

Abstract
Since failed resolution of inflammation is a major contributor to the progression of diabetic nephropathy, identifying endogenously generated molecules that promote the physiological resolution of inflammation may be a promising therapeutic approach for this disease. Annexin A1 (ANXA1), as an endogenous mediator, plays an important role in resolving inflammation. Whether ANXA1 could affect established diabetic nephropathy through modulating inflammatory states remains largely unknown. In the current study, we found that in patients with diabetic nephropathy, the levels of ANXA1 were upregulated in kidneys, and correlated with kidney function as well as kidney outcomes. Therefore, the role of endogenous ANXA1 in mouse models of diabetic nephropathy was further evaluated. ANXA1 deficiency exacerbated kidney injuries, exhibiting more severe albuminuria, mesangial matrix expansion, tubulointerstitial lesions, kidney inflammation and fibrosis in high fat diet/streptozotocin-induced-diabetic mice. Consistently, ANXA1 overexpression ameliorated kidney injuries in mice with diabetic nephropathy. Additionally, we found Ac2-26 (an ANXA1 mimetic peptide) had therapeutic potential for alleviating kidney injuries in db/db mice and diabetic Anxa1 knockout mice. Mechanistic studies demonstrated that intracellular ANXA1 bound to the transcription factor NF-κB p65 subunit, inhibiting its activation thereby modulating the inflammatory state. Thus, our data indicate that ANXA1 may be a promising therapeutic approach to treating and reversing diabetic nephropathy.
Keywords: Ac2-26, annexin A1, diabetic nephropathy, proresolution
Diabetic nephropathy (DN) is one of the common microvascular complications in diabetes and is the worldwide leading cause of end-stage kidney disease. The pathogenesis of DN is not fully clear yet. Growing evidence indicates that chronic low-grade inflammation is an important feature in DN.1,2 The failed resolution of inflammation is a major contributor to the continuous development of DN.3 It is, therefore, imperative to find endogenous mediators that can reduce inflammation to protect against the progression of DN.
Annexin A1 (ANXA1), a 37-kDa protein, is a member of the annexin superfamily.4 It has been demonstrated to be involved in a variety of cellular biological functions, including cell proliferation, differentiation, and apoptosis. ANXA1 ameliorates microvascular complications in type 1 diabetes, attenuates insulin resistance in experimental type 2 diabetes, and protects against the development of secondary complications.5,6 However, the functional role of ANXA1 in DN and whether ANXA1 could affect established nephropathy through modulating inflammatory states in DN remain largely unknown.
In this study, we assessed the levels of ANXA1 (or ANXA1 mRNA) in kidney biopsies from DN patients and evaluated their correlation with kidney function as well as outcomes in independent cohorts. We used Anxa1 knockout (KO) and Anxa1 transgenic (Tg) mice to investigate the role of ANXA1 in the progression of the kidney disease in diabetic mice. Furthermore, we explored the therapeutic potential of Ac2-26, an ANXA1 mimetic peptide, in treating established DN in db/db mice as well as Anxa1 KO mice. We also investigated the mechanism of how ANXA1 mediates kidney proresolution using a tubular epithelial cell culture model.
METHODS
Patients and samples
Clinical samples were collected from 3 individual cohorts. More details are provided in the Supplementary Methods.
Detection of ANXA1 in kidneys
Immunostaining of ANXA1 using monoclonal rabbit anti-ANXA1 antibody (ab214486; Abcam) was performed on paraffin-embedded kidney sections from Chinese cohort. Correlation studies were performed between ANXA1 and clinicopathologic parameters.
RNAscope in situ hybridization
RNAscope in situ hybridization was conducted using RNAscope BROWN kit (number 322300; Advanced Cell Diagnostics) for ANXA1 (number 465411; RNAscope Probe-Hs-ANXA1; Advanced Cell Diagnostics), according to the manufacturer’s instructions.
Experimental animals
Anxa1 KO mice were purchased from Cyagen Biosciences Inc. (China), which was created by transcription activator-like effector nucleases (TALEN) methods.7 Anxa1-Tg mice were purchased from Viewsolid Biotech (China). Anxa1-Tg mice were created in C57BL/6J with mice piggyBac Transposon method.8 The identification results of Anxa1 KO mice and Anxa1-Tg mice were presented in Supplementary Figure S1. More details are provided in the Supplementary Methods.
Male diabetic db/db (C57BL/6J-LepRdb/db) mice and their nondiabetic db/m (C57BL/6J-LepRdb/+) littermates were purchased from Department of Laboratory Animal Science, Peking University Health Science Center (Beijing, China). All animal procedures were approved by the Ethics Committee of Peking University Health Science Center, and the ethics license number is LA2011-046.
Animal studies
To determine the role of endogenous ANXA1 in a model of DN, wild-type (WT), Anxa1 KO mice, and Anxa1-Tg mice were induced to develop DN with the high-fat diet (HFD) plus streptozotocin (STZ) treatment. Ac2-26 treatment was performed on db/db mice and Anxa1 KO mice. The details of the animal studies are provided in the Supplementary Methods.
Blood and urine examination
The concentrations of urinary and plasma ANXA1, urine albumin, urine creatinine, serum creatinine, blood urea nitrogen, blood glucose, cholesterol, and triglyceride were measured following standard protocols. More details are provided in the Supplementary Methods.
Glucose tolerance test and insulin tolerance test
The glucose tolerance test and insulin tolerance test were performed following standard protocols. More details are provided in the Supplementary Methods.
Histologic analysis
Paraffin-embedded kidney tissue sections were stained with periodic acid–Schiff solution (BA-4080B; BASO) to assess glomerular mesangial matrix expansion, sclerosis, and tubulointerstitial injury, as described previously.9–11 Detailed protocols are described in the Supplementary Methods.
Immunohistochemistry and immunofluorescence
Detailed protocols are described in the Supplementary Methods.
Transmission electron microscopy
Electron microscopic sample handling and detection were performed by the Electron Microscopic Central Laboratory of Peking University First Hospital. Detailed protocols are described in the Supplementary Methods.
Real-time quantitative polymerase chain reaction analysis
Primers used for mRNA detection are listed in Supplementary Table S1. More details are provided in the Supplementary Methods.
Flow cytometry
Flow cytometry was performed following standard protocols. Additional details are provided in the Supplementary Methods.
Cell culture and treatment
Human immortalized proximal tubule epithelial HK-2 cells were purchased from American Type Culture Collection. Detailed treatment protocols are provided in the Supplementary Methods.
Western blot
Western blot was performed following standard protocols. Additional details are provided in the Supplementary Methods.
Coimmunoprecipitation
Coimmunoprecipitation was performed following the manufacturer’s instructions. Additional details are provided in the Supplementary Methods.
Nuclear factor-κB p65 activity
Following treatment, nuclear extracts were isolated using the Nuclear Extract kit, according to the manufacturer’s protocols (number 40010; Active Motif). The activity of nuclear factor (NF)-κB p65 was assessed using an enzyme-linked immunosorbent assay kit (number 43296; Active Motif).
Electrophoretic mobility shift assay
Gel shift assays were performed with the LightShift chemiluminescent electrophoretic mobility shift assay kit (number 20148; ThermoFisher Scientific). Additional details are provided in the Supplementary Methods.
Surface plasmon resonance analysis
Surface plasmon resonance measurements were performed using a Biacore T200 instrument (GE Healthcare). Detailed protocols are described in the Supplementary Methods.
Statistical analysis
Data were presented as mean ± SD or median and interquartile range, as appropriate. Analytical details are provided in the Supplementary Methods. P < 0.05 indicates statistically significant differences.
RESULTS
Kidney cortical ANXA1 is significantly elevated in DN
To detect ANXA1 protein levels in kidney tissues in human DN, we compared the levels of ANXA1 in kidney biopsies between DN patients and healthy controls from the Chinese cohort by immunohistochemical staining (Figure 1a; Supplementary Table S2 for cohort details). Kidney biopsies from DN patients showed more intense staining of ANXA1 in the glomeruli and tubulointerstitium than healthy controls (Figure 1b and c). Correlation analyses showed that the tubulointerstitial ANXA1 expression positively correlated with serum creatinine (r = 0.473, P = 0.008; Figure 1d), 24-hour urinary protein (r = 0.529, P = 0.003; Figure 1e), and tubulointerstitial inflammation (r = 0.431, P = 0.017). Tubulointerstitial ANXA1 also showed a negative correlation with estimated glomerular filtration rate (r = −0.464, P = 0.010; Figure 1f). The glomerular ANXA1 negatively correlated with the percentage of glomerulosclerosis (r = −0.453, P = 0.012; Figure 1g). To further validate these results, we evaluated ANXA1 mRNA expression in an independent cohort. Consistently, ANXA1 mRNA (log2) levels were significantly higher in patients with DN from the European Renal cDNA Biobank than those in healthy living donors in both tubulointerstitial and glomerular compartment (Supplementary Figure S2; Supplementary Table S3 for cohort details). In addition, compared with nondiabetic mice, diabetic mice demonstrated a significant increase in ANXA1 levels in kidneys (Supplementary Figure S3).
Figure 1 |. Annexin A1 (ANXA1) in kidney biopsies from patients with diabetic nephropathy (DN).
