

Abstract
c-Met is a hepatocyte growth factor receptor overexpressed in many tumors such as hepatocellular carcinoma (HCC). Therefore, c-Met may serve as a promising target for HCC immunotherapy. Modifying T cells to express c-Met-specific chimeric antigen receptor (CAR) is an attractive strategy in treating c-Met-positive HCC. This study aimed to systematically evaluate the inhibitory effects of 2nd- and 3rd-generation c-Met CAR-T cells on hepatocellular carcinoma (HCC) cells. Here, 2nd- and 3rd-generation c-Met CARs containing an anti-c-Met single-chain variable fragment (scFv) as well as the CD28 signaling domain and CD3ζ (c-Met-28-3ζ), the CD137 signaling domain and CD3ζ (c-Met-137-3ζ), or the CD28 and CD137 signaling domains and CD3ζ (c-Met-28-137-3ζ) were constructed, and their abilities to target c-Met-positive HCC cells were evaluated in vitro and in vivo. All c-Met CARs were stably expressed on T cell membrane, and c-Met CAR-T cells aggregated around c-Met-positive HCC cells and specifically killed them in vitro. c-Met-28-137-3ζ CAR-T cells secreted more interferon-gamma (IFN-γ) and interleukin 2 (IL-2) than c-Met-28-3ζ CAR-T cells and c-Met-137-3ζ CAR-T cells. Compared with c-Met low-expressed cells, c-Met CAR-T cells secreted more cytokines when co-cultured with c-Met high-expressed cells. Moreover, c-Met-28-137-3ζ CAR-T cells eradicated HCC more effectively in xenograft tumor models compared with the control groups. This study suggests that 3rd-generation c-Met CAR-T cells are more effective in inhibiting c-Met-positive HCC cells than 2nd-generation c-Met CAR-T cells, thereby providing a promising therapeutic intervention for c-Met-positive HCC.
Keywords: chimeric antigen receptor, c-Met, hepatocellular carcinoma, immunotherapy
Introduction
Hepatocellular carcinoma (HCC) is the sixth commonly diagnosed cancer and the fourth leading cause of cancer death in China[1]. At present, surgical resection is the most effective strategy for the treatment of HCC, but its postoperative recurrence rate is very high[2]. Chemotherapy, chemoembolization, radiotherapy, and transarterial embolization therapy do not improve the survival rate of most patients with advanced HCC. Therefore, other treatment options are urgently needed.
The adoptive immunotherapy technology of chimeric antigen receptor T (CAR-T) cell therapy has become the fourth treatment option just after surgery, radiotherapy, and chemotherapy. CD19-targeting CAR-T cells have achieved great advances in B-cell acute lymphoblastic leukemia and relapsed or refractory large B-cell lymphoma[3–5]. CAR-T cell therapy in treating hematological malignancies allows researchers to explore the development of this technology in solid tumors such as glioblastoma[6], breast cancer[7], and colorectal cancer[8]. However, there are still many problems in the management of solid tumors, such as the poor persistence of CAR-T cells and tumor immunosuppressive microenvironment. It is a substantial challenge to explore novel tumor antigens and optimize CAR designs.
c-Met, also known as the high-affinity receptor for the human hepatocyte growth factor (HGFR), is a cell surface protein tyrosine kinase expressed in various solid tumors, including liver cancer, lung cancer, stomach cancer and ovarian cancer[9–10]. c-Met plays an important role in tumor cell proliferation, invasion and apoptosis protection[11]. The overexpression of c-Met is an indication of increased tumor invasiveness and poor prognosis[12]. In addition, the continuous activation of the Met/HGF signaling pathway is related to the occurrence of tumors[13–14]. Due to its high expression in HCC and limited expression in normal tissues, c-Met has been used as a target for HCC immunotherapy[13].
In this study, three different c-Met CARs with co-stimulators CD28, CD137, and both CD28 and CD137 (referred to as c-Met-28-3ζ CAR-T, c-Met-137-3ζ CAR-T, and c-Met-28-137-3ζ CAR-T, respectively) were constructed, and the anti-tumor effects of corresponding c-Met CAR-T cells were evaluated in vitro and in vivo. The results indicated that 3rd-generation c-Met CAR-T cells are more effective than 2nd-generation c-Met CAR-T cells, which provides evidence for the selection of the co-stimulatory domain in the design of CAR molecules.
Materials and methods
Cell lines
Human c-Met-positive HCC cell lines (HepG2 and Bel-7402) and Lenti-X 293T cells were preserved by National Health Commission Key Laboratory of Antibody Techniques (Nanjing Medical University, China) and cultured in DMEM (Gibco, USA) supplemented with 10% fetal bovine serum (FBS; Gibco) and 1% penicillin/streptomycin (Gibco) at 37 °C in 5% CO2 (Thermo Fisher Scientific, USA). The shMet-HepG2 cells were constructed by lentiviral transduction (Public Protein/Plasmid Library, China). The shRNA sequence targeting human c-Met was as following: 5′-AATTAGTTCGCTACGATGCAA-3′. For bioluminescence assays, HepG2 cells with enforced expression of luciferase (HepG2-Luc) were generated. All cells were routinely evaluated for mycoplasma contamination.
Generation of chimeric antigen receptors
A codon-optimized gene encoding the c-Met-specific single-chain variable fragment (scFv) of human c-Met Fab was amplified[15], and the amino sequences for this scFv are provided in Supplementary Table 1 (available online). The optimized sequence contained a heavy chain variable region-(GGGS)3-light chain variable region sequence. The sequence was subcloned in-frame into lentiviral vectors containing expression cassettes encoding a CD8α hinge domain, CD8 transmembrane (TM) domain and CD28-CD3ζ, CD137-CD3ζ, or CD28-CD137-CD3ζ signaling domains. The sequence of each cloned CAR was verified by DNA sequencing (Genscript, China). The c-Met CAR fragments (c-Met-28-3ζ, c-Met-137-3ζ, and c-Met-28-137-3ζ) were generated by PCR using the following primers: forward, 5′-GGACCATCCTCTAGGGATCCATGGCCTTACCAG-3′; and reverse, 5′-AATCCGGATCGATCTCGAGGTGCATGCTAACGC-3′. They were cloned into the BamH 1 and Xho 1 sites of the lentiviral vector pCDH-CMV-MCS-EF1α-CopGFP[15]. The new vector was verified by DNA sequencing (Genscript). In addition to activated T cells, a CD19-specific CAR containing CD137 costimulatory endodomains and CD3ζ was used as a negative control, and the detailed structure has been described previously[16].
Production of the lentivirus and the generation of CAR-T cells
Lentiviral supernatants were produced by co-transfecting Lenti-X 293T cells with plasmids containing c-Met CAR, the psPAX2 plasmid encoding the envelope, and the pMD2G plasmid encoding the membrane protein using PEI transduction reagent (Polysciences, USA). The supernatants containing the lentivirus were pooled at 48 and 72 hours after transfection, then passed through a 0.45 µm filter (Merck Millipore, Germany) and concentrated 100-fold by PEG-8000 (Biofroxx, Germany).
