

Abstract
Mutations of the Amyloid Precursor Protein, from which the amyloid beta peptide Aβ42 is cleaved, are associated with familial Alzheimer’s disease. The disease-relevant familial mutations occur in Arctic (E22G), Iowa (D23N), Italian (E22K), Dutch (E22Q), Japanese (D7N), English (D6R) and Flemish (A21G) variants. A detailed mechanistic understanding of the aggregation behavior of the mutant peptides at the residue-level is, however, still lacking. We report here a study of the aggregation kinetics of these mutants in vitro by pulsed hydrogen-deuterium exchange mass spectrometry (HDX-MS) to obtain a temporally and sequence resolved picture of their self-assembly. For all variants, HDX occurs to give a bimodal distribution representing two soluble classes of aggregates, one protected and one solvent-exposed. There is no evidence of other classes of structural intermediates within the detection limits of the HDX approach. The fractional changes in the bimodal exchange profiles for several regions of Aβ42 reveal that the central and C-terminal peptides gain protection upon fibril formation, whereas the N-terminal regions remain largely solvent-accessible. For these mutants, all peptide fragments follow the same kinetics, acquiring solvent protection at the same time, further supporting that there are no significant populations of intermediate species under our experimental conditions. The results demonstrate the potential of pulsed HDX-MS for resolving the region-specific aggregation behavior of Aβ42 isoforms in solution where X-ray crystallography and solid-state NMR (ssNMR) are challenged.
Keywords: Aβ42 peptide, disease associated-mutants, pulsed hydrogen deuterium exchange, aggregation, structure, kinetics
Graphical Abstract
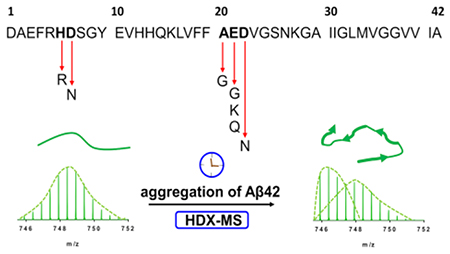
Introduction
The Aβ42 peptide plays a central role in the complex pathology of Alzheimer’s disease (AD), involving the appearance of Aβ plaques and neurofibrillary tangles composed of hyperphosphorylated tau protein. Together, these molecular aggregates act to instigate pathological changes in neurons, astrocytes, and inflammatory microglial cells1. Additional genetic components such as mutations in PSEN1 and PSEN2 and the presence of apolipoprotein (apoE) ε4 allele modulate the pathology2. Importantly, missense mutations in the Amyloid Precursor Protein (APP) gene can result in fulminant, early onset AD or cerebral amyloid angiopathy (CAA). In CAA, the cerebral vessels walls show deposition of Aβ peptide that can cause a hemorrhagic stroke. A spectrum of different point mutations has been documented: the Dutch (E22Q)3, Flemish (A21G)4, Arctic (E22K)5, Iowa (D23N)6, English (D6R)7, Japanese or Tottori (D7N)8, Piedmont (L34V)9, Osaka (E22Δ, deletion)10, Italian (E22K)11, Leuven (E11K)12, K16N13, Taiwanese (DXXH)14 and Swedish double15 mutants among others. The associated phenotypes vary and present themselves in different clinical and pathological forms spanning distinct CAA or AD pathology with amyloid plaques and neurofibrillary tangles as described previously16. For example, the Dutch (E22Q) mutation is associated mainly with hemorrhagic strokes whereas the Flemish (A21G) mutation is associated with both strokes and AD17.
Although numerous biophysical studies addressed familial Alzheimer’s disease (FAD) mutants based on their aggregation kinetics18–23, they lack consensus in ranking and rates. The discrepancies in these studies can be explained by an aggregation process that is highly sensitive to the initial peptide preparation (synthetic versus recombinantly produced), the presence of salts24, pH and temperature changes, an air/water interface and other surfaces25, and the use of agitation during aggregation.
To date, the aggregation of FAD-related Aβ variants has been studied under different experimental conditions by the most commonly employed Thioflavin-T (ThT) fluorescence17,19,20,23,26. The reaction orders and relative ranking of the mutants revealed in these reports can differ between studies, which may originate from the differences in the assay conditions, spanning from sub-micromolar to 70 μM initial concentrations and including several buffer systems, reaction vessels, and peptide sources. Moreover, the reaction orders of the fundamental processes driving the aggregation of the Aβ peptide, secondary nucleation or elongation can vary24, in particular exhibiting a change in the reaction order at higher concentrations, making extrapolation difficult from one condition to another. In other words, the rank ordering of rates can vary depending on different monomer concentrations, even if all other experimental conditions remain the same.
With respect to pathology, the induced toxicity in PC12 cells is higher for the Arctic (E22G), Italian (E22K) and Dutch (E22Q) versions, whereas the Iowa (D23N) mutant is less toxic than the wild type18. Clinically, the Arctic (E22G) mutation is characterized by a late onset (6th or 7th decade of life), presenting no hemorrhage but pronounced plaque formation in blood vessels and regional neurofibrillary tangles27. The Iowa (D23N) mutant is linked to severe CAA, with amyloid-rich meningeal vessels and abundant neurofibrillary tangles in the surroundings but with few amyloid plaques in the brain parenchyma. Carriers of the Dutch (E22Q) mutation show dementia, CAA in the cerebral vessel walls with early or diffuse plaques, and minimal neurofibrillary tangles28. The Flemish (A21G) variant is associated with presenile dementia, large plaques, and occasional cerebral hemorrhage with an onset after the 4th decade29. Interestingly, the Italian (E22K) mutant manifests in early onset AD, gives rise to CAA, and yields two kinds of fibrils depending on its environment. Although at neutral pH, it displays an anti-parallel β-sheet structure in contrast to wild type fibrils, at low pH, it forms parallel structures30. Similarly, the Iowa (D23N) mutant of Aβ40 forms either a parallel or an anti-parallel fibrillar architecture, both of which are associated with severe toxicity31,32. Furthermore, experiments with transgenic mouse models of CAA (Tg-SwDI) and overexpressing, human wild type Aβ (Tg2676) demonstrate that microvascular seeds from Tg-SwDI mice drive the wild type to aggregate into anti-parallel fibrils. This is suggestive of interaction and “crosstalk” between different strains of Aβ33. The Japanese (D7N) mutation causes an onset in the 6th decade with manifestations of dementia and vascular involvement8. One of the least characterized mutants owing to its low frequency is the English (H6R) variant that has an onset of ~55 y and exhibits characteristic AD lesions7.
A productive way to study amyloidogenic proteins is hydrogen/deuterium exchange (HDX), which occurs by a bimodal distribution34–37 similar to an EX1-like kinetics38. Quantification of the relative abundances of the two species (species1 and species2) that comprise the bimodal distribution provides a new reporter for aggregation. That reporter yields structural and temporal information of the species present during the aggregation without the addition of chemical probes that likely affect the aggregation.
Here, we compared the isoforms by pulsed HDX to probe the effects of missense mutations on Aβ42 aggregation at several time points. To ascertain inherent aggregation and assign regional effects, we fitted our peptic peptide data by using the AmyloFit software39 to afford a medium-resolution platform for discerning mutant differences in the aggregation.
This single, nonperturbing approach can achieve convincing comparisons. Although there is complex heterogeneity of species in aggregation, pulsed HDX presents a simple regional view of two species, protected and solvent-exposed, for the wild type and seven isoforms (Arctic (E22G), Iowa (D23N), Italian (E22K), Dutch (E22Q), Japanese (D7N), English (H6R), and Flemish (A21G). This discovery shows that during Aβ protein misfolding, some backbone amides become protected to elicit a tighter, more folded structure that undergoes less HDX, enabling a two-parameter comparison of the isoforms.
RESULTS AND DISCUSSION
Pulsed HDX-MS lends itself to probing both protein conformations and dynamics for aggregating proteins because it deconvolves the aggregation kinetics from the HDX kinetics while providing medium spatial resolution of the protein. Monitoring changes in solvent accessibility in this manner should report the self-association of FAD mutants compared to the wild type and reveal the roles of peptide segments on aggregation (details of isoforms studied here and some critical properties are given in Table 1). We previously described the development of this approach for aggregation of wild type Aβ42 and Aβ40 at different temperatures and in the presence or absence of Cu(II)40.
Table 1.
Wild type Aβ42 sequence (amino acid substitutions examined here in bold and peptide variants with their encoded substitutions or lack thereof, molecular weight (MW) (Da), isoelectric point (pI), number of negatively and positively charged residues and grand average hydrophobicity (GRAVY)41.
| wild type sequence: DAEFRHDSGY EVHHQKLVFF AEDVGSNKGA IIGLMVGGVV IA | ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| name | mutation | MW (Da) | pI | Number (−) residues | Number(+) residues | Hydropathicity (GRAVY) |
| Arctic | E22G | 4442.012 | 5.77 | 5 | 3 | 0.279 |
| Iowa | D23N | 4513.018 | 5.78 | 5 | 3 | 0.205 |
| Italian | E22K | 4513.151 | 6.28 | 5 | 4 | 0.195 |
| Dutch | E22Q | 4513.070 | 5.77 | 5 | 3 | 0.205 |
| English | H6R | 4533.102 | 5.41 | 6 | 4 | 0.174 |
| Japanese | D7N | 4513.091 | 5.78 | 5 | 3 | 0.205 |
| Flemish | A21G | 4500.012 | 5.41 | 6 | 3 | 0.152 |
|
| ||||||
| wild type | - | 4513.999 | 5.31 | 6 | 3 | 0.205 |
HDX-MS approach.
