

Abstract
In the endoplasmic reticulum (ER), the translocation-associated protein complex (TRAP), also called signal sequence receptor (SSR), includes four integral membrane proteins TRAPα/SSR1, TRAPβ/SSR2, and TRAPδ/SSR4 with the bulk of their extramembranous portions primarily in the ER lumen, whereas the extramembranous portion of TRAPγ/SSR3 is primarily cytosolic. Individually diminished expression of either TRAPα/SSR1, TRAPβ/SSR2, or TRAPδ/SSR4 mRNA is known in each case to lower TRAPα/SSR1 protein levels, leading to impaired proinsulin biosynthesis, whereas forced expression of TRAPα/SSR1 at least partially suppresses the proinsulin biosynthetic defect. Here, we report that diminished TRAPγ/SSR3 expression in pancreatic β-cells leaves TRAPα/SSR1 levels unaffected while nevertheless inhibiting cotranslational and posttranslational translocation of preproinsulin into the ER. Crucially, acute exposure to high glucose leads to a rapid upregulation of both TRAPγ/SSR3 and proinsulin protein without change in the respective mRNA levels, as observed in cultured rodent β-cell lines and confirmed in human islets. Strikingly, pancreatic β-cells with suppressed TRAPγ/SSR3 expression are blocked in glucose-dependent upregulation of proinsulin (or insulin) biosynthesis. Most remarkably, overexpression of TRAPγ/SSR3 in control β-cells raises proinsulin levels, even without boosting extracellular glucose. The data suggest the possibility that TRAPγ/SSR3 may fulfill a rate-limiting function in preproinsulin translocation across the ER membrane for proinsulin biosynthesis.
Introduction
Of the known human risk alleles that predispose to type 2 diabetes, many reflect gene expression specifically in pancreatic islet β-cells (1). One such risk allele is linked to the α-subunit of the complex known as translocation-associated protein (TRAP), previously named signal sequence receptor (SSR) (2). Recent evidence from us and others supports that TRAP/SSR is required in the endoplasmic reticulum (ER) for efficient biosynthesis of proinsulin and insulin (3,4). Proinsulin is the predominant translation product of pancreatic β-cells (5), and the ability to acutely upregulate its production upon physiologic stimulation (e.g., with high glucose [6], which is needed to maintain the insulin granule storage pool [7]) is essential to avoid diabetes (8). The TRAP/SSR complex contributes to this production by enhancing the efficiency of translocation of preproinsulin (and certain other proteins [9])—synthesized from ribosomes that decorate the cytosolic face of the ER membrane—into and across the Sec61 channel (see schematic in Fig. 1A). TRAP/SSR is a heterotetrameric complex that comprises TRAPα/SSR1, TRAPβ/SSR2, TRAPγ/SSR3, and TRAPδ/SSR4 (10), and optimal stability of the complex requires that all four members be present and physically interact (11–13). Excluding the peptide portions embedded within the bilayer itself, a majority of the TRAP/SSR complex, specifically including TRAPα/SSR1, TRAPβ/SSR2, and TRAPδ/SSR4, resides on the lumenal side of the ER membrane (11–13), with the exception being TRAPγ/SSR3, which has greater exposure on the cytosolic side (Fig. 1A) (14).
Figure 1.

The TRAPγ/SSR3 subunit of the TRAP/SSR complex contributes to the efficiency of recombinant proinsulin biosynthesis. A: Schematic of the TRAP/SSR complex highlights the predominant cytosolic exposure of the TRAPγ/SSR3 subunit, including a hypothetical contact with a signal peptide N-terminus during polypeptide translocation (red line). B: Control (Ctrl) (wild-type [WT] 293T cells) and SSR3-KO 293T cells were transfected with untagged human preproinsulin plasmids. At 48 h posttransfection, cell lysates were analyzed by reducing SDS-PAGE gel and immunoblotting with antiproinsulin and anti-SSR1–4 antibodies as indicated. C: Quantitation (mean ± SD) of preproinsulin, proinsulin, and TRAP/SSR subunit protein levels from four independent experiments like that shown in B (normalized to tubulin). *P < 0.05 compared with Ctrl. D: Ctrl and SSR3-KO 293T cells were transfected with human preproinsulin plasmids. At 48 h posttransfection, the cells were pulse labeled with 35S-Met/Cys for the times indicated; this is a representative pulse labeling from three identical experiments. Cell lysates (normalized to trichloroacetic acid–precipitable counts) were subjected to immunoprecipitation with anti-insulin and analyzed by reducing SDS-PAGE and phosphorimaging; both the absolute and relative amounts of recovered preproinsulin and proinsulin are shown in the graph.
Although TRAP/SSR functions as a coordinated complex, it is likely that individual subunits subserve distinct functions and may interact preferentially with various non-TRAP protein partners, and this can help to explain why defective TRAPγ/SSR3, like that for TRAPδ/SSR4, is genetically linked to the family of diseases known as congenital disorders of glycosylation with instability of the heterotetrameric TRAP complex (15,16) and dramatic developmental abnormalities (17). From cryoelectron tomography, it has been suggested that TRAPγ/SSR3 makes contact both with protein and RNA components of closely juxtaposed membrane-bound ribosomes (11). It has also been hypothesized that the N-terminal tip of the signal peptide might be close enough to interact with the cytosolic domain of TRAPγ/SSR3, potentially to help to reorient the signal peptide during transfer from the signal recognition particle to its hairpin insertion into the Sec61 translocon (18).
Recent observations suggest that TRAPβ/SSR2 and TRAPδ/SSR4, the two other lumenally disposed subunits, help to support the important activity of TRAPα/SSR1 in efficient insulin biogenesis in pancreatic β-cells (3,19). Interestingly, TRAPα/SSR1 is the subunit that can be cross-linked to nascent translocating chains, even after the signal peptide has been excised (20), which is a time when the entire 86-residue proinsulin protein would be contained within the ER lumen. This behavior would be distinct from interaction with the nascent signal peptide on the cytosolic side of the ER membrane. With these ideas in mind, we have examined whether and how TRAPγ/SSR3 may participate in proinsulin synthesis.
