. γ-Aminobutyric acid A receptor subunit mutant mice: new perspectives on alcohol actions. Biochemical pharmacology 68, 1581–1602. [DOI] [PubMed] [Google Scholar]
- Bonasera SJ, Chu H-M, Brennan TJ, Tecott LH, 2006. A null mutation of the serotonin 6 receptor alters acute responses to ethanol. Neuropsychopharmacology 31, 1801. [DOI] [PubMed] [Google Scholar]
- Bond J, Kaskutas LA, Weisner C, 2003. The persistent influence of social networks and alcoholics anonymous on abstinence. Journal of Studies on Alcohol 64, 579–588. [DOI] [PubMed] [Google Scholar]
- Bottlender M, Soyka M, 2004. Impact of craving on alcohol relapse during, and 12 months following, outpatient treatment. Alcohol Alcohol 39, 357–361. [DOI] [PubMed] [Google Scholar]
- Bouwknecht JA, Hijzen TH, van der Gugten J, Maes RA, Hen R, Olivier B, 2000. Ethanol intake is not elevated in male 5-HT(1B) receptor knockout mice. Eur J Pharmacol 403, 95–98. [DOI] [PubMed] [Google Scholar]
- Bowen MT, Carson DS, Spiro A, Arnold JC, McGregor IS, 2011. Adolescent oxytocin exposure causes persistent reductions in anxiety and alcohol consumption and enhances sociability in rats. PLoS ONE 6, e27237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen MT, Liu J, Buisman-Pijlman FTA, 2016. Oxytocin: Providing New Avenues for Treating and Understanding Problematic Drug Use, in: Preedy VR (Ed.), Neuropathology of Drug Addictions and Substance Misuse. Academic Press, San Diego, pp. 82–92. [Google Scholar]
- Bowen MT, Neumann ID, 2017. Rebalancing the Addicted Brain: Oxytocin Interference with the Neural Substrates of Addiction. Trends in neurosciences 40, 691–708. [DOI] [PubMed] [Google Scholar]
- Bowen MT, Neumann ID, 2018. The multidimensional therapeutic potential of oxytocin for the treatment of substance use disorders. Current Topics in Behavioral Neuroscience, 269–287. [DOI] [PubMed] [Google Scholar]
- Bowen MT, Peters ST, Absalom N, Chebib M, Neumann ID, McGregor IS, 2015. Oxytocin prevents ethanol actions at δ subunit-containing GABAA receptors and attenuates ethanol-induced motor impairment in rats. Proceedings of the National Academy of Sciences 112, 3104–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce-Rustay JM, Wiedholz LM, Millstein RA, Carroll J, Murphy DL, Daws LC, Holmes A, 2006. Ethanol-related behaviors in serotonin transporter knockout mice. Alcohol Clin Exp Res 30, 1957–1965. [DOI] [PubMed] [Google Scholar]
- Brady KT, Back SE, 2012. Childhood trauma, posttraumatic stress disorder, and alcohol dependence. Alcohol Res 34, 408–413. [PMC free article] [PubMed] [Google Scholar]
- Bragard E, Giorgi S, Juneau P, Curtis BL, 2021. Loneliness and Daily Alcohol Consumption During the COVID-19 Pandemic. Alcohol and Alcoholism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breese GR, Knapp DJ, 2016. Persistent adaptation by chronic alcohol is facilitated by neuroimmune activation linked to stress and CRF. Alcohol 52, 9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breese GR, Knapp DJ, Overstreet DH, 2004. Stress sensitization of ethanol withdrawal-induced reduction in social interaction: inhibition by CRF-1 and benzodiazepine receptor antagonists and a 5-HT 1A-receptor agonist. Neuropsychopharmacology 29, 470–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breese GR, Overstreet DH, Knapp DJ, 2005. Conceptual framework for the etiology of alcoholism: a “kindling”/stress hypothesis. Psychopharmacology (Berl) 178, 367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunborg GS, Norstrom T, Storvoll EE, 2018. Latent developmental trajectories of episodic heavy drinking from adolescence to early adulthood: Predictors of trajectory groups and alcohol problems in early adulthood as outcome. Drug Alcohol Rev 37, 389–395. [DOI] [PubMed] [Google Scholar]
- Buck CL, Malavar JC, George O, Koob GF, Vendruscolo LF, 2014. Anticipatory 50kHz ultrasonic vocalizations are associated with escalated alcohol intake in dependent rats. 271, 171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner JD, Schmidt NB, Lang AR, Small JW, Schlauch RC, Lewinsohn PM, 2008. Specificity of social anxiety disorder as a risk factor for alcohol and cannabis dependence. Journal of psychiatric research 42, 230–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulwa ZB, Sharlin JA, Clark PJ, Bhattacharya TK, Kilby CN, Wang Y, Rhodes JS, 2011. Increased consumption of ethanol and sugar water in mice lacking the dopamine D2 long receptor. Alcohol 45, 631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burish TG, Maisto SA, Cooper AM, Sobell MB, 1981. Effects of voluntary short-term abstinence from alcohol on subsequent drinking patterns of college students. Journal of Studies on Alcohol 42, 1013–1020. [DOI] [PubMed] [Google Scholar]
- Butler TR, Karkhanis AN, Jones SR, Weiner JL, 2016. Adolescent Social Isolation as a Model of Heightened Vulnerability to Comorbid Alcoholism and Anxiety Disorders. Alcohol Clin Exp Res 40, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camp MC, Feyder M, Ihne J, Palachick B, Hurd B, Karlsson RM, Noronha B, Chen YC, Coba MP, Grant SG, Holmes A, 2011. A novel role for PSD-95 in mediating ethanol intoxication, drinking and place preference. Addict Biol 16, 428–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll ME, Lynch WJ, Roth ME, Morgan AD, Cosgrove KP, 2004. Sex and estrogen influence drug abuse. Trends Pharmacol Sci 25, 273–279. [DOI] [PubMed] [Google Scholar]
- Caruso MJ, Seemiller LR, Fetherston TB, Miller CN, Reiss DE, Cavigelli SA, Kamens HM, 2018. Adolescent social stress increases anxiety-like behavior and ethanol consumption in adult male and female C57BL/6J mice. Sci Rep 8, 10040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassin L, Pitts SC, Prost J, 2002. Binge drinking trajectories from adolescence to emerging adulthood in a high-risk sample: predictors and substance abuse outcomes. J Consult Clin Psychol 70, 67–78. [PubMed] [Google Scholar]
- Chen AC, Manz N, Tang Y, Rangaswamy M, Almasy L, Kuperman S, Nurnberger J Jr., O’Connor SJ, Edenberg HJ, Schuckit MA, Tischfield J, Foroud T, Bierut LJ, Rohrbaugh J, Rice JP, Goate A, Hesselbrock V, Porjesz B, 2010. Single-nucleotide polymorphisms in corticotropin releasing hormone receptor 1 gene (CRHR1) are associated with quantitative trait of event-related potential and alcohol dependence. Alcohol Clin Exp Res 34, 988–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Lu RB, Chen YC, Wang MF, Chang YC, Li TK, Yin SJ, 1999. Interaction between the functional polymorphisms of the alcohol-metabolism genes in protection against alcoholism. Am J Hum Genet 65, 795–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhetri HB, Macaya-Sanz D, Kainer D, Biswal AK, Evans LM, Chen JG, Collins C, Hunt K, Mohanty SS, Rosenstiel T, Ryno D, Winkeler K, Yang X, Jacobson D, Mohnen D, Muchero W, Strauss SH, Tschaplinski TJ, Tuskan GA, DiFazio SP, 2019. Multitrait genome-wide association analysis of Populus trichocarpa identifies key polymorphisms controlling morphological and physiological traits. New Phytol 223, 293–309. [DOI] [PubMed] [Google Scholar]
- Chou K-L, Liang K, Sareen J, 2011. The association between social isolation and DSM-IV mood, anxiety, and substance use disorders: Wave 2 of the National Epidemiologic Survey on Alcohol and Related Conditions. The Journal of Clinical Psychiatry 72, 1468–1476. [DOI] [PubMed] [Google Scholar]
- Christian DT, Wang X, Chen EL, Sehgal LK, Ghassemlou MN, Miao JJ, Estepanian D, Araghi CH, Stutzmann GE, Wolf ME, 2017. Dynamic Alterations of Rat Nucleus Accumbens Dendritic Spines over 2 Months of Abstinence from Extended-Access Cocaine Self-Administration. Neuropsychopharmacology 42, 748–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu K, Koob GF, Cole M, Zorrilla EP, Roberts AJ, 2007. Dependence-induced increases in ethanol self-administration in mice are blocked by the CRF1 receptor antagonist antalarmin and by CRF1 receptor knockout. Pharmacol Biochem Behav 86, 813–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccocioppo R, Economidou D, Cippitelli A, Cucculelli M, Ubaldi M, Soverchia L, Lourdusamy A, Massi M, 2006. Genetically selected Marchigian Sardinian alcohol-preferring (msP) rats: an animal model to study the neurobiology of alcoholism. Addict Biol 11, 339–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke TK, Dempster E, Docherty SJ, Desrivieres S, Lourdsamy A, Wodarz N, Ridinger M, Maier W, Rietschel M, Schumann G, 2012. Multiple polymorphisms in genes of the adrenergic stress system confer vulnerability to alcohol abuse. Addict Biol 17, 202–208. [DOI] [PubMed] [Google Scholar]
- Clay JM, Adams C, Archer P, English M, Hyde A, Stafford LD, Parker MO, 2018. Psychosocial stress increases craving for alcohol in social drinkers: Effects of risk-taking. Drug and Alcohol Dependence 185, 192–197. [DOI] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, Marinelli M, Wolf ME, 2008. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature 454, 118–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contet C, Gardon O, Filliol D, Becker JA, Koob GF, Kieffer BL, 2011. Identification of genes regulated in the mouse extended amygdala by excessive ethanol drinking associated with dependence. Addict Biol 16, 615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo PR, Malone PS, Belsky D, Kertesz S, Pletcher M, Sloan FA, 2007. Longitudinal differences in alcohol use in early adulthood. J Stud Alcohol Drugs 68, 727–737. [DOI] [PubMed] [Google Scholar]
- Couzigou P, Coutelle C, Fleury B, Iron A, 1994. Alcohol and aldehyde dehydrogenase genotypes, alcoholism and alcohol related disease. Alcohol Alcohol Suppl 2, 21–27. [PubMed] [Google Scholar]
- Cowen MS, Schroff KC, Gass P, Sprengel R, Spanagel R, 2003. Neurobehavioral effects of alcohol in AMPA receptor subunit (GluR1) deficient mice. Neuropharmacology 45, 325–333. [DOI] [PubMed] [Google Scholar]
- Crabbe JC, Harkness JH, Spence SE, Huang LC, Metten P, 2012. Intermittent Availability of Ethanol Does Not Always Lead to Elevated Drinking in Mice. Alcohol and Alcoholism 47, 509–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabbe JC, Metten P, Rhodes JS, Yu CH, Brown LL, Phillips TJ, Finn DA, 2009. A line of mice selected for high blood ethanol concentrations shows drinking in the dark to intoxication. Biol Psychiatry 65, 662–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabbe JC, Ozburn AR, Metten P, Barkley-Levenson A, Schlumbohm JP, Spence SE, Hack WR, Huang LC, 2017. High Drinking in the Dark (HDID) mice are sensitive to the effects of some clinically relevant drugs to reduce binge-like drinking. Pharmacol Biochem Behav 160, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabbe JC, Phillips TJ, Feller DJ, Hen R, Wenger CD, Lessov CN, Schafer GL, 1996. Elevated alcohol consumption in null mutant mice lacking 5–HT1B serotonin receptors. Nature genetics 14, 98. [DOI] [PubMed] [Google Scholar]
- D’Souza MS, 2015. Glutamatergic transmission in drug reward: implications for drug addiction. Front Neurosci 9, 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannenhoffer CA, Kim EU, Saalfield J, Werner DF, Varlinskaya EI, Spear LP, 2018. Oxytocin and vasopressin modulation of social anxiety following adolescent intermittent ethanol exposure. Psychopharmacology 235, 3065–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies M, 2003. The role of GABAA receptors in mediating the effects of alcohol in the central nervous system. Journal of psychiatry & neuroscience. [PMC free article] [PubMed] [Google Scholar]
- Davis JP, Dumas TM, Berey B, Merrin GJ, Tan K, Madden DR, 2018. Poly-victimization and trajectories of binge drinking from adolescence to young adulthood among serious juvenile offenders. Drug Alcohol Depend 186, 29–35. [DOI] [PubMed] [Google Scholar]