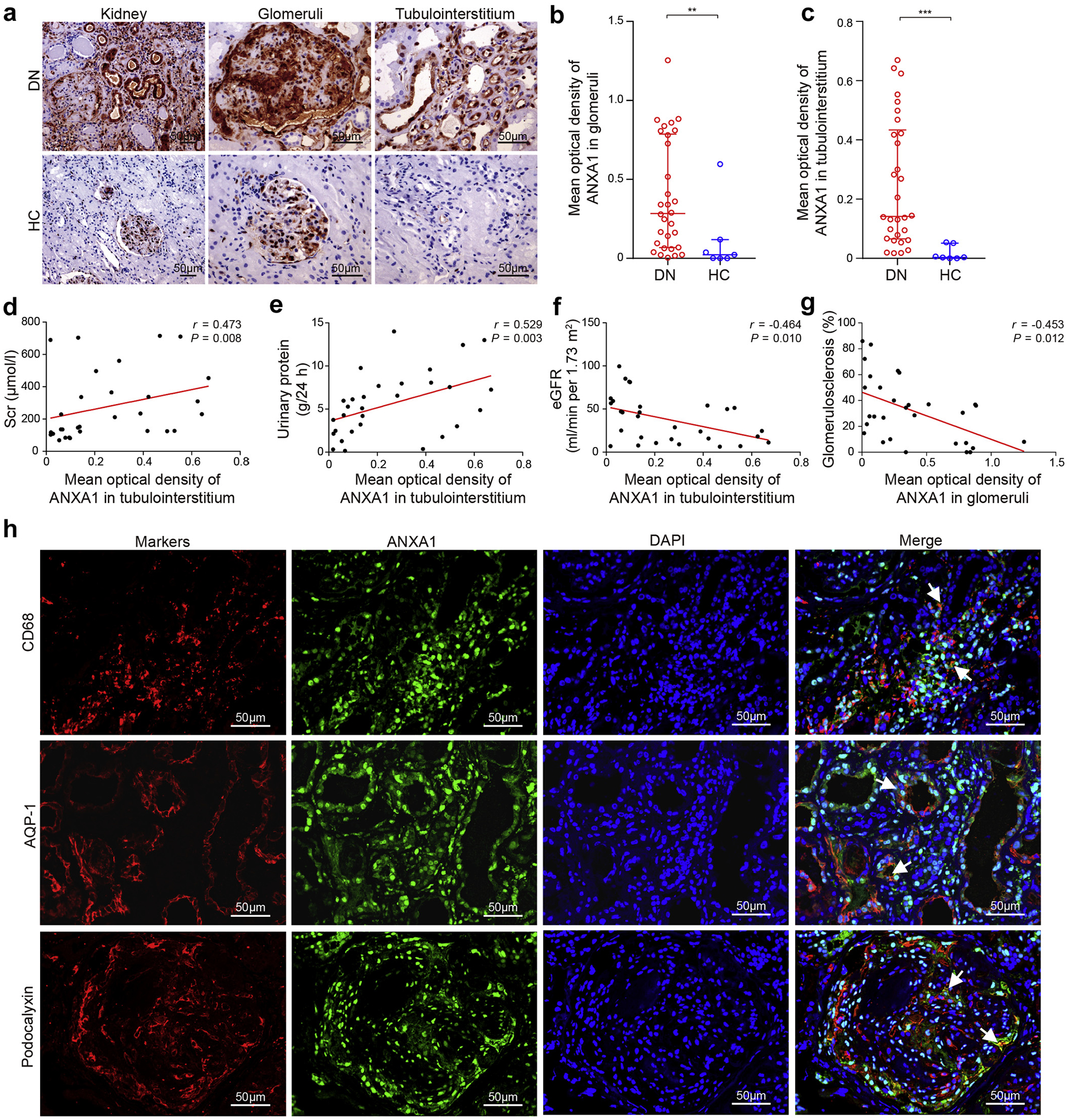
(a) Representative images of ANXA1 staining in human kidney cortical tissue, in the glomeruli and tubulointerstitium from DN and control in the Chinese cohort. Bars = 50 μm. (b) Glomerular ANXA1 staining in DN patients (n = 30) was significantly higher than that in controls (n = 7). (c) Tubulointerstitial ANXA1 staining in DN patients (n = 30) was significantly higher than that in controls (n = 7). Tubulointerstitial ANXA1 expression was positively correlated with (d) serum creatinine (Scr) and (e) 24-hour urinary protein in DN patients. (f) Tubulointerstitial ANXA1 expression was negatively correlated with estimated glomerular filtration rate (eGFR) in DN patients. (g) Glomerular ANXA1 expression was negatively correlated with the percentage of glomerulosclerosis. (h) Representative confocal microscopic images showed the expression of ANXA1 in macrophages (cluster of differentiation [CD] 68), proximal tubule epithelial cells (aquaporin 1 [AQP-1]), and podocytes (podocalyxin) in kidneys from DN. Bars = 50 μm. Data analyses were performed by Mann-Whitney U test for 2 groups. Correlations were determined by Spearman analysis. Data are expressed as median (interquartile range). **P < 0.01, and ***P < 0.001. DAPI, 4′,6-diamidino-2-phenylindole; HC, healthy control. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
To investigate the association between ANXA1 and kidney outcomes in DN patients, we used kidney biopsy samples from a Pima Indian patient population with long-term follow-up data (Supplementary Table S4 for cohort details).12 We found that lower level of ANXA1 mRNA at the time of biopsy was significantly associated with an increased risk of progression to a composite end point of end-stage kidney disease or 40% decline of baseline GFR measured by iothalamate clearance (n = 49 events) during a follow-up of 9.0 ± 4.0 years (log-rank test, P = 0.049; Supplementary Figure S4). These data suggested that higher expression of ANXA1 may be a protective factor for DN progression.
Immunofluorescence colocalization analyses showed that ANXA1 partially colocalized with tubular epithelial cells, podocytes, mesangial cells, endothelial cells, and monocytes/macrophages in kidneys of DN patients (Figure 1h and Supplementary Figure S5). To determine the major sources of ANXA1 in renal samples, in situ hybridization by RNAscope was performed. In biopsy samples from patients with DN, in situ hybridization showed that ANXA1 was mainly expressed in parietal epithelial cells and tubular epithelial cells (Supplementary Figure S6). However, there was only a minor amount of ANXA1 expression in the biopsy samples from healthy controls.
Moreover, we measured the circulating and urinary levels of ANXA1 in DN patients, diabetes mellitus patients without kidney involvement, and healthy controls (Supplementary Table S5 for general data). The level of urinary ANXA1/creatinine in DN patients was significantly higher than that in diabetes mellitus patients without kidney involvement (5.96 ± 2.42 vs. 2.30 ± 0.76 ng/mg; P < 0.001) and healthy controls (5.96 ± 2.42 vs. 1.29 ± 0.62 ng/mg; P < 0.001; Supplementary Figure S7A). The levels of urinary ANXA1/creatinine were positively correlated with serum creatinine (r = 0.454, P = 0.012) and urinary protein (r = 0.417, P = 0.022), and were negatively correlated with estimated glomerular filtration rate (r = −0.431, P = 0.017). In addition, urinary ANXA1 levels showed a positive correlation with the severity of interstitial fibrosis and tubular atrophy and tubulointerstitial inflammation (r = 0.405, P = 0.026; and r = 0.446, P = 0.013, respectively). The level of plasma ANXA1 in DN patients was significantly lower than that in diabetes mellitus patients without kidney involvement (1.22 ± 0.70 vs. 3.64 ± 2.16 ng/ml; P < 0.001) and healthy controls (1.22 ± 0.70 vs. 3.20 ± 1.35 ng/ml; P < 0.001; Supplementary Figure S7b). However, we did not find any significant correlation between plasma ANXA1 levels and clinical parameters, suggesting that urinary ANXA1/creatinine is a better reflection of kidney inflammation and kidney injuries in DN.
ANXA1 deficiency exacerbates DN in HFD/STZ-treated mice
To determine the role of endogenous ANXA1 in a model of DN, WT and Anxa1 KO mice were induced to develop DN with HFD/STZ treatment (Figure 2a).
Figure 2 |. Annexin A1 (Anxa1) knockout (KO) aggravated nephropathy in high-fat diet (HFD) plus streptozotocin (STZ)-induced diabetic nephropathy mice.
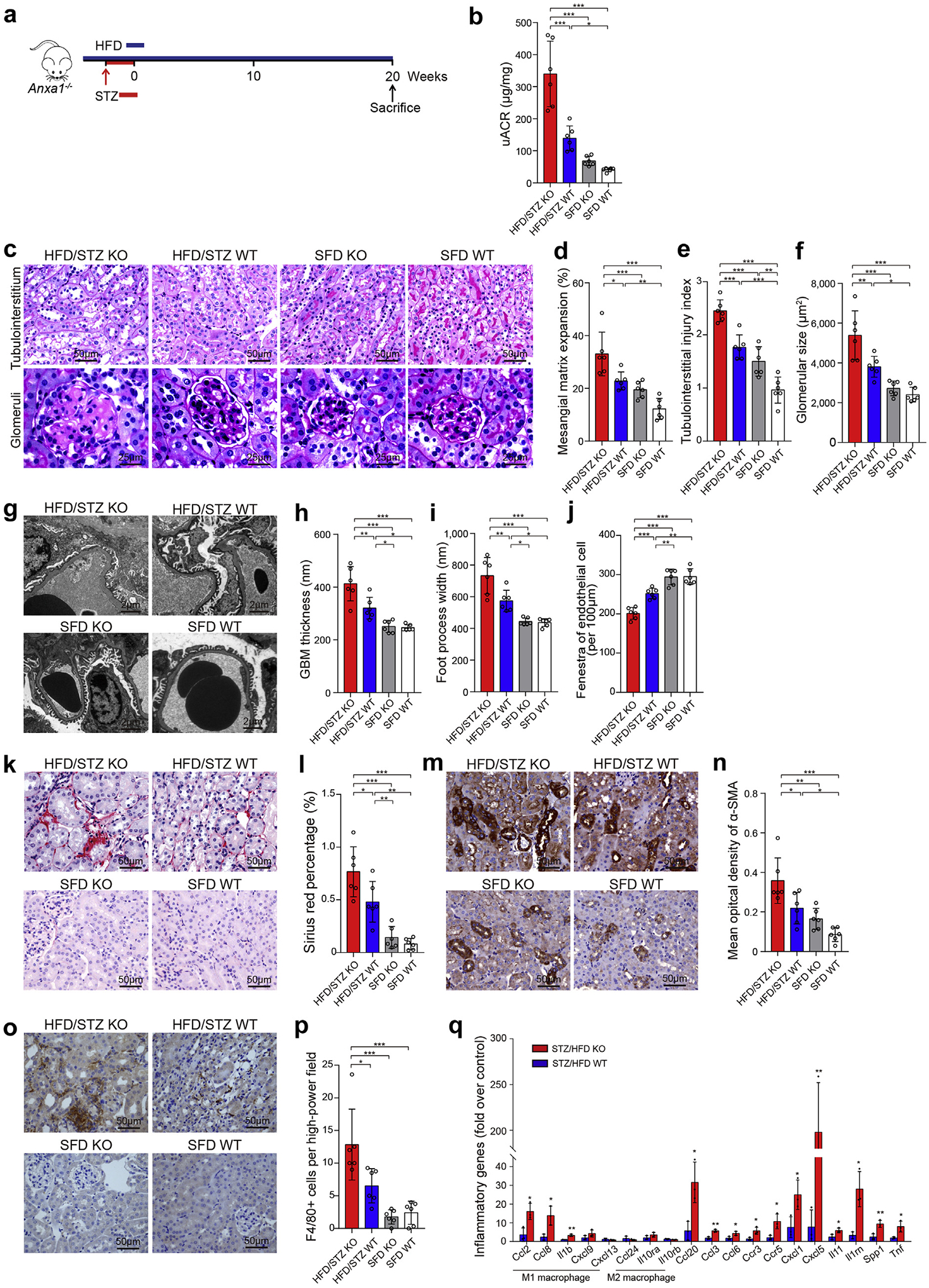
(a) Study design overview. Eight-week-old Anxa1 KO mice were fed with an HFD for 1 month, followed by i.p. injection of 50 mg/kg STZ for 5 days. Mice were maintained for 20 weeks of HFD feeding before sacrifice. n = 6 per group. (b) Four groups were evaluated for urine albumin-to-creatinine ratio (uACR). (c) Representative photomicrographs of periodic acid–Schiff staining. Bar = 50 μm for tubulointerstitium and 25 μm for glomeruli. Quantification showing (d) mesangial matrix expansion, (e) tubulointerstitial injury index, and (f) glomerular size. (g) Representative photomicrographs of transmission electron microscopy. Bars = 2 μm. Quantification of (h) mean glomerular basement membrane (GBM) thickness, (i) mean foot process width, and (j) endothelial fenestrations in 4 groups. (k) Representative photomicrographs of Sirius red. Bars = 50 μm. (l) Quantitative analysis of Sirius red. (m) Representative photomicrographs of α-smooth muscle actin (α-SMA). Bars = 50 μm. (n) Quantitative analysis of α-SMA. (o) Representative photomicrographs of F4/80 staining. Bars = 50 μm. (p) Quantitative analysis of F4/80 staining. (q) Quantification of inflammatory gene expression by polymerase chain reaction array, HFD/STZ Anxa1 KO versus HFD/STZ wild-type (WT) mice (n = 3). Throughout, data analyses were performed by Student t test for 2 groups and 2-way analysis of variance, followed by a Tukey test, for 4 groups. Data are expressed as mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001. SFD, standard fat diet. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
As shown in Supplementary Table S6, diabetic WT mice showed hyperglycemia and hyperlipemia compared with nondiabetic WT mice. Diabetic WT mice displayed increased levels of urine albumin-to-creatinine ratio (uACR), which was further increased significantly in diabetic Anxa1 KO mice (Figure 2b). Consistent with impaired kidney function, histologic changes, including glomerular size, mesangial matrix expansion, and tubulointerstitial injury, were more severe in diabetic Anxa1 KO mice than diabetic WT mice (Figure 2c–f). The glomerular basement membrane thickness and foot process width were also significantly greater in diabetic Anxa1 KO mice, along with loss of endothelial fenestrations (Figure 2g–j).