Research Design and Methods
Antibodies and Reagents
Mouse anti-rat proinsulin (CCI-17) and rabbit polyclonal anti-SSR1 antibodies (NB100-73013 and NBP1-86912, respectively) were from Novus Biologicals (Littleton, CO). Mouse anti-human proinsulin C-peptide-A chain junction, rabbit anti-SSR3 and SSR4 antibodies were made by Abmart (Shanghai, China). Rabbit anti-SSR2 (10278-1-AP) was from Proteintech (Rosemont, IL). Guinea pig anti-insulin was from Merck-Millipore (Billerica, MA). Mouse monoclonal antitubulin antibody (T5168), dithiothreitol, MG-132, and cycloheximide (CHX) were from Sigma-Aldrich (St. Louis, MO). Lipofectamine 2000, Lipofectamine RNAiMAX, NuPage gels, Opti-MEM, Met/Cys-deficient DMEM, and all other cell culture reagents were from Invitrogen. 35S-amino acid mixture was from PerkinElmer (Waltham, MA). Protein A-agarose was from Repligen (Waltham, MA). siRNAs oligo pools for rat SSR3 knockdown (KD) were synthesized commercially by Biotend (Shanghai, China) as follows:
SSR3-1: forward 5′-GGAAGAUGCUGUUUCCAAAdTdT-3′; reverse 5′-UUUGGAAACAGCAUCUUCCdTdT-3′
SSR3-2: forward 5′-AAUUUGUUCUCAAGCACAAdTdT-3′; reverse 5′-UUGUGCUUGAGAACAAAUUdTdT-3′
SSR3-3: forward 5′-GAAUGAAGUUGCCGAUUAUdTdT-3′; reverse 5′-AUAAUCGGCAACUUCAUUCdTdT-3′
Cell Culture and Transfection
293T cells (CRL-3216; ATCC) were cultured in DMEM supplemented with 10% FBS, 100 IU/mL penicillin, and 100 μg/mL streptomycin in a 37°C incubator with 5% CO2. INS832/13 rat insulinoma cells (obtained from the laboratory of Dr. C. Newgard, Duke University, Durham, NC) and INS1E cells (obtained from the laboratory of Dr. C. Wollheim, University of Geneva, Geneva, Switzerland) were cultured in RPMI medium supplemented with 10% FBS, 1 mmol/L sodium pyruvate, 10 mmol/L HEPES, and 0.05 mmol/L 2-mercaptoethanol.
293T cells and INS832/13 cells were seeded into 12-well plates 1 day before transfection. A total of 1 μg of plasmid DNA and 2.5 μL Lipofectamine 2000 were diluted into Opti-MEM and added into each well after incubation for 20 min. Cells were collected 48 h after transfection and subjected to analysis.
For SSR3 KD, INS1E cells were seeded into 12-well plates and allowed to grow to 50% confluency. Fifty picomoles rat SSR3 siRNA or scrambled siRNA and 4 μL Lipofectamine RNAiMAX were diluted into Opti-MEM and added to each well after 20-min incubation. Cells were collected 72 h after transfection and subjected to functional analysis.
Generation of TRAPγ/SSR3 Knockout 293T and INS832/13 cells
TRAPγ/SSR3 knockout (KO) 293T cells and INS832/13 cells were generated using the CRISPR/Cas9-mediated genome editing system. Single guide RNAs (sgRNAs) were designed on the Zhang laboratory website (https://zlab.bio/guide-design-resources). Nucleotide guide sequences (human sgRNA 5′-ggagctgcctttaggagcca-3′ for 293T cells; rat sgRNA-1 5′-aaatcgtcggcgctgttctt-3′ and sgRNA-2 5′-taatacttcgatgttgtaag-3′ for INS832/13 cells) were annealed and ligated into lentiCRISPR v2 vector. 293T cells or INS832/13 cells were transfected with the plasmids and allowed to grow to 90–100% confluency. Cells were replated and selected in 1 μg/mL puromycin. Puromycin-resistant pools were expanded and finally cultured in fresh normal medium without puromycin.
Human Islets
We obtained purified pancreatic islets (90% pure, 95% viable, all islet equivalents >50 μm; Prodo Laboratories) from a 54-year-old Caucasian male donor without diabetes (height 69 inches, weight 175 lb, BMI 24.8 kg/m2) who died as a result of head trauma. All routine blood tests were normal, and the donor tested negative for COVID-19; HbA1c was 5.4%. Human islets were divided into equal aliquots and treated and analyzed independently in triplicate. Islet handling is described in Fig. 6G.
Figure 6.

Overexpression of TRAPγ/SSR3 in wild-type INS832/13 cells increases the steady-state level of proinsulin. A: Wild-type INS832/13 cells were transfected with plasmids encoding empty vector (−) or Flag-tagged TRAPγ/SSR3 cDNA (+). At 48 h posttransfection, cells were lysed and analyzed in duplicate by reducing SDS-PAGE and immunoblotting with the indicated antibodies. B: Quantitation (mean ± SD) of rodent proinsulin (CCI-17 antibody) and human proinsulin (human C-A junction antibody) and each TRAP/SSR subunit as indicated (n = 4 independent experiments). C: Wild-type INS832/13 cells were transfected with plasmids encoding empty vector or each individual Flag-tagged TRAP/SSR subunit and processed as in A. The red arrows highlight expression of the Flag-tagged constructs. D: Quantitation (mean ± SD) showing increased expression of each of the TRAP/SSR subunits (top) and the impact of that overexpression on rodent proinsulin (bottom left) and human proinsulin (bottom right) levels from five independent experiments. E: Wild-type 293T cells were transfected with human wild-type preproinsulin plasmid plus plasmid encoding empty vector or each individual Flag-tagged TRAP/SSR subunit and processed as in A. F: Quantitation (mean ± SD) of individually overexpressed TRAP/SSR subunits (left) and recombinant human proinsulin (right) from four independent experiments. G: Human islets were preincubated for 90 min in RPMI medium containing 2.5 mmol/L glucose and 0.1% BSA without serum. Aliquots of 130 human islet equivalents were sedimented at 1,000g, and the preincubation medium was removed, followed by a 2-h incubation in 500 μL of fresh medium bearing either 2.5 or 25 mmol/L glucose in the absence or presence of CHX (100 μg/mL). Human islets were then lysed in RIPA buffer; lysates normalized to total protein were analyzed by SDS-PAGE and immunoblotting for the indicated proteins (tubulin was used as a loading control to confirm proper normalization of samples). *P < 0.05. EV, empty vector; PI, proinsulin.