- De Almeida Magalhães T, Correia D, De Carvalho LM, Damasceno S, Brunialti Godard AL, 2018. Maternal separation affects expression of stress response genes and increases vulnerability to ethanol consumption. Brain and Behavior 8, e00841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Guglielmo G, Crawford E, Kim S, Vendruscolo LF, Hope BT, Brennan M, Cole M, Koob GF, George O, 2016. Recruitment of a Neuronal Ensemble in the Central Nucleus of the Amygdala Is Required for Alcohol Dependence. J Neurosci 36, 9446–9453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Guglielmo G, Kallupi M, Pomrenze MB, Crawford E, Simpson S, Schweitzer P, Koob GF, Messing RO, George O, 2019. Inactivation of a CRF-dependent amygdalofugal pathway reverses addiction-like behaviors in alcohol-dependent rats. Nature Communications 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debell F, Fear NT, Head M, Batt-Rawden S, Greenberg N, Wessely S, Goodwin L, 2014. A systematic review of the comorbidity between PTSD and alcohol misuse. Social Psychiatry and Psychiatric Epidemiology 49, 1401–1425. [DOI] [PubMed] [Google Scholar]
- Deehan GA Jr., Cain ME, Kiefer SW, 2007. Differential rearing conditions alter operant responding for ethanol in outbred rats. Alcohol Clin Exp Res 31, 1692–1698. [DOI] [PubMed] [Google Scholar]
- Deehan GA Jr., Palmatier MI, Cain ME, Kiefer SW, 2011. Differential rearing conditions and alcohol-preferring rats: consumption of and operant responding for ethanol. Behav Neurosci 125, 184–193. [DOI] [PubMed] [Google Scholar]
- Delis F, Rombola C, Bellezza R, Rosko L, Grandy DK, Volkow ND, Thanos PK, 2015. Regulation of ethanol intake under chronic mild stress: roles of dopamine receptors and transporters. Frontiers in behavioral neuroscience 9, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delis F, Thanos PK, Rombola C, Rosko L, Grandy D, Wang G-J, Volkow ND, 2013. Chronic mild stress increases alcohol intake in mice with low dopamine D2 receptor levels. Behavioral neuroscience 127, 95. [DOI] [PubMed] [Google Scholar]
- DeMartini KS, Fucito LM, 2014. Variations in sleep characteristics and sleep-related impairment in at-risk college drinkers: a latent profile analysis. Health Psychol 33, 1164–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Granados JL, Graham DL, 2007. The Effects of Continuous and Intermittent Ethanol Exposure in Adolesence on the Aversive Properties of Ethanol During Adulthood. Alcoholism: Clinical and Experimental Research 31, 2020–2027. [DOI] [PubMed] [Google Scholar]
- Dick DM, Latendresse SJ, Lansford JE, Budde JP, Goate A, Dodge KA, Pettit GS, Bates JE, 2009. Role of GABRA2 in trajectories of externalizing behavior across development and evidence of moderation by parental monitoring. Arch Gen Psychiatry 66, 649–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djouma E, Card K, Lodge DJ, Lawrence AJ, 2006. The CRF1 receptor antagonist, antalarmin, reverses isolation-induced up-regulation of dopamine D2 receptors in the amygdala and nucleus accumbens of fawn-hooded rats. Eur J Neurosci 23, 3319–3327. [DOI] [PubMed] [Google Scholar]
- Dodd PR, Beckmann AM, Davidson MS, Wilce PA, 2000. Glutamate-mediated transmission, alcohol, and alcoholism. Neurochem Int 37, 509–533. [DOI] [PubMed] [Google Scholar]
- Donath C, Gräßel E, Baier D, Pfeiffer C, Karagülle D, Bleich S, Hillemacher T, 2011. Alcohol consumption and binge drinking in adolescents: comparison of different migration backgrounds and rural vs. urban residence - a representative study. BMC Public Health 11, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong L, Bilbao A, Laucht M, Henriksson R, Yakovleva T, Ridinger M, Desrivieres S, Clarke T-K, Lourdusamy A, Smolka MN, Cichon S, Blomeyer D, Treutlein J, Perreau-Lenz S, Witt S, Leonardi-Essmann F, Wodarz N, Zill P, Soyka M, Albrecht U, Rietschel M, Lathrop M, Bakalkin G, Spanagel R, Schumann G, 2011a. Effects of the Circadian Rhythm Gene Period 1 (Per1) on Psychosocial Stress-Induced Alcohol Drinking. American Journal of Psychiatry 168, 1090–1098. [DOI] [PubMed] [Google Scholar]
- Dong L, Bilbao A, Laucht M, Henriksson R, Yakovleva T, Ridinger M, Desrivieres S, Clarke TK, Lourdusamy A, Smolka MN, Cichon S, Blomeyer D, Treutlein J, Perreau-Lenz S, Witt S, Leonardi-Essmann F, Wodarz N, Zill P, Soyka M, Albrecht U, Rietschel M, Lathrop M, Bakalkin G, Spanagel R, Schumann G, 2011b. Effects of the circadian rhythm gene period 1 (per1) on psychosocial stress-induced alcohol drinking. Am J Psychiatry 168, 1090–1098. [DOI] [PubMed] [Google Scholar]
- Dong Y, Taylor JR, Wolf ME, Shaham Y, 2017. Circuit and Synaptic Plasticity Mechanisms of Drug Relapse. The Journal of neuroscience : the official journal of the Society for Neuroscience 37, 10867–10876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doremus TL, Brunell SC, Rajendran P, Spear LP, 2005. Factors influencing elevated ethanol consumption in adolescent relative to adult rats. Alcohol Clin Exp Res 29, 1796–1808. [DOI] [PubMed] [Google Scholar]
- Du Y, Nie Y, Li Y, Wan YJY, 2011. The Association Between the SLC6A3 VNTR 9-Repeat Allele and Alcoholism—A Meta-Analysis. Alcoholism: Clinical and Experimental Research 35, 1625–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Scheidt-Nave C, Knopf H, 2008. Use of Psychotropic Drugs and Alcohol among Non-Institutionalised Elderly Adults in Germany. Pharmacopsychiatry 41, 242–251. [DOI] [PubMed] [Google Scholar]
- Edenberg HJ, Dick DM, Xuei X, Tian H, Almasy L, Bauer LO, Crowe RR, Goate A, Hesselbrock V, Jones K, 2004. Variations in GABRA2, encoding the α2 subunit of the GABAA receptor, are associated with alcohol dependence and with brain oscillations. The American Journal of Human Genetics 74, 705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edenberg HJ, Gelernter J, Agrawal A, 2019. Genetics of Alcoholism. Curr Psychiatry Rep 21, 26. [DOI] [PubMed] [Google Scholar]
- Edwards S, Baynes BB, Carmichael CY, Zamora-Martinez ER, Barrus M, Koob GF, Gilpin NW, 2013. Traumatic stress reactivity promotes excessive alcohol drinking and alters the balance of prefrontal cortex-amygdala activity. Translational Psychiatry 3, e296–e296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers CL, Walker BM, Pian JP, Roth JL, Slawecki CJ, 2007. Increased alcohol drinking in isolate-housed alcohol-preferring rats. Behav Neurosci 121, 111–119. [DOI] [PubMed] [Google Scholar]
- Eisenhardt M, Leixner S, Lujan R, Spanagel R, Bilbao A, 2015. Glutamate Receptors within the Mesolimbic Dopamine System Mediate Alcohol Relapse Behavior. The Journal of neuroscience : the official journal of the Society for Neuroscience 35, 15523–15538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel S, Lyons C, Allan A, 1998. 5-HT3 receptor over-expression decreases ethanol self administration in transgenic mice. Psychopharmacology 140, 243–248. [DOI] [PubMed] [Google Scholar]
- Enoch M-A, Baghal B, Yuan Q, Goldman D, 2013. A factor analysis of global GABAergic gene expression in human brain identifies specificity in response to chronic alcohol and cocaine exposure. PLoS One 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enoch M-A, Zhou Z, Kimura M, Mash DC, Yuan Q, Goldman D, 2012. GABAergic gene expression in postmortem hippocampus from alcoholics and cocaine addicts; corresponding findings in alcohol-naive P and NP rats. PloS one 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enoch MA, Rosser AA, Zhou Z, Mash DC, Yuan Q, Goldman D, 2014. Expression of glutamatergic genes in healthy humans across 16 brain regions; altered expression in the hippocampus after chronic exposure to alcohol or cocaine. Genes Brain Behav 13, 758–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairman BJ, Simons-Morton BG, Haynie DL, Liu D, Goldstein RB, Hingson RW, Gilman SE, 2019. State alcohol policies, taxes, and availability as predictors of adolescent binge drinking trajectories into early adulthood. Addiction 114, 1173–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farris SP, Mayfield RD, 2014. RNA-Seq reveals novel transcriptional reorganization in human alcoholic brain, International review of neurobiology. Elsevier, pp. 275–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Femenia T, Manzanares J, 2012. Increased ethanol intake in prodynorphin knockout mice is associated to changes in opioid receptor function and dopamine transmission. Addict Biol 17, 322–337. [DOI] [PubMed] [Google Scholar]
- Fernandez E, Koek W, Ran Q, Gerhardt GA, France CP, Strong R, 2006. Monoamine metabolism and behavioral responses to ethanol in mitochondrial aldehyde dehydrogenase knockout mice. Alcohol Clin Exp Res 30, 1650–1658. [DOI] [PubMed] [Google Scholar]
- Finckh U, Rommelspacher H, Kuhn° S, Dufeu° P, Otto G, Heinz° A, Dettling° M, Giraldo-Velasquez M, Pelz° J, Gräf K-J, 1997. Influence of the dopamine D2 receptor (DRD2) genotype on neuroadaptive effects of alcohol and the clinical outcome of. Pharmacogenetics 7, 271–281. [DOI] [PubMed] [Google Scholar]
- Finn DA, Helms ML, Nipper MA, Cohen A, Jensen JP, Devaud LL, 2018. Sex differences in the synergistic effect of prior binge drinking and traumatic stress on subsequent ethanol intake and neurochemical responses in adult C57BL/6J mice. Alcohol 71, 33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Bonilla A, Richardson HN, 2020. Sex Differences in the Neurobiology of Alcohol Use Disorder. Alcohol Res 40, 04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox HC, Bergquist KL, Hong KI, Sinha R, 2007. Stress-induced and alcohol cue-induced craving in recently abstinent alcohol-dependent individuals. Alcohol Clin Exp Res 31, 395–403. [DOI] [PubMed] [Google Scholar]
- Fuehrlein BS, Kachadourian LK, Devylder EK, Trevisan LA, Potenza MN, Krystal JH, Southwick SM, Pietrzak RH, 2018. Trajectories of alcohol consumption in U.S. military veterans: Results from the National Health and Resilience in Veterans Study. The American Journal on Addictions 27, 383–390. [DOI] [PubMed] [Google Scholar]
- Fuke S, Suo S, Takahashi N, Koike H, Sasagawa N, Ishiura S, 2001. The VNTR polymorphism of the human dopamine transporter (DAT1) gene affects gene expression. The pharmacogenomics journal 1, 152–156. [DOI] [PubMed] [Google Scholar]
- Funk CK, Koob GF, 2007. A CRF(2) agonist administered into the central nucleus of the amygdala decreases ethanol self-administration in ethanol-dependent rats. Brain Res 1155, 172–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk CK, Zorrilla EP, Lee MJ, Rice KC, Koob GF, 2007. Corticotropin-releasing factor 1 antagonists selectively reduce ethanol self-administration in ethanol-dependent rats. Biol Psychiatry 61, 78–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriels CM, Macharia M, Weich L, 2019. Psychiatric comorbidity among alcohol-dependent individuals seeking treatment at the Alcohol Rehabilitation Unit, Stikland Hospital. S Afr J Psychiatr 25, 1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale M, Muscatello DJ, Dinh M, Byrnes J, Shakeshaft A, Hayen A, Macintyre CR, Haber P, Cretikos M, Morton P, 2015. Alcopops, taxation and harm: a segmented time series analysis of emergency department presentations. BMC Public Health 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamsby JJ, Templeton EL, Bonvini LA, Wang W, Loros JJ, Dunlap JC, Green AI, Gulick D, 2013. The circadian Per1 and Per2 genes influence alcohol intake, reinforcement, and blood alcohol levels. Behav Brain Res 249, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelernter J, Kranzler H, Sherva R, Almasy L, Koesterer R, Smith A, Anton R, Preuss U, Ridinger M, Rujescu D, 2014. Genome-wide association study of alcohol dependence: significant findings in African-and European-Americans including novel risk loci. Molecular psychiatry 19, 41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelernter J, Sun N, Polimanti R, Pietrzak RH, Levey DF, Lu Q, Hu Y, Li B, Radhakrishnan K, Aslan M, Cheung KH, Li Y, Rajeevan N, Sayward F, Harrington K, Chen Q, Cho K, Honerlaw J, Pyarajan S, Lencz T, Quaden R, Shi Y, Hunter-Zinck H, Gaziano JM, Kranzler HR, Concato J, Zhao H, Stein MB, Department of Veterans Affairs Cooperative Studies, P., Million Veteran P, 2019. Genome-wide Association Study of Maximum Habitual Alcohol Intake in >140,000 U.S. European and African American Veterans Yields Novel Risk Loci. Biol Psychiatry 86, 365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George O, Hope BT, 2017. Cortical and amygdalar neuronal ensembles in alcohol seeking, drinking and withdrawal. Neuropharmacology 122, 107–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George O, Sanders C, Freiling J, Grigoryan E, Vu S, Allen CD, Crawford E, Mandyam CD, Koob GF, 2012. Recruitment of medial prefrontal cortex neurons during alcohol withdrawal predicts cognitive impairment and excessive alcohol drinking. Proc Natl Acad Sci U S A 109, 18156–18161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghozland S, Chu K, Kieffer BL, Roberts AJ, 2005. Lack of stimulant and anxiolytic-like effects of ethanol and accelerated development of ethanol dependence in mu-opioid receptor knockout mice. Neuropharmacology 49, 493–501. [DOI] [PubMed] [Google Scholar]