Human peripheral blood mononuclear cells (PBMCs) were isolated from the peripheral blood of healthy donors provided by the Second Affiliated Hospital of Nanjing Medical University by density gradient centrifugation, and this study protocol was approved by the local institutional review board at the authors' affiliated institutions. PBMCs were stimulated with anti-CD3/anti-CD28 antibodies (eBioscience, USA; 1 μg/mL) in a 24-well plate (Corning, USA) for 48 hours. For transduction, a non-tissue culture treated 24-well plate (Corning) was pre-coated with RetroNectin (Takara, Japan, 20 μg/mL), then the T cells were transduced with lentivirus in the presence of 100 IU/mL interleukin 2 (IL-2). The transfected T cells were cultured in the complete human lymphocyte culture medium (RPMI Medium 1640 [Gibco], 10% heat inactivated FBS, 100 units/mL penicillin [Gibco], 100 μg/mL streptomycin [Gibco], 100 IU/mL recombinant human IL-2 [PeproTech, USA], 10 ng/mL recombinant human IL-7 [PeproTech] and 5 ng/mL recombinant human IL-15 [PeproTech]). After 14 days ofin vitro expansion, the genetically modified T cells could be used in the next experiment.
Flow cytometry
For all flow cytometry analyses, a FACS Calibur instrument (BD, USA) was used. The results were analyzed with FlowJo 7.6. Green fluorescent protein (GFP) encoded by CAR vector was detected by flow cytometry. The percentage of GFP-positive T cells represented the lentivirus infection efficiency. c-Met expression in tumor cell lines was detected with an APC-conjugated anti-c-Met antibody (Immunogen, China). The cells were washed twice with PBS and fixed with 70% ethanol at 4 °C for 14 hours. The cells were incubated in staining solution for 30 minutes at room temperature and evaluated by flow cytometry with a FACS Calibur. All the flow cytometry data were analyzed using FlowJo software.
Western blotting
The c-Met expression in HepG2, Bel-7402, and shMet-HepG2 cells were detected by Western blotting. Cells were collected after washed with PBS and centrifuged at 600 g for 5 minutes. RIPA cell lysate was added, then centrifuged at 12 000 g for 5 minutes to collect the protein supernatant. Proteins were quantified by BCA Protein Quantitation Kit (Thermo Fisher Scientific), and then was separated by SDS-PAGE, followed by transferring to PVDF membranes (Merck Millipore, Germany). The membranes were blocked with 5% (w/v) nonfat dried milk for 2 hours at room temperature and incubated with primary anti-c-Met (Cell Signaling, USA; 1:1000 dilution) and anti-GAPDH (Fude biological, China; 1:1000 dilution) antibodies overnight at 4 °C. Then, the membranes were washed and incubated in horseradish peroxidase-conjugated secondary antibody (Fude biological, China; 1:5000 dilution) for 1 hour at room temperature. Final detection was performed with an enhanced chemiluminescence system (Tanon, China).
Immunofluorescence
After being treated by lentiviral transduction, HepG2 cells were seeded in 12-well plates, then fixed and blocked. The cells were then immunolabeled with anti-human c-Met APC (eBioscience; 1:300 dilution) in 5% BSA overnight at 4 °C. After rinsing, the cells were incubated with an Alexa Fluor 594-conjugated AffiniPure goat anti-human IgG antibody (Jackson ImmunoResearch, USA; 1:500 dilution) for 1 hour at 37 °C in the dark. 1×106 c-Met-28-137-3ζ CAR-T cells were cocultured with the HepG2 cells for 4 hours. The nuclei were stained with Hoechst 33342 (Dojindo, Japan) for 5 minutes at room temperature. Fluorescence was detected by an IX51 microscope (Olympus, Japan).
Cytotoxicity assays
The ability of c-Met CAR-T cells to kill c-Met-positive tumor cells in vitro was tested by the cell counting Kit-8 (CCK-8) assay. The effector and target cells in each group were mixed at a ratio of 2:1, 5:1, 10:1, or 20:1 and incubated at 37 °C for 12 hours. The supernatant was then collected, and the adherent cells were washed three times and treated with 10 μL of CCK-8 and 90 μL of DMEM. Until visual color conversion occurred, the absorbance at 450 nm of each well was measured with a microplate reader (Bio-Rad, USA), and cell viability was normalized using the following formula: (final absorbance treated/final absorbance control)×100%. All experiments were performed at least three times.
Cytokine release assays
The effector cells (c-Met-28-137-3ζ, c-Met-28-3ζ, c-Met-137-3ζ, unrelated CAR-T cells (CD19 CAR-T cells) and T cells) were washed and adjusted to the density to 1×106 cells/mL. A total of 1×104 target cells were plated in a 96-well plate. The effector cells and target HepG2 cells were mixed in RPMI 1640 medium without IL-2 in a ratio of 10:1, incubated at 37 °C for 24 hours, and measured in three replicate wells. The supernatant was collected by centrifugation at 500 g for 5 minutes at room temperature. According to the manufacturer's instructions, the levels of interferon-gamma (IFN-γ) and IL-2 in the culture supernatant were measured by ELISA kit (eBioscience).
Xenograft tumor models
BALB/c nude mice (4 to 5 weeks old) were housed and treated at the Experimental Animal Center of Nanjing Medical University in specific pathogen-free conditions. All animal experiments were performed according to the protocols approved by the Animal Care and Use Committee of Nanjing Medical University (IACUC-1910013).
Mice were inoculated with 5×106 HepG2-Luc cells in the right armpit to establish the HepG2-Luc xenograft model. The size of the tumor was measured with a caliper, and tumor volume (TV) was calculated using the following equation: TV= (width)2 ×length/2. Two weeks after tumor cell injection, and when the tumor volume was about 200 to 300 mm3, the mice were randomly divided into five groups (n=6): c-Met-28-137-3ζ CAR-T cells, c-Met-28-3ζ CAR-T cells, c-Met-137-3ζ CAR-T cells, activated T cells and normal saline without T cells. The tumor size was dynamically monitored by bioluminescence imaging (BLI). On the 14th, 18th, 22nd, and 26th days after tumor cell injection, the mice were injected with D-luciferin potassium salt and anesthetized with isoflurane, then the mice were imaged to detect the luciferase signal. The BLI signal was analyzed by Xenogen IVIS 200 Imaging System. The mice then received one dose of 1×107 c-Met CAR-T cells injected subcutaneously around the tumor and received additional injections on days 18, 22 and 26. The mice were euthanized on day 33.
The isolated tumor tissues were dewaxed and rehydrated in a graded series of alcohol solutions (95%, 85%, and 75%; 5 minutes per solution). Endogenous peroxidase was inhibited using 3% hydrogen peroxide and then stained H&E and immunostained for c-Met (sc-514148; Santa Cruz Biotechnology, USA; 1:200 dilution). EnVision peroxidase kit (Dako, Agilent Technologies, USA) was used to detect the reactions.
Statistical analysis
Every group measured 3 times at least, and the data were reported as the mean±SEM. Statistical analysis was performed by two-way ANOVA to evaluate tumor burden (tumor volume). Differences in cytokine secretion and specific cytolysis were evaluated by the student's t-test. GraphPad Prism v7.0 was used for statistical calculations. P<0.05 indicated statistical significance.