Metabolic Labeling and Immunoprecipitation
293T cells were transfected to express human preproinsulin (pCDNA3.1, cytomegalovirus promoter, encoded ORF = 100% identity to human preproinsulin: 5′-ATGGCCCTGTGGATGCGCCTCCTGCCCCTGCTGGCGCTGCTGGCCCTCTGGGGACCTGACCCAGCCGCAGCCTTTGTGAACCAACAC CTGTGCGGCTCACACCTGGTGGAAGCTCTCTACCTAGT GTGCGGGGAACGAGGCTTCTTCTACACACCCAAGACC CGCCGGGAGGCAGAGGACCTGCAGGTGGGGCAGGTG GAGCTGGGCGGGGGCCCTGGTGCAGGCAGCCTGCAG CCCTTGGCCCTGGAGGGGTCCCTGCAGAAGCGTGGCA TTGTGGAACAATGCTGTACCAGCATCTGCTCCCTCTACCAGCTGGAGAACTACTGCAACTAG-3′) and pulse labeled 48 h posttransfection. To study endogenous preproinsulin synthesis, INS1E cells were seeded 1 day before labeling. Briefly, cells were incubated in Met/Cys-deficient DMEM for 30 min and pulse labeled with 35S-Met/Cys for the indicated times. Cells were lysed in 500 μL cold radioimmunoprecipitation assay (RIPA) buffer and precleared with Zysorbin for 1 h. After immunoprecipitation with anti-insulin overnight at 4°C, samples were subjected to 4–12% NuPage gradient gels under reducing and denaturing conditions.
Immunofluorescence
Cells were fixed with 4% formaldehyde for 30 min at room temperature and permeabilized with 0.2% Triton X-100 for an additional 15 min. After blocking for 2 h in the presence of 5% BSA, cells were incubated with antiproinsulin, anti-insulin, and anti-Flag antibodies at 4°C overnight, and incubated with fluorophore-conjugated secondary antibodies for 1 h at room temperature. Finally, sections were treated with ProLong Gold Antifade Mountant with DAPI and imaged with a Nikon A1 confocal microscope (Nikon Instruments, Melville, NY).
Western Blotting
Cells were lysed in RIPA buffer with protease inhibitor cocktail on ice for 15 min and centrifuged at 12,000g at 4°C for 15 min. Total protein lysates were boiled in sample buffer containing 100 mmol/L dithiothreitol for 5 min, separated by 4–12% gradient NuPage gels, and electrotransferred to nitrocellulose for blotting with primary antibodies (1:1,000, diluted in Tris-buffered saline with Tween with 5% BSA) at 4°C overnight. Horseradish peroxidase–conjugated secondary antibody (1:5,000) incubation was performed at room temperature for 1 h, with peroxidase reaction using the Clarity Western ECL Substrate.
Quantitative PCR
The relative mRNA levels were determined by quantitative RT-PCR, with specific primers listed in Table 1.
Table 1.
List of primers used for quantitative RT-PCR
| Gene | Forward | Reverse |
|---|---|---|
| SSR1 | 5′-GGCCTGCAGAACTGTATGGT-3′ | 5′-AACTCTGCGGGCCATTTACA-3′ |
| SSR2 | 5′-CCCTGGGGTGAGAACTCATT-3′ | 5′-CGAGGTGCTGGGATAGAACC-3′ |
| SSR3 | 5′-GGGAATGCGTTCATCGTGTC-3′ | 5′-GAGGGCGATGAGTCCAGATG-3′ |
| SSR4 | 5′-GAGAAGGAGGGAAGTGCGAC-3′ | 5′-AACGTCGGCATAAAGAGCCA-3′ |
| Rat INS1/2 | 5′-TCAGCAAGCAGGTCATTGTTC-3′ | 5′-CCACAAAGGTGCTGTTTGACA-3′ |
| Human INS | 5′-GGGGACCTGACCCAGCC-3′ | 5′-GCAGGTCCTCTGCCTCC-3′ |
| Actin | 5′-GACTTCGAGCAAGAGATGGCCA-3′ | 5′-CCAGACAGCACTGTGTTGGC-3′ |
Statistical Analysis
Statistical analysis was performed using Student t test and one-way ANOVA test to determine the differences between groups (GraphPad Prism 8 software). Data are presented as mean ± SD. P < 0.05 was considered statistically significant.
Data and Resource Availability
Data generated and analyzed during the current study are contained within the figures. Additional data are available from the corresponding author upon request.
Results
Effect of TRAPγ/SSR3 Deficiency on Recombinant Proinsulin Biosynthesis
As a preliminary step in determining the extent to which TRAPγ/SSR3 is needed for proinsulin biosynthesis, we used CRISPR/Cas9-mediated KO of TRAPγ/SSR3 in 293T cells and transfected these cells to express a recombinant human INS cDNA. Unlike control 293T cells, TRAPγ/SSR3-KO 293T cells expressed no detectable TRAPγ/SSR3 protein by Western blot, and with this was a partial effect on the level of other TRAP subunits (especially TRAPδ/SSR4) (Fig. 1B, with quantitation in Fig. 1C). By immunoblotting, intracellular proinsulin levels in TRAPγ/SSR3-KO 293T cells were roughly halved (Fig. 1B, light exposure), and this could not be explained by increased proinsulin secretion (observations obtained in pancreatic β-cells are described below). However, we noted a small intracellular increase in preproinsulin in TRAPγ/SSR3-KO 293T cells (Fig. 1B, dark exposure). It is known that if preproinsulin undergoes translocation but fails signal peptide cleavage (e.g., as occurs in cleavage site mutants), such preproinsulin will become oxidized within the ER lumen, which is detectable by a difference in SDS gel mobility when analyzed under reducing versus nonreducing conditions (21). However, whereas proinsulin is detectably oxidized in control cells and those with TRAPγ/SSR3-KO, preproinsulin is not (Supplementary Fig. 1), indicating that preproinsulin has not been delivered into the ER lumen. Similarly, when these cells were pulse labeled for increasingly short times with 35S-amino acids, preproinsulin was more prominent and proinsulin less prominent in TRAPγ/SSR3-KO cells (Fig. 1D). These initial data demonstrate that TRAPγ/SSR3 deficiency results in less efficient biosynthesis of recombinant proinsulin from preproinsulin in heterologous cells.
We recently reported that diminished proinsulin biosynthesis in cells with genetic defects in TRAPβ/SSR2 or TRAPδ/SSR4 could be at least partially suppressed upon overexpression of the ER lumenal partner TRAPα/SSR1 (19). We therefore further examined TRAPγ/SSR3-KO cells for the ability to rescue steady-state levels of transfected proinsulin upon forced expression of individual TRAP/SSR subunits. Whereas plasmid transfection to express epitope-tagged TRAPγ/SSR3 in TRAPγ/SSR3-KO cells resulted in a doubling in intracellular proinsulin (Supplementary Fig. 2A and B), a much smaller or nonexistent change of proinsulin level was observed in cells transfected to overexpress epitope-tagged TRAPα/SSR1, TRAPβ/SSR2, or TRAPδ/SSR4 (Supplementary Fig. 2A and B, gray bars). These data indicate that for proinsulin biosynthesis, the absence of TRAPγ/SSR3 cannot be compensated by other TRAP/SSR1 subunits.