- Gilpin NW, Richardson HN, Koob GF, 2008. Effects of CRF1-receptor and opioid-receptor antagonists on dependence-induced increases in alcohol drinking by alcohol-preferring (P) rats. Alcohol Clin Exp Res 32, 1535–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilpin NW, Weiner JL, 2017. Neurobiology of comorbid post-traumatic stress disorder and alcohol-use disorder. Genes, Brain and Behavior 16, 15–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gondré-Lewis MC, Warnock KT, Wang H, June HL, Bell KA, Rabe H, Tiruveedhula VVNPB, Cook J, Lüddens H, Aurelian L, June HL, 2016. Early life stress is a risk factor for excessive alcohol drinking and impulsivity in adults and is mediated via a CRF/GABAA mechanism. Stress 19, 235–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorwood P, Aissi F, Batel P, Ades J, Cohen-Salmon C, Hamon M, Boni C, Lanfumey L, 2002. Reappraisal of the serotonin 5-HT(1B) receptor gene in alcoholism: of mice and men. Brain Res Bull 57, 103–107. [DOI] [PubMed] [Google Scholar]
- Grahame NJ, Mosemiller AK, Low MJ, Froehlich JC, 2000. Naltrexone and alcohol drinking in mice lacking β-endorphin by site-directed mutagenesis. Pharmacology Biochemistry and Behavior 67, 759–766. [DOI] [PubMed] [Google Scholar]
- Grant KA, Samson HH, 1985a. Induction and maintenance of ethanol self-administration without food deprivation in the rat. Psychopharmacology 86, 475–479. [DOI] [PubMed] [Google Scholar]
- Grant KA, Samson HH, 1985b. Oral self administration of ethanol in free feeding rats. Alcohol 2, 317–321. [DOI] [PubMed] [Google Scholar]
- Grimm JW, Lu L, Hayashi T, Hope BT, Su TP, Shaham Y, 2003. Time-dependent increases in brain-derived neurotrophic factor protein levels within the mesolimbic dopamine system after withdrawal from cocaine: implications for incubation of cocaine craving. The Journal of neuroscience : the official journal of the Society for Neuroscience 23, 742–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grisel JE, Mogil JS, Grahame NJ, Rubinstein M, Belknap JK, Crabbe JC, Low MJ, 1999. Ethanol oral self-administration is increased in mutant mice with decreased β-endorphin expression1. Brain research 835, 62–67. [DOI] [PubMed] [Google Scholar]
- Groh D, Jason L, Keys C, 2008. Social network variables in alcoholics anonymous: A literature review. Clinical Psychology Review 28, 430–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillot A, Ren T, Jourdan T, Pawlosky RJ, Han E, Kim SJ, Zhang L, Koob GF, Gao B, 2019. Targeting liver aldehyde dehydrogenase-2 prevents heavy but not moderate alcohol drinking. Proc Natl Acad Sci U S A 116, 25974–25981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall FS, 1998. Social deprivation of neonatal, adolescent, and adult rats has distinct neurochemical and behavioral consequences. Crit Rev Neurobiol 12, 129–162. [DOI] [PubMed] [Google Scholar]
- Hall FS, 2016. Chapter 16 - Reverse Translational Implications of Genome-Wide Association Studies for Addiction Genetics, in: Preedy VR (Ed.), Neuropathology of Drug Addictions and Substance Misuse. Academic Press, San Diego, pp. 153–164. [Google Scholar]
- Hall FS, Drgonova J, Jain S, Uhl GR, 2013. Implications of genome wide association studies for addiction: are our a priori assumptions all wrong? Pharmacol Ther 140, 267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall FS, Huang S, Fong GW, Pert A, Linnoila M, 1998a. Effects of isolation-rearing on locomotion, anxiety and responses to ethanol in Fawn Hooded and Wistar rats. Psychopharmacology 139, 203–209. [DOI] [PubMed] [Google Scholar]
- Hall FS, Huang S, Fong GW, Pert A, Linnoila M, 1998b. Effects of isolation-rearing on voluntary consumption of ethanol, sucrose and saccharin solutions in Fawn Hooded and Wistar rats. Psychopharmacology (Berl) 139, 210–216. [DOI] [PubMed] [Google Scholar]
- Hall FS, Perona MT, 2012. Have studies of the developmental regulation of behavioral phenotypes revealed the mechanisms of gene-environment interactions? Physiol Behav 107, 623–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall FS, Sora I, Uhl G, 2003. Sex-dependent modulation of ethanol consumption in vesicular monoamine transporter 2 (VMAT2) and dopamine transporter (DAT) knockout mice. Neuropsychopharmacology 28, 620. [DOI] [PubMed] [Google Scholar]
- Hall FS, Sora I, Uhl GR, 2001. Ethanol consumption and reward are decreased in mu-opiate receptor knockout mice. Psychopharmacology (Berl) 154, 43–49. [DOI] [PubMed] [Google Scholar]
- Hansson AC, Koopmann A, Uhrig S, Bühler S, Domi E, Kiessling E, Ciccocioppo R, Froemke RC, Grinevich V, Kiefer F, Sommer WH, Vollstädt-Klein S, Spanagel R, 2018. Oxytocin Reduces Alcohol Cue-Reactivity in Alcohol-Dependent Rats and Humans. Neuropsychopharmacology 43, 1235–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada S, Agarwal D, Goedde H, 1981. Aldehyde dehydrogenase deficiency as cause of facial flushing reaction to alcohol in Japanese. The Lancet 318, 982. [DOI] [PubMed] [Google Scholar]
- Hardee JE, Weiland BJ, Nichols TE, Welsh RC, Soules ME, Steinberg DB, Zubieta JK, Zucker RA, Heitzeg MM, 2014. Development of impulse control circuitry in children of alcoholics. Biol Psychiatry 76, 708–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RA, McQuilkin SJ, Paylor R, Abeliovich A, Tonegawa S, Wehner JM, 1995. Mutant mice lacking the gamma isoform of protein kinase C show decreased behavioral actions of ethanol and altered function of gamma-aminobutyrate type A receptors. Proceedings of the National Academy of Sciences 92, 3658–3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haseba T, Okuda T, Maruyama M, Akimoto T, Duester G, Ohno Y, 2020. Roles of Two Major Alcohol Dehydrogenases, ADH1 (Class I) and ADH3 (Class III), in the Adaptive Enhancement of Alcohol Metabolism Induced by Chronic Alcohol Consumption in Mice. Alcohol Alcohol 55, 11–19. [DOI] [PubMed] [Google Scholar]
- Hasler BP, Pedersen SL, 2020. Sleep and circadian risk factors for alcohol problems: a brief overview and proposed mechanisms. Current Opinion in Psychology 34, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasler BP, Soehner AM, Clark DB, 2015. Sleep and circadian contributions to adolescent alcohol use disorder. Alcohol 49, 377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig M, Egli M, Crabbe JC, Becker HC, 2010. Acute withdrawal, protracted abstinence and negative affect in alcoholism: are they linked? Addiction Biology 15, 169–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendershot CS, Claus ED, Ramchandani VA, 2016. Associations of OPRM1 A118G and alcohol sensitivity with intravenous alcohol self-administration in young adults. Addict Biol 21, 125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendershot CS, Wardell JD, McPhee MD, Ramchandani VA, 2017. A prospective study of genetic factors, human laboratory phenotypes, and heavy drinking in late adolescence. Addict Biol 22, 1343–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henniger MS, Spanagel R, Wigger A, Landgraf R, Holter SM, 2002. Alcohol self-administration in two rat lines selectively bred for extremes in anxiety-related behavior. Neuropsychopharmacology 26, 729–736. [DOI] [PubMed] [Google Scholar]
- Higuchi S, 1994. Polymorphisms of ethanol metabolizing enzyme genes and alcoholism. Alcohol Alcohol Suppl 2, 29–34. [PubMed] [Google Scholar]
- Holgate JY, Garcia H, Chatterjee S, Bartlett SE, 2017. Social and environmental enrichment has different effects on ethanol and sucrose consumption in mice. Brain and Behavior 7, e00767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holter SM, Danysz W, Spanagel R, 1996. Evidence for alcohol anti-craving properties of memantine. Eur J Pharmacol 314, R1–2. [DOI] [PubMed] [Google Scholar]
- Holter SM, Danysz W, Spanagel R, 2000. Novel uncompetitive N-methyl-D-aspartate (NMDA)-receptor antagonist MRZ 2/579 suppresses ethanol intake in long-term ethanol-experienced rats and generalizes to ethanol cue in drug discrimination procedure. J Pharmacol Exp Ther 292, 545–552. [PubMed] [Google Scholar]
- Hopf FW, Chang S-J, Sparta DR, Bowers MS, Bonci A, 2010a. Motivation for Alcohol Becomes Resistant to Quinine Adulteration After 3 to 4 Months of Intermittent Alcohol Self-Administration. Alcoholism: Clinical and Experimental Research 34, 1565–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopf FW, Chang SJ, Sparta DR, Bowers MS, Bonci A, 2010b. Motivation for alcohol becomes resistant to quinine adulteration after 3 to 4 months of intermittent alcohol self-administration. Alcohol Clin Exp Res 34, 1565–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horigian VE, Schmidt RD, Feaster DJ, 2021. Loneliness, Mental Health, and Substance Use among US Young Adults during COVID-19. Journal of Psychoactive Drugs 53, 1–9. [DOI] [PubMed] [Google Scholar]
- Hwa LS, Chu A, Levinson SA, Kayyali TM, DeBold JF, Miczek KA, 2011. Persistent escalation of alcohol drinking in C57BL/6J mice with intermittent access to 20% ethanol. Alcohol Clin Exp Res 35, 1938–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwa LS, Debold JF, Miczek KA, 2013. Alcohol in excess: CRF(1) receptors in the rat and mouse VTA and DRN. Psychopharmacology (Berl) 225, 313–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwa LS, Holly EN, DeBold JF, Miczek KA, 2016. Social stress-escalated intermittent alcohol drinking: modulation by CRF-R1 in the ventral tegmental area and accumbal dopamine in mice. Psychopharmacology 233, 681–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isse T, Matsuno K, Oyama T, Kitagawa K, Kawamoto T, 2005a. Aldehyde dehydrogenase 2 gene targeting mouse lacking enzyme activity shows high acetaldehyde level in blood, brain, and liver after ethanol gavages. Alcohol Clin Exp Res 29, 1959–1964. [DOI] [PubMed] [Google Scholar]
- Isse T, Oyama T, Kitagawa K, Matsuno K, Matsumoto A, Yoshida A, Nakayama K, Nakayama K, Kawamoto T, 2002. Diminished alcohol preference in transgenic mice lacking aldehyde dehydrogenase activity. Pharmacogenetics 12, 621–626. [DOI] [PubMed] [Google Scholar]
- Isse T, Oyama T, Matsuno K, Ogawa M, Narai-Suzuki R, Yamaguchi T, Murakami T, Kinaga T, Uchiyama I, Kawamoto T, 2005b. Paired acute inhalation test reveals that acetaldehyde toxicity is higher in aldehyde dehydrogenase 2 knockout mice than in wild-type mice. J Toxicol Sci 30, 329–337. [DOI] [PubMed] [Google Scholar]
- Jenkins WJ, Thomas HC, 1981. Genetic factors in determining susceptibility to alcohol dependence and development of alcohol-induced liver disease. Clin Gastroenterol 10, 307–314. [PubMed] [Google Scholar]
