

Abstract
Cyclic (di)nucleotides act as universal second messengers endogenously produced by several pathogens. Specifically, the roles of c-di-AMP in Mycobacterium tuberculosis immunity and virulence have been largely explored, although its contribution to the safety and efficacy of live tuberculosis vaccines is less understood. In this study, we demonstrate that the synthesis of c-di-AMP is negatively regulated by the M. tuberculosis PhoPR virulence system. Accordingly, the live attenuated tuberculosis vaccine candidate M. tuberculosis vaccine (MTBVAC), based on double phoP and fadD26 deletions, produces more than 25- and 45-fold c-di-AMP levels relative to wild-type M. tuberculosis or the current vaccine bacille Calmette-Guérin (BCG), respectively. Secretion of this second messenger was exclusively detected in MTBVAC but not in M. tuberculosis or in BCG. We also demonstrate that c-di-AMP synthesis during in vitro cultivation of M. tuberculosis is a growth-phase- and medium-dependent phenotype. To uncover the role of this metabolite in the vaccine properties of MTBVAC, we constructed and validated knockout and overproducing/oversecreting derivatives by inactivating the disA or cnpB gene, respectively. All MTBVAC derivatives elicited superior interleukin-1β (IL-1β) responses compared with BCG during an in vitro infection of human macrophages. However, both vaccines failed to elicit interferon β (IFNβ) activation in this cellular model. We found that increasing c-di-AMP levels remarkably correlated with a safer profile of tuberculosis vaccines in the immunodeficient mouse model. Finally, we demonstrate that overproduction of c-di-AMP due to cnpB inactivation resulted in lower protection of MTBVAC, while the absence of c-di-AMP in the MTBVAC disA derivative maintains the protective efficacy of this vaccine in mice.
KEYWORDS: MT:therapies and applications, tuberculosis, live vaccines, cyclic dinucleotides, BCG, MTBVAC, PhoPR, innate immunity, IFN
Graphical abstract
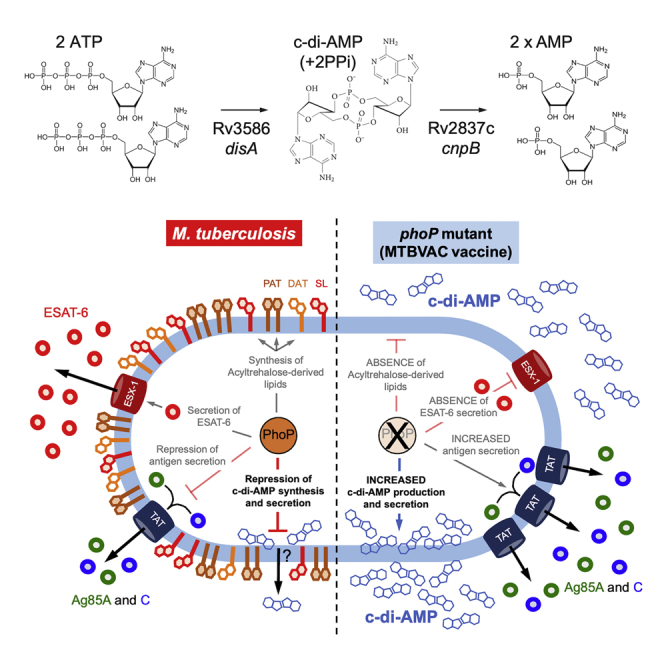
The PhoPR two-component system negatively regulates the production and secretion of the c-di-AMP second messenger in Mycobacterium tuberculosis. As a result, the MTBVAC live attenuated vaccine against tuberculosis produces and secretes elevated quantities of c-di-AMP. This phenotype influences the innate immune responses and vaccine properties of MTBVAC.
Introduction
Mycobacterium tuberculosis is the causative agent of tuberculosis (TB), which is the main historic cause of death from an infectious disease, even if antimycobacterial drugs and a centenary vaccine named bacille Calmette-Guérin (BCG) exist.1 According to the last World Health Organization report, more than 1.5 million people died from TB, and an estimated 10 million people fell ill with TB, in 2020.2 At this point, it is key to remember that poverty-related pandemic diseases, such as TB, do not resemble the unprecedented fast development of vaccines against the current coronavirus disease 2019 (COVID-19) pandemic. Rather, after more than two decades, only three TB vaccine candidates have reached efficacy trials,3 and none have been approved for emergency use authorization. Therefore, TB vaccines do not benefit from vaccine selection models such as those developed for COVID-19.4 The M. tuberculosis infectious cycle usually starts when a few bacilli are inhaled by a susceptible host and, once in the lung, are phagocytosed by alveolar macrophages. The tubercle bacillus has evolved several mechanisms to avoid the cytotoxic arsenal of macrophages, whereby the induction of phagosomal membrane rupture is thought to allow translocation from the harsh phagolysosome environment to the gentler cytosol.5,6 This process is mediated by ESAT-6, a protein with membranolytic activity,7 which is secreted by the mycobacterial type VII secretion system named ESX-1 and acts in concert with the M. tuberculosis virulence lipid phthiocerol dimycocerosate (PDIM).8 As a consequence, infected macrophages undergo apoptosis, which ultimately favors the cell-to-cell spread of M. tuberculosis.9 The TB infectious cycle continues when an infected patient expels bacilli, typically by coughing, allowing the transmission of M. tuberculosis to an uninfected individual. Deeper insights into the host-pathogen signaling activities that accompany this process are essential for a better understanding of the pathogen biology and the development of novel/alternative antimycobacterial therapies.
Nucleotide-derived molecules are produced by prokaryotic and eukaryotic organisms and serve as paradigm signaling mechanisms. Cyclic AMP (cAMP) and GMP (cGMP) have been studied for more than 50 years for their roles as second messengers, whereas the first knowledge on cyclic dinucleotides as signaling intermediates dates back to the discovery of c-di-GMP and c-di-AMP in 1987 and 2008, respectively.10,11 In recent years, the roles of c-di-AMP as a second messenger in Listeria monocytogenes, Bacillus subtilis, Staphylococcus aureus, Streptococcus pneumoniae, Streptococcus pyogenes, or M. tuberculosis and its essentiality in some of these pathogens have been gradually understood.12 Altogether, the presence of this second messenger and its essential synthesis by some bacteria highlight the importance of bacterial c-di-AMP for host-pathogen signaling. In this context, pioneering studies in L. monocytogenes demonstrated that this intracellular pathogen efficiently secreted c-di-AMP into the cytosol of host cells, which resulted in stimulation of stimulator of interferon genes (STING) and the subsequent activation of type I interferon (IFN) responses with the production of IFNβ.13,14
The synthesis of c-di-AMP in M. tuberculosis is catalyzed by the product of the Rv3586 gene (disA, also named dacA), a diadenylate cyclase that synthesizes c-di-AMP from 2 ATP molecules or, alternatively, 2 ADP molecules (Figure 1A).15 The genome of M. tuberculosis also contains the Rv2837c (cnpB, also named cdnP) gene, which encodes a c-di-AMP phosphodiesterase that cleaves c-di-AMP into 2 AMP molecules (Figure 1A).16 In sharp contrast to other pathogens, the c-di-AMP signaling mechanism is dispensable for M. tuberculosis survival since both the disA and cnpB genes have been successfully inactivated, resulting in absent or increased c-di-AMP levels, respectively.15,16 The secretion of c-di-AMP into the mycobacterial supernatant has been demonstrated only in M. tuberculosis cnpB knockout,16 probably due to the larger amounts of c-di-AMP accumulated in this mutant, which suggests that wild-type M. tuberculosis possesses molecular tools to secrete this second messenger, albeit at levels beyond the technical limit of detection.
Figure 1.

Characterization of c-di-AMP production and secretion in the live TB vaccines BCG and MTBVAC
(A) Schematic representation of c-di-AMP synthesis and degradation steps in M. tuberculosis with indications of the genes involved in each enzymatic conversion. (B) Analysis of c-di-AMP present in whole-cell extracts (left panel) or secreted into the supernatants (right panel) of M. tuberculosis MT103, MTBVAC, BCG Pasteur, or BCG Danish. n.d., not detected. (C) Chromatogram showing the peak corresponding to the c-di-AMP metabolite (represented by an arrow) in 10-fold concentrated secreted fractions of M. tuberculosis MT103 and MTBVAC. Note that both diagrams are represented with different scales, and the detection of c-di-AMP is below the limit of detection (lod) in M. tuberculosis MT103 and in BCG (data not shown). (D) Comparison of c-di-AMP production in whole-cell extracts of M. tuberculosis MT103, MTBVAC, and BCG Pasteur grown in exponential and stationary cultures. (E) Comparison of c-di-AMP produced in whole-cell extracts of M. tuberculosis MT103 and MTBVAC during stationary growth in 7H9-rich media and in Sauton minimal media. Note that graphs in (D) and (E) are represented on a logarithmic scale. Data are the mean and standard deviation from at least three biological replicates.
