

Abstract
Individuals with type 1 diabetes have an impaired glucagon counterregulatory response to hypoglycemia. Sodium—glucose cotransporter (SGLT) inhibitors increase glucagon concentrations. We evaluated whether SGLT inhibition restores the glucagon counterregulatory hormone response to hypoglycemia. Adults with type 1 diabetes (n = 22) were treated with the SGLT2 inhibitor dapagliflozin (5 mg daily) or placebo for 4 weeks in a randomized, double-blind, crossover study. After each treatment phase, participants underwent a hyperinsulinemic-hypoglycemic clamp. Basal glucagon concentrations were 32% higher following dapagliflozin versus placebo, with a median within-participant difference of 2.75 pg/mL (95% CI 1.38–12.6). However, increased basal glucagon levels did not correlate with decreased rates of hypoglycemia and thus do not appear to be protective in avoiding hypoglycemia. During hypoglycemic clamp, SGLT2 inhibition did not change counterregulatory hormone concentrations, time to recovery from hypoglycemia, hypoglycemia symptoms, or cognitive function. Thus, despite raising basal glucagon concentrations, SGLT inhibitor treatment did not restore the impaired glucagon response to hypoglycemia. We propose that clinical reduction in hypoglycemia associated with these agents is a result of changes in diabetes care (e.g., lower insulin doses or improved glycemic variability) as opposed to a direct, physiologic effect of these medications on α-cell function.
Introduction
Hypoglycemia remains a major obstacle to achieving optimal glycemic control for people living with type 1 diabetes and accounts for significant morbidity and mortality in this population (1). On average, individuals with type 1 diabetes experience multiple episodes of symptomatic hypoglycemia per week and thousands of episodes over their lifetime (2). The incidence of severe hypoglycemia (requiring the assistance of another individual) ranges from 1.0 to 4.9 episodes/patient/year (3,4). Asymptomatic hypoglycemia is also problematic, as these episodes impair the physiologic counterregulatory response to subsequent low blood glucose events and propagate hypoglycemia unawareness (5). Additionally, fear of hypoglycemia is common among people with type 1 diabetes and acts as a barrier to optimizing insulin therapy and achieving glycemic goals (3). Thus, there is a clear unmet need for treatment approaches that mitigate the harmful effects of hypoglycemia in type 1 diabetes.
One of the principal reasons for increased susceptibility to hypoglycemia among people living with type 1 diabetes is an impaired glucagon counterregulatory response (3). Under normal conditions, glucagon secreted from the α-cells of the pancreatic islets stimulates hepatic glucose production to reverse hypoglycemia. However, this glucagon rescue response is lost by most individuals shortly after diagnosis with type 1 diabetes, purportedly due to abolished β- to α-cell paracrine signaling, leading to an impaired ability to defend against, and recover from, hypoglycemia (3,6). Various approaches to address this defect have been explored, including providing patients with glucagon pens for emergencies, giving glucagon via subcutaneous infusion, and islet cell or whole pancreas transplantation (7,8). Unfortunately, these therapies all have limitations and have thus far been unable to reduce the burden of hypoglycemia for most people living with type 1 diabetes.
In recent years, there has been considerable interest in the use of sodium–glucose cotransporter (SGLT) inhibitors as adjunctive treatments to insulin, with members of the class being approved for use in type 1 diabetes in both Europe and Japan (9). The SGLT inhibitors have demonstrated an ability to reduce insulin dosing requirements and improve glycemic control without increasing, and potentially even decreasing, the risk of hypoglycemia (9). Specifically, in a post hoc analysis of pooled data from two clinical trials studying 1,362 participants with type 1 diabetes, the dual SGLT1/2 inhibitor sotagliflozin reduced hypoglycemia by ∼25% compared with placebo (10).
The reason for this reduction in hypoglycemia is not well established. However, several lines of evidence suggest that increases in circulating glucagon concentrations in response to SGLT inhibition may play a role. First, Bonner et al. (11) reported that SGLT2 is expressed in pancreatic α-cells and that treatment with the SGLT2 inhibitor dapagliflozin stimulates glucagon secretion in mice. Second, Ferrannini et al. (12) showed that in subjects with type 2 diabetes treated with an SGLT2 inhibitor, increased fasting glucagon levels were coupled with a ∼30% increase in endogenous glucose production. Finally, in a small study by Fukui et al. (13), fasting glucagon concentrations increased by 63% in participants with type 1 diabetes after treatment with the SGLT2 inhibitor ipragliflozin. Based on these findings, we hypothesized that SGLT inhibition may help restore the defective glucagon response to hypoglycemia. To test this hypothesis, we used staged euglycemia-hypoglycemia clamps to determine whether SGLT2 inhibition enhances the counterregulatory hormone response to hypoglycemia in adults with type 1 diabetes.