- Ji D, Gilpin NW, Richardson HN, Rivier CL, Koob GF, 2008. Effects of naltrexone, duloxetine, and a corticotropin-releasing factor type 1 receptor antagonist on binge-like alcohol drinking in rats. Behav Pharmacol 19, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Job MO, Tang A, Hall FS, Sora I, Uhl GR, Bergeson SE, Gonzales RA, 2007. Mu (mu) opioid receptor regulation of ethanol-induced dopamine response in the ventral striatum: evidence of genotype specific sexual dimorphic epistasis. Biol Psychiatry 62, 627–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joubert W, Nance J, Climer S, Weighill D, Jacobson D, 2019. Parallel accelerated Custom Correlation Coefficient calculations for genomics applications. Parallel Computing 84, 15–23. [Google Scholar]
- Joubert W, Weighill D, Kainer D, Climer S, Justice A, Fagnan K, Jacobson D, 2018. Attacking the Opioid Epidemic: Determining the Epistatic and Pleiotropic Genetic Architectures for Chronic Pain and Opioid Addiction, SC18: International Conference for High Performance Computing, Networking, Storage and Analysis, pp. 717–730. [Google Scholar]
- June HL, Foster KL, Eiler II WJ, Goergen J, Cook JB, Johnson N, Mensah-Zoe B, Simmons JO, June HL Jr, Yin W, 2007. Dopamine and benzodiazepine-dependent mechanisms regulate the EtOH-enhanced locomotor stimulation in the GABA A α1 subunit null mutant mice. Neuropsychopharmacology 32, 137. [DOI] [PubMed] [Google Scholar]
- Jurek B, Neumann ID, 2018. The Oxytocin Receptor: From Intracellular Signaling to Behavior. Physiological Reviews 98, 1805–1908. [DOI] [PubMed] [Google Scholar]
- Karpyak VM, Biernacka JM, Weg MWV, Stevens SR, Cunningham JM, Mrazek DA, Black JL, 2010. GENETIC STUDY: Interaction of SLC6A4 and DRD2 polymorphisms is associated with a history of delirium tremens. Addict Biol 15, 23–34. [DOI] [PubMed] [Google Scholar]
- Kaskutas LA, Bond J, Humphreys K, 2002. Social networks as mediators of the effect of Alcoholics Anonymous. Addiction 97, 891–900. [DOI] [PubMed] [Google Scholar]
- Kauhanen J, Hallikainen T, Tuomainen TP, Koulu M, Karvonen MK, Salonen JT, Tiihonen J, 2000. Association between the functional polymorphism of catechol-O-methyltransferase gene and alcohol consumption among social drinkers. Alcohol Clin Exp Res 24, 135–139. [PubMed] [Google Scholar]
- Kawakami SE, Quadros IMH, Machado RB, Suchecki D, 2013. Sex-dependent effects of maternal separation on plasma corticosterone and brain monoamines in response to chronic ethanol administration. Neuroscience 253, 55–66. [DOI] [PubMed] [Google Scholar]
- Kelai S, Aissi F, Lesch KP, Cohen-Salmon C, Hamon M, Lanfumey L, 2003. Alcohol intake after serotonin transporter inactivation in mice. Alcohol Alcohol 38, 386–389. [DOI] [PubMed] [Google Scholar]
- Kendler KS, Gardner C, Dick DM, 2011. Predicting alcohol consumption in adolescence from alcohol-specific and general externalizing genetic risk factors, key environmental exposures and their interaction. Psychol Med 41, 1507–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, 1995. Posttraumatic Stress Disorder in the National Comorbidity Survey. Archives of General Psychiatry 52, 1048. [DOI] [PubMed] [Google Scholar]
- Keyes KM, Hatzenbuehler ML, Hasin DS, 2011. Stressful life experiences, alcohol consumption, and alcohol use disorders: the epidemiologic evidence for four main types of stressors. Psychopharmacology 218, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khisti RT, Wolstenholme J, Shelton KL, Miles MF, 2006. Characterization of the ethanol-deprivation effect in substrains of C57BL/6 mice. Alcohol 40, 119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim R, Sepulveda-Orengo MT, Healey KL, Williams EA, Reissner KJ, 2018. Regulation of glutamate transporter 1 (GLT-1) gene expression by cocaine self-administration and withdrawal. Neuropharmacology 128, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimbrough A, de Guglielmo G, Kononoff J, Kallupi M, Zorrilla EP, George O, 2017a. CRF1 Receptor-Dependent Increases in Irritability-Like Behavior During Abstinence from Chronic Intermittent Ethanol Vapor Exposure. Alcohol Clin Exp Res 41, 1886–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimbrough A, Kim S, Cole M, Brennan M, George O, 2017b. Intermittent Access to Ethanol Drinking Facilitates the Transition to Excessive Drinking After Chronic Intermittent Ethanol Vapor Exposure. Alcoholism: Clinical and Experimental Research 41, 1502–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimbrough A, Lurie DJ, Collazo A, Kreifeldt M, Sidhu H, Macedo GC, D’Esposito M, Contet C, George O, 2020. Brain-wide functional architecture remodeling by alcohol dependence and abstinence. Proceedings of the National Academy of Sciences 117, 2149–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King CE, Becker HC, 2019. Oxytocin attenuates stress-induced reinstatement of alcohol seeking behavior in male and female mice. Psychopharmacology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King CE, Griffin WC, Lopez MF, Becker HC, 2021. Activation of hypothalamic oxytocin neurons reduces binge-like alcohol drinking through signaling at central oxytocin receptors. Neuropsychopharmacology 46, 1950–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King CE, Griffin WC, Luderman LN, Kates MM, McGinty JF, Becker HC, 2017. Oxytocin Reduces Ethanol Self-Administration in Mice. Alcoholism: Clinical and Experimental Research 41, 955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirshenbaum AP, Olsen DM, Bickel WK, 2009. A quantitative review of the ubiquitous relapse curve. Journal of Substance Abuse Treatment 36, 8–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhnke MD, Batra A, Kolb W, Köhnke AM, Lutz U, Schick S, Gaertner I, 2005. Association of the dopamine transporter gene with alcoholism. Alcohol and alcoholism 40, 339–342. [DOI] [PubMed] [Google Scholar]
- Koivisto T, Eriksson CJ, 1994. Hepatic aldehyde and alcohol dehydrogenases in alcohol-preferring and alcohol-avoiding rat lines. Biochem Pharmacol 48, 1551–1558. [DOI] [PubMed] [Google Scholar]
- Koob GF, 2009. Brain stress systems in the amygdala and addiction. Brain Res 1293, 61–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, 2013. Theoretical frameworks and mechanistic aspects of alcohol addiction: alcohol addiction as a reward deficit disorder. Current topics in behavioral neurosciences 13, 3–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, 2021. Drug Addiction: Hyperkatifeia/Negative Reinforcement as a Framework for Medications Development. Pharmacological Reviews 73, 163–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Le Moal M, 2005. Plasticity of reward neurocircuitry and the ‘dark side’ of drug addiction. Nature Neuroscience 8, 1442–1444. [DOI] [PubMed] [Google Scholar]
- Koob GF, Volkow ND, 2016. Neurobiology of addiction: a neurocircuitry analysis. Lancet Psychiatry 3, 760–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs KM, Szakall I, O’brien D, Wang R, Vinod KY, Saito M, Simonin F, Kieffer BL, Vadasz C, 2005. Decreased oral self-administration of alcohol in κ-opioid receptor knock-out mice. Alcoholism: Clinical and Experimental Research 29, 730–738. [DOI] [PubMed] [Google Scholar]
- Kovanen L, Saarikoski ST, Haukka J, Pirkola S, Aromaa A, Lonnqvist J, Partonen T, 2010. Circadian clock gene polymorphisms in alcohol use disorders and alcohol consumption. Alcohol Alcohol 45, 303–311. [DOI] [PubMed] [Google Scholar]
- Kranzler HR, Zhou H, Kember RL, Vickers Smith R, Justice AC, Damrauer S, Tsao PS, Klarin D, Baras A, Reid J, Overton J, Rader DJ, Cheng Z, Tate JP, Becker WC, Concato J, Xu K, Polimanti R, Zhao H, Gelernter J, 2019. Genome-wide association study of alcohol consumption and use disorder in 274,424 individuals from multiple populations. Nat Commun 10, 1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucharska-Mazur J, Grzywacz A, Pełka-Wysiecka J, Samochowiec A, Rommelspacher H, Samochowiec J, 2012. Haplotype analysis of DRD2 and ANKK1 gene polymorphisms in alcohol dependence. Archives of Psychiatry and Psychotherapy 2, 5–10. [Google Scholar]
- Kuerbis A, Treloar Padovano H, Shao S, Houser J, Muench FJ, Morgenstern J, 2018. Comparing daily drivers of problem drinking among older and younger adults: An electronic daily diary study using smartphones. Drug and Alcohol Dependence 183, 240–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBuda CJ, Sora I, Uhl GR, Fuchs PN, 2000. Stress-induced analgesia in μ-opioid receptor knockout mice reveals normal function of the δ-opioid receptor system. Brain research 869, 1–5. [DOI] [PubMed] [Google Scholar]
- Lamb RJ, Daws LC, 2013. Ethanol self-administration in serotonin transporter knockout mice: unconstrained demand and elasticity. Genes Brain Behav 12, 741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BR, Ma YY, Huang YH, Wang X, Otaka M, Ishikawa M, Neumann PA, Graziane NM, Brown TE, Suska A, Guo C, Lobo MK, Sesack SR, Wolf ME, Nestler EJ, Shaham Y, Schluter OM, Dong Y, 2013. Maturation of silent synapses in amygdala-accumbens projection contributes to incubation of cocaine craving. Nat Neurosci 16, 1644–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh-Hunt N, Bagguley D, Bash K, Turner V, Turnbull S, Valtorta N, Caan W, 2017. An overview of systematic reviews on the public health consequences of social isolation and loneliness. Public Health 152, 157–171. [DOI] [PubMed] [Google Scholar]
- Lensvelt E, Liang W, Gilmore W, Gordon E, Hobday M, Chikritzhs T, 2016. Effect of the Australian “Alcopops Tax” on Alcohol-Related Emergency Department Presentations for Injury in Two States. J Stud Alcohol Drugs 77, 730–739. [DOI] [PubMed] [Google Scholar]
- Lesscher HMB, Spoelder M, Rotte MD, Janssen MJ, Hesseling P, Lozeman v., t Klooster JG, Baars AM, Vanderschuren LJMJ, 2015. Early social isolation augments alcohol consumption in rats. Behavioural Pharmacology 26, 673–680. [DOI] [PubMed] [Google Scholar]
- Li D, Sulovari A, Cheng C, Zhao H, Kranzler HR, Gelernter J, 2014. Association of gamma-aminobutyric acid a receptor α 2 gene (GABRA2) with alcohol use disorder. Neuropsychopharmacology 39, 907–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Bian W, Dave V, Ye J-H, 2011. Blockade of GABAA receptors in the paraventricular nucleus of the hypothalamus attenuates voluntary ethanol intake and activates the hypothalamic-pituitary-adrenocortical axis. Addiction Biology 16, 600–614. [DOI] [PubMed] [Google Scholar]
- Li P, Wu P, Xin X, Fan Y-L, Wang G-B, Wang F, Ma M-Y, Xue M-M, Luo Y-X, Yang F-D, Bao Y-P, Shi J, Sun H-Q, Lu L, 2015. Incubation of alcohol craving during abstinence in patients with alcohol dependence. Addict Biol 20, 513–522. [DOI] [PubMed] [Google Scholar]
- Lindell SG, Schwandt ML, Suomi SJ, Rice KC, Heilig M, Barr CS, 2017. Intermittent Access to Ethanol Induces Escalated Alcohol Consumption in Primates. J Addict Behav Ther Rehabil 6, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo IA, Harris RA, 2008. GABAA receptors and alcohol. Pharmacology Biochemistry and Behavior 90, 90–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Lawrence AJ, 2003a. Comparative analysis of hepatic ethanol metabolism in Fawn-Hooded and Wistar-Kyoto rats. Alcohol 30, 75–79. [DOI] [PubMed] [Google Scholar]
- Lodge DJ, Lawrence AJ, 2003b. The CRF1 receptor antagonist antalarmin reduces volitional ethanol consumption in isolation-reared fawn-hooded rats. Neuroscience 117, 243–247. [DOI] [PubMed] [Google Scholar]
- Lodge DJ, Lawrence AJ, 2003c. The effect of isolation rearing on volitional ethanol consumption and central CCK/dopamine systems in Fawn-Hooded rats. Behav Brain Res 141, 113–122. [DOI] [PubMed] [Google Scholar]
- Logrip ML, Rivier C, Lau C, Im S, Vaughan J, Lee S, 2013. Adolescent alcohol exposure alters the rat adult hypothalamic-pituitary-adrenal axis responsiveness in a sex-specific manner. Neuroscience 235, 174–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue S, Chein J, Gould T, Holliday E, Steinberg L, 2014. Adolescent mice, unlike adults, consume more alcohol in the presence of peers than alone. Dev Sci 17, 79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longabaugh R, Wirtz PW, Zweben A, Stout RL, 1998. Network support for drinking, Alcoholics Anonymous and long-term matching effects. Addiction 93, 1313–1333. [DOI] [PubMed] [Google Scholar]
