

Abstract
Background and Objectives
Caregivers routinely inform medical and regulatory decision making in rare pediatric diseases. While differences in treatment preferences across caregivers and patients have been observed for Duchenne muscular dystrophy, this evidence was limited by small samples of patients and results were confounded by patient age and disease progression. We tested caregiver and patient preference concordance for treating Duchenne.
Methods
Preferences and demographic/clinical information from 115 caregivers and 107 patients were collected in an international study (response = 80%) using a previously developed discrete-choice experiment consisting of 12 experimentally controlled choice tasks. Each task presented two profiles that varied across four attributes: disease progression, drug failure probability, kidney damage risk, and fracture risk. Caregivers and patients were matched 1:1 based on patient age. We tested for concordance across each task and by comparing caregivers’ and patients’ maximum acceptable risk of drug failure, kidney damage, and fracture for a slowing of disease progression.
Results
The final analysis included 77 caregivers and 77 patients. No differences were observed in nationality (p = 0.969), disease stage (p = 0.180), or demographic/clinical factors (p = 0.093–0.857); however, patients were more optimistic (p = 0.030). Caregivers and patients chose similarly across tasks (p = 0.101–0.993). To slow disease progression by 1 year, caregivers and patients would tolerate a 9% and 11% increase in drug failure probability, respectively (p = 0.267). Alternatively, they would accept a 3% and 4% increase in the risk of kidney damage (p = 0.719) or a 15% and 20% increase in the risk of fracture (p = 0.534).
Conclusions
Caregivers and patients had concordant preferences for treating Duchenne. Providers and regulators can trust both caregiver and patient report of preferences to inform medical decision making.
Supplementary Information
The online version contains supplementary material available at 10.1007/s40271-022-00574-y.
Key Points for Decision Makers
| Regulatory agencies often use caregiver preferences to inform decision making regarding degenerative, rare, pediatric, and other conditions. |
| Previous research quantifying preferences in Duchenne muscular dystrophy indicated that caregivers and patients have different preferences, but evidence was limited by small samples and confounded by age and health status. |
| This large international study indicates that caregivers and patients have concordant treatment preferences once adjusting for patient age, an indicator of disease progression. |
| We present a methodological advancement in comparing preferences across cohorts using matching approaches. |
Introduction
Treatment advances over the past two decades have helped extend life and improve health for many patients with rare diseases [1, 2]. Even as patients increasingly live into adulthood, many still make medical decisions in conjunction with caregivers. Caregiver decision making on behalf of patients is typically centered on the best-interest standard, wherein the caregiver makes choices thought to maximize benefits and minimize harms [3]. Such determinations are highly preference sensitive, and it is unknown if patients, and the caregivers who inform decisions on their behalf, value benefits and harms in the same way. This scenario is an example of the principle-agent problem, an economics concept wherein two groups (here, caregivers and patients) have asymmetrical information (here, separate lived experience of a condition), which can result in a different willingness to assume risk in exchange for benefit [4].
Understanding the preferences and values of caregivers and patients directly affected by rare diseases is essential to improving patient-centered decision making in many contexts [5, 6]. Caregiver and patient preferences can inform treatment decisions when clinical evidence or therapeutic options are limited (as is often the case for rare diseases) [7]. There is a push to include rare disease communities in the development of care standards to ensure they are realistic and promote treatments and interventions that are worthwhile to patients [8]. Caregiver and patient input can inform patient-centered clinical trials, such as by informing the selection of relevant outcome measures [9–11]. Caregiver and patient preference information is also increasingly integrated into regulatory decision making in the USA and internationally [12–14]. The US Food and Drug Administration has endorsed the collection of patient experience data from both caregivers and patients, as well as from patient advocates [15]. Approaches for systematically integrating this information into regulatory decision making are still evolving. [16]
The Duchenne muscular dystrophy patient community and Parent Project Muscular Dystrophy have been leaders in advancing the integration of caregiver and patient perspectives into decision making [17, 18]. Parent Project Muscular Dystrophy has played a role in developing regulatory guidance, advancing the science of patient engagement, and increasing the use of preference methods to inform patient-focused drug development [19, 20]. Preference studies of caregivers and patients with rare diseases with pediatric onset have demonstrated mixed concordance, with some research suggesting the two groups have similar preferences and others indicating dissimilarities [21–26]. Such preference studies have been limited by small sample sizes of patients (i.e., generally fewer than 50), and have not been matched on patient age or other important characteristics of disease progression, introducing potential confounding. These limitations may influence inferences made about preference concordance between caregiver and patient groups.
We sought to test the concordance of caregiver and patient preferences for treating Duchenne muscular dystrophy. While we hypothesized that caregivers and patients would have the same preferences for treatment, previous evidence from a small pilot study indicated that this might not be the case [24]. There is an imperative to evaluate the concordance of rare disease caregivers’ and patients’ preferences given that both groups can inform clinical and regulatory decision making. The findings of this study have implications for clinical and regulatory decision making in Duchenne. Our study also presents a number of methodological advances in the comparison of patient and caregiver preferences that could be applied to studies of other rare diseases.
Methods
Study Participants
Duchenne muscular dystrophy is a rare genetic neuromuscular disorder for which there is currently no cure. Duchenne is characterized by progressive muscle degeneration causing functional decline and weakness beginning in the lower limbs [27]. The average lifespan of people with Duchenne is in the mid-20s, up from the teen years if left untreated [28, 29]. These gains in lifespan are mostly due to improvements in supportive pulmonary and cardiac care over the past decade [30]. Duchenne is caused by a mutation in the Duchenne gene, carried on the X-chromosome [31], and as a result almost exclusively affects male individuals. It has an incidence of 1 out of every 3500–5000 male births. [32–34]
Duchenne caregivers and patients from eight countries (Australia, Belgium, Canada, Italy, France, The Netherlands, UK, and USA) were invited to participate in an online survey. The study used a quota sampling approach wherein all countries initially agreed to enroll at least 20 patients, 20 caregivers of adult patients (aged 18 years and older), and 20 caregivers of pediatric patients (aged younger than 18 years). Country leaders identified respondents by advertising the survey through patient registries (e.g., DuchenneConnect), Duchenne patient-group social media, and word of mouth. No compensation was offered for participation.
Individuals were eligible to participate in the survey if they were: (i) aged 18 years or older; (ii) male with Duchenne OR a caregiver of a male with Duchenne; and (iii) residing in a participating country. Duchenne diagnosis was self-reported. Participants were not excluded based on any clinical characteristics, such as disease stage/ambulation status. Participants indicated their consent to participate by a one-question item on the first screen of the survey. Patients were instructed to make all answer choices on their own, even if someone else was helping them to record their answers.