Research Design and Methods
Study Protocol
Twenty-three men and women with type 1 diabetes of at least 1 year duration participated in the trial. Eligibility criteria included: aged 18 to 70 years; BMI 18.5 to 35.0 kg/m2; random C-peptide <0.7 ng/mL; and HbA1c <10.0% (86 mmol/mol). Exclusion criteria included: active use of any noninsulin antihyperglycemic medication; and diabetic ketoacidosis or severe hypoglycemic events within 3 months. All participants provided informed consent before participating in the study. During the run-in period, participants completed the eight-item Clarke hypoglycemia awareness questionnaire (14). They also recorded all insulin doses and wore a blinded professional continuous glucose monitor (CGM; FreeStyle Libre Pro; Abbott Diabetes Care, Alameda, CA) for 14 days before the intervention to assess baseline glycemic control. Participants who entered the study wearing a personal CGM were allowed to continue using the device throughout the study. Participants using multiple daily injection insulin therapy took their long-acting insulin in the mornings during the run-in period and did not take long-acting insulin the morning of a hypoglycemic clamp procedure.
Upon completion of the run-in period, participants were randomly assigned 1:1 in a double-blind fashion to receive either dapagliflozin 5 mg daily or placebo for 4 weeks (treatment A). The 5 mg dose of dapagliflozin, which is approved in Europe for use in type 1 diabetes (15), was chosen to minimize the risk of diabetic ketoacidosis. Total daily insulin doses were preemptively decreased by 10% to reduce the risk of hypoglycemia and then adjusted weekly by the investigators to target preprandial glucose levels of 80–130 mg/dL and 2-hour postprandial glucose levels of <180 mg/dL. Insulin dosing and blinded CGM data were collected during the final 14 days of treatment A. At the end of the 4-week treatment period, participants returned to complete a staged euglycemia-hypoglycemia clamp procedure.
On the day of the hypoglycemic clamp, participants arrived at the clinical research unit at 0700 h after fasting overnight for at least 8 h. Data from the blinded CGM device were reviewed, and, if the participant had hypoglycemia (glucose ≤70 mg/dL) within the previous 12 h, the hypoglycemic clamp was rescheduled. A peripheral i.v. cannula was placed for administration of insulin and dextrose. A second i.v. cannula was placed in the contralateral side to be used for the collection of blood samples. For participants using continuous subcutaneous insulin infusion, the insulin pump was suspended and disconnected. Participants then underwent a baseline, staged euglycemia-hypoglycemia clamp using a primed, continuous infusion of 30 mU/m2/min regular insulin. This dose was chosen based on prior experience that it successfully achieves hypoglycemia while maintaining serum insulin concentrations within the physiologic range. Glucose measurements (YSI 2300 Stat Plus; YSI Incorporated, Yellow Springs, OH) were collected every 5 min. Variable-rate 20% dextrose was given i.v. to clamp glucose levels at euglycemia (target 100 mg/dL) for 60 min. At the conclusion of the euglycemic stage, the i.v. dextrose infusion rate was decreased to allow serum glucose to drop to a target of 50 mg/dL over a period of no less than 20 min. Serum glucose was then similarly clamped at that level for 40 min (hypoglycemic stage). The glucose infusion rate (GIR) was recorded during euglycemia and hypoglycemia as a surrogate for insulin sensitivity. Counterregulatory hormones (glucagon, epinephrine, norepinephrine, growth hormone, and cortisol) were drawn at baseline, the final 10 min of the euglycemic stage, and every 10 min of the hypoglycemic stage until conclusion of the clamp. Upon completion of the hypoglycemic stage, the insulin infusion was stopped and the dextrose infusion was continued at the previous rate. Glucose measurements were continued every 5 min until the blood glucose level was ≥70 mg/dL. The amount of time elapsed between discontinuation of the insulin infusion and blood glucose increasing to ≥70 mg/dL was defined as the “time to recovery.” Hypoglycemia awareness was assessed using the Edinburgh Hypoglycemia Symptoms Scale, a validated self-report questionnaire that uses a 7-point Likert scale (1 = not present to 7 = very intense) to measure 11 hypoglycemic symptoms (16), at the following time points: euglycemia stage 40 and 60 min; hypoglycemia stage 0, 20, and 40 min; and upon recovery from hypoglycemia. Cognitive function was assessed using the Trail Making Test (part B) and the Digit Symbol Substitution Test (17,18) at the following time points: euglycemia stage 50 min; hypoglycemia stage 10 and 30 min; and upon recovery from hypoglycemia.
After completing treatment A, participants entered a 4-week washout period (no study drug). At the end of washout, participants crossed over into treatment B, during which they received either placebo (if they received dapagliflozin during treatment A) or dapagliflozin 5 mg (if they received placebo during treatment A). Insulin dosing and blinded CGM data were again collected. After 4 weeks of treatment, participants underwent a second hypoglycemic clamp procedure. See Supplementary Fig. 1 for study design.
