

Abstract
Reaction conditions have been developed for refractory heteroaryl–heteroaryl Suzuki–Miyaura cross-couplings. The reported method employs neopentyl heteroarylboronic esters as nucleophiles, heteroaryl bromides and chlorides as the electrophiles, and the soluble base potassium trimethylsilanolate (TMSOK) under anhydrous conditions. The addition of trimethyl borate enhances reaction rates by several mechanisms, including (1) solubilization of in situ-generated boronate complexes, (2) preventing catalyst poisoning by the heteroatomic units, and (3) buffering the inhibitory effect of excess TMSOK. The use of this method enables cross-coupling of diverse reaction partners including a broad range of π-rich and π-deficient heteroaryl boronic esters and heteroaryl bromides. Reactions proceed in good yields and short reaction times (3 h or less).
Graphical Abstract
INTRODUCTION
Heterocyclic compounds constitute important structural components of small molecules in the agrochemical and pharmaceutical industries.1 In a recent survey of all small-molecule drugs approved by the FDA between 2015 and June 2020, 88% were found to contain one or more N-heterocyclic subunits.2 Among the molecules surveyed, pyridines, piperidines, piperazines, pyrimidines, pyrazoles, and indoles are the five most represented N-heterocycles, respectively. The prevalence of nitrogen-bearing heterocycles in drug design is a consequence of the ability of these functional groups to modulate polarity, solubility, lipophilicity, and hydrogen-bonding properties of small molecules, and in turn their corresponding ADMET (absorption, distribution, metabolism, excretion, toxicity) profiles.3 Therefore, the continued development of synthetic methods to access these structures remains of considerable interest.
One class of reactions that represents an attractive approach to the incorporation of heterocycles is the Suzuki–Miyaura cross-coupling reaction owing to its predictability, functional group compatibility, late-stage amenability, and low toxicity profile for boron-derived nucleophiles.4,5 These attributes have rendered the Suzuki–Miyaura cross-coupling a powerful tool to synthetic chemists for the mild construction of carbon–carbon bonds. To this point, a 2014 study found that among 125 papers published in the J. Med. Chem., the Suzuki–Miyaura cross-coupling was the single most employed C-C bond-forming reaction.6 Moreover, advances in ligand and catalyst design have provided considerable progress to enable room-temperature cross-couplings,7 the cross-coupling of challenging electrophiles,8 as well as the ability to effect the cross-coupling of heteroaryl reaction partners.7c,9 In particular, ligands and precatalysts designed by Buchwald and co-workers have enabled access to certain heteroaryl–heteroaryl Suzuki–Miyaura cross-couplings by virtue of the activated nature of these catalyst systems (Scheme 1).9i
Scheme 1.

Heteroaryl-Heteroaryl Cross-Couplings by Buchwald and Co-workers
Despite these important advances, challenges in heteroaryl Suzuki–Miyaura cross-couplings persist. Highly polar heterocyclic compounds exhibit poor solubility in many organic solvents10 and Lewis-basic atoms in heterocyclic structures have been shown to promote catalyst deactivation under Suzuki–Miyaura conditions.9b,i,11,12 Moreover, the propensity for heteroaryl boronic acids to undergo competitive in situ protodeboronation has been well described by Lloyd-Jones and co-workers.13,14 Numerous strategies have been developed to attenuate the protodeboronation of heteroaryl boronic acids. For example, one common approach employs super-stoichiometric amounts of the labile organoboron reaction partner.15 However, this strategy is not atom-economical. More practically, masking strategies to protect the labile parent boronic acid have been developed, notably with MIDA boronates9e,f,16 and trifluoroborate salts,15,17 enabling the slow-release of the reactive boronic acid in situ. Micellar approaches have also been shown to promote heteroaryl–heteroaryl couplings in aqueous media using mild conditions.9j,k The use of copper additives has also enabled the cross-coupling of challenging heteroaromatic boron nucleophiles as a secondary transmetalation partner.9f,18,19 Additionally, Lloyd-Jones and co-workers have also demonstrated the ability of copper (and other Lewis acids) to attenuate protodeboronation pathways by binding to Lewis-basic heterocycles.13 Alternatively, the use of strictly anhydrous reaction conditions has been reported to dissuade protodeboronation pathways.20,21 However, the use of anhydrous reaction media can introduce new challenges associated with poor solubility and attendant problems associated with mass-transfer phenomena and limitations to high-throughput applications.
BACKGROUND
A 2018 report from this laboratory unambiguously demonstrated the ability of boronic esters to undergo transmetalation without prior hydrolysis to the parent boronic acid using stoichiometric amounts of arylpalladium hydroxide complexes.22 Moreover, the identity of the boronic ester significantly influences the rate of transmetalation such that many esters react faster than the parent boronic acid. Our group sought to apply this mechanistic insight into a preparative, catalytic application. This goal was realized with the disclosure of a novel method for the cross-coupling of neopentyl arylboronic esters under anhydrous, homogeneous reaction conditions using the soluble base potassium trimethylsilanolate (TMSOK) (Scheme 2).23
Scheme 2.