The current analysis was restricted to only adult patients and caregivers of adult patients. Caregivers of pediatric patients were dropped as we sought to compare preferences across caregiver and patient groups, and including caregivers of minors introduced age confounding for which we could not adjust.
Study Design
We utilized a continuous community-engaged approach to conceptualize, design, and implement a survey exploring patient-centered health outcomes for Duchenne caregivers and patients over a three year period (2018–20) [18, 35]. We follow a previously published engagement approach [36]. The study was conducted internationally, and countries varied in their experience and familiarity with patient preference research. We used several approaches to promote equitable engagement and design a patient-centered survey in collaboration with international Duchenne community partners.
First, we developed long-term engagement relationships with members of the Duchenne communities in all participating countries. We engaged 15 country leaders on the study’s advisory board, including Duchenne caregivers, patients, and other professional advocates. Engagement with country leaders revealed that patient groups internationally were interested in exploring benefit-risk trade-offs for emerging treatments [18]. They also indicated interest in exploring preferences after adjusting for patient age, as patient age is associated with health outcomes in this degenerative condition. Based on this input, we adapted a discrete-choice experiment (DCE) that had been previously developed and evaluated in a US sample of Duchenne caregivers and patients [24]. This DCE was developed following nearly a decade of engagement with the Duchenne community [18, 21, 22, 24, 35, 37–39], and intended to reflect trade-offs that caregivers and patients might face when making treatment decisions and informing regulatory medical decision making in the future.
Second, the adapted DCE was pre-tested by 18 individuals (two to three individuals in each country except for France and Italy, who joined the study after data collection had already begun). All pre-testing was done in English. The DCE was modified to specify the treatment benefit of slowing disease progression in terms of the length of the benefit, rather than qualitatively (e.g., benefit for 1, 3, or 5 years, rather than small, medium, or large benefit). This was done as individuals expressed that the qualitative labels were ambiguous, and that they would want more information about what defined the different levels of benefit in order to make meaningful trade-offs. Individuals also indicated that they selected treatment profiles based on which they thought would most likely be approved by regulators. As we sought to document preferences rather than perceptions of what regulators would approve, we also modified the DCE to instruct participants to select treatments assuming that each was “real and available to you.”
Third, we professionally translated the survey into all official national languages, and offered country leaders the opportunity to further tailor language. The survey was translated from English into French (Canadian and Belgian dialects), Dutch (The Netherlands and Flemish dialects), and Italian, and stakeholders recommended specific modifications to better reflect how the patient community described specific health phenomena.
Fourth, we offered flexibility in recruitment approaches across countries, while still striving for equal representation. In some instances, it was harder to identify participants than country leaders had anticipated, resulting in a long recruitment process (October 2018–May 2020) and the need to pursue diverse and innovative sampling approaches that varied internationally.
Fifth, we maintained open communication with country leaders before, during, and after data collection. Because of our open and frequent communication with country leaders, including bi-monthly calls and more frequent e-mails, and quarterly updates on study findings, we heard of the increased stressors brought about by the COVID-19 pandemic. As a result, it was decided to stop recruitment before meeting all sampling quotas.
Survey Instrument
Participants provided demographic and clinical information about themselves and/or the person with Duchenne including the patient’s age, nationality, history of fractures, history of kidney damage, and medications used to manage Duchenne. Two survey measures assessed disease progression. The first was the Performance of the Upper Limb Patient-Reported Outcome Measure (PUL-PROM), which measures upper limb function related to daily life. PUL-PROM is a condition-specific measure validated for use among patients with all stages of Duchenne [40, 41]. Scores range from 0 to 64, with higher scores indicating better functioning. The second measure was ambulatory status, measured via a single question and ten response options that are routinely re-categorized into the four disease stages of Duchenne: early ambulatory, late ambulatory, early non-ambulatory, and late non-ambulatory. Participants also rated themselves on several personality characteristics, including: (i) risk taking; (ii) optimism; (iii) health seeking; and (iv) control seeking, along a five-point Likert scale (strongly disagree-strongly agree) [24]. Responses were dichotomized to agree/strongly agree vs neutral/disagree/strongly disagree.
The adapted DCE included the following four attributes with three levels each: (i) slowing disease progression (1, 3, 5 years); (ii) chance of drug benefit (25%, 50%, 75% chance); (iii) added risk of kidney damage (none, 10%, 20%); and (iv) added risk of fracture (none, 10%, 20%). We used Ngene [42] to create a balanced, D-efficient experimental design with zero priors and 12 choice tasks, each of which compared two profiles that varied on the level of the attributes. Attribute levels for the DCE were selected based on a literature review and expert guidance of what drugs for Duchenne might look like in the future [3]. It was not explicitly indicated that a treatment failure would imply no benefit but still all the risks. The attribute of slowing disease progression was meant to reflect prolonging the amount of time someone was in a given disease stage, rather than explicitly increasing overall survival. Participants were asked, “If both drugs were real and available to you, which would you choose?” We did not include an opt-out choice in the DCE as we intended to report trade-offs rather than thresholds or utility specification. Caregivers and patients received identical versions of the DCE, and both groups were asked to report on their own personal preferences.
Several efforts were made to maximize and evaluate participant understanding of the DCE. These included: (i) the use of pictograms and text to visualize and explain each attribute and level; (ii) a “warm-up” task wherein participants rated the importance of each attribute after having read a description about it; and (iii) an example DCE task. Participants also evaluated the DCE according to a set of debriefing items that assessed how relevant, easy to understand, easy to answer, and consistent with their preferences the instrument was [24]. Responses to these items helped to assess internal validity.
Sample Size
We conducted a post-hoc sample size calculation to demonstrate that we were statistically powered to identify a difference between groups. Sample size for DCEs are typically justified using rules of thumb, parametric approaches, or previous literature [43–45]. A formal analytic method has emerged to identify minimum sample size required to test specific hypotheses of DCEs, and requires specification of significance level (), statistical power (), statistical model (e.g., conditional logit, mixed logit), initial belief about parameter estimates, and DCE experimental design [46]. We calculated an estimated sample size using this approach by specifying an = 0.05, = 0.15, and using post-hoc parameter estimates derived from conditional models estimated aggregately for caregivers and patients. According to these specifications, the resulting minimum sample size for caregiver and patient groups was 64 respondents. Additional respondents may be needed to power a mixed logit model.