Biochemical Analysis
Blood samples for glucagon were collected in prechilled EDTA tubes and for epinephrine and norepinephrine in Na-Hep tubes. After centrifugation at 4°C, plasma was collected and frozen at −80°C for subsequent analysis at Quest Diagnostics (San Juan Capistrano, CA). Glucagon was measured using ELISA (Mercodia AB, Uppsala, Sweden). Epinephrine was measured using high-performance liquid chromatography (LC)/electrochemical detection and norepinephrine by chromatography/electrochemical detection. Blood samples for insulin, growth hormone, total cortisol, and nonesterified fatty acid (NEFA) were collected in tubes with no additive and allowed to clot for 30 min. After centrifugation, serum was collected and frozen at −80°C for subsequent analysis at Quest Diagnostics. Insulin was measured by immunocapture-LC/tandem mass spectrometry, growth hormone by immunoassay, cortisol by equilibrium dialysis/LC-mass spectrometry, and NEFA by enzymatic spectrophotometry.
Calculations
To quantify the counterregulatory hormone response to the hypoglycemic phase of the clamp study, we used trapezoidal approximation to construct the areas under the curve (AUCs) for each hormone during the 20-min euglycemic control period and during the 40-min hypoglycemic period. After normalizing the euglycemic control period and hypoglycemic AUCs to the same time interval, we subtracted each counterregulatory hormone’s AUC during hypoglycemia minus the AUC during the euglycemic control period to yield the change in AUC. This change in AUC quantifies the response to controlled hypoglycemia for each counterregulatory hormone.
Statistics
The Wilcoxon matched-pairs signed rank test was used in Prism version 9.1.0 (GraphPad Software, San Diego, CA) to assess statistically significant differences in diabetes-related parameters between the dapagliflozin and placebo interventions. Linear mixed-effects models were used to assess differences in repeated-measures assessments of cognitive function and hypoglycemia between the two interventions. Speakman rank correlation was used in SPSS version 27.0 (IBM Corporation, Armonk, NY) to measure bivariate association between variables of interest. Data are summarized as medians and 95% CIs unless otherwise indicated.
Data and Resource Availability
The data sets generated and/or analyzed during the current study are available from the corresponding author upon reasonable request. No novel resources were generated or analyzed during the current study.
Results
Participant Characteristics
Twenty-three participants were randomly assigned. One participant withdrew due to fatigue and nausea after 2 weeks of treatment with placebo. Table 1 summarizes baseline clinical characteristics of participants. Elements to highlight include baseline HbA1c of 6.6% (49 mmol/mol), high baseline rates of hypoglycemia as measured by CGM, and average Clarke score of 3 with an interquartile range of 1–5, indicating a mixed hypoglycemia-aware population.
Table 1.
Baseline participant characteristics (n = 23)
| Male sex, % (n) | 43.5 (10) |
| Age (years) | 39 (28–52) |
| Weight (kg) | 77.9 (65.3–90.4) |
| BMI (kg/m2) | 25.2 (23.3–29.1) |
| Type 1 diabetes duration (years) | 19 (9–28) |
| HbA1c (%) | 6.6 (6.1–7.2) |
| HbA1c (mmol/mol) | 49 (43–55) |
| Average CGM glucose (mg/dL) | 125.5 (108.3–156.5) |
| CGM time <70 mg/dL (%) | 14 (8–24) |
| CGM time <54 mg/dL (%) | 4 (2–9) |
| Clarke survey score* | 3 (1–5) |
Continuous data are presented as medians (interquartile range).
Score range 0 to 7. A score ≥4 implies impaired awareness of hypoglycemia; ≤2 suggests normal hypoglycemia awareness; and 3 is indeterminate.
Diabetes-Related Parameters During Treatment
During the two 4-week intervention periods, participants’ total daily dose of insulin was 11% lower on dapagliflozin versus placebo (P = 0.015) (Fig. 1A and E). Median CGM glucose was 10 mg/dL lower on dapagliflozin versus placebo, but the difference was not statistically significant (123 vs. 133 mg/dL; P = 0.15) (Fig. 1B and F). Likewise, no appreciable differences for percent time in hypoglycemia (P = 0.40) (Fig. 1C and G) and change in weight (P = 0.63) (Fig. 1D) were observed between interventions.
Figure 1.

Differences in total daily dose of insulin (A), average CGM glucose (B), percent of time CGM glucose was <70 mg/dL (C), and change in weight (D) while participants in the crossover study were taking placebo (red circles) or dapagliflozin (Dapa; blue circles) for 4 weeks. Median values are annotated above each column and represented by the bar. Dots depict individual values for each participant. P values determined by Wilcoxon matched-pairs signed rank tests are indicated above brackets. Within-participant differences (placebo minus dapagliflozin) for total daily dose of insulin (E), average (Avg) CGM glucose (F), and percent of time CGM glucose was <70 mg/dL (G). Black vertical lines signify the median differences for the entire study cohort.