Effect of TRAPγ/SSR3 Deficiency on Endogenous Proinsulin Biosynthesis
We next attempted siRNA-mediated KD of TRAPγ/SSR3 (SSR3i) in INS1E rat pancreatic β-cells (this maneuver does not diminish cellular levels of Ins1 + Ins2 mRNA) (Supplementary Fig. 3A). KD of TRAPγ/SSR3 resulted in a halving of steady-state levels of endogenous proinsulin (Fig. 2A–C). To determine whether this behavior in β-cells is mechanistically similar to that in 293T cells (Fig. 1 and Supplementary Fig. 2), we metabolically labeled control and TRAPγ/SSR3-KD cells for increasingly short pulse periods and immunoprecipitated the newly synthesized proinsulin for analysis by SDS-PAGE and quantitative phosphorimaging (Fig. 2D and E). On average, TRAPγ/SSR3-KD β-cells synthesized ∼40% less proinsulin than control INS1E cells, indicating that much of the decreased steady-state level of proinsulin (Fig. 2C) could be accounted for by diminished proinsulin biosynthesis.
Figure 2.

Effect of TRAPγ/SSR3 deficiency on endogenous proinsulin biosynthesis in β-cells. A: INS1E cells were transfected with scrambled oligo or TRAPγ/SSR3-KD (SSR3i). At 72 h posttransfection, cells were lysed and analyzed by reducing SDS-PAGE and immunoblotted with antiproinsulin and anti-SSR3 antibodies. B and C: Quantitation (mean ± SD) of TRAPγ/SSR3 protein and proinsulin protein from three independent experiments (normalized to tubulin). D: At 72 h posttransfection, control (Ctrl) and TRAPγ/SSR3-KD (SSR3i) INS1E cells were pulse labeled with 35S-Met/Cys for the indicated times. Cell lysates (normalized to trichloroacetic acid–precipitable counts) were immunoprecipitated with anti-insulin and analyzed by reducing SDS-PAGE and phosphorimaging. E: Quantitation (mean ± SD) of newly synthesized proinsulin from five independent experiments like that shown in D, with the proinsulin band from Ctrl INS1E cells at 10 min labeling set to 1. *P < 0.05 compared with Ctrl.
INS832/13 β-cells express human preproinsulin (22) and are relatively flat and thus favorable for immunofluorescence. All INS832/13 cells express SSR3 (Supplementary Fig. 4), but after siRNA transfection for TRAPγ/SSR3-KD, it was apparent that some INS832/13 cells had nearly complete loss of TRAPγ/SSR3, while other cells in the population had little or no KD (Fig. 3A, red). Interestingly, β-cells with obvious TRAPγ/SSR3-KD also had loss of immunostainable insulin (Fig. 3A, green). At the biochemical level in the overall cell population, this acute TRAPγ/SSR3-KD in INS832/13 cells also resulted in a decrease in the steady-state level of proinsulin (Fig. 3B, top panels) with no decrease of TRAPα/SSR1, a small decrease of TRAPβ/SSR2, and an ∼50% decrease in TRAPδ/SSR4 (Fig. 3B, quantified in Fig. 3C). Moreover, while proteasome inhibition with MG132 (4 h) did not significantly increase human proinsulin levels (Fig. 3B, quantified in Fig. 3D), this treatment augmented human preproinsulin levels (Fig. 3B, quantified in Fig. 3E), thereby raising the preproinsulin-to-proinsulin ratio in TRAPγ/SSR3-KD cells (Fig. 3F) and suggesting that inefficiently translocated human preproinsulin leads to its proteasomal disposal. Finally, along with the detected decrease of proinsulin in TRAPγ/SSR3-KD cells was a marked decrease of stored processed insulin (Fig. 3G).
Figure 3.

TRAPγ/SSR3 deficiency results in a decrease in the steady-state level of stored, processed insulin in β-cells. INS832/13 cells were transfected with TRAPγ/SSR3-targeted siRNA. A: Immunofluorescence with anti-TRAPγ/SSR3 (red) and anti-insulin (green) in INS832/13 cells at 72 h posttransfection. Nuclei were counterstained with DAPI (blue). B: Cells transfected as in A were treated with or without MG132 (10 μmol/L) for 4 h. Cells were lysed and analyzed by reducing SDS-PAGE and immunoblotting with either anti-human-(pre)proinsulin or a battery of antibodies, as indicated. C: Quantitation (mean ± SD, n = 8 independent experiments) of each of the TRAP/SSR subunits in control (Ctrl) cells and TRAPγ/SSR3-KD (SSR3i). D: Quantitation of human proinsulin levels from five independent experiments like that shown in B. E: Quantitation of human preproinsulin levels from five independent experiments like that shown in B. F: Human preproinsulin-to-proinsulin ratios in MG132-treated INS832/13 cells were calculated from the experiments shown in D and E (n = 5). G: Quantitation (mean ± SD) of insulin protein levels from three independent experiments like that shown in B. *P < 0.05 compared with Ctrl. h, human; PI, proinsulin; PPI, preproinsulin.
Altogether, these data point to a role for TRAPγ/SSR3 in the translocation of preproinsulin in the ER as well as the size of the insulin storage pool (7), which is the most well-recognized marker of β-cell identity. In considering this further, we note that physiological regulation of proinsulin biosynthesis is highly attuned to extracellular glucose levels (23). We therefore examined the proinsulin response of control INS1E cells or TRAPγ/SSR3-KD cells that are normally grown in the presence of 11.1 mmol/L glucose. We exposed the cells for 4 h to different glucose concentrations, an exposure time too brief to cause significant change of INS mRNA levels or the mRNA levels of the TRAP/SSR subunits (Supplementary Fig. 3B). TRAPγ/SSR3-KD cells exhibited the expected decrease of TRAPγ/SSR3 protein levels, but at 2.5 mmol/L glucose, there was no appreciable decrease of proinsulin levels (Fig. 4A, quantified in gray bars and open bars of Fig. 4B). However, as the β-cells were incubated at higher glucose levels, the abundance of TRAPγ/SSR3 protein significantly increased (Fig. 4A, left, quantified in blue bars of Fig. 4B). Importantly, the increase of TRAPγ/SSR3 protein was accompanied by a rise in proinsulin (Fig. 4A, quantified in gray bars of Fig. 4B), a result that was also observed in cells cotreated with the transcription blocker actinomycin D. However, in TRAPγ/SSR3-KD cells, the effect of higher glucose levels on TRAPγ/SSR3 protein was blocked (Fig. 4A, left, quantified in Fig. 4B), and this severely blunted the glucose-dependent rise in proinsulin levels (Fig. 4A and B). The blunted increase of intracellular mature insulin levels in TRAPγ/SSR3-KD β-cells in response to high-glucose treatment (Fig. 4A, top left, and Fig. 4B) could not be ascribed to enhanced insulin release into the medium; indeed, insulin secretion in TRAPγ/SSR3-KD β-cells was actually decreased compared with that from control cells (Fig. 4A, bottom left). In addition, the limited increase of intracellular proinsulin in TRAPγ/SSR3-KD β-cells could not be accounted for by any discernible increase of misfolded proinsulin disulfide-linked dimers or higher-molecular-weight complexes (24), as analyzed by nonreducing SDS-PAGE and Western blotting (Fig. 4A, right). Thus, the data indicate that the inability to upregulate TRAPγ/SSR3 protein blunts the ability of β-cells to increase intracellular proinsulin (and insulin) levels in response to high glucose (Fig. 4A and B).