Results
Construction of a c-Met CAR
A c-Met-specific scFv was cloned in frame into lentiviral expression vectors containing CAR expression cassettes with CD28 and CD3ζ (c-Met-28-3ζ), CD137 and CD3ζ (c-Met-137-3ζ), or CD28, CD137, and CD3ζ (c-Met-28-137-3ζ) endodomains (Fig. 1A). For the production of c-Met CAR lentivirus, c-Met CAR expression vector, plasmid psPAX2, and plasmid pMD2G were co-transfected into X-293T cells using polyethylenimine transfection reagent (Invitrogen, USA). The supernatant containing lentivirus was collected and concentrated 100 times by ultracentrifugation (Amicon Ultra 100 kD, USA). The titer of the lentivirus was counted by the LaSRT method and observed by fluorescence microscopy.
Figure 1.

Generation of c-Met CAR-T cells.
A: Schematic representations for constructions of 2nd and 3rd-generation c-Met CAR. B: White and GFP photomicrographs (scale bar, 100 μm) of c-Met-28-137-3ζ CAR-T cells revealing GFP-positive clusters. C: Expressions of c-Met CAR were detected by flow cytometry. CAR-T cells are shown in blue and activated T cells in red. Data were the mean±SEM from triplicates. CD8TM: CD8 transmembrane domain; CAR-T: chimeric antigen receptor T-cell; GFP: green fluorescent protein.
Generation of c-Met CAR-T cells
T cells were activated by anti-CD3/anti-CD28 antibodies and transduced with the c-Met CAR lentivirus. The GFP expression of c-Met CAR-T cells was observed by fluorescence microscope after 48 hours of infection (Fig. 1B). On the 4th day after infection, GFP expression was assessed by flow cytometry to measure the transduction efficiency of different CARs in human T cells. The transduction efficiencies of c-Met-28-137-3ζ CAR-T cells, c-Met-137-3ζ CAR-T cells, and c-Met-28-3ζ CAR-T cells were 46.3%, 57.0%, and 47.4%, respectively (Fig. 1C). All CARs were stably expressed on the surface of T cells with no significant difference in infection efficiency. It was demonstrated that the T cells transduced with the c-Met CAR lentivirus could express the c-Met CARs correctly.
Cytotoxicity of c-Met CAR-T cells against c-Met-positive HCC cells in vitro
The c-Met expression levels of HepG2, Bel-7402, and shMet-HepG2 cells were assessed by flow cytometry and Western blotting (Fig. 2A and B). c-Met was highly expressed in HepG2 and Bel-7402 cells but lowly in shMet-HepG2 cells. To test whether c-Met CAR-T cells could specifically recognize and kill c-Met-positive HCC cells, c-Met-positive HepG2 cells were immunofluorescently labeled and co-cultured with c-Met-28-137-3ζ CAR-T cells for 12 hours. Fluorescence microscopy showed that the c-Met-28-137-3ζ CAR-T cells were able to recognize and aggregate around the c-Met-positive HepG2 cells and the HepG2 cell numbers were reduced significantly (Fig. 2C).
Figure 2.
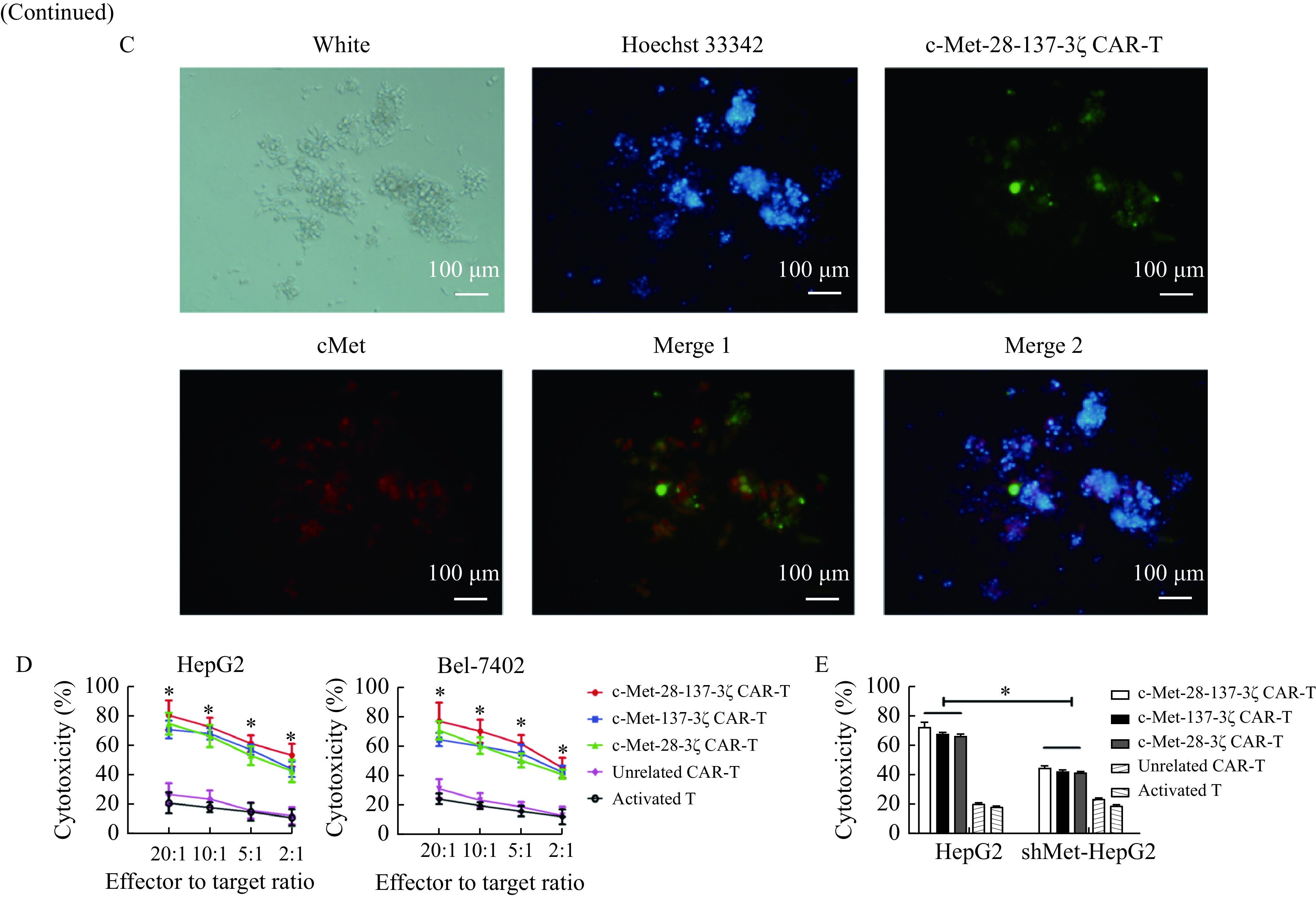
c-Met CAR-T cells recognize and kill c-Met-positive HCC cells.