TMSOK-Promoted Coupling of Neopentyl Boronic Esters
The anhydrous nature of this cross-coupling method precluded hydrolysis of the boronic ester coupling partner. Accordingly, the increases in transmetalation rate observed under stoichiometric conditions were successfully translated to other catalytic systems. When this method was compared with three existing Suzuki–Miyaura cross-coupling examples from the literature,24 a >10-fold decrease in reaction time was observed in all cases (Scheme 3). Notably, the only change in conditions from the original reports are the use of a boronic ester in place of the boronic acid, and the use of potassium trimethylsilanolate as the base under anhydrous conditions. Thus, boronic ester identity was established as a new parameter for optimization.
Scheme 3.

Rate Enhancements of Known Reactions Using TMSOK
Despite the generality of the previously described method, only a select number of heteroaromatic nucleophiles and electrophiles reacted successfully under the optimized conditions. These examples include thienyl, furanyl, 6-substituted pyridyl, and Boc-protected pyrrolyl nucleophiles, as well as benzoyl-protected indolyl and pyridyl electrophiles. In view of the challenges associated with catalyst deactivation, it was assumed that more Lewis-basic heterocycles led to deleterious coordination of the palladium catalyst. This hypothesis was corroborated by the addition of 0.5 equiv of pyridine or 1-methylimidazole to a previously optimized cross-coupling leading to a loss in reactivity (Scheme 4). Longer reaction times or increases in temperature did not restore reactivity.
Scheme 4. Catalyst Deactivation by Addition of Pyridine or 1-Methylimidazole.

aYields determined by 19F NMR spectroscopy using 1.0 equiv of 1,2-difluorobenzene as an internal standard.
RESEARCH PLAN
In 2007, Hartwig and co-workers demonstrated the beneficial effect of triethyl- and triphenylboranes as additives for palladium-catalyzed aminations of azines.25 This study demonstrated that the ability of boron additives to coordinate to Lewis-basic azine substrates both electronically activated the aryl ring for reductive elimination and mitigated competitive catalyst deactivation. We speculated whether other Lewis-acidic additives could afford similar benefits under the anhydrous Suzuki–Miyaura coupling conditions. Specifically, could other soluble Lewis acids enhance reactivity by: (1) promoting solubilization of challenging heterocyclic substrates, particularly under anhydrous conditions, (2) attenuating catalyst deactivation processes by coordination of the Lewis acid to Lewis-basic heteroatoms, and (3) buffering the inhibitory effect of TMSOK observed in the previous communication.23 Additionally, employing anhydrous reaction conditions in concert with a soluble Lewis acid was envisaged to attenuate competitive protodeboronation. With these goals in mind, a program was formulated to explore the effect of soluble Lewis acid additives with the original reaction conditions to allow the cross-coupling of refractory reaction partners.
RESULTS
Preliminary Investigations of Trimethyl Borate.
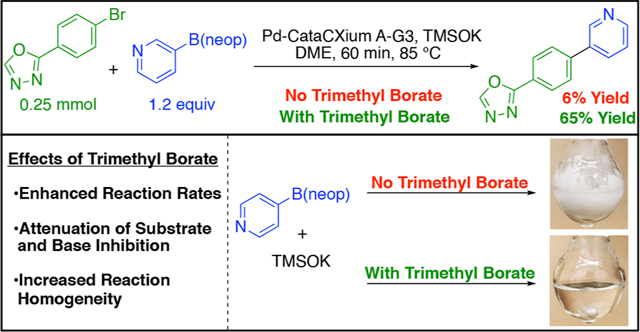
Initial studies explored the effect of the Lewis acid trimethyl borate as a reaction additive owing to its chemically benign character compared with the pyrophoricity and radical reactivity associated with alkylboranes.26 The initial investigation began by evaluating the effect of trimethyl borate on the Suzuki–Miyaura cross-coupling of 2-(4-bromophenyl)-1,3,4-oxadiazole 1b. This heteroaromatic electrophile was previously found to be unreactive under our optimized reaction conditions when paired with neopentyl 4-fluorophenylboronic ester 2a. Heating this reaction mixture at reflux for 6 h yielded only a trace of cross-coupled product by 1H NMR analysis (Scheme 5A). However, addition of 3.0 equiv of trimethyl borate afforded a 21% yield of cross-coupled product under otherwise identical conditions (Scheme 5B).
Scheme 5. Initial Investigation of Trimethyl Borate Under Original Reaction Conditions.

aYields determined by 1H NMR spectroscopy using 1.0 equiv of 1,3,5-trimethoxybenzene as an internal standard.
The initial reactivity change observed in Scheme 5 inspired an evaluation of the effect of trimethyl borate on the previously unreactive heteroaromatic nucleophile neopentyl 5-pyrimidyl-boronic ester 2b (Table 1). To our delight, quantitative coupling was observed in the presence of trimethyl borate. Coupling of boronic ester 2b in 1,4-dioxane with 1-bromo-4-fluorobenzene 1c using a 1,1'-bis(diphenylphosphino)-ferrocene Buchwald precatalyst exhibited “on/off” reactivity in the presence/absence of trimethyl borate (Table 1). Notably, no cross-coupling was observed in the absence of trimethyl borate after 1 h. Trimethyl borate loadings ranging from 0.6 to 2.4 equiv were observed to be equally effective in promoting the desired cross-coupling. Additionally, a control experiment was carried using 0.6 equiv of tris(trimethylsilyl) borate in place of trimethyl borate to preclude the in situ generation of potassium methoxide as the operative base (Scheme 6). Under otherwise identical reaction conditions to those shown in Table 1, the reaction proceeded quantitatively as observed with 0.6 equiv of trimethyl borate.