Similar to L. monocytogenes, the synthesis of c-di-AMP by M. tuberculosis elicits IFNβ responses by host macrophages in a STING-dependent manner.17 Notably, the type I IFN responses were dependent on the bacterial c-di-AMP levels since the infection of macrophages with disA-knockout and disA-overexpressing mutants resulted in reduced or increased IFN responses, respectively. The abolition of bacterial c-di-AMP synthesis resulted in increased M. tuberculosis virulence, while c-di-AMP overexpression resulted in reduced virulence in murine infection models.17 In addition, the production of IFNβ was independent of the host cyclic GMP-AMP synthase (cGAS), indicating that macrophages are able to sense the c-di-AMP produced by this pathogen when released into the cytosol.17 The inactivation, or chemical inhibition, of cnpB resulted in virulence attenuation and higher type I IFN responses compared with the wild-type strain.16,18 In addition to their ability to sense the c-di-AMP produced by intracellular pathogens, eukaryotic cells also are able to detect other microbial products, which then initiate cytosolic innate immune responses. It has been demonstrated that cytosolic sensing of M. tuberculosis DNA also drives type I IFN responses, and this phenotype is dependent on a functional ESX-1 system.19,20
Although the roles of c-di-AMP in the virulence or induction of innate immune responses against M. tuberculosis have been recently studied,16, 17, 18, 19, 20 the contribution of mycobacterial c-di-AMP to the efficacy and level of attenuation of TB vaccines has been less explored. It is important to remember that of the different TB vaccine candidates in the pipeline, c-di-AMP is exclusively synthesized by live vaccines, which are limited to BCG (either as recombinant BCG or BCG revaccination strategies) or the attenuated M. tuberculosis vaccine MTBVAC.3 The BCG vaccine was obtained a century ago after the in vitro culture passaging of a Mycobacterium bovis strain, the causative agent of bovine TB. During this in vitro subcultivation, BCG lost more than a hundred genes relative to M. tuberculosis.21 The main genetic determinant of BCG attenuation is linked to the absence of RD1,22 which corresponds to an ∼9 kb deletion in the ESX-1 locus, resulting in the loss of esxA (encoding the 6 kDa early secretory antigenic target ESAT-6) and flanking genes. On the other hand, MTBVAC was rationally attenuated by the deletion of the phoP and fadD26 genes,23 where PhoP is part of the two-component system PhoP-PhoR (PhoPR), which regulates M. tuberculosis virulence through three regulatory circuits.24 Consequently, M. tuberculosis phoP and phoPR mutants (1) fail to secrete ESAT-6,25,26 (2) do not synthesize acyltrehalose-derived lipids and sulfolipids,27,28 and (3) secrete higher amounts of immunodominant antigens.29,30 FadD26 is the first enzyme in a virulence gene cluster involved in the biosynthesis of PDIM.31 Accordingly, the MTBVAC vaccine lacks PhoP- and PDIM-dependent virulence phenotypes.
Some studies have explored the role of c-di-AMP in BCG in relation to the interaction of this vaccine with immune cells and reported that a BCG strain overexpressing c-di-AMP induced higher IFNβ responses in murine macrophages than wild-type BCG did,17 although the overall amounts of IFNβ measured in this assay were much lower than those reported from other IFNβ-induction studies using THP-1 macrophage-like cells.19,32 Based on this observed difference, the authors tested the vaccine efficacy of wild-type BCG and disA-overexpressing BCG in a guinea pig vaccination model and found that guinea pigs vaccinated with BCG overexpressing disA showed lower pathology and lower bacterial loads upon challenge with M. tuberculosis compared with BCG-vaccinated animals.33 In another study using BCG overexpressing disA, the authors found increased immune responses after mice were challenged with M. tuberculosis, even if the bacterial burdens in mouse organs were comparable between BCG- and BCG-disA-vaccinated animals.34 These findings suggested a role for c-di-AMP in the vaccine properties of live attenuated vaccines, and accordingly, we sought to study the regulation and impact of the c-di-AMP second messenger on the attenuation and protective efficacy of the MTBVAC vaccine candidate.
Results
The c-di-AMP second messenger is more highly produced and exclusively secreted by the MTBVAC vaccine compared with M. tuberculosis or BCG
We analyzed the presence of c-di-AMP in bacterial lysates of MTBVAC23 and its parental strain named M. tuberculosis MT103, which is a clinical isolate belonging to the “modern” lineage 4.35 As controls, we used two representative BCG strains: BCG Danish, which is licensed in Europe as a TB vaccine, and BCG Pasteur, the most widely used strain as a laboratory reference.21 As testing conditions, we used exponential growth cultures using routine culture media for mycobacteria. Our results using whole-cell fractions demonstrated that even if M. tuberculosis MT103 and BCG produced c-di-AMP levels in the range of 5–8 ng/mL, the MTBVAC vaccine produced ∼225 ng/mL of this metabolite, which represents a 25- and 45-fold increase relative to M. tuberculosis and BCG, respectively (Figure 1B). Since the whole-cell lysates used in our experiments exclusively contain metabolites from bacterial pellets, we aimed to analyze the presence of c-di-AMP in bacterial supernatants, indicating efficient secretion of this second messenger. We failed to detect c-di-AMP above the limit of detection in M. tuberculosis and BCG, while we observed ∼50 ng/mL in the MTBVAC secreted fractions (Figure 1B). The absence of c-di-AMP secretion in M. tuberculosis and BCG was subsequently confirmed after concentrating 10-fold the bacterial supernatants prior to c-di-AMP measurements (Figure 1C). Previous studies also failed to detect the secretion of this second messenger in M. tuberculosis, even if they used an ELISA-based approach, in contrast to our spectrometric measurements.16,36 Collectively, it is unclear whether yet unknown technical limitations exist for the detection of this metabolite in the secreted fractions or whether M. tuberculosis exclusively secretes c-di-AMP under certain conditions. It is also possible that only specific M. tuberculosis strains are able to secrete c-di-AMP in detectable amounts. We tried to discard this latter assumption by assaying different M. tuberculosis strains in our study, but we should be aware that they do not represent the whole variability of M. tuberculosis. Nevertheless, our results suggest a direct correlation between intracellular production and the secretion of this molecule and agree with previous findings using an M. tuberculosis H37Rv cnpB mutant.16,36 Notably, the detection of higher c-di-AMP secretion in MTBVAC was unexpected since this vaccine strain does not contain mutations in the c-di-AMP biosynthetic or degradation genes. Next, we analyzed the specific in vitro conditions leading to c-di-AMP synthesis and compared the c-di-AMP production in whole-cell fractions of exponential- or stationary-grown cultures. Both M. tuberculosis and MTBVAC produced higher amounts of this second messenger as bacterial growth progressed (Figure 1D), a finding also reproduced in M. tuberculosis CDC1551.17 However, the BCG vaccine maintained equivalent c-di-AMP production irrespective of exponential or stationary growth. Second, we compared c-di-AMP production in rich (7H9) versus defined (Sauton) culture media during stationary growth. M. tuberculosis and MTBVAC produced lower levels of c-di-AMP in Sauton media than in 7H9 broth (Figure 1D). Altogether, these results suggest that the synthesis of c-di-AMP in the M. tuberculosis complex is a strain-, growth-phase-, and growth-medium-dependent phenotype.
Synthesis of c-di-AMP is a novel PhoPR-regulated phenotype in M. tuberculosis
The unexpected results of exacerbated amounts of c-di-AMP produced or secreted by MTBVAC relative to its parental M. tuberculosis MT103 (Figure 1) led us to study the mechanism responsible for this phenotype. MTBVAC carries two well-defined genetic deletions in the phoP and fadD26 genes relative to its parental M. tuberculosis MT103 strain. To our knowledge, the fadD26-encoded gene product is exclusively involved in PDIM biosynthesis, and it seems unlikely to affect the synthesis of the c-di-AMP second messenger. However, PhoP acts as a transcription factor of the PhoPR two-component virulence system, which controls approximately 4% of the coding capacity of M. tuberculosis,29 and we postulated that c-di-AMP synthesis might represent another PhoPR-regulated circuit. We used an M. tuberculosis phoPR mutant and a phoPR-complemented strain constructed in the H37Rv genetic background to confirm our hypothesis. Confirming the finding of higher c-di-AMP production in the MTBVAC vaccine, we observed higher amounts of c-di-AMP in whole-cell fractions of the phoPR mutant relative to the wild-type and complemented strains (Figure 2A). This result was also reproduced in the secreted fractions of the abovementioned strains (Figure 2B), indicating that c-di-AMP synthesis and secretion are negatively regulated by PhoP in M. tuberculosis. Intriguingly, most of the previously described PhoP-dependent phenotypes (ESAT-6 secretion, synthesis of acyltrehalose-derived lipids, or the control of a noncoding RNA that impacts antigen secretion) are positively regulated by PhoP, and accordingly, this novel phenotype represents an exception to this rule. We also tried to identify the precise molecular mechanism underlying c-di-AMP regulation by PhoP. However, neither the disA nor the cnpB gene appeared differentially regulated in the H37Rv phoPR mutant or in the MTBVAC vaccine relative to their parental strains (Figure 2C). To examine whether the PhoPR system activates the transcription of proteins or other molecules that either degrade or inhibit the translation of enzymes related to c-di-AMP metabolism, we measured DisA and CnpB protein levels in M. tuberculosis H37Rv and its phoPR mutant by targeted multiple-reaction monitoring/mass spectrometry (MRM-MS). We analyzed and quantified five specific peptides of each protein to ensure the specific identification of both enzymes. Unexpectedly, DisA and CnpB protein levels showed nonsignificant differences between the wild type and the phoPR mutant, with average fold changes (WT/phoPR) of 0.88 and 0.87 for DisA and CnpB, respectively (Figures 2D and 2E). Taken together, our results rule out a PhoPR-dependent transcriptional or posttranscriptional mechanism over c-di-AMP metabolic enzymes. To our knowledge, there are no other genes described that are involved in c-di-AMP metabolism in M. tuberculosis, which may indicate that PhoPR regulates c-di-AMP synthesis and/or degradation by a yet-unknown mechanism. Interrogation of those PhoPR-regulated genes with hypothetical or unknown functions might help to identify novel players in the c-di-AMP metabolism of M. tuberculosis.
Figure 2.