Insulin Concentrations, GIRs, Glucose Concentrations, and NEFA Concentrations During Clamp Studies
Median basal arterialized serum insulin concentrations were comparable following each intervention (29.5 µU/mL with dapagliflozin vs. 35.5 µU/mL with placebo, median difference 5.0 µU/mL [95% CI −2.0 to 13.0]). During the euglycemic and hypoglycemic phases of the clamp study, serum concentrations of insulin, plasma concentrations of glucagon, and GIRs were not statistically different between dapagliflozin and placebo interventions (Fig. 2A–C). The median basal blood NEFA concentration was 0.37 mmol/L after dapagliflozin versus 0.16 mmol/L after placebo (median within-participant difference of 0.06 mmol/L [95% CI 0.03–0.22]; P = 0.016) (Fig. 2D). Insulin infusion during the euglycemic stage of the clamp suppressed NEFA levels similarly between dapagliflozin and placebo studies, and NEFA levels were not statistically different between interventions during the hypoglycemic stage.
Figure 2.
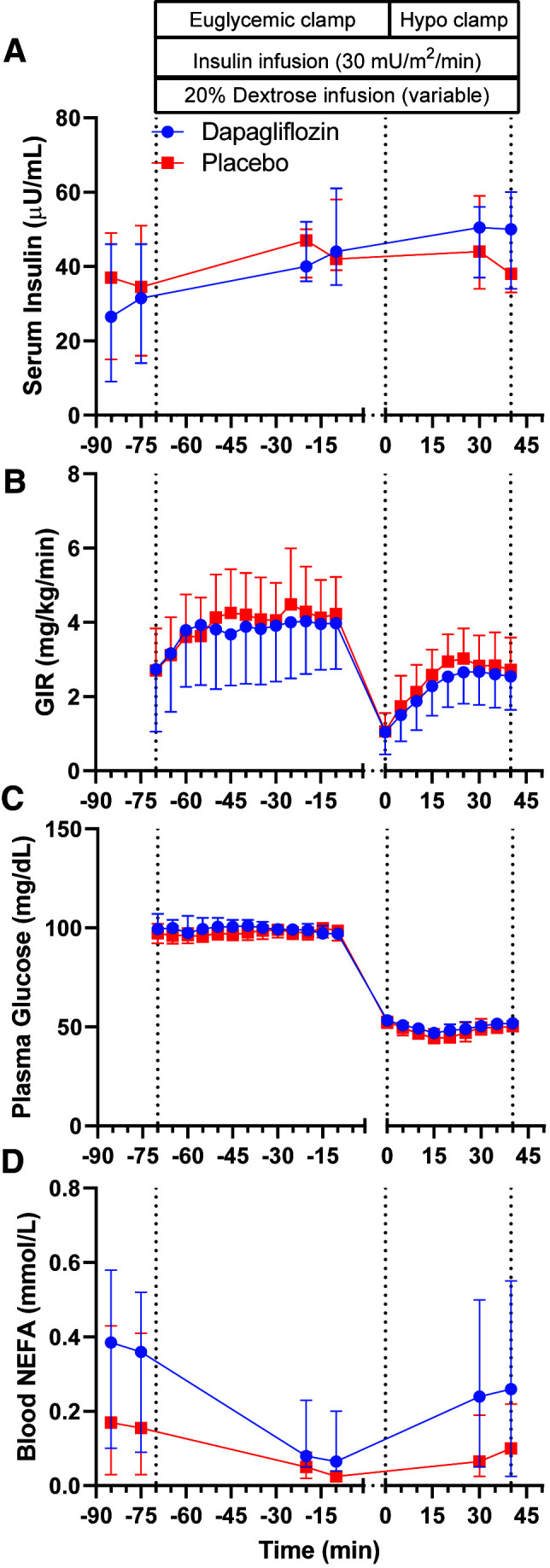
Arterialized plasma concentrations of insulin (A), GIR (B), arterialized plasma concentration of glucose (C), and blood NEFA levels (D) during hyperinsulinemic-hypoglycemic clamp studies after 4 weeks of placebo (red squares) or dapagliflozin (blue circles) in the crossover study. Data are summarized as medians and 95% CIs.