Sirius red staining showed an increased deposition of collagen I and III in diabetic WT mice, whereas kidneys from diabetic Anxa1 KO mice had significantly increased staining, especially in the interstitium and around the vasculature (Figure 2k and l). A similar trend in α-smooth muscle actin accumulation was detected (Figure 2m and n). Moreover, our results showed a significantly higher level of macrophage infiltration in diabetic Anxa1 KO mice than in diabetic WT mice (Figure 2o and p).
We used polymerase chain reaction array to compare the expression of inflammatory cytokines and receptor genes in kidneys between diabetic Anxa1 KO and diabetic WT group (Supplementary Table S7). The expression of inflammatory cytokines was significantly upregulated (P < 0.05) in diabetic Anxa1 KO mice, including chemokine (C-C motif) ligand 2, 3, 6, 8, and 20; chemokine receptor 3 and 5; chemokine (C-X-C motif) ligand 1 and 5; interleukin 11, 1β, and 1rn (Il-11, Il-1β, and Il-1rn); secreted phosphoprotein 1 (Spp1); and tumor necrosis factor (Tnf), compared with diabetic WT mice (Figure 2q). Among these inflammatory cytokine genes, the expression of M1 macrophage markers (Ccl2, Ccl8, and Il-1β) was upregulated significantly in diabetic Anxa1 KO mice compared with diabetic WT mice, although there was no significant difference in the expression of M2 macrophage markers (Il-10 and Ccl24) between these 2 groups. Moreover, flow cytometry analyses further confirmed that the number of M1 macrophages increased in the kidneys in diabetic Anxa1 KO mice compared with diabetic WT mice, although the number of M2 macrophages did not have a significant difference between these 2 groups, suggesting that ANXA1 deletion increased M1 polarization in DN (Supplementary Figure S8).
ANXA1 deficiency causes kidney injuries in HFD-treated mice
We utilized the murine model of HFD-induced diabetes to confirm the protective role of ANXA1 in the kidney (Figure 3a). Mice treated with HFD for 4 months presented with obesity, impaired glucose tolerance, and acquired insulin resistance (Supplementary Figure S9). Mice treated with HFD did not develop obvious kidney injury on the C57BL/6J background, as revealed by periodic acid–Schiff staining (Figure 3b–d), which is consistent with the previous studies.13,14 Ultrastructural analysis demonstrated that HFD-treated Anxa1 KO mice developed more severe kidney injuries, as evidenced by glomerular basement membrane thickness and podocyte foot process width, compared with the other 3 groups (Figure 3e–g). Podocyte injuries in HFD-treated Anxa1 KO mice were further confirmed by a significant loss of key podocyte differentiation markers, including nephrin and podocin (Figure 3h–j). In addition, macrophage infiltration marginally increased (Figure 3k and l), and Il-1β mRNA levels significantly increased, in diabetic Anxa1 KO mice compared with diabetic WT mice (Figure 3m). These results further suggested that ANXA1 deficiency may cause kidney injuries in diabetes.
Figure 3 |. Annexin A1 (Anxa1) knockout (KO) caused kidney injuries in high-fat diet (HFD)–induced diabetic mice.
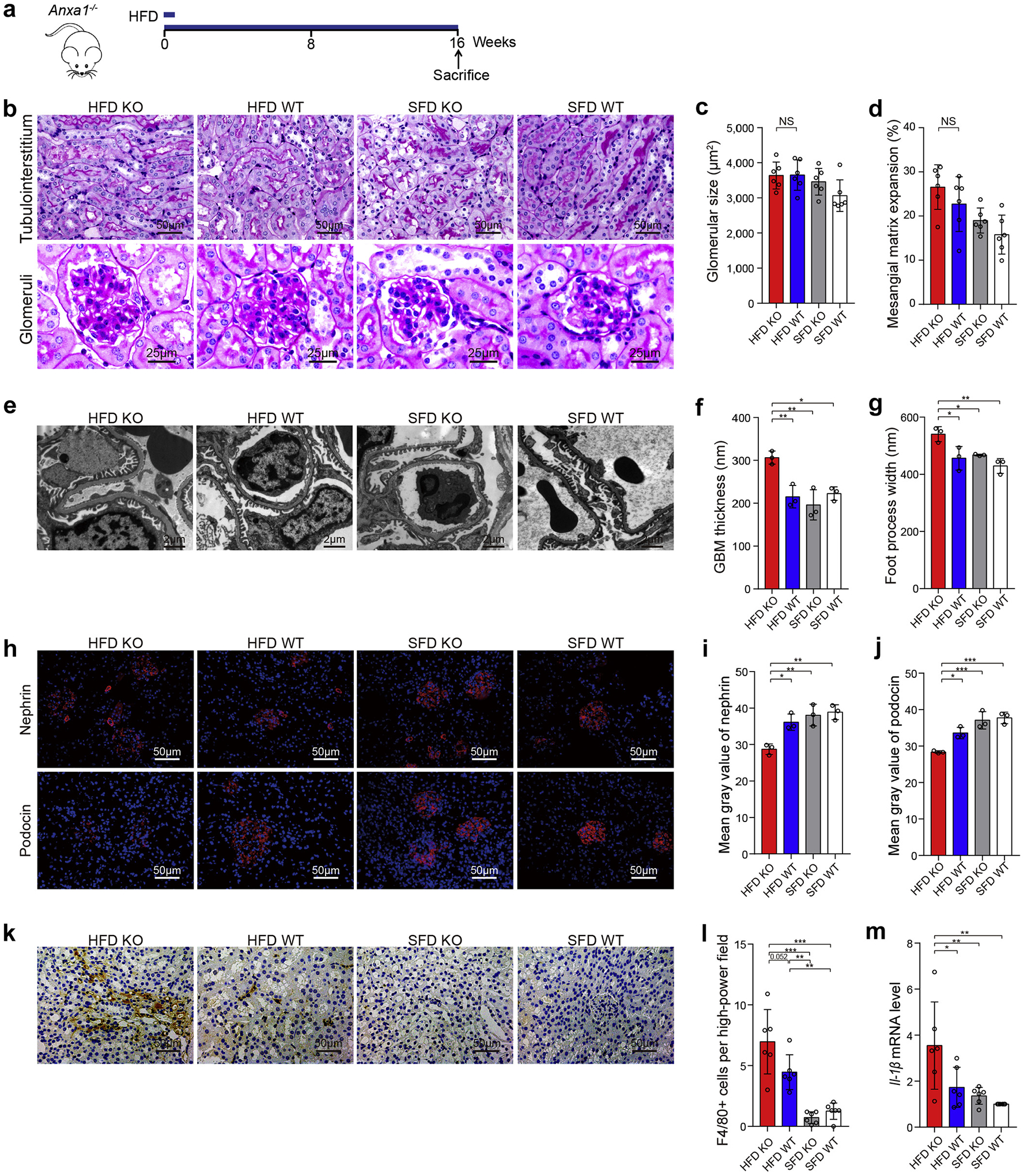
(a) Study design overview. Anxa1 KO mice were maintained for 16 weeks of HFD feeding. (b) Representative photomicrographs showing typical tubulointerstitial (bars = 50 μm) and glomerular structure (bars = 25 μm) in control and diabetic mice. Quantification showing (c) glomerular size and (d) mesangial matrix expansion (n = 6). (e) Representative transmission electron microscopy images in kidney cortex tissue isolated from diabetic Anxa1 KO and wild-type (WT) mice as well as nondiabetic mice. Bars = 2 μm. Quantification of (f) mean glomerular basement membrane (GBM) thickness and (g) mean foot process width in 4 groups (n = 3). (h) Representative microscopic images showing the expressions of nephrin and podocin in the kidney from different groups of mice. Bars = 50 μm. Quantitative analysis of (i) nephrin and (j) podocin (n = 3). (k) Representative photomicrographs of F4/80 staining. Bars = 50 μm. (l) Quantitative analysis of F4/80 staining. (m) Quantification of Il-1β gene expression by real-time polymerase chain reaction among 4 groups (n = 6). Throughout, data analyses were performed by 2-way analysis of variance, followed by a Tukey test, for 4 groups and expressed as mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001. SFD, standard fat diet. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
Overexpression of ANXA1 ameliorates DN in HFD/STZ-treated mice
To assess the protective role of endogenous ANXA1, we treated Anxa1-Tg mice and WT mice with HFD/STZ to promote kidney injuries of diabetes (Figure 4a). There was no significant difference between diabetic Anxa1-Tg mice and diabetic WT mice in blood glucose, body weight, or kidney weight (data not shown). Marked decrease in uACR was observed in diabetic Anxa1-Tg mice compared with diabetic WT mice (Figure 4b). HFD/STZ treatment caused increases in glomerular area, mesangial expansion, and tubulointerstitial injuries in WT mice, as revealed by periodic acid–Schiff staining, and the severity of these injuries was markedly attenuated in diabetic Anxa1-Tg mice (Figure 4c–e). In addition, HFD/STZ-induced kidney tubulointerstitial fibrosis (Figure 4f–i) and inflammation, including macrophage infiltration (Figure 4j and k) and proinflammatory cytokine expression (Supplementary Figure S10A), were significantly attenuated in Anxa1-Tg mice. Anxa1 overexpression also improved ultrastructural changes, including foot process width and glomerular basement membrane thickness in HFD/STZ-treated mice (Supplementary Figure S10B). These findings suggested that overexpression of Anxa1 protected the kidney against DN, possibly through regulation of inflammatory and fibrotic processes in the kidney.
Figure 4 |. Overexpression of annexin A1 (Anxa1) suppresses diabetic nephropathy.