The WU laboratory previously implemented a pulsed HDX-MS methodology40. In this method, various samples are collected during self-assembly, pulse-labeled with deuterium, quenched, desalted, digested on an on-line pepsin column and eluted for detection by MS. Adaptation and further refinement of this workflow and its interpretation has now positioned us to monitor the self-assembly of disease-related mutants in vitro, capturing information about their time-resolved aggregation and region specificity at the peptide-level. Recently, the original method was also applied for the analysis of a multifunctional inhibitor of aggregation42.
Sample preparation plays a major role in in vitro studies of Aβ42 aggregation25,43. Here, we reduced the initial monomer concentration from 50 μM in the original paper to 25 μM, which is more comparable with orthologue biophysical studies. Furthermore, we monomerized the synthetic peptides by treating them with hexafluoro-2-propanol (HFIP) instead of DMSO (dimethyl sulfoxide), subjected them to sodium hydroxide (NaOH) treatment, sonication, and filtration on spin filters prior to aggregation to remove residual pre-formed aggregates. We also extended the linear gradient segment for peptide separation to give better coverage.40 We confirmed the purity and mass of the variants by using high resolution MS (Fig. S2 – 9).
Reducing the initial monomer concentration permitted us to observe that HDX occurs via a two-component exchange pattern, remarkable for this highly disperse system of many oligomers, protofibrils, and fibrils. One component represents a readily exchanging species and the other a protected one that becomes gradually more solvent-inaccessible and increasingly buried as the aggregation proceeds (Fig. S10). This two-component behavior allows straightforward comparisons over aggregation time in a single experimental protocol. For most regions of the variants, a unimodal distribution occurs at the beginning and during early phases of aggregation, indicating an open conformation that undergoes rapid HDX. At the starting points and during the lag phases of aggregation, the concentrations of fibrillar species are low. As aggregation occurs, these open structures adopt a new structure shielded from solvent and HDX, affording bimodal distributions in their HDX-patterns. The two main exchange components are resolvable as separate isotopic clusters, representing two species of Aβ42, one more protected, undergoing less HDX, and reporting on the aggregation. We modeled the isotopic clusters and calculated the relative abundance of each species, expressing them as a fractional species (see Methods). The solvent-accessible species (species2) decreases over time, trading off with a solvent-shielded species1. An important feature of this approach is the deuterium uptake of each peptide does not vary significantly with time of aggregation, but the relative abundances of species1 and species2 change, region-by-region over the length of Aβ in a manner that permits a simple internal comparison as a ratio of species abundances.
We obtained full sequence coverage of each Aβ42 isoforms, with peptides spanning 1-9 (2+), 1-19 (2+), 20-34 (2+) and 35-42 (1+) peptic peptides. Subtle differences are seen in some of the peptic peptides obtained for the Iowa (D23N) mutant, encompassing peptides 11-19 (2+) and 21-34 (2+). This fortunate outcome indicates that pepsin, a widely-used, marginally specific proteolytic enzyme44 cleaves Aβ42 and its variants nearly the same.
Pulsed HDX reveals three distinct aggregating forms of Aβ isoforms.
The point mutations discussed here are situated in two key regions near salt bridges (amino acids 21, 22 and 23) or at the N-terminus (amino acids 6 and 7). These regions are responsible for vital electrostatic intra- and intermolecular interactions (Fig. S1) where the several substitutions affect aggregation rates. The rates can be categorized into three groups: fast, intermediate, and slow.
The aggregation of FAD mutants is influenced by local electrostatic repulsions, global changes in net charge, and other structural determinants45 (e.g., alterations in salt bridge formations and long-range interactions of the native structure46–48). The Arctic (E22G), Iowa (D23N), Dutch (E22Q) and Japanese (D7N) mutations undergo a shift of net charge from −3 to −2 (one negatively charged residue is substituted for a neutral one), decreasing electrostatic repulsion and increasing aggregation. Moreover, the Italian (E22K) mutant’s net charge changes from −3 to −1 owing to substitution of a negative charge on E with a positive charge on K, further reducing electrostatic repulsions and accelerating aggregation of this variant to be the fastest of all18. Clearly, alterations in electrostatic repulsion account in part for increased aggregation23,24, and they may offer an opportunity for inhibitor design.
Alterations clustering in the turn region at residues 21-23 (e.g., Arctic (E22G), Iowa (D23N), Italian (E22K), Dutch (E22Q), Flemish (A21G), and Osaka (E22Δ) variants) change the rates of aggregation48. In agreement with this notion, MD simulations of the wild type and of mutant monomers49 show that for Arctic (E22K) and Dutch (E22Q), the peptide’s α-helicity in the region of 20-23 is modified, whereas for Dutch (E22Q), Arctic (E22G) and Iowa (D23N), the helicity of the 33-36 region of the monomer is reduced. It is important to consider local conformational changes, exemplified by the Flemish (A21G) mutant, which aggregates slower because salt-bridge formation with N-terminal R5 is significantly reduced, resulting in increased solvent accessibility47. Notably, the English (H6R) and Japanese or Tottori (D7N) isoforms whose mutations reside in the disordered region of Aβ42 must influence the electrostatic interactions of the N-terminus. All atom MD simulations show that the English (H6R) variant has a reduced negative charge, weakening interactions of side chains that participate in self-assembly48. Our work delineates differences that occur in several regions of the isoforms and assesses the overall differences in the aggregation kinetics of FAD mutants.
Fast aggregators.
Changes in electrostatics reduce the charge of Aβ42 to promote enhanced aggregation. Five variants that have reduced negative net charge with respect to the wild type have less intermolecular electrostatic repulsion than the wild type. Three of these five, Arctic (E22G), Iowa (D23N) and Italian (E22K), behave similarly and undergo fast aggregation without a visible lag phase (Fig. 1A, B and C), although experiments at a higher time resolution23 suggest that the lag time may be simply too short to be detected in our measurements rather than completely absent.
Figure 1.
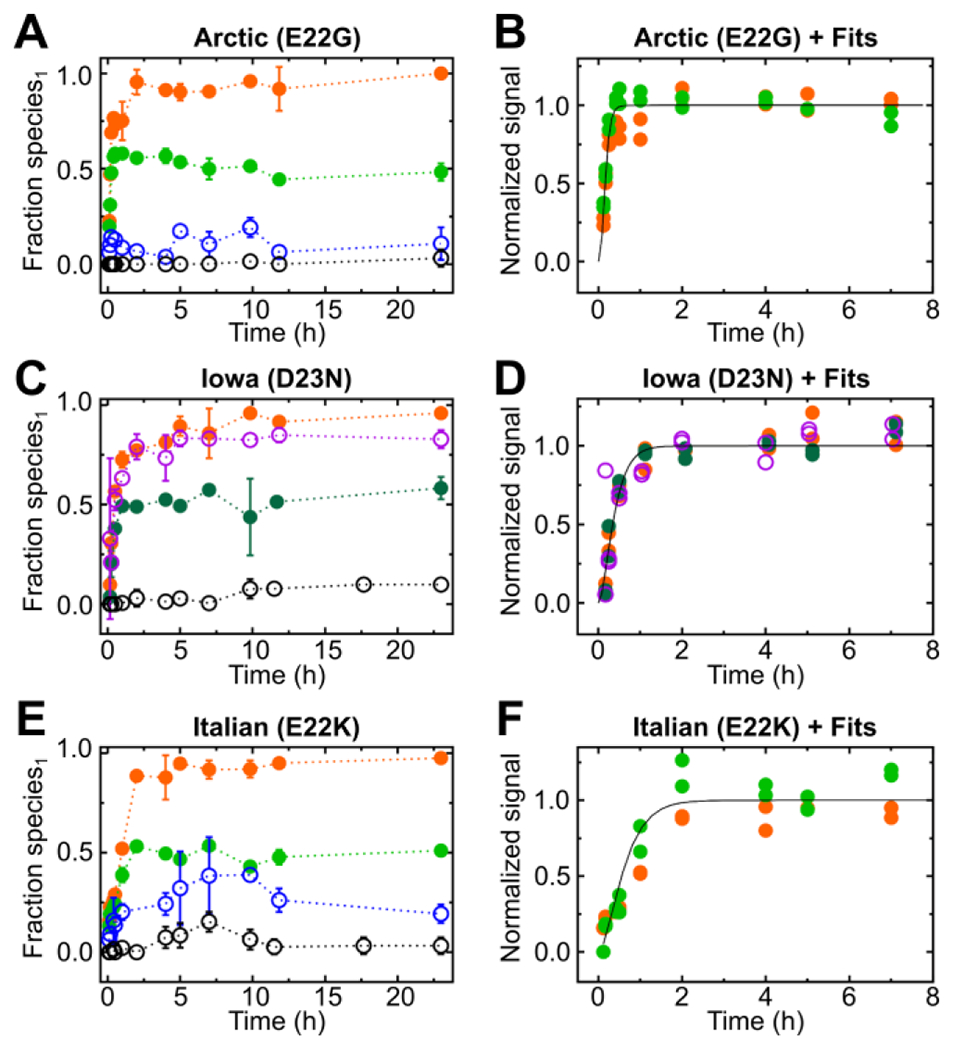
Fractional species1 from pulsed HDX-MS for different regions of Aβ peptic peptides and their kinetic fits for each peptide showing a plateau in HDX, respectively. Time-dependent fractional species1 from peptic peptides following HDX-MS for the Arctic (E22G) mutant (A) and kinetic fits for normalized fractional species1 (protected species) that showed a plateau phase by HDX-MS (B). HDX-MS data showing fractional species1 of peptides obtained from the Iowa (D23N) mutant (C) and kinetic fits of those peptides for normalized fractional species1 that showed a plateau by HDX-MS (D). Fractional species1 of various HDX-MS peptides from the Italian (E22K) (E) mutant with corresponding kinetic fits for normalized fractional species1 (F) that exhibited a plateau in HDX-MS. The peptic peptides are color coded: 1-9 (2+) black open circle, 1-19 (2+) blue open circle, 20-34 (2+) green closed circle, 21-34 (2+) dark green closed circle, and 35-42 (1+) orange closed circle.