Counterregulatory Hormone Response
Median basal glucagon concentrations were higher following dapagliflozin (15.5 pg/mL [95% CI 12.8–29.1]) versus placebo (11.75 pg/mL [95% CI 8.54–19.7 pg/mL]; P = 0.041) with a median within-participant difference of 2.75 pg/mL (95% CI 1.38–12.6). Basal glucagon concentrations correlated inversely with basal insulin concentrations following dapagliflozin (ρ = −0.595; P = 0.003) and placebo (ρ = −0.490; P = 0.021) and with CGM average glucose (ρ = −0.492; P = 0.020) and time above range (ρ = −0.471; P = 0.027) following dapagliflozin but not placebo. Basal glucagon concentrations directly correlated with basal β-hydroxybutyrate (ρ = 0.554; P = 0.007) and basal NEFA levels (ρ = 0.518; P = 0.014) following dapagliflozin but not placebo. Significant bivariate correlations with basal glucagon were not observed for CGM time in range or below range, weight change, or average total daily dose of insulin following either intervention. Median basal cortisol was slightly higher following dapagliflozin (9.63 µg/dL [95% CI 9.09–12.1]) versus placebo (8.93 µg/dL [95% CI 7.75–10.3]; P = 0.029), with a median within-participant difference of 1.65 µg/dL (95% CI 0.157–2.91). Significant differences between interventions were not observed for basal concentrations of epinephrine (P = 0.096), norepinephrine (P = 0.924), and growth hormone (P = 0.058).
During the hypoglycemic phase of the clamp study, there were no significant differences in the counterregulatory hormone responses between interventions (Fig. 3A–E). The median within-individual differences for counterregulatory hormone AUC between interventions (placebo minus dapagliflozin [i.e., positive value denotes higher AUC for placebo, and negative value denotes higher AUC following dapagliflozin]) were 0.75 pg/mL/min (95% CI −3.1 to 3.8) for glucagon, −4.82 pg/mL/min (95% CI −55.8 to 28.7) for epinephrine, −18.0 pg/mL/min (95% CI −79.1 to 24.4) for norepinephrine, 0.12 ng/mL/min (95% CI −2.8 to 1.8) for growth hormone, and −1.13 µg/dL/min (95% CI −2.66 to 0.0918) for cortisol (Fig. 3F–I). The median time to recovery from hypoglycemia (i.e., glucose level ≥70 mg/dL) was 32 min for placebo (95% CI 31–45) versus 40 min for dapagliflozin (95% CI 36–44), with a median within-participant difference of 6 min (95% CI −3 to 9; P = 0.139) (Supplementary Fig. 2).
Figure 3.

Arterialized plasma concentrations of glucagon (A), epinephrine (B), norepinephrine (C), growth hormone (D), and cortisol (E) during hyperinsulinemic-hypoglycemic clamp studies after 4 weeks of placebo (red squares) or dapagliflozin (Dapa; blue circles) in the crossover study. Data are summarized as medians and 95% CIs. Within-participant differences (placebo minus dapagliflozin) for counterregulatory hormones AUC in response to hypoglycemia for glucagon (F), epinephrine (G), norepinephrine (H), growth hormone (GH; I), and cortisol (J). Black vertical lines signify the median differences for the entire study cohort.
To analyze a potential effect of antecedent hypoglycemia, a subgroup analysis grouped participants into tertiles based on their prerandomization CGM time below range (Supplementary Fig. 3A–C). Within-tertile differences in time below range between the two interventions were minimal (Supplementary Fig. 3D–I). During the hypoglycemic clamp, the counterregulatory responses of glucagon (Supplementary Fig. 3J–O) and epinephrine (Supplementary Fig. 3P–U) were minimally different between interventions within each tertile.
Hypoglycemia awareness during the study, as assessed by the Edinburgh Hypoglycemia Symptoms Scale, did not differ appreciably following the two treatments (P = 0.641) (Supplementary Fig. 4A). Similarly, cognitive function, quantified using the Trail Making Test (part B) and the Digit Symbol Substitution Test, differed minimally (P = 0.550 and P = 0.962, respectively) (Supplementary Fig. 4B and C).
Discussion
In this trial, 22 participants with type 1 diabetes received 4 weeks of dapagliflozin 5 mg daily and 4 weeks of placebo in a randomized, double-blind, crossover design to evaluate the effects of SGLT2 inhibition on the counterregulatory response to hypoglycemia. This study builds upon prior work showing that SGLT inhibitors increase glucagon concentrations in vitro (11,19–21) and in vivo (12,13), as well as clinical trials of SGLT inhibitors in type 1 diabetes showing either no increase in hypoglycemia or a decrease in hypoglycemia with concurrent glucose lowering (9). The primary outcome of our study was glucagon response to hypoglycemia during treatment with either dapagliflozin or placebo for 4 weeks. Although we found a 32% higher basal glucagon concentration following dapagliflozin treatment, SGLT2 inhibition did not restore the glucagon counterregulatory response to hypoglycemia.