Data Analysis
Matching is an approach to reduce bias and can help isolate potential sources of outcome variation [47]. Caregiver and patients were matched based on patient age. Patient age is related to health status in this degenerative condition, and is a high fidelity and low-burden measure. We used a subset, fixed 1:1 matching without replacement for the primary analysis [48, 49]. This approach created a subset of observations wherein there were an equal number of caregiver and patient respondents, and the two groups had identical patient age structures; i.e., if there were three patients aged 25 years in the dataset, then there were also three caregivers reporting on patients aged 25 years in the dataset. We used a random selection process facilitated by a random number generator to determine which respondents were included in the subset.
Differences in the clinical, demographic, and personality characteristics of caregiver and patient groups were assessed using t tests and chi-square/Fisher exact tests. Caregiver and patient preference concordance were explored in several ways. First, bivariate analysis using chi-square/Fisher exact tests was conducted to assess whether the proportion of caregivers and patients who chose either given profile in the DCE (A or B, for tasks 1–12) were the same. Second, preferences for caregivers and patients were estimated using a mixed logistic regression. Mixed logistic regressions allow for individual-specific variation in preferences [50]. Model-level differences in preferences for caregivers and patients were assessed using a Swait and Louvier test [51]. Third, whether any preference differences could be observed after adjusting for any difference in scale across caregivers and patients was explored using a heteroskedastic multinomial logistic regression [52]. Fourth, maximum acceptable risk (MAR) percentages using the willingness-to-pay approach [53, 54] were derived from mixed logit models and compared across caregiver and patient groups using Z-tests. Maximum acceptable risk identifies how much risk participants will accept in exchange for a given benefit, and is routinely used to explore trade-offs between potential therapeutic benefits and risks [55, 56]. We separately estimated caregivers’ and patients’ MAR of an increase in probability of drug failure, risk of kidney damage, and risk of fracture in exchange for a 1-year slowing of disease progression. Probability of drug failure was derived by reverse coding the chance of benefit attribute presented in the DCE in an approach consistent with standard analytic practice [24, 57]. Maximum acceptable risk confidence intervals were estimated using the delta method.
We used chi-square tests to compare how caregivers and patients evaluated the DCE using dichotomized versions of the debriefing items. Percent endorsement of items as agree/strongly agree was also evaluated in comparison to an a priori 75% threshold using a two-sided Z-test [24]. All analyses were performed using Stata at a significance level of = 0.05.
Sensitivity Analysis
We conducted three sensitivity analyses to test the robustness of our results. The first sensitivity analysis explored preference concordance for caregivers and patients when truncated by patient age, and only including responses regarding patients who were aged 19–35 years. This analysis was anticipated to validate results from the primary analysis, as it reduced differences in age observed across groups. The second sensitivity analysis explored preference differences across the full samples without any censoring or age matching. This analysis was conducted to meet good practices of preference research, which indicate that all respondents should be included in the analysis [58]. The third sensitivity analysis explored preference differences across those participants who were excluded from the primary dataset. This analysis was intended to demonstrate whether preference differences could be ascertained at much lower sample sizes when groups continued to vary on important clinical characteristics for Duchenne such as patient age.
All protocols for data collection and analysis were approved as research exempt from further human subjects review by the Johns Hopkins Bloomberg School of Public Health (Institutional Review Board [IRB] No. 8175) and The Ohio State University College of Medicine (IRB No. 2019E0113). We worked with country leaders to determine if an additional, country-specific IRB review was needed. Each country came to the independent decision that a further ethical review was not necessary.
Results
A total of 115 caregivers and 107 patients were eligible for this analysis. Eighty percent of individuals identified by international patient groups completed the survey. After age matching, the caregiver and patient groups each contained 77 participants with identical patient age structures (Fig. 1) and a mean patient age of 25 years (standard deviation = 6). These 154 participants comprised the primary sample that is reported on in this paper (Table 1). The two groups did not vary in terms of nationality (p = 0.969). They were similar in clinical characteristics including ambulatory stage of Duchenne (p = 0.180), PUL-PROM score (p = 0.093), history of fracture (p = 0.183), history of kidney damage (p = 0.649), and treatments used to manage Duchenne, including generic steroids (p = 0.857), Duchenne-specific steroids (p = 0.253), and exon-skipping therapies (p = 0.720). The groups were equally likely to self-report as being risk taking (p = 0.431), health seeking (p = 0.836), and control seeking (p = 0.679). Seventy-four percent of patients reported they were optimistic as compared with 57% of caregivers (p = 0.030).
Fig. 1.
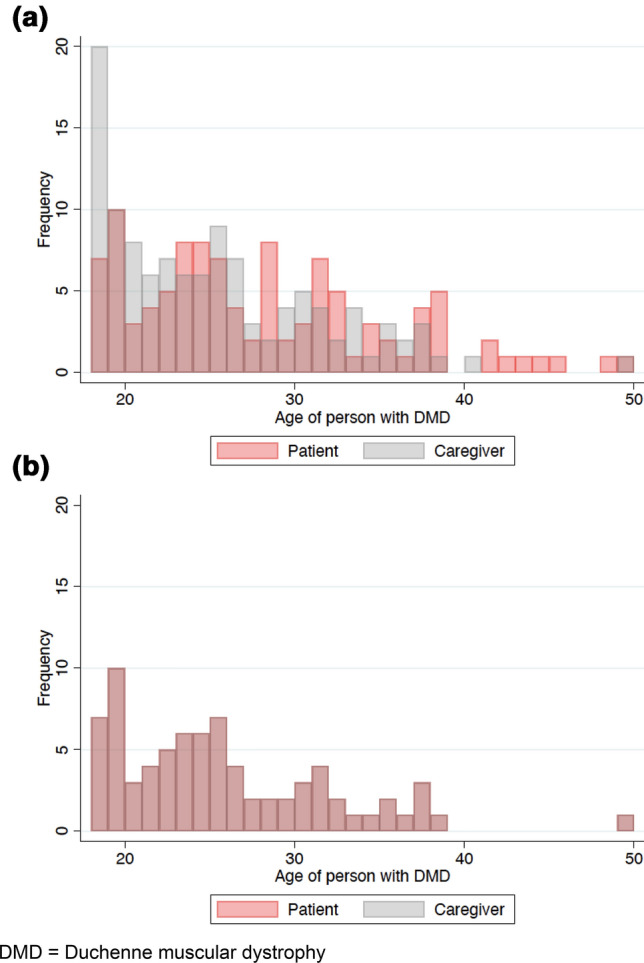
Participant age distributions, a for full sample prior to age matching and b after age matching
Table 1.