Table 1.
Effect of Trimethyl Borate Loading on the Cross-Coupling of 2b
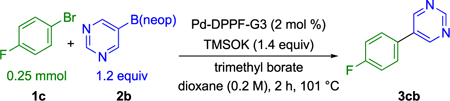
| ||
|---|---|---|
| entry | trimethyl borate (equiv) | yield (%)a |
| 1 | 0.0 | 0 |
| 2 | 0.6 | 100 |
| 3 | 1.2 | 100 |
| 4 | 2.4 | 100 |
Yields determined by 19F NMR spectroscopy using 1.0 equiv of 1-fluoronaphthalene as an internal standard.
Scheme 6. Control Experiment Using Tris(trimethylsilyl) Borate.

aYields determined by 19F NMR spectroscopy using 1.0 equiv of 1-fluoronaphthalene as an internal standard.
Optimization of Boronate Solubility.
The preliminary identification of operative reaction conditions led to evaluation of reaction homogeneity. Heteroaromatic substrates often present challenges with solubility (vide infra), particularly in anhydrous reaction media. It was observed experimentally that the primary limitation for solubility arose from the in situ generation of four-coordinate boronate complexes by the treatment of neopentyl heteroarylboronic esters with TMSOK. Among the initial neopentyl heteroarylboronic esters examined, neopentyl 4-pyridylboronic ester 2c was exceptionally challenging in this regard. Therefore, homogeneity optimization efforts were designed around boronic ester 2c.
The effect of different boronic esters on reaction homogeneity was first explored (Scheme 7A). 4-Pyridylboronic esters derived from neopentyl glycol 2c, tetrahydrofuran-3,4-diol 5, cis-cyclopentane-1,2-diol 6, and cis-cyclohexane-1,2-diol 7 were evaluated in the presence of 2.4 equiv of trimethyl borate in 1,4-dioxane at reflux. Each ester was treated with 1.4 equiv of TMSOK and the resulting solubility was established visually. Notably, only the neopentyl-derived boronic ester 2c and the cis-cyclopentane-1,2-diol derived ester 6, afforded homogeneous reaction mixtures. Therefore, in view of the availability of neopentyl boronic esters, 2c was chosen as the preferred candidate for subsequent optimization.
Scheme 7.

Optimization of Boronate Solubility
Next, four borate additives were tested under similar conditions using boronic ester 2c (Scheme 7B). Tris(trimethylsilyl) borate, triisopropyl borate, and tri-n-butyl borate all affoorded heterogeneous reaction profiles for the in situ-generated boronate complex. Only trimethyl borate afforded a homogeneous reaction mixture. Therefore, the additive trimethyl borate was selected for further optimization. Additionally, six reaction solvents were tested using the optimal boronic ester and borate additive (Scheme 7C). In accordance with previous reports, acetonitrile was found to react with TMSOK.27 Diglyme and NMP were also found to interact unfavorably with the soluble base. Although toluene and 1,4-dioxane were compatible with TMSOK, only 1,2-dimethyoxyethane was found to enable entirely homogeneous reaction conditions and was thus chosen as the solvent of choice.
Ligand Optimization.
Guided by the previous optimizations, eight, third-generation Buchwald precatalysts were then surveyed to identify the optimal ligand for reactivity in reaction between 1c and 2c (Table 2). The precatalyst bearing the CataCXium A ligand was found to be uniquely competent at promoting Suzuki–Miyaura cross-coupling under these reaction conditions. Further optimization of the continuous variables was carried out by a design of experiment (DoE) campaign (see Supporting Information) to maximize yield. The optimized reaction conditions employ 3 mol% of Pd-CataCXium A-G3 precatalyst, 3.0 equiv of trimethyl borate, 1.1 equiv of neopentyl heteroarylboronic ester, and 1.2 equiv of TMSOK.
Table 2.
Precatalyst Survey Using Buchwald Third-Generation Precatalysts
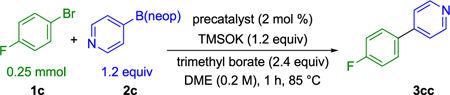
| ||
|---|---|---|
| entry | precatalyst | yield (%)a |
| 1 | Pd-P(Cy)3-G3 | 25 |
| 2 | Pd-P(t-Bu)3-G3 | 4 |
| 3 | Pd-CataCXium A-G3 | 100 |
| 4 | Pd-dppb-G3 | 17 |
| 5 | Pd-dppf-G3 | 34 |
| 6 | Pd-JosiPhos-G3 | 0 |
| 7 | Pd-SPhos-G3 | 10 |
| 8 | Pd-XPhos-G3 | 32 |
Yields determined by 19F NMR spectroscopy using 1.0 equiv of 1-fluoronaphthalene as an internal standard.
Evaluation of Reaction Scope.
