

Abstract
Genetic background often influences the phenotypic consequences of mutations, resulting in variable expressivity. How standing genetic variants collectively cause this phenomenon is not fully understood. Here, we comprehensively identify loci in a budding yeast cross that impact the growth of individuals carrying a spontaneous missense mutation in the nuclear-encoded mitochondrial ribosomal gene MRP20. Initial results suggested that a single large effect locus influences the mutation’s expressivity, with 1 allele causing inviability in mutants. However, further experiments revealed this simplicity was an illusion. In fact, many additional loci shape the mutation’s expressivity, collectively leading to a wide spectrum of mutational responses. These results exemplify how complex combinations of alleles can produce a diversity of qualitative and quantitative responses to the same mutation.
Keywords: genetic background, background effects, spontaneous mutation, expressivity, complex traits, genetic architecture
Genetic background effects impact the contributions of mutations to traits of biomedical, evolutionary, and agricultural significance. The main cause of background effects is likely epistasis between mutations and genetic polymorphisms, but the architectures of these genetic interactions are not fully understood. Here, Schell et al. show that a large number of genetic variants interact with a spontaneous mutation in a yeast cross, causing the mutation’s phenotypic effect to range from lethal to near zero.
Introduction
Mutations frequently exhibit different phenotypic effects across individuals in the same population or species (“background effects”) (Nadeau 2001; Chandler et al. 2013; Riordan and Nadeau 2017). Individuals with the same mutation may differ in whether they show an effect (“incomplete penetrance”) or in their degrees of response (“variable expressivity”) (Griffiths et al. 2020). For example, people with the same disease-causing mutation often vary in whether they express the associated disorder and in their manifestation of symptoms (Cooper et al. 2013). Background effects can arise due to a myriad of reasons, including genetic interactions between a mutation and segregating loci (“epistasis”), dominance in individuals heterozygous for a mutation, and variability in environmental conditions experienced by individuals (Nadeau 2001), as well as differences in microbiome composition among individuals (Wagner et al. 2021) and stochastic noise (Raj et al. 2010). Multiple of these factors may jointly cause a mutation to show a background effect (Lee et al. 2016, 2019).
In this study, we focus on the role of epistasis in background effects, an important topic for evolution and inheritance (Gibson and Dworkin 2004; Jarosz et al. 2010; Paaby and Rockman 2014; Siegal and Leu 2014). Genetic interactions with loci can impact the evolutionary trajectories of mutations, influencing selection for and against beneficial and deleterious mutations, respectively (Weinreich et al. 2005; Lang et al. 2011; Kryazhimskiy et al. 2014; Johnson et al. 2019). Such epistasis can also have bearing on genotype–phenotype relationships, as a mutation may alter the phenotypic effects of interacting loci (Dworkin et al. 2003; Jarosz et al. 2010; Geiler-Samerotte et al. 2016; Schell et al. 2016; Mullis et al. 2018). In extreme cases, a mutation may mask genetically interacting loci or convert them from cryptic to phenotypically visible (Gibson and Hogness 1996; Rutherford and Lindquist 1998; Queitsch et al. 2002; Jarosz and Lindquist 2010).
Epistasis between mutations and loci is also relevant to modern genetic applications. In medicine, loci that genetically interact with mutations are referred to as “modifiers” (Nadeau 2001). Knowing the modifiers of a disease mutation may enable personalized prediction of disease severity and treatment (Riordan and Nadeau 2017). This knowledge may also be necessary to ensure potentially curative genome editing therapies, such as those for sickle cell disease and beta thalassemia (Frangoul et al. 2021), have their intended consequences. In agriculture, genome editing will likely become a common crop improvement strategy (Nasti and Voytas 2021), but its success depends on edits having their predicted phenotypic effects.
Studies in diverse species have found a large fraction of mutations show background effects due to epistasis with loci (Dowell et al. 2010; Chari and Dworkin 2013; Paaby et al. 2015; Vu et al. 2015; Mullis et al. 2018; Galardini et al. 2019; Johnson et al. 2019). Yet, questions about the number of loci that typically show epistasis with a mutation and the architecture of genetic interactions between mutations and loci remain (Goldstein and Ehrenreich 2021). Addressing these questions is difficult because natural populations harbor substantial genetic diversity, which presents a technical challenge for genetic mapping and enables complex forms of epistasis between mutations and multiple loci (Dowell et al. 2010; Chari and Dworkin 2013; Chandler et al. 2014, 2017; Taylor and Ehrenreich 2014, 2015b; Paaby et al. 2015; Vu et al. 2015; Lee et al. 2016; Taylor et al. 2016; Mullis et al. 2018; Galardini et al. 2019; Hou et al. 2019; Lee et al. 2019; Parts et al. 2021).
Lab crosses can overcome these challenges (Dowell et al. 2010; Chandler et al. 2014; Taylor and Ehrenreich 2014, 2015b; Mullis et al. 2018; Hou et al. 2019; Parts et al. 2021). Crosses can be grown in controlled environments and have balanced allele and multilocus genotype frequencies providing statistical power to detect epistatic loci (Ehrenreich 2017). In inbred or haploid crosses, dominance will be absent, eliminating another potential concern. To date, crosses have shown that background effects often involve multiple loci that may genetically interact not only with a focal mutation, but also each other (Dowell et al. 2010; Chandler et al. 2014; Taylor and Ehrenreich 2014, 2015b; Lee et al. 2016, 2019; Taylor et al. 2016; Mullis et al. 2018; Hou et al. 2019; Parts et al. 2021). However, these studies mainly focused on mutations that cause binary phenotypes and show incomplete penetrance. Similar work is needed on mutations with quantitative phenotypic effects and variable expressivity.
In this study, we employ a series of crosses in the budding yeast Saccharomyces cerevisiae to determine how loci cause a spontaneous mutation to show variable expressivity. We focus on a missense mutation in MRP20, a nuclear-encoded subunit of the mitochondrial ribosome that is essential for cellular respiration (Fearon and Mason 1992). This mutation occurred by chance in a previously generated cross between the reference strain BY4716 (“BY”) and the clinical isolate 322134S (“3S”) (Mullis et al. 2018). Here, we identify the MRP20 mutation and demonstrate its variable expressivity among haploid BYx3S cross progeny (“segregants”). We then determine how loci in the BYx3S cross individually and collectively cause this variable expressivity. These data show that expressivity can be shaped by many loci that show a spectrum of effect sizes and act in a predominantly additive manner.
Materials and methods
Generation of segregants
The mrp20-A105E mutation occurred spontaneously in a BY/3S HOS3/hos3Δ::KanMX diploid produced in Mullis et al. (2018) (Fig. 1a). As described in Fig. 1, this mutant strain was generated through targeted disruption of 1 copy of the HOS3 gene in a BY/3S diploid by transformation of a KanMX cassette with homology tails (Wach et al. 1994). The BY/3S diploid used in this transformation had been produced by mating a BY MATa ho can1Δ::STE2pr-SpHIS5 his3Δ haploid and a 3S MATα ho::HphMX his3Δ::NatMX haploid, resulting in a BY/3S MATa/MATα CAN1/can1Δ::STE2pr-SpHIS5 his3Δ/his3Δ ho/ho::HphMX diploid. This diploid genotype made it possible to obtain MATa haploid F2 segregants by sporulating the initial diploid strain or its HOS3/hos3Δ::KanMX derivative and selecting for random spores on His- plates containing canavanine (Tong and Boone 2006). Haploid segregants from the HOS3/hos3Δ::KanMX diploid also went through a second selection for the hos3Δ mutation on G418. The mrp20-A105E mutation was heterozygous in all BY/3S HOS3/hos3Δ::KanMX cells, implying it was likely present in the original cell in which 1 HOS3 copy had been deleted. Following discovery of mrp20-A105E, we sporulated the BY/3S HOS3/hos3Δ::KanMX again and performed tetrad dissection, avoiding any marker selections. This made it possible to obtain all 4 products from each meiotic event, which was necessary to distinguish the phenotypic effects of the mrp20-A105E and hos3Δ mutations and to test whether these 2 mutations genetically interact.
Fig. 1.
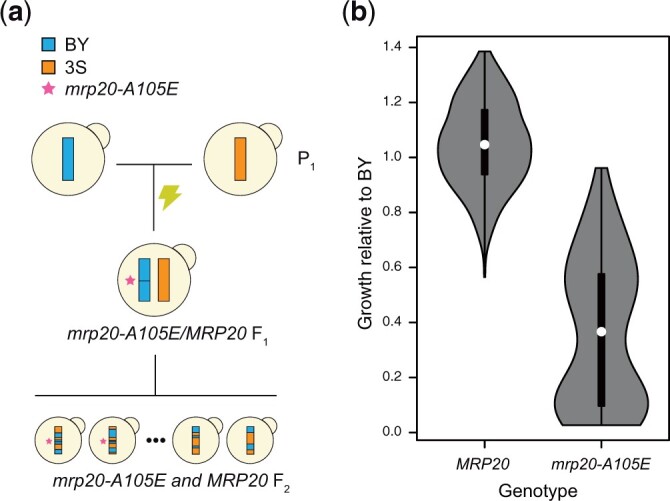
The mrp20-A105E mutation occurred spontaneously, increasing phenotypic variance in the BYx3S cross. a) A spontaneous mutation in a BY/3S diploid gave rise to a BYx3S segregant population in which mrp20-A105E segregated. b) The mrp20-A105E segregants exhibited increased phenotypic variance and a bimodal distribution of phenotypes. Throughout the study, blue and orange are used to denote BY and 3S genetic material, respectively. All growth data presented in the paper are measurements of colonies on agar plates containing rich medium with ethanol as the carbon source.
To fine map the Chromosomes XIV locus harboring MKT1, we produced haploid F3 segregants by mating 2 mrp20-A105E F2 segregants from Mullis et al. (2018), one with the BY allele of the locus and the other with the 3S allele (Fig. 3a). Because these segregants were both MATa, we mating-type switched the segregant with the 3S allele of the Chromosome XIV locus. To enable mating-type switching, we first deleted URA3 from the segregant using a HphMX cassette with homology tails (Goldstein and McCusker 1999). We then mating-type switched the strain by transforming it with a counterselectable URA3 plasmid containing an inducible HO endonuclease (Herskowitz and Jensen 1991), inducing HO, and plating single cells. After mating the 2 haploids, the resulting diploid was sporulated and random haploid F3 segregants were obtained by plating on His- media.
