

Abstract
Double-allylation reagents allow for the construction of highly complex molecules in an expedient fashion. We have developed an efficient, modular, and enantioselective approach towards accessing novel variants of these reagents through Cu/Pd-catalyzed alkenylboration of alkenylboron derivatives. Importantly, we demonstrate novel use of an allylBdan directly in a stereocontrolled allylation without initial deprotection to the boronic ester. These allylation products are employed in a second intermolecular allylation to access complex diol motifs, which has yet to be shown with these types of double-allylation reagents. Overall, the modularity of this approach and the ease in which complex structural motifs can be accessed in a rapid manner signify the importance and utility of this method.
Graphical Abstract
“Make it a Double” A strategy for the synthesis of novel double allylation reagents based on the alkenylboration of alkenylboronates is presented. The ease and modularity with which the reagents can be prepared allows rapid access to a variety of complex diol motifs. In the context of these studies, we also demonstrate the first examples of the use of boronamides in allylation reactions.
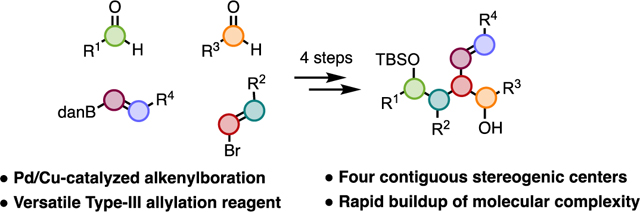
Double-allylation reagents constitute unique molecular frameworks that permit iterative allylation reactions, wherein the first allylation unveils a second allylation reagent. These compounds, sometimes referred to as bifunctional or bimetallic allylation reagents, allow for rapid generation of molecular complexity. Their usage facilitates access to a variety of diols that are of value in synthesis and found in many biologically interesting molecules.1 The structural variations found in these reagents have been delineated into three categories by Hall.2 Type I and Type II reagents (Scheme 1A) have been employed successfully by a variety of research groups.3 Despite their less common usage, Type III reagents have also been utilized effectively in synthesis (Scheme 1B). This includes work of Hall, who prepared a chiral Type III reagent and, through iterative stereoselective allylations, was able to access complex, enantioenriched tetrahydrofuran derivatives.2 Additionally, Morken disclosed an enantioselective diboration of dienes, which formed highly substituted 1,4-cyclohexanediol derivatives after reaction with a tethered dicarbonyl.4 In both cases, the double-allylation reagent has a single stereocenter, requiring the second allylation to be performed in an intramolecular manner in order to achieve a stereocontrolled reaction. Therefore, synthesis of acyclic complex diol scaffolds with Type III allylation reagents is not known.
Scheme 1.

Type III Double Allylation Reagents.
With these examples in mind, we considered the feasibility of accessing a hitherto unknown Type III reagent that bears multiple stereogenic centers, particularly at the α-position of both metallic sites (Scheme 1C). After the first allylation, we would be uniquely positioned to carry out a second, stereocontrolled intermolecular allylation, allowing rapid access to molecular complexity in a modular fashion. Given our extensive work in the realm of Cu/Pd-catalyzed alkene arylboration,5,6,7 we envisioned adapting our system to the alkenylboration of alkenylboron derivatives to prepare the desired Type III reagent. At the outset of our studies, only limited work on arylboration of vinylboron derivatives had been demonstrated by our lab.5g However, nearing the completion of our studies (which were greatly delayed due to the COVID pandemic), Yun reported an arylboration of alkenylborons to generate only one diastereomer of product.8 In one example, an alkenylboration was demonstrated; however, low enantioselectivity was observed. In addition, the utility of the products was not demonstrated. Herein, we report an enantioselective and stereodivergent Cu/Pd-catalyzed alkenylboration of alkenylboron derivatives to generate novel Type III double-allylation reagents, which are capable of iterative stereoselective allylations that allow for access to acyclic complex diol motifs in a modular fashion.
To assess the feasibility of our initial aims, we probed various 1,2-disubstituted alkenes as coupling partners for Cu/Pd alkenylboration (Scheme 2A). Disubstituted alkenylsilanes were not competent under the reaction conditions (product (±)-1), and while alkenylboronic esters demonstrated higher reactivity, their reactions proved to be poorly diastereoselective (product (±)-2) despite attempted optimization. However, by switching to vinylboronamide derivatives we were able to overcome this low diastereoselectivity while maintaining good yield (product (±)-3).9 Analysis via X-ray crystallography revealed that the anti-diastereomer is the major product. A proposed catalytic cycle is depicted in Scheme 2B, in which a borylated copper species (intermediate B) undergoes migratory insertion with the alkenylboron to generate alkylcopper intermediate C.10 Simultaneously, the palladium(0) species D (formed from base activation of the Buchwald pre-catalyst)11,12 undergoes oxidative addition with the alkenyl electrophile to access complex E. Stereoinvertive transmetallation between intermediates C and E provide alkylpalladium complex F, which upon reductive elimination generates the desired product.