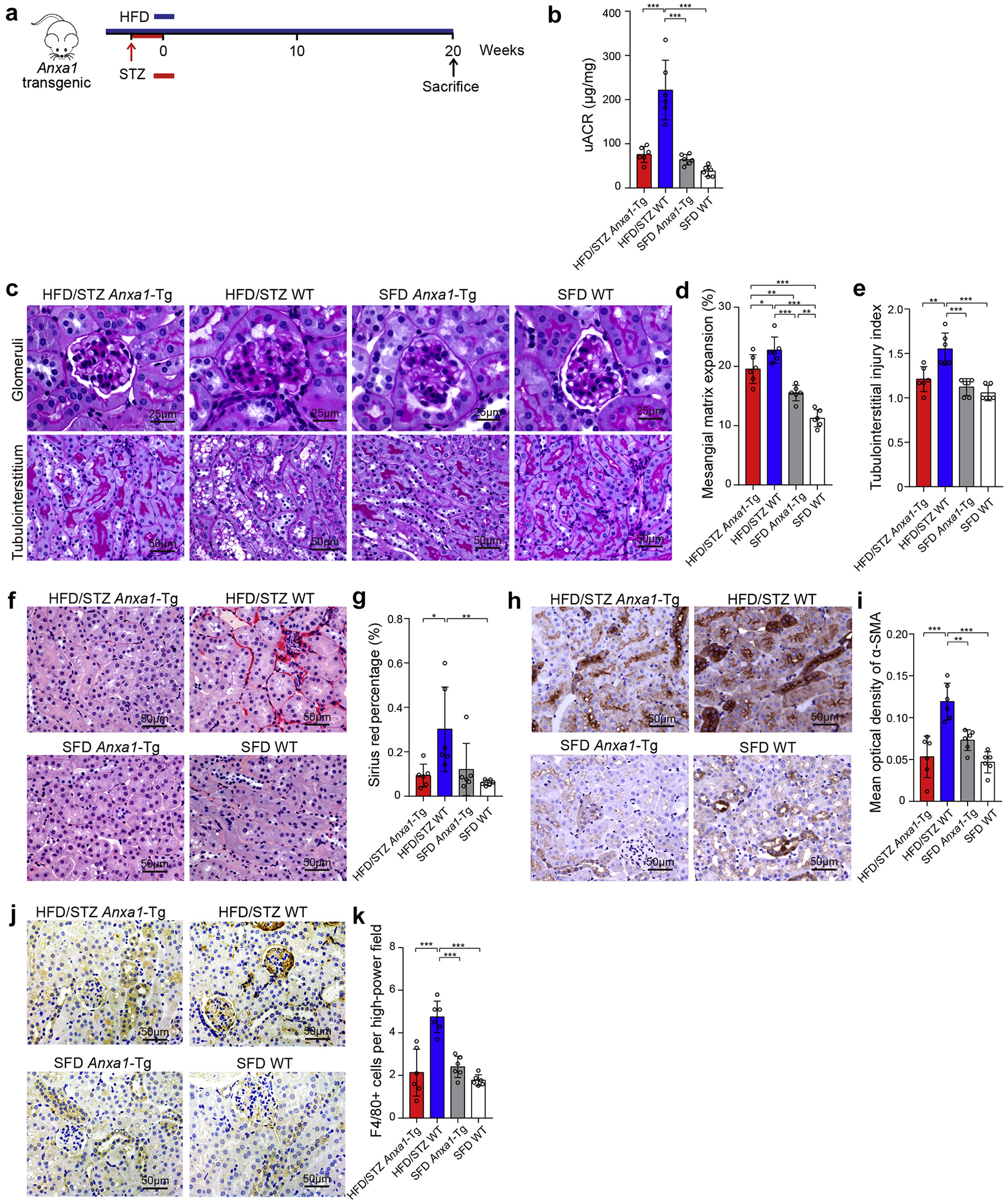
(a) Study design overview. Anxa1 transgenic (Anxa1-Tg) mice and wild-type (WT) mice were fed with a high-fat diet (HFD) for 1 month, followed by a daily i.p. injection of 50 mg/kg streptozotocin (STZ) for 5 days. Mice were maintained for 20 weeks on an HFD before sacrifice. n = 6 per group. (b) Urine albumin-to-creatinine ratio (uACR) in 4 groups. (c) Representative photomicrographs of periodic acid–Schiff staining. Bars = 50 μm for tubulointerstitium and 25 μm for glomeruli. Quantification showing (d) mesangial matrix expansion and (e) tubulointerstitial injury index. (f) Representative photomicrographs of Sirius red. Bars = 50 μm. (g) Quantitative analysis of Sirius red. (h) Representative photomicrographs of α-smooth muscle actin (α-SMA). Bars = 50 μm. (i) Quantitative analysis of α-SMA. (j) Representative photomicrographs of F4/80 staining. Bars = 50 μm. (k) Quantitative analysis of F4/80 staining. Data represent mean ± SD. Data analyses were performed by 2-way analysis of variance, followed by a Tukey test, for 4 groups. *P < 0.05, **P < 0.01, and ***P < 0.001. SFD, standard fat diet. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
Ac2-26 attenuates established nephropathy in a genetic model of diabetes
At the age of 10 weeks, all db/db mice had already developed diabetes, with significantly higher body weight and blood glucose than nondiabetic db/m mice. These diabetic mice also developed significantly higher levels of uACR compared with db/m mice (361.04 ± 83.84 vs. 58.33 ± 4.15 μg/mg; P < 0.001; n = 6). To investigate whether Ac2-26 could attenuate established nephropathy in diabetes, we randomly divided db/db mice into treatment arms receiving different doses of Ac2-26. Db/db mice were treated with Ac2-26 for 10 consecutive weeks (Figure 5a). The level of uACR in vehicle-treated db/db mice increased compared with the baseline, whereas Ac2-26 treatment groups presented with significant decrease in uACR compared with the baseline (Figure 5b). In addition, diabetes-induced mesangial matrix expansion and tubulointerstitial injuries were significantly attenuated by Ac2-26 (Figure 5c–e). We assessed kidney inflammation and found an increased level of macrophage infiltration in kidney tissues in vehicle-treated db/db mice compared with vehicle-treated db/m mice. Ac2-26 treatment in db/db mice significantly attenuated the degree of macrophage infiltration, compared with vehicle-treated db/db mice (Figure 5f and g). Furthermore, Ac2-26 treatment groups showed decreased levels of proinflammatory cytokines and chemokines, including Mcp-1, Il-1α, and Tnf-α, in kidneys (Figure 5h–j). Sirius red staining showed that medium and high dose of Ac2-26 treatment could attenuate kidney fibrosis (Figure 5k and l). Transmission electron microscopy analysis showed that Ac2-26 treatment groups alleviated ultrastructural changes, including glomerular basement membrane thickness, podocyte foot process width, and endothelial fenestrations, compared with vehicle-treated db/db mice (Figure 5m–p).
Figure 5 |. Ac2-26 administration treats established nephropathy in db/db mice.
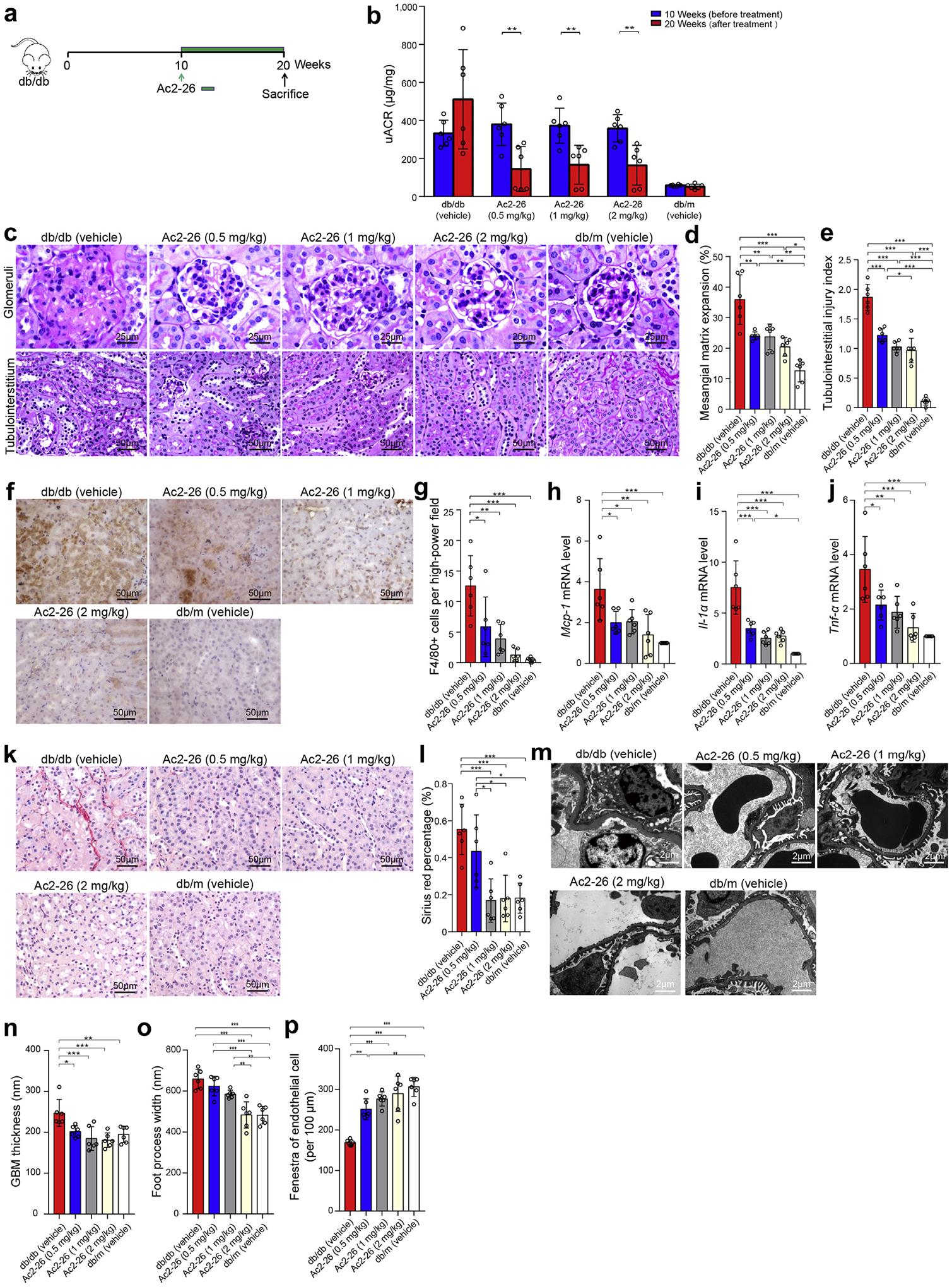
(a) Ac2-26 treatment protocol for db/db mice. Diabetic db/db mice were allowed to progress for 10 weeks, after which the subgroups of diabetic mice were administered different concentrations of Ac2-26 (0.5, 1, or 2 mg/kg), or phosphate-buffered saline every 2 days i.p. from weeks 10 to 20. n = 6 per group. (b) The levels of urine albumin-to-creatinine ratio (uACR) at 10 and 20 weeks (n = 6). (c) Representative photomicrographs of periodic acid–Schiff staining. Bars = 50 μm for tubulointerstitium and 25 μm for glomeruli. Quantification showing (d) mesangial matrix expansion and (e) tubulointerstitial injury index. (f) Representative photomicrographs of F4/80 staining. Bars = 50 μm. (g) Quantitative analysis of F4/80 staining. Quantification of (h) Mcp-1, (i) Il-1α, and (j) Tnf-α gene expression by real-time polymerase chain reaction among 4 groups. (k) Representative photomicrographs of Sirius red. Bars = 50 μm. (l) Quantitative analysis of Sirius red. (m) Representative transmission electron microscopy images. Bars = 2 μm. Quantification of (n) mean glomerular basement membrane (GBM) thickness, (o) foot process width, and (p) endothelial fenestrations (n = 6). Data analyses were performed by Student t test for 2 groups and 1-way analysis of variance, followed by a Tukey test, for multiple groups. *P < 0.05, **P < 0.01, and ***P < 0.001. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
To determine whether an exogenous supply of ANXA1 mimic peptide could reverse the kidney injuries in diabetic Anxa1 KO mice, Ac2-26 treatment was performed on HFD/STZ-treated diabetic Anxa1 KO mice. Compared with diabetic Anxa1 KO mice without Ac2-26 administration, diabetic Anxa1 KO mice with Ac2-26 administration had significant improvements in albuminuria and kidney pathologic changes (Supplementary Figure S11). These results were consistent with the above-mentioned data of the protective effect of Ac2-26 treatment in db/db mice.