A: Expressions of c-Met (in blue) on HepG2, Bel-7402, and shMet-HepG2 cells were detected by FACS analysis. B: Western blotting analysis of the c-Met expression in HepG2, Bel-7402, and shMet-HepG2 cells. C: The recognition of c-Met-28-137-3ζ CAR-T cells on HepG2 cells were detected by the immunofluorescence assay. The c-Met-28-137-3ζ CAR-T cells were shown in green, the c-Met in HepG2 cells was stained red, and the nucleus was stained blue. D: In vitro cytotoxic activity of c-Met CAR-T cells against HepG2 or Bel-7402 cells at the indicated effector to target ratios. E: Cytotoxicity of the engineered T cells incubated with indicated target cells at a ratio of 10:1 for 12 hours. Data were the mean±SEM from triplicates. Statistical analyses were performed by two-way ANOVA for comparisons between multiple groups with multiple variables (D) and one-way ANOVA for comparisons between multiple groups with a single variable (E). *P<0.05. CAR-T: chimeric antigen receptor T-cell; shMet-HepG2 cells: HepG2 cells with c-Met knocked down. Merge 1: c-Met-28-137-3ζ CAR-T and cMet; Merge 2: c-Met-28-137-3ζ CAR-T, Hoechst 33342 and cMet.
Figure 2.
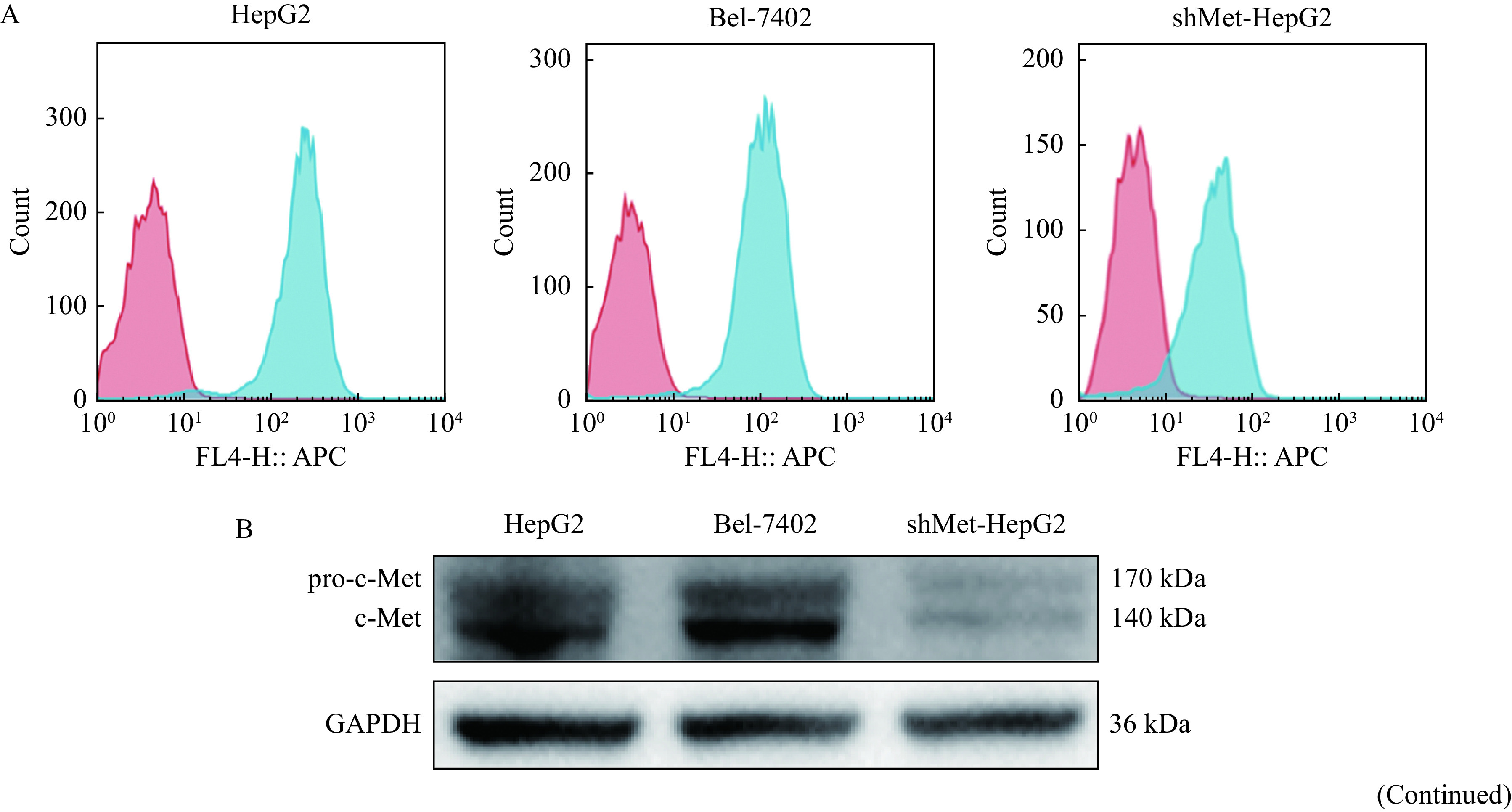
In order to assess the killing effects of c-Met CAR-T cells on HCC cells, CCK-8 cytotoxicity assays were performed using HepG2, Bel-7402, and shMet-HepG2 cells as target cells. The results indicated that when the effector to target ratios were 20:1, 10:1, 5:1, or 2:1, c-Met-28-137-3ζ, c-Met-28-3ζ, and c-Met-137-3ζ CAR-T cells could efficiently lyse the c-Met-positive HCC cells (Fig. 2D). However, when c-Met level on the surface of HepG2 cells was knocked down, the lysis ability of c-Met CAR-T cells was significantly decreased (Fig. 2E). The results indicated that the cytolytic activity of c-Met CAR-T cells was positively correlated with the level of c-Met in HCC cells. In contrast, the control effector cells including unrelated CAR-T cells and activated T cells could not lyse c-Met-positive HCC cells specifically.
c-Met CAR-T cells produced cytokines in response to c-Met-positive HCC cells
c-Met CAR-T cells were shown to recognize c-Met-positive HCC cells in an antigen-specific manner. We then assessed the ability of cytokine secretion by cytokine production assays. HepG2 cells and shMet-HepG2 cells were evaluated in the same panel for ELISA assays. Target cells were cocultured with c-Met CAR-T cells in a ratio of 10:1 for 24 hours, and the culture supernatants were collected, and the levels of cytokines (IL-2 and IFN-γ) were examined by ELISA kits.
In the cytokine release assay, c-Met-positive target cells induced cytokine production in c-Met CAR-T cells but not in unrelated CAR-T cells or activated T cells. C-Met-28-137-3ζ, c-Met-137-3ζ or c-Met-28-3ζ CAR-T cells displayed augmented IL-2 and IFN-γ secretion compared with unrelated CAR-T cells or activated T cells. More IFN-γ was secreted by the c-Met-28-137-3ζ and c-Met-137-3ζ CAR-T cells than by the c-Met-28-3ζ CAR-T cells (P<0.05,Fig. 3A). More IL-2 was secreted by the c-Met-28-137-3ζ CAR-T cells than by the c-Met-28-3ζ and c-Met-137-3ζ CAR-T cells (P<0.05,Fig. 3B). In a word, c-Met-28-137-3ζ CAR-T cells could secrete more IFN-γ and IL-2 than c-Met-28-3ζ CAR-T cells and c-Met-137-3ζ CAR-T cells. When c-Met CAR-T cells were cocultured with shMet-HepG2 cells, cytokine production declined (Fig. 3C and D). The expression level of c-Met on target cells was positively correlated with cytokine production. But the IL-2 and IFN-γ secretion of c-Met CAR-T cells cocultured with shMet-HepG2 was higher than that of unrelated CAR-T cells and activated T cells.