Figure 4.

The ability of β-cells to acutely increase proinsulin (and insulin) level in response to high-glucose stimulation depends on TRAPγ/SSR3. A: INS1E cells were transfected with scrambled oligo or TRAPγ/SSR3-KD (SSR3i). At 72 h posttransfection, cells were preincubated in RPMI medium containing 2.5 mmol/L glucose for 1.5 h followed by a 4-h incubation at either 2.5, 11.1, or 25 mmol/L glucose, respectively. Cells were lysed and analyzed by SDS-PAGE under reducing conditions (left) or nonreducing conditions (right) for immunoblotting with the antibodies indicated. The 4-h media were collected and similarly analyzed by SDS-PAGE and immunoblotting with anti-insulin (bottom left). B: Quantitation (mean ± SD) of proinsulin, insulin, and each TRAP/SSR subunit from five independent experiments. *P < 0.05. Ctrl, control.
The foregoing data demonstrating TRAPγ/SSR3 dependence on acute changes of proinsulin level at different extracellular glucose levels would seem to imply a primary effect of TRAPγ/SSR3 on proinsulin biosynthesis. On the other hand, as an alternative, low proinsulin levels in starved β-cells have been proposed to be caused by lysosomal fusion with nascent proinsulin-rich immature secretory granules to promote their degradation (25). To more carefully assess the requirement of TRAPγ/SSR3 in the maintenance of intracellular proinsulin levels in pancreatic β-cells, we created INS832/13 cells with CRISPR/Cas9-mediated TRAPγ/SSR3-KO and then used plasmid-mediated transfection in an attempt to restore TRAPγ/SSR3 function. TRAPγ/SSR3-KO cells exhibited zero detectable TRAPγ/SSR3 protein (Fig. 5A, quantified in Fig. 5B), which was accompanied by a significant lowering of TRAPδ/SSR4 (Fig. 5A, quantified in white bar of Fig. 5B) but no major change of TRAPα/SSR1 (Fig. 5A, quantified in white bar of Fig. 5B). In such cells, steady-state proinsulin levels dropped by 75% (Fig. 5A, quantified in white bar of Fig. 5B). Conventional lipofectamine-mediated plasmid transfection in INS832/13 cells is rather inefficient; nevertheless, transient expression of epitope-tagged TRAPγ/SSR3-KO resulted in the appearance of detectable TRAPγ/SSR3-KO protein (Fig. 5A, quantified in cross-hatched bar of Fig. 5B) accompanied by increased proinsulin levels (Fig. 5A, quantified in cross-hatched bar of Fig. 5B). Proinsulin (Fig. 5C, quantified in white bar of Fig. 5D) increased selectively in TRAPγ/SSR3-KO cells that re-expressed TRAPγ/SSR3. Moreover, in INS832/13 cells bearing KD of either TRAPα/SSR1 or TRAPγ/SSR3, the re-expression of (Flag-tagged) SSR3 only effectively restored proinsulin levels in cells deficient for TRAPγ/SSR3 but not in cells deficient for TRAPα/SSR1 (Supplementary Fig. 5), indicating that SSR3 function (and its own protein stability) depends on other members of the TRAP/SSR complex. As TRAP/SSR acts in preproinsulin translocation into the ER (Figs. 1 and 2), the foregoing data, taken together, suggest that regulation of proinsulin steady-state level in β-cells incubated in lower versus higher extracellular glucose may be tightly linked to TRAP/SSR-supported preproinsulin translocation for proinsulin biosynthesis, which in turn is required for insulin biosynthesis (Fig. 5E).
Figure 5.

TRAPγ/SSR3 re-expression rescues proinsulin protein levels in TRAPγ/SSR3-KO β-cells. TRAPγ/SSR3-KO INS832/13 cells were transfected with empty vector (−) or plasmids encoding Flag-tagged TRAPγ/SSR3 cDNA (+). Wild-type (WT) INS832/13 cells were also transfected with empty vector as control. A: At 48 h posttransfection, cells were lysed and analyzed by reducing SDS-PAGE and immunoblotting for anti-rodent proinsulin and the additionally indicated antibodies. B: Quantitation (mean ± SD) of proinsulin and each TRAP/SSR subunit from four independent experiments like that shown in A; expression levels in WT control cells were set to 1.0. C: TRAPγ/SSR3-KO INS832/13 cells transfected as in A were examined by immunofluorescence with anticalnexin (red), anti-Flag (Flag-SSR3, purple), and anti-rodent proinsulin (green); a merged anti-Flag/antiproinsulin image is shown. D: Quantitation (mean ± SD) of rodent proinsulin–positive TRAPγ/SSR3-KO cells that either re-express or do not re-express TRAPγ/SSR3 protein from three independent experiments like that shown in C. E: Quantitation (mean ± SD) of insulin-positive TRAPγ/SSR3-KO cells that either re-express or do not re-express TRAPγ/SSR3 protein from four independent experiments. *P < 0.05.
