

Summary
Heart failure (HF) is the ultimate outcome of a variety of heart diseases, including restrictive cardiomyopathy (RCM), ischemic heart disease (IHD), and valvular heart disease (VHD). To date, accumulating evidence has suggested an important role of noncoding RNAs (ncRNAs) in HF. We performed RNA-sequencing studies with myocardial mRNAs/lncRNAs/circRNAs/miRNAs from non-failing hearts (donor heart tissue from heart transplantation) and three groups of patients with HF (RCM, IHD, and VHD). HF-related gene regulatory networks and gene co-expression networks were constructed based on the interaction relationship and expression profiles of differentially expressed mRNAs/ncRNAs. Our results indicated that HF with different etiologies is regulated by complex lncRNA/circRNA/miRNA/mRNA regulatory networks, comprising common pathways that are shared by all HF types as well as distinct pathways that are enriched in specific HF types. In addition, the HF biomarkers identified in our study have an important clinical application value in HF staging and HF type diagnosis.
Subject areas: Pathophysiology, Transcriptomics
Graphical abstract
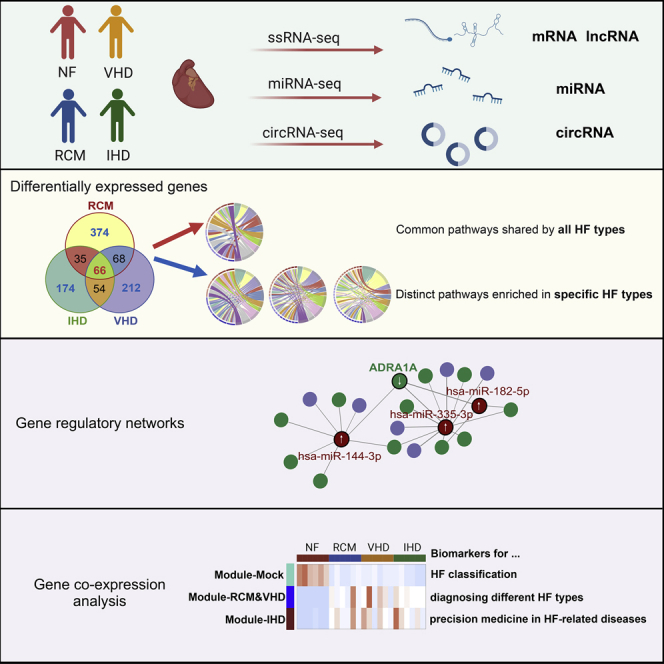
Highlights
-
•
Identified common pathways that are shared by all HF types
-
•
Identification of the distinct pathways that are enriched in specific HF types
-
•
LncRNA/circRNA-miRNA-mRNA co-expression networks and regulatory networks construction
-
•
Identification of potential biomarkers for HF with distinct etiology
Pathophysiology; Transcriptomics
Introduction
Heart failure (HF) is the ultimate outcome of a variety of heart diseases, including restrictive cardiomyopathy (RCM), ischemic heart disease (IHD), and valvular heart disease (VHD). RCM is characterized by the limited filling of the left and/or right ventricles, and diastolic volume reduction; it is mainly divided into three categories, such as cardiac amyloidosis, cardiac sarcoidosis, and cardiac hemochromatosis (Muchtar et al., 2017). IHD is mainly caused by the progressive narrowing of the coronary arteries that supply oxygen to the myocardium, which results in regional myocardial ischemia when the body's oxygen demands increase (Braunwald and Nicholls, 2015). The main pathogenic factor of IHD is atherosclerosis, a chronic inflammatory thickening of the arterial wall (Wirtz and von Kanel, 2017). VHD is a result of the dysfunction of heart valves, namely mitral valve, tricuspid valve, aortic valve, and pulmonary valve, due to congenital or acquired problems (Mrsic et al., 2018). Defective valves affect normal blood flow, leading to abnormal heart workload and eventually HF. Clearly, despite their common end-stage disease, these heart diseases have distinct characteristics and pathogenesis (Bauleo, 2002). However, it remains unknown whether there is a shared mechanism underlying pathogenesis and progression of HF in these diseases.
Currently, the diagnosis of HF mainly relies on biomarkers. Among them, B-type natriuretic peptide (BNP) and N-terminal B-type natriuretic peptide precursor (NT-proBNP) are the most used clinical biomarkers in the prediction model of acute HF (Gandhi et al., 2015). Recently, accumulating evidence has suggested an important role of noncoding RNAs (ncRNAs), including micro-RNAs (miRNAs), long noncoding RNAs (lncRNAs), and circular RNAs (circRNAs), in HF (Dangwal et al., 2017).
miRNA refers to small, uncompiled RNA that binds to the complementary sequence of the 3′ untranslated regions (3′-UTR) of messenger RNAs (mRNAs) to inhibit the translation and/or promote the degradation of targeted mRNA, thereby regulating gene expression (Bartel, 2009). It has been reported that not only do miRNAs (i.e., miR-208) regulate cardiac development but they also play an important role in the pathological remodeling of the heart in various diseases (Gama-Carvalho et al., 2014; Luo et al., 2015; Thum et al., 2007; van Rooij et al., 2007). In addition, miRNAs are effective biomarkers for diagnosis of HF (Santaguida et al., 2014). LncRNAs can act as a sponge of miRNAs to interact with corresponding miRNAs and regulate expression of mRNAs (Furio-Tari et al., 2016; Klattenhoff et al., 2013; Wang et al., 2014). For example, lncRNA cardiac hypertrophy-related factor (Chrf) can downregulate the expression of miR-489 to mediate cardiomyocyte hypertrophy and apoptosis (Wang et al., 2014). As such, lncRNAs also play an important role in cardiovascular development (Fatima et al., 2015; Schonrock et al., 2012). Finally, circRNAs have been identified as new biomarkers and possible therapeutic targets in HF (Devaux et al., 2017). In a mouse model, heart-related circRNA (Hrcr) has been suggested to regulate myocardial hypertrophy by interacting with miR-223 (Wang et al., 2016).
In the present study, we performed transcriptomics analysis of myocardial samples of non-failing hearts and three groups of patients with HF (RCM, IHD, and VHD). We showed that HF with different etiologies is regulated by complex lncRNA/circRNA/miRNA/mRNA regulatory networks, which comprise common pathways that are shared by all HF types as well as distinct pathways that are enriched in specific HF types. These findings have potential for the clinical diagnosis and treatment of HF.
Results
Common regulatory genes for HF with different etiology
We performed RNA-seq studies with myocardial mRNAs from non-failing hearts (heart transplant donor hearts failed to be implanted after heart acquisition) and three groups of patients with HF (RCM, IHD, and VHD). In total, 983 mRNAs were differentially expressed in at least one of the HF groups. Among them, 66 mRNAs were differentially expressed in all three HF groups (Figure 1A), indicating a common response to all HF conditions. Strikingly, all HF-associated genes followed the same trend in all of the HF groups and there were no “bidirectional” genes. Compared with the control group, 32 mRNAs were upregulated and 34 mRNAs were downregulated in the HF groups (Figure 1B). In addition, the principal components analysis (PCA) (Figure S1) and correlation analysis (Figure S2) of differentially expressed mRNAs/miRNAs/lncRNAs/circRNAs in at least one group revealed that the correlation among the three HF groups was higher than that with the control group. Taken together, these data suggest that the three types of HF share a common set of disease-related genes.