The aggregation introduces protection that is regionally dependent. For example, peptide 1-9 (+2) represents, for the three fast-aggregating isoforms, a largely disordered region that remains solvent-accessible during aggregation. In contrast, peptides 20-34 (+2) and 35-42 (+1) show a large gain in protection and are involved in aggregation presumably because the regions they represent span the hydrophobic core/salt bridge/hydrophobic patch50 motifs of Aβ42. For fitting the kinetic data for peptides that reached a clear plateau, the data were normalized individually to reveal that fits overlap for each mutant (Figs. 1D, E, and F), indicating that the structural changes in different regions of the peptide occur simultaneously.
Noteworthy are peptide 35-42 (1+), for all three mutants, and peptide 11-19 (2+) for the Iowa (D23N) mutant that become buried in aggregation, adopting a solvent-shielded state (fractional species1 ≈ 1.0) at late stages of aggregation. In contrast, fractional species2 (solvent accessible) approaches ~0.5 late in the aggregation for regions represented by peptides 20-34 (2+)/21-34 (2+). That the backbone hydrogens in these regions retain their solvent accessibility during assembly of protofibrils and fibrils may point to either a structure alternately pointing inward and outward or the presence of two distinct polymorphs.
Another key finding is that peptides 20-34 (2+)/21-34 (2+) and 35-42 (1+) follow the same kinetics when rescaled to their final plateau value. This strongly suggests that the reorganization of regions of Aβ represented by these peptides takes place simultaneously during self-assembly, on the timescale of our measurements, rather than consecutively as was previously proposed40.
Lazo et al.51 concluded that the so-called protease-resistant region (residues 21-30) harbors a nucleation core (residues 24-28) and becomes altered owing to introduction of missense mutations. The substitutions clustered in this region potentially destabilize or remodel long-range intramolecular Coulombic interactions of K28 with E22 and D23, and perturb hydrophobic interactions of V24 and K28 side chains of the Aβ monomer51, corroborating the enhanced aggregation of the Arctic (E22G), Iowa (D23N) and Italian (E22K) mutants. Further support comes from clear differences in HDX in the most aggregation-prone regions, 20-34 (2+)/21-34 (2+) and 35-42 (1+) (see SI for pulsed HDX-MS patterns of each mutant for peptides 20-34 (2+)/21-34 (2+) situated in an aggregation-prone hydrophobic core region (Fig. S11–18). The maximum fractional species1 occurs for peptides 20-34 (2+)/21-34 (2+) (Fig. S19, Table S1) and 35-42 (2+) (Fig. S20, Table S2) at the last aggregation time point taken for each mutant.
Intermediate aggregators
The Dutch (E22Q), Japanese (D7N) and wild type variants comprise this class and aggregate more slowly. For the Dutch (E22Q), the substitution of a negatively charged amino acid with an uncharged residue reduces electrostatic repulsions. As a result, the inflection point of the sharply increasing curves is pushed forward to ~ 7 h (Fig. 2A and B). Pulsed HDX-MS for aggregation of the wild type displayed a well-defined lag phase followed by a plateau phase (Fig. 2C and D), especially for regions 20-34 (+2) and 35-42 (+1). For the Japanese (D7N) variant, peptides 20-34 (2+) and 35-42 (1+) interestingly show a gradual increase of fractional species1 at the early time points without a clear lag phase (Fig. 2E and F), unlike the two peptic peptides representing regions nearer the N-terminus. Furthermore, the fractional species for region 1-19, as revealed by its (2+) peptide, is delayed (11.7 h) compared to all variants. Substitution of a negatively charged residue by an uncharged group near the N-terminus likely remodels some of the interactions. Both mutants reached a marginally higher fractional species1 than the wild type in the hydrophobic core regions of 20-34 (2+) (Fig. S19, Table S1) and 35-42 (1+) (Fig. S20, Table S2).
Figure 2.
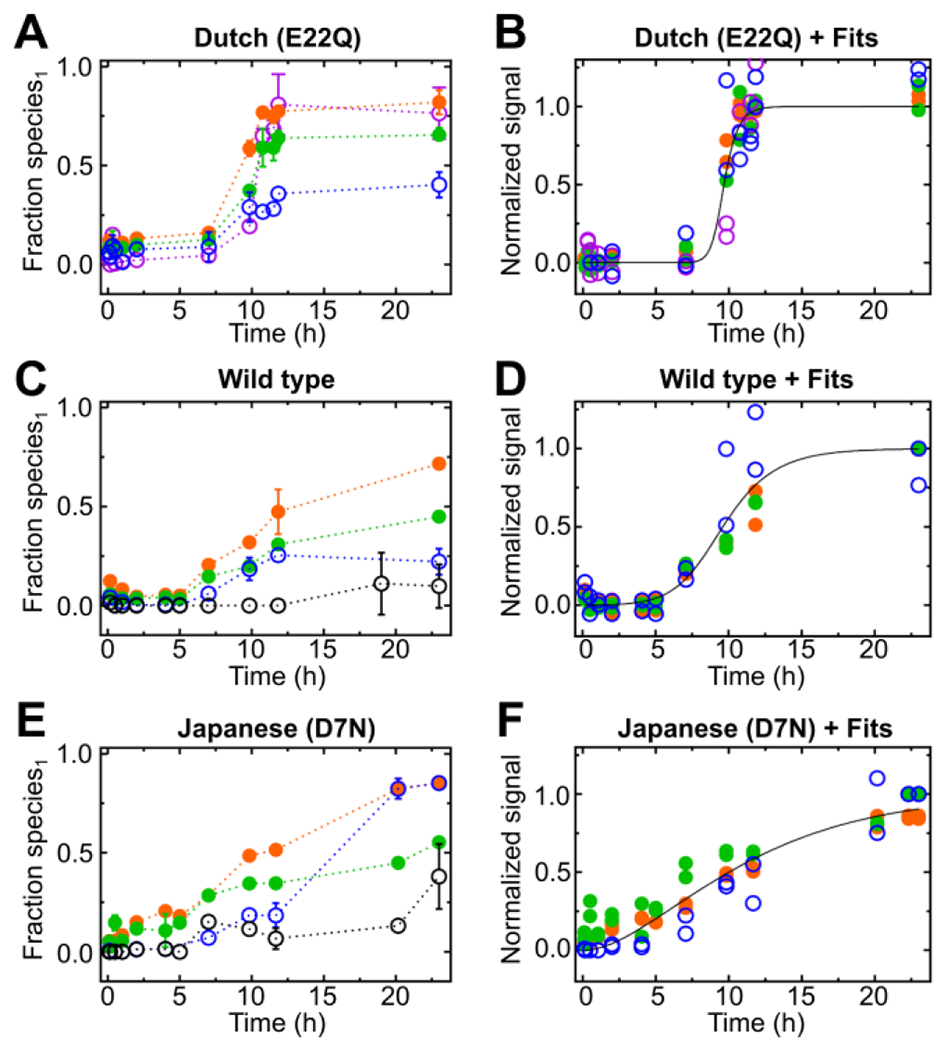
Fractional species1 taken from pulsed HDX-MS for various regions of Aβ peptic peptides and their kinetic fits for each peptide showing a plateau in HDX, respectively. HDX-MS peptide of the Dutch (E22Q) mutant (A) and their kinetic fits for normalized fractional species1 (protected species) that reached a plateau in the time-resolved HDX-MS (B), HDX-MS peptides of wild type Aβ42 (C) and corresponding kinetic fits for normalized fractional species1 (protected species), reaching a plateau (D) Fractional species1 of pulsed HDX-MS peptides obtained from the Japanese (D7N) mutant (E) and kinetic fits for normalized fractional species1 showing a plateau in the time-resolved HDX-MS (F). The peptic peptides are color coded: peptide 1-9 (2+) black open circle, peptide 1-19 (2+) blue open circle, peptide 11-19 (2+) purple open circle, peptide 20-34 (2+) green closed circle and peptide 35-42 (1+) orange closed circle.
Slow aggregators.
English (H6R) and Flemish (A21G) mutants comprise this class. The English mutant (H6R) undergoes aggregation, however, there are no clear inflections and more scatter than in the other mutants (Fig. 3A and B). The Flemish (A21G) mutant exhibits the slowest aggregation behavior of all, with an extended lag phase (Fig. 3C and D), in agreement with previous reports18,19,26 Although the sequence variation is modest, substitution of A for G can increase flexibility and slow assembly of an ordered region that is needed for the aggregate (see below). It was shown that the Flemish mutant has reduced protofibril formation and fibril elongation both by ThT26,52 and ion mobility MS53. For both the English and Flemish mutants, the corresponding maximum fractional species1 for peptides 20-34 (2+) and 35-41 (1+) are similar to or lower than that of the wild type (Fig. S19).
Figure 3.