Figure 3.
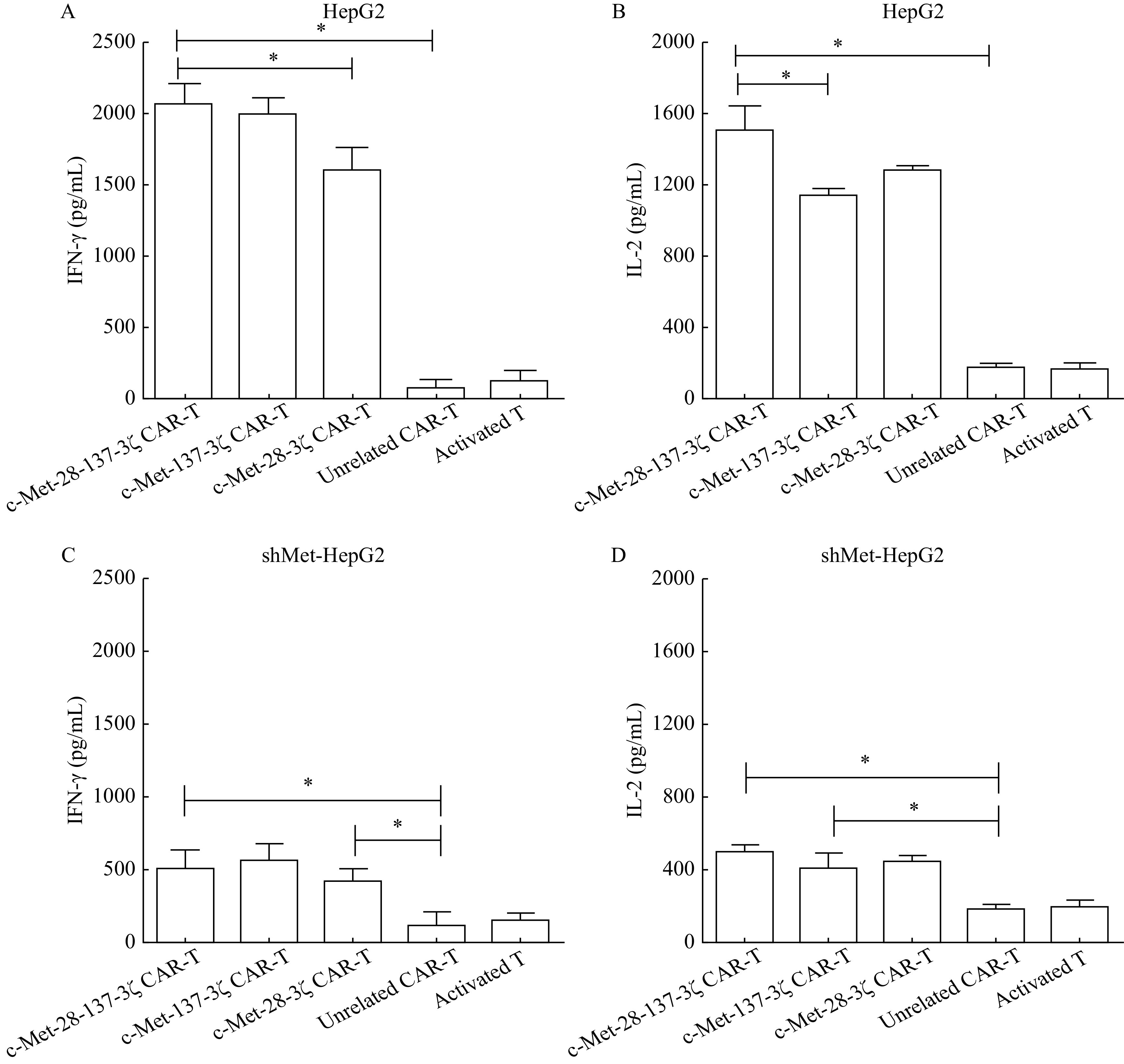
c-Met CAR-T cells produced IFN-γ and IL-2 upon stimulation with HepG2 and shMet-HepG2 cells.
A and B: The secretion of IFN-γ (A) and IL-2 (B) of the engineered T cells following coculture with HepG2 cells for 24 hours at a 10:1 ratio was determined by ELISA. C and D: The secretion of IFN-γ (C) and IL-2 (D) of the engineered T cells following 24 hours coculture with shMet-HepG2 cells at a 10:1 ratio was determined by ELISA. Data were the mean±SEM from triplicates. Statistical analyses were performed by one-way ANOVA for comparisons between multiple groups with a single variable. *P<0.05. IFN-γ: interferon-gamma; IL-2: interleukin-2; ELISA: enzyme-linked immunosorbent assay.
c-Met CAR-T cells suppressed the growth of c-Met-positive HCC in vivo
BALB/c nude mice with established HepG2-Luc xenograft were used to further explore the anti-tumor effects of c-Met CAR-T cells. A total of 5×106 HepG2 Luc cells were injected subcutaneously into the right armpit of the mice. On days 14, 18, 22, 26, c-Met-28-3ζ CAR-T cells, c-Met-137-3ζ CAR-T cells or c-Met-28-137-3ζ CAR-T cells were injected subcutaneously around the tumor. Mice injected with activated T cells or normal saline served as controls (Fig. 4A).
Figure 4.