Figure 1.

Identification of differentially expressed mRNAs in HF-related diseases such as RCM, IHD, and VHD
(A) Venn diagrams of the number of differentially expressed mRNAs in RCM, IHD, and VHD groups.
(B) Heatmap of differentially expressed mRNAs in all HF groups, including RCM, IHD, and VHD. mRNAs marked with red stars have been reported to be related to HF.
(C) GO chord plot of differentially expressed mRNAs in all groups, including RCM, IHD, and VHD.
To gain insight into the functional relevance, we performed GO enrichment analysis on those common HF genes (Figure 1C). In the upregulated gene set, GO terms were enriched in “positive regulation of tumor necrosis factor (TNF)-mediated signaling pathway” and “positive regulation of cytokine-mediated signaling pathway.” In the downregulated gene set, GO terms were enriched in “negative regulation of phosphatase activity” and “negative regulation of dephosphorylation.” It is well known that TNF and other inflammatory cytokines are related to HF. Furthermore, phosphorylation and dephosphorylation mediate multiple cardiac HF-related signaling pathways, such as GP130, angiotensin, and adrenergic receptors. In addition, previous studies have shown that inhibiting phosphodiesterase increases cAMP levels, and cAMP plays an important role in the maintenance of myocardial function. The cAMP-PKA signaling pathway is also involved in myocardial infarction (Feng et al., 2016).
In addition, we validated the common differentially expressed mRNAs and their enrichment pathways in several public datasets, such as GSE46224 and GSE1145, which were obtained from Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) (Figure 2A). Four of the differentially expressed mRNAs, including two upregulated mRNAs (CNTNAP3 and THY1) and two downregulated mRNAs (FOSL1 and FAM166A), were also differentially expressed in at least one public dataset (Figures 2A and 2B). In the differentially expressed gene set of the ICM group in GSE46224 and GSE1145, GO terms were also enriched in TNF-related pathways, cytokine-related pathways, and phosphorylation/dephosphorylation-related pathways (Figure 2A). These results indicated the consistency between our mRNA-sequencing data and public data.
Figure 2.

Differential expression analysis and GO enrichment analysis in public datasets
(A) Differential expression analysis and GO enrichment analysis of mRNA dataset of GSE46224 and GSE1145. P values in scatter plots were determined by edgeR package (edgeR-3.26.5). P values in bar plots were determined by Fisher exact test. P values in scatter plots were determined by edgeR package (edgeR-3.26.5). P value in bar plots were determined by Fisher exact test.
(B) Differential expression analysis of miRNA dataset of GSE104150 and GSE46224. P values in scatter plots were determined by edgeR package (edgeR-3.26.5).
Disease type-specific genes in HF regulation
Then, we performed GO enrichment analysis on the 374, 174, and 212 genes that were differentially expressed in RCM, IHD, and VHD groups, respectively (Figure 3). First, we noted that GO terms “protein-containing complex localization” and “purine nucleotide biosynthetic process” were enriched only in RCM samples (Figure 3A), indicating proteomic stress (from amyloidosis or hemochromatosis) or active proliferation of infiltrating cells (sarcoidosis) in the myocardium. There is no evidence that the pathogenesis of RCM is related to hypoxic remodeling or mechanical stimulation; indeed, such GO terms were not enriched in RCM. Next, the VHD group showed GO term enrichment in “positive regulation of blood circulation,” “positive regulation of vasoconstriction,” “sensory perception of mechanical stimulus,” “detection of mechanical stimulus involved in sensory perception of pain,” and “detection of mechanical stimulus involved in sensory perception” (Figure 3B). Clearly, these GO terms are related to blood flow and mechanical stimuli, indicating that myocardial responses to abnormal hemodynamic stress likely resulted from valve dysfunction. Finally, the IHD group showed GO term enrichment in “I-κB kinase/NF-κB signaling” and “positive regulation of I-κB kinase/NF-κB signaling” (Figure 3C). Given that NF-κB is a key regulator of inflammation and a direct modulator of HIF1a expression, these GO enrichment results are consistent with the inflammatory and hypoxic nature of IHD (Rius et al., 2008). Moreover, the GO pathway specifically enriched in the IHD group of our data was also specifically enriched in the IHD group of public datasets GSE46224 and GSE1145 (Figure 3D). Collectively, these GO enrichment analyses demonstrated that each type of HF triggers a unique transcriptional response and that the type-specific gene sets may underscore the distinct pathogenesis of different cardiac diseases.
Figure 3.

GO enrichment analysis of uniquely differentially expressed mRNA in RCM, IHD, and VHD groups
(A–C) GO chord plot of differentially expressed mRNAs in only RCM group (A), IHD group (B), or VHD group (C).
(D) GO bar plot of differentially expressed mRNAs in ICM group of GSE1145 and GSE46224. GO pathways showing specific enrichment in the RCM group, IHD group, or VHD group in our RNA-seq data are labeled with different colors. P values in bar plots were determined by Fisher exact test.
LncRNAs target HF-related genes
From the lncRNA-seq study, we found a total of 764 differentially expressed lncRNAs, of which 205, 143, and 185 were differentially expressed in the RCM, IHD, and VHD groups, respectively. A total of 75 lncRNAs were differentially expressed in all three groups. Compared with the control group, 47 lncRNAs were upregulated, while 28 were downregulated (Figures 4A and 4B). Among these 75 lncRNAs, AL121908.1, AC087289.6, AC092069.1, AC009119.3, and AL033519.4 were mapped to the vicinity of PHACTR3, AC087289.1, RAD23A, MLYCD, and FKBP5, which were identified as differentially expressed genes in one or more disease groups (Figures 4C and 4D). In particular, significant upregulation of MLYCD has been shown in patients with mitral regurgitation (MR)-based HF (Chen et al., 2016), indicating a potential role of this gene in mitral valve insufficiency and pathogenesis of VHD. In this study, we found that MLYCD was significantly upregulated in both the IHD and VHD groups, and this process may be regulated by lncRNA. To date, there is no literature on these lncRNAs in HF.
Figure 4.

Differentially expressed lncRNAs and their corresponding strand differentially expressed mRNAs
(A) Heatmap of differentially expressed lncRNAs in all groups, including RCM, IHD, and VHD.
(B) Venn diagrams of the number of differentially expressed lncRNAs in RCM, IHD, and VHD groups.
(C) Heatmap of five mRNAs that were differentially expressed in one or more disease groups and located on the reverse strand of corresponding differentially expressed lncRNA.
(D) Gene structure of differentially expressed lincRNAs and their corresponding differentially expressed mRNAs.
Key circRNAs interact with HF-related miRNA and proteins
In the circRNA-seq, we identified 2402 differentially expressed circRNAs, among which 165 circRNAs (112 upregulated and 53 downregulated) were shared by all of the groups (Figures 5A and 5B). In addition, 374, 1120, and 378 circRNAs were differentially expressed only in RCM, IHD, and VHD groups, respectively.
Figure 5.

Differentially expressed circRNAs and their differentially expressed RNA binding proteins
(A) Venn diagrams of the number of differentially expressed circRNAs in RCM, IHD, and VHD groups.
(B) Heatmap of differentially expressed circRNAs in all HF groups, including RCM, IHD, and VHD, for which corresponding mRNAs have been reported to be related to HF.
(C) RNA-binding proteins and the number of their binding sites of differentially expressed circRNA in all HF groups, including RCM, IHD, and VHD, for which corresponding mRNA have been reported to be related to HF.