- Lopez MF, Doremus-Fitzwater TL, Becker HC, 2011. Chronic social isolation and chronic variable stress during early development induce later elevated ethanol intake in adult C57BL/6J mice. Alcohol 45, 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love TM, Cranford JA, Burmeister M, Wojnar M, Zucker RA, J. Brower K, 2018. Oxytocin Genotype Moderates the Impact of Social Support on Psychiatric Distress in Alcohol-Dependent Patients. Alcohol and Alcoholism 53, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery-Gionta EG, Marcinkiewcz CA, Kash TL, 2015. Functional alterations in the dorsal raphe nucleus following acute and chronic ethanol exposure. Neuropsychopharmacology 40, 590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Koya E, Zhai H, Hope BT, Shaham Y, 2006. Role of ERK in cocaine addiction. Trends in neurosciences 29, 695–703. [DOI] [PubMed] [Google Scholar]
- Lu L, Uejima JL, Gray SM, Bossert JM, Shaham Y, 2007. Systemic and central amygdala injections of the mGluR(2/3) agonist LY379268 attenuate the expression of incubation of cocaine craving. Biol Psychiatry 61, 591–598. [DOI] [PubMed] [Google Scholar]
- Lu L, Wang X, Wu P, Xu C, Zhao M, Morales M, Harvey BK, Hoffer BJ, Shaham Y, 2009. Role of ventral tegmental area glial cell line-derived neurotrophic factor in incubation of cocaine craving. Biol Psychiatry 66, 137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucht MJ, Kuehn KU, Schroeder W, Armbruster J, Abraham G, Schattenberg A, Gaensicke M, Barnow S, Tretzel H, Herrmann FH, 2001. Influence of the dopamine D2 receptor (DRD2) exon 8 genotype on efficacy of tiapride and clinical outcome of alcohol withdrawal. Pharmacogenetics and Genomics 11, 647–653. [DOI] [PubMed] [Google Scholar]
- Ludwig AM, Wikler A, 1974. “Craving” and relapse to drink. Q J Stud Alcohol 35, 108–130. [PubMed] [Google Scholar]
- Ludwig AM, Wikler A, Stark LH, 1974. The first drink: psychobiological aspects of craving. Arch Gen Psychiatry 30, 539–547. [DOI] [PubMed] [Google Scholar]
- Luis C, Cannella N, Spanagel R, Kohr G, 2017. Persistent strengthening of the prefrontal cortex - nucleus accumbens pathway during incubation of cocaine-seeking behavior. Neurobiol Learn Mem 138, 281–290. [DOI] [PubMed] [Google Scholar]
- Ma YY, Lee BR, Wang X, Guo C, Liu L, Cui R, Lan Y, Balcita-Pedicino JJ, Wolf ME, Sesack SR, Shaham Y, Schluter OM, Huang YH, Dong Y, 2014. Bidirectional modulation of incubation of cocaine craving by silent synapse-based remodeling of prefrontal cortex to accumbens projections. Neuron 83, 1453–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFadyen K, Loveless R, DeLucca B, Wardley K, Deogan S, Thomas C, Peris J, 2016. Peripheral oxytocin administration reduces ethanol consumption in rats. Pharmacol Biochem Behav 140, 27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makhijani VH, Franklin JP, Van Voorhies K, Fortino B, Besheer J, 2021. The synthetically produced predator odor 2,5-dihydro-2,4,5-trimethylthiazoline increases alcohol self-administration and alters basolateral amygdala response to alcohol in rats. Psychopharmacology 238, 67–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Fardon R, Weiss F, 2013. Modeling relapse in animals. Curr Top Behav Neurosci 13, 403–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita S, Higuchi S, 2014. Genetic differences in response to alcohol. Handb Clin Neurol 125, 617–627. [DOI] [PubMed] [Google Scholar]
- McCarthy MJ, Fernandes M, Kranzler HR, Covault JM, Welsh DK, 2013. Circadian clock period inversely correlates with illness severity in cells from patients with alcohol use disorders. Alcohol Clin Exp Res 37, 1304–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCool BA, Chappell AM, 2009. Early social isolation in male Long-Evans rats alters both appetitive and consummatory behaviors expressed during operant ethanol self-administration. Alcohol Clin Exp Res 33, 273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon JE, Wang X, Tseng KY, Wolf ME, Marinelli M, 2011. Calcium-permeable AMPA receptors are present in nucleus accumbens synapses after prolonged withdrawal from cocaine self-administration but not experimenter-administered cocaine. The Journal of neuroscience : the official journal of the Society for Neuroscience 31, 5737–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGregor IS, Bowen MT, 2012. Breaking the loop: Oxytocin as a potential treatment for drug addiction. Hormones and Behavior 61, 331–339. [DOI] [PubMed] [Google Scholar]
- McGregor IS, Bowen MT, 2013. Oxytocin and addiction: recent preclinical advances and future clinical potential, in: Choleris E, Pfaff DW, Kavaliers M (Eds.), Oxytocin, Vasopressin and Related Peptides in the Regulation of Behavior. Cambridge University Press, Cambridge UK, pp. 270–287. [Google Scholar]
- McGregor IS, Schrama L, Ambermoon P, Dielenberg RA, 2002. Not all ‘predator odours’ are equal: cat odour but not 2,4,5 trimethylthiazoline (TMT; fox odour) elicits specific defensive behaviours in rats. Behavioural Brain Research 129, 1–16. [DOI] [PubMed] [Google Scholar]
- Melendez RI, 2011. Intermittent (every-other-day) drinking induces rapid escalation of ethanol intake and preference in adolescent and adult C57BL/6J mice. Alcohol Clin Exp Res 35, 652–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melendez RI, Middaugh LD, Kalivas PW, 2006. Development of an alcohol deprivation and escalation effect in C57BL/6J mice. Alcohol Clin Exp Res 30, 2017–2025. [DOI] [PubMed] [Google Scholar]
- Merline A, Jager J, Schulenberg JE, 2008. Adolescent risk factors for adult alcohol use and abuse: stability and change of predictive value across early and middle adulthood. Addiction 103 Suppl 1, 84–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz AV, Chynoweth J, Allan AM, 2006. Influence of genetic background on alcohol drinking and behavioral phenotypes of 5-HT3 receptor over-expressing mice. Pharmacology Biochemistry and Behavior 84, 120–127. [DOI] [PubMed] [Google Scholar]
- Meyers J, McCutcheon VV, Pandey AK, Kamarajan C, Subbie S, Chorlian D, Salvatore J, Pandey G, Almasy L, Anokhin A, Bauer L, Bender A, Dick DM, Edenberg HJ, Hesselbrock V, Kramer J, Kuperman S, Agrawal A, Bucholz K, Porjesz B, 2019. Early Sexual Trauma Exposure and Neural Response Inhibition in Adolescence and Young Adults: Trajectories of Frontal Theta Oscillations During a Go/No-Go Task. J Am Acad Child Adolesc Psychiatry 58, 242–255 e242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihalek RM, Bowers BJ, Wehner JM, Kralic JE, VanDoren MJ, Morrow AL, Homanics GE, 2001. GABAA-receptor δ subunit knockout mice have multiple defects in behavioral responses to ethanol. Alcoholism: Clinical AND Experimental Research 25, 1708–1718. [PubMed] [Google Scholar]
- Mitchell JM, Arcuni PA, Weinstein D, Woolley JD, 2016. Intranasal Oxytocin Selectively Modulates Social Perception, Craving, and Approach Behavior in Subjects With Alcohol Use Disorder. Journal of addiction medicine 10, 182–189. [DOI] [PubMed] [Google Scholar]
- Mojica-Perez Y, Callinan S, Livingston M, 2020. Examining beverage-specific trends in youth drinking in Australia before and after the implementation of the alcopops tax. Drug and Alcohol Review 39, 246–254. [DOI] [PubMed] [Google Scholar]
- Molander A, Vengeliene V, Heilig M, Wurst W, Deussing JM, Spanagel R, 2012. Brain-specific inactivation of the Crhr1 gene inhibits post-dependent and stress-induced alcohol intake, but does not affect relapse-like drinking. Neuropsychopharmacology 37, 1047–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CF, Lynch WJ, 2015. Alcohol preferring (P) rats as a model for examining sex differences in alcohol use disorder and its treatment. Pharmacol Biochem Behav 132, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moos RH, Brennan PL, Schutte KK, Moos BS, 2010. Social and Financial Resources and High-Risk Alcohol Consumption Among Older Adults. 34, 646–654. [DOI] [PubMed] [Google Scholar]
- Morales M, McGinnis MM, McCool BA, 2015. Chronic ethanol exposure increases voluntary home cage intake in adult male, but not female, Long-Evans rats. Pharmacol Biochem Behav 139, 67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morice E, Denis C, Giros B, Nosten-Bertrand M, 2010. Evidence of long-term expression of behavioral sensitization to both cocaine and ethanol in dopamine transporter knockout mice. Psychopharmacology (Berl) 208, 57–66. [DOI] [PubMed] [Google Scholar]
- Moriya Y, Kasahara Y, Hall FS, Sakakibara Y, Uhl GR, Tomita H, Sora I, 2015. Sex differences in the effects of adolescent social deprivation on alcohol consumption in μ-opioid receptor knockout mice. Psychopharmacology 232, 1471–1482. [DOI] [PubMed] [Google Scholar]
- Nakamura A, Inada T, Kitao Y, Katayama Y, 2001. Association between catechol-O-methyltransferase (COMT) polymorphism and severe alcoholic withdrawal symptoms in male Japanese alcoholics. Addict Biol 6, 233–238. [DOI] [PubMed] [Google Scholar]
- Nealon-Woods MA, Ferrari JR, Jason LA, 1995. Twelve-step program use among Oxford house residents: Spirituality or social support in sobriety? Journal of Substance Abuse 7, 311–318. [DOI] [PubMed] [Google Scholar]
- Nelson BS, Sequeira MK, Schank JR, 2018. Bidirectional relationship between alcohol intake and sensitivity to social defeat: association with Tacr1 and Avp expression. Addict Biol 23, 142–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nennig SE, Fulenwider HD, Eskew JE, Whiting KE, Cotton MR, McGinty GE, Solomon MG, Schank JR, 2020. Intermittent Ethanol Access Increases Sensitivity to Social Defeat Stress. Alcohol Clin Exp Res 44, 600–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EL, Albrechet-Souza L, Andrew PM, Auld JG, Burk KC, Hwa LS, Zhang EY, Debold JF, Miczek KA, 2018a. Persistent escalation of alcohol consumption by mice exposed to brief episodes of social defeat stress: suppression by CRF-R1 antagonism. Psychopharmacology 235, 1807–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EL, Covington HE, Leonard MZ, Burk K, Miczek KA, 2021. Hypoactive Thalamic Crh+ Cells in a Female Mouse Model of Alcohol Drinking After Social Trauma. Biological Psychiatry 90, 563–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EL, Leonard MZ, Arena DT, De Almeida RMM, Miczek KA, 2018b. Social defeat stress and escalation of cocaine and alcohol consumption: Focus on CRF. Neurobiology of Stress 9, 151–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen DA, Goldman D, Virkkunen M, Tokola R, Rawlings R, Linnoila M, 1994. Suicidality and 5-hydroxyindoleacetic acid concentration associated with a tryptophan hydroxylase polymorphism. Arch Gen Psychiatry 51, 34–38. [DOI] [PubMed] [Google Scholar]
- Nielsen DA, Virkkunen M, Lappalainen J, Eggert M, Brown GL, Long JC, Goldman D, Linnoila M, 1998. A tryptophan hydroxylase gene marker for suicidality and alcoholism. Arch Gen Psychiatry 55, 593–602. [DOI] [PubMed] [Google Scholar]
- Nikolova YS, Knodt AR, Radtke SR, Hariri AR, 2016. Divergent responses of the amygdala and ventral striatum predict stress-related problem drinking in young adults: possible differential markers of affective and impulsive pathways of risk for alcohol use disorder. Molecular Psychiatry 21, 348–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell PJ, 1984. The abstinence violation effect and circumstances surrounding relapse as predictors of outcome status in male alcoholic outpatients. J Psychol 117, 257–262. [DOI] [PubMed] [Google Scholar]
- Okuda T, Haseba T, Katsuyama M, Maruyama M, Akimoto T, Igarashi T, Ohno Y, 2018. Metabolic pharmacokinetics of early chronic alcohol consumption mediated by liver alcohol dehydrogenases 1 and 3 in mice. J Gastroenterol Hepatol 33, 1912–1919. [DOI] [PubMed] [Google Scholar]
