

Abstract
The genus Orthohantavirus (family Hantaviridae, order Bunyavirales) consists of numerous genetic and pathologically distinct viral species found within rodent and mammalian insectivore populations world-wide. Although reservoir hosts experience persistent asymptomatic infection, numerous rodent-borne orthohantaviruses cause severe disease when transmitted to humans, with case-fatality rates up to 40%. The first isolation of an orthohantavirus occurred in 1976 and, since then, the field has made significant progress in understanding the immune correlates of disease, viral interactions with the human innate immune response, and the immune kinetics of reservoir hosts. Much still remains elusive regarding the molecular mechanisms of orthohantavirus recognition by the innate immune response and viral antagonism within the reservoir host, however. This review provides a summary of the last 45 years of research into orthohantavirus interaction with the host innate immune response. This summary includes discussion of current knowledge involving human, non-reservoir rodent, and reservoir innate immune responses to viruses which cause hemorrhagic fever with renal syndrome and hantavirus cardio-pulmonary syndrome. Review of the literature concludes with a brief proposition for the development of novel tools needed to drive forward investigations into the molecular mechanisms of innate immune activation and consequences for disease outcomes in the various hosts for orthohantaviruses.
Keywords: hantavirus, viral immunity, RNA virus, zoonosis, viral hemorrhagic fever, hantavirus cardio-pulmonary syndrome, hemorrhagic fever with renal syndrome, interferon, viral reservoir
Introduction
Orthohantaviruses are a family of zoonotic RNA viruses within the order Bunyaviridae. Orthohantaviral viral genomes consist of 3 segments of negative-sense, single-stranded RNA, encoding four or five proteins. Although the prototypical hantavirus, Hantaan virus (HTNV), was only recently isolated and identified in 1976, Hemorrhagic Fever with Renal Syndrome (HFRS) had been described decades earlier in China and the Korean peninsula [1]. During the Korean War in the 1950s, more than 3000 American and Korean troops reportedly fell ill with kidney failure and shock, later attributed to HTNV[2, 3]. Orthohantaviruses have been identified in rodent and other mammalian insectivore populations throughout the world, many of which may be non-pathogenic in humans[4–6]. However, many rodent-borne orthohantaviruses do cause significant morbidity and mortality, up to 40% case-fatality rates in some cases, in human infection.
Within the orthohantavirus family, two unique groups emerge due to differences in geography and disease manifestation. New World (NW) orthohantaviruses, predominately found in wild rodent and insectivore populations throughout the Americas, cause Hantavirus Cardio-Pulmonary Syndrome (HCPS). These include Andes virus (ANDV), Sin Nombre virus (SNV), Prospect Hill virus (PHV) and New York 1 virus (NY-1V), among others. Old World (OW) orthohantaviruses, circulating in Europe, Russia and Southeast Asia, are responsible for HFRS. These include HTNV, Seoul virus (SEOV), Pumaala virus (PUUV), and Tula Virus (TULV). While both syndromes have unique tissue tropism for manifestation of acute disease (lung/heart for NW and kidney for OW), the mechanism of disease at each tissue site is remarkably similar. In general, hantavirus disease is characterized by strongly induced inflammatory signaling, vascular leakage, and thrombocytopenia. Infection in both rodent reservoir and non-reservoir humans primarily targets endothelial cells throughout the body[7]. Because endothelial infection is non-lytic, vascular damage and disease is thought to be primarily immune-mediated. This topic and current hypotheses regarding mechanisms of vascular dysfunction have been thoroughly reviewed previously [8–14].