In the HF common circRNA set, 16 circRNAs had corresponding mRNA related to HF. Furthermore, 12 of them contained more than 100 bp (Figure S3) and had miRNA response elements (MREs) (Figure S3), indicating that they may act as miRNA sponges.
Next, bioinformatic analysis showed that 39 RNA-binding proteins can bind to the above 16 circRNAs, and their corresponding mRNAs were differentially expressed in at least one of the HF groups (Figure 5C). There were 26, 11, and 12 mRNA-binding proteins with corresponding mRNAs differentially expressed in the RCM, VHD, and IHD groups, respectively. Among the RNA-binding proteins, AR, CEBPB, and EP300 proteins contained more binding sites than other RNA-binding proteins. The AR/CEBPB/EP300 mRNAs were only differentially expressed in the RCM group (Figure 5C), indicating that RCM is more likely to be regulated by circRNAs than VHD and IHD. The upregulated circRNAs (hsa-BAALC_0012, hsa-STC2_0003, and hsa-VEGFC_0030) and the downregulated circRNA (hsa-F13A1_0031) could interact with more proteins than other circRNAs (Figure 5C). We speculate that the upregulated and downregulated circRNAs could form a balance and jointly regulate HF-related diseases. Of note, 31 out of 39 RNA-binding proteins were only differentially expressed in one of the HF groups (Figure 5C), indicating that circRNA may be involved in disease-specific regulation. However, although FOSL1 and MTA1 have few RNA-binding sites, their mRNAs were downregulated in all of the HF groups (RCM/VHD/IHD) (Figure 5C), suggesting their critical roles in HF.
The miRNA regulatory network in HF
Our miRNA-seq data revealed 120 differentially expressed miRNAs, of which 25, 17, and 34 were differentially expressed in the RCM, IHD, and VHD groups, respectively (Figure 6A). Compared with the control group, 15 miRNAs were differentially expressed in all three groups, with 12 upregulated and three downregulated. Interestingly, similar to the results from mRNA-seq and lncRNA-seq studies, all miRNAs exhibited the same trend of differential expression in all of the HF groups and none was “bidirectional” (Figure 6B).
Figure 6.

Differentially expressed miRNAs and gene regulatory networks for HF-related diseases
(A) Venn diagrams of the number of differentially expressed miRNAs in RCM, IHD, and VHD groups.
(B) Heatmap of differentially expressed miRNAs in all HF groups, including RCM, IHD, and VHD.
(C) Gene regulatory networks for HF-related diseases. MiRNAs, mRNAs, lncRNAs, and circRNAs in the networks are differentially expressed in all HF groups, including RCM, IHD, and VHD.
Then, we validated our common differentially expressed miRNAs using several public datasets, such as GSE104150 and GSE46224, which were obtained from GEO database (https://www.ncbi.nlm.nih.gov/geo/) (Figure 2B). Seven of the upregulated mRNAs, namely hsa-miR-183-5p, hsa-miR-451a, hsa-miR-144-3p, hsa-miR-182-5p, hsa-miR-4732-3p, hsa-miR-144-5p, and hsa-miR-31-5p, were also upregulated in at least one public dataset. These results indicated the consistency between our miRNA-sequencing data and public data.
We integrated prediction results of miRDB database, DIANA tools database, and miRanda software to map the miRNA regulatory network in HF (Figure 6C), with focus on the genes regulated by multiple miRNAs. For example, ADRA1A appears to be a target gene of miR-144-3p, miR-335-3p, and miR-182-5p. ADRA1A encodes the α1A adrenergic receptor, which is critical to normal cardiac function and hypertensive heart disease (HHD) (He and Huang, 2017). We identified that ADRA1A is relevant to all three types of HF (Figure 1B). Therefore, these three miRNAs may participate in all HF disease types. Indeed, it has been reported that miRNA-144 is a cytoprotective miRNA in ischemia/reperfusion injury models (Li et al., 2018), that miR-335-3p induces pulmonary arterial hypertension (PAH) through inhibition of APJ (Fan et al., 2020), and that miR-182 is a potential prognostic marker for chronic congestive heart failure (CHF) (Cakmak et al., 2015). It is plausible that different HF-related diseases could be regulated by ADRA1A through different miRNAs, such as miR-144-3p, miR-335-3p, and miR-182-5p. We obtained the sequence information of miR-144-3p, miR-335-3p, and miR-182-5p from the miRDB database and NCBI database, and performed alignment analysis. As shown in Figure 7A, miR-144-3p is conserved across humans, rats, and mice (Figure 7A). Thus, we next verified whether miR-144-3p could target ADRA1A mRNA in neonatal rat ventricle myocytes (NRVMs) by transient transfection with miR-144-3p inhibitor or mimics. We examined the changes in ADRA1A protein level by western blot (WB) in NRVMs after the transfection. The results demonstrated that miR-144-3p negatively regulated the protein expression of ADRA1A (Figure 7B).
Figure 7.

MiR-144-3p could target ADRA1A mRNA in NRVM
(A) The sequences of miR-144-3p, miR-182-5p, and miR-335-3p in humans, rats, and mice.
(B) The relative miR-144-3p level in NRVMs transfected with miR-144-3p inhibitor or mimics (left), and the protein expression of ADRA1A in NRVMs transfected with miR-144-3p inhibitor or mimics (right). Data in barplot are shown as mean ±SEM.
In addition, CORIN, a cardiac protease that cleaves pronatriuretic peptides, was downregulated in all disease samples (Figure 6C). The CORIN gene promoter shares many of the transcription binding sites with ANP and BNP precursors (such as GATA-4.2) (Ngo et al., 2013). It has been shown that corin expression in the mouse heart is reduced in a dilated cardiomyopathy model, and cardiac overexpression of corin protects mice from HF and death (Gladysheva et al., 2013). According to our results, CORIN has a potential regulatory relationship to miR-187 (Figure 6C), and miR-187 is also downregulated in all of these disease samples (Figure 6B). Hitherto, miR-187 has never been reported to be associated with HF.
The gene co-expression networks for HF-related diseases
Based on the mRNAs/miRNAs/lncRNAs/circRNAs that were differentially expressed in all of the HF groups, we constructed a co-expression network of HF-related diseases. The differentially expressed genes were clustered into three modules, including Module-Mock, Module-RCM&VHD, and Module-IHD (Figure 8A). Among them, Module-Mock had a higher correlation with the control group, but had a lower correlation with all HF disease groups, indicating that the genes in Module-Mock had an impact on all three HF-related diseases (Figure 8A). However, Module-RCM&VHD and Module-IHD had the lowest correlation with the control samples, while the correlation with other samples was not consistent; this indicates that although the genes in Module-RCM&VHD and Module-IHD were differentially expressed in the three HF-related disease samples, the genes in Module-RCM&VHD and Module-IHD may only affect certain patients in the disease groups (Figure 8A). This is consistent with the clinical notion that, although patients with end-staged RCM/IHD/VHD have similar symptoms, their pathogenesis is not the same. For example, Module-RCM&VHD showed a high correlation with RCM5 sample (cor = 0.47, p value = 0.02) and VHD2 sample (cor = 0.64, p value = 8 × 10−4), and Module-IHD and IHD1 samples showed a high correlation (cor = 0.57, p value = 3 × 10−3). This demonstrates that the genes in Module-RCM&VHD may only affect some patients with RCM and VHD, while Module-IHD may only affect some patients with IHD. These findings also suggest that the causes of HF-related diseases are very complex at the genetic level, and that different genes may cause the same phenotype.