- Olfson E, Bierut LJ, 2012. Convergence of genome-wide association and candidate gene studies for alcoholism. Alcohol Clin Exp Res 36, 2086–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive MF, Koenig HN, Nannini MA, Hodge CW, 2002. Elevated extracellular CRF levels in the bed nucleus of the stria terminalis during ethanol withdrawal and reduction by subsequent ethanol intake. Pharmacol Biochem Behav 72, 213–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive MF, Mehmert KK, Koenig HN, Camarini R, Kim JA, Nannini MA, Ou CJ, Hodge CW, 2003. A role for corticotropin releasing factor (CRF) in ethanol consumption, sensitivity, and reward as revealed by CRF-deficient mice. Psychopharmacology (Berl) 165, 181–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oreland L, Lagravinese G, Toffoletto S, Nilsson KW, Harro J, Robert Cloninger C, Comasco E, 2018. Personality as an intermediate phenotype for genetic dissection of alcohol use disorder. J Neural Transm (Vienna) 125, 107–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornelas LC, Tyler RE, Irukulapati P, Paladugu S, Besheer J, 2021. Increased alcohol self-administration following exposure to the predator odor TMT in active coping female rats. Behavioural Brain Research 402, 113068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterndorff-Kahanek E, Ponomarev I, Blednov YA, Harris RA, 2013. Gene expression in brain and liver produced by three different regimens of alcohol consumption in mice: comparison with immune activation. PLoS One 8, e59870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet DH, Knapp DJ, Angel RA, Navarro M, Breese GR, 2006. Reduction in repeated ethanol-withdrawal-induced anxiety-like behavior by site-selective injections of 5-HT 1A and 5-HT 2C ligands. Psychopharmacology 187, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AA, Sharpe AL, Burkhart-Kasch S, McKinnon CS, Coste SC, Stenzel-Poore MP, Phillips TJ, 2004. Corticotropin-releasing factor overexpression decreases ethanol drinking and increases sensitivity to the sedative effects of ethanol. Psychopharmacology (Berl) 176, 386–397. [DOI] [PubMed] [Google Scholar]
- Pastor R, McKinnon CS, Scibelli AC, Burkhart-Kasch S, Reed C, Ryabinin AE, Coste SC, Stenzel-Poore MP, Phillips TJ, 2008. Corticotropin-releasing factor-1 receptor involvement in behavioral neuroadaptation to ethanol: a urocortin1-independent mechanism. Proc Natl Acad Sci U S A 105, 9070–9075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul P, Dahale A, Kishore B, Chand P, Benegal V, Jain S, Murthy P, Purushottam M, 2017. Association of N-Methyl-D-Aspartate receptor 2B Subunit (GRIN2B) polymorphism with earlier age at onset of withdrawal symptoms in Indian alcohol dependent subjects. J Addict Dis 36, 48–52. [DOI] [PubMed] [Google Scholar]
- Pedersen CA, Smedley KL, Leserman J, Jarskog LF, Rau SW, Kampov-Polevoi A, Casey RL, Fender T, Garbutt JC, 2012. Intranasal Oxytocin Blocks Alcohol Withdrawal in Human Subjects. Alcohol Clin Exp Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng G-S, Yin S-J, 2009. Effect of the allelic variants of aldehyde dehydrogenase ALDH2* 2 and alcohol dehydrogenase ADH1B* 2on blood acetaldehyde concentrations. Human genomics 3, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters S, Slattery DA, Flor PJ, Neumann ID, Reber SO, 2013. Differential effects of baclofen and oxytocin on the increased ethanol consumption following chronic psychosocial stress in mice. Addiction Biology 18, 66–77. [DOI] [PubMed] [Google Scholar]
- Peters ST, Bowen MT, Bohrer K, McGregor IS, Neumann ID, 2017. Oxytocin inhibits ethanol consumption and ethanol-induced dopamine release in the nucleus accumbens. Addict Biol 22, 702–711. [DOI] [PubMed] [Google Scholar]
- Petrakis IL, Trevisan L, Boutros NN, Limoncelli D, Cooney NL, Krystal JH, 2001. Effect of tryptophan depletion on alcohol cue-induced craving in abstinent alcoholic patients. Alcohol Clin Exp Res 25, 1151–1155. [PubMed] [Google Scholar]
- Pfefferbaum A, Kwon D, Brumback T, Thompson WK, Cummins K, Tapert SF, Brown SA, Colrain IM, Baker FC, Prouty D, De Bellis MD, Clark DB, Nagel BJ, Chu W, Park SH, Pohl KM, Sullivan EV, 2018. Altered Brain Developmental Trajectories in Adolescents After Initiating Drinking. Am J Psychiatry 175, 370–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierucci-Lagha A, Covault J, Feinn R, Nellissery M, Hernandez-Avila C, Oncken C, Morrow AL, Kranzler HR, 2005. GABRA2 alleles moderate the subjective effects of alcohol, which are attenuated by finasteride. Neuropsychopharmacology 30, 1193–1203. [DOI] [PubMed] [Google Scholar]
- Pinel JP, Huang E, 1976. Effects of periodic withdrawal on ethanol and saccharin selection in rats. Physiol Behav 16, 693–698. [DOI] [PubMed] [Google Scholar]
- Plemenitas A, Kores Plesnicar B, Kastelic M, Porcelli S, Serretti A, Dolzan V, 2015. Genetic variability in tryptophan hydroxylase 2 gene in alcohol dependence and alcohol-related psychopathological symptoms. Neurosci Lett 604, 86–90. [DOI] [PubMed] [Google Scholar]
- Ponomarev I, Wang S, Zhang L, Harris RA, Mayfield RD, 2012. Gene coexpression networks in human brain identify epigenetic modifications in alcohol dependence. The Journal of neuroscience : the official journal of the Society for Neuroscience 32, 1884–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popova NK, Vishnivetskaya GB, Ivanova EA, Skrinskaya JA, Seif I, 2000. Altered behavior and alcohol tolerance in transgenic mice lacking MAO A: a comparison with effects of MAO A inhibitor clorgyline. Pharmacol Biochem Behav 67, 719–727. [DOI] [PubMed] [Google Scholar]
- Portero-Tresserra M, Gracia-Rubio I, Cantacorps L, Pozo OJ, Gómez-Gómez A, Pastor A, López-Arnau R, De La Torre R, Valverde O, 2018. Maternal separation increases alcohol-drinking behaviour and reduces endocannabinoid levels in the mouse striatum and prefrontal cortex. European Neuropsychopharmacology 28, 499–512. [DOI] [PubMed] [Google Scholar]
- Preuss UW, Wurst FM, Ridinger M, Rujescu D, Fehr C, Koller G, Bondy B, Wodarz N, Soyka M, Zill P, 2013. Association of functional DBH genetic variants with alcohol dependence risk and related depression and suicide attempt phenotypes: results from a large multicenter association study. Drug Alcohol Depend 133, 459–467. [DOI] [PubMed] [Google Scholar]
- Preuss UW, Zill P, Koller G, Bondy B, Hesselbrock V, Soyka M, 2006. Ionotropic glutamate receptor gene GRIK3 SER310ALA functional polymorphism is related to delirium tremens in alcoholics. Pharmacogenomics J 6, 34–41. [DOI] [PubMed] [Google Scholar]
- Priddy BM, Carmack SA, Thomas LC, Vendruscolo JC, Koob GF, Vendruscolo LF, 2017. Sex, strain, and estrous cycle influences on alcohol drinking in rats. Pharmacol Biochem Behav 152, 61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purgianto A, Loweth JA, Miao JJ, Milovanovic M, Wolf ME, 2016. Surface expression of GABAA receptors in the rat nucleus accumbens is increased in early but not late withdrawal from extended-access cocaine self-administration. Brain Res 1642, 336–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putzke J, Spanagel R, Tolle TR, Zieglgansberger W, 1996. The anti-craving drug acamprosate reduces c-fos expression in rats undergoing ethanol withdrawal. Eur J Pharmacol 317, 39–48. [DOI] [PubMed] [Google Scholar]
- Quintanilla ME, Israel Y, Sapag A, Tampier L, 2006. The UChA and UChB rat lines: metabolic and genetic differences influencing ethanol intake. Addict Biol 11, 310–323. [DOI] [PubMed] [Google Scholar]
- Radouco-Thomas S, Garcin F, Laperriere A, Marquis PA, Lambert J, Denver J, Lacerte M, Lacroix D, Radouco-Thomas C, 1979. Genetic epidemiology and the prevention of functional mental disorders and alcoholism: family study and biological predictors. Prog Neuropsychopharmacol 3, 165–189. [DOI] [PubMed] [Google Scholar]
- Raeder H, Holter SM, Hartmann AM, Spanagel R, Moller HJ, Rujescu D, 2008. Expression of N-methyl-d-aspartate (NMDA) receptor subunits and splice variants in an animal model of long-term voluntary alcohol self-administration. Drug Alcohol Depend 96, 16–21. [DOI] [PubMed] [Google Scholar]
- Ramachandra V, Kang F, Kim C, Nova AS, Bajaj A, Hall FS, Uhl GR, Gonzales RA, 2011. The mu opioid receptor is not involved in ethanol-stimulated dopamine release in the ventral striatum of C57BL/6J mice. Alcohol Clin Exp Res 35, 929–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray LA, Bujarski S, Squeglia LM, Ashenhurst JR, Anton RF, 2014. Interactive effects of OPRM1 and DAT1 genetic variation on subjective responses to alcohol. Alcohol and Alcoholism 49, 261–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray LA, Hutchison KE, 2004. A polymorphism of the μ-opioid receptor gene (OPRM1) and sensitivity to the effects of alcohol in humans. Alcoholism: Clinical and Experimental Research 28, 1789–1795. [DOI] [PubMed] [Google Scholar]
- Ray LA, Miranda R Jr, Tidey JW, McGeary JE, MacKillop J, Gwaltney CJ, Rohsenow DJ, Swift RM, Monti PM, 2010. Polymorphisms of the μ-opioid receptor and dopamine D4 receptor genes and subjective responses to alcohol in the natural environment. Journal of abnormal psychology 119, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray LA, Sehl M, Bujarski S, Hutchison K, Blaine S, Enoch MA, 2013. The CRHR1 gene, trauma exposure, and alcoholism risk: a test of G x E effects. Genes Brain Behav 12, 361–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond JS, Wilson BB, Tan O, Gururajan A, Bowen MT, 2019. Acute alcohol exposure dose-dependently alleviates social avoidance in adolescent mice and inhibits social investigation in adult mice. Psychopharmacology. [DOI] [PubMed] [Google Scholar]
- Rhodes JS, Best K, Belknap JK, Finn DA, Crabbe JC, 2005. Evaluation of a simple model of ethanol drinking to intoxication in C57BL/6J mice. Physiol Behav 84, 53–63. [DOI] [PubMed] [Google Scholar]
- Rhodes JS, Ford MM, Yu CH, Brown LL, Finn DA, Garland T Jr., Crabbe JC, 2007. Mouse inbred strain differences in ethanol drinking to intoxication. Genes Brain Behav 6, 1–18. [DOI] [PubMed] [Google Scholar]
- Risinger FO, Bormann NM, Oakes RA, 1996. Reduced sensitivity to ethanol reward, but not ethanol aversion, in mice lacking 5-HT1B receptors. Alcohol Clin Exp Res 20, 1401–1405. [DOI] [PubMed] [Google Scholar]
- Risinger FO, Doan AM, Vickrey AC, 1999. Oral operant ethanol self-administration in 5-HT1b knockout mice. Behav Brain Res 102, 211–215. [DOI] [PubMed] [Google Scholar]
- Rivera-Meza M, Quintanilla ME, Tampier L, Mura CV, Sapag A, Israel Y, 2010. Mechanism of protection against alcoholism by an alcohol dehydrogenase polymorphism: development of an animal model. FASEB J 24, 266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivier C, Bruhn T, Vale W, 1984. Effect of ethanol on the hypothalamic-pituitary-adrenal axis in the rat: role of corticotropin-releasing factor (CRF). J Pharmacol Exp Ther 229, 127–131. [PubMed] [Google Scholar]
- Roberts AJ, McDonald JS, Heyser CJ, Kieffer BL, Matthes HW, Koob GF, Gold LH, 2000. mu-Opioid receptor knockout mice do not self-administer alcohol. J Pharmacol Exp Ther 293, 1002–1008. [PubMed] [Google Scholar]
- Roberts SP, Siegel MB, DeJong W, Naimi TS, Jernigan DH, 2015. Brand Preferences of Underage Drinkers Who Report Alcohol-Related Fights and Injuries. Substance Use & Misuse 50, 619–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodd-Henricks ZA, McKinzie DL, Murphy JM, McBride WJ, Lumeng L, Li TK, 2000. The expression of an alcohol deprivation effect in the high-alcohol-drinking replicate rat lines is dependent on repeated deprivations. Alcohol Clin Exp Res 24, 747–753. [PubMed] [Google Scholar]
- Romanus G, 2000. Alcopops in Sweden--a supply side initiative. Addiction 95 Suppl 4, S609–619. [DOI] [PubMed] [Google Scholar]
- Rose JH, Karkhanis AN, Chen R, Gioia D, Lopez MF, Becker HC, McCool BA, Jones SR, 2016. Supersensitive kappa opioid receptors promotes ethanol withdrawal-related behaviors and reduce dopamine signaling in the nucleus accumbens. International Journal of Neuropsychopharmacology 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenwasser AM, Fixaris MC, Crabbe JC, Brooks PC, Ascheid S, 2013. Escalation of intake under intermittent ethanol access in diverse mouse genotypes. Addiction biology 18, 496–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossheim ME, Thombs DL, 2013. Multiple fruit-flavored alcoholic drinks in a can (MFAC): an overlooked class of potentially harmful alcohol products. The American Journal of Drug and Alcohol Abuse 39, 280–283. [DOI] [PubMed] [Google Scholar]