Fig. 3.

MKT1 genetically interacts with mrp20-A105E. a) To improve mapping resolution of the Chromosome XIV locus, we crossed 2 mrp20-A105E F2 segregants with different alleles of the locus and gathered a panel of F3 segregants. b) Linkage mapping in the F3 segregants identified the Chromosome XIV locus at high resolution, with a peak at position 467,219. Tick marks denote every 100,000 bases along the chromosome. c) Recombination breakpoints in the F3 segregants delimited the Chromosome XIV locus to a single SNP in MKT1 at position 467,219. Vertical dashed line highlights the delimited causal polymorphism, while small vertical lines along the x-axis indicate different SNPs in the window that is shown. d) Engineering the BY allele into mrp20-A105E XIV3S segregants changed growth (left), while substitutions at the nearest upstream and downstream variants did not (right).
In addition, to better understand the genetic basis of mrp20-A105E’s expressivity, we generated new haploid F2 segregant panels, 1 in which all individuals were mrp20-A105E MKT1BY and 1 in which all individuals were mrp20-A105E MKT13S (Fig. 5a). To facilitate this, we engineered mrp20-A105E, as well as the 3S and BY causal variants at MKT1 position 467,219 into BY and 3S, respectively, using a 2-step CRISPR/Cas9 strategy described later. BY mrp20-A105E was mated to 3S mrp20-A105E MKT1BY twice independently, the resulting diploids were sporulated, and BYx3S mrp20-A105E MKT1BY haploid segregants were obtained by tetrad dissection. The same process was followed with BY mrp20-A105E MKT13S and 3S mrp20-A105E strains to obtain BYx3S mrp20-A105E MKT13S haploid segregants.
Fig. 5.

Additional loci govern response to the mutation. a) We generated BY × 3S crosses in which all segregants carried mrp20-A105E. Two crosses were performed: 1 in which all segregants carried MKT1BY and 1 in which all segregants carried MKT13S. Tetrads were dissected and spores were phenotyped for growth on ethanol. b) Each of the new crosses showed increased growth that extended from inviable to wild type, differing from the more qualitative bimodal phenotypes seen among the original mrp20-A105E MKT1 segregant populations. c) Linkage mapping identified a total of 16 loci that influenced growth. After 4 iterations of a forward regression, no additional loci were identified.
Genotyping
Genotyping of previously generated F2 segregants is described in Mullis et al. (2018) and was performed in the same manner as segregants produced in the current study. All haploid F2 and F3 segregants generated in this study were genotyped by low coverage whole genome sequencing, using previously reported methods that we summarize here (Bloom et al. 2013; Taylor and Ehrenreich 2014; Lee et al. 2016, 2019; Mullis et al. 2018). Strains were inoculated into liquid overnight cultures and grown to stationary phase at 30°C on a shaker. DNA was extracted from cell pellets using Qiagen 96-well DNeasy kits. Sequencing libraries were prepared using the Illumina Nextera Kit and custom barcoded adapters. Segregants from each respective cross were pooled in equimolar fractions into multiplexed libraries, run on a gel, size selected, and purified with the Qiagen Gel Extraction kit. 150 × 150-bp paired-end sequencing was done on an Illumina HiSeq4000 with Novogene.
All sequence processing and analyses were conducted in bash and R, and specific programs and functions used are listed in Supplementary Table 1. Sequencing reads were mapped against the S288C reference genome (version S288C_reference_sequence_R64-2-1_20150113.fsa from the Saccharomyces Genome Database https://www.yeastgenome.org (accessed 28 January 2022)) using BWA version 0.7.7-r44 (Li and Durbin 2009). Note, BY is a close derivative of S288C, the main reference strain of yeast. Samtools v1.9 was then used to create a pileup file for each segregant (Li et al. 2009). For both BWA and Samtools, default settings were employed. Base calls and coverages were gathered for 44,429 SNPs that segregate in the cross and are spaced approximately every 300 bp (Taylor et al. 2016). Low coverage individuals (<0.7× average per site coverage) were removed from analyses. Ninety percentage of segregants had an average per site coverage of 2.5 or more across all SNPs and the median per site coverage across all segregants in this study was 12. Diploid and contaminated individuals were identified by abnormal patterns of heterozygosity or sequencing coverage and excluded from all analyses.
For each segregant, a raw genotype vector was determined based on the percentage of 3S calls at each marker. We then used a Hidden Markov Model (HMM) implemented in the “HMM” package v 1.0 in R to correct each raw genotype vector. The HMM was used both to impute genotype at markers lacking direct sequencing information and to correct for sequencing errors at low coverage sites. Both types of corrections were rare. The following probability matrices (Rabiner 1989) were used, transitionProbabilitiy = matrix{c[0.(\d+),.0001,.0001,.9999],2} and emissionProbability = matrix{c[0.(\d+).25,0.75,0.75,0.25],2}. These values were taken from past work (Taylor and Ehrenreich 2014; Lee et al. 2016, 2019; Mullis et al. 2018); their appropriateness here was confirmed by visual comparison of HMM outputs to raw genotype vectors.
Aneuploidies were identified based on segregants showing elevated sequencing coverage across entire chromosomes. Specifically, we employed the normalmixEM() function from the mixtools library v 1.2.0 (Benaglia et al. 2009) in R to each segregant, identifying chromosomes that had unusually high coverage relative to the rest of the nuclear genome. A Chromosome II duplication in a subset of BYx3S mrp20-A105E MKT1BY and BYx3S mrp20-A105E MKT13S segregants was the only detected aneuploidy. The coverage of Chromosome II in aneuploid individuals was roughly double the rest of the genome, suggesting the presence of 1 extra copy of that chromosome.
Phenotyping
Segregants were inoculated into broth containing 1% yeast extract, 2% peptone, and 2% dextrose (“YPD”). Cultures were grown to stationary phase at 30°C over 2 days and were then pinned onto YP plates containing ethanol (“YPE”) and 2% agar. YPE is 1% yeast extract, 2% peptone, and 2% ethanol. Each pinned colony results from clonal growth of a genetically distinct strain. Plates were grown at 30°C for 2 days. Growth assays were conducted in a minimum of 3 replicates across 3 plates, with randomized positioning of strains. On each plate, a BY parental strain was included as a control. Plates were imaged with the BioRAD Gel Doc XR+ Molecular Imager at a standard size of 11.4 × 8.52 cm2 (width × length) and imaged with epi-illumination using an exposure time of 0.5 s. Images were saved as 600 dpi tiffs. ImageJ (http://rsbweb.nih.gov/ij/ (accessed 28 January 2022)) was used to quantify pixel intensity of each colony through the Plate Analysis JRU v1 plugin (https://research.stowers.org/imagejplugins/zipped_plugins.html (accessed 28 January 2022)), as discussed in a previous study (Matsui and Ehrenreich 2016). In brief, each colony was normalized to the BY control grown on the same plate to account for batch effects. Then, the 3 normalized biological replicates for a given strain were averaged to produce a single growth value for each genetically distinct segregant. We used replicates to calculate the broad sense heritability of growth on ethanol. Specifically, all replicates of the 749 segregants engineered to carry mrp20-A105E and specific MKT1 alleles were utilized as data points in the fixed effects linear model growth ∼ strain + error. From this model, we took the Sum of Squares of the strain term and divided it by the Sum of Squares Total. This produced a broad sense heritability estimate of 0.93.
Comparison of phenotypic variance between mutant and wild-type segregants
To compare phenotypic variance between wild-type and mutant segregants, we used the leveneTest() function in the car library (v 3.0.6) in R (Fox and Weisberg 2018). We employed the default settings for this function, which use the median as the center for each group rather than the mean, providing a more robust and conservative test (Fox and Weisberg 2018).
Linkage mapping
Initial discovery of the spontaneous mrp20-A105E mutation was done by performing linkage mapping with 385 haploid F2 segregants (164 wild type and 221 hos3Δ) generated in Mullis et al. (2018). The hos3Δ segregants had been engineered to lack the gene encoding the histone deacetylase Hos3. As discussed in the “Generation of segregants” section of the Materials and Methods, this gene deletion had been introduced by transformation of PCR tailed KanMX cassette into a BY/3S diploid. To identify mrp20-A105E, we employed the fixed effects linear model growth ∼ hos3Δ + locus + hos3Δxlocus + error, from which the hos3Δxlocus interaction term was used to identify loci that differentially explained growth in hos3Δ segregants. Examination of the hos3Δxlocus interaction term led to discovery of the spontaneous mrp20-A105E mutation, which was present in the BY copy of MRP20 in only the hos3Δ segregants.
Following the identification of mrp20-A105E, we attempted to identify genetically interacting loci using the fixed effects linear model growth ∼ MRP20 + locus + MRP20xlocus + error with only hos3Δ segregants. From this scan, we examined the MRP20xlocus interaction term. Only a single locus was identified in this scan at a −log10(P) exceeding 4 and it passed even the most conservative thresholds, including a Bonferroni threshold (Bland and Altman 1995) accounting for all SNPs in the cross. Three hundred and sixty-one mrp20-A105E F3 segregants were then used to better resolve the Chromosome XIV locus. In the experiment, we employed the fixed effects linear model growth ∼ locus + error and examined the locus term, focusing on the Chromosome XIV locus, whose significance also exceeded a Bonferroni threshold.
To map loci affecting growth among mrp20-A105E segregants, we generated new populations of mrp20-A105E MKT1BY (353) and mrp20-A105E MKT13S (396) haploid segregants. The combined 749 mrp20-A105E segregants were used in linkage mapping with a forward regression approach. We obtained residuals (“residuals1”) from the fixed effects linear model growth ∼ MKT1 + error and then implemented a first genome-wide scan using the model residuals1 ∼ locus + error. We examined the locus term and called loci significant if they exceeded the 95th quantile of maximal −log10(P-values) from 1,000 permutations (Churchill and Doerge 1994). Permutations were implemented by randomly shuffling the residuals1 vector and rerunning the genome-wide scan. In the first scan, we only called the single most significant locus on each chromosome.