Orthohantaviruses are transmitted horizontally within reservoir populations, likely through biting, fecal-oral, or other close-contact interactions [15, 16]. One of the most interesting facets of orthohantaviruses is that disease outcomes for infections are host-specific (Figure 1). The course of infection in reservoir hosts is largely asymptomatic, despite lifelong viral infection and at times high viral loads [17, 18]. In contrast, experimental infection of non-reservoir adult rodents, such as Mus musculus, results in an asymptomatic acute phase and rapid viral clearance, with one notable exception discussed later in this review. Phylogenetic clustering analysis suggests that orthohantavirus species have co-evolved closely with their reservoir host, although evidence of host-switching has also been found recently [19–23]. This long evolutionary relationship may explain how orthohantaviruses can persistently infect their reservoir hosts without overt signs of disease. The role of the innate immune response in disease outcome is largely unclear and the impetus for this review.
Figure 1.
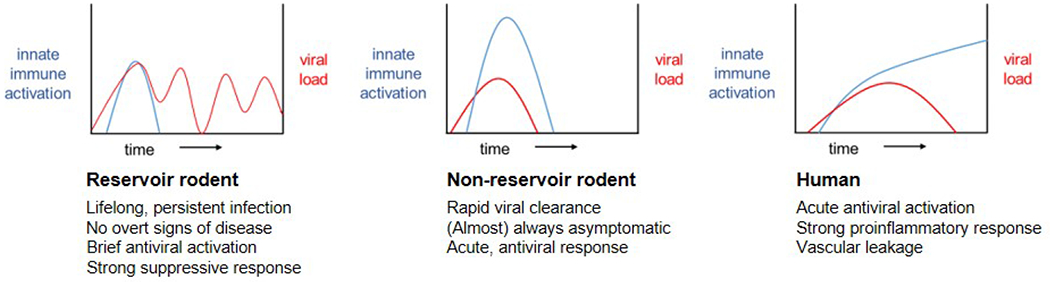
Schematic representation infection dynamics for orthohantavirus in various hosts.
Innate immunity: first responders
At the outset of a viral infection, recognition by the invaded cell is paramount to triggering a swift and decisive counter offense, preventing the virus from spreading throughout the tissue and systemically. Generally, the term innate immunity encompasses effector molecules such as α-defensins expressed at basal levels, ready to interact with invading pathogens, all the way up to innate immune cells such as neutrophils, eosinophils and natural killer cells. The central factor differentiating innate immunity from adaptive immune responses is the intrinsic nature of innate immunity, relying on pre-existing factors that are present or induced in response to stimuli but do not change, or adapt, to specific environmental insults and provide a more generalist approach to counteract the invasion. Here, we will focus on cell intrinsic factors that interact directly with viral invaders or interact with recruited innate immune cells to the site of infection to initiate a broader, regional antiviral response.
An antiviral response begins with recognition of non-self pathogen associated molecular patterns (PAMPS). PAMPs are evolutionarily conserved molecular signatures presented during pathogen encounter that differ from the host, such as non-capped RNA, components of bacterial and fungal cell walls, dsRNA, and viral capsids. Cellular factors such as membrane-bound Toll-like receptors (TLR) and cytosolic RIG-I-like receptors (RLR) bind to PAMPs and initiate a signaling cascade through protein-protein interactions that ultimately results in transcriptional regulation of type I and type III interferons (IFN) as well as many antiviral genes under the control of interferon regulatory factors (IRF), IRF1, IRF3, or IRF7. IFN is produced and secreted from the infected cell. IFNs can signal in an autocrine fashion to drive further amplification of antiviral signaling in the infected cell or in a paracrine fashion to induce antiviral signaling in nearby cells to defend against viral spread. IFN signaling through canonical Jak/STAT activation pathways leads to the induction of thousands of genes associated with antiviral, proliferation, metabolism, and apoptotic pathways. All of this is comprehensively reviewed elsewhere, and likely in this special issue [24–27]. Figure 2 shows an abbreviated schematic of the signaling cascades downstream of TLRs and RLRs that have been reported to recognize viral antigens. Importantly, these molecular interactions at the first moment of viral invasion may be sufficient to eliminate most pathogens before a full infection is established. But in cases where the innate immune response is insufficient to clear a pathogen quickly, the innate response will recruit components of the cellular innate immune response and adaptive arms of immunity to program a larger systemic response. It is likely that cell intrinsic responses to viral infection define the type and magnitude of systemic responses and that these interactions may determine disease outcomes. For example, the unique transcriptional signatures and regulatory overlap of NF-κB and IRF3 in response to RLR and TLR stimulation suggest potential skewing of gene expression dependent on the types of signals received [28, 29]. Therefore, the mechanism of viral recognition and the feedback loop created within tissues may have profound effects on the progression of disease and outcome of viral infection. How these programmatic differences may explain host-specific disease outcomes for orthohantavirus infection is unknown and further discussed in the final section of this review.