Identification of PhoPR as a novel regulator of c-di-AMP synthesis and secretion in M. tuberculosis
(A) Production of c-di-AMP in whole-cell extracts of M. tuberculosis H37Rv, its ΔphoPR mutant, and the mutant strain complemented (comp) with the phoPR genes. (B) Secretion of c-di-AMP into the supernatant of strains depicted in (A). Data are the mean and standard deviation from at least three biological replicates. (C) RNA sequencing (RNA-seq) profiles of three different genetic regions in M. tuberculosis MT103, MTBVAC, H37Rv, and the H37Rv ΔphoPR mutant. The disA, cnpB, and pks2 gene expression is indicated with a dashed box. Note that disA and cnpB show equivalent transcription levels in MTBVAC and the H37Rv ΔphoPR mutant relative to their wild-type strains. In contrast, the pks2 gene, which is a well-known PhoPR-regulated gene, shows downregulation in MTBVAC and the H37Rv ΔphoPR mutant. (D) Quantification of DisA and CnpB protein levels in H37Rv and the H37Rv ΔphoPR mutant by MRM-MS. Chromatograms show the identification of three different transitions from a specific peptide. Bars represent the area under the curve for every transition in H37Rv and the H37Rv ΔphoPR mutant. Equivalent results were obtained for the DisA peptides ANVQLVPDPSIPTDESGTR, HVLTDSATILSR, ANQAIATLER, and VFGYPTTTEAQDSTLSPR and for the CnpB peptides VEVSFAAPATLPESLR, LGALGDLTDSGR, VLGSAQLVSEAVGGR, and TVNLAAVASGFGGGGHR (data not shown).
Genetic inactivation of disA and cnpB genes results in variable expression and secretion of c-di-AMP in the MTBVAC vaccine
To study the contribution of c-di-AMP synthesis to the vaccine phenotypes of MTBVAC, we constructed disA and cnpB knockouts in the MTBVAC vaccine strain by using the bacterial artificial chromosome (BAC)-recombineering strategy37 to obtain ΔdisA::Km and ΔcnpB::Km mutants (Figures 3A and 3B). After PCR confirmation of the recombinant colonies (Figures 3A and 3B), we analyzed the synthesis of c-di-AMP in these MTBVAC derivatives relative to the MTBVAC parent strain. Notably, the production of c-di-AMP in the whole-cell fraction of the cnpB mutant was as high as 23 μg/mL, compared with 0.3 μg/mL in the parental MTBVAC, which represents more than a 75-fold increase. As expected, we failed to detect c-di-AMP production in the MTBVAC disA mutant (Figure 3C). These phenotypes were then also evaluated for the secretion of c-di-AMP into the supernatant. As expected, we did not detect the secretion of c-di-AMP in the MTBVAC disA mutant, and the MTBVAC cnpB mutant secreted approximately 10 times more c-di-AMP than MTBVAC into the supernatant (Figure 3D). Accordingly, we obtained a set of MTBVAC vaccine candidate mutants that produced and secreted differential levels of c-di-AMP, which were used to characterize the role of this molecule in MTBVAC vaccine efficacy.
Figure 3.

Construction and characterization of MTBVAC disA and MTBVAC cnpB mutants
(A) Schematic representation of the construction of the MTBVAC disA mutant indicating the primers used to confirm the genetic deletion of disA by PCR. The lower panel shows PCR bands of ΔdisA::Km colonies indicative of a specific allelic exchange. (B) Schematic representation of the construction of the MTBVAC cnpB mutant indicating the primers used to confirm the genetic deletion of cnpB by PCR. The lower panel shows PCR bands of ΔcnpB::Km colonies indicative of a specific allelic exchange. (C) Production of c-di-AMP in MTBVAC and its disA and cnpB mutant derivatives. (D) Secretion of c-di-AMP in MTBVAC and its disA and cnpB mutant derivatives. n.d., not detected. Data are the mean and standard deviation from three biological replicates.
MTBVAC and BCG fail to induce an IFNβ response, but they successfully produce IL-1β in infected macrophages
Since bacterial-derived c-di-AMP is a well-known stimulator of STING,13,14 we were prompted to study the type I IFN response elicited by the MTBVAC derivatives described in the previous section. Accordingly, we infected macrophages with MTBVAC, their disA and cnpB mutants, and BCG, and we measured the IFNβ produced by the cells. The existing literature indicates that mycobacteria must reach the cytosol to properly stimulate type I IFN responses. In this context, we should remember that BCG lacks a functional ESX-1 system due to RD1 deletion and that MTBVAC fails to secrete ESAT-6 because of phoP inactivation; accordingly, both vaccine strains and the disA or cnpB mutants are not expected to establish contact with the host cell cytosol. Consequently, we did not observe IFNβ induction upon infection with these vaccine strains (Figure 4A). Then, we used wild-type M. tuberculosis MT103 and H37Rv strains, which are known to rupture the phagosome, to confirm that both elicited a proper IFNβ response in our model (Figure 4A). Next, we demonstrated that macrophages infected with M. tuberculosis H37Rv ΔRD1 (lacking a functional ESX-1 system due to a 9 kb deletion)38 failed to induce IFNβ production, confirming the hypothesis that a functional ESX-1 system is essential to mount type I IFN responses against M. tuberculosis (Figure 4A). These results suggest that cytosolic contact is a prerequisite to stimulate STING,19,32,39 whereby these findings disagree with the results of the previously mentioned study that reported IFNβ responses are elicited by BCG,17 albeit at a low quantitative level. Taken together, these findings open the question of whether the contribution to STING stimulation exclusively depends on bacterial DNA sensing via cGAS, as previously demonstrated,19,20 or whether the endogenous c-di-AMP second messenger produced by Mycobacterium spp.17 can also contribute to cytosolic responses against this pathogen.
Figure 4.

IFNβ and IL-1β responses during THP-1 macrophage infections with different MTBVAC derivatives
(A) IFNβ produced by macrophages infected with the vaccines MTBVAC, its disA and cnpB mutants, and BCG Pasteur. M. tuberculosis MT103 and H37Rv strains and the M. tuberculosis ΔRD1 mutant served as positive and negative controls of IFNβ production, respectively. (B) IL-1β produced after macrophage infection with the strains denoted in (A). Bars represent the mean and standard deviation from three cellular infections. Statistical analysis was performed using one-way ANOVA followed by Turkey’s post-test. Asterisks indicate the following p values: ∗ 0.05 > p > 0.01; ∗∗ 0.01 > p > 0.0005; ∗∗∗∗ 0.0001 > p.
In parallel, we analyzed the IL-1β responses in macrophages infected with the mycobacterial strains described above. Induction of IL-1β after BCG vaccination is associated with protective effects against related or unrelated bacterial and fungal pathogens40 and hence constitutes a plausible mechanism to generate trained innate immunity in BCG and MTBVAC vaccines.40,41 Remarkably, MTBVAC and its derived mutants elicited equivalent IL-1β responses to wild-type M. tuberculosis, and these responses were comparatively higher relative to BCG or M. tuberculosis H37Rv ΔRD1 (Figure 4B). This result indicates that the absence of the ESX-1-encoding RD1 region in either BCG or M. tuberculosis H37Rv negatively impacts triggering complete IL-1β innate responses. Concerning the MTBVAC-derived strains, MTBVAC cnpB elicited lower IL-1β responses, while MTBVAC disA produced slightly higher IL-1β responses than MTBVAC (Figure 4B). This result might imply that type I IFN responses antagonize IL-1β production, as has been previously demonstrated during experimental infection with M. tuberculosis.42
Differential levels of c-di-AMP produced by MTBVAC influence vaccine attenuation and protective efficacy
A recent study demonstrated that disA overexpression in BCG and the concomitant increase in c-di-AMP levels improved vaccine efficacy by triggering superior type I IFN responses in guinea pigs.33 On the other hand, it is also possible that high c-di-AMP levels might be detrimental for the survival of a live vaccine by triggering a strong innate immune response. Considering the exacerbated c-di-AMP levels produced by the MTBVAC cnpB mutant compared with MTBVAC and the null production of c-di-AMP by the disA mutant (Figures 3C and 3D), we were prompted to check whether differential c-di-AMP synthesis might influence the vaccine properties of MTBVAC in the mouse model. We first measured the impact on virulence attenuation, a parameter related to the safety of a vaccine, by enumerating bacterial load in the lungs of severe combined immunodeficient (SCID) mice infected with BCG or the MTBVAC vaccine set. BCG resulted in a less attenuated vaccine, MTBVAC and MTBVAC disA showed intermediate attenuation, and MTBVAC cnpB was more attenuated (Figure 5A). Considering the absence of adaptive immunity in the SCID mice, these findings seem to support the hypothesis that the induction of potent innate immune responses, mediated by c-di-AMP cytosolic signaling, results in strong vaccine attenuation. In line with this observation, a previous study correlated BCG vaccine attenuation with protective efficacy, demonstrating that the more attenuated BCG strains resulted in less protection and vice versa.43 Then, we analyzed the protective efficacy of these vaccines using a challenge model with a virulent M. tuberculosis Beijing isolate. All vaccines protected against M. tuberculosis, but MTBVAC and MTBVAC disA showed better protection than BCG in the mouse model used. Conversely, MTBVAC cnpB displayed equivalent protection compared with BCG in our experimental model (Figure 5B). These results correlate with the attenuation profile of the MTBVAC set since MTBVAC and MTBVAC disA showed equivalent attenuation compared with the more attenuated MTBVAC cnpB mutant (Figure 5A). Altogether, we have started to elucidate the roles of c-di-AMP in TB vaccine safety and protective efficacy, but further work is needed in additional animal models to ascertain the precise contribution of this metabolite to the vaccine phenotypes of MTBVAC. In addition, nonspecific effects shown by BCG,44 such as treatment of bladder cancer, protection against heterologous infections, therapeutic treatment of asthma, or potentiation of different immune responses, could also benefit from the modulation of c-di-AMP levels in MTBVAC-based vaccines.
Figure 5.

Characterization of the impact of differential c-di-AMP production on MTBVAC vaccine attenuation and efficacy
(A) Bacterial loads in the lungs of SCID mice after 4 weeks infected with BCG Pasteur, MTBVAC, and the MTBVAC disA and cnpB mutants. (B) Protective efficacy of BCG Pasteur, MTBVAC, and the MTBVAC disA and cnpB mutants in the C3H/HeNRj mouse model. Mice were immunized with the previously mentioned strains, left unvaccinated as a control, and challenged 8 weeks later with the M. tuberculosis Beijing W4 strain. Bacterial loads in the lungs were enumerated 4 weeks after the challenge as a measure of vaccine efficacy. All data are mean ± SEM. Statistical analysis was performed by one-way ANOVA and the Bonferroni post hoc test.