Following the first scan, we accounted for all detected loci using the fixed effects linear model residuals1 ∼ locus 1 + locus 2 + … locus n + error and obtained the residuals (“residuals2”). These new residuals were used in another genome-wide scan with the fixed effects linear model residuals2 ∼ locus + error. Permutation thresholds were calculated anew for this second stage scan in the same manner as the first scan and again a maximum of 1 significant locus per chromosome was called. This process was repeated for 3 additional iterations, at which point no additional loci were detectable using a permutation-based significance threshold. Chromosome II was excluded from linkage mapping due to the presence of its aneuploidy in a subset of individuals. The Chromosome II duplication was tested for significance using the model growth ∼ MKT1 + Chromosome II + error, from which the Chromosome II term was examined.
We then conducted genome-wide scans in mutant segregants for pairwise interactions with detected loci, similar to Brem et al. (2005) and Storey et al. (2005). We used the fixed effects linear model growth ∼ detected_locus + new_locus + detected_locusxnew_locus + error. In this model, detected_Locus corresponded to the peak marker at one of the loci detected in the forward scan and new_Locus represented a different marker. From each test, the P-value of the detected_locusxnew_locus term was extracted. Significance thresholds were determined in each scan using the same permutation strategy described above, with different permutations conducted for each of the previously detected loci. No genetic interactions exceeded the scan-specific permutation thresholds. We also explicitly tested for epistasis between MKT1 and the newly detected locus on Chromosome XIV that we refer to as “SAL1” using the fixed effects linear model growth ∼ MKT1 + SAL1 + MKT1xSAL1 + error. We examined the P-value for the MKT1xSAL1 interaction term, which was greater than 0.05.
All linkage mapping was performed in R. Fixed effects linear models were implemented using the lm() function in base R. Although we had genotype calls for 44,429 SNPs, we only used a subset of these markers in genetic mapping. The difference between the total and tested number of markers reflects the fact that certain linked markers had exactly the same genotype calls across all segregants. Such markers containing the same genetic information will produce identical test results. To avoid unnecessarily inflating the number of tests, we randomly selected 1 marker in each set and used it in linkage mapping. Because each genetic mapping population contained different numbers of segregants and different recombination breakpoints, the number of markers with unique information varied among them, from a minimum of 7,025 in the F2 population used in identifying mrp20-A105E to 18,246 in the F2 populations genetically engineered to carry mrp20-A105E and particular MKT1 alleles.
In all scans, we required that the most significant site (“peak marker”) be a minimum of 150,000 kb away from any other locus. We also required peaks to be more than 20 kb from the edge of a chromosome. We report confidence intervals using established protocols based on drops in the logarithm of the odds (“LOD”), here approximated by −log10(P) values, surrounding a peak marker at a locus; here, we use conventional 2 LOD drops (Lander and Botstein 1989).
Classification of inviable segregants
Initial discovery of the genetic interaction between mrp20-A105E and MKT1 suggested that expressivity of mrp20-A105E was largely determined by variation at MKT1. All mrp20-A105E MKT1BY segregants exhibited very poor growth, while all mrp20-A105E MKT13S segregants showed higher levels of growth. We termed this initial mrp20-A105E MKT1BY segregant population as “inviable.” Figures 7 and 8 include a gray dashed line to denote the highest growth value observed among the original inviable segregants.
Fig. 7.

Mitochondrial genome instability partially underlies the expressivity of mrp20-A105E. We measured petite formation frequency, which estimates the proportion of cells within a clonal population capable of respiratory growth. Higher petite frequency is a proxy for greater mitochondrial genome instability. a) We examined MRP20 and mrp20-A105E versions of the BY and 3S parent strains. For each, average values and 95% bootstrapped confidence intervals are shown. BY showed elevated mitochondrial genome instability in the presence of mrp20-A105E, while 3S showed no change. b) We examined 16 BYx3S MRP20 segregants. These segregants were randomly selected and spanned the range of growth values for MRP20 segregants. c) 45 BYx3S mrp20-A105E segregants. Poorer growing segregants tended to exhibit higher mitochondrial genome instability, though some exhibited wild-type levels of mitochondrial genome instability. The gray dashed line indicates the threshold used to call inviability.
Fig. 8.

Detected loci quantitatively and qualitatively explain mutant phenotypes. a) We fit a linear model accounting for the effects of all detected loci and the aneuploidy on the growth of mrp20-A105E segregants. This model not only explained the growth of the new BYx3S mrp20-A105E crosses generated in this study, but also accurately predicted the phenotypes of the mutant parents and previously generated segregants. b) We examined growth relative to the sum of detrimental alleles carried by a segregant. This relationship shows how collections of loci produce a quantitative spectrum of phenotypes, including instances of qualitative phenotypic responses. This relationship explains the full range of responses, from inviable to wild-type growth, across MKT1-SAL1 genotypes. The gray dashed line indicates the threshold used to call inviability.
Resolving loci and identifying candidate genes
To help resolve loci, we utilized all 44,429 SNPs to analyze recombination breakpoints within loci containing mrp20-A105E, MKT1, and SAL1/PMS1. In each case, we split the appropriate segregants into 2 groups depending on whether they carried the BY or the 3S allele at the peak marker. Segregants’ haplotypes across the adjacent genomic window were then examined. The likely causal regions were determined by identifying the SNPs fixed for BY among all BY individuals and fixed for 3S among all 3S individuals. All recombination breakpoints were confirmed by visual inspection of raw Illumina sequencing using the view function of BWA version 0.7.7-r44 (Li and Durbin 2009). Previously generated F2 segregants (Mullis et al. 2018), newly generated F3 segregants, and newly generated genetically engineered segregants were used to localize MRP20, MKT1, and SAL1/PMS1, respectively. Delimiting these loci in this manner was only possible because of their large phenotypic effects. For other loci with smaller effects, we identified all markers showing peak significance and listed any genes containing these markers as “candidates.” These loci and the candidate genes in them are provided in Table 1 and Supplementary Tables 3 and 4.
Table 1.
Candidate genes at detected loci have diverse cellular functions.
| Candidate gene(s) | Summary of associated function(s) |
|---|---|
| AFR1 | Pheromone-induced projection (shmoo) formation; Septin architecture during mating |
| HOS2 | Histone deacetylase, subunit of Set3 and Rpd3L complexes |
| YGL193C | Haploid specific gene |
| IME4 | Methyltransferase, conditionally essential for meiosis |
| SDH1 | Flavoprotein involved in TCA cycle in mitochondria |
| BPT1 | Vacuolar transmembrane protein |
| RNH203 | Ribonuclease H2 subunit, ribonucleotide excision repair |
| BOP2 | Unknown |
| ECM7 | Putative integral membrane protein with a role in calcium uptake |
| YMR090W | Unknown |
| NPL6 | Component of the RSC chromatin remodeling complex |
| ECM5 | Subunit of Snt2C complex involved in gene regulation in response to oxidative stress |
| AEP2 | Mitochondrial protein; likely involved in translation |
| PBR1 | Putative oxidoreductase; required for cell viability |
| SAL1 | ADP/ATP transporter in mitochondria |
| PMS1 | ATP-binding protein required for mismatch repair; required for both mitosis and meiosis |
| BRX1 | Nucleolar protein involved in rRNA processing |
| YOR008C-A | Unknown, potential transmembrane domain |
| TIR4 | Cell wall mannoprotein; required for anaerobic growth |
| ISN1 | Ionosine metabolism |
| ISW2 | ATP-dependent DNA translocase involved in chromatin remodeling |
Candidate genes were identified by looking at all genes within an interval that contained markers showing maximal significance. For this analysis, we considered all SNPs within an interval, not just the subset used in linkage mapping. Candidate genes in these intervals are listed with brief annotations for the candidates are provided here from the Saccharomyces Genome Database (Cherry et al. 2012). More information about these loci is provided in Supplementary Tables 3 and 4.
Reciprocal hemizygosity experiments
Four hos3Δ F2MATa segregants were used in all reciprocal hemizygosity (RH) experiments focused on the Chromosome IV locus (“IV”) (Steinmetz et al. 2002): 2 were hos3Δ IVBY XIVBY and 2 were hos3Δ IV3S XIVBY. The 4 segregants were first mating-type switched to enable mating of these segregants to produce homozygous IVBY/IVBY, homozygous IV3S/IV3S, or heterozygous IVBY/IV3S diploids. Each pairwise mating was performed and confirmed by plating on mating-type tester plates. These diploid strains were then phenotyped on YPE plates, which verified that IVBY has an effect in diploids and acts in a recessive manner. Using the haploid MATa and MATα versions of these 4 segregants, we individually engineered premature stop codons into DIT1, MRP20, and PDR15 using CRISPR-mediated targeted gene disruption and lithium acetate transformations (Gietz and Woods 2002). Plasmid-based CRISPR-Cas9 was employed to target the beginning of each coding region and 20-bp repair templates which contained a premature stop codon followed by 1-bp deletions were incorporated. Each sgRNA and repair template was designed so that only the first 15 (of 537), 26 (of 264), and 33 (of 1,530) amino acids would be translated for DIT1, MRP20, and PDR15, respectively. Engineered strains were confirmed by PCR and Sanger sequencing. After confirmation, wild-type and knockout strains for each gene were then mated in particular combinations to produce reciprocal hemizygotes that were otherwise isogenic. A minimum of 2 distinct hemizygotes were generated for each allele of each gene.
Construction of nucleotide replacement strains
Single nucleotide replacement strains were generated for MRP20 and MKT1 using a CRISPR/Cas9-mediated approach. For a given replacement, the appropriate strain was first transformed with a modified version of pML104 that constitutively expresses Cas9 using LiAc transformation (Gietz and Woods 2002; Laughery et al. 2015). We then inserted the KanMX gene using cotransformation of a double-stranded DNA containing KanMX with 30-bp upstream and 30-bp downstream homology tails and gRNAs targeting the region containing the site of interest (Kannan et al. 2016). DNA oligos and PCR were used to construct custom sgDNA templates which included crRNA and tracrRNA in a single molecule. Next, we employed T7 RNA Polymerase to express sgDNA templates in vitro. DNase treatment and phenol extraction were used to obtain purified sgRNAs. Transformants were selected on media containing G418, and KanMX integration was confirmed by PCR. Next, KanMX was replaced with the nucleotide of interest. To do this, integrants were cotransformed with 4 gRNAs targeting KanMX, a 60-bp single-stranded DNA repair oligo, and a marker plasmid expressing either HphMX or NatMX using electroporation (Thompson et al. 1998). Marker plasmids were constructed by Gibson assembly with HphMX or NatMX and pRS316 (Sikorski and Hieter 1989; Gibson et al. 2009). Repair constructs were 60-bp ssDNA oligos ordered from Integrated DNA technologies that included upstream homology, the desired nucleotide at the site of interest, and downstream homology. Transformants were selected on media containing either hygromycin or nourseothricin, depending on what marker plasmid was used. Replacement strains were then confirmed by Sanger sequencing.