Demographic and clinical characteristics of participants
| Characteristics | Patients (n = 77) | Caregivers (n = 77) | P value |
|---|---|---|---|
| Patient age, years, mean (range) | 25.3 (18–50) | 25.3 (18–50) | 1.000 |
| Residence, n (%) | 0.969 | ||
| Australia | 4 (5) | 4 (5) | |
| Belgium | 6 (8) | 5 (6) | |
| Canada | 5 (6) | 10 (10) | |
| France | 11 (14) | 8 (10) | |
| Italy | 12 (16) | 10 (13) | |
| The Netherlands | 8 (10) | 10 (13) | |
| UK | 18 (23) | 17 (22) | |
| USA | 13 (17) | 15 (19) | |
| Ambulatory stage, n (%) | 0.180 | ||
| Early ambulatory | (0) | 1 (1) | |
| Late ambulatory | 8 (10) | 2 (3) | |
| Early non-ambulatory | 40 (52) | 45 (58) | |
| Late non-ambulatory | 29 (38) | 29 (38) | |
| PUL-PROM, mean (range) | 23.36 (0–64) | 17.93 (0–61) | 0.093 |
| History of fracture, n (%) | 52 (68) | 44 (57) | 0.183 |
| History of kidney damage, n (%) | 3 (4) | 2 (3) | 0.649 |
| Treatments used, n (%) all that apply | |||
| Generic steroid | 42 (55) | 42 (56) | 0.857 |
| Duchenne-specific steroid | 23 (30) | 29 (39) | 0.253 |
| Exon skipping | 5 (6) | 6 (8) | 0.720 |
| Personality, n (%) | |||
| Risk taking | 26 (35) | 20 (29) | 0.431 |
| Optimistic | 55 (74) | 40 (57) | 0.030 |
| Health seeking | 54 (73) | 50 (71) | 0.836 |
| Control seeking | 56 (76) | 55 (79) | 0.679 |
PUL-PROM Performance of the Upper Limb Patient-Reported Outcome Measure
Caregivers and patients had the same likelihood of selecting a given treatment profile across each of the 12 choice tasks (p = 0.101–0.993). For both groups and across all tasks, the most preferred profile was one that described a treatment that slowed progression for 3 years, had a drug failure rate of 25%, had a 10% extra kidney damage risk, and no extra fracture risk. Ninety-nine percent of participants preferred this profile as compared with the alternate profile, which described a treatment that slowed progression for 1 year, had a drug failure rate of 75%, had a 20% extra kidney damage risk, and 20% extra fracture risk (Fig. 2).
Fig. 2.

Discrete choice experiment treatment profile selection (caregiver n = 77, patient n = 77)
Mixed logit preference models for both groups were overall similar (p = 0.428) and groups displayed no differences in scale (p = 0.469; Appendix Table 1 of the Electronic Supplementary Material [ESM]). Caregivers and patients were willing to accept similar levels of risk in exchange for treatments that slowed disease progression (Fig. 3). To slow disease progression by 1 year, caregivers and patients would tolerate a 9% and 11% increase in drug failure probability, respectively (p = 0.267). Alternatively, they would accept a 3% and 4% increase in kidney damage risk (p = 0.719) or a 15% and 20% increase in fracture risk (p = 0.534) to slow disease progression by 1 year.
Fig. 3.
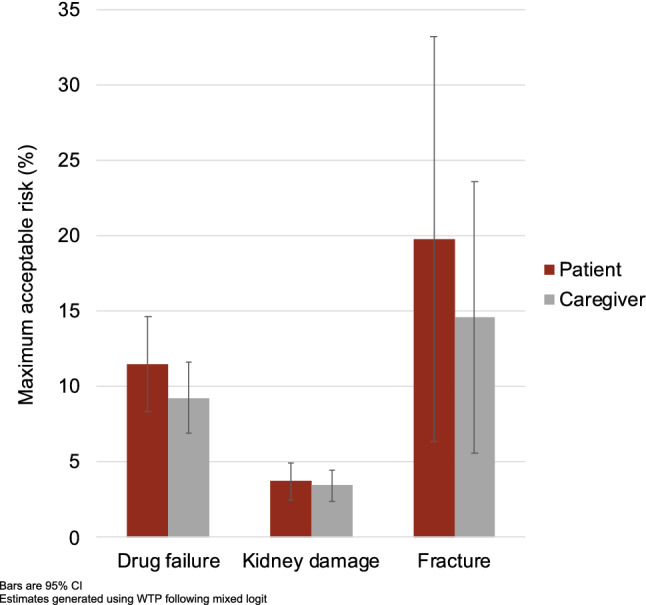
Maximum acceptable risk for treatments slowing Duchenne progression (caregiver n = 77, patient n = 77)
Caregivers and patients were equally likely to endorse the DCE as relevant (p = 0.737), easy to understand (p = 0.132), easy to answer (p = 0.229), and consistent with their preferences (p = 0.616) in debriefing items. Both groups met the a priori 75% acceptability benchmark for these four items, indicating that their evaluation of the DCE was consistent with normative expectations.
Sensitivity Analysis
A total of 87 caregivers and 82 patients were included in the first sensitivity analysis restricted to those reporting on patients aged 19–35 years. The two groups were similar across all characteristics (Appendix Table 2 and Fig. 1 of the ESM). Preference models for both groups were overall similar (p = 0.562) and groups demonstrated no differences in scale (p = 0.420). Caregivers and patients expressed no difference in the likelihood of selecting a given treatment profile for any of the 12 choice tasks (p = 0.120–0.783). There was also no difference in the MAR of drug failure probability (p = 0.497), increase in kidney damage risk (p = 0.758), or increase in fracture risk (p = 0.678) that caregivers and patients would assume to slow disease progression for 1 year (Appendix Fig. 2).
The full sample of 115 caregivers and 107 patients was included in the second sensitivity analysis. Patients in this full sample were on average 28 years old while caregivers were reporting on patients who were on average 25 years old (p < 0.001; Appendix Table 2). Preference models for both groups were overall similar (p = 0.548) and demonstrated no differences in scale (p = 0.145). Patients self-reporting were less likely to report using a Duchenne-specific steroid than those reported on by caregivers (p = 0.014). There was no difference in the likelihood of selecting a given treatment profile for any of the 12 choice tasks (p = 0.153–0.954). There was also no difference in the MAR of drug failure probability (p = 0.178), increase in kidney damage risk (p = 0.562), or increase in fracture risk (p = 0.686) that caregivers and patients would assume to slow disease progression for 1 year (Appendix Fig. 3).