Figure 2.
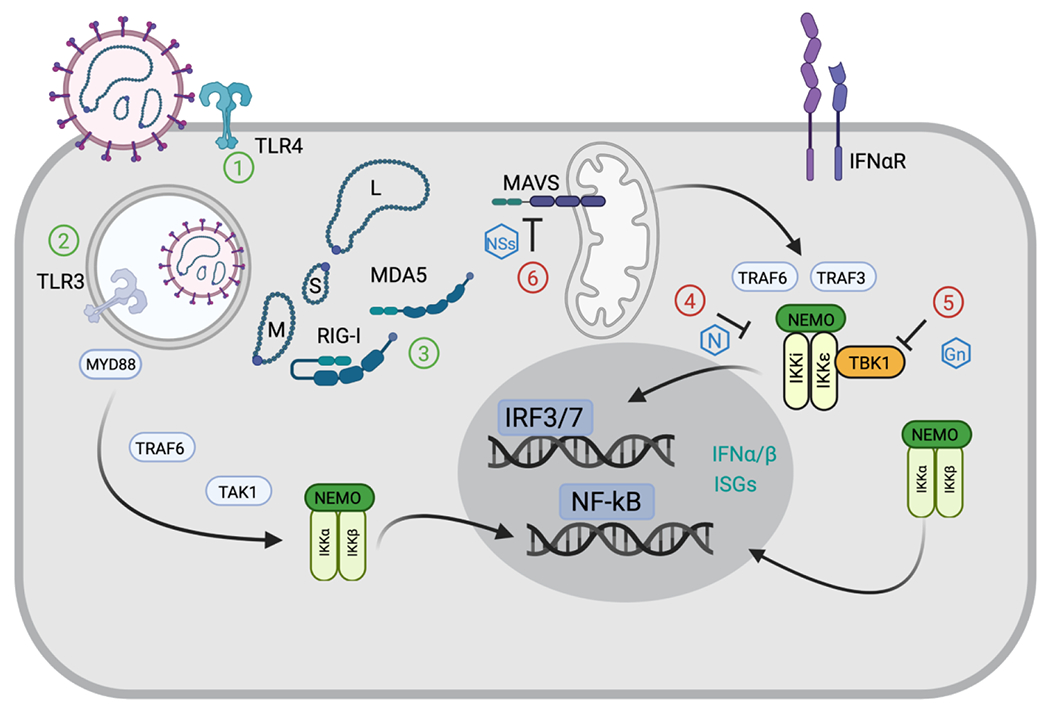
Summary of proposed molecular interactions between orthohantaviruses and human innate immune pathways in an infected endothelial cell. 1) Recognition of external viral products by TLR4 has been proposed[44]. 2) Viral entry through the endocytic pathway provides a potential mechanism for viral RNA recognition by TLR3[40]. 3) Activation of RIG-I and MDA5-dependent signaling has been reported, likely through recognition of cytoplasmic viral RNA species [31, 58]. Viral recognition and activation of each of these PRR leads to IRF3 and/or NF-κB transcription factor activation, with subsequent expression of type I IFN and ISGs to mount an antiviral defense. Antagonism of IRF3 activation downstream of dsRNA stimulation has been attributed to the nucleoprotein (4), the Gn protein (5), and the NSs (6) of certain orthohantaviruses [51, 62] [47, 63].