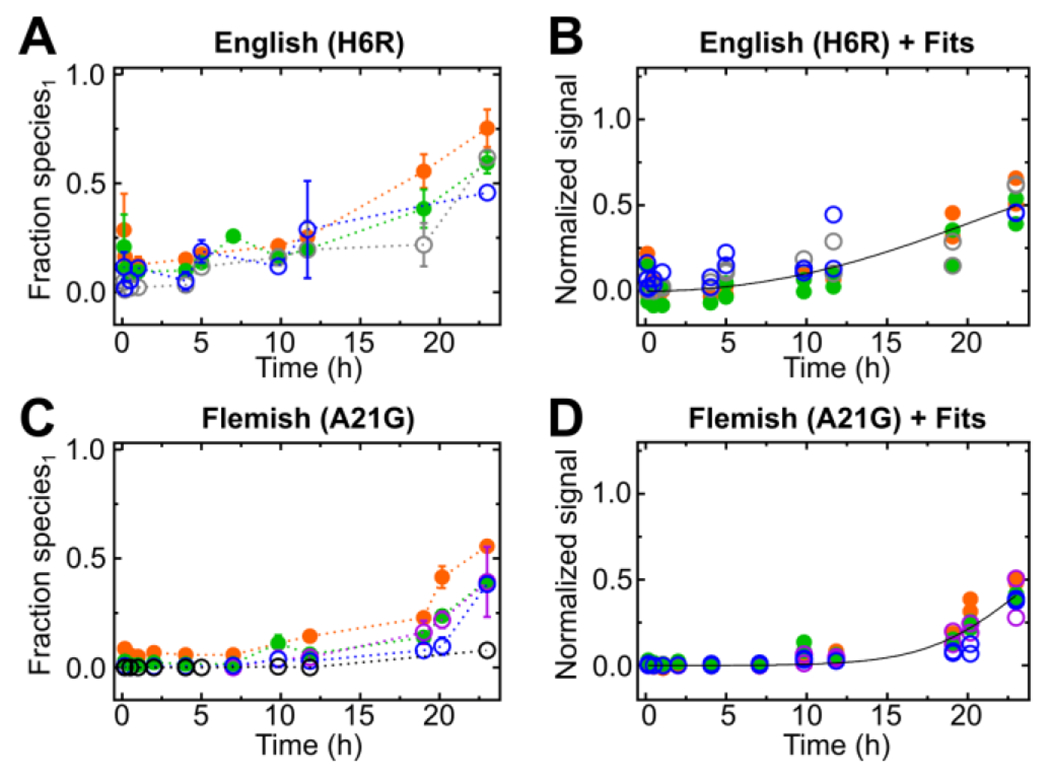
Fractional species1 of pulsed HDX-MS peptic peptides and kinetic fits of fractional species1 are shown respectively for the English (H6R) (A and B) and Flemish (A21G) (C and D) mutants as function of time. The peptic peptides are color coded: peptide 1-9 (2+) black open circle, peptide 1-19 (2+) blue open circle, peptide 4-19 (2+) gray open circle, peptide 20-34 (2+) green closed circle and peptide 35-42 (1+) orange closed circle.
Rates of aggregation.
We fit the data to kinetic equations, as detailed in the Methods and elsewhere39, to estimate the rates of aggregation. We used the model by Cohen et al.54 that described wild type aggregation. We did not explicitly include a saturation term for secondary nucleation, as was done in previous work23, because this term can only be accurately fit if the kinetics are measured over a range of monomer concentrations. Our analysis yields two effective rates: λ, denoting the rate at which new fibril mass is produced via elongation and primary nucleation, and κ, which describes the rate at which new fibril mass is produced via elongation and secondary nucleation55. These rates allow the comparison of models that include saturation with those that do not (i.e., data at a single monomer concentration) (see Meisl et al.39) (Fig. S21). The corresponding rates are given in Table 2.
Table 2.
Rates of the primary process, λ, and the secondary process, κ, as well as the critical seed concentration above which secondary processes become the main source of new aggregates. In the fast aggregating mutants, the lag phase is so short that an assumption about lambda had to be made, based on previously measured rates (starred entries in the table), to obtain sufficient constraints.
| name | mutation | lambda (λ) (s−1) | kappa (κ) (s−1) | crit. seed conc. (M) |
|---|---|---|---|---|
| Arctic | E22G | 7.47E-04* | 1.01E-02 | 1.36E-07 |
| Iowa | D23N | 3.59E-04* | 6.52E-03 | 7.56E-08 |
| Italian | E22K | 4.10E-04* | 2.09E-03 | 9.63E-07 |
| Dutch | E22Q | 4.35E-09 | 7.19E-04 | 9.17E-16 |
| English | H6R | 1.29E-05 | 4.07E-05 | 2.50E-06 |
| wild type | - | 1.06E-05 | 1.98E-04 | 7.10E-08 |
| Japanese | D7N | 4.57E-05 | 1.89E-05 | 1.46E-04 |
| Flemish | A21G | 2.19E-06 | 9.84E-05 | 1.24E-08 |
Although the conditions with respect to the use of synthetic Aβ42 versus recombinant source, buffer conditions, and agitated versus quiescent samples were different to those published before23, kinetic analyses of the fast aggregating mutants (i.e., Arctic (E22G), Iowa (D23N) and Italian (E22K)) are consistent with previously published values23. The rates of the Dutch (E22Q), the wild type and the Flemish (A21G) mutant calculated here were, however, lower than those reported in previous work by Linse and coauthors23. The preparation and handling conditions of Aβ42 are major determinants during the aggregation and can be responsible for such differences. In addition to λ and κ, we also calculated the concentration of seeds at which the number of nuclei produced by secondary nucleation exceeds that produced by primary (Table 2). This is micromolar or below for all mutants except the Japanese (D7N), suggesting that the kinetics for this latter variant are best described by a mechanism in which primary nucleation is dominant for the entire reaction, and secondary processes are not relevant. This is also evident in the Japanese (D7N) aggregation curve shapes that show a gradual increase over the entire time course, with no pronounced upward curvature.
Mechanism for aggregation.
From fitting the pulsed HDX results via modeling, we can propose that for primary nucleation, monomeric subunits first come together to form small aggregates (see Fig. 4 for a cartoon representation). These subunits elongate by addition of monomeric protein to their ends. As the number of fibrils increases, they also catalyze the nucleation of new fibrils by adding monomers on their surface in a secondary nucleation process. Although primary nucleation is the main source of new fibrils at the beginning of an unseeded reaction, secondary nucleation quickly takes over once fibrillar material starts to accumulate. Elongation and secondary nucleation couple together in a positive feedback loop that gives rise to the characteristic, initially exponential, increase in fibril mass.
Fig 4.
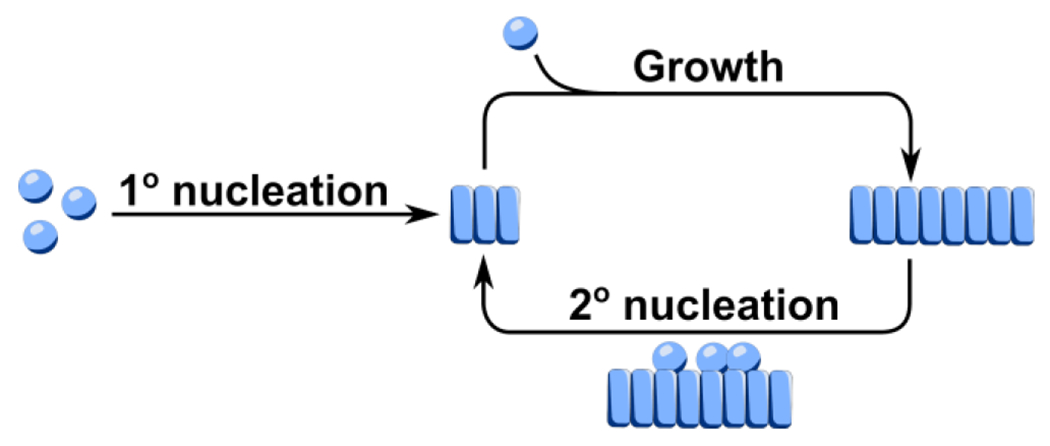
Schematic of the aggregation reaction. Monomers (spheres) self-assemble into initial aggregates (rectangles) via primary (1°) nucleation. Elongation proceeds by the addition of monomers to the end of aggregates. Nucleation of new fibrils can also occur by the assembly of monomers on the surface of existing fibrils in a secondary (2°) nucleation process. A positive feedback loop between elongation and secondary nucleation leads to the accelerated formation of fibril mass.
Morphological and Structural insights.
Considering first the morphology, we addressed this by negatively stained transmission electron microscopy for all the Aβ42 variants. They all displayed protofibrillar/fibrillar morphology at the end point of aggregation as seen in the electron microscopy images (Fig. S22). A more detailed or regional picture, however, emerges from a consideration of the pulsed HDX results.
On the basis of our HDX-MS data, peptide 1-9 (2+) remains nearly solvent accessible, with a low fractional species1 throughout the aggregation of the FAD mutants. This low protection indicates that the very N-terminus remains flexible and unstructured throughout aggregation. An exception is the Japanese (D7N) which gives a maximum fractional species1 ≈ 0.38. For this variant, the amino acid substitution near the N terminus changes a water-soluble carboxylate (Asp) to a less soluble Asn, decreasing its solvent accessibility during aggregation. The adjoining/overlapping peptides 4-19 (2+), 1-19 (2+) and 11-19 (+) showed a fractional species1 that increases with time, confirming that this variant became more structured during aggregation. Interestingly, both the Japanese (D7N) and English (H6R) mutations, harboring the missense mutation on the N-terminus, reached the highest fractional species1 for fragment 1-19 (2+) as compared to the other variants (Fig. 5 A).