A total of 38 caregivers and 30 patients were included in the third sensitivity analysis comprising those dropped from the full sample in order to create the primary age-matched analytic sample. Patients in this sample were on average 10 years older than patients reported on by caregivers (p < 0.001; Appendix Table 3 and Fig. 4). Patients also had poorer upper limb functioning (p < 0.001) and were less likely to have used Duchenne-specific steroids (p = 0.003). Preference models for both groups were significantly different (p = 0.021) but groups demonstrated no differences in scale (p = 0.712). Caregivers and patients also differed in their likelihood of selecting a given treatment profile for one of the 12 choice tasks (task 6, p = 0.0308, other tasks p = 0.082–1.00). There was no difference in the MAR of drug failure probability (p = 0.520), increase in kidney damage risk (p = 0.206), or increase in fracture risk (p = 0.437) that caregivers and patients would assume to slow disease progression for 1 year (Appendix Fig. 5).
Discussion
Patients with Duchenne are more likely to live into adulthood than ever before. This research answers an important call to understand whether the perspectives of patients vary from those of caregivers, who have been the traditional decision makers in Duchenne and other rare diseases. Results are drawn from an age-matched sample of 77 patients and 77 caregivers spanning eight countries and collected using a survey with an 80% response rate. This is among the largest and most diverse samples of caregivers and patients yet to be included in a preference study for a single rare condition.
Caregivers and patients with Duchenne frequently make choices that weigh the benefits of treatments in comparison to both side effects and risks of uncertainty. At present, treatment options for Duchenne in the USA include the use of corticosteroids for all people with Duchenne muscular dystrophy, and several exon-skipping therapies for particular mutations [59], with other potential treatments in the pipeline [60]. These treatment decisions are not specific to those in the USA, but also are made by patients internationally. Results demonstrate that both caregivers and patients are willing to accept risks of treatment in exchange for a potential benefit of slowing disease progression.
In Duchenne specifically, caregiver and patient preference concordance has been previously compared with mixed results, and without taking into account fundamental differences in the disease experiences across the two groups. Only four prior quantitative patient preference studies in Duchenne have included patients to our knowledge. Two of these studies suggested similar preferences among caregivers and patients, specifically regarding prioritization of quality-of-life outcomes [22] and preferences for treatments with pulmonary benefits [21]. A third study found some variation in prioritization of endpoints for gene therapy trials, particularly between caregivers and patients with non-ambulatory patients as compared with caregivers reporting on younger ambulatory patients [23]. The fourth study, from which the current DCE is derived, concluded that caregiver and patient preferences for disease-slowing therapies differed, as patients were more risk adverse than caregivers or professionals with regards to their willingness to accept the risk of fractures, risk of kidney damage, or drug failure in exchange for a treatment that slowed progression [24]. Each of the two studies demonstrating preference discordance were limited by a small sample of patients (n = 27 and n = 9, respectively), and conclusions as to preference discordance across groups may have been biased by the confounding of patient/caregiving status with patient age and health status.
Self-reported optimism was the only characteristic upon which caregiver and patient groups differed after adjusting for patient age. Previous research in Duchenne has suggested that therapeutic optimism may influence preferences and decision making regarding the use of experimental therapies, specifically by making individuals more likely to assume risks and uncertainty in exchange for the option to try new therapies [61, 62]. Although patients in the current study indicated that they were more optimistic than caregivers, preferences did not vary across the two groups. Further, caregivers and patients reported similar levels of burden as demonstrated through their being equally likely to endorse the DCE as easy to understand, easy to answer, relevant, and consistent with their preferences during debriefing.
Our application of matching methods to reduce bias and confounding is innovative in preference research. To date, few preference studies have used these methods, and general considerations for using matching techniques and sensitivity analysis in preference research have only recently begun to emerge [63–65]. The current analysis makes a methodological contribution to the preference measurement literature by demonstrating several approaches for using matching to explore differences across caregiver and patient groups that are not confounded by other experiential factors of the condition. Even when restricting to this age-matched sample, the sample size still exceeded that required to identify statistical significance based on sample size calculations. Further, differences in preferences were observed at much lower sample sizes than estimated by sample size calculations in the sensitivity analysis. This indicates that the absence of differences observed in the primary analysis was not the result of insufficient sample or statistical power. A natural next step for advancing matching approaches in preference studies is to match on multiple characteristics. Because of the large heterogeneity of patient characteristics in many rare diseases, matching upon more than one factor will require a much larger sample size than might be feasibly collected through traditional survey research, and instead may require the use of innovative prospective and registry-based studies.
Our sustained engagement activities that sought to promote equity across international partners is a strength of the study. Doing so ultimately ensured the relevance of the research topic and appropriateness of the study design to international members of the Duchenne community. Many of the traditional metrics of research success, such as a high response rate and a large sample size, are attributable to the preliminary focus our research team placed on establishing long-lasting, mutually beneficial partnerships with international Duchenne patient communities. The benefit of engaging international partners extends far beyond increasing recruitment, and also informed the conceptualization, analysis, and dissemination of study findings. We included international stakeholders in the research development, pre-tested the survey in many countries, conducted formal translation, and used adaptive approaches to recruit participants depending on the resources available to the international patient groups.
We focused on exploring differences among caregivers and patients given our practical interest in knowing whether caregivers are good decision-making agents on behalf of patients. It is possible that other demographic and clinical characteristics may also influence preferences. We conducted several post-hoc analyses to explore preferences across clinical characteristics and found that preference models did not vary by history of fracture, history of kidney damage, or history of steroid use. While preferences did not vary across patients and caregivers for MAR of fractures, we did observe substantial preference heterogeneity for fractures within both caregiver and patient groups. We heard from country leaders that preferences for avoiding fractures may vary due to the differing impacts that a fracture may have on quality of life for patients at different disease stages. Finally, we would also naturally expect characteristics such as respondent age and sex to systematically vary across caregiver and patient groups, and it is possible that these characteristics influence underlying preferences.
Several features of the current study limit the scope of our conclusions. First, the study is not intended to inform individual-level clinical decision making. Although the choice tasks asked specifically about treatments for Duchenne, results presented here are subject to ecological bias if interpreted at the individual level. Second, the current study does not provide evidence regarding whether caregivers are good proxies for patients with Duchenne muscular dystrophy, but rather seeks to understand whether the two groups have similar preferences, independent of the other. Caregivers were asked to report on their own preferences, which were ultimately similar to those of patients. It also is unknown whether caregivers’ perception of patient preferences are the same as patients’ own preferences, that is, we do not know whether the results would be the same if caregivers had been asked to select the treatment options that they thought would be preferred by the patient. It is possible that caregivers’ preferences may not be independent of the preferences of the patient they care for, given that they do share some experiences with the condition.