Discussion
Cyclic (di)nucleotides constitute key second messengers in the microbial world, allowing not only intra- and interbacterial communication but also intracellular signaling in infected host cells.12,45 In this study, we specifically focused on the role of c-di-AMP in M. tuberculosis in the context of vaccine attenuation and efficacy. When comparing the production of c-di-AMP in the existing two live vaccines in the TB vaccine pipeline, BCG and MTBVAC, we observed that MTBVAC unexpectedly produced higher c-di-AMP levels than BCG (Figure 1B). This phenotype was also translated to c-di-AMP secretion (Figures 1B and 1D), which revealed that M. tuberculosis possesses the mechanisms required to export this second messenger outside the bacterium, in agreement with previous studies.16,36 Nevertheless, whether the c-di-AMP molecule is secreted by passive diffusion throughout the mycobacterial membrane or whether it is exported by a specific transporter remains to be elucidated.
Exploration of the molecular mechanism responsible for the increased production of c-di-AMP in MTBVAC demonstrated that the PhoPR two-component system negatively regulates the production of this metabolite in an M. tuberculosis clinical isolate (MT103) and in a widely used laboratory strain (H37Rv) since both the M. tuberculosis H37Rv phoPR mutant and the MTBVAC vaccine produced higher c-di-AMP levels than their respective wild-type strains (Figures 1 and 2). In addition, these results indicate that PhoPR-dependent synthesis of c-di-AMP is not related to the genetic background since MTBVAC and M. tuberculosis phoPR knockouts were constructed in unrelated parental strains. Furthermore, successful PhoPR complementation of c-di-AMP synthesis and secretion demonstrates that PhoPR, and not an unrelated mutation, is specifically responsible for this phenotype. Further work is needed to decipher the precise PhoPR-dependent mechanism leading to c-di-AMP regulation since both the synthetic and degrading enzymes of this metabolite are not differentially expressed in phoP or phoPR mutants (Figure 2C). In a previous study, we demonstrated that the secretion of immunodominant antigens in M. tuberculosis phoPR mutants30 is indirectly regulated by a posttranscriptional mechanism involving the PhoPR-dependent noncoding RNA mcr7.29 Thus, it is possible that mcr7, or other yet-unknown PhoPR-regulated noncoding RNAs, might exert posttranscriptional regulation over c-di-AMP synthesis in M. tuberculosis.
Since the M. tuberculosis phoP and phoPR mutants, as well as the MTBVAC vaccine, are attenuated in animal models,23,27,28,46,47 we can hypothesize that the high production of c-di-AMP in these strains might partly account for their attenuation. Indeed, M. tuberculosis cnpB mutants, known to produce and secrete high c-di-AMP levels, were reported to be more attenuated in murine infection models than the wild-type strain.16,18 In this same line of evidence, the M. tuberculosis disA overexpression strain also produces increased amounts of c-di-AMP levels and shows a higher attenuation in mice than the parental strain.17 Our findings that an MTBVAC cnpB mutant was more attenuated than MTBVAC in SCID mice (Figure 5A) further support the conclusion that c-di-AMP overproduction might contribute to stronger attenuation of M. tuberculosis phoPR mutants. We should remember that the PhoPR two-component system regulates many key virulence phenotypes in M. tuberculosis, including the secretion of ESAT-6. Thus, elucidating the precise contribution of increased c-di-AMP production to the attenuation of phoP and phoPR mutants is technically challenging. However, it has been demonstrated that MTBVAC elicits a clear dose-response induction of IL-1β, IL-6, IL-10, and tumor necrosis factor alpha (TNF-α) in human monocytes,41 and similar responses have also been observed upon infection with the attenuated strains M. tuberculosis cnpB mutant or M. tuberculosis overexpressing disA,17,18 suggesting that a proper induction of innate immunity by high c-di-AMP amounts might result in better control of bacterial multiplication and consequently virulence attenuation.
Since the induction of IFNβ responses upon infection with BCG or BCG overexpressing disA has been previously demonstrated,17,33 it is intriguing that we did not detect a dose-response induction of IFNβ responses upon infection of THP-1-differentiated macrophages with BCG, MTBVAC, MTBVAC disA, or MTBVAC cnpB mutants (Figure 4A).32 However, it should be noted that different macrophages were used in both studies. While Dey et al. used mouse RAW264.7 cells and bone-marrow-derived macrophages, we used the THP1 macrophage cell line. In THP1 macrophages, virulent M. tuberculosis efficiently induced IFNβ responses, but this induction was strictly dependent on the presence of a completely functional RD1 region (Figure 5A). This result indicates that phagosomal escape might be essential to trigger the cytosolic surveillance pathway in these macrophages. However, the higher cytotoxic activity of RAW264.7 cells and bone-marrow-derived macrophages might result in IFNβ responses even in the absence of an intact RD1 region. A side-to-side comparison of IFNβ responses using different macrophage lines or primary cells would help to elucidate the precise roles of c-di-AMP and the RD1 region in triggering cytosolic surveillance responses.
Despite the macrophage infections with MTBVAC, and the MTBVAC disA or the MTBVAC cnpB mutants not resulting in differential IFNβ immune responses (Figure 4), the production and secretion of variable c-di-AMP levels by our vaccine set have an impact on vaccine attenuation and efficacy (Figure 5). The exacerbated c-di-AMP amounts produced by the MTBVAC cnpB mutant otherwise results in higher vaccine attenuation (Figure 5A). One possible explanation for this phenotype could be that the robust induction of intracellular surveillance responses otherwise results in better control of bacterial multiplication in the lung. In agreement with a previous observation,43 the overattenuation of a vaccine is associated with lower protective efficacy, as observed with the MTBVAC cnpB mutant (Figure 5B). Nevertheless, it was previously demonstrated that BCG overexpressing disA exhibits superior efficacy to BCG in the guinea pig challenge model.33 In terms of c-di-AMP production, both the MTBVAC cnpB mutant and the recombinant BCG overexpressing disA are able to produce increased c-di-AMP levels relative to their parental strains, and consequently, the opposing results in protective efficacy upon vaccination with both strains is unexpected. Several explanations exist for this discrepancy. First, different animal models were used in both experiments, and the mouse model does not necessarily reflect the more sensitive guinea pig model used in Dey et al. Second, different challenge strains were used in these studies. While Dey et al. used the M. tuberculosis H37Rv laboratory strain, we used M. tuberculosis W4, which is known to belong to the Beijing hypervirulent family. Third, it remains to be determined whether the MTBVAC cnpB mutant and the BCG overexpressing disA produce equivalent c-di-AMP levels since we should remember that MTBVAC and MTBVAC cnpB produce much greater quantities of this second messenger relative to BCG. Fourth, to our knowledge, there is no evidence of c-di-AMP secretion in BCG since a BCG cnpB mutant failed to reproduce secretion of this metabolite compared with an M. tuberculosis cnpB mutant.36 Taken together, these technical differences might very well account for the different results in vaccine efficacy.
In the context of TB vaccines, an unresolved question regarding host cytosolic signaling refers to the access of the c-di-AMP produced by live vaccines to the host cell cytosol to stimulate STING. Previous studies demonstrated that M. tuberculosis efficiently stimulates STING in a process dependent on functional ESX-1.19,20 However, it is key to remember that neither BCG nor MTBVAC vaccines secrete ESAT-6, and consequently, these vaccines do not have access to the cytosol, as M. tuberculosis does.5 Thus, the induction of IFNβ responses by BCG or BCG overexpressing disA is puzzling.17 It is possible that endogenous c-di-AMP produced by BCG freely traverses the eukaryotic membrane of the phagolysosome, as has been reported with other cyclic dinucleotides.48,49 This hypothesis would explain a direct STING stimulation by vaccine-produced c-di-AMP in the absence of a functional ESX-1 system and would pave the way to improving live TB vaccines.
The question of whether induction of the host cytosolic surveillance with production of IFNβ is beneficial or detrimental to viral or bacterial pathogens is still a matter of debate.50,51 The factors that determine whether this response is protective or pathogenic have yet to be defined, and frequently, the answer depends on the experimental model, the pathogen itself, or the specific stage at which IFNβ responses are produced. Considering the coexistence of c-di-AMP synthetic and degradation pathways in M. tuberculosis, this might represent an evolutionary mechanism to manipulate the host response in favor of the pathogen. While c-di-AMP endogenously produced by M. tuberculosis triggers the host cytosolic surveillance pathway, which is probably detrimental for bacterial survival, the TB bacillus also possesses a c-di-AMP phosphodiesterase that contributes to alleviating this detrimental effect. We should remember that the M. tuberculosis PhoPR virulence system activates many phenotypes required for M. tuberculosis virulence. PhoPR allows ESAT-6 secretion25 and subsequent cytosolic escape,5 which ultimately favors the cell-to-cell spread of M. tuberculosis.9 In addition, PhoPR controls the synthesis of acyltrehalose-derived lipids and sulfolipids,27,47 which inhibits the innate immune response52 and induces a productive cough,53 facilitating transmission to uninfected individuals. Finally, PhoPR activates transcription of a noncoding RNA that downregulates secretion of immunodominant antigens,29 thus decreasing immune recognition (Figure 6).30 In line with these observations, our demonstration that c-di-AMP synthesis is repressed by the PhoPR virulence system reinforces the hypothesis that production of this second messenger is detrimental to M. tuberculosis. Therefore, it is tempting to speculate that M. tuberculosis has evolved two complementary pathways to restrain c-di-AMP levels: the capacity to degrade this metabolite though CnpB phosphodiesterase and the repression of c-di-AMP synthesis by PhoPR. Both mechanisms would contribute to diminished recognition of M. tuberculosis by the host cytosolic surveillance system (Figure 6).