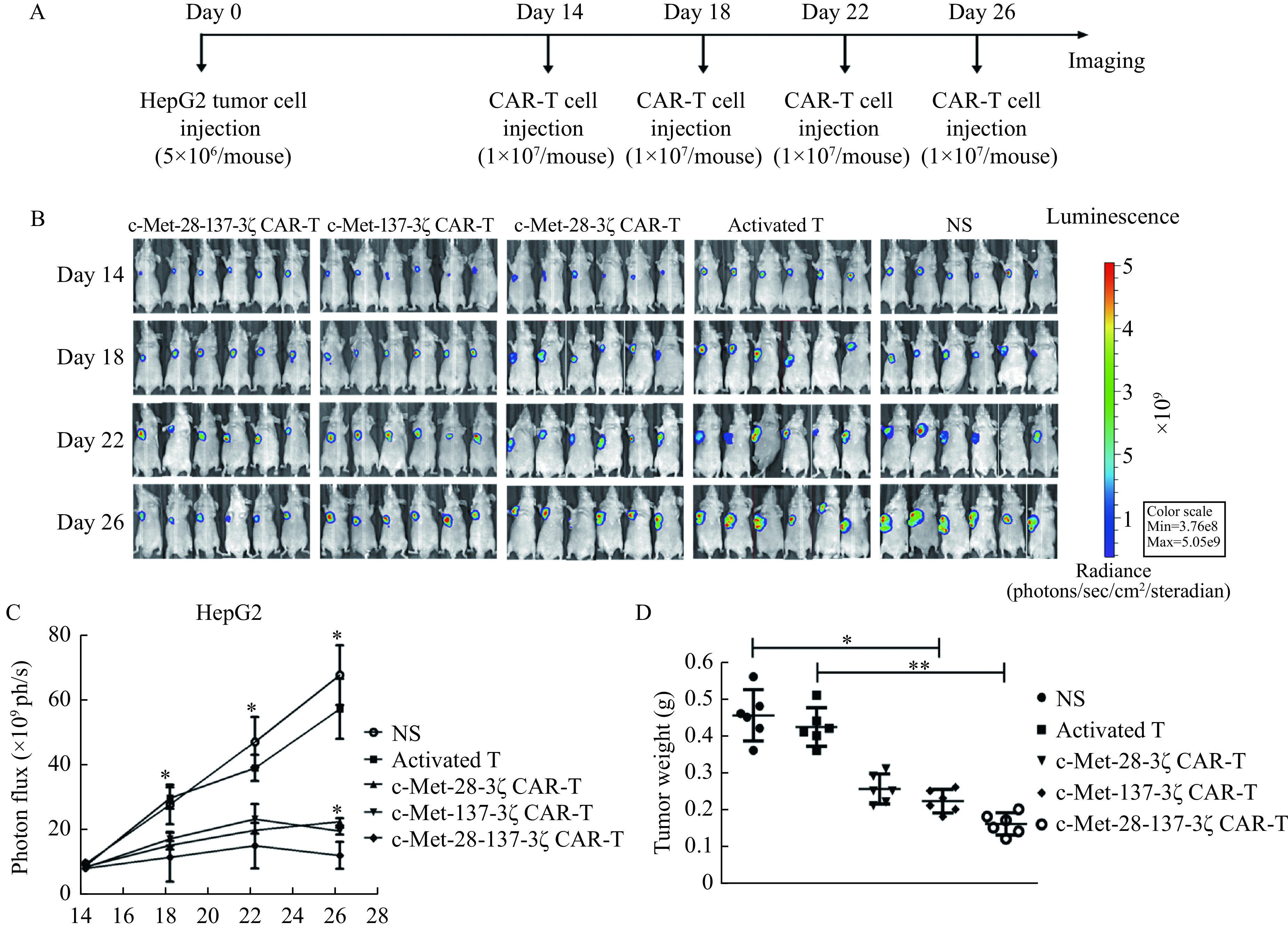
Anti-tumor efficacy of c-Met CAR-T cells in human HCC xenograft model.
A: Schematic representation the subcutaneous implantation of HepG2-Luc cells and treatment of c-Met CAR-T cells. BALB/c nude mice (n=6 for each group) were injected with 5×106 HepG2-Luc subcutaneously into the right underarm. A total of 1×107 c-Met CAR-T cells or activated T cells were injected subcutaneously around the tumor on days14, 18, 22 and 26, respectively. B: Serial tumor bioluminescence imaging of mice on the 14th, 18th, 22nd, and 26th days after the tumor cell injection. C: The tumor burden was assessed by total bioluminescence signals. D: At the end of the experiment, the tumor tissue was isolated and weighed. Statistical analyses were performed by one-way ANOVA for comparisons between multiple groups with a single variable (C and D). *P<0.05;**P<0.01. HCC: hepatocellular carcinoma; HepG2-Luc: HepG2 cells expressing luciferase. NS: 0.9% NaCl.
Compared with the control group, treatment with c-Met-28-3ζ, c-Met-137-3ζ, or c-Met-28-137-3ζ CAR-T cells significantly reduced the tumor burden, as judged by tumor bioluminescence over time. According to the bioluminescence results, the antitumor effects of c-Met-28-3ζ CAR-T cells, c-Met-137-3ζ CAR-T cells and c-Met-28-137-3ζ CAR-T cells were markedly higher than those of normal saline and activated T cells after day 18 (normal saline, [29.6×109±14.6×109] ph/s; activated T cells, [27.6×109±5.7×109] ph/s; c-Met-28-3ζ, [15.1×109±2.9×109] ph/s; c-Met-137-3ζ, [17.1×109±5.2×109] ph/s; and c-Met-28-137-3ζ, [11.4×109±7.6×109] ph/s; all P<0.05). Injection of c-Met-28-137-3ζ CAR-T cells resulted in a significantly greater reduction in tumor burden on day 26 compared to the injection of c-Met-28-3ζ or c-Met-137-3ζ CAR-T cells. There were no significant differences between c-Met-28-3ζ and c-Met-137-3ζ CAR-T cells regarding antitumor activity (c-Met-28-3ζ, [17.3×109±3.5×109] ph/s; c-Met-137-3ζ, [14.6×109±4.5×109] ph/s; and c-Met-28-137-3ζ, [12.0×109±5.3×109] ph/s, Fig. 4B and C).
The mice were euthanized at the end of the experiment (day 33). The average tumor weight and standard deviation were calculated. The tumor weight of mice receiving c-Met-28-137-3ζ CAR-T cell treatment was significantly lower than that of the control group (Fig. 4D). The tumor inhibition rates of the activated T cell group, c-Met-28-3ζ CAR-T cell group, c-Met-137-3ζ CAR-T cell group, and c-Met-28-137-3ζ CAR-T cell group were 6.7%, 43.1%, 49.8%, and 65.3%, respectively. The antitumor effects in the c-Met CAR-T cell groups were significantly higher than those of the control groups. These results indicated that c-Met CAR-T cells could specifically inhibit the growth of HepG2-Luc cells in vivo.
The histological structure of tumor tissue samples treated with c-Met CAR-T cells changed dramatically. HE results showed that the c-Met-28-137-3ζ cells group had more tumor tissue necrosis compared with other control groups. The immunohistochemistry staining results of human CD3 showed that compared with the activated T cell treatment group, mice treated with c-Met CAR-T cells had more T cells infiltrated in the tumor tissue, and there were no infiltrating T cells in the tumor tissue of the mice injected with normal saline (Fig. 5). It was found that c-Met-137-3ζ, c-Met-137-3ζ, and c-Met-28-137-3ζ CAR-T cells were more likely to infiltrate into tumors and exert tumor-killing effects than activated T cells.
Figure 5.

H&E and immunohistochemistry staining of the tumor tissues.
Tumor samples from the mice treated with c-Met CAR-T cells were stained for human CD3 by H&E and IHC staining to detect the human T cell infiltration. A: Representative pictures of H&E and IHC staining analysis of the human T cell infiltration in tumor samples for anti-CD3. Magnification, 200×; Scale bar, 50 μm. B: Quantitative analysis of CD3 positive staining region. Data were the mean±SEM from triplicates. Statistical analyses were performed by one-way ANOVA for comparisons between multiple groups with a single variable.*P<0.05. H&E: hematoxylin and eosin; IHC: immunohistochemistry. The arrow indicates the infiltrating T cells. NS: 0.9% NaCl.
Discussion
The approval of CD19 CAR-T cell products in patients with B-cell leukemia or lymphoma allows researchers to further explore their applications in solid tumors[3]. However, CAR-T cells still face many problems in treating solid tumors. Clinical response is limited by the poor persistence of CAR-T cells[17], therefore, optimizing CAR design is the focus of current research.