Conclusions
Advances in treatment and supportive care have helped many patients with rare diseases with pediatric onset live into adulthood, raising important questions about the concordance of patient and caregiver preferences for treatments. This large international study conducted in partnership with the Duchenne muscular dystrophy community indicates that caregivers and patients have concordant treatment preferences after adjusting for patient age, an important indicator of the disease experience for this degenerative condition. Both caregivers and patients were willing to accept risk in exchange for a treatment that would potentially slow the disease. The findings of this study provide insight into risk tolerance in Duchenne muscular dystrophy, which can be used to inform regulatory benefit-risk determinations, and inform conversations about treatment decision making with patients with Duchenne muscular dystrophy in clinical settings. Our study also presents a number of methodological advances in the comparison of subgroups, which can be applied to preference studies comparing caregiver and patient preferences across other conditions.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
We thank members of the Duchenne community who participated in this research. We also thank the following organizations for participating on the stakeholder advisory council: Save Our Sons Duchenne Organization, Australia; Parent Project, Belgium; Jesse’s Journey, Canada; Duchenne Parent Project, The Netherlands; Duchenne UK. The following groups also provided support in recruiting participants to the survey: Canadian Neuromuscular Disease Registry, Muscular Dystrophy Canada, and LaForce DMD. An earlier version of this paper was awarded the Lee B Lusted Anne Stiggelbout Award for Outstanding Presentation in Patient and Stakeholder Preferences and Engagement at the 2020 annual meeting of the Society for Medical Decision Making.
Declarations
Funding
Financial support for this study was provided entirely by a contract with Parent Project Muscular Dystrophy. Parent Project Muscular Dystrophy receives financial support from Pfizer to conduct benefit-risk research. The funding agreement ensured the authors’ independence in designing the study, interpreting the data, writing, and publishing the report. The authors and Parent Project Muscular Dystrophy are open to collaborations involving additional analysis and extensions of this survey.
Conflict of interest
Ryan Fischer and Pat Furlong are employees of Parent Project Muscular Dystrophy. Norah Crossnohere, Elizabeth Vroom, and John Bridges have no conflicts to declare.
Ethics approval
Protocols for data collection and analysis were approved as research exempt from further human subject review (Johns Hopkins Bloomberg School of Public Health IRB No. 8175, The Ohio State University College of Medicine IRB No. 2019E0113). Protocols for engagement were deemed non-human subject research (Johns Hopkins Bloomberg School of Public Health IRB No. 7984).
Consent to participate
Participants provided informed consent.
Consent for publication
Not applicable.
Availability of data and material
The authors and PPMD are open to collaborations involving additional analysis and extensions of this survey.
Code availability
The authors and PPMD are open to sharing code for collaborations involving additional analysis and extensions of this survey.
Author contributions
NC, JB, RF contributed to study design. NC, JB participated in data analysis. NC drafted the manuscript. NC, RF, PF, EV, JB provided critical revisions.
References
- 1.Passamano L, Taglia A, Palladino A, et al. Improvement of survival in Duchenne muscular dystrophy: retrospective analysis of 835 patients. Acta Myologica. 2012;31(2):121. [PMC free article] [PubMed] [Google Scholar]
- 2.Mendell JR, Al-Zaidy S, Shell R, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377(18):1713–1722. doi: 10.1056/NEJMoa1706198. [DOI] [PubMed] [Google Scholar]
- 3.Kopelman LM. The best-interests standard as threshold, ideal, and standard of reasonableness. J Med Philosophy. 1997;22(3):271–289. doi: 10.1093/jmp/22.3.271. [DOI] [PubMed] [Google Scholar]
- 4.Grossman SJ, Hart OD. An analysis of the principal-agent problem. In: Dionne G, Harrington SE, editors. Foundations of insurance economics. Dordrecht: Springer; 1992. pp. 302–40. [Google Scholar]
- 5.Pinto Taylor E, Doolittle B. Caregiver decision-making for terminally ill children: a qualitative study. J Palliat Care. 2020;35(3):161–166. doi: 10.1177/0825859719885947. [DOI] [PubMed] [Google Scholar]
- 6.Hopkins KA, Ott MA, Salih Z, Bosslet GT, Lantos J. When adolescent and parents disagree on medical plan, who gets to decide? Pediatrics. 2019;144(2):e20190291. doi: 10.1542/peds.2019-0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Montori VM, Brito JP, Murad MH. The optimal practice of evidence-based medicine: incorporating patient preferences in practice guidelines. JAMA. 2013;310(23):2503–2504. doi: 10.1001/jama.2013.281422. [DOI] [PubMed] [Google Scholar]
- 8.Khodyakov D, Kinnett K, Grant S, et al. Engaging patients and caregivers managing rare diseases to improve the methods of clinical guideline development: a research protocol. JMIR Res Protoc. 2017;6(4):e57. doi: 10.2196/resprot.6902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsai J-H, Crossnohere NL, Strong T, Bridges JF. Measuring meaningful benefit-risk tradeoffs to promote patient-focused drug development in Prader-Willi Syndrome: a discrete-choice experiment. MDM Policy Pract. 2021;6(2):23814683211039457. doi: 10.1177/23814683211039457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gaasterland C, Jansen-van der Weide M, Vroom M, et al. The POWER-tool: recommendations for involving patient representatives in choosing relevant outcome measures during rare disease clinical trial design. Health Policy. 2018;122(12):1287–1294. doi: 10.1016/j.healthpol.2018.09.011. [DOI] [PubMed] [Google Scholar]
- 11.Morel T, Cano SJ. Measuring what matters to rare disease patients: reflections on the work by the IRDiRC taskforce on patient-centered outcome measures. Orphanet J Rare Dis. 2017;12(1):1–13. doi: 10.1186/s13023-017-0718-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.21st Century Cures Act, H.R. 34, 114th Congress (2016).