Figure 8.

Gene co-expression networks for HF-related diseases
(A) Three modules for all differentially expressed genes in all HF groups, including RCM, IHD, and VHD, and the correlation coefficients between modules and samples. P values in parentheses were determined by Pearson correlation test.P-vlues in parentheses were determined by Pearson correlation test.
(B) Gene co-expression networks based on each module for HF-related diseases.
In further analysis of the co-expression network of HF-related diseases, we found that ADRA1A had a very high correlation with the expression of multiple differentially expressed genes, including differentially expressed lncRNAs and circRNAs, suggesting a core regulatory role of ADRA1A in the development of HF-related diseases (Figure 8B). Kyoto Encyclopedia of Genes and Genomes (KEGG) database also suggested that many signaling pathways involving ADRA1A are related to HF-related diseases, including hsa0415: AMPK signaling pathway, hsa0426: adrenergic signaling in cardiomyocytes, hsa0402: cGMP-PKG signaling pathway, hsa0497: salivary secretion, hsa0402: calcium signaling pathway, and hsa0427: vascular smooth muscle contraction. AMP-activated protein kinase, the core of the AMPK signaling pathway, controls a large number of metabolic pathways and plays a key role in regulating the metabolism of myocardial tissue (Kim and Dyck, 2015). Regulating the epinephrine signaling pathway to cardiomyocytes has always been one of the most critical goals of HF therapy (Fujita and Ishikawa, 2011). cGMP usually stimulates the production of cardioprotective protein (protein kinase G, PKG), which is known to protect the heart muscle from stress damage caused by disease (Lee et al., 2015). Previous studies have shown that patients with HF have dysfunction of salivary gland secretion (Klimiuk et al., 2020). The regulation of vascular smooth muscle cell (VSMC) contraction is calcium-dependent. Vasoconstrictors increase intracellular Ca2+ levels to induce VSMC contraction (Touyz et al., 2018).
Discussion
HF remains an unmet clinical challenge because all cardiovascular diseases eventually lead to HF, and there is not yet a cure. Therefore, identification of novel pathogenic pathways and biomarkers in HF is of great scientific and clinical importance. Here, we reported the results of the comprehensive transcriptomic study on human subjects with three different types of HF, namely RCM, IHD, and VHD, which are well known to have distinct etiology. By integrating the bioinformatic data from mRNA, miRNA, lncRNA, and circRNA sequencing, we identified the common and distinct pathways related to HF with different etiologies, and explored the interactions among different RNA species. Furthermore, certain small RNA molecules identified in this study were associated with a specific HF type and thereby have potential to serve as novel biomarkers in clinical setting.
To the best of our knowledge, we were first to systematically demonstrate how pathogenesis of HF is regulated by multiple mechanisms at the mRNA, miRNA, lncRNA, and circRNA levels. As described below, these HF-related coding and noncoding RNAs can be divided into three categories, namely the HF common set, the HF type-specific set, and the HF individual-specific set. First, the HF common set RNAs were differentially expressed in all patients with HF, indicating a shared pathological mechanism or end-stage HF condition. Through the gene co-expression network and gene regulatory network, we found that the common regulatory genes related to different causes of HF play a central role in the network, such as genes in Module-Mock (Figures 6C, 8A, and 8B). Second, the HF type-specific set RNAs were only differentially expressed in one type of HF. Notably, even in end-stage HF, these genes were differentially expressed, and GO pathway analysis suggested that they may regulate distinct pathogenesis pathways triggered by different causes (Figures 2A–2C). Third, the HF individual-specific set RNAs were only differentially expressed in certain subjects regardless of HF types, such as genes in Module-RCM&VHD and Module-IHD (Figure 8A). This set of RNAs may be associated with individual genetic variants. Mechanistically, GO enrichment analysis revealed that the HF common set mRNAs were enriched in the signaling pathways related to TNF and cAMP, which is consistent with the inflammatory nature of HF; the HF type-specific set mRNAs were enriched in pathways related to energy metabolism/inflammation and hypoxia/hemodynamic pressure, which is closely related to the specific causes of the corresponding diseases. Furthermore, we provided evidence indicating the regulatory role of lncRNA and the molecular sponge role of circRNA in HF. Finally, we integrated all data to construct the co-expression network and the gene regulation network to comprehensively illustrate the possible transcriptional and post-transcriptional regulatory mechanisms in the pathogenesis of HF.
Another key finding of our study is related to the identification of novel HF biomarkers that may have important clinical application value in HF staging and HF type diagnosis. At present, the staging of HF in clinical practice mainly relies on clinical symptoms, with a handful of biomarkers such as NT-proBNP, cardiac troponin (cTn) (Gommans et al., 2021), growth stimulation expressed gene 2 (ST2) (Dieplinger and Mueller, 2015), galectin 3 (GAL3) (McCullough et al., 2011), growth differentiation factor 15 (GDF15) (Anand et al., 2010; Havranek and Marek, 2021), and copeptin (Maisel et al., 2011). These HF biomarkers could evaluate different stages of HF through different ways and mechanisms, and improve the sensitivity and specificity of the diagnosis, treatment, and prognosis. However, there are limitations to the use of these biomarkers. For example, ST2 has higher accuracy in the short term, but its predictive accuracy drops significantly as disease progress (Gaggin et al., 2014; Ky et al., 2011); the expression level of GAL3 may be interfered by renal function of patients (Anand et al., 2013); and the increase in GDF15 expression is not specific to HF but may be affected by damage to various systems (Kempf et al., 2006). Therefore, identification of novel HF biomarkers is of clinical importance. In our study, we identified three sets of HF genes. Among them, the HF common set of genes may be suitable as biomarkers for HF classification or therapeutic targets to ameliorate HF symptoms; the HF type-specific set could be HF type-specific biomarkers to diagnose different HF types and provide basis for specific HF treatment plans; and the individual-specific set is potentially useful for the realization of precision medicine in HF-related diseases.
Another important aspect of our work is related to the identification of HF regulatory network formed by mRNA, miRNA, lncRNA, and circRNA. Based on the expression levels and nucleic acid sequence, we constructed the gene co-expression network and the gene regulatory network with transcriptomic data from all four RNA species (Figures 6C and 8B). We found that lncRNAs appeared less enriched in the gene regulatory network, while miRNAs appeared less enriched in the gene co-expression network, indicating their unique regulatory mechanisms. In the gene regulatory network, miRNAs act as key nodes to connect mRNAs, lncRNAs, and circRNAs, mainly by nucleic acid sequence-based targeting regardless of miRNA expression levels. Only two lncRNAs showed sequence interactions with miRNAs, indicating that the predominant role of lncRNAs in gene expression is independent of miRNAs. Theoretically, lncRNA could bind to homologous DNA/RNA sequences, fold into a complex secondary structure, and bind to a variety of proteins (Ponting et al., 2009). In the gene co-expression network, the interaction of lncRNA with mRNA, miRNA, and circRNA is mainly reflected in the correlation of their expression levels, implying a regulatory relationship between lncRNA and mRNA/circRNA in pathogenesis of HF.