Following this strategy, the mrp20-A105E nucleotide was engineered into 2 hos3Δ IV3S XIVBY segregants, and 2 hos3Δ IVBY XIVBY segregants were restored to MRP20. Similarly, at MKT1 the causal, nearest upstream and downstream SNPs were engineered into 2 hos3Δ IVBY XIV3S segregants. Similarly, we generate BY mrp20-A105E, BY MKT13S, 3S mrp20-A105E, and 3S MKT1BY strains in this manner. Each single nucleotide parental replacement strain was then backcrossed to its own progenitor. Each subsequent diploid was sporulated and tetrad dissected, and we confirmed haploid genotypes by Sanger sequencing. The same approach was used to generate 3S mrp20-A105E MKT1BY haploids by crossing 3S mrp20-A105E and 3S MKT1BY strains. However, this strategy could not be followed to generate BY mrp20-A105E MKT13S haploids, because, crossing BY mrp20-A105E and BY MKT13s strains failed to produce any tetrads with 4 viable spores. Instead, we took BY MKT13S strains and converted MRP20 to mrp20-A105E.
Mitochondrial genome instability experiments
Budding yeast are facultative anaerobes and can survive without functional mitochondria (Contamine and Picard 2000). Cells with defective mtDNA or no mtDNA are referred to as “petites” because they form small colonies relative to cells with mtDNA which form normal-sized “grande” colonies (Ephrussi et al. 1949; Dujon 1981). Mitochondrial genome stability can be quantitatively measured as the frequency of spontaneous petites in a population of cells (Sherman 2002; Dimitrov et al. 2009). We performed petite frequency assays as described in Dimitrov et al. (2009). In brief, freezer stocks were streaked onto solid YPD media and grown for 2 days at 30°C. Single colonies were then resuspended in PBS, plated across dilutions onto YPDG plates (1% yeast extract, 2% peptone, 0.1% glucose, and 3% glycerol) and grown for 5 days at 30°C. Plates were then imaged with the BioRAD Gel Doc XR+ Molecular Imager at a standard size of 12.4 × 8.9 cm2 (width × length) and imaged with epi-illumination using an exposure time of 0.5 s. Images were saved as 600 dpi tiffs. ImageJ (http://rsbweb.nih.gov/ij/ (accessed 28 January 2022)) was used to examine growth and quantitate colony size as discussed in Dimitrov et al. (2009). Colonies were then classified as petite and grande using a threshold defined as the maximum colony diameter of observed petites among BY and 3S wild-type strains. Petite frequency is the ratio of small colonies to total colonies.
Modeling growth and examining the model in additional segregant populations
We modeled growth for mrp20-A105E segregants from the BYx3S crosses fixed for mrp20-A105E and engineered at MKT1. We incorporated MKT1, the 16 detected loci, and the Chromosome II aneuploidy into a fixed effects linear model growth ∼ MKT1 + locus1 + locus2 + … locus16 + Chromosome II + error. This model was used to generate predicted growth values. We then compared our observed growth values to these predictions. We also obtained predictions for the original mutant population and cloned parent strains from this linear model. These predictions were compared to observed growth measurements, shown in Fig. 8a. In addition, a ten-fold cross-validation approach was employed in which we fit the model to distinct 9/10th subsets of data and then used it to obtain phenotypic predictions for the remaining 1/10ths of data. We then compared the predicted values to the observed growth values and obtained Pearson correlations. We also fit a genomic best linear unbiased prediction (“gBLUP”) model to these data in which genetic relatedness was used to predict phenotype (Henderson 1975). Specifically, we used the A.mat() function from the “sommer” library (v.4.0.9) in R to generate the genetic relatedness matrix and the mmer() function to fit this matrix to our growth data and obtain phenotypic predictions (Henderson 1975; Endelman and Jannink 2012; Covarrubias-Pazaran 2016).
Relationship between detrimental alleles, growth, and inviability
At each detected locus influencing response to mrp20-A105E, we determined the allele associated with worse growth (“detrimental allele”). Next, we counted the number of detrimental alleles carried by each mrp20-A105E segregant and examined how phenotypic response to mrp20-A105E related to it. The MKT1 and SAL1 loci were not included when counting detrimental alleles, so that this relationship could be examined across different MKT1-SAL1 genotype classes.
Results and discussion
A spontaneous mutation increases phenotypic variance in the BYx3S cross
In a BY/3S diploid, a spontaneous mutation occurred in a core domain of Mrp20 that is conserved from bacteria to humans (Fig. 1a, Supplementary Fig. 2, and Supplementary Table 2) (Fearon and Mason 1992; Koc et al. 2001). This mutation resulted in an alanine to glutamic acid substitution at amino acid 105 (mrp20-A105E). We sporulated this diploid, unaware of the mutation, and obtained haploid BYx3S F2 segregants, some of which possessed mrp20-A105E. We then phenotyped the segregants for colony growth on agar plates containing glucose, a fermentable carbon source, or ethanol, a respirable carbon source. In yeast, colony growth assays are commonly used to measure the phenotypic effects of mutations (e.g. Dowell et al. 2010; Costanzo et al. 2016), as well as to characterize phenotypic differences among genetically diverse strains (e.g. Bloom et al. 2013, 2015; Matsui and Ehrenreich 2016). Here, cells from each segregant were pinned onto agar plates and colony size was measured at a common endpoint, with final size a proxy for the number of mitotic divisions that occurred.
The mrp20-A105E mutation was discovered because some haploid BYx3S segregants showed near-zero growth (“inviability”) on ethanol but not glucose. On respirable carbon sources specifically, loss-of-function mutations in MRP20 should cause poor growth by disrupting translation in the mitochondrial compartment and impairing cellular respiration. Unexpectedly, many mrp20-A105E segregants exhibited high levels of growth on ethanol, with some possessing phenotypes similar to wild-type segregants. The BYx3S mrp20-A105E segregants exhibited substantially elevated phenotypic variance relative to wild-type segregants (Fig. 1b; Levene’s test using median, P = 5.9 × 10−22). Among the mrp20-A105E segregants, there were 2 modes, centered on 10% and 57% growth relative to the haploid BY parent (bimodal fit log likelihood = 30; Supplementary Fig. 3). This increase in phenotypic variance, as well as certain segregants showing a lack of impairment by mrp20-A105E, suggested the mutation displays variable expressivity on ethanol, our focal environment hereafter.
A large effect locus shows epistasis with mrp20-A105E
Multiple lines of evidence suggest that epistasis with segregating loci in the BYx3S cross causes mrp20-A105E’s variable expressivity. The same polymorphisms segregated among MRP20 and mrp20-A105E segregants, which were derived from a common diploid progenitor. All segregants were haploid and phenotyped under the same conditions, ruling out dominance and environmental effects as explanations, respectively. Also, we measured the growth of yeast colonies, which contain hundreds of thousands to millions of cells, making stochastic phenotypic differences among cells an unlikely explanation as well.
To identify loci that genetically interact with mrp20-A105E, we performed linkage mapping using all MRP20 and mrp20-A105E segregants, seeking loci that show pairwsie epistasis with the mutation. In a genome-wide scan, we detected a single locus on Chromosome XIV (interaction term in a full-factorial 2-way ANOVA, P = 4.3 × 10−16; Fig. 2a). This locus (“XIV”) exceeded even a conservative threshold based on a Bonferroni correction (Bland and Altman 1995) accounting for all 44,429 genotyped SNPs in the cross. Individuals with the XIVBY allele grew worse than individuals with XIV3S allele in both MRP20 and mrp20-A105E segregants, but to a greater extent among the mutants (Fig. 2b). XIV explained 79% of the phenotypic variance within mrp20-A105E segregants (ANOVA, P = 3.2 × 10−31) and XIVBY was present in all inviable segregants (Fig. 2b).
Fig. 2.

Epistasis between MRP20 and Chr XIV appears to mostly explain response to mrp20-A105E. a) Linkage mapping in the BYx3S segregants shown in Fig. 1 identified a locus on Chromosome XIV that exhibits a 2-way genetic interaction with MRP20. b) The Chromosome XIV locus had effects in both MRP20 and mrp20-A105E segregants but had a greater effect among mrp20-A105E segregants.
Yeast is a highly recombinogenic organism, with 1 centimorgan in genetic map distance roughly equal to 2.5 kb in physical map distance (Cherry et al. 1997; Mancera et al. 2008). Thus, we performed another cross, with the goal of using yeast’s high recombination rate to help resolve the causal polymorphism. We mated 2 mrp20-A105E segregants that differed at XIV (mrp20-A105E XIVBY × mrp20-A105E XIV3S; Fig. 3a and Supplementary Table 2). From this advanced intercross, 361 F3mrp20-A105E progeny were sequenced and phenotyped. Linkage mapping with these data reidentified XIV at a P-value of 2.50 × 10−43 (ANOVA; Fig. 3b and Supplementary Fig. 4). This locus also far exceeded a Bonferroni threshold. Analysis of the peak marker and recombination breakpoints surrounding it delimited XIV to a single SNP in the coding region of MKT1 (Fig. 3c). This SNP causes a substitution at amino acid 30, with BY and 3S encoding glycine and serine, respectively, and was validated by nucleotide replacement in mrp20-A105E segregants (Fig. 3d). Notably, this specific SNP was previously shown to play a role in mitochondrial genome stability (Dimitrov et al. 2009).