Figure 5.
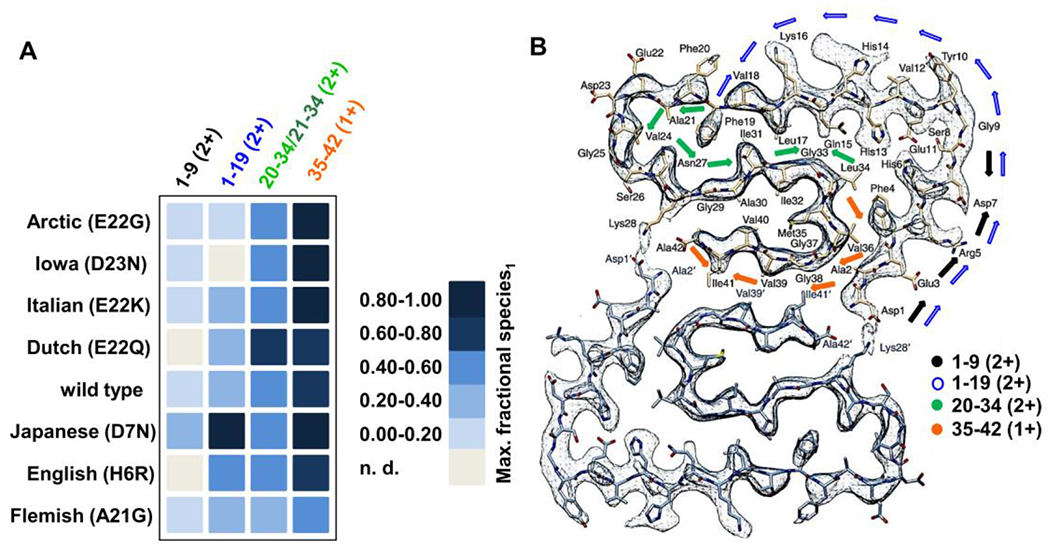
Maximum experimental fractional species1 reached within ~23 hr of aggregation for the main fragments. Abbreviation n.d. denotes that a particular peptide was not found (no data) (A). Cryo-EM image of a wild type Aβ42 fibril58 showing regions interrogated with peptic peptides and HDX. Arrows are color coded denoting the positions of the most commonly shared peptic peptides across the different mutants mapped onto the cryo-EM image (B) CryoEM structure of Aβ42 fibril (adapted and reprinted with permission from reference 58.
By way of contrast, significant growth in structure occurs upon aggregation for all variants in regions 20-34 (2+)/21-34 (2+) (Fig. S19, Table S1 and Fig. 5A) and particularly 35-42 (1+) (Fig. S20, Table S2 and Fig. 5A). For the mutants, the fractional species1 plateaued between 0.5-1.0 except for the Flemish (A21G) variant.
A key finding of this research is that only two fractional species terms are required to characterize the aggregation of Aβ42. All regions follow the same kinetics; that is, there are no regions in which protection maximizes significantly earlier than for other regions. Together, these results indicate that the population of species is not structurally diverse during the aggregation of any of the Aβ42 variants under our conditions. Although oligomeric species have been suggested as intermediates in aggregation and potential toxic agents in vivo, they either have the same structural features as all other small or large oligomers or, if different, exist at a level below our detection limit.
Furthermore, the variants that reach the highest fractional species1 (exceeding that of the wild type) for regions 20-34 and 35-42, as represented by a (1+) peptide, have a net charge of 0 or −1 (ignoring His protonation). These charge states represent loss of negative charge with respect to the wild type. Diminishment of charge should increase the aggregation propensity of the monomer. The central role of residues encompassing regions 20-34 and 35-42 of Aβ42 in amyloid fibril formation is consistent with other MS-based footprinting results56 and other higher resolution structural data57,58. These regions that become highly protected must constitute the structural cores of the aggregates, in which the shielded amide hydrogens participate in hydrogen bonding to form β-sheet-rich structures. It is important to note that these conclusions pertain to soluble species during aggregation, not to solid aggregates that are the end points of aggregation. Denaturation with 3 M urea 1% TFA during the quench step and a brief on-line pepsin digestion may introduce a bias toward resolving HDX signals derived from soluble intermediates. Inclusion of an internal standard addressing any potential loss of signal intensity over the course of aggregation, as shown for the curli protein Csg A36, would be highly beneficial in future studies of Aβ42.
Solid-state NMR of Aβ42 also reveals a very dynamic N-terminus with weak signals for amino acids 1-10 and a highly organized, rigid C-terminus. The wild type atomic fibril structure was characterized by a triple parallel β-sheet structure, consisting of three β-sheets formed by residues 12-18, 24-33 and 36-4059. A high-resolution cryo-electron microscopy (cryo-EM) structure of the wild type fibril58 enables an even more detailed structural interpretation of the HDX fractional species1. Wild type fibrils prepared under acidic conditions have a twisted protofibrillar architecture consisting of two filaments facing one another, each with a so-called “LS-shaped” assembly58 (Fig. 5B). Peptide 1-9 (2+) maps onto the L portion of this topology, a region with considerable solvent accessibility according to our HDX-MS data. Peptide 1-19 (2+) covers the entirety of the bent “L-shape” described as the “groove end” and transitions into the beginning of the bent “S-shape”. The turn region, residues 20-25, in the cryo-EM structure is where most of the FAD mutants are found. Regions 20-34 (2+)/21-34 (2+) span a large portion of this bend, a region that affords a fractional species1 ≈ 0.5 for most mutants at the longest times of aggregation. For a structure of this nature, it is plausible that almost half of the amide backbones remain exchangeable, maintaining solvent exposure even for advanced aggregation, whereas the other half becomes more shielded.
Moreover, in the cryo-EM topology, residues 27-33 form a “ridge end” with considerable organization. The staggered dimeric interface of growing protofibrils, centered on the S-shapes in a parallel, in-register cross-β structure suggested by the solid state cryo-EM data58 corroborate well the highly protected nature of the last, C-terminal peptic peptide. Nevertheless, alternative anti-parallel arrangements of the Iowa (D23N) Aβ4031 and Italian (E22K) Aβ4230 were reported by ssNMR, warranting research into the topology of the fibrils formed by the FAD mutants.
CONCLUSIONS
Disease-associated missense mutations of Aβ42 lead to strikingly different time-dependent aggregation as seen by pulsed HDX-MS of FAD linked variants and associated kinetic modeling. Despite differences in time of aggregation, the structural features of the soluble aggregates are similar. Remarkably, this highly complex and polydisperse aggregation can be characterized by only two fractional species manifested as bimodal isotopic distributions in the pulsed HDX and measured regionally over the sequence of Aβ42. A time-dependent fractional species1 indicates subtle variations between the mutants, allowing classification in three main groups. The Arctic (E22G), Iowa (D23N) and Italian (E22K) mutants aggregate rapidly, without a detectable lag phase under the conditions employed here. The Dutch (E22Q) mutant aggregates somewhat slower than the other point mutants that have the mutation at residue 22 and gives a sharper rise in protection than other members of this intermediate class. The N-terminal Japanese (D7N) mutant displayed a slowly increasing fractional species1 like the wild type’s, with a more pronounced increase in protection at later time points. The English (H6R) variant aggregates slowly, not reaching a plateau. The Flemish (A21G) mutant was distinctly different, showing the slowest aggregation kinetics, in agreement with previous literature18,19,23. For all mutants, regions 20-34/21-34 (2+) and 35-42 (1+) drive the aggregation, becoming solvent-inaccessible during the aggregation progresses and participating in building the core of solvent shielded cross-β structure.
There is a methodological advance underlying these biochemical conclusions. Previous biophysical studies addressed the inherent aggregation propensities of a subset of mutants focusing on both the shorter Aβ40 and more toxic Aβ42 forms18,19,53. More recently, important underlying principles were outlined about the aggregation kinetics of Aβ42 mutations in the vicinity of amino acids 21-2323 based on ThT assays that afford high throughput, utilize lower initial monomeric concentrations, and are amenable to seeding experiments. The limitations of fluorescent assays, however, are that they are unable to provide regional data and their interpretation often is accompanied by the uncertainty of whether addition of a probe perturbs structure. Gaussian fitting of global HDX-MS of wild type Aβ42, without peptic digestion, examined the solvent accessibility of wild type Aβ42 over an extended period up to 11 days. Up to three distinct structural features could be resolved by the later stages of aggregation60. The first one was characterized by little or no protection up to day one, from day 1 to day 5 the second feature became apparent with moderate protection and following day 9 to day 11 significant protection was observed. Cellular toxicity correlated with the presence of protected oligomers. High resolution methods (e.g. X-ray crystallography, ssNMR31,59, HDX NMR57, high field magic angle spinning NMR61 and cryo-EM58,58) provide insights on Aβ at the beginning and end of aggregation but at higher concentrations than employed here and with more effort. Bridging these techniques is pulsed HDX-MS that affords new insights at the peptide or regional level in solution and during the aggregation of Aβ42 mutants as a function of time.
The HDX data and its processing forecast new opportunities to probe the inhibitory effects of promising therapeutic interventions by antibodies or small molecules (natural products) that arrest or redirect aggregation or that “cap” toxic intermediates. Combined with top-down fragmentation (e.g., Electron Transfer or Capture Dissociation62) or with other protein footprinting56,63 may increase the spatial resolution even to the residue level. A wide variety of lipids and lipid rafts are found in the brain and its blood vessels providing a critical microenvironment for studying these hereditary mutants64–66 and their modulatory roles.