In our previous research, the human anti-c-Met Fab antibody was screened, identified[15], and conjugated with doxorubicin (Dox) to prepare c-Met Fab-DOX, then its binding activity to HCC cells and its cytotoxic effects on HCC cells were detected[18]. The application for the c-Met Fab-DOX patent has been approved in our previous work (ZL201110065209.2). The c-Met scFv in this study was derived from the anti-c-Met Fab antibody, which had been confirmed to specifically bind to human HCC cells expressing c-Met and had significant inhibitory effects on tumor growth. The above research results suggest that this high-affinity anti-c-Met Fab antibody can be used for further research on CAR-T technology.
T cell activation can be initiated by human leukocyte antigen-restricted T cell receptors or the CARs. CD28, a member of the immunoglobulin superfamily, can effectively enhance the proliferation and differentiation of naive T cells induced by T cell receptors[19]. Studies have shown that adding CD28 to the structure of CAR will promote the secretion of IL-2 and the expansion of T cells in vitro[20]. It is reported that CD137 is a potent mitigator of exhaustion in chronically stimulated CAR-T cells, thus enhancing the persistence of CAR-T cells[21–23]. Li et al also demonstrated that the incorporation of CD137 in CARs may endow T cells with long-lasting survival ability, thus improving the long-term anti-tumor effect of CAR-T cells[24]. Because CD28 and CD137 signaling activate different pathways in T cells, combining them in a single third-generation CAR may provide additional benefits and overcome the limitations of each individual costimulatory domain. Thus, c-Met-28-3ζ, c-Met-137-3ζ, or c-Met-28-137-3ζ CAR-T cells were prepared in this study, and their anti-tumor effects were detected in vivo andin vitro.
In the current study, the c-Met-targeted CAR-T cells specifically killed c-Met-positive HCC cells in an antigen-dependent manner, which means a positive correlation between the cytolytic activity of c-Met CAR-T cells and the level of c-Met in tumor cells. The shMet-HepG2 cells did not exert significant cytotoxicity, suggesting that the three types of c-Met CAR cells do not trigger consequential tonic signaling, and CAR engagement by HepG2 cells can induce effective killing by c-Met CAR-T cells but not by the control groups. Studies have shown that CAR structures with costimulatory domain enhance the proliferation and cytokine production of CAR-T cells compared to CAR structures without costimulatory domains[23]. Similar to cytotoxicity assay, c-Met CAR-T cells produced higher levels of IL-2 and IFN-γ compared with the control groups (Fig. 3A and B), and 3rd-generation c-Met CAR-T cells could secrete more cytokines than 2nd-generation c-Met CAR-T cells, which is consistent with the results of Zhong et al[23] that the incorporation of the CD28 and CD137 component results in the secretion of a variety of cytokines. In in vivo experiment, mice treated with c-Met-28-137-3ζ, c-Met-28-3ζ or c-Met-137-3ζ CAR-T cells showed significant tumor reduction, while the tumor volume of mice in the two control groups increased obviously (Fig. 4B–D), which suggested that c-Met could be used as a target for HCC immunotherapy. Compared with the c-Met-28-3ζ and c-Met-137-3ζ CAR-T cell groups, the mice injected with c-Met-28-137-3ζ CAR-T cells had a significantly lower tumor burden on day 26, which indicated that the 3rd-generation c-Met CAR-T cells had stronger tumor-suppressing effects than the 2nd-generation c-Met CAR-T cells in vivo. Similarly, a clinical trial showed that third-generation CD19.CARTs have greater expansion and longer persistence than the second-generation CD19.CARTs. This difference was most significant in five patients with low disease burden and few circulating normal B cells, and the results proved that low-dose third-generation CAR-T cells were effective and safe. CAR-T cells produced anti-tumor activity while causing manageable toxicities[25].
The migration of CAR-T cells to the tumor site is a prerequisite for cytolytic effects. Local CAR-T cell delivery in tumors can maximize the accumulation of CAR-T cells at the tumor site, improve safety, and limit their systemic biodistribution. In the current tumor model, the subcutaneous injection around the tumor was used to reduce the incidence of off-target effects and improve the efficacy of c-Met CAR-T cells. A previous study proved that peritumoral delivery of CAR-T cells demonstrated improved antitumor efficacy compared to intravenous delivery in xenograft models targeting mesothelin[25], because it can significantly improve early-stage CAR-T cell infiltration into tumor tissues. In addition, clinical trials of local CAR-T cell delivery in tumors have been completed, for example, anti-CEA CAR-T cells delivered through percutaneous hepatic artery infusion in six patients with CEA-expressing adenocarcinoma liver metastases were safe and promoted sustained stabilization of disease in one patient, who was alive at 23 months follow-up[26]. However, regional administration has certain risks, so its effectiveness and safety still need further study.
c-Met is also expressed in various epithelial tissues (liver, pancreas, prostate, kidney, muscle, and bone-marrow) during embryogenesis[27], and c-Met is found on several other normal cell types, including endothelial cells, hepatocytes, neurons and hematopoietic cells[28–29]. Thus, injection of c-Met CAR-T cells may cause on-target off-tumor effects. In this study, specific adverse events could not be assessed due to the use of xenograft mouse models. An early-phase clinical trial has been completed to evaluate the safety and feasibility of treating metastatic breast cancer with intratumoral injection of mRNA c-Met-CAR T cells (NCT01837602). The results showed that the patients were well tolerated after injection, and there were no drug-related adverse events exceeding grade 1[29]. Nevertheless, some measures can be considered to improve the safety of CAR-T cells, such as using bispecific c-Met/PD-L1 CAR-T cells[16], or reducing the number of CAR-T cells.
Cytokines are important factors for T cell development and homeostasis and can promote the proliferation and differentiation of T cells. During the in vitro culture of CAR-T cells in this study, IL-2, IL-7, and IL-15 were added to the culture medium. IL-2 is the main cytokine used for T cell culture in vitro, as it can stimulate T cells to enter the separation cycle and can enhance the activity of antigen-induced cytotoxic T cellsin vivo. IL-7 plays a key role in the development and maturation of T cells. It promotes the generation of naive and central memory T cell subsets and regulates their homeostasis. IL-15 mediates the formation and homeostasis of CD8 memory T cells. It is said that the combined application of IL-2, IL-7, and IL-15 provides the strongest stimulation for T cell expansion, and the obtained T cells have the highest survival rate and the strongest differentiation ability.
However, our study also has some limitations. In animal model experiments, due to the subcutaneous tumor formation of local tumors, the lack of an immunosuppressive tumor microenvironment and other factors, the biological characteristics of HCC patients cannot be mimicked[30]. Therefore, research into the cytotoxic effects of c-Met CAR-T cells on HCC is needed, and ultimately, decisive answers through clinical trials are required.
Taken together, third-generation c-Met-targeting CAR-T cells that express both CD28 and CD137 costimulatory domains have superior anti-tumor effects, which potentially provides objective insights that may facilitate the development of optimal CAR-T cell-based immunotherapies. These findings establish the basis for subsequent research, such as retrofitting CARs with a "suicide switch" to enhance safety or preparing a bivalent CAR to optimize the treatment of solid tumors[16,31].