Figure 6.

Schematic representation of the PhoPR-related phenotypes in M. tuberculosis (left side) and MTBVAC (right side) in the context of the macrophage environment
Until the publication of this manuscript, three virulence networks were documented to be regulated by PhoPR in M. tuberculosis: the synthesis of di- and poli-acyltrehaloses (DAT and PAT, respectively) and sulfolipid (SL), the latter lipid being involved in the inhibition of innate immunity and the production of cough; the secretion of ESAT-6, which mediates phagosomal rupture and the consequent cytosolic escape; and the downregulation of TatC, known to be an essential constituent of the twin arginine translocation (TAT), which results in restrained antigen secretion. These phenotypes are inactivated in MTBVAC because of a phoP-inactivating mutation and result in a lack of PhoPR-regulated lipids, the absence of ESAT-6 secretion, and increased secretion of TAT substrates. We demonstrate here that, in addition to the aforementioned phenotypes, c-di-AMP synthesis and secretion are negatively regulated by PhoPR in M. tuberculosis. Accordingly, the high c-di-AMP levels in the MTBVAC vaccine result in enhanced innate immune responses.
A previous study demonstrated that a tuberculosis subunit vaccine adjuvant with a synthetic cyclic-dinucleotide analog conferred enhanced protection and superior Th1 and Th17 responses compared with BCG vaccination.54 This finding highlights the relevance of cyclic dinucleotides as vaccine adjuvants and opens perspectives to rationally engineer live vaccines. In this study, we attempted to provide knowledge to unresolved questions concerning the role of this second messenger in the TB vaccine field. Inactivation of phoP in MTBVAC results in unrestrained c-di-AMP synthesis and enhanced stimulation of innate immunity relative to BCG. Altogether, our research has paved the way to modulate endogenous c-di-AMP production by live attenuated TB vaccines as a strategy to treat not only infectious diseases but also diseases with an immunological component.
Material and methods
Bacterial strains and growth conditions
We used the M. tuberculosis strain H37Rv, its phoPR mutant and the complemented strain Mt103, and H37Rv ΔRD1. We also used the vaccine strain MTBVAC and its derivatives MTBVAC ΔcnpB::Km and MTBVAC ΔdisA::Km, as well as BCG Pasteur and BCG Danish. These strains are further described in Table 1. Mycobacteria were cultured in Middlebrook 7H9 liquid medium (Difco) containing 0.05% Tween 80 and supplemented with 10% (vol/vol) ADC or dextrose/NaCl. For solid media, Middlebrook 7H10 broth containing 10% (vol/vol) ADC was used. When necessary, 20 μg/mL hygromycin (Hyg) or 20 μg/mL kanamycin (Km) was added to the media. Escherichia coli was cultured in liquid media Luria-Bertani (LB) broth or in solid media LB agar. When required, media were supplemented with 100 μg/mL ampicillin (Amp), 12.5 μg/mL chloramphenicol (Cm), 20 μg/mL Km, or 50 μg/mL Hyg.
Table 1.
Mycobacterial strains used in this study
| Strain designation | Description | Reference |
|---|---|---|
| Mt103 | clinical isolate of M. tuberculosis lineage 4 | Jackson et al.55 |
| H37Rv | reference laboratory strain of M. tuberculosis | Cole et al.56 |
| H37Rv ΔphoPR | mutant of phoP and phoR genes in H37Rv | Gonzalo-Asensio et al.47 |
| H37Rv ΔphoPR complemented | complemented phoPR genes in H37Rv ΔphoPR strain | Gonzalo-Asensio et al.47 |
| H37Rv ΔRD1 | H37Rv deleted RD1 region | Pym et al.22 |
| BCG Pasteur | current vaccine against TB, attenuated from M. bovis | Brosch et al.21 |
| BCG Danish | current vaccine against TB, attenuated from M. bovis | Brosch et al.21 |
| MTBVAC::hyg (Mt103 ΔfadD26 ΔphoP::hyg) | double marked mutant in phoP with res-Ωhyg-res cassette | Arbues et al.23 |
| MTBVAC | double unmarked mutant of phoP and fadD26 in Mt103 strain | Arbues et al.23 |
| MTBVAC ΔcnpB::km | marked mutant of cnpB gene in MTBVAC | this study |
| MTBVAC ΔdisA::km | marked mutant of disA gene in MTBVAC | this study |
Construction of cnpB and disA mutants in MTBVAC
Deletion of the cnpB or disA genes in MTBVAC was obtained using the BAC-rec strategy.37 An M. tuberculosis H37Rv BAC library was constructed in the pBeloBAC11 vector and contained in E. coli DH10B.57 The thermosensitive plasmid pKD46 (containing the red recombinase from lambda phage) was transformed into the DH10B clone carrying Rv404 (containing the cnpB gene) and Rv222 (containing the disA gene).58 E. coli strains carrying the BAC Rv404 or Rv222 and pKD46 plasmids were cultured in media in the presence of 0.15% arabinose and transformed with a PCR product containing a Km-resistant cassette from pKD4 flanked by 40 bp identity arms to target the gene of interest. PCR products were obtained using cnpB-P1-Fw/cnpB-P2-Rv and disA-P1-Fw/disA-P2. Recombinants were selected in LB plates containing Km, and gene deletion in the BAC was confirmed by PCR using specific primers (Table 2).
Table 2.
Primers used in this study
| Primers | Sequence 5–3 |
|---|---|
| cnpB-P1-Fw | GGGTGCGGCAAGCGGGTAGAGGTC AGCTTTGCCGCGCCGGGTGT AGGCTGGAGCTGCTTC |
| cnpB-P2-Rv | GCGTCGTCGATCGAGCCGG TGGTCGTATACCCCGCGGC CACATATGAATATCCTCCTTAGT |
| KO-cnpB-Fw | GGGCGTTGTTCCGGTCTTCG |
| KO-cnpB-Rv | CCTAGCCCTAACGCCGCTG |
| Confirm-KO-cnpB-Fw | GCGGTGATCATCGGTTTCAATGTGCG |
| Confirm-KO-cnpB-Rv | CAACGCACTCGCCACCGTC |
| cnpB-I1 | GGGCGATCTAACTGATTCCGGGC |
| cnpB-I2 | CGACCTCCTTGAACACCGCCG |
| disA-P1-Fw | GCACGCTGTGACTCGTCCGACCCTGC GTGAGGCTGTCGCCGTGTAGGCT GGAGCTGCTTC |
| disA-P2-Rv | CGTCCGACAGGGCGTCCAGTTCGT CCAGGGTGGCATTGATCATATGAAT ATCCTCCTTAGT |
| KO-disA-Fw | CTCGCCGCACAGTCCGAC |
| KO-disA-Rv | GATCGCGGTGGCCATCGTCATC |
| Confirm-KO-disA-Fw | GCGCGCTCTATGTCTCTGGTGAG |
| Confirm-KO-disA-Rv | CGTTTGCGTGACCTGGCCCTAC |
| disA-I1 | GATGGTGGCTTCTCCCTCGATGTC |
| disA-I2 | CACCACCGTCATCACATCGCG |
| P1-inv | GAAGCAGCTCCAGCCTACAC |
| P2-inv-long | CTTCGGAATAGGAACTAAG GAGGATATTCATATG |
Allelic exchange substrates (AESs) containing a Km-resistant cassette flanked by 1.1 kb identity arms for site-specific recombination were obtained by PCR using BAC Rv404-ΔcnpB::Km and Rv222-ΔdisA::Km templates and cnpB-KO-Fw/cnpB-KO-Rv and disA-KO-Fw/disA-KO-Rv primers. AESs were transformed in MTBVAC carrying pJV53H and cultured in the presence of 0.2% acetamide.59 Recombinants were selected in 7H10 plates containing Km and confirmed using specific primers. The sequences of the primers used for knockout construction and verification are provided in Table 2.
Metabolite extraction
Mycobacterial strains were cultured until log-phase (OD600 nm–0.6) in 7H9 medium supplemented with ADC or dextrose/NaCl to measure c-di-AMP production or secretion, respectively. To quantify c-di-AMP in the secreted fraction, cultures were centrifuged, and supernatants were filtered through a 0.22 μm pore-size filter and used for metabolite quantification. To quantify c-di-AMP produced by bacteria in the whole-cell fraction, bacterial pellets were resuspended in 300 μL extraction solution (acetonitrile/methanol/water, 2/2/1, vol/vol/vol, high-performance liquid chromatography [HPLC] grade) and transferred to tubes containing glass beads (MP Biomedicals). Mycobacterial suspensions were disrupted by FastPrep (6.5 m/s, 45 s), incubated on ice for 15 min, and heated at 95°C for 10 min. Suspensions were cooled on ice, and supernatants were transferred into a new vial after centrifugation at 14,000 RPM at 4°C for 10 min. Two additional extractions were performed by the addition of 200 μL to the tubes containing glass beads. Suspensions were disrupted by FastPrep (6.5 m/s, 45 s) and incubated on ice for 15 min, and supernatants were combined into vials after centrifugation (14,000 RPM, at 4°C for 10 min). Vials containing the supernatants were stored overnight at −20°C. The day after, vials were centrifuged at 14,000 RPM for 10 min at 4°C, and supernatants were filtered through a 0.22 μm pore-size filter to quantify c-di-AMP in whole-cell lysates.