Epistasis between MRP20 and MKT1 differs in cross parents and segregants
Yeast is also highly amenable to genetic engineering (Schindler 2020), making it possible to validate results from linkage mapping using allele replacement. To validate the epistasis between mrp20-A105E and MKT1, we introduced all 4 possible combinations of the causal nucleotides at these genes into BY and 3S haploids using CRISPR/Cas9-mediated genetic engineering (Fig. 4). The mrp20-A105E mutation affected growth in both parent strains (t-tests, P in BY strain = 4.3 × 10−24 and P in 3S strain = 4.0 × 10−4). However, the magnitude of the phenotypic effect differed between the 2: mrp20-A105E caused inviability in BY but had a more modest effect in 3S. In addition, response to mrp20-A105E was modified by MKT1 in a parent background-dependent manner. Specifically, response to the mutation was more severe in the presence of MKT1BY than MKT13S in 3S (ANOVA, P = 0.01) but not BY [ANOVA, P = 0.99], presumably because BY mrp20-A105E strains were generally inviable.
Fig. 4.
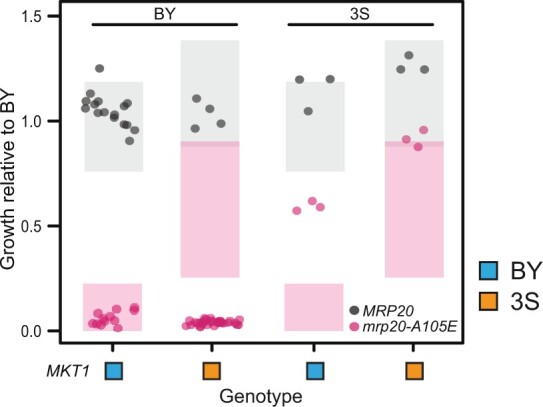
Genetic engineering of parents revealed unexpected responses to mrp20-A105E. We engineered all combinations of MRP20 and MKT1 into the BY and 3S cross parents. Expected phenotypes are shown as shaded boxes denoting 95% confidence interval based on the originally obtained segregant phenotypes.
Although linkage mapping initially suggested that MKT1 explains most of mrp20-A105E’s variable expressivity, our genetic engineering results clearly showed this was not the case. Specifically, BY mrp20-A105E MKT13S, 3S mrp20-A105E MKT1BY, and 3S mrp20-A105E MKT13S all exhibited different phenotypes than expected based on mrp20-A105E segregants (Fig. 4). These results imply additional loci must also genetically interact with mrp20-A105E and contribute to its variable expressivity.
Many additional loci affect the expressivity of mrp20-A105E
To enable the identification of other loci influencing the expressivity of mrp20-A105E, we generated 2 new BYx3S haploid F2 crosses using genetically engineered BY and 3S parents (Fig. 5a and Supplementary Table 2). Both crosses were designed so that all segregants in the same cross would carry mrp20-A105E and the same MKT1 allele, either MKT1BY or MKT13S. By engineering the crosses in this way, we increased the chance of detecting additional loci contributing to the variable expressivity of mrp20-A105E. From these engineered crosses, 749 total segregants were obtained through tetrad dissection, ensuring these individuals would have balanced allele frequencies at all loci and random multilocus genotypes. These segregants were then sequenced and phenotyped for growth on ethanol.
In contrast to the bimodal phenotypic distribution observed in the original mrp20-A105E segregants, these new crosses exhibited continuous ranges of phenotypes, as well as higher phenotypic variance (Fig. 5b). This finding suggests that genetically modifying the cross parents altered the phenotypic effects of loci that genetically interact with mrp20-A105E or MKT1, uncoupled linked loci that also genetically interact with mrp20-A105E from MKT1, or both. In both the MKT1BY and MKT13S crosses, mrp20-A105E segregants ranged from inviable to nearly wild type. The distributions of phenotypes in the 2 crosses differed in a manner consistent with their MKT1 alleles, with the mean of the MKT1BY segregants lower than the MKT13S segregants (t-test, P = 4.8 × 10−34). These data show that regardless of the MKT1 allele present, additional loci can cause mrp20-A105E to show phenotypic effects ranging from lethal to benign.
We next mapped these other loci influencing the expressivity of mrp20-A105E. Excluding MKT1, which explained 18% of the phenotypic variance in the new crosses, linkage mapping identified 16 new loci (Fig. 5c, Supplementary Fig. 5, and Supplementary Table 3). We did not detect genetic interactions among the loci, suggesting any epistasis between them is weak. Of the new loci, the BY allele was inferior at 10 and superior at 6. These loci individually explained between 0.79% and 14% of the phenotypic variance in the new crosses. Thirteen of these loci resided on a subset of chromosomes but were distantly linked: 4 on Chromosomes XII, 3 on XIII, 2 on XIV, and 4 on XV. The 3 remaining loci were detected on Chromosomes IV, VII, and XI. We delimited these other loci to small genomic intervals spanning 1 (12 loci), 2 (3 loci), or 3 (1 locus) candidate genes based on markers showing peak significance (Supplementary Table 4). The candidate genes in these intervals functioned in many compartments of the cell and implicated multiple pathways rather than a single molecular process, suggesting the mechanistic basis of mrp20-A105E’s variable expressivity is complex (Table 1).
The Chromosome XIV locus contains multiple causal variants
Among the newly detected loci, the largest effect (14% phenotypic variance explained) was on Chromosome XIV near the first locus we had detected at MKT1. The position of maximal significance at this new site was 2 genes away from the end of MKT1, with a confidence interval that did not encompass the causal variant in MKT1 (Supplementary Table 4). Thus, the originally identified large effect XIV locus in fact represents multiple distinct closely linked nucleotides that both genetically interact with MRP20 and occur in different genes (Fig. 6a).
Fig. 6.

Analysis of MKT1 and SAL1 genotypes reveals an aneuploidy also contributes to mrp20-A105E’s variable expressivity. a) Inviable segregants were present among all mrp20-A105E MKT1 SAL1 genotype classes. b) Aneuploid individuals with duplicated Chromosome II showed reduced growth. Aneuploid individuals were not evenly detected across the different MKT1-SAL1 genotype classes.
The new locus on Chromosome XIV was delimited to 2 genes, one of which was SAL1, encoding a mitochondrial ADP/ATP transporter that physically interacts with Mrp20 (Singh et al. 2020). A SNP in SAL1 that segregates in this cross was previously linked to increased mitochondrial genome instability in BY (Dimitrov et al. 2009), suggesting it is likely also causal in our study. For this reason, we refer to this additional Chromosome XIV locus as “SAL1.” We found no evidence for epistasis between MKT1 and SAL1 (interaction term in a full-factorial 2-way ANOVA, P = 0.77). Although the MKT1-SAL1 locus had a large effect, it explained a minority of the phenotypic variance among mrp20-A105E segregants in a model including all detected loci (32% for MKT1-SAL1 vs 36% for all other loci collectively). Thus, by enabling MKT1 and SAL1 to segregate independently through genetic engineering and examining a large number of mrp20-A105E segregants with different MKT1-SAL1 genotypes, we observed a greater diversity of phenotypes among individuals with the mutation than was originally seen and detected many additional loci contributing to the mutation’s expressivity.
Aneuploidy also contributes to the expressivity of mrp20-A105E
Despite the fact that the identified loci explain most of mrp20-A105E’s expressivity, some individuals exhibited growth that was appreciably lower than expected, suggesting yet another unidentified genetic factor was present (Fig. 6b). This finding led to the detection of a Chromosome II duplication that reduced growth (ANOVA, 1.2 × 10−48). The aneuploidy was common among mrp20-A105E segregants, with a higher prevalence when MKT13S was also present (Fisher’s exact test, P = 1.5 × 10−43; Supplementary Table 5). The Chromosome II aneuploidy was not seen among wild-type segregants. These data suggest that mrp20-A105E increases the rate of aneuploidization and that genetic variation in MKT1 influences the degree to which mrp20-A105E segregants duplicate Chromosome II. The aneuploidy’s contribution to phenotypic variation was relatively minor, explaining 5% of phenotypic variance among mrp20-A105E segregants in a model also including all identified loci.
Multiple cellular mechanisms underlie poor growth in the presence of mrp20-A105E
Evidence suggests mitochondrial genome instability contributes to the variable expressivity of mrp20-A105E. First, mitochondrial genome instability is known to cause poor growth on respirable carbon sources, such as ethanol (Shadel 1999; Lipinski et al. 2010). Second, the exact variants that segregate in our cross at MKT1 and SAL1 were previously linked to mitochondrial genome instability (Dimitrov et al. 2009). Third, both Mrp20 and Sal1 function in the mitochondria (Koc et al. 2001; Kucejova et al. 2008). Fourth, 2 other candidate genes in the newly detected loci encode proteins that function in the mitochondria (Table 1).
To examine whether mitochondrial genome instability contributes to the variable expressivity of mrp20-A105E, we quantified petite formation, a measure of spontaneous mitochondrial genome loss (Fig. 7) (Ephrussi and Slonimski 1955; Sherman 2002; Dimitrov et al. 2009). This phenotype can be examined by plating individual cells on media containing a respirable carbon source and inspecting the resulting colonies; cells with defective mitochondria will produce petite colonies, while cells with functional mitochondria will produce normal-sized colonies (Ephrussi et al. 1949; Dujon 1981; Dimitrov et al. 2009). The proportion of petite and normal colonies produced by a strain provides an estimate of its mitochondrial genome instability. Petite formation and colony growth are distinct but related phenotypes: a high rate of petite formation is one, but not the only, reason a strain might grow poorly on ethanol.
In the petite assays, we examined MRP20 and mrp20-A105E BY and 3S parent strains, as well as 16 MRP20 segregants and 42 mrp20-A105E segregants. Despite causing reduced growth in both parents, mrp20-A105E only led to elevated mitochondrial genome instability in BY (t-test, P = 0.013 in BY and P = 0.39 in 3S; Fig. 7a). Also, although mrp20-A105E segregants exhibited increased mitochondrial genome instability relative to MRP20 segregants (Wilcoxon rank-sum test, P = 0.023), especially at lower levels of growth, a subset of inviable segregants did not show elevated petite formation (Fig. 7, b and c). These results show that mitochondrial genome instability is 1 cellular process contributing to mrp20-A105E’s variable expressivity, but also imply that other mechanisms are involved as well. This finding agrees with our mapping results, which suggested that a number of distinct molecular pathways influence the phenotypic effect of mrp20-A105E (Table 1).