Subunits within the heterotetrameric TRAP/SSR complex exist in a 1:1:1:1 stoichiometry, but the stoichiometry of their biosynthesis is unknown. As glucose-dependent upregulation of proinsulin biosynthesis requires upregulation of TRAPγ/SSR3 protein (Fig. 4), we wondered whether forced expression of TRAPγ/SSR3 might enhance proinsulin production even in control β-cells that have no induced defect in expression of any of the four endogenous TRAP/SSR subunits. Such an effect might especially be anticipated if the biosynthetic expression of TRAPγ/SSR3 at normoglycemic levels lags stoichiometrically that of one or more of the other TRAP complex subunits. Remarkably, a very modest 19% increase of expression of TRAPγ/SSR3 protein increased the steady-state levels of both the endogenous (rat) proinsulin and the transfected (human) proinsulin protein expressed in INS832/13 cells (Fig. 6A and B). This effect of TRAPγ/SSR3 in INS832/13 cells could not be emulated by overexpression of any other TRAP/SSR subunits (Fig. 6C and D) or even after a fivefold increase of TRAPα/SSR1 upon adenoviral-mediated overexpression (Supplementary Fig. 6), despite the established importance of TRAPα/SSR1 in proinsulin biosynthesis (3,4,19). The same selective phenotype upon increased TRAPγ/SSR3 expression (but not other TRAP/SSR subunits) was also observed to increase the level of recombinant proinsulin expressed heterologously in 293T cells (Fig. 6E and F). Indeed, in human nondiabetic islets, a 2-h exposure increasing the extracellular glucose from 2.5 to 25 mmol/L caused little effect on TRAP/SSR subunits except TRAPγ/SSR3 protein levels, which rose by 94% (1.48 ± 0.08 to 2.87 ± 0.04) while proinsulin levels jumped by 112% (1.08 ± 0.16 to 2.29 ± 0.37) (Supplementary Fig. 7). Notably, the increase of TRAPγ/SSR3 and proinsulin protein were both totally blocked when the protein synthesis inhibitor CHX was included during the 2-h high-glucose incubation (Fig. 6G and Supplementary Fig. 7). Altogether, the data in Fig. 6 strongly suggest that the glucose-regulated protein expression of endogenous TRAPγ/SSR3 is a critical, limiting factor that affects the functionality of the TRAP complex both for endogenous and recombinant proinsulin biosynthesis in β-cell lines and for proinsulin translation/translocation endogenous to human pancreatic islets.
In recent years, it has been recognized that whereas the major fraction of preproinsulin translocation is likely to be cotranslational, a smaller subfraction may be translocated posttranslationally (7,8). As TRAPγ/SSR3 topology involves exposure on the cytosolic side of the ER membrane (Fig. 1A), we wondered whether TRAPγ/SSR3 might also participate in the posttranslational translocation of preproinsulin. It is technically challenging to study posttranslational translocation of wild-type preproinsulin; therefore, we examined translocation of the diabetogenic preproinsulin-R6C mutant, which has lost positive charge in the N-terminal segment of the signal peptide, rendering it defective for cotranslational translocation and shows an increased fraction of posttranslational ER entry (26,27). In TRAPγ/SSR3-KO 293T cells, we observed a defect in steady-state proinsulin levels in cells expressing preproinsulin-R6C (Fig. 7A, lane 1 vs. 3). It was also clear that in both control 293T cells and TRAPγ/SSR3-KO cells, a significant fraction of untranslocated mutant preproinsulin-R6C was routed to proteasomal degradation, as the preproinsulin level was greatly increased in the presence of MG132 (Fig. 7A, lanes 2 and 4); however, in TRAPγ/SSR3-KO cells, augmentation of preproinsulin levels upon MG132 treatment did not restore translocation-dependent conversion to proinsulin (Fig. 7A, lane 4). Moreover, in control cells pulse labeled with 35S-amino acids and chased in the presence of both MG132 and CHX (to block postpulse incorporation of labeled amino acids into newly synthesized protein), there was a posttranslational increase of newly synthesized proinsulin 30 min after the initial synthesis of preproinsulin-R6C (Fig. 7B, lane 3). However, in SSR3-KO cells, newly synthesized proinsulin not only was diminished immediately postpulse (lane 4) but also did not increase during the course of a 30-min chase (Fig. 7B). These data suggest that in addition to supporting cotranslational preproinsulin translocation, TRAPγ/SSR3 may also contribute to the efficiency of preproinsulin posttranslational translocation.
Figure 7.
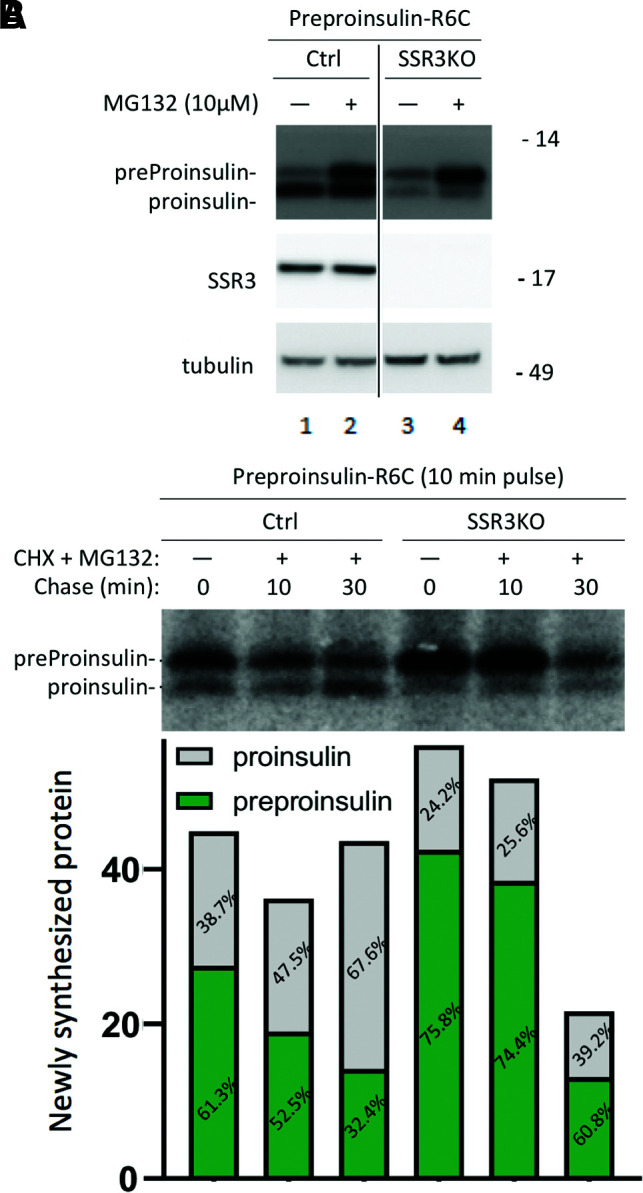
Effects of TRAPγ/SSR3 on the posttranslational translocation of mutant preproinsulin-R6C. Control (Ctrl) 293T cells and SSR3-KO 293T cells were transfected to express untagged human preproinsulin-R6C. A: At 48 h posttransfection, cells were treated with or without MG132 (10 μmol/L) for 2 h. The cells were lysed and analyzed by reducing SDS-PAGE and immunoblotting with the indicated antibodies. B: After transfection as in A, cells were pulse labeled with 35S-Met/Cys for 10 min followed by a 0-, 10-, or 30-min chase in the presence of CHX (10 μg/mL) and MG132 (10 μmol/L). Cell lysates (normalized to trichloroacetic acid–precipitable counts) were subjected to immunoprecipitation with anti-insulin and analyzed by reducing SDS-PAGE and phosphorimaging; both the absolute and the relative amounts of recovered preproinsulin and proinsulin are shown below each lane of the gel.