c-di-AMP quantification
Quantification of total c-di-AMP and c-di-GMP of the H37Rv, H37Rv ΔphoPR, H37Rv ΔphoPR complemented strain, Mt103, and MTBVAC strains was performed in collaboration with the Institute of Pharmacology, Hannover Medical School, Hannover (Germany) as described in Corrigan et al.60 These measurements were performed using isotope-labeled 13C15N of c-di-AMP. The same extractions were also quantified in the proteomics facility of Servicios Científico Técnicos of CIBA (IACS-Universidad de Zaragoza) using c-di-GMP as an internal control because our previous finding demonstrated a lack of production of this metabolite in M. tuberculosis. Briefly, c-di-AMP quantification (LC–ESI–MS/MS) was performed on an Agilent 1200 HPLC and a 4000 QTRAP mass spectrometer (SCIEX), column Clarity 3 μm Oligo-RP 100 × 2 mm (Phenomenex). Eluents used included: Phase A, 50 mM TEAA, and eluent B methanol. c-di-GMP was used as the internal standard. A calibration curve was obtained from 1 to 3,000 ng/mL. Samples were incubated at 45°C for 90 min and resuspended in a specific volume of 10 mM TEAA and c-di-GMP (400 ng/mL). Solutions were vortexed, sonicated for 5 min, vortexed again, and filtered through a 0.45 μm pore size. The injection volume was 25 μL, and the flow rate was 200 μL/min throughout the chromatographic run. Also, 95% A and 5% B were used from 0 to 5 min, followed by a linear gradient from 95% A to 30% A for 9 min, followed by 9 min until 95% A and 5% B. The detection method was selected reaction monitoring (SRM). All secreted fractions were evaluated in collaboration with Fundación MEDINA, Granada (Spain). The equipment used was an LC Agilent 1290 coupled to an API 4000 mass spectrometer (SCIEX) and an automatated CTC PAL injector. The chromatographic column was Xbridge BEH amide, with dimensions of 100 × 2.1 mm and 3.5 μm (Waters). The injection volume was 7 μL, and the chromatographic run was 7.1 min. Mobile phase A consisted of deionized water (0.1% ammonia), and mobile phase B consisted of acetonitrile (0.1% ammonia) with a gradient flow rate of 300 μL/min. The gradient elution was performed as follows: t = 0.0–0.50 min 95% B; t = 0.50–4.50 min 30% B; t = 4.50–5.70 min 30% B; t = 5.70–5.80 min 95% B; t = 5.80–7.10 min 95% B. For the analysis, c-di-GMP was used as the internal standard. The standards for the calibration curve were obtained by adding c-di-AMP (14.7 to 1,890 ng/mL). In the samples, the internal standard was also added (100 ng/mL), and 200 μL of acetonitrile 0.1% formic acid was added, vortexed, and centrifuged (5 min, 3,700 RPM at 4°C). The concentration of the analyte was obtained using the calibration curve and the relation of the analyte area and the internal standard area. The limit of detection (lod) of c-di-AMP in the technique was 5 ng/mL, equivalent to 7.6 nM of pure compound.
Protein identification by MRM/MS
The MRM assay approach was applied for the measurement of specific DisA and CnpB peptides in whole-cell extracts of H37Rv and its phoPR mutant. Bacterial cultures (20 mL) were grown to an OD (600 nm) of 0.6 and pelleted. Bacterial pellets were resuspended in 2 mL Tris-HCl 100 mM and disrupted in FastPrep (2 cycles, 45 s at speed 6.5 m/s, samples were cooled on ice between cycles). Tubes were centrifuged, and the aqueous phases containing whole-cell protein were filtered through a 0.22 μm filter. For in-solution digestion, protein was resuspended in denaturing buffer (6 M urea, 100 mM Tris-HCl pH 7.8). Cysteines were reduced with 200 mM DTT for 30 min at 37°C and alkylated with 200 mM iodoacetamide for 30 min in the dark. Unreacted iodoacetamide was consumed by adding an excess of the reducing agent (200 mM DTT) for 30 min at room temperature. Samples were diluted with 50 mM ammonium bicarbonate to a final concentration of less than 1 M urea. Protein digestion was carried out with trypsin (Trypsin Gold, Promega) at a 1:20 ratio (enzyme/protein) for 18 h at 37°C. The reaction was stopped by adding concentrated formic acid (Sigma). Samples were dried in a vacuum concentrator and reconstituted in 98% H2O, 2% acetonitrile, and 0.1% formic acid.
Protein identification was performed on a triple quadrupole/linear ion trap mass spectrometer QTRAP 6500+ (SCIEX, Foster City, CA, USA) coupled to nano/micro-HPLC (Eksigent LC 425, SCIEX). Sample preconcentration and desalting of tryptic digests were performed on a C18 column (Luna 0.3 mm id, 20 mm, 5 μm particle size, Phenomenex, Torrance, CA, USA). Tryptic peptides were then separated using a C18 column (Luna Omega Polar 0.3 mm id, 150 mm, 3 μm particle size, Phenomenex, Torrance, CA, USA) at a flow rate of 5 μL/min, with a 30 min linear gradient from 5 to 35% acetonitrile in 0.1% formic acid. The mass spectrometer was interfaced with a microspray source (Turbo V) equipped with an uncoated fused silica emitter tip (25 μm inner diameter, 10 μm tip, New Objective, Woburn, MA, USA) and was operated in positive ion mode. MS source parameters were as follows: capillary voltage 5,000 V, source temperature 150°C, declustering potential (DP) 85 V, curtain, ion source gas (nitrogen) 25 psi, and collision gas (nitrogen) 15 psi. Analyses were performed using an information-dependent acquisition (IDA) method with the following steps: single enhanced mass spectra (EMS, 400-1,400 m/z), from which the most intense peaks were subjected to an enhanced product ion (EPI [MS/MS]) scan. Peptide identity was confirmed using an MRM-initiated detection and sequencing (MIDAS) algorithm using Mascot (v.2.3, Matrix Science, UK) and Swiss-Prot. The identified and quantified peptides were as follows: for DisA, ANVQLVPDPSIPTDESGTR, HVLTDSATILSR, ANQAIATLER, LQLDELLGGNDTAR, and VFGYPTTTEAQDSTLSPR and for CnpB, VEVSFAAPATLPESLR, LGALGDLTDSGR, LVEIGVDNATVSR, VLGSAQLVSEAVGGR, and TVNLAAVASGFGGGGHR.
Mycobacterial infection of THP-1
THP-1 cells were grown in RPMI 1640 GlutaMAX medium supplemented with 10% (vol/vol) decomplemented and filtrated fetal bovine serum (FBS). THP-1 monocytes were differentiated into macrophages by adding 10 ng/mL PMA for 72 h. Cells were distributed in a 24-well plate (500,000 cells per well), and mycobacteria strains were added at a multiplicity of infection (MOI) of 5:1. For bacteria preparation, cultures were centrifuged, and pellets were resuspended in 5 mL of 1× DPBS. OD was measured to calculate the volume required. Bacteria were added to THP-1 cells and incubated at 37°C and 5% CO2. Supernatants were collected after 24 h and filtered through a 0.22 μm pore-size filter. IL-1β and IFNβ released were detected by ELISA. Human IL-1β/IL-1F2 and IFNβ released by THP-1 macrophages were quantified using the DY201-05 (R&D Systems) and DY814-05 (R&D Systems) kits, respectively.
Mouse experiments
All mice were observed and kept under controlled conditions. For attenuation experiments, MTBVAC, MTBVAC ΔcnpB::Km, MTBVAC ΔdisA::Km, or BCG Pasteur was inoculated with 105 CFU contained in 40 μL of PBS by the intranasal route in immunocompromised SCID mice. The bacterial burden in the lungs was assessed 4 weeks postinoculation by enumerating colonies grown in 7H10-ADC plates. For protection experiments, C3H/HeNRj female mice were subcutaneously vaccinated with 106 CFU in 100 μL PBS containing each of the abovementioned strains or left unvaccinated as a control group. Eight weeks later, the mice were challenged by the intranasal route with 200 CFU in 40 μL PBS of the M. tuberculosis W4 strain. Four weeks postchallenge, the bacterial burden was evaluated in the lungs by inoculating serial dilutions onto 7H10-ADC plates.
Ethics statement
Experimental animal studies were performed in agreement with European and national directives for the protection of animals for experimental purposes. All procedures were carried out under Project License PI46/18, approved by the Ethics Committee for Animal Experiments from the University of Zaragoza.
Acknowledgments
The authors would like to acknowledge Prof. Volkhard Kaever from the Hannover Medical School and Irene Orera from the Proteomics Platform of Servicios Científico Técnicos del CIBA (IACS-Universidad de Zaragoza), ProteoRed-ISCIII member, for helping us with c-di-AMP quantification. We also acknowledge the use of Servicio General de Apoyo a la Investigación-SAI, Universidad de Zaragoza. Finally, we offer our special recognition to Prof. Iñigo Lasa from Navarrabiomed for his guidance during this project. This work was supported by a grant from the Spanish Ministry of Science and Innovation to J.G.-A. (reference PID2019-104690RB-I00). I.P. was a recipient of a “DGA-Fondo Social Europeo” grant. I.P. also was a recipient of a “Programa CAI-Ibercaja Estancias de Investigación” grant (CM3/18) and a “FEMS Research and Training” grant (FEMS-GO-2018-118) to perform an internship at the Integrated Mycobacterial Pathogenomics Unit at the Institut Pasteur of Paris. MEDINA authors disclosed the receipt of financial support from Fundación MEDINA, a public–private partnership of Merck Sharp & Dohme de España S.A./Universidad de Granada/Junta de Andalucía (PIN-0474-2016).
Author contributions
Conceptualization, J.G.-A., C.M., and I.P.; methodology, J.G.-A., C.M., R.B., N.A., C.D., F.V., F.S., S.U., and I.P.; experimental research, J.G.-A., C.D., F.S., S.U., E.C.-P., and I.P.; writing – original draft, J.G.-A. and I.P.; writing – review & editing, J.G.-A.; funding acquisition, J.G.-A.; supervision, J.G-A., C.M., R.B., and F.V. All authors reviewed the final report.