- 13.van Overbeeke E, Forrester V, Simoens S, Huys I. Use of patient preferences in health technology assessment: perspectives of canadian, belgian and German HTA representatives. Patient. 2021;14(1):119–128. doi: 10.1007/s40271-020-00449-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Bekker-Grob EW, Berlin C, Levitan B, et al. Giving patients’ preferences a voice in medical treatment life cycle: the PREFER Public-Private Project. Patient. 2017;10(3):263–266. doi: 10.1007/s40271-017-0222-3. [DOI] [PubMed] [Google Scholar]
- 15.US Food and Drug Administration . Patient-focused drug development: collecting comprehensive and representative input. Guidance for industry, Food and Drug Administration staff, and other stakeholders. Rockville: US Food and Drug Administration; 2020. [Google Scholar]
- 16.Assessment of the use of patient experience data in regulatory decision-making. Eastern Research Group, Inc. Lexington, MA. 2021. https://www.fda.gov/media/150405/download.
- 17.Furlong P, Bridges J, Charnas L, et al. How a patient advocacy group developed the first proposed draft guidance document for industry for submission to the US Food and Drug Administration. Orphanet J Rare Dis. 2015;10(1):82. doi: 10.1186/s13023-015-0281-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crossnohere NL, Fischer R, Crossley E, Vroom E, Bridges JF. The evolution of patient-focused drug development and Duchenne muscular dystrophy. Expert Rev Pharmacoecon Outcomes Res. 2020;20(1):57–68. doi: 10.1080/14737167.2020.1734454. [DOI] [PubMed] [Google Scholar]
- 19.US Food and Drug Administration. Duchenne muscular dystrophy and related dystrophinopathies: developing drugs for treatment. Rockville, MD: Guidance for industry. 2018. https://www.fda.gov/media/92233/download.
- 20.US Food and Drug Administration. Director's Corner: working with patient advocacy groups. 2015. Podcast. https://www.fda.gov/drugs/news-events-human-drugs/transcript-directors-corner-working-patient-advocacy-groups. Accessed 2 Feb 2022.
- 21.Hollin IL, Peay H, Apkon SD, Bridges J. Patient-centered benefit-risk assessment in Duchenne muscular dystrophy. Muscle Nerve. 2017;55(5):626–634. doi: 10.1002/mus.25411. [DOI] [PubMed] [Google Scholar]
- 22.Hollin IL, Peay H, Fischer R, Janssen EM, Bridges JFP. Engaging patients and caregivers in prioritizing symptoms impacting quality of life for Duchenne and Becker muscular dystrophy. Qual Life Res. 2018;27(9):2261–2273. doi: 10.1007/s11136-018-1891-7. [DOI] [PubMed] [Google Scholar]
- 23.Paquin RS, Fischer R, Mansfield C, et al. Priorities when deciding on participation in early-phase gene therapy trials for Duchenne muscular dystrophy: a best-worst scaling experiment in caregivers and adult patients. Orphanet J Rare Dis. 2019;14(1):1–9. doi: 10.1186/s13023-019-1069-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bridges JF, Tsai J-H, Janssen E, Crossnohere NL, Fischer R, Peay H. How do members of the Duchenne and Becker muscular dystrophy community perceive a discrete-choice experiment incorporating uncertain treatment benefit? An application of research as an event. Patient. 2019;12(2):247–257. doi: 10.1007/s40271-018-0330-8. [DOI] [PubMed] [Google Scholar]
- 25.Su J, Li N, Joshi N, et al. Patient and caregiver preferences for haemophilia A treatments: a discrete choice experiment. Haemophilia. 2020;26(6):e291–e299. doi: 10.1111/hae.14137. [DOI] [PubMed] [Google Scholar]
- 26.Cruz R, Belter L, Wasnock M, Nazarelli A, Jarecki J. Evaluating benefit-risk decision-making in spinal muscular atrophy: a first-ever study to assess risk tolerance in the SMA patient community. Clin Ther. 2019;41(5):943–60.e4. doi: 10.1016/j.clinthera.2019.03.012. [DOI] [PubMed] [Google Scholar]
- 27.National Institutes of Health. About Duchenne muscular dystrophy. https://www.genome.gov/Genetic-Disorders/Duchenne-Muscular-Dystrophy. 2013. Accessed 8 Mar 2020.
- 28.Darras BT, Jones HR, Ryan MM, Darryl C. Neuromuscular disorders of infancy, childhood, and adolescence: a clinician's approach. London: Elsevier; 2014. [Google Scholar]
- 29.Leigh F, Ferlini A, Biggar D, et al. Neurology care, diagnostics, and emerging therapies of the patient with Duchenne muscular dystrophy. Pediatrics. 2018;142(Suppl. 2):S5–16. doi: 10.1542/peds.2018-0333C. [DOI] [PubMed] [Google Scholar]
- 30.Eagle M, Baudouin SV, Chandler C, Giddings DR, Bullock R, Bushby K. Survival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul Disord. 2002;12(10):926–929. doi: 10.1016/S0960-8966(02)00140-2. [DOI] [PubMed] [Google Scholar]
- 31.Hoffman EP, Brown RH, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51(6):919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 32.Emery AE. Population frequencies of inherited neuromuscular diseases: a world survey. Neuromuscul Disord. 1991;1(1):19–29. doi: 10.1016/0960-8966(91)90039-U. [DOI] [PubMed] [Google Scholar]
- 33.Mah JK, Korngut L, Dykeman J, Day L, Pringsheim T, Jette N. A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul Disord. 2014;24(6):482–491. doi: 10.1016/j.nmd.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 34.Romitti PA, Zhu Y, Puzhankara S, et al. Prevalence of Duchenne and Becker muscular dystrophies in the United States. Pediatrics. 2015;135(3):513–521. doi: 10.1542/peds.2014-2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crossnohere NL, Fischer R, Lloyd A, Prosser LA, Bridges JFP. Assessing the appropriateness of the EQ-5D for Duchenne muscular dystrophy: a patient-centered study. Med Decis Mak. 2021;41(2):209–221. doi: 10.1177/0272989X20978390. [DOI] [PubMed] [Google Scholar]
- 36.Hollin IL, Young C, Hanson C, Bridges JF, Peay H. Developing a patient-centered benefit-risk survey: a community-engaged process. Value Health. 2016;19(6):751–757. doi: 10.1016/j.jval.2016.02.014. [DOI] [PubMed] [Google Scholar]
- 37.Hollin I, Peay H, Bridges J. Caregiver preferences for emerging duchenne muscular dystrophy treatments: a comparison of best-worst scaling and conjoint analysis. Patient. 2015;8(1):19–27. doi: 10.1007/s40271-014-0104-x. [DOI] [PubMed] [Google Scholar]
- 38.Peay H, Hollin I, Bridges J. Prioritizing parental worry associated with Duchenne muscular dystrophy using best-worst scaling. J Genet Couns. 2016;25(2):305–313. doi: 10.1007/s10897-015-9872-2. [DOI] [PubMed] [Google Scholar]
- 39.Peay H, Hollin I, Fischer R, Bridges J. A community-engaged approach to quantifying caregiver preferences for the benefits and risks of emerging therapies for Duchenne muscular dystrophy. Clin Ther. 2014;36(5):624–637. doi: 10.1016/j.clinthera.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 40.Klingels K, Mayhew AG, Mazzone ES, et al. Development of a patient-reported outcome measure for upper limb function in Duchenne muscular dystrophy: DMD upper limb PROM. Dev Med Child Neurol. 2017;59(2):224–231. doi: 10.1111/dmcn.13277. [DOI] [PubMed] [Google Scholar]
- 41.Mayhew A, Mazzone ES, Eagle M, et al. Development of the performance of the upper limb module for Duchenne muscular dystrophy. Dev Med Child Neurol. 2013;55(11):1038–1045. doi: 10.1111/dmcn.12213. [DOI] [PubMed] [Google Scholar]
- 42.Ngene. ChoiceMetrics. http://www.choice-metrics.com/download.html. Accessed 2019.