Scheme 2.

Initial Findings. Reaction performed on a 0.5 mmol scale. Yield of isolated purified product, represents the average of two independent experiments.
Having established a set of reaction conditions that allows for good yield and high diastereoselectivity, we began exploring the scope (Scheme 3). Different substitution of the alkenyl boronamide such as Ph (product (±)-5) and Cy (product (±)-4) functioned well. In addition, reaction of monosubstituted alkenylboronamides allowed for product formation (products (±)-6 and (±)-7). Synthesis of product (±)-7 is notable in that a sterically demanding boron-substituted quaternary carbon is generated. A variety of alkenylbromides were competent coupling partners, including E- and Z-vinylbromides (products (±)-12 and (±)-13) and those with increasing steric congestion (products (±)-8 and (±)-9). Cyclic vinyltriflates performed well (product (±)-10), along with simple vinylbromide (product (±)-11). While the focus of this work is on alkenylbromides/triflates, PhBr could also be used to deliver product (±)-15 in good yield and dr. Furthermore, the reaction was carried out on gram-scale with minor reduction in yield to afford a bench-stable solid (product (±)-4). Finally, it was found that use of the corresponding Z-alkene led to formation of the same diastereomer as that generated from the E-alkene (product (±)-12).
Scheme 3.

Cu/Pd-Catalyzed anti-Alkenylboration: Scope. Reaction performed on 0.5 mmol scale. Yield is of isolated purified product and represents the average of two independent experiments. a Reaction with alkenyltriflate b solvent ratio is 5.7:1 tol:THF.
With a route to the anti-diastereomer secured, efforts were then directed towards preparation of the syn-diastereomer, as this would allow access to other stereoisomers after allylation. On the basis of prior reports from our lab, modification of the Pd-catalyst can impact the transmetallation event to favor either stereoretention or inversion of the alkyl-Cu complex, thus allowing for stereodivergent synthesis.5b,c While attempts were made to selectively prepare the syn-diastereomer from the E-alkenylBdan (E-16), conditions could not be identified (Scheme 4A, entry 1). Therefore, we elected to explore reactions of the Z-alkenylBdan (Z-16), itself readily prepared in one step from the corresponding alkyne.13 Evaluation of catalyst combinations led to the finding that use of IPrCuCl in combination with [Pd-APhos] did allow for formation of the syn-diastereomer (±)-17 (Scheme 4A, entry 2). Further evaluation of conditions and catalyst revealed that use of [Pd-XPhos] allowed for formation of (±)-17 in 17:1 dr, albeit in 47% yield (Scheme 4A, entry 3). The yield of the reaction could be increased if the catalyst loading was doubled (Scheme 4A, entry 4). Finally, it is interesting to note that reaction of E-16 generated anti-diastereomer (±)-5 in 17:1 dr (Scheme 4A, entry 5). Thus, the IPrCuCl/XPhosPdG3 conditions are stereospecific, whereas the SIMesCuCl/APhosPdG3 are not (see Scheme 3, product (±)-4).
Scheme 4.

Cu/Pd-Catalyzed syn-Alkenylboration: Scope. a Reaction with 10 mol % IPrCuCl and 5 mol % XPhos-PdG3. b Reaction performed on 0.25 mmol scale. Yield of isolated purified product and represents the average of two independent experiments. Yield/dr in parentheses was determined by 1H NMR analysis of the unpurified reaction mixture with an internal standard.
Under the optimized set of conditions, additional examples were explored and revealed that high selectivity was maintained regardless of alkenylbromide substitution pattern (products (±)-18 to (±)-22) or if PhBr was used (product (±)-25). In addition, while reaction with the n-Bu-alkenylBdan (product (±)-23) was effective, reaction with the Cy-variant (product (±)-24) resulted in low yield.