Moreover, many questions remain concerning the processing and release of Aβ isoforms, their immediate environment and regulated clearance. While some of the FAD mutations are associated with elevated levels of Aβ (e.g. Flemish (A21G)67), others show decreased secretion (e.g., Arctic (E22G), Italian (E22K) and Dutch (E22Q) besides the Osaka (E22Δ) and A2T mutations45). The Dutch (E22Q) has reduced clearance from the cerebrospinal fluid, and its transport from the central nervous system into blood is hindered as compared to wild type Aβ4068. Thus, the primary concentration of Aβ and the respective aggregation propensities alone do not define pathology, indicating that the complexity of disease etiology warrants interdisciplinary studies for successful therapeutic interventions wherein this methodology can play a role.
MATERIALS AND METHODS
To obtain characteristic peptide parameters shown in Table 1., fasta sequence files were used for each variant and input into ExPASy ProtParam41, outputs such as isoelectric point, number of positive and negative residues, grand average hydropathicity (GRAVY) were then tabulated for the summary table.
Aggregation assay.
Aliquots of 42 μL of 25 μM Aβ42 in phosphate buffered saline were set up in replicates in low binding Eppendorf tubes and incubated at 37 °C, at 200 rpm with continuous shaking in a BioShake (Scientific Innovations) benchtop shaker, sealed with parafilm to minimize evaporation. Aliquots were withdrawn at time points, centrifuged for 5 min at 14,000 x g in a benchtop centrifuge (Eppendorf 5418, Hamburg, Germany) at 4 °C, and the top-half and bottom-half of the solution were separated as previously described40. The bottom-half (20 μL) of the solution was exchanged with ice-cold D2O for a 1 min pulse on ice at 1:1 ( vol/vol), and quenched with 1:1.2 (vol/vol) 3 M urea and 1% TFA at pH 2.5, snap-frozen in liquid nitrogen, and stored at −80 °C for ≤ 2 days for HDX-MS analysis.
Peptide mapping and HDX-MS analysis.
On-line pepsin digestion and high-performance liquid chromatography (HPLC) separation were as previously described with minor modifications40. Protein samples were digested over a custom-packed online pepsin column. The resulting peptides were trapped and desalted on a C8 trap column (Zorbax XDB, Agilent Inc., Santa Clara, CA). Separation of the peptide mixtures was on a 1.9 μM reversed–phase C18 column (Hypersil Gold Thermo Fisher Scientific, Waltham, MA) with a 9.5 min linear gradient at 100 μL/min flow rate. Solvent A was water with 0.1% formic acid, and solvent B was 80% acetonitrile with 0.1% formic acid. The gradient was stepped for elution and re-equilibration: 0.0 min 95% A and 5% B to 0.3 min 85% A and 15% B, ramped to 5.5 min 50% A and 50% B, from 6.0 min 100% B held to 7.5 min, and from 7.6 min 95% A and 5% B held to 9.5 min. All experiments were performed in duplicate. A minimum of two blanks were injected after each duplicate to reduce carry-over.
MS analysis of the eluted peptides was with a linear trap quadrupole Fourier transform ion cyclotron resonance LTQ-FTICR (Thermo Scientific, Santa Clara, CA) in the positive-ion ESI mode. The ESI spray voltage was 5 kV, temperature 275 °C, capillary voltage 38 V, sheath gas flow rate 10 L/min, and nebulizer gas flow rate 8 L/min.
Identification of peptic peptides was achieved by submitting undeuterated samples to collision-induced dissociation (CID) on the same LTQ FTICR instrument. Product-ion spectra were then submitted to MassMatrix (version 2.4.2)69 for identification. HDX data were inspected and processed with MathCAD (v14, Parametric Technology Corp., MA). Each peptide spectral isotopic pattern was fitted with a mixture of two binomial distributions, one for each of the two components (modes), which have been labeled as species1 and species2. The binomial distributions’ order was the number of peptide residues, proline not counted, minus two. For illustration, four fits are shown in Fig. S23.
Kinetic Modeling.
The data were normalized and fit by a model including primary nucleation, secondary nucleation, and elongation, following the procedure detailed in Meisl et al.39 The reaction orders of primary and secondary nucleation were both assumed to be 2, based on what was found for the wild type protein by ThT kinetics. These values cannot be constrained because the present dataset does not contain measurements at different concentrations. Given that the concentration of protein in our measurements was considerably higher than in ThT experiments, it was likely that saturation effects in secondary nucleation and possibly elongation would lead to a lower effective reaction order. Therefore, rates, λ and κ, rather than rate constants were used as these can be compared across systems without specific knowledge of the reaction orders.
Supplementary Material
Fig. S1. FAD associated mutations shown on pdb structure of Aβ42
Fig. S2. Mass spectrum of the Arctic (E22G) mutant under denaturing conditions
Fig. S3. Mass spectrum of the Iowa (D23N) mutant
Fig. S4. Mass spectrum of the Italian (E22K) mutant
Fig. S5. Mass spectrum of the Dutch (E22Q) mutant
Fig. S6. Mass spectrum of wild type Aβ42
Fig. S7. Mass spectrum of the Japanese mutant (D7N)
Fig. S8. Mass spectrum of the English mutant (H6R)
Fig. S9. Mass spectrum of the Flemish mutant (A21G)
Fig. S10. Examples for the development of fractional species1 and species2
Fig. S11. Pulsed HDX-MS exchange pattern of the 20-34 peptide of the Arctic (E22G) mutant
Fig. S12. Pulsed HDX-MS exchange pattern of the of the 21-34 peptide of the Iowa (D23N) mutant
Fig. S13. Pulsed HDX-MS exchange pattern of the 20-34 peptide of the Italian (E22K) mutant
Fig. S14. Pulsed HDX-MS exchange pattern of the 20-34 peptide of the Dutch (E22Q) mutant
Fig. S15. Pulsed HDX-MS exchange pattern of the 20-34 peptide of the wild type Aβ42
Fig. S16. Pulsed HDX-MS exchange pattern of the 20-34 peptide of the Japanese (D7N) variant
Fig. S17. Pulsed HDX-MS exchange pattern of the 20-34 peptide of the English (H6R) mutant
Fig. S18. Pulsed HDX-MS exchange pattern of the 20-34 peptide of the Flemish (A21G) mutant
Fig. S19. Maximum fractional species1 for peptides 20-34 (2+)/21-34 (2+) for each mutant
Table S1. Values of one-way Anova with Sidak’s post-hoc test for peptides 20-34/21-34
Fig. S20. Maximum fractional species1 for peptide 35-42 (1+) for each variant
Table S2. Values of one-way Anova with Sidak’s post-hoc test for peptides 35-42
Fig. S21. Rates of the primary process, λ, plotted against rates of the secondary process, κ
Fig. S22. Transmission electron microscopy images of the different Aβ42 mutants
Fig. S23. Example intensity patterns of mass spectrum isotopic profiles
Acknowledgements
We thank Dr Henry Rohrs for help with HDX-MS instrumentation. We acknowledge the Nano Research Facility at Washington University in St Louis for access to TEM. We thank the NIH (Grants P41GM103422 R24GM136766), BrightFocus (A201427S), and Ramon Jenkins Research Fellowship, Sidney Sussex College, Cambridge (GM) for funding.
Footnotes
Conflicts of interest
M.L.G. is on the scientific advisory boards of Protein Metrics, Inc and Gen Next, two companies involved in structural mass spectrometry.