This work was supported by grants from National Natural Science Foundation of China (81773268), Collaborative Innovation Center for Cancer Personalized Medicine, China (JX21817902/005).
Acknowledgments
This work was supported by grants from National Natural Science Foundation of China (81773268), Collaborative Innovation Center for Cancer Personalized Medicine, China (JX21817902/005).
Footnotes
CLC number: R730.51, Ducument code: A
The authors reported no conflict of interests.
Contributor Information
Qi Tang, Email: qitang@njmu.edu.cn.
Zhenqing Feng, Email: fengzhenqing@njmu.edu.cn.
References
- 1.Bray F, Ferlay J, Soerjomataram I, et al Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Altekruse SF, McGlynn KA, Reichman ME Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J Clin Oncol. 2009;27(9):1485–1491. doi: 10.1200/JCO.2008.20.7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kochenderfer JN, Dudley ME, Kassim SH, et al Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33(6):540–549. doi: 10.1200/JCO.2014.56.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Edwards BK, Ward E, Kohler BA, et al Annual report to the nation on the status of cancer, 1975-2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer. 2010;116(3):544–573. doi: 10.1002/cncr.24760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown CE, Alizadeh D, Starr R, et al Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. 2016;375(26):2561–2569. doi: 10.1056/NEJMoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun M, Shi H, Liu C, et al Construction and evaluation of a novel humanized HER2-specific chimeric receptor. Breast Cancer Res. 2014;16(3):R61. doi: 10.1186/bcr3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guest RD, Kirillova N, Mowbray S, et al Definition and application of good manufacturing process-compliant production of CEA-specific chimeric antigen receptor expressing T-cells for phase I/II clinical trial. Cancer Immunol Immunother. 2014;63(2):133–145. doi: 10.1007/s00262-013-1492-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Preusser M, Streubel B, Berghoff AS, et al Amplification and overexpression of CMET is a common event in brain metastases of non-small cell lung cancer . Histopathology. 2014;65(5):684–692. doi: 10.1111/his.12475. [DOI] [PubMed] [Google Scholar]
- 10.Chae YK, de Melo Gagliato D, Pai SG, et al The association between EGFR and cMET expression and phosphorylation and its prognostic implication in patients with breast cancer. PLoS One. 2016;11(4):e0152585. doi: 10.1371/journal.pone.0152585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pérol M Negative results of METLung study: an opportunity to better understand the role of MET pathway in advanced NSCLC. Transl Lung Cancer Res. 2014;3(6):392–394. doi: 10.3978/j.issn.2218-6751.2014.09.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sakai K, Aoki S, Matsumoto K Hepatocyte growth factor and Met in drug discovery. J Biochem. 2015;157:271–284. doi: 10.1093/jb/mvv027. [DOI] [PubMed] [Google Scholar]
- 13.Kondo S, Ojima H, Tsuda H, et al Clinical impact of c-Met expression and its gene amplification in hepatocellular carcinoma. Int J Clin Oncol. 2013;18(2):207–213. doi: 10.1007/s10147-011-0361-9. [DOI] [PubMed] [Google Scholar]
- 14.Jin H, Yang R, Zheng Z, et al MetMAb, the one-armed 5D5 anti-c-met antibody, inhibits orthotopic pancreatic tumor growth and improves survival. Cancer Res. 2008;68(11):4360–4368. doi: 10.1158/0008-5472.CAN-07-5960. [DOI] [PubMed] [Google Scholar]
- 15.Jiao Y, Zhao P, Zhu J, et al Construction of human naïve Fab library and characterization of anti-met fab fragment generated from the library. Mol Biotechnol. 2005;31(1):41–54. doi: 10.1385/MB:31:1:041. [DOI] [PubMed] [Google Scholar]
- 16.Jiang W, Li T, Guo J, et al Bispecific c-Met/PD-L1 CAR-T cells have enhanced therapeutic effects on hepatocellular carcinoma. Front Oncol. 2021;11:546586. doi: 10.3389/fonc.2021.546586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guedan S, Ruella M, June CH Emerging cellular therapies for cancer. Annu Rev Immunol. 2019;37:145–171. doi: 10.1146/annurev-immunol-042718-041407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen X, Ding G, Gao Q, et al A human anti-c-met fab fragment conjugated with doxorubicin as targeted chemotherapy for hepatocellular carcinoma. PLoS One. 2013;8(5):e63093. doi: 10.1371/journal.pone.0063093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Acuto O, Michel F CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat Rev Immunol. 2003;3(12):939–951. doi: 10.1038/nri1248. [DOI] [PubMed] [Google Scholar]
- 20.Sadelain M, Brentjens R, Rivière I The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol. 2009;21(2):215–223. doi: 10.1016/j.coi.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Long AH, Haso WM, Shern JF, et al 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21(6):581–590. doi: 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Y, Pullambhatla M, Banerjee SR, et al Synthesis and biological evaluation of low molecular weight fluorescent imaging agents for the prostate-specific membrane antigen. Bioconjug Chem. 2012;23(12):2377–2385. doi: 10.1021/bc3003919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhong X, Matsushita M, Plotkin J, et al Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication . Mol Ther. 2010;18(2):413–420. doi: 10.1038/mt.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li S, Tao Z, Xu Y, et al CD33-specific chimeric antigen receptor T cells with different co-stimulators showed potent anti-leukemia efficacy and different phenotype. Hum Gene Ther. 2018;29(5):626–639. doi: 10.1089/hum.2017.241. [DOI] [PubMed] [Google Scholar]
- 25.Ramos CA, Rouce R, Robertson CS, et al In vivo fate and activity of second- versus third-generation CD19-specific CAR-T cells in B cell non-hodgkin's lymphomas . Mol Ther. 2018;26(12):2727–2737. doi: 10.1016/j.ymthe.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lv J, Zhao R, Wu D, et al Mesothelin is a target of chimeric antigen receptor T cells for treating gastric cancer. J Hematol Oncol. 2019;12(1):18. doi: 10.1186/s13045-019-0704-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Organ SL, Tsao MS. An overview of the c-MET signaling pathway[J]. Ther Adv Med Oncol, 2011, 3(1 Suppl): S7–S19.
- 28.Cui JJ Targeting receptor tyrosine kinase MET in cancer: small molecule inhibitors and clinical progress. J Med Chem. 2014;57(11):4427–4453. doi: 10.1021/jm401427c. [DOI] [PubMed] [Google Scholar]
- 29.Tchou J, Zhao Y, Levine BL, et al Safety and efficacy of intratumoral injections of chimeric antigen receptor (CAR) T cells in metastatic breast cancer. Cancer Immunol Res. 2017;5(12):1152–1161. doi: 10.1158/2326-6066.CIR-17-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dotti G, Gottschalk S, Savoldo B, et al Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev. 2014;257(1):107–126. doi: 10.1111/imr.12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khaleghi S, Rahbarizadeh F, Ahmadvand D, et al A caspase 8-based suicide switch induces apoptosis in nanobody-directed chimeric receptor expressing T cells. Int J Hematol. 2012;95(4):434–444. doi: 10.1007/s12185-012-1037-6. [DOI] [PubMed] [Google Scholar]