Limitations of the study
In our study, we found that the common pathways shared by all HF types and the distinct pathways enriched in specific HF types together constitute a complex lncRNA/circRNA-miRNA-mRNA regulatory network that is related to the occurrence of HF with different etiologies. In addition, we provided novel HF biomarkers with an important clinical application value in HF staging and HF type diagnosis. Our results need to be further verified by experiment combined with clinical data.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Critical commercial assays | ||
| TRIzol reagent | Invitrogen | Cat#15596-018 |
| TRIzol reagent | Shanghai Promega | LS1040 |
| RNAiMAX reagent | Invitrogen | Cat#13778-150 |
| RIPA buffer | Beyotime Biotechnology | P0013B |
| Protease inhibitor cocktails | Roche | Cat#04693132001 |
| Phosphatase inhibitor cocktails | Roche | Cat#04906845001 |
| Anti-GAPDH polyclonal antibody | Utibody | UM4002 |
| Anti-ADRA1A polyclonal antibody | Proteintech | 19777-1-AP |
| Agilent 2100 Bioanalyzer | Agilent | SD-UF0000050 |
| Ribo-Zero™ rRNA Removal Kit | Epicentre | RZH1046 |
| TruSeq PE Cluster Kit v3-cBot-HS | Illumina | PE-401-3001 |
| Illumina HiSeq X sequencing system | Illumina | 15050091 v07 |
| 14-30 ssRNA Ladder Marker | Takara | H3416 |
| QIAquick Gel Extraction Kit | QIAGEN | Cat#28704 |
| BGISEQ-500 platform | BGI-Shenzhen | N/A |
| Deposited data | ||
| Human reference genome NCBI build 38, GRCh38 | ENSEMBL | http://ftp.ensembl.org/pub/release-97/fasta/homo_sapiens/dna/ |
| Human gene annotation file (Ensembl version 97) | ENSEMBL | http://ftp.ensembl.org/pub/release-97/gff3/homo_sapiens/ |
| Public data, Non-coding RNA profiling by high throughput sequencing | GEO | GEO: GSE46224 |
| Public data, Non-coding RNA profiling by array | GEO | GEO: GSE104150 |
| Public data, Expression profiling by array | GEO | GEO: GSE1145 |
| Raw data | This paper | HRA001366; https://ngdc.cncb.ac.cn/gsa-human/s/E0neHkH8 |
| Software and algorithms | ||
| FastQC | Babraham Bioinformatics | http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| STAR-v2.5.1b | Dobin et al., 2013 | https://github.com/alexdobin/STAR/archive/refs/tags/2.5.1b.tar.gz |
| Rsubread-1.32.3 | Liao et al., 2019 | http://www.bioconductor.org/packages/release/bioc/html/Rsubread.html |
| circAtlas 2.0 database | Wu et al., 2020 | http://circatlas.biols.ac.cn/ |
| bowtie-1.1.2 | Langmead et al., 2009 | http://bowtie-bio.sourceforge.net/index.shtml |
| mirdeep-2.0.0.8 | Mackowiak, 2011 | https://github.com/rajewsky-lab/mirdeep2/archive/refs/tags/v0.0.8.tar.gz |
| miRBase 22 database | Kozomara et al., 2019 | http://www.mirbase.org/ |
| DESeq2-1.24.0 | Love et al., 2014 | http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html |
| edgeR-3.26.5 | Robinson et al., 2010 | http://www.bioconductor.org/packages/release/bioc/html/edgeR.html |
| clusterProfiler-3.12.0 | Yu et al., 2012 | http://www.bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| miRDB database | Chen and Wang, 2020; Liu and Wang, 2019 | http://mirdb.org/ |
| DIANA LncBase Predicted v.2 tools | Paraskevopoulou et al., 2016 | http://carolina.imis.athena-innovation.gr/diana_tools/web/index.php?r=lncbasev2/index-predicted |
| miRanda-3.3a | Betel et al., 2010 | https://anaconda.org/bioconda/miranda |
| WGCNA-1.69 | Langfelder and Horvath, 2008 | https://cran.r-project.org/web/packages/WGCNA/index.html |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Nianguo Dong (dongnianguo@hotmail.com).
Materials availability
This work did not generate new unique reagents.
Experimental model and subject details
This study contained a total of 24 samples in four groups, including non-failing hearts (heart transplant donor heart failed to be implanted after heart acquisition), RCM patients, IHD patients, and VHD patients, with six samples in each group (Table S1). Non-failing hearts were free of any cardiac disease. HF groups were diagnosed with end-stage HF of the corresponding etiology (Table S1). This study was conducted in accordance with the Declaration of Helsinki and was approved by the local ethics committees of all participating hospitals. Written informed consent was obtained from each patient.
Methods details
RNA extraction
Total RNA was extracted from the left ventricular myocardium of RCM/IHD/VHD patients and non-failing hearts using TRIzol reagent following the manufacturer’s instructions (Invitrogen, Carlsbad, CA, USA). Briefly, approximately 60 mg of tissue was ground in liquid nitrogen, dissolved in 1.5 mL of TRIzol reagent, and subjected to RNA extraction. Subsequently, RNA samples were quantified using Nano Drop, and the quality of RNA was analyzed using Agilent 2100 bioanalyzer (Agilent, USA).
mRNA/lncRNA and circRNA RNA-seq
To prepare RNA-seq libraries, 5 μg of total RNA per sample was subjected to DNA digestion and rRNA removal (Ribo-Zero rRNA Removal Kit) processes to eliminate contamination of DNA and rRNA, respectively. For circRNA libraries, RNA samples were further treated with RNase R digestion to remove linear RNA and followed by fragmentation. Next, the fragmented circRNA and mRNA/lncRNA samples were subjected to reverse transcription and PCR to generate double-strand cDNA libraries. For quality control (QC), the concentration and size range of DNA fragments were determined by real-time PCR and Agilent 2100 Bioanalyzer (Agilent, USA). Then, the libraries were denatured into single strand by adding NaOH, hybridized with the adapters on Flowcell, and subjected to bridged PCR amplification using the TruSeq PE Cluster Kit v3-cBot-HS (Illumina, USA). Finally, paired-end 150 bases reads were generated on Illumina HiSeq X sequencing system.
MiRNA library construction
To isolate miRNA, 1 μg of total RNA was subjected to electrophoresis on a 15% urea denaturing polyacrylamide gel electrophoresis (PAGE) gel, and the gel regions corresponding to 18–30 nt RNA (14–30 ssRNA Ladder Marker, TAKARA) were excised to recover and purify miRNA. After ligation of 5′- and 3’-adaptors, miRNA samples were subjected to reverse transcription and PCR amplification. The PCR products were screened by agarose gel electrophoresis, and the target fragments of 100–120 bp were purified using QIAquick Gel Extraction Kit (QIAGEN, Valencia, CA). After the same QC processes as for mRNA, miRNA cDNA libraries were sequenced using a BGISEQ-500 platform (BGI-Shenzhen, China).