References
- 1.Skaper SD; Facci L; Zusso M; Giusti P An Inflammation-Centric View of Neurological Disease: Beyond the Neuron. Front. Cell. Neurosci 2018, 12, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Strooper B; Karran E The Cellular Phase of Alzheimer’s Disease. Cell 2016, 164, 603–615. [DOI] [PubMed] [Google Scholar]
- 3.Levy E; Carman MD; Fernandez-Madrid IJ; Power MD; Lieberburg I; van Duinen SG; Bots GT; Luyendijk W; Frangione B Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 1990, 248, 1124–1126. [DOI] [PubMed] [Google Scholar]
- 4.Hendriks L; van Duijn CM; Cras P; Cruts M; Van Hul W; van Harskamp F; Warren A; McInnis MG; Antonarakis SE; Martin JJ Presenile dementia and cerebral haemorrhage linked to a mutation at codon 692 of the beta-amyloid precursor protein gene. Nat. Genet 1992, 1, 218–221. [DOI] [PubMed] [Google Scholar]
- 5.Kamino K; Orr HT; Payami H; Wijsman EM; Alonso ME; Pulst SM; Anderson L; O’dahl S; Nemens E; White JA Linkage and mutational analysis of familial Alzheimer disease kindreds for the APP gene region. Am. J. Hum. Genet 1992, 51, 998–1014. [PMC free article] [PubMed] [Google Scholar]
- 6.Grabowski TJ; Cho HS; Vonsattel JP; Rebeck GW; Greenberg SM Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. Ann. Neurol 2001, 49, 697–705. [DOI] [PubMed] [Google Scholar]
- 7.Janssen JC; Beck JA; Campbell TA; Dickinson A; Fox NC; Harvey RJ; Houlden H; Rossor MN; Collinge J Early onset familial Alzheimer’s disease: Mutation frequency in 31 families. Neurology 2003, 60, 235–239. [DOI] [PubMed] [Google Scholar]
- 8.Wakutani Y; Watanabe K; Adachi Y; Wada-Isoe K; Urakami K; Ninomiya H; Saido TC; Hashimoto T; Iwatsubo T; Nakashima K Novel amyloid precursor protein gene missense mutation (D678N) in probable familial Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1039–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Obici L; Demarchi A; de Rosa G; Bellotti V; Marciano S; Donadei S; Arbustini E; Palladini G; Diegoli M; Genovese E, et al. A novel AbetaPP mutation exclusively associated with cerebral amyloid angiopathy. Ann. Neurol 2005, 58, 639–644. [DOI] [PubMed] [Google Scholar]
- 10.Tomiyama T; Nagata T; Shimada H; Teraoka R; Fukushima A; Kanemitsu H; Takuma H; Kuwano R; Imagawa M; Ataka S, et al. A new amyloid beta variant favoring oligomerization in Alzheimer’s-type dementia. Ann. Neurol 2008, 63, 377–387. [DOI] [PubMed] [Google Scholar]
- 11.Bugiani O; Giaccone G; Rossi G; Mangieri M; Capobianco R; Morbin M; Mazzoleni G; Cupidi C; Marcon G; Giovagnoli A, et al. Hereditary cerebral hemorrhage with amyloidosis associated with the E693K mutation of APP. Arch. Neurol 2010, 67, 987–995. [DOI] [PubMed] [Google Scholar]
- 12.Zhou L; Brouwers N; Benilova I; Vandersteen A; Mercken M; Van Laere K; Van Damme P; Demedts D; Van Leuven F; Sleegers K, et al. Amyloid precursor protein mutation E682K at the alternative beta-secretase cleavage beta’-site increases Abeta generation. EMBO Mol. Med 2011, 3, 291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaden D; Harmeier A; Weise C; Munter LM; Althoff V; Rost BR; Hildebrand PW; Schmitz D; Schaefer M; Lurz R, et al. Novel APP/Abeta mutation K16N produces highly toxic heteromeric Abeta oligomers. EMBO Mol. Med 2012, 4, 647–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lan MY; Liu JS; Wu YS; Peng CH; Chang YY A novel APP mutation (D678H) in a Taiwanese patient exhibiting dementia and cerebral microvasculopathy. J. Clin. Neurosci 2014, 21, 513–515. [DOI] [PubMed] [Google Scholar]
- 15.Mullan M; Crawford F; Axelman K; Houlden H; Lilius L; Winblad B; Lannfelt L A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of beta-amyloid. Nat. Genet 1992, 1, 345–347. [DOI] [PubMed] [Google Scholar]
- 16.Di Fede G; Giaccone G; Tagliavini F Hereditary and sporadic beta-amyloidoses. Front. Biosci. (Landmark Ed) 2013, 18, 1202–1226. [DOI] [PubMed] [Google Scholar]
- 17.Kumar-Singh S; Julliams A; Nuydens R; Ceuterick C; Labeur C; Serneels S; Vennekensh K; Van Osta P; Geerts H; De Strooper B, et al. In vitro studies of Flemish, Dutch and wild-type beta-amyloid provide evidence for two-stage neurotoxicity. Neurobiol. Dis 2002, 11, 330–340. [DOI] [PubMed] [Google Scholar]
- 18.Murakami K; Irie K; Morimoto A; Ohigashi A; Shindo M; Nagao M; Shimizu T; Shirasawa T Neurotoxicity and physicochemical properties of Abeta mutant peptides from cerebral amyloid angiopathy: implication for the pathogenesis of cerebral amyloid angiopathy and Alzheimer’s disease. Journal of Biological Chemistry 2003, 278, 46179–46187. [DOI] [PubMed] [Google Scholar]
- 19.Betts V; Leissring MA; Dolios G; Wang R; Selkoe DJ; Walsh DM Aggregation and catabolism of disease-associated intra-Aβ mutations: reduced proteolysis of AβA21G by neprilysin. Neurobiology of disease 2008, 31, 442–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vandersteen A; Hubin E; Sarroukh R; De Baets G; Schymkowitz J; Rousseau F; Subramaniam V; Raussens V; Wenschuh H; Wildemann D, et al. A comparative analysis of the aggregation behavior of amyloid-β peptide variants. FEBS Lett. 2012, 586, 4088–4093. [DOI] [PubMed] [Google Scholar]
- 21.Clements A; Allsop D; Walsh DM; Williams CH Aggregation and metal-binding properties of mutant forms of the amyloid A beta peptide of Alzheimer’s disease. J. Neurochem 1996, 66, 740–747. [DOI] [PubMed] [Google Scholar]
- 22.Meisl G; Yang X; Hellstrand E; Frohm B; Kirkegaard JB; Cohen SI; Dobson CM; Linse S; Knowles TP Differences in nucleation behavior underlie the contrasting aggregation kinetics of the Abeta40 and Abeta42 peptides. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 9384–9389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang X; Meisl G; Frohm B; Thulin E; Knowles TPJ; Linse S On the role of side chain size and charge in the aggregation of Abeta42 with familial mutations. Proc. Natl. Acad. Sci. U. S. A 2018, 115, E5849–E5858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meisl G; Yang X; Dobson CM; Linse S; Knowles TPJ Modulation of electrostatic interactions to reveal a reaction network unifying the aggregation behaviour of the Abeta42 peptide and its variants. Chem. Sci 2017, 8, 4352–4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meisl G; Yang X; Frohm B; Knowles TPJ; Linse S Quantitative analysis of intrinsic and extrinsic factors in the aggregation mechanism of Alzheimer-associated Aβ-peptide. Sci. Rep 2016, 6, 18728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hatami A; Monjazeb S; Milton S; Glabe CG Familial Alzheimer’s Disease Mutations within the Amyloid Precursor Protein Alter the Aggregation and Conformation of the Amyloid-β Peptide. J. Biol. Chem 2017, 292, 3172–3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Basun H; Bogdanovic N; Ingelsson M; Almkvist O; Näslund J; Axelman K; Bird TD; Nochlin D; Schellenberg GD; Wahlund LO, et al. Clinical and neuropathological features of the arctic APP gene mutation causing early-onset Alzheimer disease. Arch. Neurol 2008, 65, 499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Natté R; Maat-Schieman ML; Haan J; Bornebroek M; Roos RA; van Duinen SG Dementia in hereditary cerebral hemorrhage with amyloidosis-Dutch type is associated with cerebral amyloid angiopathy but is independent of plaques and neurofibrillary tangles. Ann. Neurol 2001, 50, 765–772. [DOI] [PubMed] [Google Scholar]
- 29.Brooks WS; Kwok JB; Halliday GM; Godbolt AK; Rossor MN; Creasey H; Jones AO; Schofield PR Hemorrhage is uncommon in new Alzheimer family with Flemish amyloid precursor protein mutation. Neurology 2004, 63, 1613–1617. [DOI] [PubMed] [Google Scholar]
- 30.Hubin E; Deroo S; Kaminski C; Serpell L; Subramaniam V; van Nuland N; Broersen K; Raussens V; Saroukh R Two distinct beta-sheet structure in Italian-mutant amyloid-beta fibrils: A potential link to different clinical phenotypes. Cell. Mol. Life Sci 2015, 72, 4899–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qiang W; Yau WM; Luo Y; Mattson MP; Tycko R Antiparallel β-sheet architecture in Iowa-mutant β-amyloid fibrils. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 4443–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alred EJ; Scheele EG; Berhanu WM; Hansmann UH Stability of Iowa mutant and wildtype Aβ-peptide aggregates. Journal of Chemical Physics 2014, 141, 175101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu F; Fu Z; Dass S; Kotarba AE; Davis J; Smith SO; Van Nostrand WE Cerebral vascular amyloid seeds drive amyloid beta-protein fibril assembly with a distinct anti-parallel structure. Nat. Commun 2016, 7, 13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singh J; Sabareesan AT; Mathew MK; Udgaonkar JB Development of the structural core and of conformational heterogeneity during the conversion of oligomers of the mouse prion protein to worm-like amyloid fibrils. J. Mol. Biol 2012, 423, 217–231. [DOI] [PubMed] [Google Scholar]
- 35.Paslawski W; Mysling S; Thomsen K; Jorgensen TJ; Otzen DE Co-existence of two different alpha-synuclein oligomers with different core structures determined by hydrogen/deuterium exchange mass spectrometry. Angew. Chem. Int. Ed Engl 2014, 53, 7560–7563. [DOI] [PubMed] [Google Scholar]
- 36.Wang H; Shu Q; Rempel DL; Frieden C; Gross ML Understanding curli amyloid-protein aggregation by hydrogen–deuterium exchange and mass spectrometry. International Journal of Mass Spectrometry 2016, 420, 16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Illes-Toth E; Rempel DL; Gross ML Pulsed Hydrogen-Deuterium Exchange Illuminates the Aggregation Kinetics of alpha-Synuclein, the Causative Agent for Parkinson’s Disease. ACS Chem. Neurosci 2018, 9, 1469–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ferraro DM; Lazo N; Robertson AD EX1 hydrogen exchange and protein folding. Biochemistry 2004, 43, 587–594. [DOI] [PubMed] [Google Scholar]