Genetic underpinnings of mrp20-A105E’s expressivity in segregants and parents
We determined the extent to which the identified loci explained phenotypic variability among mutants. We fit a fixed effects linear model of growth as a function of all identified loci and the aneuploidy. This model accounted for most (78%) of the broad sense heritability among mrp20-A105E segregants. Furthermore, phenotypic predictions for segregants based on this model were strongly correlated with their observed phenotypes (Pearson’s r = 0.85, P = 4.4 × 10−209; Fig. 8a). A ten-fold cross-validation analysis found this result was robust, with the ten-fold producing correlations between observed and predicted phenotypes that ranged from 0.78 to 0.91 (mean Pearson’s r = 0.84). In addition, genomic best linear unbiased predictions (gBLUPs) had a Pearson’s r of 0.82 with observed phenotypes, which was roughly equal to the correlation obtained using predictions from the fixed effects linear model. These results suggest we have explained nearly all of the genetic basis of mrp20-A105E’s variable expressivity.
These results show that the variable expressivity of mrp20-A105E is driven by many loci. Confirming this point, the fixed effects linear model including all detected loci was also effective at predicting the phenotypes of genotypes that were not present in the new engineered crosses, but had been generated throughout the course of this work. For example, the model accurately predicted the phenotypes of the original mrp20-A105E segregant population (Pearson’s r = 0.90, P = 1.6 × 10−39), as well as the phenotypes of cross parents engineered to carry mrp20-A105E (Fig. 8a). Moreover, the model explained both qualitative and quantitative variation within and between the 2 XIV classes that were originally seen among mrp20-A105E segregants.
Finally, we examined how diverse combinations of loci collectively produced similar phenotypic responses to mrp20-A105E. We examined the relationship between growth and the total number of detrimental alleles carried by mrp20-A105E segregants, keeping track of each individual’s genotype at MKT1 and SAL1, the largest effect loci (Fig. 8b). The number of detrimental alleles carried by a segregant showed a strong negative relationship with growth, which was not observed in wild-type segregants (Supplementary Fig. 6). Furthermore, regardless of genotype at MKT1 and SAL1, the phenotypic effect of mrp20-A105E ranged from lethal to benign in a manner dependent on the number of detrimental alleles present at other loci. These findings demonstrate that many segregating loci beyond the large effect MKT1-SAL1 locus influence the expressivity of mrp20-A105E and enable different genotypes in the cross to exhibit a broad range of responses to the mutation, including inviability.
Conclusion
We have provided a detailed genetic characterization of the expressivity of a spontaneous mutation, mrp20-A105E. Response to this mutation in a budding yeast cross is influenced by at least 18 genetic factors in total, with the largest effect due to 2 closely linked variants. However, at least 15 additional loci segregate and jointly exert greater effects than the largest 2, MKT1 and SAL1. Different combinations of alleles across these loci produce a continuous spectrum of phenotypic responses to the mutation. Due to tight linkage between MKT1 and SAL1 in the original cross parents, the full extent of this continuum was not originally observed, leading to an initial understanding of the expressivity of the mrp20-A105E mutation that was simplistic. It was only once we disrupted linkage disequilibrium between MKT1 and SAL1 through genetically engineering the parent strains and producing new segregants that the full extent of mrp20-A105E’s variable expressivity was visible.
The identified loci largely explain mrp20-A105E’s variable expressivity and thus make it possible to answer practical and theoretical questions about background effects. For example, our work helps to connect the manifestation of discrete, qualitative mutational responses to their quantitative genetic underpinnings. Whether responses to a mutation appear qualitative may depend on the combinations of responsive alleles in examined individuals. In our case, tight linkage between the largest effect loci, MKT1 and SAL1, made it appear at first that response to mrp20-A105E might be more discrete in nature, as 2 phenotypic modes were observed among mutants such that segregants with the MKT1BY allele were often inviable (Fig. 1b). Yet, later work showed these results were misleading and that segregants in all 4 genotype classes involving MKT1 and SAL1 in fact exhibit continuous responses to mrp20-A105E. Our data suggest this is because of the large number of additional loci that contribute to variable expressivity in our study, which despite having small effects individually are able to exert a large influence over segregants’ responses to mrp20-A105E collectively.
In addition, our findings imply that different individuals may show similar responses to the same mutation due to distinct molecular and cellular mechanisms. When we examined mitochondrial genome instability, we found that most, but not all, mrp20-A105E segregants that were inviable on ethanol exhibited substantially elevated instability. However, a minority of the inviable mrp20-A105E segregants had highly stable mitochondrial genomes, implying they grew poorly on respirable carbon sources because of a different cellular mechanism. The cause of inviability in these individuals is less clear, as identified candidate genes functioned in many different processes (Table 1). This finding likely reflects the fact that growth is a composite phenotype shaped by many cellular processes. Supporting this point, gene–gene deletion studies in yeast have shown genes in diverse cellular processes can genetically interact to affect growth (Costanzo et al. 2016), illustrating the distinction between genetic interactions and direct functional relationships between gene products (Boone et al. 2007). With this said, we note that in other work on discrete traits (Taylor and Ehrenreich 2014, 2015b; Lee et al. 2016, 2019; Taylor et al. 2016), we found a different outcome: identified genes underlying background effects functioned in common pathways and impacted the regulation of a single key gene. These findings illustrate how the genetic basis of a background effect may depend on the mechanisms giving rise to the affected phenotype.
Another major area of interest regarding background effects is their impact on evolution. Within a population, background effects may influence whether beneficial alleles enable adaptation or deleterious alleles are purged (Lang et al. 2011; Kryazhimskiy et al. 2014; Johnson et al. 2019). Regarding the latter, in the case of mrp20-A105E, a highly deleterious allele in respiratory conditions, we found that some segregants with the mutation exhibited wild-type levels of growth. This finding demonstrates how variable expressivity that is caused by epistatic loci may enable some individuals to avoid phenotypic or fitness impairments in the presence of deleterious mutations (Siegal and Leu 2014), potentially muting purifying selection on these alleles. Depending on the prevalence of background effects across different mutations, epistasis between mutations and segregating loci could have a substantial impact on the persistence of deleterious mutations within populations over time.
Our results also inform efforts to understand variable expressivity in other systems, including humans. For example, there is interest in determining why people who carry highly penetrant alleles known to cause disease do not develop pathological conditions (Chen et al. 2016; Narasimhan et al. 2016; Riordan and Nadeau 2017). Such resilience could provide insights into potential therapies, but our work indicates it is likely to involve numerous loci. This speaks to the complicated and unexpected epistasis that can arise between mutations and segregating loci in genetically diverse populations (Carlborg and Haley 2004; Shao et al. 2008; Dowell et al. 2010; Chari and Dworkin 2013; Mackay 2014; Chandler et al. 2014, 2017; Taylor and Ehrenreich 2014, 2015a, 2015b; Paaby et al. 2015; Vu et al. 2015; Lee et al. 2016, 2019; Taylor et al. 2016; Forsberg et al. 2017; Campbell et al. 2018; Mullis et al. 2018; Hou et al. 2019). At the same time, our study also provides reason for optimism. Despite the highly complex genetic basis of mrp20-A105E’s variable expressivity, a gBLUP model lacking any explicitly defined loci explained the mutation’s expressivity as well as a model explicitly including all identified loci. This implies that directly mapping the epistatic loci causing variable expressivity may not be necessary to achieve a predictive understanding of genotype–phenotype relationships among individuals carrying particular mutations. These insights illustrate how characterizing background effects in genetically diverse populations is immediately relevant to inheritance, disease, and evolution.
Data availability
All raw and processed data used in this work is publicly available. Processed data tables and all code used for analyses are included in Supplementary materials S1–S9 and are available on figshare: https://doi.org/10.25386/genetics.18737102 (accessed 28 January 2022). Raw sequencing data can be accessed through Bioproject accession PRJNA739014 in the NCBI Short Read Archive.
Acknowledgments
The authors thank A. Dudley and G. Cromie for input at an early stage of this project, A. Mahableshwarkar and J. Sloan for assistance with experiments, and A. Coradini, I. Goldstein, and C. Hull for feedback during the implementation and writing phases of this study. They also thank the anonymous reviewers of this manuscript for their valuable feedback, Steven Finkel and his lab for allowing use of laboratory equipment, and Oscar Aparicio for sharing the counterselectable HO plasmid.
Funding
This work was funded by grants R01GM110255 and R35GM130381 from the National Institutes of Health (Ian M. Ehrenreich). Rachel Schell and Martin N. Mullis were supported by Research Enhancement Fellowships from the University of Southern California Graduate School. Joseph J. Hale was supported by the National Institutes of Health training grant T32GM118289.
Conflicts of interest
None declared.