Studies of SGLT inhibitors as adjunct to insulin therapy in type 1 diabetes have consistently shown no increase in hypoglycemia risk, with some data suggesting clinically relevant hypoglycemia is actually decreased with SGLT inhibitor use (9,10). This may be related to the observation that fasting concentrations of the counterregulatory hormone glucagon are increased with SGLT inhibition in both type 1 and type 2 diabetes (12,13), a finding replicated in the current study. Indeed, we found that the median fasting glucagon concentration following dapagliflozin therapy was 32% higher than following placebo. Debate continues regarding the primary physiologic mechanism underlying this observation. Earlier data suggested that increased glucagon secretion is due to a direct effect of SGLT inhibition on pancreatic α-cells (11,19,20). A subsequent study suggested an intraislet effect, by which SGLT inhibition decreases somatostatin secretion from δ-cells, relieving α-cells of paracrine suppression and increasing glucagon release (21). However, more recent work has challenged these hypotheses by reporting very low or absent SGLT2 protein expression in human islets and no effect of SGLT inhibition on the secretion of somatostatin or glucagon from isolated perfused rat or mouse pancreases, human islets in vitro, or human islets transplanted to mice (22–24). These data favor an indirect effect of SGLT inhibitors on α-cell activity in which falling blood glucose, due to glycosuria, stimulates glucagon release to increase endogenous glucose production (22,23).
Just as the mechanism of the SGLT inhibitor glucagonotropic effect remains controversial, so, too, does the relevance of this phenomenon in reducing clinical hypoglycemia. To date, there is a paucity of human data exploring the relationship between SGLT inhibition and glucagon secretion during hypoglycemia. To our knowledge, the current study is the first to investigate whether this class of medications can restore the counterregulatory hormone response. The results of this trial demonstrate that SGLT2 inhibition has no significant effect on the glucagon counterregulatory response to insulin-induced hypoglycemia in adults with type 1 diabetes.
We also found no difference between the dapagliflozin and placebo interventions in hypoglycemia awareness, cognitive function, or time to recovery (blood glucose ≥70 mg/dL) during hyperinsulinemic-hypoglycemic clamp studies. It remains possible that the SGLT inhibitor–induced increase in basal glucagon, by itself, helps protect against hypoglycemia. However, we found no difference between the dapagliflozin and placebo interventions in percent of time in hypoglycemia (CGM <70 mg/dL), and we detected no significant correlation between basal glucagon concentrations and percent of time in hypoglycemia in either intervention. This finding is consistent with data from previous clinical trials in type 1 diabetes that showed rates of hypoglycemia were similar among study participants who received dapagliflozin 5 mg, dapagliflozin 10 mg, or placebo (25–27). To study the effects of SGLT inhibition while minimizing the risk of adverse events, such as diabetic ketoacidosis, we chose the lower available dose of dapagliflozin (5 mg), which is also the dose approved for use in type 1 diabetes in Europe (15). It is possible that the higher dose of dapagliflozin or a different SGLT inhibitor medication would lead to different results. Given differences in study design, investigational product, and treatment duration, we do not believe our results contradict those of Danne et al. (10), whose post hoc analysis of 1,362 participants with type 1 diabetes found a reduction in hypoglycemia among those taking the dual SGLT1/2 inhibitor sotagliflozin.
This study has some limitations. First, participants had very good glycemic control at baseline, with median HbA1c 6.6% (49 mmol/mol) and interquartile range 6.1–7.2% (43–55 mmol/mol). Thus, the ability to generalize our findings to individuals with significantly higher HbA1c is limited; SGLT inhibitor therapy may have different effects on average blood glucose, insulin requirements, weight loss, and fasting glucagon concentrations in this population. Second, participants had high rates of ambulatory hypoglycemia by CGM, with an average of 14% time <70 mg/dL at baseline and 10–13.5% time <70 mg/dL during treatment. This finding was surprising, given most participants did not meet criteria for impaired awareness of hypoglycemia based on Clarke scores, and none had recent severe hypoglycemia (exclusion criteria). Because of the increased rates of hypoglycemia and greater potential for hypoglycemia-associated autonomic failure (HAAF), we conducted subgroup analyses to ascertain whether a potential difference in the counterregulatory response was blunted in the tertile with more frequent hypoglycemia but present in a tertile with less frequent hypoglycemia. We found no substantial differences between interventions in the counterregulatory response whether antecedent hypoglycemia was lower or higher. This finding implies HAAF did not meaningfully blunt a difference in the counterregulatory response that would have otherwise existed had HAAF been absent. Unfortunately, a recent publication by Galindo et al. (28) shows that the CGM system used in our study, FreeStyle Libre Pro, reports lower mean daily glucose and higher rates of hypoglycemia (especially nocturnal and prolonged hypoglycemia) when compared with point-of-care capillary glucose testing. These data raise the distinct possibility that CGM time in hypoglycemia was overestimated in our study.