Collectively, these data suggested that Ac2-26 administration could attenuate kidney injuries in diabetes.
ANXA1 binds to the p65 subunit of NF-κB and inhibits its activation in HK-2 cells
Because we found that ANXA1 colocalized with the tubular epithelial cell in kidney specimens of DN patients, we stimulated proximal tubular epithelial HK-2 cells with high glucose plus palmitic acid (HGPA) to simulate the in vivo environment of diabetes. Compared with HK-2 cells without treatment, the ANXA1 mRNA expression increased in cells exposed to HGPA (Supplementary Figure S12). ANXA1 knockdown (small, interfering RNA [siRNA] against ANXA1 [siANXA1]) could upregulate the proinflammatory factors in response to HGPA treatment, including IL-1β, TGF-β, and TNF (Figure 6a–c).
Figure 6 |. Annexin A1 (ANXA1) binds to the p65 subunit of nuclear factor (NF)–κB and inhibits its activation in HK-2 cells.
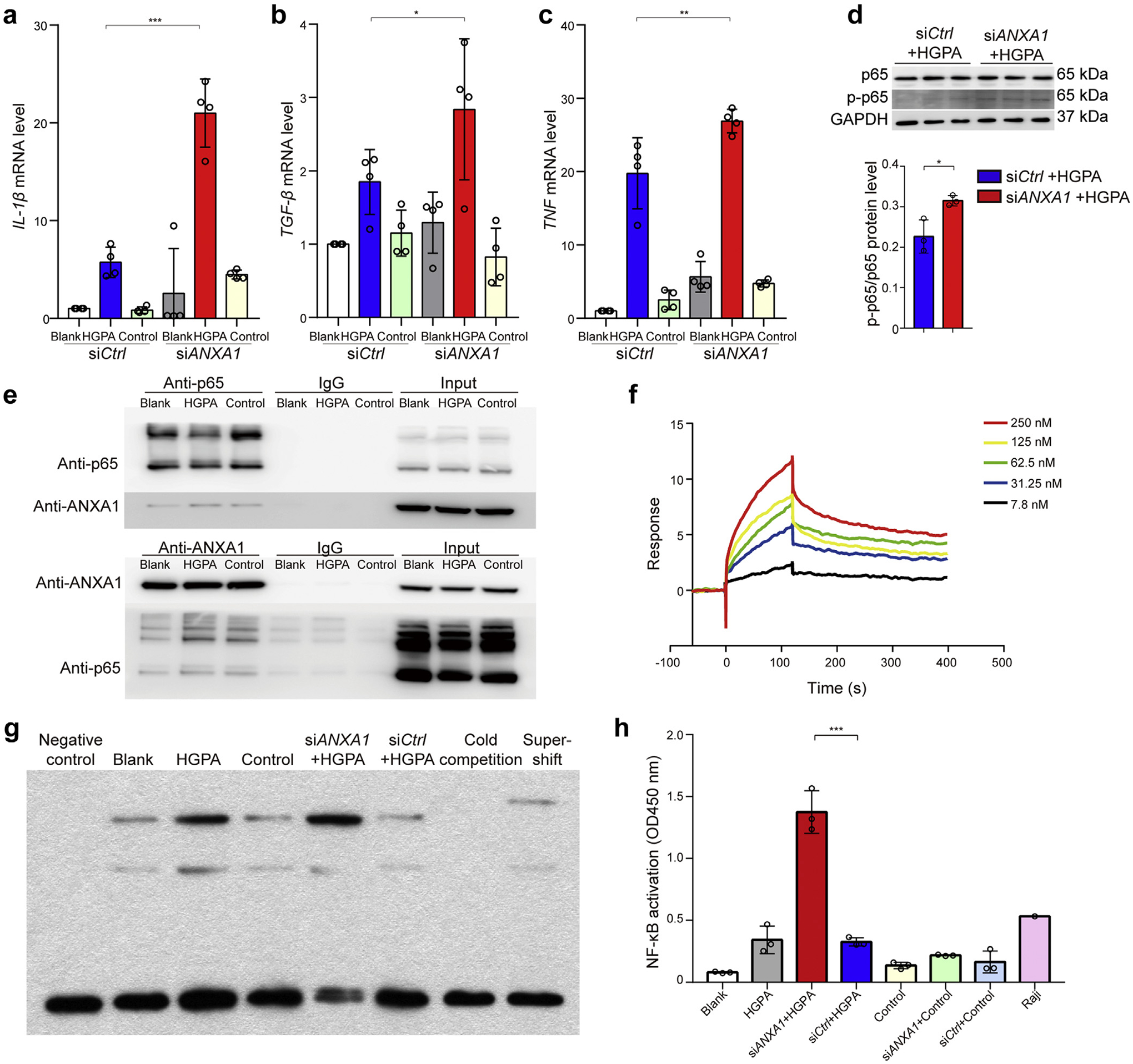
Quantitative real-time polymerase chain reaction analyzed (a) Il-1β, (b) TGF-β, and (c) TNF mRNA level in HK-2 cells transfected with ANXA1 small, interfering RNA (siRNA) under high glucose plus palmitic acid (HGPA) conditions. n = 4 per group. (d) Top: Representative Western blot images. Bottom: Analyses of phosphorylated p65 (p-p65) and p65 in HK-2 cells with treatment. n = 3 per group. (e) Top: Total protein lysates of HK-2 cells treated with HGPA or siRNA against ANXA1 (siANXA1) were immunoprecipitated using an antibody against the p65 subunit of NF-κB and immunoblotted (IB) against ANXA1. Bottom: Total protein lysates of HK-2 cells treated with HGPA or siANXA1 were immunoprecipitated using an antibody against ANXA1 and IB against the p65 subunit of NF-κB. (f) The surface plasmon resonance (SPR) curves generated from ANXA1 binding to p65. The ANXA1 protein concentration ranged from 7.8 to 250 nM. The apparent dissociation constants for the interaction were obtained from fitting the SPR response curves to a simple 1:1 Langmuir binding model. (g) Electrophoretic mobility shift assay showed the binding of NF-κB to the DNA probe in HK-2 cells under different treatments. (h) Enzyme-linked immunosorbent assay showed the NF-κB activity as indicated. n = 3 per group. Data analysis was performed by Student t test for 2 groups and 2-way analysis of variance, followed by a Tukey test, for multiple groups and expressed as mean ± SD. The blank group served as the normal control group. The control group served as the isotonic solvent control group using mannitol and endotoxin-free bovine serum albumin. *P < 0.05, **P < 0.01, and ***P < 0.001. GAPDH, glyceraldehyde 3-phosphate dehydrogenase; siCtrl, negative control siRNA.
The transcription factor NF-κB plays an important role in the process of inflammation.15,16 Western blot analysis suggested that siANXA1 led to an increase in the expression of NF-κB phosphorylated p65 in HK-2 cells treated with HGPA (Figure 6d), consistent with the in vivo results in mouse models (Supplementary Figure S13). However, the phosphorylation level of IκBα was not significantly increased in siANXA1 group treated with HGPA compared with negative control siRNA group treated with HGPA (Supplementary Figure S14). To explore the binding of ANXA1 and p65, we performed coimmunoprecipitation of ANXA1 and p65 in HK-2 cells as well as in kidney tissues of HFD/STZ WT mice. The results showed that ANXA1 physically interacted with p65 both in vitro and in vivo (Figure 6e and Supplementary Figure S15). Moreover, surface plasmon resonance analysis demonstrated that ANXA1 exhibited a high binding affinity for p65 (equilibrium dissociation constant [KD] = 1.299 × 10−8 mol/L; Figure 6f). To determine whether there was any functional consequence to the coexpression, the electrophoretic mobility shift assay test was performed using nuclear extracts from HK-2 cells. As shown in Figure 6g, HGPA increased the binding of NF-κB to the κB probe. Compared with the negative control siRNA group treated with HGPA, the binding activity of NF-κB to the κB oligomer was higher in the siANXA1 group treated with HGPA. Consistent with the electrophoretic mobility shift assay analysis, the NF-κB activity, assessed by enzyme-linked immunosorbent assay, was significantly increased in the siANXA1 group treated with HGPA compared with negative control siRNA group treated with HGPA (Figure 6h). Altogether, these findings suggested that ANXA1 could associate with p65 to prevent p65 phosphorylation and NF-κB activation in HK-2 cells.
In addition, we explored the potential protection of ANXA1 mimetic peptide Ac2-26 in HK-2 cells. We found Ac2-26 (15 μmol/L) decreased HGPA-induced cytokine release (Supplementary Figure S16A). Ac2-26, as an exogenous administration of peptide, is not able to cross the cell membrane and is thus unlikely to bind to NF-κB p65, like ANXA1 protein, which was consistent with surface plasmon resonance analysis (Supplementary Figure S16B). To determine the potential pharmacologic targeting of Ac2-26, lipoxin A(4)/formyl peptide receptor 2 (ALX/FPR2) selective antagonist Trp-Arg-Trp-Trp-Trp-Trp (WRW4), selective FPR1 antagonist N-(tert-butoxycarbonyl)-Met-Leu-Phe (Boc-MLF) (1 μmol/L), or pan-FPR antagonist Boc-MLF (10 μmol/L) was added before Ac2-26 and HGPA. We found that it was mainly FPR1, not ALX/FPR2, that plays the role of mediating Ac2-26 effects on proresolution in HK-2 cells (Supplementary Figure S16A). We found that the NF-κB activation increased in HK-2 cells treated with HGPA compared with that without HGPA. However, HGPA-triggered NF-κB activation was reduced by Ac2-26 treatment (Supplementary Figure S16C).
DISCUSSION
Over the last decade, numerous studies have demonstrated that inflammation is involved in the cascade of events that lead to DN.17 Therefore, promoting the resolution of inflammation is a potential therapeutic approach for alleviating diabetes and its complications.18 ANXA1 promotes the production of anti-inflammatory cytokine interleukin-10 and plays an important role in the resolution of inflammation.19 Although numerous studies have focused on the profitable role of ANXA1 in disease statuses, the role of ANXA1 in DN is not yet fully clear. Herein, we identified upregulation of ANXA1 in kidneys of DN patients and reported the correlation between the level of ANXA1 in kidneys and kidney function as well as kidney outcomes in independent cohorts for the first time. We further evaluated the potential role of ANXA1 in DN, showing that genetic ablation of Anxa1 exacerbated kidney injury and overexpression of Anxa1 ameliorated kidney injury in DN mice. In addition to endogenous ANXA1, we evaluated the therapeutic potential of Ac2-26 (ANXA1 mimetic peptide) for DN. Finally, we demonstrated the corresponding protective mechanisms of ANXA1 in tubular epithelial cells in vitro.