Based on the studies outlined in Schemes 2-4, conditions have been identified for the synthesis of either diastereomer, which was found to be dependent on both alkene geometry and catalyst combination. To interrogate the reaction mechanism, we chose to study reactions that were stereospecific (synthesis of (±)-5 and (±)-17 from E- or Z-16, respectively) and non-stereospecific (synthesis of (±)-4 from E- or Z-30).
In the case of the stereospecific reactions, treatment of E- or Z-16 with in situ-generated IPrCuBpin led to formation of Cu-complexes (±)-26 and (±)-28, respectively, by a likely syn-addition (Scheme 5A).10 Subjection of these complexes to the catalytic coupling conditions with XPhos-PdG3 allowed for formation of the expected products (±)-5 and (±)-17. Based on the stereochemistry of product, it is suggested that the cross-coupling involves a stereoinvertive transmetallation via 27 and 29. However, when the analogous study was conducted with in situ-generated SIMesCuBpin and E- or Z-30, conversion to the same Cu-complex 31 was observed (Scheme 5B). At this time, the stereochemistry of the Cu-complex could not be identified. Despite this, cross-coupling of the Cu-complex with APhos-PdG3 led to formation of the expected product (±)-4. To summarize, the reactions with IPrCu/XPhosPd are stereospecific due a configurationally stable Cu-complex, whereas the processes with SIMesCu/APhosPd are non-stereospecific because of a configurationally labile Cu-complex that converges to preferential formation of one diastereomer (Scheme 5C). At this stage, it is not clear why the IPr derived Cu-complexes are configurationally stable and the SIMes ones are not, and this will be the focus of future studies.
Scheme 5.

Mechanistic Investigation.
With methods available to access both the syn- and anti-diastereomers as racemates, attention was turned towards development of an enantioselective variant. Based on prior work from our lab,5c,f chiral NHC-Cu-complex 3214 was evaluated with Z-16.15 While high enantioselectivity was observed, the diastereoselectivity was poor (Scheme 6A, entry 1). In prior reports,5b,c [Pd-RuPhos] was deemed optimal; however, poor selectivity and yield were observed (Scheme 6A, entry 2). Further evaluation of Pd-complexes revealed that JackiePhos-NMe2 allowed for formation of (+)-5 in 13:1 dr and 93:7 er (Scheme 6A, entry 6).16 At this stage, it is not clear what is responsible for the high levels of diastereoselectivity, as using ligands in which any component of JackiePhos-NMe2 is changed resulted in significantly lower dr (Scheme 6A, entry 3–5). With this result in hand, other alkenylbromides were evaluated with uniformly high selectivities observed (products (+)-33 and (+)-34). However, when Cy-substituted alkene was subjected to the reaction the desired product was observed with reduced levels of diastereoselectivity (product (+)-4). Despite the lower selectivity, the diastereomers were easily separable by column chromatography. Finally, to access the syn-diastereomer, the conditions analogous to those outlined in Scheme 6A, entry 5 were not applicable as only 1:1 dr was observed (Scheme 6A, entry 4). However, it was identified that use of [Pd-Pi-Bu3] was competent for synthesis of (+)-17 (Scheme 6A, entry 7 and 8).5b
Scheme 6.

Enantioselective Reaction Development. a Reaction run with 4:1 tol:THF. b Reaction performed on 0.25 mmol scale. Yield/dr in parentheses was determined by 1H NMR analysis of the unpurified reaction mixture with an internal standard. Yield is of isolated purified product and represents the average of two independent experiments.
Scheme 8.

Allylation with Boronamide: Scope. Yield is of isolated purified product and represents the average of two independent experiments.
On the basis of the observed enantiomer of (+)-33, a model (35) that rationalizes the observed selectivity is provided in Scheme 6C. Here, the Bdan unit is positioned away from the proximal Mes moiety and opposite the Ph of the imidazole ring that projects towards the Cu-atom. After borylcupration, the alkyl-Cu-complex can undergo either stereoretentive transmetallation with [Pd-JackiePhos-NMe2] or stereoinvertive transmetallation with [Pd-Pi-Bu3] to provide the products.5b
Based on the cumulative efforts outlined in Schemes 2-6, we have established methods for enantioselective and racemic synthesis of both diastereomers of the alkenylboration products. This provides the end-user maximum flexibility in choosing a set of conditions to meet their needs. In addition, the results highlight the subtle interplay between the Cu- and Pd-complexes and their influence on diastereoselectivity. Importantly, for examples that lie beyond the scope illustrated here, evaluation of both Cu- and Pd-complexes give the practitioner greater opportunity for reaction optimization. Finally, achieving a stereoselective synthesis of all diastereomers and enantiomers of these Type III double-allylation reagents advances the fundamental aim of this work, as it allows access to more diverse stereochemical configurations upon carrying out the allylation sequences.