Transcriptome alignment
After using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) to perform QC on raw data, we applied STAR (STAR-v2.5.1b) (Dobin et al., 2013) to map lncRNA-seq data and circRNA-seq data to the human reference genome GRCh38.p10/hg38. Then, for lncRNA-seq data, Rsubread package (Rsubread-1.32.3) (Liao et al., 2019) and gene annotation file (Ensembl version 97) were applied to quantify the transcripts. According to the biotype provided in the Ensembl annotation file, mRNA and lncRNA were extracted for subsequent analysis. For circRNA-seq data, Rsubread package and circRNA annotation file in the circAtlas 2.0 database (http://circatlas.biols.ac.cn/) (Wu et al., 2020) were applied to quantify the transcripts. For miRNA-seq data, bowtie (bowtie-1.1.2) (Langmead et al., 2009) was employed to map miRNA-seq data to the human reference genome GRCh38.p10/hg38. Then, miRDeep2 (mirdeep-2.0.0.8) (Mackowiak, 2011) and miRBase database (miRBase 22, http://www.mirbase.org/) (Kozomara et al., 2019) were used to quickly quantify human reported miRNAs. Sequencing information for mRNA-seq and lncRNA-seq, miRNA-seq, and circRNA-seq is displayed in Tables S2–S4.
Differential gene expression analysis
We utilized DESeq2 package (DESeq2-1.24.0) (Love et al., 2014) to examine differential gene expression. Our definition of a differentially expressed gene was as follows: |log2fold change| ≥1 and FDR ≤0.05. Next, the rpkm function in the edgeR package (edgeR-3.26.5) (Robinson et al., 2010) was applied to calculate RPKM (Reads Per Kilobase per Million mapped reads) of each transcript. Heat maps of the differential genes were drawn using the pheatmap package (pheatmap-1.0.12) according to RPKM of the corresponding differential genes. The Venn diagram of differential genes was drawn using the Vennerable package (Vennerable-3.0). For the differential genes in each sample, the correlation of their expression levels was calculated. Then, principal components analysis (PCA) was applied for dimensionality reduction analysis of differential genes in each sample.
For public data sets, GSE46224, GSE1145, and GSE104150 were obtained from GEO database. We utilized the edgeR package (edgeR-3.26.5) to determine differential gene expression in the above public data sets. Our definition of a differentially expressed gene was as follows: |log2fold change| ≥0.8 and FDR ≤0.05.
Gene ontology enrichment analysis
Gene ontology (GO) enrichment analysis assigns functional annotations to selected gene categories or related genes. We used the R language clusterProfiler package (Yu et al., 2012) (clusterProfiler-3.12.0) and human genome annotation information package org.Hs.eg.db (org.Hs.eg.db-3.8.2) to extract GO annotation information of the differentially expressed genes. The GO enrichment analysis of the differentially expressed genes and corresponding GO annotation information was performed by hypergeometric test. We defined the enriched GO pathway as that with a p-value ≤0.05. Then, the GOplot package (GOplot-1.0.2) was applied to draw chord diagrams for the differentially expressed genes and enriched GO pathways. We applied the same method for GO enrichment analysis of the public data sets GSE46224 and GSE1145.
Gene regulatory networks construction
Gene regulatory networks were constructed with genes that were differentially expressed in RCM, IHD, and VHD samples. For the interaction between mRNAs and miRNAs, the target genes corresponding to human miRNAs were predicted with miRDB database (http://mirdb.org/) (Chen and Wang, 2020; Liu and Wang, 2019). For the interaction between miRNAs and lncRNAs, miRNA recognition elements (MREs) on human lncRNAs were predicted with DIANA LncBase Predicted v.2 tools (Paraskevopoulou et al., 2016). The interaction between miRNAs and circRNAs was predicted with miRanda software (miRanda-3.3a) (Betel et al., 2010).
Gene co-expression network construction
The WGCNA package (WGCNA-1.69) (Langfelder and Horvath, 2008) was applied to perform weighted gene co-expression network analysis (WGCNA) on genes that were differentially expressed in RCM, IHD, and VHD samples to describe the gene association patterns between different samples. Then, we used the formula amn=|cmn|β (amn: adjacent value between gene m and gene n, cmn: Pearson correlation coefficient between gene m and gene n expression, β: soft threshold) to create a weighted adjacency matrix. We selected gene pairs with adjacency value greater than 0.25 to draw a gene co-expression network.
Cell culture and transfection
Neonatal rat ventricle myocytes (NRVMs) were isolated and cultured as described previously (Liao et al., 2015). The cell transfection was carried out accordance to the manufacturer’s instructions. Briefly, the mimics, inhibitors RNA oligos from GenePharma (100nm) were transfected into NRVMs by Lipofectamine RNAiMAX Reagent (Invitrogen, 13778-150). rno-miR-144-3p mimics: UACAGUAUAGAUGAUGUACU; rno-miR-144-3p inhibitor: AGUACAUCAUCUAUACUGUA.
RNA extraction and qPCR
The cDNA was synthesized using a cDNA synthesis kit, and a qRT-PCR kit was used for quantification (GenePharma). The relative abundance of miRNA was calculated using the 2-ΔΔCt method, and the relative expression levels were normalized to the expression of U6.
Protein extraction and western blot
Protein from NRVMs was extracted using RIPA buffer (Beyotime Biotechnology, P0013B) supplemented with protease and phosphatase inhibitor cocktails (Roche, 04693132001 and 04906845001). Protein lysates were subjected to SDS–PAGE and transferred to nitrocellulose membranes. The membranes were then blocked and incubated with appropriate primary antibodies followed by HRP-conjugated secondary antibodies. The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression was used as internal control for normalization. Anti-GAPDH polyclonal antibody (Utibody, UM4002); anti-ADRA1A polyclonal antibody (Proteintech, 19777-1-AP).
Statistical analysis
R version 4.0.0 were employed to all statistical analysis and data visualization. All figures were produced with ggplot2 (v3.3.1). Significance levels were defined as following: ns, P > 0.05, ∗P < 0.05 and ∗∗P < 0.01 for gene ontology enrichment analysis with Fisher’s exact test.
Acknowledgments
This work was financially supported by the grant from the National Key R&D Program of China [2016YFA0101100 to Nianguo Dong], the grant from the National Natural Science Foundation of China [81600354 to Chao Zhang], the grant from the National Natural Science Foundation of China [grant number 81873496 to Xudong Liao] and the grant from the Fundamental Research Funds for the Central Universities, HUST [NO. 2021GCRC073 to Ximiao He]. We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Author contributions
N.D., X.H., and X.L. designed the research. M.Z., C.Z., and Z.Z. collected and analyzed the data. M.Z., C.Z., X.L., R.L., and S.L. drafted the manuscript. X.L. and D.R. performed the qPCR and WB experiment. All authors read and approved the final manuscript.
Declaration of interests
The authors declare that they have no competing interests.
Published: March 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.103935.
Contributor Information
Ximiao He, Email: ximiaohe@hust.edu.cn.
Nianguo Dong, Email: dongnianguo@hotmail.com.