In the present study, we detected ANXA1 in protein levels and ANXA1 mRNA levels in kidneys in different DN cohorts and characterized the expression profile of ANXA1 in various types of cells in kidneys. The expression of ANXA1 in kidneys was increased in patients with DN compared with healthy controls. Moreover, there was a significant negative correlation between the level of ANXA1 in kidneys and renal function. These data suggested that in the context of DN, the upregulation of ANXA1 in kidneys may be a possible attempt to attenuate the local inflammatory response. However, we found glomerular ANXA1 negatively correlated with glomerulosclerosis. It may be caused by a decrease in glomerular cells and an increased accumulation of extracellular matrix during the progression of glomerulosclerosis in DN.20,21 We found that plasma ANXA1 level was decreased in DN patients. ANXA1 is mainly produced by a variety of immune cells, including monocytes, macrophages, and neutrophils.22,23 DN is characterized by the infiltration of macrophages and the increased production of cytokines in kidneys. The decrease of circulating ANXA1 in DN may be caused by the increase of ANXA1 in the target inflammatory sites (i.e., kidneys). A similar phenomenon was also found in exacerbated asthmatics.24 In addition, we demonstrated that renal epithelial cells, including podocytes and tubular cells, also secreted ANXA1 in response to kidney inflammation. Moreover, we found that the urinary level of ANXA1 was increased, and there was a significant correlation between urinary ANXA1 and tubulointerstitial injuries in DN. Ka et al. found that the urinary ANXA1 was detectable as early as the stage of normoalbuminuria in diabetes mellitus patients.25 Based on the previous studies and our data, we speculate that ANXA1 may enter the urine by either secretion from renal epithelial cells or direct release from immune cells. However, the results did not exclude the possibility that some of the excreted ANXA1 may be derived from glomerular filtration.
Our studies further explored the functional role of ANXA1 in DN mouse models. We found that Anxa1 KO worsened diabetes-induced kidney injury. These findings suggested that once there was Anxa1 deficiency, proresolving activities were obviously insufficient to counteract proinflammatory signaling in HFD/STZ diabetic model, leading to persistent inflammation and more severe kidney injury. Moreover, flow cytometry and polymerase chain reaction array results suggested that ANXA1 could influence macrophage polarization. Lack of ANXA1 is able to lead to an increase of M1 macrophages and aggravation of inflammation in kidneys in DN, which is consistent with the previous study.26 In addition, our results showed that overexpression of Anxa1 alleviated HFD/STZ-induced kidney injury, suggesting the proresolving activation mediated by ANXA1 could counteract the proinflammatory response.
To further explore the therapeutic effects of exogenous ANXA1, db/db mice were administrated different doses of Ac2-26.27 Treatment with Ac2-26 is considered to have fewer adverse effects due to its ability to mimic or induce natural pathways mediating the resolution of inflammation.28 Renoprotective effects were observed in db/db mice administered Ac2-26. Consistently, we also found the protective effects of Ac2-26 in diabetic Anxa1 KO mice.
In the present study, we further found that ANXA1 knockdown could upregulate the proinflammatory and profibrotic factors in vitro, showing ANXA1 could directly target renal tubular epithelial cells in kidneys to prevent the injury from HGPA stimulation. In addition to the well-recognized pathway of extracellular ANXA1 binding to FPRs,29 intracellular ANXA1 associating with NF-κB to suppress its transcriptional activity in cancer cells could be of relevance.30 Because NF-κB is one of the crucial transcription factors relevant to the progression of DN,15,16 we assessed whether intracellular ANXA1 plays a role in DN by modulating NF-κB activity. Both in vitro and in vivo studies showed the interactions between ANXA1 and p65, and the binding of ANXA1 to NF-κB further inhibited the activation of NF-κB signaling. Somewhat surprisingly, the phosphorylation levels of IκBα were not altered significantly after ANXA1 silencing. Thus, we proposed that ANXA1 interacts with p65 and inhibits its phosphorylation, instead of affecting the degradation of IκBs and the release of p65 (i.e., ANXA1 affects the downstream IκBα degradation). Consistent with this hypothesis, increased phosphorylated p65 was observed in siANXA1 group treated with HGPA compared with negative control siRNA group treated with HGPA.
According to the comprehensive work by Purvis et al., Anxa1 gene deletion in mice treated with HFD led to severe diabetic phenotype. It was reported that ANXA1 could protect against excessive mitochondrial proton leak by activating ALX/FPR2 under hyperglycemic conditions in human hepatocytes.5 Another study from the same group demonstrated that Anxa1 KO aggravated kidney injuries in STZ-induced mouse model of type 1 diabetes by regulating v-akt murine thymoma viral oncogene homolog (Akt) signaling.6 The current study further confirmed ANXA1 in restricting kidney inflammation in DN. Moreover, we simulated the in vivo environment of diabetes and explored the direct role of intracellular ANXA1 in renal tubular epithelial cells.
In conclusion, our findings demonstrate that endogenous ANXA1 protects against nephropathy in diabetes. Intracellular ANXA1 may suppress NF-κB signaling to modulate kidney inflammation in DN. Our preclinical studies suggest that ANXA1 may be a promising therapeutic approach to treat DN. These observations also indicate that promoting endogenous resolution of inflammation may be an effective therapeutic paradigm for DN.
Supplementary Material
Supplementary Methods. Patients and samples, experimental animals, animal studies, blood and urine examination, glucose tolerance test (GTT) and insulin tolerance test (ITT), histologic analysis, immunohistochemistry and immunofluorescence, transmission electron microscopy, real-time quantitative polymerase chain reaction (qPCR) analysis, flow cytometry, cell culture and treatment, Western blot, co-Immunoprecipitation (Co-IP), electrophoretic mobility shift assay (EMSA); surface plasmon resonance (SPR) analysis, and statistical analysis.
Figure S1. Identification of annexin A1 (Anxa1) knockout mice and Anxa1 transgenic mice. (A) The DNA sequencing results of WT mice (up) and Anxa1 knockout heterozygote mice (below). The double peak showed a 10 bases deletion (CTTCAATGTA) in one strand. (B) The agarose gel electrophoresis of Anxa1 transgenic mice (the first and third lines) and WT mice (the second and fourth lines). The positive transgenic marker is 461 base pairs. (C) Western blot demonstrated ANXA1 expression was not detected in Anxa1 KO mice, and overexpressed in Anxa1 transgenic mice. (D) RT-qPCR analysis showing Anxa1 mRNA levels in kidneys in SFD Anxa1 KO and SFD WT mice. n = 6. (E) RT-qPCR analysis showing Anxa1 mRNA levels in kidneys in SFD Anxa1-Tg mice and SFD WT mice. n = 6. *P < 0.05, **P < 0.01, and ***P < 0.001, determined by Student t test. Anxa1-Tg, Anxa1 transgenic; KO, knockout; SFD, standard fat diet; WT, wild type.
Figure S2. Annexin A1 (ANXA1) mRNA in kidney biopsies from patients with diabetic nephropathy (DN). (A) ANXA1 mRNA (log2) levels in the tubulointerstitial compartment in advanced DN patients’ samples from ERCB (n = 17) and healthy controls (n = 21). (B) ANXA1 mRNA (log2) levels in the glomerular compartment in advanced DN patients’ samples from ERCB (n = 12, who had glomerular data among 17 DN patients) and healthy controls (n = 21). *P < 0.05, **P < 0.01, and ***P < 0.001. FC, fold change; HC, healthy control.
Figure S3. Western blot and RT-qPCR were used to detect the expression of ANXA1 in experimental animals. (A) Western blot and (B) RT-qPCR analyzing the ANXA1 levels in kidneys of SFD WT mice (left), HFD/STZ WT mice (mid), and HFD WT mice (right). (C) Western blot and (D) RT-qPCR analyzing the ANXA1 levels in kidneys of db/db mice (left) and db/m mice (right). Data analyses were performed by Student t test for 2 groups and 1-way ANOVA followed by a Tukey test for 3 groups. Data expressed as mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001.
Figure S4. ANXA1 mRNA levels associated with kidney outcomes. According to tertiles, Kaplan-Meier survival curves depicting kidney survival in low (T1), medium (T2), and high (T3) ANXA1 mRNA groups (log-rank test) in the Pima Indians cohort during a follow-up of 9.0 ± 4.0 years.
Figure S5. Immunofluorescence colocalization analyses. (A) Representative fluorescent images showed the expression of ANXA1 in mesangial cells (integrin α8) and endothelial cells (CD31) in the kidneys from DN patients. (B) For colocalization experiments, the negative control without primary antibody was used and no background fluorescence was observed. Bar = 50 μm.
Figure S6. In situ hybridization (ISH) analyses were performed to detect the source of ANXA1. (Top) ISH showed that ANXA1 was mainly expressed in parietal epithelial cells and tubular epithelial cells in kidneys from DN patients (red arrow). (Bottom) There was only a minor amount of ANXA1 expression in kidneys from healthy controls (HCs). Bar = 100 μm.
Figure S7. Urine and plasma levels of ANXA1 in patients with DN. (A) The level of urinary ANXA1/Cr in DN patients was significantly higher than that in DM patients without kidney involvement and healthy controls. (B) The level of plasma ANXA1 in DN patients was significantly lower than that in DM patients without kidney involvement and healthy controls. Data analyses were performed by 1-way ANOVA followed by a Tukey test for 3 groups and expressed as mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001.
Figure S8. Flow cytometry testing M1 and M2 macrophages in HFD/STZ Anxa1 KO mice and HFD/STZ WT mice. Representative flow cytometry results of (A) isotype control (ISO), (B) HFD/STZ WT mice, and (C) HFD/STZ Anxa1 KO mice. (D) Statistical data for flow cytometry results. Data analyses were performed by Student t test. ***P < 0.001.
Figure S9. HFD-induced diabetic mice had impaired glucose tolerance and insulin tolerance. (A) Body weight in SFD WT mice and HFD WT mice (n = 6). (B) Glucose tolerance in SFD WT mice and HFD WT mice (n = 6). (C) Insulin tolerance in WT mice and HFD mice (n = 6). *P < 0.05, **P < 0.01, and ***P < 0.001, determined by Student t test. HFD, high-fat diet; SFD, standard fat diet.
Figure S10. Overexpression of ANXA1 reduced the expression of inflammatory factors. (A) RT-qPCR analyzed the mRNA expression level of interleukin-6 (Il-6), tumor necrosis factor-α (Tnf-α), and interleukin-1β (Il-1β) in Anxa1-Tg mice and WT mice (n = 6 per group). (B) Representative transmission electron microscopy images from WT and Anxa1-Tg mice. Bar = 2 μm. Quantification of mean foot process width and quantification of mean glomerular basement membrane (GBM) thickness. *P < 0.05, **P < 0.01, and ***P < 0.001, determined by 2-way ANOVA, followed by the Tukey test. Anxa1-Tg, Anxa1 transgenic; SFD, standard fat diet.