At this point, we were interested in exploring the further reactivity of the Type III allylation reagents as this is the primary goal of this study. Realizing that Bdan derivatives are notoriously reluctant to undergo reaction, we initially investigated conversion to the Bpin. Following conditions from Hall,17 who demonstrated deprotection of an alkenylboronamide derivative prior to allylation, we attempted to access the corresponding allylboronic ester from (±)-4. However, our attempts resulted in either no conversion or transformation to an undesired elimination byproduct (Scheme 7A). Since the overall goal was to access a Type III allylation reagent and demonstrate its efficacy, and because functionalizations of boronamides themselves are rare,18 we focused on achieving allylation directly with the allylboronamide (Scheme 7B). To our knowledge, this type of reaction has not been reported. Initially, we attempted the reaction under conditions commonly used for allylboronic esters; however, no reaction occurred and only starting material was recovered. The lack of reactivity of the allylBdan is likely due to the poor Lewis acidity of the boron atom, which prohibits reaction via a six-membered ring transition state. Therefore, it was reasoned that addition of a Lewis acid might result in coordination to the nitrogen atom of the boronamide and restore the reactivity, as is seen in similar boronic ester systems.19,20,21 Evaluation of Lewis acids led to the finding that BF3OEt2 promoted the reaction to provide (±)-38 in 63% yield in >20:1 dr (Scheme 7B). Optimization of the reaction led to formation of the (±)-38 in 76% NMR yield (63% isolated yield) (Scheme 7B). Importantly, the allylation product was further elaborated through oxidation and esterification to establish its relative configuration via X-ray crystallography (product (±)-39) (Scheme 7B). In addition, (+)-4 was used to generate the product (+)-38 without change in enantiomeric ratio.22
Scheme 7.

Allylation with Boronamide: Optimization.
With development of optimized allylation conditions complete, we turned to investigating the scope of the allylation (Scheme 8A). Reaction with trisubstituted alkenes of the allylboronamide performed well, affording quaternary carbon centers in good yield and as a single observable diastereomer (products (±)-40 and (±)-41). Use of other arylaldehydes (products (±)-43 and (±)-44) or alkylaldehydes (products (±)-45 and (±)-46) also proved to be competent electrophiles, affording products in good yields and high diastereoselectivites. A more complex aldehyde that contained an NBoc group allowed for formation of (±)-47. Additionally, cyclic allylboronamide derivatives formed the corresponding allylation products (product (±)-42; relative configuration established via X-ray crystallography). The use of syn-diastereomer (±)-17 was also tolerated to generate (±)-48. Finally, we sought to examine the nature of the transition state of the reaction. Since this type of allylation has not been demonstrated with an allylboronamide directly, there is no precedent for an open versus closed transition state in this system. While the high diastereoselectivity observed across numerous substrates implies a closed transition state, we believe comparison of the allylation to form products (±)-49 and (±)-51 provides the strongest evidence (Scheme 8B). E- and Z-allylboronamides provided the allylation products as a single diastereomer. Relative configuration of product (±)-49 was determined via functionalization to the ferrocene ester and subsequent analysis by X-ray crystallography (product (±)-50). Since the reaction of the allylboronamide isomers results in high diastereoselectivity for orthogonal diastereomers, this suggests that the allylation proceeds through a closed transition state as depicted in Scheme 8B, with the Lewis acid activating the boronamide by making it more Lewis acidic for allylation. 19,20,21
Having successfully achieved an initial stereocontrolled allylation with these Type III reagents, we sought to demonstrate their use in a second, intermolecular allylation (Scheme 9). To our knowledge, this has yet to be done with this class of double-allylation reagents. After protecting the secondary alcohol (product (±)-52), allylation with benzaldehyde in the absence of Lewis acid provided the desired product in good yield as a single observable diastereomer (product (±)-53). This was functionalized to the biphenyl ester in order to analyze the relative stereochemistry via X-ray crystallography (product (±)-54). The observed stereochemistry supports allylation through a closed transition state, as is typical for allylboronic esters. Furthermore, through the synthesis of (±)-55 and (±)-56 it is demonstrated that multiple stereotetrads can be prepared with precise control of stereochemistry. Ultimately, this highlights the significance of being able to synthesize and utilize these Type III double-allylation reagents in intermolecular allylations to access linear diol motifs.