The generality of the optimized reaction conditions was explored in more complex heteroaryl–heteroaryl Suzuki–Miyaura cross-coupling reactions using a variety of π-rich and π-deficient nucleophiles and electrophiles (Table 3). π-Rich neopentyl 2-furyl- and n-Bocpyrrolyl-2-boronic esters coupled with heterocyclic electrophiles in which the bromine atom was attached to one or more heteroatom-bearing carbons (81–89% yield). Both 1,3- (3fh, 3gh) and 1,4-diazines (3dg, 3dc) performed successfully as reaction electrophiles. Notably, highly protodeboronation-prone13,14 neopentyl 1-methylpyrazolyl- and 3,5-dimethylisox-azolylboronic esters were productively coupled under the anhydrous reaction conditions.28 Neopentyl 1-methylpyrazo-lylboronic esters also coupled with both quinoline (3ii) and isoquinoline (3ji) substrates. 6-Bromoquinoline electrophiles were highly reactive partners for both π-rich (3ii) and π-deficient nucleophiles (3nb), affording the desired products in 5–10 min. The electrophile for product 3kj, derived from nicotinic acid, highlights the compatibility of tert-butyl esters using this method. However, TMSOK was found to cleave methyl and ethyl esters, consistent with previous reports.29 Additionally, 2-aminopyridine electrophiles are suitable under the optimized reaction conditions (3lj), despite their known ability to deactivate transition metal catalysts.30
Table 3.
Overview of Reaction Scopea
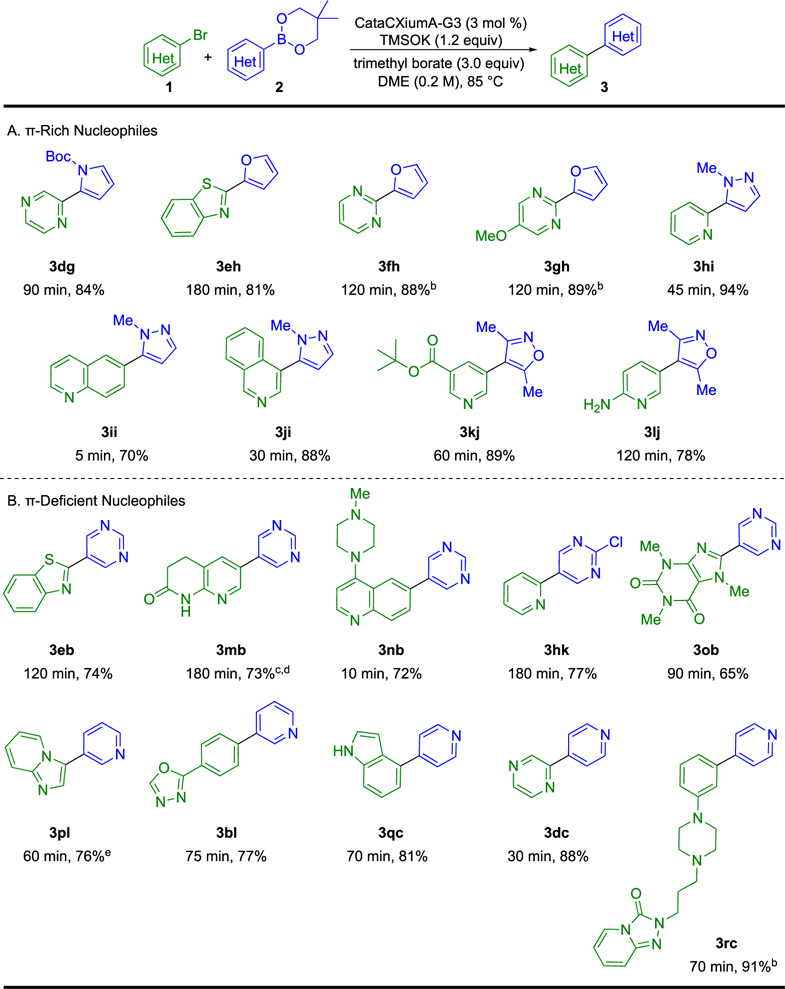
|
Reactions run with 1.00 mmol of aryl halide and 1.1 equiv of boronic ester. Yields of isolated product after purification.
Aryl chloride used in place of aryl bromide.
Product was further purifed to analytical purity, see the Supporting Information.
1,4-Dioxane used.
Pd-PPh3-G3 used.
π-Deficient neopentyl 5-pyrimidylboronic esters are well suited as nucleophiles affording coupling product in good isolated yields (70–77%). The quinazolinone electrophile corresponding to product 3mb was highly insoluble in DME but running the reaction in 1,4-dioxane afforded slightly better solubility and conversion. Neopentyl 2-chloro-5-pyrimidylboronic esters do not suffer from competitive side-reactivity (3hk). Nitrogen rich 8-bromocaffeiene was also successfully coupled under the optimized reaction conditions (3ob). 3- and 4-pyridylboronic esters reacted smoothly with a variety of electrophiles. However, the highly labile pinacol 2-pyridylboronic ester underwent apparent protodeboronation. Interestingly, the imidazopyridyl electrophile leading to product 3pl reacted more efficiently using Pd-PPh3-G3 as the precatalyst in place of Pd-CataCXiumA-G3. Oxadiazole (3bl) and unprotected indole (3qc) were also compatible with the reaction conditions. However, unprotected imidazoles were observed to confer no reactivity and methyl-protected imidazoles reacted in poor yields. Additionally, some electron-deficient pyrimidyl electrophiles (e.g., 2-chloro-5-nitropyrimidine) underwent competitive SNAr with TMSOK. A table of problematic substrate pairs is included in the Supporting Information.