Figure S11. Ac2-26 administration ameliorated inflammation in diabetic Anxa1 KO mice. (A) Study design overview. Eight-week-old Anxa1 KO mice were fed with a high-fat diet for 1 month, followed by i.p. injection of 50 mg/kg STZ for 5 days. After STZ injection, mice were maintained for 16 weeks of HFD feeding with Ac2-26 treatment or with vehicle (PBS) before sacrifice. n = 5 to 6 per group. (B) Four groups were evaluated for urine albumin-to-creatinine ratio (uACR). (C) Representative photomicrographs of periodic acid–Schiff (PAS) staining (bar = 50 μm for tubulointerstitium, and bar 25 μm for glomeruli), F4/80 staining (bar = 50 μm), and α-SMA (bar = 50 μm). (D) Quantification of mesangial matrix expansion. (E) Quantification of tubulointerstitial injury index. (F) Quantitative analysis of F4/80 staining. (G) Quantitative analysis of α-SMA expression. (H) Representative transmission electron microscopy (TEM) images in kidney cortex tissues isolated from diabetic Anxa1 KO mice with and without Ac2-26 treatment. Quantification of mean glomerular basement membrane (GBM) thickness and mean foot process width in 2 groups (n = 3). Bar = 2 μm. (I) Immunohistochemical staining for F4/80 with the negative control in kidney tissues from HFD/STZ-induced diabetic mice. (J) Immunohistochemical staining for α-SMA with the negative control in kidney tissues from HFD/STZ-induced diabetic mice. Throughout, data analyses were performed by Student t test for 2 groups and 2-way ANOVA followed by a Tukey test for 4 groups. Data expressed as mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001. KO, knockout; WT, wild type.
Figure S12. RT-qPCR and Western blot were used to detect the expression of ANXA1 in HK-2 cells. (A) ANXA1 mRNA levels were analyzed by RT-qPCR in HK-2 cells with HGPA treatment (n = 3). (B) RT-qPCR and (C) Western blot analyzing the expression level of ANXA1 in HK-2 cells (n = 3). The blank group was served as a normal control group. Mannitol and endotoxin-free BSA served as an isotonic solvent control group. Data analyses were performed by Student t test for 2 groups and 1-way ANOVA followed by a Tukey test for multiple groups. *P < 0.05, **P < 0.01, and ***P < 0.001. HGPA, high glucose plus palmitic acid; siANXA1, siRNA against ANXA1; siCtrl, negative control siRNA.
Figure S13. The phosphorylation levels of NF-κB p65. Representative Western blot images and the analyses of phosphorylated p65 and p65 in HFD/STZ WT mice and HFD/STZ Anxa1 KO mice. **P < 0.01, determined by Student t test.
Figure S14. The degradation of IκBα in siANXA1 group treated with HGPA. Western blot showed that the phosphorylation levels of IκBα were not significantly altered in siANXA1 group treated with HGPA compared with siCtrl group treated with HGPA (by Student t test). HGPA, high glucose plus palmitic acid; NS, no significance; siANXA1, siRNA against ANXA1; siCtrl, negative control siRNA.
Figure S15. Coimmunoprecipitation of ANXA1 and p65 in kidney tissues. Total protein lysates of kidney tissues from HFD/STZ WT mice were immunoprecipitated using an antibody against ANXA1 and immunoblotting against the p65.
Figure S16. Ac2-26 played the proresolving role via FPR1 in HK-2 cells. (A) The mRNA expression level of Il-1β, MCP-1, TGF-β, and TNF in HK-2 cells treated with Ac2-26, WRW4, Boc-MLF (1 μmol/L), and Boc-MLF (10 μmol/L) by RT-qPCR. (B) The SPR response curves generated from Ac2-26 binding to p65. The results suggested that Ac2-26 did not interact with p65 directly. (C) The activation of NF-κB in HK-2 cells treated with different conditions. Data analyses were performed by 1- or 2-way ANOVA followed by a Tukey test and expressed as mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001.
Table S1. Primer sequences used in real-time PCR analysis.
Table S2. General data of patients from China for measuring intrarenal ANXA1 expression.
Table S3. General data of patients from European Renal cDNA Bank with late DN for measuring intrarenal ANXA1 mRNA expression.
Table S4. General data of American Indians with early DN for measuring intrarenal ANXA1 mRNA expression.
Table S5. General data of patients for measuring urinary and circulating ANXA1 levels.
Table S6. Physical and biochemical parameters of experimental animals.
Table S7. Quantification of mRNA by PCR array.
Translational Statement.
Diabetic nephropathy (DN) is the leading cause of end-stage kidney disease worldwide. Novel strategies for DN therapy are needed urgently. Our data demonstrate that annexin A1 (ANXA1), as a proresolving molecule, is a promising therapeutic target for treating DN. We show that ANXA1 deficiency exacerbates kidney injuries in DN, whereas ANXA1 overexpression or treatment with Ac2-26, an ANXA1 mimetic peptide, protects against the progression of DN by reducing kidney tubular injuries, kidney inflammation, and fibrosis. Our findings suggest a potential therapeutic approach based on inflammation resolution to treat DN effectively.
ACKNOWLEDGEMENTS
SE reports grants from National Institutes of Health (NIH)–National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and National Center for Advancing Translational Sciences, during the conduct of the study. MK reports grants from the NIH during the conduct of the study. This study is supported by a grant from National Key Research and Development Program (no. 2016YFC1305405), grants from the National Natural Science Foundation (nos. 82090021, 82070748, 91639108, 81770272, and 81970425), a grant from Interdisciplinary Clinical Research Project of Peking University First Hospital, a grant by the University of Michigan Health System and Peking University Health Sciences Center Joint Institute for Translational and Clinical Research (no. BMU2017JI001), a grant from Peking University Medicine Seed Fund for Interdisciplinary Research (no. BMU2018MX025), and Innovation Fund for Outstanding Doctoral Candidates of Peking University Health Science Center (no. 71013Y2029). Additional support was provided by the Applied Systems Biology Core at the University of Michigan George M. O’Brien Kidney Translational Core Center (P30 DK081943) and by the Intramural Research Program of the NIDDK.
DISCLOSURE
SE reports grants from AstraZeneca, Gilead Sciences, NovoNordisk, Janssen, and Eli Lilly and Company, outside the submitted work. MK reports nonfinancial support from University of Michigan during the conduct of the study; grants from JDRF, AstraZeneca, NovoNordisk, Eli Lilly, Gilead, Goldfinch Bio, Merck, Janssen, Boehringer-Ingelheim, Moderna, European Union Innovative Medicine Initiative, Chan Zuckerberg Initiative, Certa, Chinook, amfAR, Angion Pharmaceuticals, RenalytixAI, Travere Therapeutics, Regeneron, and IONIS Pharmaceuticals; other from Astellas; other from Poxel; and grants from Shire; outside the submitted work. In addition, MK has a licensed patent PCT/EP2014/073413, “Biomarkers and Methods for Progression Prediction for Chronic Kidney Disease.” WJ has a licensed patent PCT/EP2014/073413, “Biomarkers and Methods for Progression Prediction for Chronic Kidney Disease.” All the other authors declared no competing interests.
Footnotes
SUPPLEMENTARY MATERIAL
REFERENCES
- 1.Pradhan AD, Manson JE, Rifai N, et al. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA. 2001;286:327–334. [DOI] [PubMed] [Google Scholar]
- 2.Hu FB, Meigs JB, Li TY, et al. Inflammatory markers and risk of developing type 2 diabetes in women. Diabetes. 2004;53:693–700. [DOI] [PubMed] [Google Scholar]
- 3.Serhan CN, Yacoubian S, Yang R. Anti-inflammatory and proresolving lipid mediators. Annu Rev Pathol. 2008;3:279–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flower RJ, Blackwell GJ. Anti-inflammatory steroids induce biosynthesis of a phospholipase A2 inhibitor which prevents prostaglandin generation. Nature. 1979;278:456–459. [DOI] [PubMed] [Google Scholar]
- 5.Purvis GSD, Collino M, Loiola RA, et al. Identification of AnnexinA1 as an endogenous regulator of RhoA, and its role in the pathophysiology and experimental therapy of type-2 diabetes. Front Immunol. 2019;10:571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Purvis GSD, Chiazza F, Chen J, et al. Annexin A1 attenuates microvascular complications through restoration of Akt signalling in a murine model of type 1 diabetes. Diabetologia. 2018;61:482–495. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7.Panda SK, Wefers B, Ortiz O, et al. Highly efficient targeted mutagenesis in mice using TALENs. Genetics. 2013;195:703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Z, Michael IP, Zhou D, et al. Simple piggyBac transposon-based mammalian cell expression system for inducible protein production. Proc Natl Acad Sci U S A. 2013;110:5004–5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tervaert TW, Mooyaart AL, Amann K, et al. Pathologic classification of diabetic nephropathy. J Am Soc Nephrol. 2010;21:556–563. [DOI] [PubMed] [Google Scholar]
- 10.Wu P, Wang Y, Davis ME, et al. Store-operated Ca2+ channels in mesangial cells inhibit matrix protein expression. J Am Soc Nephrol. 2015;26:2691–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kelly DJ, Wilkinson-Berka JL, Allen TJ, et al. A new model of diabetic nephropathy with progressive renal impairment in the transgenic (mRen-2)27 rat (TGR). Kidney Int. 1998;54:343–352. [DOI] [PubMed] [Google Scholar]
- 12.Nair V, Komorowsky CV, Weil EJ, et al. A molecular morphometric approach to diabetic kidney disease can link structure to function and outcome. Kidney Int. 2018;93:439–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Breyer MD, Böttinger E, Brosius FC, et al. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2005;16:27–45. [DOI] [PubMed] [Google Scholar]
- 14.Brosius FC, Alpers CE, Bottinger EP, et al. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2009;20:2503–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanchez AP, Sharma K. Transcription factors in the pathogenesis of diabetic nephropathy. Expert Rev Mol Med. 2009;11:e13. [DOI] [PubMed] [Google Scholar]
- 16.Schmid H, Boucherot A, Yasuda Y, et al. Modular activation of nuclear factor-kappaB transcriptional programs in human diabetic nephropathy. Diabetes. 2006;55:2993–3003. [DOI] [PubMed] [Google Scholar]
- 17.Navarro-González JF, Mora-Fernández C, Muros de Fuentes M, et al. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat Rev Nephrol. 2011;7:327–340. [DOI] [PubMed] [Google Scholar]
- 18.Brennan EP, Mohan M, McClelland A, et al. Lipoxins regulate the early growth response-1 network and reverse diabetic kidney disease. J Am Soc Nephrol. 2018;29:1437–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferlazzo V, D’Agostino P, Milano S, et al. Anti-inflammatory effects of annexin-1: stimulation of IL-10 release and inhibition of nitric oxide synthesis. Int Immunopharmacol. 2003;3:1363–1369. [DOI] [PubMed] [Google Scholar]
- 20.Ziyadeh FN, Wolf G. Pathogenesis of the podocytopathy and proteinuria in diabetic glomerulopathy. Curr Diabetes Rev. 2008;4:39–45. [DOI] [PubMed] [Google Scholar]
- 21.Nakagawa T, Sato W, Glushakova O, et al. Diabetic endothelial nitric oxide synthase knockout mice develop advanced diabetic nephropathy. J Am Soc Nephrol. 2007;18:539–550. [DOI] [PubMed] [Google Scholar]
- 22.Perretti M, Croxtall JD, Wheller SK, et al. Mobilizing lipocortin 1 in adherent human leukocytes downregulates their transmigration. Nat Med. 1996;2:1259–1262. [DOI] [PubMed] [Google Scholar]
- 23.Oliani SM, Damazo AS, Perretti M. Annexin 1 localisation in tissue eosinophils as detected by electron microscopy. Mediators Inflamm. 2002;11:287–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee SH, Lee PH, Kim BG, et al. Annexin A1 in plasma from patients with bronchial asthma: its association with lung function. BMC Pulm Med. 2018;18:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ka SM, Tsai PY, Chao TK, et al. Urine annexin A1 as an index for glomerular injury in patients. Dis Markers. 2014;2014:854163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McArthur S, Juban G, Gobbetti T, et al. Annexin A1 drives macrophage skewing to accelerate muscle regeneration through AMPK activation. J Clin Invest. 2020;130:1156–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perretti M, Dalli J. Exploiting the Annexin A1 pathway for the development of novel anti-inflammatory therapeutics. Br J Pharmacol. 2009;158:936–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li C, Zhao Y, Cheng J, et al. A proresolving peptide nanotherapy for site-specific treatment of inflammatory bowel disease by regulating proinflammatory microenvironment and gut microbiota. Adv Sci. 2019;6: 1900610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ansari J, Kaur G, Gavins FNE. Therapeutic potential of Annexin A1 in ischemia reperfusion injury. Int J Mol Sci. 2018;19:1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Z, Huang L, Zhao W, et al. Annexin 1 induced by anti-inflammatory drugs binds to NF-kappaB and inhibits its activation: anticancer effects in vitro and in vivo. Cancer Res. 2010;70:2379–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods. Patients and samples, experimental animals, animal studies, blood and urine examination, glucose tolerance test (GTT) and insulin tolerance test (ITT), histologic analysis, immunohistochemistry and immunofluorescence, transmission electron microscopy, real-time quantitative polymerase chain reaction (qPCR) analysis, flow cytometry, cell culture and treatment, Western blot, co-Immunoprecipitation (Co-IP), electrophoretic mobility shift assay (EMSA); surface plasmon resonance (SPR) analysis, and statistical analysis.