Scheme 9.

Second Allylation. Yield is of isolated purified product and represents the average of two independent experiments.
In conclusion, we have demonstrated the synthesis of novel Type III double-allylation reagents and their use in intermolecular iterative allylations. Using Cu/Pd catalysis as a platform to make these Type III reagents, we produced a variety of them in a stereodivergent and enantioselective fashion. Initial struggles with the first allylation culminated in the discovery of an allylation system that utilized the boronamide moiety directly without initial deprotection. Additionally, we demonstrated further use of the Type III reagent via a second intermolecular allylation; its significance is derived from the fact that this type of reactivity with Type III double-allylation reagents is unknown. Ultimately, this method leads to the generation of stereochemically complex diol motifs in a rapid manner.
Supplementary Material
Acknowledgements
We thank Indiana University and the NIH (R35GM131755) for financial support. This project was partially funded by the Vice Provost for Research through the Research Equipment Fund and the NSF MRI program, CHE-1726633 and CHE-1920026. We thank Dr. Maren Pink and Dr. Veronica Carta of the IU Molecular Structure Center for acquisition of X-ray crystal structure data. Support for the acquisition of the Bruker Venture D8 diffractometer through the Major Scientific Research Equipment Fund from the President of Indiana University and the Office of the Vice President for Research is gratefully acknowledged. NSF’s ChemMatCARS Sector 15 is supported by the Divisions of Chemistry (CHE) and Materials Research (DMR), National Science Foundation, under grant number NSF/CHE- 1834750. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357
References
- 1).(a) Hunt JA, Roush WR. J. Org. Chem. 1997, 62, 1112–1124. [Google Scholar]; (b) Barrett AGM, Braddock DC, de Koning PD, White AJP, Williams DJ. J. Org. Chem. 2000, 65, 375–380. [DOI] [PubMed] [Google Scholar]; (c) Williams DR, Claeboe CD, Liang B, Zorn N, Chow NSC. Org. Lett. 2012, 14, 3866–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2).Peng F, Hall DG. J. Am. Chem. Soc. 2007, 129, 3070–3071. [DOI] [PubMed] [Google Scholar]
- 3).(a) Along with the references in [1], see:Roush WR, Grover P. Tetrahedron 1990, 31, 7567–7570. [Google Scholar]; (b) Barrett AGM, Malecha JW. J. Org. Chem. 1991, 56, 5243–5245. [Google Scholar]; (c) Hoffmann RW, Schlapbach A.Tetrahedron 1992, 48, 1959–1968. [Google Scholar]; (d) Roush WR, Pinchuk AN, Micalizio GC. Tetrahedron Lett. 2000, 41, 9413–9417. [Google Scholar]; (e) Flamme EM, Roush WR. J. Am. Chem. Soc. 2002, 124, 13644–13645. [DOI] [PubMed] [Google Scholar]; (f) González AZ, Román JG, Alicea E, Canales E, Soderquist JA. J. Am. Chem. Soc. 2009, 131, 1269–1273. [DOI] [PubMed] [Google Scholar]; (g) Liu J, Gao S, Chen M. Org. Process Res. Dev. 2019, 23, 1659–1662. [Google Scholar]
- 4).Ferris GE, Hong K, Roundtree IA, Morken JP. J. Am. Chem. Soc. 2013, 135, 2501–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5).(a) Smith KB, Logan KM, You W, Brown MK. Chem. Eur. J. 2014, 20, 12032–12036. [DOI] [PubMed] [Google Scholar]; (b) Logan KM, Smith KB, Brown MK. Angew. Chem. Int. Ed. 2015, 54, 5228–5231. [DOI] [PubMed] [Google Scholar]; (c) Logan KM, Brown MK. Angew. Chem. Int. Ed. 2016, 56, 851–855. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Smith KB, Brown MK, J. Am. Chem. Soc. 2017, 139, 7721–7724. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Sardini SR, Brown MK, J. Am. Chem. Soc. 2017, 139, 9823–9826. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Bergmann AM, Dorn SK, Smith KB, Logan KM, Brown MK. Angew. Chem. Int. Ed. 2019, 58, 1719–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Huang Y, Brown MK. Angew. Chem. Int. Ed. 2019, 58, 6048–6052. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Bergmann AM, Sardini SR, Smith KB, Brown MK. Isr. J. Chem. 2019, 60, 394–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).(a) For contribution from other labs, see:Semba K, Nakao Y, J. Am. Chem. Soc. 2014, 136, 7567–7570. [DOI] [PubMed] [Google Scholar]; (b) Chen B, Cao P, Yin X, Liao Y, Jiang L, Ye J, Wang M, Liao J. ACS Catal. 2017, 7, 2425–2429. [Google Scholar]; (c) Liao Y, Yin X, Wang X, Yu W, Fang D, Hu L, Wang M, Liao J, Angew. Chem. Int. Ed. 2020, 59, 1176–1180. [DOI] [PubMed] [Google Scholar]
- 7).(a) For reviews, see:Semba K, Nakao Y, Tetrahedron 2019, 75, 709–719. [Google Scholar]; (b) Xue W, Oestreich M. ACS Cent Sci 2020, 6, 1070–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Whyte A, Torelli A, Mirabi B, Zhang A, Lautens M. ACS Catal. 2020, 10, 11578–11622. [Google Scholar]
- 8).Our studies were greatly delayed due to the COVID-19 pandemic. Lee H, Lee S, J. Yun. ACS Catal 2020, 10, 2069–2073. [Google Scholar]
- 9).See the SI for optimization details.