- 43.Lancsar E, Louviere J. Conducting discrete choice experiments to inform healthcare decision making. Pharmacoeconomics. 2008;26(8):661–677. doi: 10.2165/00019053-200826080-00004. [DOI] [PubMed] [Google Scholar]
- 44.Orme B. Sample size issues for conjoint analysis studies. Sequim: Sawtooth Software Technical Paper; 1998.
- 45.Johnson R, Orme B. Getting the most from CBC. Sequim: Sawtooth Software Research Paper Series, Sawtooth Software; 2003.
- 46.de Bekker-Grob EW, Donkers B, Jonker MF, Stolk EA. Sample size requirements for discrete-choice experiments in healthcare: a practical guide. Patient. 2015;8(5):373–384. doi: 10.1007/s40271-015-0118-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stuart EA. Matching methods for causal inference: a review and a look forward. Stat Sci. 2010;25(1):1–21. doi: 10.1214/09-STS313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zubizarreta JR, Paredes RD, Rosenbaum PR. Matching for balance, pairing for heterogeneity in an observational study of the effectiveness of for-profit and not-for-profit high schools in Chile. Ann Appl Stat. 2014;8(1):204–231. doi: 10.1214/13-AOAS713. [DOI] [Google Scholar]
- 49.Austin PC. Statistical criteria for selecting the optimal number of untreated subjects matched to each treated subject when using many-to-one matching on the propensity score. Am J Epidemiol. 2010;172(9):1092–1097. doi: 10.1093/aje/kwq224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hauber AB, González JM, Groothuis-Oudshoorn CG, et al. Statistical methods for the analysis of discrete choice experiments: a report of the ISPOR Conjoint Analysis Good Research Practices Task Force. Value Health. 2016;19(4):300–315. doi: 10.1016/j.jval.2016.04.004. [DOI] [PubMed] [Google Scholar]
- 51.Swait J, Louviere J. The role of the scale parameter in the estimation and comparison of multinomial logit models. J Market Res. 1993;305–14.
- 52.Vass CM, Wright S, Burton M, Payne K. Scale heterogeneity in healthcare discrete choice experiments: a primer. Patient. 2018;11(2):167–173. doi: 10.1007/s40271-017-0282-4. [DOI] [PubMed] [Google Scholar]
- 53.Hole AR. Fitting mixed logit models by using maximum simulated likelihood. Stata J. 2007;7(3):388–401. doi: 10.1177/1536867X0700700306. [DOI] [Google Scholar]
- 54.Train KE. Discrete choice methods with simulation. Cambridge: Cambridge University Press; 2009. [Google Scholar]
- 55.Ho MP, Gonzalez JM, Lerner HP, et al. Incorporating patient-preference evidence into regulatory decision making. Surg Endosc. 2015;29(10):2984–2993. doi: 10.1007/s00464-014-4044-2. [DOI] [PubMed] [Google Scholar]
- 56.Hauber AB, Fairchild AO, Johnson FR. Quantifying benefit–risk preferences for medical interventions: an overview of a growing empirical literature. Appl Health Econ Health Policy. 2013;11(4):319–329. doi: 10.1007/s40258-013-0028-y. [DOI] [PubMed] [Google Scholar]
- 57.Gonzalez JM, Boeri M. The impact of the risk functional form assumptions on maximum acceptable risk measures. Patient. 2021;14(6):827–836. doi: 10.1007/s40271-021-00518-y. [DOI] [PubMed] [Google Scholar]
- 58.Bridges JF, Hauber AB, Marshall D, et al. Conjoint analysis applications in health: a checklist: a report of the ISPOR Good Research Practices for Conjoint Analysis Task Force. Value Health. 2011;14(4):403–413. doi: 10.1016/j.jval.2010.11.013. [DOI] [PubMed] [Google Scholar]
- 59.National Institutes of Health, Genetic and Rare Disease Information Center. Duchenne muscular dystrophy. https://rarediseases.info.nih.gov/diseases/6291/duchenne-muscular-dystrophy. 2021. Accessed 18 June 2021.
- 60.Duchenne Drug Development Pipeline. Parent Project Muscular Dystrophy. https://www.parentprojectmd.org/duchenne-drug-development-pipeline/. 2020. Accessed 8 Mar 2021.
- 61.Peay HL, Tibben A, Fisher T, Brenna E, Biesecker BB. Expectations and experiences of investigators and parents involved in a clinical trial for Duchenne/Becker muscular dystrophy. Clin Trials. 2014;11(1):77–85. doi: 10.1177/1740774513512726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Peay HL, Scharff H, Tibben A, et al. “Watching time tick by…”: decision making for Duchenne muscular dystrophy trials. Contemp Clin Trials. 2016;46:1–6. doi: 10.1016/j.cct.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu T, Tsang W, Xie Y, et al. Preferences for artificial intelligence clinicians before and during the COVID-19 pandemic: discrete choice experiment and propensity score matching study. J Med Internet Res. 2021;23(3):e26997. doi: 10.2196/26997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vass C, Boeri M, Poulos C, Turner A. Matching and weighting in health preference research. In: Paper presented at: 12th Meeting of the International Academy of Health Preference Research, 23-24 June 2021. Virtual. 2021.
- 65.Lavelle TA, Crossnohere NL, Bridges JF. Quantifying the burden of hyperphagia in Prader-Willi syndrome using quality-adjusted life-years. Clin Ther. 2021. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.