- Ruby CL, Prosser RA, DePaul MA, Roberts RJ, Glass JD, 2009. Acute ethanol impairs photic and nonphotic circadian phase resetting in the Syrian hamster. Am J Physiol Regul Integr Comp Physiol 296, R411–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs BD, Salahi AA, Caron MG, 2014. Congenital brain serotonin deficiency leads to reduced ethanol sensitivity and increased ethanol consumption in mice. Neuropharmacology 77, 177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salimov RM, Salimova NB, 1993. L-glutamate abolishes differential responses to alcohol deprivation in mice. Alcohol 10, 251–257. [DOI] [PubMed] [Google Scholar]
- Salvatore JE, Han S, Farris SP, Mignogna KM, Miles MF, Agrawal A, 2019. Beyond genome-wide significance: integrative approaches to the interpretation and extension of GWAS findings for alcohol use disorder. Addict Biol 24, 275–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson HH, 1986. Initiation of Ethanol Reinforcement using a Sucrose-Substitution Procedure in Food- and Water-Sated Rats. 10, 436–442. [DOI] [PubMed] [Google Scholar]
- Sanchez-Roige S, Palmer AA, Clarke TK, 2020. Recent Efforts to Dissect the Genetic Basis of Alcohol Use and Abuse. Biol Psychiatry 87, 609–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchis-Segura C, Borchardt T, Vengeliene V, Zghoul T, Bachteler D, Gass P, Sprengel R, Spanagel R, 2006. Involvement of the AMPA receptor GluR-C subunit in alcohol-seeking behavior and relapse. The Journal of neuroscience : the official journal of the Society for Neuroscience 26, 1231–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander T, Harms H, Lesch KP, Dufeu P, Kuhn S, Hoehe M, Rommelspacher H, Schmidt LG, 1997. Association analysis of a regulatory variation of the serotonin transporter gene with severe alcohol dependence. Alcoholism: Clinical and Experimental Research 21, 1356–1359. [PubMed] [Google Scholar]
- Savelieva KV, Caudle WM, Findlay GS, Caron MG, Miller GW, 2002. Decreased ethanol preference and consumption in dopamine transporter female knock-out mice. Alcohol Clin Exp Res 26, 758–764. [PubMed] [Google Scholar]
- Schenk S, Gorman K, Amit Z, 1990. Age-Dependent Effects of Isolation Housing on the Self-Administration of Ethanol in Laboratory Rats. Alcohol 7, 321–326. [DOI] [PubMed] [Google Scholar]
- Scheyer AF, Christian DT, Wolf ME, Tseng KY, 2018. Emergence of Endocytosis-Dependent mGlu1 LTD at Nucleus Accumbens Synapses After Withdrawal From Cocaine Self-Administration. Front Synaptic Neurosci 10, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt HD, Sangrey GR, Darnell SB, Schassburger RL, Cha JH, Pierce RC, Sadri-Vakili G, 2012. Increased brain-derived neurotrophic factor (BDNF) expression in the ventral tegmental area during cocaine abstinence is associated with increased histone acetylation at BDNF exon I-containing promoters. J Neurochem 120, 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuckit MA, 1985. Studies of populations at high risk for alcoholism. Psychiatr Dev 3, 31–63. [PubMed] [Google Scholar]
- Schuckit MA, 2009. Alcohol-use disorders. Lancet 373, 492–501. [DOI] [PubMed] [Google Scholar]
- Schuckit MA, 2018. A Critical Review of Methods and Results in the Search for Genetic Contributors to Alcohol Sensitivity. Alcohol Clin Exp Res 42, 822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra S, Brunetti G, Vacca G, Lobina C, Carai MA, Gessa GL, Colombo G, 2003. Stable preference for high ethanol concentrations after ethanol deprivation in Sardinian alcohol-preferring (sP) rats. Alcohol 29, 101–108. [DOI] [PubMed] [Google Scholar]
- Sery O, Didden W, Mikes V, Pitelova R, Znojil V, Zvolsky P, 2006. The association between high-activity COMT allele and alcoholism. Neuro Endocrinol Lett 27, 231–235. [PubMed] [Google Scholar]
- Shin CB, Serchia MM, Shahin JR, Ruppert-Majer MA, Kippin TE, Szumlinski KK, 2016. Incubation of cocaine-craving relates to glutamate over-flow within ventromedial prefrontal cortex. Neuropharmacology 102, 103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin CB, Templeton TJ, Chiu AS, Kim J, Gable ES, Vieira PA, Kippin TE, Szumlinski KK, 2018. Endogenous glutamate within the prelimbic and infralimbic cortices regulates the incubation of cocaine-seeking in rats. Neuropharmacology 128, 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shurman J, Koob GF, Gutstein HB, 2010. Opioids, Pain, the Brain, and Hyperkatifeia: A Framework for the Rational Use of Opioids for Pain. Pain Medicine 11, 1092–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberman Y, Matthews RT, Winder DG, 2013. A corticotropin releasing factor pathway for ethanol regulation of the ventral tegmental area in the bed nucleus of the stria terminalis. The Journal of neuroscience : the official journal of the Society for Neuroscience 33, 950–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simms JA, Bito-Onon JJ, Chatterjee S, Bartlett SE, 2010. Long-Evans Rats Acquire Operant Self-Administration of 20% Ethanol Without Sucrose Fading. Neuropsychopharmacology 35, 1453–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simms JA, Steensland P, Medina B, Abernathy KE, Chandler LJ, Wise R, Bartlett SE, 2008. Intermittent access to 20% ethanol induces high ethanol consumption in Long-Evans and Wistar rats. Alcohol Clin Exp Res 32, 1816–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons RM, Hahn AM, Simons JS, Murase H, 2017. Emotion dysregulation and peer drinking norms uniquely predict alcohol-related problems via motives. Drug Alcohol Depend 177, 54–58. [DOI] [PubMed] [Google Scholar]
- Simpson S, Shankar K, Kimbrough A, George O, 2020. Role of corticotropin-releasing factor in alcohol and nicotine addiction. Brain Res 1740, 146850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair JD, 1971. The alcohol-deprivation effect in monkeys. Psychonomic Science 25, 21–22. [Google Scholar]
- Sinclair JD, 1979. Alcohol-deprivation effect in rats genetically selected for their ethanol preference. Pharmacol Biochem Behav 10, 597–602. [DOI] [PubMed] [Google Scholar]
- Sinclair JD, Tiihonen K, 1988. Lack of alcohol-deprivation effect in AA rats. Alcohol 5, 85–87. [DOI] [PubMed] [Google Scholar]
- Sinha R, Li CS, 2007. Imaging stress- and cue-induced drug and alcohol craving: association with relapse and clinical implications. Drug Alcohol Rev 26, 25–31. [DOI] [PubMed] [Google Scholar]
- Sittig LJ, Carbonetto P, Engel KA, Krauss KS, Barrios-Camacho CM, Palmer AA, 2016. Genetic Background Limits Generalizability of Genotype-Phenotype Relationships. Neuron 91, 1253–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith NDL, Cottler LB, 2018. The Epidemiology of Post-Traumatic Stress Disorder and Alcohol Use Disorder. Alcohol Res 39, 113–120. [PMC free article] [PubMed] [Google Scholar]
- Sora I, Elmer G, Funada M, Pieper J, Li XF, Hall FS, Uhl GR, 2001. Mu opiate receptor gene dose effects on different morphine actions: evidence for differential in vivo mu receptor reserve. Neuropsychopharmacology 25, 41–54. [DOI] [PubMed] [Google Scholar]
- Spanagel R, Bartsch D, Brors B, Dahmen N, Deussing J, Eils R, Ende G, Gallinat J, Gebicke-Haerter P, Heinz A, Kiefer F, Jager W, Mann K, Matthaus F, Nothen M, Rietschel M, Sartorius A, Schutz G, Sommer WH, Sprengel R, Walter H, Wichmann E, Wienker T, Wurst W, Zimmer A, 2010. An integrated genome research network for studying the genetics of alcohol addiction. Addict Biol 15, 369–379. [DOI] [PubMed] [Google Scholar]
- Spanagel R, Holter SM, Allingham K, Landgraf R, Zieglgansberger W, 1996. Acamprosate and alcohol: I. Effects on alcohol intake following alcohol deprivation in the rat. Eur J Pharmacol 305, 39–44. [DOI] [PubMed] [Google Scholar]
- Spanagel R, Pendyala G, Abarca C, Zghoul T, Sanchis-Segura C, Magnone MC, Lascorz J, Depner M, Holzberg D, Soyka M, Schreiber S, Matsuda F, Lathrop M, Schumann G, Albrecht U, 2005. The clock gene Per2 influences the glutamatergic system and modulates alcohol consumption. Nat Med 11, 35–42. [DOI] [PubMed] [Google Scholar]
- Sparta DR, Hopf FW, Gibb SL, Cho SL, Stuber GD, Messing RO, Ron D, Bonci A, 2013. Binge ethanol-drinking potentiates corticotropin releasing factor R1 receptor activity in the ventral tegmental area. Alcohol Clin Exp Res 37, 1680–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear LP, 2015. Adolescent alcohol exposure: Are there separable vulnerable periods within adolescence? Physiology & Behavior 148, 122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear LP, 2020. Timing eclipses amount: The critical importance of intermittency in alcohol exposure effects. Alcoholism: Clinical and Experimental Research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spoelder M, Hesseling P, Baars AM, Lozeman-van ‘t Klooster JG, Rotte MD, Vanderschuren LJ, Lesscher HM, 2015. Individual Variation in Alcohol Intake Predicts Reinforcement, Motivation, and Compulsive Alcohol Use in Rats. Alcohol Clin Exp Res 39, 2427–2437. [DOI] [PubMed] [Google Scholar]
- Stefanik MT, Milovanovic M, Werner CT, Spainhour JCG, Wolf ME, 2018. Withdrawal From Cocaine Self-administration Alters the Regulation of Protein Translation in the Nucleus Accumbens. Biol Psychiatry 84, 223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens DN, Pistovcakova J, Worthing L, Atack JR, Dawson GR, 2005. Role of GABA A α5-containing receptors in ethanol reward: the effects of targeted gene deletion, and a selective inverse agonist. European journal of pharmacology 526, 240–250. [DOI] [PubMed] [Google Scholar]
- Sternberg A, Pandika D, Elam KK, Chassin L, 2018. The relation of parent alcohol disorder to young adult drinking outcomes mediated by parenting: Effects of developmentally limited versus persistent parent alcohol disorder. Drug Alcohol Depend 188, 224–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson JR, Wenner SM, Freestone DM, Romaine CC, Parian MC, Christian SM, Bohidar AE, Ndem JR, Vogel IR, O’Kane CM, 2017. Oxytocin reduces alcohol consumption in prairie voles. Physiology & Behavior 179, 411–421. [DOI] [PubMed] [Google Scholar]
- Stohs ME, Schneekloth TD, Geske JR, Biernacka JM, Karpyak VM, 2019. Alcohol Craving Predicts Relapse After Residential Addiction Treatment. Alcohol Alcohol 54, 167–172. [DOI] [PubMed] [Google Scholar]
- Suh J, Ressler KJ, 2018. Common Biological Mechanisms of Alcohol Use Disorder and Post-Traumatic Stress Disorder. Alcohol Res 39, 131–145. [PMC free article] [PubMed] [Google Scholar]
- Susser M, 1985. Separating heredity and environment. Am J Prev Med 1, 5–23. [PubMed] [Google Scholar]
- Szumlinski KK, Shin CB, 2018. Kinase interest you in treating incubated cocaine-craving? A hypothetical model for treatment intervention during protracted withdrawal from cocaine. Genes Brain Behav 17, e12440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi JS, Hong HK, Ko CH, McDearmon EL, 2008. The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat Rev Genet 9, 764–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawa EA, Hall SD, Lohoff FW, 2016. Overview of the Genetics of Alcohol Use Disorder. Alcohol Alcohol 51, 507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele TE, Crabbe JC, Boehm SL 2nd, 2014. “Drinking in the Dark” (DID): a simple mouse model of binge-like alcohol intake. Curr Protoc Neurosci 68, 9 49 41–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele TE, Navarro M, 2014. “Drinking in the dark” (DID) procedures: A model of binge-like ethanol drinking in non-dependent mice. Alcohol 48, 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiihonen J, Hallikainen T, Lachman H, Saito T, Volavka J, Kauhanen J, Salonen JT, Ryynanen OP, Koulu M, Karvonen MK, Pohjalainen T, Syvalahti E, Hietala J, 1999. Association between the functional variant of the catechol-O-methyltransferase (COMT) gene and type 1 alcoholism. Mol Psychiatry 4, 286–289. [DOI] [PubMed] [Google Scholar]