- 10).Laitar DS, Tsui EY, Sadighi JP, Organometallics 2006, 25, 2405–2408. [Google Scholar]
- 11).Guram AS, King AO, Allen JG, Wang X, Schenkel LB, Chan J, Bunel EE, Faul MM, Larsen RD, Martinelli MJ, Reider PJ. Org. Lett. 2006, 8, 1787–1789. [DOI] [PubMed] [Google Scholar]
- 12).Bruno NC, Tudge MT, Buchwald SL. Chem. Sci. 2013, 4, 916–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13).Ohmura T, Yamamoto Y, Miyaura N. J. Am. Chem. Soc. 2000, 122, 4990–4991. [Google Scholar]
- 14).(a) Park JK, Lackey HH, Rexford MD, Kovnir K, Shatruk M, McQuade DT. Org. Lett. 2010, 12, 5008–5011. [DOI] [PubMed] [Google Scholar]; (b) Park JK, McQuade DT. Angew. Chem. Int. Ed. 2012, 51, 2717–2721. [DOI] [PubMed] [Google Scholar]; (c) Park J, McQuade D. Synthesis 2012, 44, 1485–1490. [Google Scholar]
- 15).Reaction with the E-alkenylboronamide and Cu-complex 32 was attempted, however, low enantioselectivity was observed. See the SI for details.
- 16).Park NH, Vinogradova EV, Surry DS, Buchwald SL. Angew. Chem. Int. Ed. 2015, 54, 8259–8262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17).Lee JCH, McDonald R, Hall DG. Nat. Chem. 2011, 3, 894–899. [DOI] [PubMed] [Google Scholar]
- 18).(a) Kamio S, Yoshida H. Adv. Synth. Catal. 2021, early view. [Google Scholar]; (b) Tsuchimoto T, Utsugi H, Sugiura T, Horio S. Adv. Synth. Catal. 2015, 357, 77–82. [Google Scholar]; (c) Tani T, Sawatsugawa Y, Sano Y, Hirataka Y, Takahashi N, Hashimoto S, Sugiura T, Tsuchimoto T. Adv. Synth. Catal. 2019, 361, 1815–1834. [Google Scholar]; (d) Yamamoto K, Mohara Y, Mutoh Y, Saito S. J. Am. Chem. Soc. 2019, 141, 17042–17047. [DOI] [PubMed] [Google Scholar]; (e) Yoshida H, Seki M, Kamio YS, Tanaka H, Izumi Y, Li J, Osaka I, Abe M, Andoh H, Yajima T, Tani T, Tsuchimoto T. ACS Catal. 2020, 10, 346–351. [Google Scholar]; (f) Mutoh Y, Yamamoto K, Saito S. ACS Catal. 2020, 10, 352–357. [Google Scholar]
- 19).Chen M, Roush WR, Org. Lett. 2010, 12, 2706–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20).(a) Rauniyar V, Hall DG. J. Am. Chem. Soc. 2004, 126, 4518–4519. [DOI] [PubMed] [Google Scholar]; (b) D. Hall. Synlett 2007, 2007, 1644–1655. [Google Scholar]
- 21).Sakata K, Fujimoto H. J. Am. Chem. Soc. 2008, 130, 12519–12526. [DOI] [PubMed] [Google Scholar]
- 22).See the SI for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.