Literature cited
- Benaglia T, Cahauveau D, Hunter DR, Young DS.. mixtools: an R package for analyzing mixture models. J Stat Softw. 2009;32:1–29. [Google Scholar]
- Bland JM, Altman DG.. Multiple significance tests: the Bonferroni method. BMJ. 1995;310(6973):170.doi: 10.1136/bmj.310.6973.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom JS, Ehrenreich IM, Loo WT, Lite T-LV, Kruglyak L.. Finding the sources of missing heritability in a yeast cross. Nature. 2013;494(7436):234–237. doi: 10.1038/nature11867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom JS, Kotenko I, Sadhu MJ, Treusch S, Albert FW, Kruglyak L.. Genetic interactions contribute less than additive effects to quantitative trait variation in yeast. Nat Commun. 2015;6:8712.doi: 10.1038/ncomms9712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone C, Bussey H, Andrews BJ.. Exploring genetic interactions and networks with yeast. Nat Rev Genet. 2007;8(6):437–449. doi: 10.1038/nrg2085. [DOI] [PubMed] [Google Scholar]
- Brem RB, Storey JD, Whittle J, Kruglyak L.. Genetic interactions between polymorphisms that affect gene expression in yeast. Nature. 2005;436(7051):701–703. doi: 10.1038/nature03865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell RF, McGrath PT, Paaby AB.. Analysis of epistasis in natural traits using model organisms. Trends Genet. 2018;34(11):883–898. doi: 10.1016/j.tig.2018.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlborg O, Haley CS.. Epistasis: too often neglected in complex trait studies? Nat Rev Genet. 2004;5(8):618–625. doi: 10.1038/nrg1407. [DOI] [PubMed] [Google Scholar]
- Chandler CH, Chari S, Dworkin I.. Does your gene need a background check? How genetic background impacts the analysis of mutations, genes, and evolution. Trends Genet. 2013;29(6):358–366. doi: 10.1016/j.tig.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler CH, Chari S, Tack D, Dworkin I.. Causes and consequences of genetic background effects illuminated by integrative genomic analysis. Genetics. 2014;196(4):1321–1336. doi: 10.1534/genetics.113.159426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler CH, Chari S, Kowalski A, Choi L, Tack D, DeNieu M, Pitchers W, Sonnenschein A, Marvin L, Hummel K, et al. How well do you know your mutation? Complex effects of genetic background on expressivity, complementation, and ordering of allelic effects. PLoS Genet. 2017;13(11):e1007075. doi: 10.1371/journal.pgen.1007075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chari S, Dworkin I.. The conditional nature of genetic interactions: the consequences of wild-type backgrounds on mutational interactions in a genome-wide modifier screen. PLoS Genet. 2013;9(8):e1003661.doi: 10.1371/journal.pgen.1003661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Shi L, Hakenberg J, Naughton B, Sklar P, Zhang J, Zhou H, Tian L, Prakash O, Lemire M, et al. Analysis of 589,306 genomes identifies individuals resilient to severe Mendelian childhood diseases. Nat Biotechnol. 2016;34(5):531–538. doi: 10.1038/nbt.3514. [DOI] [PubMed] [Google Scholar]
- Cherry JM, Ball C, Weng S, Juvik G, Schmidt R, Adler C, Dunn B, Dwight S, Riles L, Mortimer RK, et al. Genetic and physical maps of Saccharomyces cerevisiae. Nature. 1997;387(6632 Suppl.):67–73. [PMC free article] [PubMed] [Google Scholar]
- Cherry JM, Hong EL, Amundsen C, Balakrishnan R, Binkley G, Chan ET, Christie KR, Costanzo MC, Dwight SS, Engel SR, et al. Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res. 2012;40:D700–D705. doi: 10.1093/nar/gkr1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill GA, Doerge RW.. Empirical threshold values for quantitative trait mapping. Genetics. 1994;138(3):963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contamine V, Picard M.. Maintenance and Integrity of the Mitochondrial Genome: a Plethora of Nuclear Genes in the Budding Yeast. Microbiol Mol Biol Rev. 2000;64(2):281–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DN, Krawczak M, Polychronakos C, Tyler-Smith C, Kehrer-Sawatzki H.. Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum Genet. 2013;132(10):1077–1130. doi: 10.1007/s00439-013-1331-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, VanderSluis B, Koch EN, Baryshnikova A, Pons C, Tan G, Wang W, Usaj M, Hanchard J, Lee SD, et al. A global genetic interaction network maps a wiring diagram of cellular function. Science. 2016;353(6306):aaf1420. doi: 10.1126/science.aaf1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covarrubias-Pazaran G. Genome-assisted prediction of quantitative traits using the R package sommer. PLoS One. 2016;11(6):e0156744). doi: 10.1371/journal.pone.0156744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrov LN, Brem RB, Kruglyak L, Gottschling DE.. Polymorphisms in multiple genes contribute to the spontaneous mitochondrial genome instability of Saccharomyces cerevisiae S288C strains. Genetics. 2009;183(1):365–383. doi: 10.1534/genetics.109.104497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowell RD, Ryan O, Jansen A, Cheung D, Agarwala S, Danford T, Bernstein DA, Rolfe PA, Heisler LE, Chin B, et al. Genotype to phenotype: a complex problem. Science. 2010;328(5977):469. doi: 10.1126/science.1189015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujon B. Mitochondrial genetics and functions. In: Strathern JN, Jones EW, Broach JR, editors. The Molecular Biology of the Yeast Saccharomyces: Life Cycle and Inheritance. Cold Spring Harbor (NY: ): Cold Spring Harbor Laboratory; 1981. p. 505–635. [Google Scholar]
- Dworkin I, Palsson A, Birdsall K, Gibson G.. Evidence that Egfr contributes to cryptic genetic variation for photoreceptor determination in natural populations of Drosophila melanogaster. Curr Biol. 2003;13(21):1888–1893. doi: 10.1016/j.cub.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Ehrenreich IM. Epistasis: searching for interacting genetic variants using crosses. Genetics. 2017;206(2):531–535. doi: 10.1534/genetics.117.203059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endelman JB, Jannink J-L.. Shrinkage estimation of the realized relationship matrix. G3 (Bethesda). 2012;2:1405–1413. doi: 10.1534/g3.112.004259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ephrussi B, Hottinguer H, Chimenes Y.. Action de l’acriflavine sur les levures. I. La mutation “petite colonie.” Ann Inst Pasteur. 1949;76:351–367. [Google Scholar]
- Ephrussi B, Slonimski PP.. Subcellular units involved in the synthesis of respiratory enzymes in yeast. Nature. 1955;176(4495):1207–1208. doi: 10.1038/1761207b0. [DOI] [PubMed] [Google Scholar]
- Fearon K, Mason TL.. Structure and function of MRP20 and MRP49, the nuclear genes for two proteins of the 54 S subunit of the yeast mitochondrial ribosome. J Biol Chem. 1992;267(8):5162–5170. [PubMed] [Google Scholar]
- Forsberg SKG, Bloom JS, Sadhu MJ, Kruglyak L, Carlborg Ö.. Accounting for genetic interactions improves modeling of individual quantitative trait phenotypes in yeast. Nat Genet. 2017;49(4):497–503. doi: 10.1038/ng.3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox J, Weisberg S.. An R Companion to Applied Regression. Los Angeles (CA): Sage Publishing; 2018. [Google Scholar]
- Frangoul H, Altshuler D, Cappellini MD, Chen Y-S, Domm J, Eustace BK, Foell J, de la Fuente J, Grupp S, Handgretinger R, et al. CRISPR-Cas9 gene editing for sickle cell disease and β-Thalassemia. N Engl J Med. 2021;384(3):252–260. doi: 10.1056/NEJMoa2031054. [DOI] [PubMed] [Google Scholar]
- Galardini M, Busby BP, Vieitez C, Dunham AS, Typas A, Beltrao P.. The impact of the genetic background on gene deletion phenotypes in Saccharomyces cerevisiae. Mol Syst Biol. 2019;15(12):e8831.doi: 10.15252/msb.20198831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiler-Samerotte KA, Zhu YO, Goulet BE, Hall DW, Siegal ML.. Selection Transforms the Landscape of Genetic Variation Interacting with Hsp90. PLoS Biol. 2016;14(10):e2000465. doi: 10.1371/journal.pbio.2000465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson G, Hogness DS.. Effect of polymorphism in the Drosophila regulatory gene Ultrabithorax on homeotic stability. Science. 1996;271(5246):200–203. doi: 10.1126/science.271.5246.200. [DOI] [PubMed] [Google Scholar]
- Gibson G, Dworkin I.. Uncovering cryptic genetic variation. Nat Rev Genet. 2004;5(9):681–690. doi: 10.1038/nrg1426. [DOI] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, Smith HO.. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6(5):343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- Gietz RD, Woods RA.. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzym. 2002;350:87–96. doi:10.1016/s0076-6879(02)50957-5. [DOI] [PubMed] [Google Scholar]
- Goldstein AL, McCusker JH.. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast. 1999;15(14):1541–1553. doi:. [DOI] [PubMed] [Google Scholar]
- Goldstein I, Ehrenreich IM.. The complex role of genetic background in shaping the effects of spontaneous and induced mutations. Yeast. 2021;38(3):187–196. doi: 10.1002/yea.3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths AJF, Doebley SR, Peichel JC, Wasserman DA.. 2020. Introduction to Genetic Analysis. New York (NY: ): Macmillian Publishers. [Google Scholar]
- Henderson CR. Best linear unbiased estimation and prediction under a selection model. Biometrics. 1975;31(2):423–447. doi: 10.2307/2529430. [DOI] [PubMed] [Google Scholar]
- Herskowitz I, Jensen RE.. [8] Putting the HO gene to work: practical uses for mating-type switching. In: Guthrie C, Fink GR, editors. Methods in Enzymology. Cambridge, MA: Academic Press; 1991. p. 132–146. [DOI] [PubMed] [Google Scholar]
- Hou J, Tan G, Fink GR, Andrews BJ, Boone C.. Complex modifier landscape underlying genetic background effects. Proc Natl Acad Sci USA. 2019;116(11):5045–5054. doi: 10.1073/pnas.1820915116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarosz DF, Taipale M, Lindquist S.. Protein homeostasis and the phenotypic manifestation of genetic diversity: principles and mechanisms. Annu Rev Genet. 2010;44:189–216. doi:10.1146/annurev.genet.40.110405.090412. [DOI] [PubMed] [Google Scholar]