Discussion
The TRAP/SSR complex is believed to assist most in the translocation of a subset of proteins that are said to have a weak signal peptide (9). Of such proteins, preproinsulin translocation has been found to depend on the TRAP complex, especially TRAPα/SSR1 (3,4,19). Interestingly, earlier classic studies showed that TRAPα/SSR1 can be cross-linked to nascent chains that have already had their signal peptide cleaved by signal peptidase (20), and for a molecule the size of proinsulin, such cleavage would essentially be concurrent with the protein having been fully translocated across the ER membrane (28). This makes sense, given that the bulk of the extramembranous TRAP/SSR complex, including TRAPα/SSR1, resides on the lumenal side of the ER membrane (Fig. 1A). Thus, although we do not understand precisely the assistance that TRAPα/SSR1 provides to preproinsulin, these observations taken together (11–13) suggest that TRAPα/SSR1 may interact on the lumenal side of the ER membrane, perhaps contributing to pulling forces involved in translocation (28). Our recent studies suggest that TRAPβ/SSR2 and TRAPδ/SSR4, whose extramembranous domains are also primarily lumenal, are important to the stability of the TRAPα/SSR1 subunit, and to some extent, the defect in proinsulin biosynthesis caused by deficiency of TRAPβ/SSR2 and TRAPδ/SSR4 can be suppressed by simple overexpression of TRAPα/SSR1 (19).
In contrast, structural studies suggest that the extramembranous portion of TRAPγ/SSR3 provides a primary function on the cytosolic side of the ER membrane (11,14); perhaps even making direct contact with the N-terminal tip of the signal peptide to facilitate hairpin insertion into the Sec61 translocon (18). Importantly, unlike the situation for the other TRAP/SSR subunits (19), we find that loss of TRAPγ/SSR3 does not result in significant depletion of TRAPα/SSR1 (Supplementary Fig. 2 and Figs. 3B and C, 4A and B, and 5A and B). Yet in spite of this, TRAPγ/SSR3 deficiency impairs proinsulin biosynthesis with a defect in preproinsulin translocation that is at least partially cotranslational (Figs. 1 and 2) and might be partially posttranslational (Fig. 7). Restoration of proinsulin levels in TRAPγ/SSR3-KO cells cannot be achieved by overexpression of TRAPα/SSR1 (Supplementary Fig. 2) but can be selectively rescued by re-expression of TRAPγ/SSR3 in pancreatic β-cells, which is accompanied by rescued insulin levels (Fig. 5).
Webb et al. (6) found that after a 24-h exposure to 25 vs. 5.5 mmol/L glucose, the mRNA encoding TRAPγ/SSR3 was the single most differentially upregulated transcript of all secretory pathway gene products, speaking to its potential physiological significance. However, we observed that even after a brief exposure to high glucose, before any significant change in the mRNA levels of INS or TRAP/SSR subunits (Supplementary Fig. 3B), the TRAPγ/SSR3 protein level is increased more than other TRAP subunits (Fig. 4), and this is also apparent in human nondiabetic pancreatic islets (Fig. 6G), suggesting important acute translational regulation of TRAPγ/SSR3.
What is crucial from these considerations is that almost all the glucose-induced increase in proinsulin level within 4 h requires the ability to upregulate the level of TRAPγ/SSR3 protein (Fig. 4). The data strongly suggest that acute upregulation of TRAPγ/SSR3 protein levels (above and beyond those found in β-cells at lower prevailing glucose concentrations) is required for the upregulation of proinsulin biosynthesis in response to acute high-glucose exposure. These observations are certainly consistent with the idea that acute changes in proinsulin biosynthesis are translationally regulated (7,8,29,30). Additionally, because much preproinsulin translocation is cotranslational (31), there may be a tight relationship between proinsulin translational regulation and preproinsulin translocation, in which case, TRAPγ/SSR3 protein expression may be a rate-limiting factor for formation of the functional TRAP/SSR complexes that are needed for proinsulin biosynthesis.
With these ideas in mind, we are excited to observe that simple overexpression of TRAPγ/SSR3 in cells that are not deficient for any TRAP/SSR subunit can immediately increase proinsulin levels in β-cells (Fig. 6). This phenotype cannot be re-created by expression of TRAPα/SSR1, even to high levels (Supplementary Fig. 6). Thus, we posit that efficient acute modulation of proinsulin levels by extracellular glucose in pancreatic β-cells depends on changes in TRAPγ/SSR3 protein levels that help to shift the efficiency of the preproinsulin translation product away from proteasomal destruction and toward translocation into the ER such that simple overexpression of TRAPγ/SSR3 can, at least in part, bypass the high-glucose effect to stimulate intracellular proinsulin levels. Additionally, we offer suggestive evidence that TRAPγ/SSR3 may help to facilitate posttranslational translocation of preproinsulin (Fig. 7). In conclusion, factors that upregulate TRAPγ/SSR3 protein expression in pancreatic β-cells may deserve consideration as a means to enhance insulin biogenesis in states of insulin insufficiency, including states of diminished β-cell mass or increased insulin resistance requiring compensatory upregulation of β-cell insulin production.
Article Information
Funding. This collaborative work was supported in the U.S. by the National Institutes of Health (R01-DK-111174 and R01-DK-48280) and in China by the National Natural Science Foundation of China (81830025, 81620108004, 82000796, and 81700699), the National Key R&D Program of China (2019YFA0802502), the PUMC Youth Fund supported by the Fundamental Research Funds for the Central Universities (3332020080), and the Tianjin Municipal Science and Technology Commission (17ZXMFSY00150 and 18JCQNJC82100). We acknowledge the University of Michigan Protein Folding Diseases Initiative.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. X.X., Y.H., and X.L. conducted the investigation, generated and reviewed research data, and contributed to discussions. P.A. and M.L. designed the research project, supervised the study, and acquired funding. All authors contributed to writing, editing, and reviewing the manuscript. P.A. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Prior Presentation. Parts of this study were presented as a symposium talk at the 81st Scientific Sessions of the American Diabetes Association, 25–29 June 2021.