Supplemental information
Data and code availability
Our data had been uploaded to the National Genomics Data Center (https://ngdc.cncb.ac.cn/gsa-human/), under accession number NGDC: HRA001366 (https://ngdc.cncb.ac.cn/gsa-human/s/E0neHkH8). The data are available upon request by contacting lead contact, Nianguo Dong. No new code was generated during this study. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Anand I.S., Kempf T., Rector T.S., Tapken H., Allhoff T., Jantzen F., Kuskowski M., Cohn J.N., Drexler H., Wollert K.C. Serial measurement of growth-differentiation factor-15 in heart failure: relation to disease severity and prognosis in the Valsartan Heart Failure Trial. Circulation. 2010;122:1387–1395. doi: 10.1161/CIRCULATIONAHA.109.928846. [DOI] [PubMed] [Google Scholar]
- Anand I.S., Rector T.S., Kuskowski M., Adourian A., Muntendam P., Cohn J.N. Baseline and serial measurements of galectin-3 in patients with heart failure: relationship to prognosis and effect of treatment with valsartan in the Val-HeFT. Eur. J. Heart Fail. 2013;15:511–518. doi: 10.1093/eurjhf/hfs205. [DOI] [PubMed] [Google Scholar]
- Bartel D.P. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauleo A. [Interview of Armando Bauleo by Alejandro Vainer] Vertex. 2002;13:301–306. [PubMed] [Google Scholar]
- Betel D., Koppal A., Agius P., Sander C., Leslie C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010;11:R90. doi: 10.1186/gb-2010-11-8-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunwald E., Nicholls M. Leaders in cardiovascular medicine. Eugene Braunwald MD: an icon of the 20th century still going strong. Eur. Heart J. 2015;36:1350–1351. doi: 10.1093/eurheartj/ehv101. [DOI] [PubMed] [Google Scholar]
- Cakmak H.A., Coskunpinar E., Ikitimur B., Barman H.A., Karadag B., Tiryakioglu N.O., Kahraman K., Vural V.A. The prognostic value of circulating microRNAs in heart failure: preliminary results from a genome-wide expression study. J. Cardiovasc. Med. (Hagerstown) 2015;16:431–437. doi: 10.2459/JCM.0000000000000233. [DOI] [PubMed] [Google Scholar]
- Chen M.C., Chang J.P., Lin Y.S., Pan K.L., Ho W.C., Liu W.H., Chang T.H., Huang Y.K., Fang C.Y., Chen C.J. Deciphering the gene expression profile of peroxisome proliferator-activated receptor signaling pathway in the left atria of patients with mitral regurgitation. J. Transl. Med. 2016;14:157. doi: 10.1186/s12967-016-0871-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Wang X. miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020;48:D127–D131. doi: 10.1093/nar/gkz757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dangwal S., Schimmel K., Foinquinos A., Xiao K., Thum T. Noncoding RNAs in heart failure. Handb. Exp. Pharmacol. 2017;243:423–445. doi: 10.1007/164_2016_99. [DOI] [PubMed] [Google Scholar]
- Devaux Y., Creemers E.E., Boon R.A., Werfel S., Thum T., Engelhardt S., Dimmeler S., Squire I. Circular RNAs in heart failure. Eur. J. Heart Fail. 2017;19:701–709. doi: 10.1002/ejhf.801. [DOI] [PubMed] [Google Scholar]
- Dieplinger B., Mueller T. Soluble ST2 in heart failure. Clin. Chim. Acta. 2015;443:57–70. doi: 10.1016/j.cca.2014.09.021. [DOI] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J., Fan X., Guang H., Shan X., Tian Q., Zhang F., Chen R., Ye F., Quan H., Zhang H., et al. Upregulation of miR-335-3p by NF-kappaB transcriptional regulation contributes to the induction of pulmonary arterial hypertension via APJ during hypoxia. Int. J. Biol. Sci. 2020;16:515–528. doi: 10.7150/ijbs.34517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatima R., Akhade V.S., Pal D., Rao S.M. Long noncoding RNAs in development and cancer: potential biomarkers and therapeutic targets. Mol. Cell. Ther. 2015;3:5. doi: 10.1186/s40591-015-0042-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G., Yan Z., Li C., Hou Y. microRNA-208a in an early stage myocardial infarction rat model and the effect on cAMP-PKA signaling pathway. Mol. Med. Rep. 2016;14:1631–1635. doi: 10.3892/mmr.2016.5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita T., Ishikawa Y. Apoptosis in heart failure. -The role of the beta-adrenergic receptor-mediated signaling pathway and p53-mediated signaling pathway in the apoptosis of cardiomyocytes- Circ. J. 2011;75:1811–1818. doi: 10.1253/circj.cj-11-0025. [DOI] [PubMed] [Google Scholar]
- Furio-Tari P., Tarazona S., Gabaldon T., Enright A.J., Conesa A. spongeScan: a web for detecting microRNA binding elements in lncRNA sequences. Nucleic Acids Res. 2016;44:W176–W180. doi: 10.1093/nar/gkw443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaggin H.K., Szymonifka J., Bhardwaj A., Belcher A., De Berardinis B., Motiwala S., Wang T.J., Januzzi J.J. Head-to-head comparison of serial soluble ST2, growth differentiation factor-15, and highly-sensitive troponin T measurements in patients with chronic heart failure. JACC Heart Fail. 2014;2:65–72. doi: 10.1016/j.jchf.2013.10.005. [DOI] [PubMed] [Google Scholar]
- Gama-Carvalho M., Andrade J., Bras-Rosario L. Regulation of cardiac cell fate by microRNAs: implications for heart regeneration. Cells. 2014;3:996–1026. doi: 10.3390/cells3040996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi P.U., Testani J.M., Ahmad T. The current and potential clinical relevance of heart failure biomarkers. Curr. Heart Fail. Rep. 2015;12:318–327. doi: 10.1007/s11897-015-0268-2. [DOI] [PubMed] [Google Scholar]
- Gladysheva I.P., Wang D., McNamee R.A., Houng A.K., Mohamad A.A., Fan T.M., Reed G.L. Corin overexpression improves cardiac function, heart failure, and survival in mice with dilated cardiomyopathy. Hypertension. 2013;61:327–332. doi: 10.1161/HYPERTENSIONAHA.112.193631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gommans D., Cramer G.E., Fouraux M.A., Heijmans S., Michels M., Timmermans J., Verheugt F., de Boer R.A., Kofflard M., Brouwer M.A. Usefulness of high-sensitivity cardiac troponin T to predict long-term outcome in patients with hypertrophic cardiomyopathy. Am. J. Cardiol. 2021;152:120–124. doi: 10.1016/j.amjcard.2021.04.040. [DOI] [PubMed] [Google Scholar]
- Havranek S., Marek J. Biomarker GDF-15 in cardiology. Vnitr. Lek. 2021;67:11–14. [PubMed] [Google Scholar]
- He L., Huang C. MiR-19b and miR-16 cooperatively signaling target the regulator ADRA1A in hypertensive heart disease. Biomed. Pharmacother. 2017;91:1178–1183. doi: 10.1016/j.biopha.2017.04.041. [DOI] [PubMed] [Google Scholar]
- Kempf T., Eden M., Strelau J., Naguib M., Willenbockel C., Tongers J., Heineke J., Kotlarz D., Xu J., Molkentin J.D., et al. The transforming growth factor-beta superfamily member growth-differentiation factor-15 protects the heart from ischemia/reperfusion injury. Circ. Res. 2006;98:351–360. doi: 10.1161/01.RES.0000202805.73038.48. [DOI] [PubMed] [Google Scholar]
- Kim T.T., Dyck J.R. Is AMPK the savior of the failing heart? Trends Endocrinol. Metab. 2015;26:40–48. doi: 10.1016/j.tem.2014.11.001. [DOI] [PubMed] [Google Scholar]