DISCUSSION
Purity of TMSOK.
The purity of TMSOK was found to influence the course of the reaction. Some batches of TMSOK significantly lowered the rate of cross-coupling, resulting in reaction stalling.31 Quantitation of the potassium content of a problematic batch of TMSOK by ICP analysis showed a significantly lower value than theoretical (see Supporting Information). Moreover, samples from this batch were found to be largely insoluble in diethyl ether. These observations are consistent with an early report by Tatlock and Rochow suggesting the presence of potassium hydroxide as a contaminant.32 While the stock solutions of TMSOK used in this report were prepared under atmospheric conditions, it is recommended that bulk TMSOK be stored in a dry atmosphere and sourced in high purity.31
Role of Trimethyl Borate on Solubility.
Control experiments investigating the effect of trimethyl borate on boronic ester solubility were carried out using neopentyl 4-pyridyl- (2c) and 1-methylpyrazolylboronic esters (2i). The addition of 1.2 equiv of TMSOK to a boronic ester solution in DME, with and without trimethyl borate, produced significantly different solubility profiles (Scheme 8). For each boronic ester, the presence of 3.0 equiv of trimethyl borate led to homogeneous reaction mixtures. Notably, reaction homogeneity was maintained upon cooling to room temperature. Conversely, in the absence of trimethyl borate, highly heterogeneous reaction mixtures were observed with concomitant loss of stirring. The improvements in solubility are likely attributable to N-to-B coordination of the TMSOK· boronate complex with trimethyl borate as well as changes in the corresponding aggregation state.
Scheme 8.

Effect of Trimethyl Borate on Reaction Solubility
Role of Trimethyl Borate on Base Deactivation.
The heteroaryl–heteroaryl coupling of 2-(4-bromophenyl)-1,3,4-oxadiazole 1b with neopentyl 3-pyridylboronic ester 2l was chosen as a model system to investigate an additional benefit of trimethyl borate as a reaction additive. The original disclosure from this laboratory for the TMSOK-promoted, Suzuki–Miyaura cross-coupling of neopentyl arylboronic esters with aryl halides found a strong dependence on base loading.23 Employing super-stoichiometric amounts of TMSOK for slow cross-coupling reactions resulted in catalyst deactivation and incomplete conversion. A protocol was therefore developed that employed two additions of base over time. To investigate the effect of super-stoichiometric amounts of TMSOK in this system, 1.2 equiv of TMSOK was added in one addition at the outset of the reaction in the presence and absence of trimethyl borate (Scheme 9). Remarkably, only a trace of cross-coupled product was observed by 1H NMR analysis after 60 min in the absence of trimethyl borate, consistent with the deactivation observed previously. However, when 3.0 equiv of trimethyl borate was present, the reaction afforded a 65% yield under otherwise identical conditions. This buffering of the inhibitory effect of excess TMSOK was found to be a general phenomenon across all substrate pairs allowing for a single addition of TMSOK in all entries shown in Table 3.
Scheme 9. Effect of Trimethyl Borate on Base Inhibition.

aYields determined by 1HNMR spectroscopy using 1.0 equiv of 1,3,5-trimethoxybenzene as an internal standard.
Generality of the Effect of Trimethyl Borate.
To demonstrate the generality of the reaction additive, three additional substrate pairs from Table 3 were monitored by 1H NMR analysis in the presence and absence of trimethyl borate (Scheme 10). Each substrate combination was found to give a higher yield of cross-coupled product in the presence of trimethyl borate. Cross-coupling to give product 3lj was significantly inhibited in the absence of trimethyl borate. However, the effect of base inhibition was found to be less pronounced in the formation of cross-coupled product 3ji. Notably, the beneficial effect of trimethyl borate was preserved when the ligand PPh3 was used in place of CataCXium A for the formation of cross-coupled product 3pl.
Scheme 10. Generality of Trimethyl Borate on Base Inhibition.

aYields determined by 1H NMR spectroscopy using 1.0 equiv of 1,3,5-trimethoxybenzene as an internal standard.
Effect of Trimethyl Borate on Reaction Rates.
The model coupling reaction was also investigated using sub-stoichiometric amounts of TMSOK. By precluding the inhibitory effect of excess TMSOK, the intrinsic rate increase associated with the presence of trimethyl borate was assessed. When 0.9 equiv of TMSOK was used in the absence of trimethyl borate, an 13% yield was observed by 1H NMR analysis after 60 min (Scheme 11). However, when 3.0 equiv of trimethyl borate was employed, a 81% yield was observed by 1H NMR analysis after 60 min. The ~6 times increase in reaction rate is potentially attributable to N-to-B coordination with trimethyl borate, mitigating catalyst inhibition and enabling turnover. The increase in substrate solubility enabled by trimethyl borate is also likely operative in the observed rate increase. The slightly greater reactivity observed using sub-stoichiometric loadings of TMSOK with trimethyl borate, in comparison to super-stoichiometric loadings with trimethyl borate, highlights the competing nature of excess base under the optimized reaction conditions. While the influence of each of these three parameters (i.e., solubility, buffering of base, and attenuation of Lewis-basic functional groups) varies among different substrate combinations, the use of trimethyl borate as a reaction additive has been found to be broadly beneficial.