In summary, SGLT2 inhibitor treatment increased median basal glucagon concentrations but did not enhance the counterregulatory hormone response to insulin-induced hypoglycemia among participants with type 1 diabetes. While the ultimate effects of SGLT2 inhibition on α-cell function remain controversial, we believe that our study provides definitive evidence that, when used in clinically relevant doses, SGLT2 inhibition does not alter the physiological response to hypoglycemia, improve recovery from hypoglycemia, or improve cognitive function during episodes of hypoglycemia. Thus, we propose that any reductions in hypoglycemia associated with SGLT inhibitor use are due to other variables, such as lower insulin dosing or decreased glycemic excursions, rather than a direct pharmacological effect of the medication during hypoglycemia.
Our mechanistic findings do not detract from the potential clinical benefit of SGLT inhibitors in this population. Indeed, we believe this study sheds light on and provides hope for the use of adjunctive therapies in type 1 diabetes in general. Lowering the risk of clinical hypoglycemia may be possible by reducing insulin doses and improving glucose control (i.e., reducing glycemic variability) rather than requiring a specific alteration in glucose counterregulation. In support of this theory, our own work with a glucagon receptor antagonist as adjunct to insulin in participants with type 1 diabetes has shown that this therapy improves glycemic control, reduces insulin doses, and does not increase rates of hypoglycemia (29). Though further study in this area is needed, treatments that reduce insulin requirements, whether by directly lowering glucose or by addressing insulin resistance, may mitigate the adverse effects of insulin therapy including weight gain and hypoglycemia.
Article Information
Acknowledgments. The authors thank the late Robert R. Henry for his support and contribution to the conception of this study. Dr. Henry was an enthusiastic and selfless mentor to J.H.P., S.C.B., and countless others in the field of diabetes.
Funding. Research in this publication was supported by a grant from JDRF under award number 2-SRA-2018-606-M-B (awarded to J.H.P.) and by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under award number K23DK123392 (to J.M.G.). J.M.G. was supported by a JDRF Career Development Award (5-ECR-2020-950-A-N).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the JDRF.
Duality of Interest. J.M.G. reports consulting fees from InClinica and advisory board fees from Eli Lilly and Company, Medtronic, Mannkind Corporation, Dompe, and vTv Therapeutics. J.H.P. reports consulting fees from Diasome Pharmaceuticals, Eli Lilly and Company, MannKind Corporation, Novo Nordisk, and Sanofi. No other potential conflicts of interest were reported.
Author Contributions. S.C.B. assisted in study design, acquired the data, assisted in data analysis, and wrote the manuscript. J.M.G. analyzed the data, prepared the figures, and helped write the manuscript. E.R.G. helped acquire the data. J.H.P. conceived of and designed the study, supervised the project, and helped write the manuscript. All authors critically reviewed and approved the final version of the manuscripts. J.H.P. is the guarantor of this work and, as such, had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Prior Presentation. Parts of this study were presented in abstract form at the 81st Scientific Sessions of the American Diabetes Association, 25–29 June 2021.
Footnotes
S.C.B. and J.M.G. contributed equally to this work.
Clinical trial reg. no. NCT03704818, clinicaltrials.gov
This article contains supplementary material online at https://doi.org/10.2337/figshare.17064497.
References
- 1. Frier BM. Morbidity of hypoglycemia in type 1 diabetes. Diabetes Res Clin Pract 2004;65(Suppl. 1):S47–S52 [DOI] [PubMed] [Google Scholar]
- 2. Cryer PE. The barrier of hypoglycemia in diabetes. Diabetes 2008;57:3169–3176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McCrimmon RJ, Sherwin RS. Hypoglycemia in type 1 diabetes. Diabetes 2010;59:2333–2339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aronson R, Galstyan G, Goldfracht M, Al Sifri S, Elliott L, Khunti K. Direct and indirect health economic impact of hypoglycaemia in a global population of patients with insulin-treated diabetes. Diabetes Res Clin Pract 2018;138:35–43 [DOI] [PubMed] [Google Scholar]