Footnotes
X.X. and Y.H. contributed equally to this work.
This article contains supplementary material online at https://doi.org/10.2337/figshare.17064434.
References
- 1. Mahajan A, Taliun D, Thurner M, et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet 2018;50:1505–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mahajan A, Go MJ, Zhang W, et al.; DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium; Asian Genetic Epidemiology Network Type 2 Diabetes (AGEN-T2D) Consortium; South Asian Type 2 Diabetes (SAT2D) Consortium; Mexican American Type 2 Diabetes (MAT2D) Consortium; Type 2 Diabetes Genetic Exploration by Next-generation sequencing in multi-Ethnic Samples (T2D-GENES) Consortium . Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet 2014;46:234–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li X, Itani OA, Haataja L, et al. Requirement for translocon-associated protein (TRAP) α in insulin biogenesis. Sci Adv 2019;5:eaax0292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kriegler T, Kiburg G, Hessa T. Translocon-associated protein complex (TRAP) is crucial for co-translational translocation of pre-proinsulin. J Mol Biol 2020;432:166694. [DOI] [PubMed] [Google Scholar]
- 5. Schuit FC, In’t Veld PA, Pipeleers DG. Glucose stimulates proinsulin biosynthesis by a dose-dependent recruitment of pancreatic beta cells. Proc Natl Acad Sci U S A 1988;85:3865–3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Webb GC, Akbar MS, Zhao C, Steiner DF. Expression profiling of pancreatic beta cells: glucose regulation of secretory and metabolic pathway genes. Proc Natl Acad Sci U S A 2000;97:5773–5778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu M, Huang Y, Xu X, et al. Normal and defective pathways in biogenesis and maintenance of the insulin storage pool. J Clin Invest 2021;131:e142240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu M, Weiss MA, Arunagiri A, et al. Biosynthesis, structure, and folding of the insulin precursor protein. Diabetes Obes Metab 2018;20(Suppl. 2):28–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fons RD, Bogert BA, Hegde RS. Substrate-specific function of the translocon-associated protein complex during translocation across the ER membrane. J Cell Biol 2003;160:529–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rapoport TA. Protein transport across the endoplasmic reticulum membrane: facts, models, mysteries. FASEB J 1991;5:2792–2798 [DOI] [PubMed] [Google Scholar]
- 11. Pfeffer S, Dudek J, Schaffer M, et al. Dissecting the molecular organization of the translocon-associated protein complex. Nat Commun 2017;8:14516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lang S, Nguyen D, Pfeffer S, Förster F, Helms V, Zimmermann R. Functions and mechanisms of the human ribosome-translocon complex. Subcell Biochem 2019;93:83–141 [DOI] [PubMed] [Google Scholar]
- 13. Gemmer M, Förster F. A clearer picture of the ER translocon complex. J Cell Sci 2020;133:jcs231340. [DOI] [PubMed] [Google Scholar]
- 14. Bañó-Polo M, Martínez-Garay CA, Grau B, Martínez-Gil L, Mingarro I. Membrane insertion and topology of the translocon-associated protein (TRAP) gamma subunit. Biochim Biophys Acta Biomembr 2017;1859:903–909 [DOI] [PubMed] [Google Scholar]
- 15. Ng BG, Lourenço CM, Losfeld M-E, et al.; University of Washington Center for Mendelian Genomics . Mutations in the translocon-associated protein complex subunit SSR3 cause a novel congenital disorder of glycosylation. J Inherit Metab Dis 2019;42:993–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Phoomak C, Cui W, Hayman TJ, et al. The translocon-associated protein (TRAP) complex regulates quality control of N-linked glycosylation during ER stress. Sci Adv 2021;7:eabc6364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dittner-Moormann S, Lourenco CM, Reunert J, et al. TRAPγ-CDG shows asymmetric glycosylation and an effect on processing of proteins required in higher organisms. J Med Genet 2021;58:213–216 [DOI] [PubMed] [Google Scholar]
- 18. Nguyen D, Stutz R, Schorr S, et al. Proteomics reveals signal peptide features determining the client specificity in human TRAP-dependent ER protein import. Nat Commun 2018;9:3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huang Y, Xu X, Arvan P, Liu M. Deficient endoplasmic reticulum translocon-associated protein complex limits the biosynthesis of proinsulin and insulin. FASEB J 2021;35:e21515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Görlich D, Prehn S, Hartmann E, et al. The signal sequence receptor has a second subunit and is part of a translocation complex in the endoplasmic reticulum as probed by bifunctional reagents. J Cell Biol 1990;111:2283–2294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu M, Lara-Lemus R, Shan SO, et al. Impaired cleavage of preproinsulin signal peptide linked to autosomal-dominant diabetes. Diabetes 2012;61:828–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hohmeier HE, Mulder H, Chen G, Henkel-Rieger R, Prentki M, Newgard CB. Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes 2000;49:424–430 [DOI] [PubMed] [Google Scholar]
- 23. Vasiljević J, Torkko JM, Knoch K-P, Solimena M. The making of insulin in health and disease. Diabetologia 2020;63:1981–1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Arunagiri A, Haataja L, Pottekat A, et al. Proinsulin misfolding is an early event in the progression to type 2 diabetes. eLife 2019;8:e44532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goginashvili A, Zhang Z, Erbs E, et al. Insulin granules. Insulin secretory granules control autophagy in pancreatic β cells. Science 2015;347:878–882 [DOI] [PubMed] [Google Scholar]
- 26. Guo H, Xiong Y, Witkowski P, et al. Inefficient translocation of preproinsulin contributes to pancreatic β cell failure and late-onset diabetes. J Biol Chem 2014;289:16290–16302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guo H, Sun J, Li X, et al. Positive charge in the n-region of the signal peptide contributes to efficient post-translational translocation of small secretory preproteins. J Biol Chem 2018;293:1899–1907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kriegler T, Magoulopoulou A, Amate Marchal R, Hessa T. Measuring endoplasmic reticulum signal sequences translocation efficiency using the Xbp1 arrest peptide. Cell Chem Biol 2018;25:880–890.e3 [DOI] [PubMed] [Google Scholar]
- 29. Lee EK, Kim W, Tominaga K, et al. RNA-binding protein HuD controls insulin translation. Mol Cell 2012;45:826–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Itoh N, Okamoto H. Translational control of proinsulin synthesis by glucose. Nature 1980;283:100–102 [DOI] [PubMed] [Google Scholar]
- 31. Wolin SL, Walter P. Discrete nascent chain lengths are required for the insertion of presecretory proteins into microsomal membranes. J Cell Biol 1993;121:1211–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]