Scheme 11. Effect of Trimethyl Borate on Reaction Rates.

aYields determined by 1H NMR spectroscopy using 1.0 equiv of 1,3,5-trimethoxybenzene as an internal standard.
CONCLUSIONS AND OUTLOOK
In summary, a novel method for the anhydrous, Suzuki–Miyaura cross-coupling of heteroaryl–heteroaryl reaction partners has been described. A broad range of previously inaccessible π-rich and π-deficient heterocycles are demonstrated to be compatible under the new reaction conditions using TMSOK as the base. The novelty of this methods stems from the use of the innocuous reaction additive trimethyl borate. Trimethyl borate influences the course of the reaction by: (1) promoting the solubilization of in situ-generated boronate complexes, (2) buffering base-promoted catalyst inhibition, and (3) attenuating the Lewis-basicity of heteroatomic subunits, thereby mitigating catalyst poisoning. Employing anhydrous reaction conditions in concert with trimethyl borate enables the coupling of many labile heteroaromatic nucleophiles susceptible to protodeboronation. The application of this method is therefore anticipated to be of value for the coupling of challenging boron-containing heteroaromatic nucleophiles. Additionally, the soluble nature of both TMSOK and trimethyl borate are envisioned to be highly amenable to high-throughput experimentation applications.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the National Institutes of Health (Grant GM R35 127010) for generous financial support. We also thank the UIUC SCS support facilities (microanalysis, mass spectrometry, and NMR spectroscopy) for their assistance. Dr. Gerald Larson (Gelest) is thanked for a generous gift of TMSOK used for this study.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c06419.
Experimental procedures and characterization data for all new compounds along with copies of the NMR spectra (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.1c06419
Contributor Information
Vincent M. Kassel, Roger Adams Laboratory, Department of Chemistry, University of Illinois, Urbana, Illinois 61801, United States
Christopher M. Hanneman, Roger Adams Laboratory, Department of Chemistry, University of Illinois, Urbana, Illinois 61801, United States
Connor P. Delaney, Roger Adams Laboratory, Department of Chemistry, University of Illinois, Urbana, Illinois 61801, United States
Scott E. Denmark, Roger Adams Laboratory, Department of Chemistry, University of Illinois, Urbana, Illinois 61801, United States.
REFERENCES
- (1).(a) Lamberth C Heterocyclic Chemistry in Crop Protection. Pest Manage. Sci 2013, 69, 1106–1114. [DOI] [PubMed] [Google Scholar]; (b) Vitaku E; Smith DT; Njardarson JT Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]; (c) Heravi MM; Zadsirjan V Prescribed Drugs Containing Nitrogen Heterocycles: An Overview. RSC Adv 2020, 10, 44247–44311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Bhutani P; Joshi G; Raja N; Bachhav N; Rajanna PK; Bhutani H; Paul AT; Kumar RUS FDA Approved Drugs from 2015-June 2020: A Perspective. J. Med. Chem. 2021, 64, 2339–2381. [DOI] [PubMed] [Google Scholar]
- (3).(a) Dalvie D; Kang P; Loi C-M; Goulet L; Nair S Influence of Heteroaromatic Rings on ADME Properties of Drugs. Metabolism, Pharmacokinetics and Toxicity of Functional Groups: Impact of Chemical Building Blocks on ADMET; The Royal Society of Chemistry, 2010; Chap. 7, pp 328–369. [Google Scholar]; (b) Gomtsyan A Heterocycles in Drugs and Drug Discovery. Chem. Heterocycl. Compd. 2012, 48, 7–10. [Google Scholar]; (c) Jampilek J Heterocycles in Medicinal Chemistry. Molecules 2019, 24, 3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Lennox AJJ; Lloyd-Jones GC Selection of Boron Reagents for Suzuki–Miyaura Coupling. Chem. Soc. Rev. 2014, 43, 412–443. [DOI] [PubMed] [Google Scholar]
- (5).Blakemore D Suzuki–Miyaura Coupling. Synthetic Methods in Drug Discovery; The Royal Society of Chemistry, 2016; Vol. 1, Chap. 1, pp 1–69. [Google Scholar]
- (6).Brown DG; Boström J Analysis of Past and Present Synthetic Methodologies on Medicinal Chemistry: Where Have All the New Reactions Gone? J. Med. Chem. 2016, 59, 4443–4458. [DOI] [PubMed] [Google Scholar]