- Jarosz DF, Lindquist S.. Hsp90 and environmental stress transform the adaptive value of natural genetic variation. Science. 2010;330(6012):1820–1824. doi: 10.1126/science.1195487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MS, Martsul A, Kryazhimskiy S, Desai MM.. Higher-fitness yeast genotypes are less robust to deleterious mutations. Science. 2019;366(6464):490–493. doi: 10.1126/science.aay4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan K, Tsvetanova B, Chuang R-Y, Noskov VN, Assad-Garcia N, Ma L, Hutchison Iii CA, Smith HO, Glass JI, Merryman C, et al. One step engineering of the small-subunit ribosomal RNA using CRISPR/Cas9. Sci Rep. 2016;6:30714. doi: 10.1038/srep30714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koc EC, Burkhart W, Blackburn K, Moyer MB, Schlatzer DM, Moseley A, Spremulli LL.. The large subunit of the mammalian mitochondrial ribosome. Analysis of the complement of ribosomal proteins present. J Biol Chem. 2001;276(47):43958–43969. doi: 10.1074/jbc.M106510200. [DOI] [PubMed] [Google Scholar]
- Kryazhimskiy S, Rice DP, Jerison ER, Desai MM.. Global epistasis makes adaptation predictable despite sequence-level stochasticity. Science. 2014;344(6191):1519–1522. doi: 10.1126/science.1250939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucejova B, Li L, Wang X, Giannattasio S, Chen XJ.. Pleiotropic effects of the yeast Sal1 and Aac2 carriers on mitochondrial function via an activity distinct from adenine nucleotide transport. Mol Genet Genomics. 2008;280(1):25–39. doi: 10.1007/s00438-008-0342-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Botstein D.. Mapping mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics. 1989;121(1):185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang GI, Botstein D, Desai MM.. Genetic variation and the fate of beneficial mutations in asexual populations. Genetics. 2011;188(3):647–661. doi: 10.1534/genetics.111.128942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughery MF, Hunter T, Brown A, Hoopes J, Ostbye T, Shumaker T, Wyrick JJ.. New vectors for simple and streamlined CRISPR-Cas9 genome editing in Saccharomyces cerevisiae. Yeast. 2015;32(12):711–720. doi: 10.1002/yea.3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JT, Taylor MB, Shen A, Ehrenreich IM.. Multi-locus genotypes underlying temperature sensitivity in a mutationally induced trait. PLoS Genet. 2016;12(3):e1005929.doi: 10.1371/journal.pgen.1005929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JT, Coradini ALV, Shen A, Ehrenreich IM.. Layers of cryptic genetic variation underlie a yeast complex trait. Genetics. 2019;211(4):1469–1482. doi: 10.1534/genetics.119.301907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski KA, Kaniak-Golik A, Golik P.. Maintenance and expression of the S. cerevisiae mitochondrial genome—from genetics to evolution and systems biology. Biochim Biophys Acta. 2010;1797(6–7):1086–1098. doi: 10.1016/j.bbabio.2009.12.019. [DOI] [PubMed] [Google Scholar]
- Mackay TFC. Epistasis and quantitative traits: using model organisms to study gene–gene interactions. Nat Rev Genet. 2014;15(1):22–33. doi: 10.1038/nrg3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancera E, Bourgon R, Brozzi A, Huber W, Steinmetz LM.. High-resolution mapping of meiotic crossovers and non-crossovers in yeast. Nature. 2008;454(7203):479–485. doi: 10.1038/nature07135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Ehrenreich IM.. Gene-environment interactions in stress response contribute additively to a genotype-environment interaction. PLoS Genet. 2016;12(7):e1006158.doi:10.1371/journal.pgen.1006158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullis MN, Matsui T, Schell R, Foree R, Ehrenreich IM.. The complex underpinnings of genetic background effects. Nat Commun. 2018;9(1):3548.doi: 10.1038/s41467-018-06023-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau JH. Modifier genes in mice and humans. Nat Rev Genet. 2001;2(3):165–174. doi: 10.1038/35056009. [DOI] [PubMed] [Google Scholar]
- Narasimhan VM, Hunt KA, Mason D, Baker CL, Karczewski KJ, Barnes MR, Barnett AH, Bates C, Bellary S, Bockett NA, et al. Health and population effects of rare gene knockouts in adult humans with related parents. Science. 2016;352(6284):474–477. doi: 10.1126/science.aac8624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasti RA, Voytas DF.. Attaining the promise of plant gene editing at scale. Proc Natl Acad Sci USA. 2021;118(22):e2004846117.doi: 10.1073/pnas.2004846117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paaby AB, Rockman MV.. Cryptic genetic variation: evolution’s hidden substrate. Nat Rev Genet. 2014;15(4):247–258. doi: 10.1038/nrg3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paaby AB, White AG, Riccardi DD, Gunsalus KC, Piano F, Rockman MV.. Wild worm embryogenesis harbors ubiquitous polygenic modifier variation. Elife. 2015;4:e09178. doi: 10.7554/eLife.09178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parts L, Batté A, Lopes M, Yuen MW, Laver M, San Luis B-J, Yue J-X, Pons C, Eray E, Aloy P, et al. Natural variants suppress mutations in hundreds of essential genes. Mol Syst Biol. 2021;17(5):e10138. doi: 10.15252/msb.202010138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queitsch C, Sangster TA, Lindquist S.. Hsp90 as a capacitor of phenotypic variation. Nature. 2002;417(7):618–624. [DOI] [PubMed] [Google Scholar]
- Rabiner LR. A tutorial on hidden Markov models and selected applications in speech recognition. Proc IEEE. 1989;77(2):257–286. [Google Scholar]
- Raj A, Rifkin SA, Andersen E, van Oudenaarden A.. Variability in gene expression underlies incomplete penetrance. Nature. 2010;463(7283):913–918. doi: 10.1038/nature08781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riordan JD, Nadeau JH.. From peas to disease: modifier genes, network resilience, and the genetics of health. Am J Hum Genet. 2017;101(2):177–191. doi: 10.1016/j.ajhg.2017.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford S, Lindquist S.. HSP90 as a capacitor for morphological evolution. Nature. 1998;396(6709):336–342. doi: 10.1038/24550. [DOI] [PubMed] [Google Scholar]
- Schell R, Mullis M, Ehrenreich IM.. Modifiers of the genotype–phenotype map: hsp90 and beyond. PLoS Biol. 2016;14(11):e2001015.doi: 10.1371/journal.pbio.2001015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler D. Genetic engineering and synthetic genomics in yeast to understand life and boost biotechnology. Bioengineering. 2020;7(4):137.doi: 10.3390/bioengineering7040137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shadel GS. Yeast as a model for human mtDNA replication. Am J Hum Genet. 1999;65(5):1230–1237. doi: 10.1086/302630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao H, Burrage LC, Sinasac DS, Hill AE, Ernest SR, O'Brien W, Courtland H-W, Jepsen KJ, Kirby A, Kulbokas EJ, et al. Genetic architecture of complex traits: large phenotypic effects and pervasive epistasis. Proc Natl Acad Sci USA. 2008;105(50):19910–19914. doi: 10.1073/pnas.0810388105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman F. Getting started with yeast. In: Guthrie C, Fink GR, editors.. Methods in Enzymology, Guide to Yeast Genetics and Molecular and Cell Biology—Part B. Cambridge, MA: Academic Press; 2002. p. 3–41. [DOI] [PubMed] [Google Scholar]
- Siegal ML, Leu JY.. On the nature and evolutionary impact of phenotypic robustness mechanisms. Annu Rev Ecol Evol Syst. 2014;45:496–517. doi: 10.1146/annurev-ecolsys-120213-091705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P.. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122(1):19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AP, Salvatori R, Aftab W, Aufschnaiter A, Carlström A, Forne I, Imhof A, Ott M.. Molecular connectivity of mitochondrial gene expression and OXPHOS biogenesis. Mol Cell. 2020;79(6):1051–1065.e10. doi: 10.1016/j.molcel.2020.07.024. [DOI] [PubMed] [Google Scholar]
- Steinmetz LM, Sinha H, Richards DR, Spiegelman JI, Oefner PJ, McCusker JH, Davis RW.. Dissecting the architecture of a quantitative trait locus in yeast. Nature. 2002;416(6878):326–330. doi: 10.1038/416326a. [DOI] [PubMed] [Google Scholar]
- Storey JD, Akey JM, Kruglyak L.. Multiple locus linkage analysis of genomewide expression in yeast. PLoS Biol. 2005;3(8):e267. doi: 10.1371/journal.pbio.0030267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MB, Ehrenreich IM.. Genetic interactions involving five or more genes contribute to a complex trait in yeast. PLoS Genet. 2014;10(5):e1004324.doi: 10.1371/journal.pgen.1004324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MB, Ehrenreich IM.. Higher-order genetic interactions and their contribution to complex traits. Trends Genet. 2015a;31(1):34–40. doi: 10.1016/j.tig.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MB, Ehrenreich IM.. Transcriptional derepression uncovers cryptic higher-order genetic interactions. PLoS Genet. 2015b;11(10):e1005606.doi: 10.1371/journal.pgen.1005606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MB, Phan J, Lee JT, McCadden M, Ehrenreich IM.. Diverse genetic architectures lead to the same cryptic phenotype in a yeast cross. Nat. Commun. 2016;7:11669. doi: 10.1038/ncomms11669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JR, Register E, Curotto J, Kurtz M, Kelly R.. An improved protocol for the preparation of yeast cells for transformation by electroporation. Yeast. 1998;14(6):565–571. doi:10.1002/(SICI)1097-0061(19980430)14:6<565::AID-YEA251>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Tong AH, Boone C.. Synthetic genetic array analysis in Saccharomyces cerevisiae. Methods Mol Biol. 2006;313:171–192. doi: 10.1385/1-59259-958-3:171. [DOI] [PubMed] [Google Scholar]
- Vu V, Verster AJ, Schertzberg M, Chuluunbaatar T, Spensley M, Pajkic D, Hart GT, Moffat J, Fraser AG.. Natural variation in gene expression modulates the severity of mutant phenotypes. Cell. 2015;162(2):391–402. doi: 10.1016/j.cell.2015.06.037. [DOI] [PubMed] [Google Scholar]
- Wach A, Brachat A, Pöhlmann R, Philippsen P.. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast. 1994;10(13):1793–1808. doi: 10.1002/yea.320101310. [DOI] [PubMed] [Google Scholar]
- Wagner MR, Tang C, Salvato F, Clouse KM, Bartlett A, Vintila S, Phillips L, Sermons S, Hoffmann M, Balint-Kurti PJ, et al. Microbe-dependent heterosis in maize. Proc Natl Acad Sci USA. 2021;118:e2021965118. doi: 10.1073/pnas.2021965118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreich DM, Watson RA, Chao L.. Perspective: sign epistasis and genetic constraint on evolutionary trajectories. Evolution. 2005;59(6):1165–1174. doi: 10.1554/04-272. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All raw and processed data used in this work is publicly available. Processed data tables and all code used for analyses are included in Supplementary materials S1–S9 and are available on figshare: https://doi.org/10.25386/genetics.18737102 (accessed 28 January 2022). Raw sequencing data can be accessed through Bioproject accession PRJNA739014 in the NCBI Short Read Archive.