- 5. Cryer PE. Hypoglycemia in type 1 diabetes mellitus. Endocrinol Metab Clin North Am 2010;39:641–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Arbelaez AM, Xing D, Cryer PE, et al.; Diabetes Research in Children Network (DirecNet) Study Group . Blunted glucagon but not epinephrine responses to hypoglycemia occurs in youth with less than 1 yr duration of type 1 diabetes mellitus. Pediatr Diabetes 2014;15:127–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Russell SJ, El-Khatib FH, Sinha M, et al. Outpatient glycemic control with a bionic pancreas in type 1 diabetes. N Engl J Med 2014;371:313–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rickels MR, Fuller C, Dalton-Bakes C, et al. Restoration of glucose counterregulation by islet transplantation in long-standing type 1 diabetes. Diabetes 2015;64:1713–1718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boeder S, Edelman SV. Sodium-glucose co-transporter inhibitors as adjunctive treatment to insulin in type 1 diabetes: A review of randomized controlled trials. Diabetes Obes Metab 2019;21(Suppl. 2):62–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Danne T, Pettus J, Giaccari A, et al. Sotagliflozin added to optimized insulin therapy leads to lower rates of clinically relevant hypoglycemic events at any HbA1c at 52 weeks in adults with type 1 diabetes. Diabetes Technol Ther 2019;21:471–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bonner C, Kerr-Conte J, Gmyr V, et al. Inhibition of the glucose transporter SGLT2 with dapagliflozin in pancreatic alpha cells triggers glucagon secretion. Nat Med 2015;21:512–517 [DOI] [PubMed] [Google Scholar]
- 12. Ferrannini E, Muscelli E, Frascerra S, et al. Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J Clin Invest 2014;124:499–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fukui T, Ohara M, Yamagishi SI. Glucagon in type 1 diabetes patients receiving SGLT2 inhibitors: a friend or foe? Diabetes Metab Res Rev 2021;37:e3415. [DOI] [PubMed] [Google Scholar]
- 14. Clarke WL, Cox DJ, Gonder-Frederick LA, Julian D, Schlundt D, Polonsky W. Reduced awareness of hypoglycemia in adults with IDDM. A prospective study of hypoglycemic frequency and associated symptoms. Diabetes Care 1995;18:517–522 [DOI] [PubMed] [Google Scholar]
- 15. European Medicines Agency . Forxiga, 2020. Accessed 26 October 2021. Available from https://www.ema.europa.eu/en/medicines/human/EPAR/forxiga
- 16. Deary IJ, Hepburn DA, MacLeod KM, Frier BM. Partitioning the symptoms of hypoglycaemia using multi-sample confirmatory factor analysis. Diabetologia 1993;36:771–777 [DOI] [PubMed] [Google Scholar]
- 17. Jaeger J. Digit symbol substitution test. J Clin Psychopharmacol 2018;38:513–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gaudino EA, Geisler MW, Squires NK. Construct validity in the Trail Making Test: what makes Part B harder? J Clin Exp Neuropsychol 1995;17:529–535 [DOI] [PubMed] [Google Scholar]
- 19. Pedersen MG, Ahlstedt I, El Hachmane MF, Göpel SO. Dapagliflozin stimulates glucagon secretion at high glucose: experiments and mathematical simulations of human A-cells. Sci Rep 2016;6:31214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Solini A, Sebastiani G, Nigi L, Santini E, Rossi C, Dotta F. Dapagliflozin modulates glucagon secretion in an SGLT2-independent manner in murine alpha cells. Diabetes Metab 2017;43:512–520 [DOI] [PubMed] [Google Scholar]
- 21. Vergari E, Knudsen JG, Ramracheya R, et al. Insulin inhibits glucagon release by SGLT2-induced stimulation of somatostatin secretion. Nat Commun 2019;10:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kuhre RE, Ghiasi SM, Adriaenssens AE, et al. No direct effect of SGLT2 activity on glucagon secretion. Diabetologia 2019;62:1011–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dai C, Walker JT, Shostak A, et al. Dapagliflozin does not directly affect human α or β cells. Endocrinology 2020;161:bqaa080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chae H, Augustin R, Gatineau E, et al. SGLT2 is not expressed in pancreatic α- and β-cells, and its inhibition does not directly affect glucagon and insulin secretion in rodents and humans. Mol Metab 2020;42:101071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dandona P, Mathieu C, Phillip M, et al. Efficacy and safety of dapagliflozin in patients with inadequately controlled type 1 diabetes (DEPICT-1): 24 week results from a multicentre, double-blind, phase 3, randomised controlled trial. Lancet Diabetes Endocrinol 2017;5:864–876 [DOI] [PubMed] [Google Scholar]
- 26. Dandona P, Mathieu C, Phillip M, et al.; DEPICT-1 Investigators . Efficacy and safety of dapagliflozin in patients with inadequately controlled type 1 diabetes: the DEPICT-1 52-week study. Diabetes Care 2018;41:2552–2559 [DOI] [PubMed] [Google Scholar]
- 27. Mathieu C, Dandona P, Gillard P, et al.; DEPICT-2 Investigators . Efficacy and safety of dapagliflozin in patients with inadequately controlled type 1 diabetes (the DEPICT-2 study): 24-week results from a randomized controlled trial. Diabetes Care 2018;41:1938–1946 [DOI] [PubMed] [Google Scholar]
- 28. Galindo RJ, Migdal AL, Davis GM, et al. Comparison of the FreeStyle Libre Pro Flash continuous glucose monitoring (CGM) system and point-of-care capillary glucose testing in hospitalized patients with type 2 diabetes treated with basal-bolus insulin regimen. Diabetes Care 2020;43:2730–2735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pettus J, Reeds D, Cavaiola TS, Boeder S, Levin M, Tobin G, et al. Effect of a glucagon receptor antibody (REMD-477) in type 1 diabetes: a randomized controlled trial. Diabetes Obes Metab 2018;20:1302–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]