- (7).(a) Old DW; Wolfe JP; Buchwald SL A Highly Active Catalyst for Palladium-Catalyzed Cross-Coupling Reactions: Room-Temperature Suzuki Couplings and Amination of Unactivated Aryl Chlorides. J. Am. Chem. Soc. 1998, 120, 9722–9723. [Google Scholar]; (b) Littke AF; Dai C; Fu GC Versatile Catalysts for the Suzuki Cross-Coupling of Arylboronic Acids with Aryl and Vinyl Halides and Triflates under Mild Conditions. J. Am. Chem. Soc. 2000, 122, 4020–4028. [Google Scholar]; (c) Kinzel T; Zhang Y; Buchwald SL A New Palladium Precatalyst Allows for the Fast Suzuki–Miyaura Coupling Reactions of Unstable Polyfluorophenyl and 2-Heteroaryl Boronic Acids. J. Am. Chem. Soc. 2010, 132, 14073–14075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Zhang C; Huang J; Trudell ML; Nolan SP Palladium–Imidazol-2-Ylidene Complexes as Catalysts for Facile and Efficient Suzuki Cross-Coupling Reactions of Aryl Chlorides with Arylboronic Acids. J. Org. Chem. 1999, 64, 3804–3805. [Google Scholar]; (b) Tobisu M; Xu T; Shimasaki T; Chatani N Nickel-Catalyzed Suzuki–Miyaura Reaction of Aryl Fluorides. J. Am. Chem. Soc. 2011, 133, 19505–19511. [DOI] [PubMed] [Google Scholar]; (c) Yadav MR; Nagaoka M; Kashihara M; Zhong R-L; Miyazaki T; Sakaki S; Nakao Y The Suzuki–Miyaura Coupling of Nitroarenes. J. Am. Chem. Soc. 2017, 139, 9423–9426. [DOI] [PubMed] [Google Scholar]
- (9).(a) Kudo N; Perseghini M; Fu GC A Versatile Method for Suzuki Cross-Coupling Reactions of Nitrogen Heterocycles. Angew. Chem., Int. Ed. 2006, 45, 1282–1284. [DOI] [PubMed] [Google Scholar]; (b) Billingsley KL; Anderson KW; Buchwald SL A Highly Active Catalyst for Suzuki–Miyaura Cross-Coupling Reactions of Heteroaryl Compounds. Angew. Chem., Int. Ed. 2006, 45, 3484–3488. [DOI] [PubMed] [Google Scholar]; (c) Billingsley K; Buchwald SL Highly Efficient Monophosphine-Based Catalyst for the Palladium-Catalyzed Suzuki–Miyaura Reaction of Heteroaryl Halides and Heteroaryl Boronic Acids and Esters. J. Am. Chem. Soc. 2007, 129, 3358–3366. [DOI] [PubMed] [Google Scholar]; (d) Billingsley KL; Buchwald SL A General and Efficient Method for the Suzuki–Miyaura Coupling of 2-Pyridyl Nucleophiles. Angew. Chem., Int. Ed. 2008, 47, 4695–4698. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Knapp DM; Gillis EP; Burke MD A General Solution for Unstable Boronic Acids: Slow-Release Cross-Coupling from Air-Stable MIDA Boronates. J. Am. Chem. Soc. 2009, 131, 6961–6963. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Dick GR; Woerly EM; Burke MD A General Solution for the 2-Pyridyl Problem. Angew. Chem., Int. Ed. 2012, 51, 2667–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Ge S; Hartwig JF Highly Reactive, Single-Component Nickel Catalyst Precursor for Suzuki–Miyuara Cross-Coupling of Heteroaryl Boronic Acids with Heteroaryl Halides. Angew. Chem., Int. Ed. 2012, 51, 12837–12841. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Oberli MA; Buchwald SL A General Method for Suzuki-Miyaura Coupling Reactions Using Lithium Triisopropyl Borates. Org. Lett. 2012, 14, 4606–4609. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Düfert MA; Billingsley KL; Buchwald SL Suzuki–Miyaura Cross-Coupling of Unprotected, Nitrogen-Rich Heterocycles: Substrate Scope and Mechanistic Investigation. J. Am. Chem. Soc. 2013, 135, 12877–12885. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Isley NA; Wang Y; Gallou F; Handa S; Aue DH; Lipshutz BH A Micellar Catalysis Strategy for Suzuki–Miyaura Cross-Couplings of 2-Pyridyl MIDA Boronates: No Copper, in Water, Very Mild Conditions. ACS Catal 2017, 7, 8331–8337. [Google Scholar]; (k) Guo P; Zhang H; Zhou J; Gallou F; Parmentier M; Wang H Micelle-Enabled Suzuki–Miyaura Cross-Coupling of Heteroaryl Boronate Esters. J. Org. Chem. 2018, 83, 7523–7527. [DOI] [PubMed] [Google Scholar]
- (10).Noël T; Musacchio AJ Suzuki–Miyaura Cross-Coupling of Heteroaryl Halides and Arylboronic Acids in Continuous Flow. Org. Lett. 2011, 13, 5180–5183. [DOI] [PubMed] [Google Scholar]
- (11).Slagt VF; de Vries AHM; de Vries JG; Kellogg RM Practical Aspects of Carbon-Carbon Cross-Coupling Reactions Using Heteroarenes. Org. Process Res. Dev. 2010, 14, 30–47. [Google Scholar]
- (12).Whelligan DK; Thomson DW; Taylor D; Hoelder S Two-Step Synthesis of Aza- and Diazaindoles from Chloroamino-N-Heterocycles Using Ethoxyvinylborolane. J. Org. Chem. 2010, 75, 11–15. [DOI] [PubMed] [Google Scholar]
- (13).Cox PA; Leach AG; Campbell AD; Lloyd-Jones GC Protodeboronation of Heteroaromatic, Vinyl, and Cyclopropyl Boronic Acids: pH–Rate Profiles, Autocatalysis, and Disproportionation. J. Am. Chem. Soc. 2016, 138, 9145–9157. [DOI] [PubMed] [Google Scholar]