- Timko C, Halvorson M, Kong C, Moos RH, 2015. Social processes explaining the benefits of Al-Anon participation. Psychol Addict Behav 29, 856–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolliver GA, Sadeghi KG, Samson HH, 1988. Ethanol preference following the sucrose-fading initiation procedure. 5, 9–13. [DOI] [PubMed] [Google Scholar]
- Tomie A, Azogu I, Yu L, 2013. Effects of naltrexone on post-abstinence alcohol drinking in C57BL/6NCRL and DBA/2J mice. Prog Neuropsychopharmacol Biol Psychiatry 44, 240–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomie A, Miller WC, Dranoff E, Pohorecky LA, 2006. Intermittent presentations of ethanol sipper tube induce ethanol drinking in rats. Alcohol Alcohol 41, 225–230. [DOI] [PubMed] [Google Scholar]
- Toth I, Neumann ID, Slattery DA, 2012. Social fear conditioning: a novel and specific animal model to study social anxiety disorder. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 37, 1433–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Town T, Abdullah L, Crawford F, Schinka J, Ordorica PI, Francis E, Hughes P, Duara R, Mullan M, 1999. Association of a functional μ-opioid receptor allele (+ 118A) with alcohol dependency. American journal of medical genetics 88, 458–461. [PubMed] [Google Scholar]
- Treutlein J, Kissling C, Frank J, Wiemann S, Dong L, Depner M, Saam C, Lascorz J, Soyka M, Preuss UW, Rujescu D, Skowronek MH, Rietschel M, Spanagel R, Heinz A, Laucht M, Mann K, Schumann G, 2006. Genetic association of the human corticotropin releasing hormone receptor 1 (CRHR1) with binge drinking and alcohol intake patterns in two independent samples. Mol Psychiatry 11, 594–602. [DOI] [PubMed] [Google Scholar]
- Trezza V, Baarendse PJJ, Vanderschuren LJMJ, 2014. On the interaction between drugs of abuse and adolescent social behavior. Psychopharmacology 231, 1715–1729. [DOI] [PubMed] [Google Scholar]
- Trudell JR, Messing RO, Mayfield J, Harris RA, 2014. Alcohol dependence: molecular and behavioral evidence. Trends in pharmacological sciences 35, 317–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunstall BJ, Kirson D, Zallar LJ, McConnell SA, Vendruscolo JCM, Ho CP, Oleata CS, Khom S, Manning M, Lee MR, Leggio L, Koob GF, Roberto M, Vendruscolo LF, 2019. Oxytocin blocks enhanced motivation for alcohol in alcohol dependence and blocks alcohol effects on GABAergic transmission in the central amygdala. PLOS Biology 17, e2006421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhart M, Weerts EM, McCaul ME, Guo X, Yan X, Kranzler HR, Li N, Wand GS, 2013. GABRA2 markers moderate the subjective effects of alcohol. Addict Biol 18, 357–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underwood MD, Kassir SA, Bakalian MJ, Galfalvy H, Dwork AJ, Mann JJ, Arango V, 2018. Serotonin receptors and suicide, major depression, alcohol use disorder and reported early life adversity. Transl Psychiatry 8, 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varlinskaya EI, Spear LP, 2004. Changes in sensitivity to ethanol-induced social facilitation and social inhibition from early to late adolescence. Ann N Y Acad Sci 1021, 459–461. [DOI] [PubMed] [Google Scholar]
- Varlinskaya EI, Spear LP, 2006. Differences in the social consequences of ethanol emerge during the course of adolescence in rats: social facilitation, social inhibition, and anxiolysis. Dev Psychobiol 48, 146–161. [DOI] [PubMed] [Google Scholar]
- Varlinskaya EI, Truxell EM, Spear LP, 2015a. Ethanol intake under social circumstances or alone in sprague-dawley rats: impact of age, sex, social activity, and social anxiety-like behavior. Alcohol Clin Exp Res 39, 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varlinskaya EI, Truxell EM, Spear LP, 2015b. Sex differences in sensitivity to the social consequences of acute ethanol and social drinking during adolescence. Behav Brain Res 282, 6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendruscolo LF, Roberts AJ, 2014. Operant alcohol self-administration in dependent rats: Focus on the vapor model. 48, 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vengeliene V, Bachteler D, Danysz W, Spanagel R, 2005. The role of the NMDA receptor in alcohol relapse: a pharmacological mapping study using the alcohol deprivation effect. Neuropharmacology 48, 822–829. [DOI] [PubMed] [Google Scholar]
- Vengeliene V, Bilbao A, Spanagel R, 2014. The alcohol deprivation effect model for studying relapse behavior: A comparison between rats and mice. Alcohol 48, 313–320. [DOI] [PubMed] [Google Scholar]
- Vengeliene V, Cannella N, Takahashi T, Spanagel R, 2016. Metabolic shift of the kynurenine pathway impairs alcohol and cocaine seeking and relapse. Psychopharmacology (Berl) 233, 3449–3459. [DOI] [PubMed] [Google Scholar]
- Vengeliene V, Heidbreder CA, Spanagel R, 2007. The effects of lamotrigine on alcohol seeking and relapse. Neuropharmacology 53, 951–957. [DOI] [PubMed] [Google Scholar]
- Vengeliene V, Leonardi-Essmann F, Perreau-Lenz S, Gebicke-Haerter P, Drescher K, Gross G, Spanagel R, 2006. The dopamine D3 receptor plays an essential role in alcohol-seeking and relapse. FASEB J 20, 2223–2233. [DOI] [PubMed] [Google Scholar]
- Vengeliene V, Leonardi-Essmann F, Sommer WH, Marston HM, Spanagel R, 2010. Glycine transporter-1 blockade leads to persistently reduced relapse-like alcohol drinking in rats. Biol Psychiatry 68, 704–711. [DOI] [PubMed] [Google Scholar]
- Vengeliene V, Siegmund S, Singer MV, Sinclair JD, Li TK, Spanagel R, 2003. A comparative study on alcohol-preferring rat lines: effects of deprivation and stress phases on voluntary alcohol intake. Alcohol Clin Exp Res 27, 1048–1054. [DOI] [PubMed] [Google Scholar]
- Verhulst B, Neale MC, Kendler KS, 2015. The heritability of alcohol use disorders: a meta-analysis of twin and adoption studies. Psychol Med 45, 1061–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba K, Attonito J, Mendy A, Devieux JG, Gasana J, Dorak TM, 2015. A meta-analysis of the associations between the SLC6A4 promoter polymorphism (5HTTLPR) and the risk for alcohol dependence. Psychiatr Genet 25, 47–58. [DOI] [PubMed] [Google Scholar]
- Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA, Yang J, 2017. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am J Hum Genet 101, 5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrettou M, Nilsson KW, Tuvblad C, Rehn M, Aslund C, Andershed AK, Wallen-Mackenzie A, Andershed H, Hodgins S, Nylander I, Comasco E, 2019. VGLUT2 rs2290045 genotype moderates environmental sensitivity to alcohol-related problems in three samples of youths. Eur Child Adolesc Psychiatry 28, 1329–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walcott AT, Ryabinin AE, 2020. Assessing effects of oxytocin on alcohol consumption in socially housed prairie voles using radio frequency tracking. Addiction Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walden B, Iacono WG, McGue M, 2007. Trajectories of change in adolescent substance use and symptomatology: impact of paternal and maternal substance use disorders. Psychol Addict Behav 21, 35–43. [DOI] [PubMed] [Google Scholar]
- Weera MM, Schreiber AL, Avegno EM, Gilpin NW, 2020. The role of central amygdala corticotropin-releasing factor in predator odor stress-induced avoidance behavior and escalated alcohol drinking in rats. Neuropharmacology 166, 107979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weighill D, Tschaplinski TJ, Tuskan GA, Jacobson D, 2019. Data Integration in Poplar: ‘Omics Layers and Integration Strategies. Front Genet 10, 874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzman ER, Chen Y-Y, 2005. Risk modifying effect of social capital on measures of heavy alcohol consumption, alcohol abuse, harms, and secondhand effects: national survey findings. Journal of Epidemiology and Community Health 59, 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner CT, Murray CH, Reimers JM, Chauhan NM, Woo KK, Molla HM, Loweth JA, Wolf ME, 2017. Trafficking of calcium-permeable and calcium-impermeable AMPA receptors in nucleus accumbens medium spiny neurons co-cultured with prefrontal cortex neurons. Neuropharmacology 116, 224–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner DF, Blednov YA, Ariwodola OJ, Silberman Y, Logan E, Berry RB, Borghese CM, Matthews DB, Weiner JL, Harrison NL, 2006. Knockin mice with ethanol-insensitive α1-containing γ-aminobutyric acid type A receptors display selective alterations in behavioral responses to ethanol. Journal of Pharmacology and Experimental Therapeutics 319, 219–227. [DOI] [PubMed] [Google Scholar]
- Whitfield JB, 1997. Meta-analysis of the effects of alcohol dehydrogenase genotype on alcohol dependence and alcoholic liver disease. Alcohol Alcohol 32, 613–619. [DOI] [PubMed] [Google Scholar]
- Whitfield JB, 2002. Alcohol dehydrogenase and alcohol dependence: variation in genotype-associated risk between populations. Am J Hum Genet 71, 1247–1250; author reply 1250–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills TA, Knapp DJ, Overstreet DH, Breese GR, 2009. Sensitization, duration, and pharmacological blockade of anxiety-like behavior following repeated ethanol withdrawal in adolescent and adult rats. Alcoholism: Clinical and Experimental Research 33, 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise RA, 1973. Voluntary ethanol intake in rats following exposure to ethanol on various schedules. Psychopharmacologia 29, 203–210. [DOI] [PubMed] [Google Scholar]
- Wolf ME, Tseng KY, 2012. Calcium-permeable AMPA receptors in the VTA and nucleus accumbens after cocaine exposure: when, how, and why? Front Mol Neurosci 5, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolffgramm J, 1990. Free choice ethanol intake of laboratory rats under different social conditions. Psychopharmacology (Berl) 101, 233–239. [DOI] [PubMed] [Google Scholar]
- Xia Y, Ma D, Hu J, Tang C, Wu Z, Liu L, Xin F, 2012. Effect of metabotropic glutamate receptor 3 genotype on N-acetylaspartate levels and neurocognition in non-smoking, active alcoholics. Behav Brain Funct 8, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JW, Winstanley CA, Brady AM, Hall FS, 2017. Research Domain Criteria versus DSM V: How does this debate affect attempts to model corticostriatal dysfunction in animals? Neurosci Biobehav Rev 76, 301–316. [DOI] [PubMed] [Google Scholar]
- Zghoul T, Abarca C, Sanchis-Segura C, Albrecht U, Schumann G, Spanagel R, 2007. Ethanol self-administration and reinstatement of ethanol-seeking behavior in Per1(Brdm1) mutant mice. Psychopharmacology (Berl) 190, 13–19. [DOI] [PubMed] [Google Scholar]
- Zhang X, Gainetdinov RR, Beaulieu JM, Sotnikova TD, Burch LH, Williams RB, Schwartz DA, Krishnan KR, Caron MG, 2005. Loss-of-function mutation in tryptophan hydroxylase-2 identified in unipolar major depression. Neuron 45, 11–16. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Karlsson C, Liang T, Xiong W, Kimura M, Tapocik JD, Yuan Q, Barbier E, Feng A, Flanigan M, Augier E, Enoch MA, Hodgkinson CA, Shen PH, Lovinger DM, Edenberg HJ, Heilig M, Goldman D, 2013. Loss of metabotropic glutamate receptor 2 escalates alcohol consumption. Proc Natl Acad Sci U S A 110, 16963–16968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Yuan Q, Mash DC, Goldman D, 2011. Substance-specific and shared transcription and epigenetic changes in the human hippocampus chronically exposed to cocaine and alcohol. Proceedings of the National Academy of Sciences 108, 6626–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Xie Q, Zhang M, Zhang C, Zhao G, Jin M, Yu L, 2009. Chronic alcohol consumption from adolescence-to-adulthood in mice--effect on growth and social behavior. Drug and alcohol dependence 104, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]