- 39.Meisl G; Kirkegaard JB; Arosio P; Michaels TC; Vendruscolo M; Dobson CM; Linse S; Knowles TP Molecular mechanisms of protein aggregation from global fitting of kinetic models. Nat. Protoc 2016, 11, 252–272. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Y; Rempel DL; Zhang J; Sharma AK; Mirica LM; Gross ML Pulsed hydrogen-deuterium exchange mass spectrometry probes conformational changes in amyloid beta (Abeta) peptide aggregation. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 14604–14609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gasteiger E; Hoogland C; Gattiker A; Duvaud S; Wilkins MR; Appel RD; Bairoch A Protein identification and Analysis on the ExPASy Server; The Proteomics Protocol Handbook. In 2005; pp 571–607. [Google Scholar]
- 42.Cho H; Sharma AK; Zhang Y; Gross ML; Mirica LM A Multifunctional Chemical Agent as an Attenuator of Amyloid Burden and Neuroinflammation in Alzheimer’s Disease. ACS Chem. Neurosci 2020, 11, 1471–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ryan TM; Caine J; Mertens HDT; Kirby N; Nigro J; Breheney K; Waddington LJ; Streltsov VA; Curtain C; Masters CL, et al. Ammonium hydroxide treatment of Aβ produces an aggregate free solution suitable for biophysical and cell culture characterization Peer J. 2013, i:e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dunn BM Overview of pepsin-like aspartic peptidases. Curr. Protoc. Protein Sci 2001, Chapter 21, Unit 21.3. [DOI] [PubMed] [Google Scholar]
- 45.Attar A; Meral D; Urbanc B; Bitan G Assembly of Amyloid-Beta-Protein Variants Containing Familial Alzheimer’s Disease Linked Amino Acid Substitutions. Bio-Imaging 2013, 38, 429–442. [Google Scholar]
- 46.Krone MG; Baumketner A; Bernstein SL; Wyttenbach T; Lazo ND; Teplow DB; Bowers MT; Shea JE Effects of familial Alzheimer’s mutations on the folding nucleation of amyloid beta-protein. J. Mol. Biol 2008, 381, 221–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chong S; Jim J; Ham S Structural heterogeneity in familal Alzheimer’s disease mutants of amyloid-beta peptides. Mol. Biosyst 2013, 9, 997–1003. [DOI] [PubMed] [Google Scholar]
- 48.Viet MH; Nguyen PH; Derreumaux P; Li MS Effect of the English familial disease mutation (H6R) on the monomers and dimers of Abeta40 and Abeta42. ACS Chem. Neurosci. 2014, 5, 646–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin YS; Pande VS Effects of familial mutations on the monomeric structure of Abeta42. Biophys. J 2012, 103, L47–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Han X; He G Toward a Rational Design to Regulate beta-Amyloid Fibrillation for Alzheimer’s Disease Treatment. ACS Chem. Neurosci 2018, 9, 198–210. [DOI] [PubMed] [Google Scholar]
- 51.Lazo ND; Grant MA; Condron MC; Rigby AC; Teplow DB On the nucleation of amyloid beta-protein monomer folding. Protein Sci. 2005, 14, 1581–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murakami K; Irie K; Morimoto A; Ohigashi H; Shindo M; Nagao M; Shimizu T; Shirasawa T Synthesis, aggregation, neurotoxicity, and secondary structure of various A beta 1–42 mutants of familial Alzheimer’s disease at positions 21–23. Biochem. Biophys. Res. Commun 2002, 294, 5–10. [DOI] [PubMed] [Google Scholar]
- 53.Gessel MM; Bernstein S; Kemper M; Teplow DB; Bowers MT Familial Alzheimer’s disease mutations differentially alter amyloid beta-protein oligomerization. ACS Chem. Neurosci 2012, 3, 909–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cohen SI; Linse S; Luheshi LM; Hellstrand E; White DA; Rajah L; Otzen DE; Vendruscolo M; Dobson CM; Knowles TP Proliferation of amyloid-beta42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 9758–9763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cohen SIA; Vendruscolo M; Dobson CM; Knowles TPJ Nucleated polymerization with secondary pathways. II. Determination of self-consistent solutions to growth processes described by non-linear master equations. J. Chem. Phys 2011, 135, 065106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li KS; Rempel DL; Gross ML Conformational-Sensitive Fast Photochemical Oxidation of Proteins and Mass Spectrometry Characterize Amyloid Beta 1–42 Aggregation. J. Am. Chem. Soc 2016, 138, 12090–12098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Luhrs T; Ritter C; Adrian M; Riek-Loher D; Bohrmann B; Dobeli H; Schubert D; Riek R 3D structure of Alzheimer’s amyloid-beta(1–42) fibrils. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 17342–17347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gremer L; Scholzel D; Schenk C; Reinartz E; Labahn J; Ravelli RBG; Tusche M; Lopez-Iglesias C; Hoyer W; Heise H, et al. Fibril structure of amyloid-beta(1–42) by cryo-electron microscopy. Science 2017, 358, 116–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xiao Y; Ma B; McElheny D; Parthasarathy S; Long F; Hoshi M; Nussinov R; Ishii Y Aβ(1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nature Structural & Molecular Biology 2015, 22, 499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Serra-Vidal B; Pujadas L; Rossi D; Soriano E; Madurga S; Carulla N Hydrogen/deuterium exchange-protected oligomers populated during Aβ fibril formation correlate with neuronal cell death. ACS Chem. Biol 2014, 9, 2678–2685. [DOI] [PubMed] [Google Scholar]
- 61.Colvin MT; Silvers R; Ni QZ; Can TV; Sergeyev I; Rosay M; Donovan KJ; Michael B; Wall J; Linse S, et al. Atomic Resolution Structure of Monomorphic Abeta42 Amyloid Fibrils. J. Am. Chem. Soc 2016, 138, 9663–9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rand KD; Pringle SD; Morris M; Engen JR; Brown JM ETD in a traveling wave ion guide at tuned Z-spray ion source conditions allows for site-specific hydrogen/deuterium exchange measurements. J. Am. Soc. Mass Spectrom 2011, 22, 1784–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mendoza VL; Vachet RW Protein surface mapping using diethylpyrocarbonate with mass spectrometric detection. Anal. Chem 2008, 80, 2895–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yamamoto N; Hirabayashi Y; Amari M; Yamaguchi H; Romanov G; Van Nostrand WE; Yanagisawa K Assembly of hereditary amyloid beta-protein variants in the presence of favorable gangliosides. FEBS Lett. 2005, 579, 2185–2190. [DOI] [PubMed] [Google Scholar]
- 65.Yamamoto N; Van Nostrand WE; Yanagisawa K Further evidence of local ganglioside-dependent amyloid beta-protein assembly in brain. Neuroreport 2006, 17, 1735–1737. [DOI] [PubMed] [Google Scholar]
- 66.Pifer PM; Yates EA; Legleiter J Point mutations in Abeta result in the formation of distinct polymorphic aggregates in the presence of lipid bilayers. PLoS One 2011, 6, e16248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Van Nostrand WE; Melchor JP; Cho HS; Greenberg SM; Rebeck GW Pathogenic effects of D23N Iowa mutant amyloid beta-protein. Journal of Biological Chemistry 2001, 276, 32860–32866. [DOI] [PubMed] [Google Scholar]
- 68.Monro OR; Mackic JB; Yamada S; Segal MB; Ghiso J; Maurer C; Calero M; Frangione B; Zlokovic BV Substitution at codon 22 reduces clearance of Alzheimer’s amyloid-beta peptide from the cerebrospinal fluid and prevents its transport from the central nervous system into blood. Neurobiol. Aging 2002, 23, 405–412. [DOI] [PubMed] [Google Scholar]
- 69.Xu H; Freitas MA MassMatrix: a database search program for rapid characterization of proteins and peptides from tandem mass spectrometry data. Proteomics 2009, 9, 1548–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. FAD associated mutations shown on pdb structure of Aβ42
Fig. S2. Mass spectrum of the Arctic (E22G) mutant under denaturing conditions
Fig. S3. Mass spectrum of the Iowa (D23N) mutant
Fig. S4. Mass spectrum of the Italian (E22K) mutant
Fig. S5. Mass spectrum of the Dutch (E22Q) mutant
Fig. S6. Mass spectrum of wild type Aβ42
Fig. S7. Mass spectrum of the Japanese mutant (D7N)
Fig. S8. Mass spectrum of the English mutant (H6R)
Fig. S9. Mass spectrum of the Flemish mutant (A21G)
Fig. S10. Examples for the development of fractional species1 and species2
Fig. S11. Pulsed HDX-MS exchange pattern of the 20-34 peptide of the Arctic (E22G) mutant
Fig. S12. Pulsed HDX-MS exchange pattern of the of the 21-34 peptide of the Iowa (D23N) mutant
Fig. S13. Pulsed HDX-MS exchange pattern of the 20-34 peptide of the Italian (E22K) mutant
Fig. S14. Pulsed HDX-MS exchange pattern of the 20-34 peptide of the Dutch (E22Q) mutant
Fig. S15. Pulsed HDX-MS exchange pattern of the 20-34 peptide of the wild type Aβ42
Fig. S16. Pulsed HDX-MS exchange pattern of the 20-34 peptide of the Japanese (D7N) variant
Fig. S17. Pulsed HDX-MS exchange pattern of the 20-34 peptide of the English (H6R) mutant
Fig. S18. Pulsed HDX-MS exchange pattern of the 20-34 peptide of the Flemish (A21G) mutant
Fig. S19. Maximum fractional species1 for peptides 20-34 (2+)/21-34 (2+) for each mutant
Table S1. Values of one-way Anova with Sidak’s post-hoc test for peptides 20-34/21-34
Fig. S20. Maximum fractional species1 for peptide 35-42 (1+) for each variant
Table S2. Values of one-way Anova with Sidak’s post-hoc test for peptides 35-42
Fig. S21. Rates of the primary process, λ, plotted against rates of the secondary process, κ
Fig. S22. Transmission electron microscopy images of the different Aβ42 mutants
Fig. S23. Example intensity patterns of mass spectrum isotopic profiles