- (14).Cox PA; Reid M; Leach AG; Campbell AD; King EJ; Lloyd-Jones GC Base-Catalyzed Aryl-B(OH)2 Protodeboronation Revisited: From Concerted Proton Transfer to Liberation of a Transient Aryl Anion. J. Am. Chem. Soc. 2017, 139, 13156–13165. [DOI] [PubMed] [Google Scholar]
- (15).Molander GA; Canturk B; Kennedy LE Scope of the Suzuki–Miyaura Cross-Coupling Reactions of Potassium Heteroaryltrifluoroborates. J. Org. Chem. 2009, 74, 973–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Dick GR; Knapp DM; Gillis EP; Burke MD General Method for Synthesis of 2-Heterocyclic N-Methyliminodiacetic Acid Boronates. Org. Lett. 2010, 12, 2314–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Molander GA Organotrifluoroborates: Another Branch of the Mighty Oak. J. Org. Chem. 2015, 80, 7837–7848. [DOI] [PubMed] [Google Scholar]
- (18).Hodgson PB; Salingue FH The Preparation of a Stable 2-Pyridylboronate and Its Reactivity in the Suzuki–Miyaura Cross-Coupling Reaction. Tetrahedron Lett. 2004, 45, 685–687. [Google Scholar]
- (19).Deng JZ; Paone DV; Ginnetti AT; Kurihara H; Dreher SD; Weissman SA; Stauffer SR; Burgey CS Copper-Facilitated Suzuki Reactions: Application to 2-Heterocyclic Boronates. Org. Lett. 2009, 11, 345–347. [DOI] [PubMed] [Google Scholar]
- (20).Cammidge AN; Crépy KVL Application of the Suzuki Reaction as the Key Step in the Synthesis of a Novel Atropisomeric Biphenyl Derivative for Use as a Liquid Crystal Dopant. J. Org. Chem. 2003, 68, 6832–6835. [DOI] [PubMed] [Google Scholar]
- (21).Altenhoff G; Goddard R; Lehmann CW; Glorius F Sterically Demanding, Bioxazoline-Derived N-Heterocyclic Carbene Ligands with Restricted Flexibility for Catalysis. J. Am. Chem. Soc. 2004, 126, 15195–15201. [DOI] [PubMed] [Google Scholar]
- (22).Thomas AA; Zahrt AF; Delaney CP; Denmark SE Elucidating the Role of the Boronic Esters in the Suzuki–Miyaura Reaction: Structural, Kinetic, and Computational Investigations. J. Am. Chem. Soc. 2018, 140, 4401–4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Delaney CP; Kassel VM; Denmark SE Potassium Trimethylsilanolate Enables Rapid, Homogeneous Suzuki–Miyaura Cross-Coupling of Boronic Esters. ACS Catal 2020, 10, 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).(a) Landa A; Minkkilä A; Blay G; Jørgensen KA Bis(Oxazoline) Lewis Acid Catalyzed Aldol Reactions of Pyridine N-Oxide Aldehydes—Synthesis of Optically Active 2-(1-Hydroxyalkyl)-Pyridine Derivatives: Development, Scope, and Total Synthesis of an Indolizine Alkaloid. Chem. - Eur. J 2006, 12, 3472–3483. [DOI] [PubMed] [Google Scholar]; (b) Paunescu E; Matuszak N; Melnyk P Suzuki-Miyaura Cross-Coupling Reaction as the Key Step for the Synthesis of Some New 4'-Aryl and Alkyl Substituted Analogues of Amodiaquine and Amopyroquine. Tetrahedron 2007, 63, 12791–12810. [Google Scholar]; (c) Kotian PL; Babu YS; Wu M; Chintareddy VR; Kumar VS; Zhang W Human Plasma Kallikrein Inhibitors. WO2015134998A1, 2015. [Google Scholar]
- (25).Shen Q; Hartwig JF Lewis Acid Acceleration of C-N Bond-Forming Reductive Elimination from Heteroarylpalladium Complexes and Catalytic Amidation of Heteroaryl Bromides. J. Am. Chem. Soc. 2007, 129, 7734–7735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Yamamoto Y; Yoshimitsu T; Wood JL; Schacherer LN Triethylborane. Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons, Ltd.: Hoboken, NJ, 2007. DOI: 10.1002/047084289X.rt219.pub3 [DOI] [Google Scholar]
- (27).Merchant KJ Potassium Trimethylsilanolate Mediated Hydrolysis of Nitriles to Primary Amides. Tetrahedron Lett. 2000, 41, 3747–3749. [Google Scholar]
- (28). Technically, the protodeboronation of these heterocycles was demonstrated on the parent boronic acids, not the neopentyl esters. [Google Scholar]
- (29).Laganis ED; Chenard BL Metal Silanolates: Organic Soluble Equivalents for O–2. Tetrahedron Lett. 1984, 25, 5831–5834. [Google Scholar]
- (30).Itoh T; Mase T Direct Synthesis of Hetero-Biaryl Compounds Containing an Unprotected NH2 Group via Suzuki–Miyaura Reaction. Tetrahedron Lett. 2005, 46, 3573–3577. [Google Scholar]
- (31). TMSOK sourced from Gelest was found to be optimal and was used throughout this study. [Google Scholar]
- (32).Tatlock WS; Rochow EG The Preparation and Hydrolysis of Some Organosilanolates. J. Org. Chem. 1952, 17, 1555–1563. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.