Declaration of interests
C.M. and J.G.-A. are coinventors on the patent “Tuberculosis vaccine”. N.A., C.M., and J.G.-A. are coinventors on the patent “Compositions for use as a prophylactic agent to those at risk of infection of tuberculosis, or as secondary agents for treating infected tuberculosis patients”. Both patents were filed by the University of Zaragoza.
References
- 1.Paulson T. Epidemiology: a mortal foe. Nature. 2013;502:S2–S3. doi: 10.1038/502S2a. [DOI] [PubMed] [Google Scholar]
- 2.WHO . 2021. Global Tuberculosis Report. [Google Scholar]
- 3.Martin C., Aguilo N., Marinova D., Gonzalo-Asensio J. Update on TB vaccine pipeline. Appl. Sci. 2020;10:15. doi: 10.3390/app10072632. [DOI] [Google Scholar]
- 4.Abdelwahab S.F., Issa U.H., Ashour H.M. A novel vaccine selection decision-making model (VSDMM) for COVID-19. Vaccines (Basel) 2021;9 doi: 10.3390/vaccines9070718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van der Wel N., Hava D., Houben D., Fluitsma D., van Zon M., Pierson J., Brenner M., Peters P.J. M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell. 2007;129:1287–1298. doi: 10.1016/j.cell.2007.05.059. [DOI] [PubMed] [Google Scholar]
- 6.Simeone R., Sayes F., Lawaree E., Brosch R. Breaching the phagosome, the case of the tuberculosis agent. Cell Microbiol. 2021;23:e13344. doi: 10.1111/cmi.13344. [DOI] [PubMed] [Google Scholar]
- 7.Houben D., Demangel C., van Ingen J., Perez J., Baldeon L., Abdallah A.M., Caleechurn L., Bottai D., van Zon M., de Punder K., et al. ESX-1-mediated translocation to the cytosol controls virulence of mycobacteria. Cell Microbiol. 2012;14:1287–1298. doi: 10.1111/j.1462-5822.2012.01799.x. [DOI] [PubMed] [Google Scholar]
- 8.Augenstreich J., Arbues A., Simeone R., Haanappel E., Wegener A., Sayes F., Le Chevalier F., Chalut C., Malaga W., Guilhot C., et al. ESX-1 and phthiocerol dimycocerosates of Mycobacterium tuberculosis act in concert to cause phagosomal rupture and host cell apoptosis. Cell Microbiol. 2017;19 doi: 10.1111/cmi.12726. [DOI] [PubMed] [Google Scholar]
- 9.Aguilo J.I., Alonso H., Uranga S., Marinova D., Arbues A., de Martino A., Anel A., Monzon M., Badiola J., Pardo J., et al. ESX-1-induced apoptosis is involved in cell-to-cell spread of Mycobacterium tuberculosis. Cell Microbiol. 2013;15:1994–2005. doi: 10.1111/cmi.12169. [DOI] [PubMed] [Google Scholar]
- 10.Ross P., Weinhouse H., Aloni Y., Michaeli D., Weinberger-Ohana P., Mayer R., Braun S., de Vroom E., van der Marel G.A., van Boom J.H., Benziman M. Regulation of cellulose synthesis in Acetobacter xylinum by cyclic diguanylic acid. Nature. 1987;325:279–281. doi: 10.1038/325279a0. [DOI] [PubMed] [Google Scholar]
- 11.Witte G., Hartung S., Buttner K., Hopfner K.P. Structural biochemistry of a bacterial checkpoint protein reveals diadenylate cyclase activity regulated by DNA recombination intermediates. Mol. Cel. 2008;30:167–178. doi: 10.1016/j.molcel.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 12.Corrigan R.M., Grundling A. Cyclic di-AMP: another second messenger enters the fray. Nat. Rev. Microbiol. 2013;11:513–524. doi: 10.1038/nrmicro3069. [DOI] [PubMed] [Google Scholar]
- 13.Woodward J.J., Iavarone A.T., Portnoy D.A. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science. 2010;328:1703–1705. doi: 10.1126/science.1189801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sauer J.D., Sotelo-Troha K., von Moltke J., Monroe K.M., Rae C.S., Brubaker S.W., Hyodo M., Hayakawa Y., Woodward J.J., Portnoy D.A., Vance R.E. The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect. Immun. 2011;79:688–694. doi: 10.1128/IAI.00999-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bai Y., Yang J., Zhou X., Ding X., Eisele L.E., Bai G. Mycobacterium tuberculosis Rv3586 (DacA) is a diadenylate cyclase that converts ATP or ADP into c-di-AMP. PloS one. 2012;7:e35206. doi: 10.1371/journal.pone.0035206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang J., Bai Y., Zhang Y., Gabrielle V.D., Jin L., Bai G. Deletion of the cyclic di-AMP phosphodiesterase gene (cnpB) in Mycobacterium tuberculosis leads to reduced virulence in a mouse model of infection. Mol. Microbiol. 2014;93:65–79. doi: 10.1111/mmi.12641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dey B., Dey R.J., Cheung L.S., Pokkali S., Guo H., Lee J.H., Bishai W.R. A bacterial cyclic dinucleotide activates the cytosolic surveillance pathway and mediates innate resistance to tuberculosis. Nat. Med. 2015;21:401–406. doi: 10.1038/nm.3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dey R.J., Dey B., Zheng Y., Cheung L.S., Zhou J., Sayre D., Kumar P., Guo H., Lamichhane G., Sintim H.O., Bishai W.R. Inhibition of innate immune cytosolic surveillance by an M. tuberculosis phosphodiesterase. Nat. Chem. Biol. 2017;13:210–217. doi: 10.1038/nchembio.2254. [DOI] [PubMed] [Google Scholar]
- 19.Wassermann R., Gulen M.F., Sala C., Perin S.G., Lou Y., Rybniker J., Schmid-Burgk J.L., Schmidt T., Hornung V., Cole S.T., Ablasser A. Mycobacterium tuberculosis differentially activates cGAS- and inflammasome-dependent intracellular immune responses through ESX-1. Cell host & microbe. 2015;17:799–810. doi: 10.1016/j.chom.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 20.Watson R.O., Bell S.L., MacDuff D.A., Kimmey J.M., Diner E.J., Olivas J., Vance R.E., Stallings C.L., Virgin H.W., Cox J.S. The cytosolic sensor cGAS detects Mycobacterium tuberculosis DNA to induce type I interferons and activate autophagy. Cell host & microbe. 2015;17:811–819. doi: 10.1016/j.chom.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brosch R., Gordon S.V., Garnier T., Eiglmeier K., Frigui W., Valenti P., Dos Santos S., Duthoy S., Lacroix C., Garcia-Pelayo C., et al. Genome plasticity of BCG and impact on vaccine efficacy. Proc. Natl. Acad. Sci. United States America. 2007;104:5596–5601. doi: 10.1073/pnas.0700869104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pym A.S., Brodin P., Brosch R., Huerre M., Cole S.T. Loss of RD1 contributed to the attenuation of the live tuberculosis vaccines Mycobacterium bovis BCG and Mycobacterium microti. Mol. Microbiol. 2002;46:709–717. doi: 10.1046/j.1365-2958.2002.03237.x. [DOI] [PubMed] [Google Scholar]
- 23.Arbues A., Aguilo J.I., Gonzalo-Asensio J., Marinova D., Uranga S., Puentes E., Fernandez C., Parra A., Cardona P.J., Vilaplana C., et al. Construction, characterization and preclinical evaluation of MTBVAC, the first live-attenuated M. tuberculosis-based vaccine to enter clinical trials. Vaccine. 2013;31:4867–4873. doi: 10.1016/j.vaccine.2013.07.051. [DOI] [PubMed] [Google Scholar]
- 24.Broset E., Martin C., Gonzalo-Asensio J. Evolutionary landscape of the Mycobacterium tuberculosis complex from the viewpoint of PhoPR: implications for virulence regulation and application to vaccine development. mBio. 2015;6 doi: 10.1128/mBio.01289-15. 01289–01215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frigui W., Bottai D., Majlessi L., Monot M., Josselin E., Brodin P., Garnier T., Gicquel B., Martin C., Leclerc C., et al. Control of M. tuberculosis ESAT-6 secretion and specific T cell recognition by PhoP. PLoS Pathog. 2008;4:e33. doi: 10.1371/journal.ppat.0040033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Solans L., Aguilo N., Samper S., Pawlik A., Frigui W., Martin C., Brosch R., Gonzalo-Asensio J. A specific polymorphism in Mycobacterium tuberculosis H37Rv causes differential ESAT-6 expression and identifies WhiB6 as a novel ESX-1 component. Infect. Immun. 2014;82:3446–3456. doi: 10.1128/IAI.01824-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gonzalo Asensio J., Maia C., Ferrer N.L., Barilone N., Laval F., Soto C.Y., Winter N., Daffe M., Gicquel B., Martin C., Jackson M. The virulence-associated two-component PhoP-PhoR system controls the biosynthesis of polyketide-derived lipids in Mycobacterium tuberculosis. J. Biol. Chem. 2006;281:1313–1316. doi: 10.1074/jbc.C500388200. [DOI] [PubMed] [Google Scholar]
- 28.Walters S.B., Dubnau E., Kolesnikova I., Laval F., Daffe M., Smith I. The Mycobacterium tuberculosis PhoPR two-component system regulates genes essential for virulence and complex lipid biosynthesis. Mol. Microbiol. 2006;60:312–330. doi: 10.1111/j.1365-2958.2006.05102.x. [DOI] [PubMed] [Google Scholar]
- 29.Solans L., Gonzalo-Asensio J., Sala C., Benjak A., Uplekar S., Rougemont J., Guilhot C., Malaga W., Martin C., Cole S.T. The PhoP-dependent ncRNA Mcr7 modulates the TAT secretion system in Mycobacterium tuberculosis. PLoS Pathog. 2014;10:e1004183. doi: 10.1371/journal.ppat.1004183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sayes F., Blanc C., Ates L.S., Deboosere N., Orgeur M., Le Chevalier F., Groschel M.I., Frigui W., Song O.R., Lo-Man R., et al. Multiplexed quantitation of intraphagocyte Mycobacterium tuberculosis secreted protein effectors. Cell Rep. 2018;23:1072–1084. doi: 10.1016/j.celrep.2018.03.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Camacho L.R., Ensergueix D., Perez E., Gicquel B., Guilhot C. Identification of a virulence gene cluster of Mycobacterium tuberculosis by signature-tagged transposon mutagenesis. Mol. Microbiol. 1999;34:257–267. doi: 10.1046/j.1365-2958.1999.01593.x. [DOI] [PubMed] [Google Scholar]