Figure S1. Identification of annexin A1 (Anxa1) knockout mice and Anxa1 transgenic mice. (A) The DNA sequencing results of WT mice (up) and Anxa1 knockout heterozygote mice (below). The double peak showed a 10 bases deletion (CTTCAATGTA) in one strand. (B) The agarose gel electrophoresis of Anxa1 transgenic mice (the first and third lines) and WT mice (the second and fourth lines). The positive transgenic marker is 461 base pairs. (C) Western blot demonstrated ANXA1 expression was not detected in Anxa1 KO mice, and overexpressed in Anxa1 transgenic mice. (D) RT-qPCR analysis showing Anxa1 mRNA levels in kidneys in SFD Anxa1 KO and SFD WT mice. n = 6. (E) RT-qPCR analysis showing Anxa1 mRNA levels in kidneys in SFD Anxa1-Tg mice and SFD WT mice. n = 6. *P < 0.05, **P < 0.01, and ***P < 0.001, determined by Student t test. Anxa1-Tg, Anxa1 transgenic; KO, knockout; SFD, standard fat diet; WT, wild type.
Figure S2. Annexin A1 (ANXA1) mRNA in kidney biopsies from patients with diabetic nephropathy (DN). (A) ANXA1 mRNA (log2) levels in the tubulointerstitial compartment in advanced DN patients’ samples from ERCB (n = 17) and healthy controls (n = 21). (B) ANXA1 mRNA (log2) levels in the glomerular compartment in advanced DN patients’ samples from ERCB (n = 12, who had glomerular data among 17 DN patients) and healthy controls (n = 21). *P < 0.05, **P < 0.01, and ***P < 0.001. FC, fold change; HC, healthy control.
Figure S3. Western blot and RT-qPCR were used to detect the expression of ANXA1 in experimental animals. (A) Western blot and (B) RT-qPCR analyzing the ANXA1 levels in kidneys of SFD WT mice (left), HFD/STZ WT mice (mid), and HFD WT mice (right). (C) Western blot and (D) RT-qPCR analyzing the ANXA1 levels in kidneys of db/db mice (left) and db/m mice (right). Data analyses were performed by Student t test for 2 groups and 1-way ANOVA followed by a Tukey test for 3 groups. Data expressed as mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001.
Figure S4. ANXA1 mRNA levels associated with kidney outcomes. According to tertiles, Kaplan-Meier survival curves depicting kidney survival in low (T1), medium (T2), and high (T3) ANXA1 mRNA groups (log-rank test) in the Pima Indians cohort during a follow-up of 9.0 ± 4.0 years.
Figure S5. Immunofluorescence colocalization analyses. (A) Representative fluorescent images showed the expression of ANXA1 in mesangial cells (integrin α8) and endothelial cells (CD31) in the kidneys from DN patients. (B) For colocalization experiments, the negative control without primary antibody was used and no background fluorescence was observed. Bar = 50 μm.
Figure S6. In situ hybridization (ISH) analyses were performed to detect the source of ANXA1. (Top) ISH showed that ANXA1 was mainly expressed in parietal epithelial cells and tubular epithelial cells in kidneys from DN patients (red arrow). (Bottom) There was only a minor amount of ANXA1 expression in kidneys from healthy controls (HCs). Bar = 100 μm.
Figure S7. Urine and plasma levels of ANXA1 in patients with DN. (A) The level of urinary ANXA1/Cr in DN patients was significantly higher than that in DM patients without kidney involvement and healthy controls. (B) The level of plasma ANXA1 in DN patients was significantly lower than that in DM patients without kidney involvement and healthy controls. Data analyses were performed by 1-way ANOVA followed by a Tukey test for 3 groups and expressed as mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001.
Figure S8. Flow cytometry testing M1 and M2 macrophages in HFD/STZ Anxa1 KO mice and HFD/STZ WT mice. Representative flow cytometry results of (A) isotype control (ISO), (B) HFD/STZ WT mice, and (C) HFD/STZ Anxa1 KO mice. (D) Statistical data for flow cytometry results. Data analyses were performed by Student t test. ***P < 0.001.
Figure S9. HFD-induced diabetic mice had impaired glucose tolerance and insulin tolerance. (A) Body weight in SFD WT mice and HFD WT mice (n = 6). (B) Glucose tolerance in SFD WT mice and HFD WT mice (n = 6). (C) Insulin tolerance in WT mice and HFD mice (n = 6). *P < 0.05, **P < 0.01, and ***P < 0.001, determined by Student t test. HFD, high-fat diet; SFD, standard fat diet.
Figure S10. Overexpression of ANXA1 reduced the expression of inflammatory factors. (A) RT-qPCR analyzed the mRNA expression level of interleukin-6 (Il-6), tumor necrosis factor-α (Tnf-α), and interleukin-1β (Il-1β) in Anxa1-Tg mice and WT mice (n = 6 per group). (B) Representative transmission electron microscopy images from WT and Anxa1-Tg mice. Bar = 2 μm. Quantification of mean foot process width and quantification of mean glomerular basement membrane (GBM) thickness. *P < 0.05, **P < 0.01, and ***P < 0.001, determined by 2-way ANOVA, followed by the Tukey test. Anxa1-Tg, Anxa1 transgenic; SFD, standard fat diet.
Figure S11. Ac2-26 administration ameliorated inflammation in diabetic Anxa1 KO mice. (A) Study design overview. Eight-week-old Anxa1 KO mice were fed with a high-fat diet for 1 month, followed by i.p. injection of 50 mg/kg STZ for 5 days. After STZ injection, mice were maintained for 16 weeks of HFD feeding with Ac2-26 treatment or with vehicle (PBS) before sacrifice. n = 5 to 6 per group. (B) Four groups were evaluated for urine albumin-to-creatinine ratio (uACR). (C) Representative photomicrographs of periodic acid–Schiff (PAS) staining (bar = 50 μm for tubulointerstitium, and bar 25 μm for glomeruli), F4/80 staining (bar = 50 μm), and α-SMA (bar = 50 μm). (D) Quantification of mesangial matrix expansion. (E) Quantification of tubulointerstitial injury index. (F) Quantitative analysis of F4/80 staining. (G) Quantitative analysis of α-SMA expression. (H) Representative transmission electron microscopy (TEM) images in kidney cortex tissues isolated from diabetic Anxa1 KO mice with and without Ac2-26 treatment. Quantification of mean glomerular basement membrane (GBM) thickness and mean foot process width in 2 groups (n = 3). Bar = 2 μm. (I) Immunohistochemical staining for F4/80 with the negative control in kidney tissues from HFD/STZ-induced diabetic mice. (J) Immunohistochemical staining for α-SMA with the negative control in kidney tissues from HFD/STZ-induced diabetic mice. Throughout, data analyses were performed by Student t test for 2 groups and 2-way ANOVA followed by a Tukey test for 4 groups. Data expressed as mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001. KO, knockout; WT, wild type.
Figure S12. RT-qPCR and Western blot were used to detect the expression of ANXA1 in HK-2 cells. (A) ANXA1 mRNA levels were analyzed by RT-qPCR in HK-2 cells with HGPA treatment (n = 3). (B) RT-qPCR and (C) Western blot analyzing the expression level of ANXA1 in HK-2 cells (n = 3). The blank group was served as a normal control group. Mannitol and endotoxin-free BSA served as an isotonic solvent control group. Data analyses were performed by Student t test for 2 groups and 1-way ANOVA followed by a Tukey test for multiple groups. *P < 0.05, **P < 0.01, and ***P < 0.001. HGPA, high glucose plus palmitic acid; siANXA1, siRNA against ANXA1; siCtrl, negative control siRNA.
Figure S13. The phosphorylation levels of NF-κB p65. Representative Western blot images and the analyses of phosphorylated p65 and p65 in HFD/STZ WT mice and HFD/STZ Anxa1 KO mice. **P < 0.01, determined by Student t test.
Figure S14. The degradation of IκBα in siANXA1 group treated with HGPA. Western blot showed that the phosphorylation levels of IκBα were not significantly altered in siANXA1 group treated with HGPA compared with siCtrl group treated with HGPA (by Student t test). HGPA, high glucose plus palmitic acid; NS, no significance; siANXA1, siRNA against ANXA1; siCtrl, negative control siRNA.
Figure S15. Coimmunoprecipitation of ANXA1 and p65 in kidney tissues. Total protein lysates of kidney tissues from HFD/STZ WT mice were immunoprecipitated using an antibody against ANXA1 and immunoblotting against the p65.
Figure S16. Ac2-26 played the proresolving role via FPR1 in HK-2 cells. (A) The mRNA expression level of Il-1β, MCP-1, TGF-β, and TNF in HK-2 cells treated with Ac2-26, WRW4, Boc-MLF (1 μmol/L), and Boc-MLF (10 μmol/L) by RT-qPCR. (B) The SPR response curves generated from Ac2-26 binding to p65. The results suggested that Ac2-26 did not interact with p65 directly. (C) The activation of NF-κB in HK-2 cells treated with different conditions. Data analyses were performed by 1- or 2-way ANOVA followed by a Tukey test and expressed as mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001.
Table S1. Primer sequences used in real-time PCR analysis.
Table S2. General data of patients from China for measuring intrarenal ANXA1 expression.
Table S3. General data of patients from European Renal cDNA Bank with late DN for measuring intrarenal ANXA1 mRNA expression.
Table S4. General data of American Indians with early DN for measuring intrarenal ANXA1 mRNA expression.
Table S5. General data of patients for measuring urinary and circulating ANXA1 levels.
Table S6. Physical and biochemical parameters of experimental animals.
Table S7. Quantification of mRNA by PCR array.