- Klattenhoff C.A., Scheuermann J.C., Surface L.E., Bradley R.K., Fields P.A., Steinhauser M.L., Ding H., Butty V.L., Torrey L., Haas S., et al. Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell. 2013;152:570–583. doi: 10.1016/j.cell.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimiuk A., Zalewska A., Sawicki R., Knapp M., Maciejczyk M. Salivary oxidative stress increases with the progression of chronic heart failure. J. Clin. Med. 2020;9:769. doi: 10.3390/jcm9030769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara A., Birgaoanu M., Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic Acids Res. 2019;47:D155–D162. doi: 10.1093/nar/gky1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ky B., French B., McCloskey K., Rame J.E., McIntosh E., Shahi P., Dries D.L., Tang W.H., Wu A.H., Fang J.C., et al. High-sensitivity ST2 for prediction of adverse outcomes in chronic heart failure. Circ. Heart Fail. 2011;4:180–187. doi: 10.1161/CIRCHEARTFAILURE.110.958223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D.I., Zhu G., Sasaki T., Cho G.S., Hamdani N., Holewinski R., Jo S.H., Danner T., Zhang M., Rainer P.P., et al. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature. 2015;519:472–476. doi: 10.1038/nature14332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Cai S.X., He Q., Zhang H., Friedberg D., Wang F., Redington A.N. Intravenous miR-144 reduces left ventricular remodeling after myocardial infarction. Basic Res. Cardiol. 2018;113:36. doi: 10.1007/s00395-018-0694-x. [DOI] [PubMed] [Google Scholar]
- Liao X., Zhang R., Lu Y., Prosdocimo D.A., Sangwung P., Zhang L., Zhou G., Anand P., Lai L., Leone T.C., et al. Kruppel-like factor 4 is critical for transcriptional control of cardiac mitochondrial homeostasis. J. Clin. Invest. 2015;125:3461–3476. doi: 10.1172/JCI79964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Smyth G.K., Shi W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019;47:e47. doi: 10.1093/nar/gkz114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W., Wang X. Prediction of functional microRNA targets by integrative modeling of microRNA binding and target expression data. Genome Biol. 2019;20:18. doi: 10.1186/s13059-019-1629-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X., Yang B., Nattel S. MicroRNAs and atrial fibrillation: mechanisms and translational potential. Nat. Rev. Cardiol. 2015;12:80–90. doi: 10.1038/nrcardio.2014.178. [DOI] [PubMed] [Google Scholar]
- Mackowiak S.D. Identification of novel and known miRNAs in deep-sequencing data with miRDeep2. Curr. Protoc. Bioinformatics. 2011;12:10–12. doi: 10.1002/0471250953.bi1210s36. [DOI] [PubMed] [Google Scholar]
- Maisel A., Xue Y., Shah K., Mueller C., Nowak R., Peacock W.F., Ponikowski P., Mockel M., Hogan C., Wu A.H., et al. Increased 90-day mortality in patients with acute heart failure with elevated copeptin: secondary results from the biomarkers in Acute Heart Failure (BACH) study. Circ. Heart Fail. 2011;4:613–620. doi: 10.1161/CIRCHEARTFAILURE.110.960096. [DOI] [PubMed] [Google Scholar]
- McCullough P.A., Olobatoke A., Vanhecke T.E. Galectin-3: a novel blood test for the evaluation and management of patients with heart failure. Rev. Cardiovasc. Med. 2011;12:200–210. doi: 10.3909/ricm0624. [DOI] [PubMed] [Google Scholar]
- Mrsic Z., Hopkins S.P., Antevil J.L., Mullenix P.S. Valvular heart disease. Prim. Care. 2018;45:81–94. doi: 10.1016/j.pop.2017.10.002. [DOI] [PubMed] [Google Scholar]
- Muchtar E., Blauwet L.A., Gertz M.A. Restrictive cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ. Res. 2017;121:819–837. doi: 10.1161/CIRCRESAHA.117.310982. [DOI] [PubMed] [Google Scholar]
- Ngo D.T., Horowitz J.D., Sverdlov A.L. Heart failure: a corin-deficient state? Hypertension. 2013;61:284–285. doi: 10.1161/HYPERTENSIONAHA.112.196253. [DOI] [PubMed] [Google Scholar]
- Paraskevopoulou M.D., Vlachos I.S., Karagkouni D., Georgakilas G., Kanellos I., Vergoulis T., Zagganas K., Tsanakas P., Floros E., Dalamagas T., et al. DIANA-LncBase v2: indexing microRNA targets on non-coding transcripts. Nucleic Acids Res. 2016;44:D231–D238. doi: 10.1093/nar/gkv1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponting C.P., Oliver P.L., Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136:629–641. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Rius J., Guma M., Schachtrup C., Akassoglou K., Zinkernagel A.S., Nizet V., Johnson R.S., Haddad G.G., Karin M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453:807–811. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida P.L., Don-Wauchope A.C., Ali U., Oremus M., Brown J.A., Bustamam A., Hill S.A., Booth R.A., Sohel N., McKelvie R., et al. Incremental value of natriuretic peptide measurement in acute decompensated heart failure (ADHF): a systematic review. Heart Fail. Rev. 2014;19:507–519. doi: 10.1007/s10741-014-9444-9. [DOI] [PubMed] [Google Scholar]
- Schonrock N., Harvey R.P., Mattick J.S. Long noncoding RNAs in cardiac development and pathophysiology. Circ. Res. 2012;111:1349–1362. doi: 10.1161/CIRCRESAHA.112.268953. [DOI] [PubMed] [Google Scholar]
- Thum T., Galuppo P., Wolf C., Fiedler J., Kneitz S., van Laake L.W., Doevendans P.A., Mummery C.L., Borlak J., Haverich A., et al. MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation. 2007;116:258–267. doi: 10.1161/CIRCULATIONAHA.107.687947. [DOI] [PubMed] [Google Scholar]
- Touyz R.M., Alves-Lopes R., Rios F.J., Camargo L.L., Anagnostopoulou A., Arner A., Montezano A.C. Vascular smooth muscle contraction in hypertension. Cardiovasc. Res. 2018;114:529–539. doi: 10.1093/cvr/cvy023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooij E., Sutherland L.B., Qi X., Richardson J.A., Hill J., Olson E.N. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007;316:575–579. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- Wang K., Liu F., Zhou L.Y., Long B., Yuan S.M., Wang Y., Liu C.Y., Sun T., Zhang X.J., Li P.F. The long noncoding RNA CHRF regulates cardiac hypertrophy by targeting miR-489. Circ. Res. 2014;114:1377–1388. doi: 10.1161/CIRCRESAHA.114.302476. [DOI] [PubMed] [Google Scholar]
- Wang K., Long B., Liu F., Wang J.X., Liu C.Y., Zhao B., Zhou L.Y., Sun T., Wang M., Yu T., et al. A circular RNA protects the heart from pathological hypertrophy and heart failure by targeting miR-223. Eur. Heart J. 2016;37:2602–2611. doi: 10.1093/eurheartj/ehv713. [DOI] [PubMed] [Google Scholar]
- Wirtz P.H., von Kanel R. Psychological stress, inflammation, and coronary heart disease. Curr. Cardiol. Rep. 2017;19:111. doi: 10.1007/s11886-017-0919-x. [DOI] [PubMed] [Google Scholar]
- Wu W., Ji P., Zhao F. CircAtlas: an integrated resource of one million highly accurate circular RNAs from 1070 vertebrate transcriptomes. Genome Biol. 2020;21:101. doi: 10.1186/s13059-020-02018-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Our data had been uploaded to the National Genomics Data Center (https://ngdc.cncb.ac.cn/gsa-human/), under accession number NGDC: HRA001366 (https://ngdc.cncb.ac.cn/gsa-human/s/E0neHkH8). The data are available upon request by contacting lead contact, Nianguo Dong. No new code was generated during this study. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.