- 32.Groschel M.I., Sayes F., Shin S.J., Frigui W., Pawlik A., Orgeur M., Canetti R., Honore N., Simeone R., van der Werf T.S., et al. Recombinant BCG expressing ESX-1 of Mycobacterium marinum combines low virulence with cytosolic immune signaling and improved TB protection. Cell Rep. 2017;18:2752–2765. doi: 10.1016/j.celrep.2017.02.057. [DOI] [PubMed] [Google Scholar]
- 33.Dey R.J., Dey B., Singh A.K., Praharaj M., Bishai W. Bacillus calmette-guerin overexpressing an endogenous stimulator of interferon genes agonist provides enhanced protection against pulmonary tuberculosis. J. Infect. Dis. 2020;221:1048–1056. doi: 10.1093/infdis/jiz116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ning H., Wang L., Zhou J., Lu Y., Kang J., Ding T., Shen L., Xu Z., Bai Y. Recombinant BCG with bacterial signaling molecule cyclic di-AMP as endogenous adjuvant induces elevated immune responses after Mycobacterium tuberculosis infection. Front. Immunol. 2019;10:1519. doi: 10.3389/fimmu.2019.01519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perez I., Uranga S., Sayes F., Frigui W., Samper S., Arbues A., Aguilo N., Brosch R., Martin C., Gonzalo-Asensio J. Live attenuated TB vaccines representing the three modern Mycobacterium tuberculosis lineages reveal that the Euro-American genetic background confers optimal vaccine potential. EBioMedicine. 2020;55:102761. doi: 10.1016/j.ebiom.2020.102761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y., Yang J., Bai G. Cyclic di-AMP-mediated interaction between Mycobacterium tuberculosis DeltacnpB and macrophages implicates a novel strategy for improving BCG vaccination. Pathog. Dis. 2018;76 doi: 10.1093/femspd/fty008. [DOI] [PubMed] [Google Scholar]
- 37.Aguilo N., Gonzalo-Asensio J., Alvarez-Arguedas S., Marinova D., Gomez A.B., Uranga S., Spallek R., Singh M., Audran R., Spertini F., Martin C. Reactogenicity to major tuberculosis antigens absent in BCG is linked to improved protection against Mycobacterium tuberculosis. Nat. Commun. 2017;8:16085. doi: 10.1038/ncomms16085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsu T., Hingley-Wilson S.M., Chen B., Chen M., Dai A.Z., Morin P.M., Marks C.B., Padiyar J., Goulding C., Gingery M., et al. The primary mechanism of attenuation of bacillus Calmette-Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc. Natl. Acad. Sci. United States America. 2003;100:12420–12425. doi: 10.1073/pnas.1635213100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stanley S.A., Johndrow J.E., Manzanillo P., Cox J.S. The Type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J. Immunol. 2007;178:3143–3152. doi: 10.4049/jimmunol.178.5.3143. [DOI] [PubMed] [Google Scholar]
- 40.Kleinnijenhuis J., Quintin J., Preijers F., Joosten L.A., Ifrim D.C., Saeed S., Jacobs C., van Loenhout J., de Jong D., Stunnenberg H.G., et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc. Natl. Acad. Sci. U S A. 2012;109:17537–17542. doi: 10.1073/pnas.1202870109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tarancon R., Dominguez-Andres J., Uranga S., Ferreira A.V., Groh L.A., Domenech M., Gonzalez-Camacho F., Riksen N.P., Aguilo N., Yuste J., et al. New live attenuated tuberculosis vaccine MTBVAC induces trained immunity and confers protection against experimental lethal pneumonia. PLoS Pathog. 2020;16:e1008404. doi: 10.1371/journal.ppat.1008404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mayer-Barber K.D., Andrade B.B., Barber D.L., Hieny S., Feng C.G., Caspar P., Oland S., Gordon S., Sher A. Innate and adaptive interferons suppress IL-1alpha and IL-1beta production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity. 2011;35:1023–1034. doi: 10.1016/j.immuni.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang L., Ru H.W., Chen F.Z., Jin C.Y., Sun R.F., Fan X.Y., Guo M., Mai J.T., Xu W.X., Lin Q.X., Liu J. Variable virulence and efficacy of BCG vaccine strains in mice and correlation with genome polymorphisms. Mol. Ther. 2016;24:398–405. doi: 10.1038/mt.2015.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martin C., Marinova D., Aguilo N., Gonzalo-Asensio J. MTBVAC, a live TB vaccine poised to initiate efficacy trials 100 years after BCG. Vaccine. 2021;39:7277–7285. doi: 10.1016/j.vaccine.2021.06.049. [DOI] [PubMed] [Google Scholar]
- 45.Jenal U., Reinders A., Lori C. Cyclic di-GMP: second messenger extraordinaire. Nat. Rev. Microbiol. 2017;15:271–284. doi: 10.1038/nrmicro.2016.190. [DOI] [PubMed] [Google Scholar]
- 46.Perez E., Samper S., Bordas Y., Guilhot C., Gicquel B., Martin C. An essential role for phoP in Mycobacterium tuberculosis virulence. Mol. Microbiol. 2001;41:179–187. doi: 10.1046/j.1365-2958.2001.02500.x. [DOI] [PubMed] [Google Scholar]
- 47.Gonzalo-Asensio J., Malaga W., Pawlik A., Astarie-Dequeker C., Passemar C., Moreau F., Laval F., Daffe M., Martin C., Brosch R., Guilhot C. Evolutionary history of tuberculosis shaped by conserved mutations in the PhoPR virulence regulator. Proc. Natl. Acad. Sci. U S A. 2014;111:11491–11496. doi: 10.1073/pnas.1406693111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li L., Yin Q., Kuss P., Maliga Z., Millan J.L., Wu H., Mitchison T.J. Hydrolysis of 2'3'-cGAMP by ENPP1 and design of nonhydrolyzable analogs. Nat. Chem. Biol. 2014;10:1043–1048. doi: 10.1038/nchembio.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ablasser A., Schmid-Burgk J.L., Hemmerling I., Horvath G.L., Schmidt T., Latz E., Hornung V. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature. 2013;503:530–534. doi: 10.1038/nature12640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Devaux L., Kaminski P.A., Trieu-Cuot P., Firon A. Cyclic di-AMP in host-pathogen interactions. Curr. Opin. Microbiol. 2018;41:21–28. doi: 10.1016/j.mib.2017.11.007. [DOI] [PubMed] [Google Scholar]
- 51.McNab F., Mayer-Barber K., Sher A., Wack A., O'Garra A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015;15:87–103. doi: 10.1038/nri3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blanc L., Gilleron M., Prandi J., Song O.R., Jang M.S., Gicquel B., Drocourt D., Neyrolles O., Brodin P., Tiraby G., et al. Mycobacterium tuberculosis inhibits human innate immune responses via the production of TLR2 antagonist glycolipids. Proc. Natl. Acad. Sci. U S A. 2017;114:11205–11210. doi: 10.1073/pnas.1707840114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ruhl C.R., Pasko B.L., Khan H.S., Kindt L.M., Stamm C.E., Franco L.H., Hsia C.C., Zhou M., Davis C.R., Qin T., et al. Mycobacterium tuberculosis sulfolipid-1 activates nociceptive neurons and induces cough. Cell. 2020;181:293–305.e11. doi: 10.1016/j.cell.2020.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Van Dis E., Sogi K.M., Rae C.S., Sivick K.E., Surh N.H., Leong M.L., Kanne D.B., Metchette K., Leong J.J., Bruml J.R., et al. STING-activating adjuvants elicit a Th17 immune response and protect against Mycobacterium tuberculosis infection. Cell Rep. 2018;23:1435–1447. doi: 10.1016/j.celrep.2018.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jackson M., Raynaud C., Laneelle M.A., Guilhot C., Laurent-Winter C., Ensergueix D., Gicquel B., Daffe M. Inactivation of the antigen 85C gene profoundly affects the mycolate content and alters the permeability of the Mycobacterium tuberculosis cell envelope. Mol. Microbiol. 1999;31:1573–1587. doi: 10.1046/j.1365-2958.1999.01310.x. [DOI] [PubMed] [Google Scholar]
- 56.Cole S.T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S.V., Eiglmeier K., Gas S., Barry C.E., 3rd, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 57.Brosch R., Gordon S.V., Billault A., Garnier T., Eiglmeier K., Soravito C., Barrell B.G., Cole S.T. Use of a Mycobacterium tuberculosis H37Rv bacterial artificial chromosome library for genome mapping, sequencing, and comparative genomics. Infect. Immun. 1998;66:2221–2229. doi: 10.1128/iai.66.5.2221-2229.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Datsenko K.A., Wanner B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U S A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van Kessel J.C., Hatfull G.F. Recombineering in Mycobacterium tuberculosis. Nat. Methods. 2007;4:147–152. doi: 10.1038/nmeth996. [DOI] [PubMed] [Google Scholar]
- 60.Corrigan R.M., Abbott J.C., Burhenne H., Kaever V., Grundling A. c-di-AMP is a new second messenger in Staphylococcus aureus with a role in controlling cell size and envelope stress. PLoS Pathog. 2011;7:e1002217. doi: 10.1371/journal.ppat.1002217. [DOI] [PMC free article] [PubMed] [Google Scholar]