In this review, we will first focus predominately on the current knowledge of innate immune responses to orthohantaviruses in humans and then touch on our limited understanding in reservoir hosts. This review will conclude with a discussion on the tools needed to be developed for orthohantaviruses to better understand how innate immune responses drive disease outcomes.
Innate immunity in non-reservoir hosts:
Viral lifecycle exposes PAMPS/Innate immune recognition of orthohantaviruses
The process of viral infection and genome replication provides specific opportunities for host recognition and innate immune activation. Through investigations into the viral life cycle and host cell requirements for replication, the field has gained an understanding of specific vulnerabilities for recognition and mechanisms of antiviral defense to orthohantaviruses.
The biochemical nature of the orthohantavirus genome is thought to prevent genome recognition from the cytosolic recognition receptors RIG-I and MDA5[30]. Specifically, orthohantaviruses undergo 5’ terminus “trimming” to remove the 5’ppp motif from newly synthesized genomic RNA, likely to avoid detection by RIG-I. Further, very small levels of dsRNA have been detected in orthohantavirus-infected cells, suggesting they have developed a strategy to avoid triggering MDA5 activation and type I IFN induction. In spite of these attempts, decades of research suggests that orthohantavirus infection is sensed by the infected cell and does initiate an innate immune response in human cells [31–37]. Early work studying transcriptional upregulation of ISGs in HTNV infection demonstrated a strong induction of innate immune signaling in A549 type II alveolar epithelial cell line[33, 38, 39]. Subsequent studies have recapitulated these results in various cells including hepatocytes[40], astrocytes[41], macrophages[36, 42], endothelial cells[43, 44] [31, 38, 45, 46], and keratinocytes[32]. Activation of both IRF3 and NF-κB following infection with New World and Old World viruses has been observed [43, 44, 47–52].
To date, several host proteins have been proposed to be involved in orthohantavirus attachment and entry. The central receptor for orthohantaviruses in human endothelial cells has long been proposed to be β3-integrins [53, 54]. Recently, PCDH-1 was identified to be a required host factor for New World orthohantavirus entry into human pulmonary endothelial cells, but not required for Old World orthohantavirus entry [55]. Using CRISPR genetic knockouts in a human endothelial cell line, Dieterle et al. tested the reliance of New World and Old World orthohantaviruses on β3 integrin, β1 integrin, protocadherin-1 (PCDH1), and decay-accelerating factor (DAF) for cellular entry [56]. The authors confirm their previous observation that PCDH-1 is required for entry of New World orthohantaviruses, but found no significant decrease in entry of Old World viruses in the absence of any of the receptors examined. It has been postulated that Old World orthohantaviruses are internalized via clatherin-dependent endocytosis while New World orthohantaviruses may enter cells via dynamin-dependent uptake or micropinocytosis. This process was recently reviewed in depth[57]. It remains unclear however, if endosomal recognition pathways or receptor activation can trigger innate immune signaling during entry. Interestingly, it has been proposed that TLR4 may be involved in activation of NF-kB and IRF3 following HTNV infection of endothelial-like cells[44]. However, the mechanism of action and PAMP recognized by the outer cell membrane-bound TLR4 have yet to be defined.
Upon fusion of the viral and endosomal membranes, the viral genome is released into the cytosol. Each of the three negative sense single stranded genomic segments in the virion is adorned with a viral RNA-dependent RNA polymerase (L) and coated with viral nucleoprotein (N). It has been reported that TLR3, a PRR present in the endosome and responsive to dsRNA, is involved in initiating an antiviral response in human hepatocyte cell lines, but the mechanism allowing for viral RNA release into the endosome for TLR3 binding is unknown[40]. The subsequent steps which lead to viral uncoating, trafficking to sites of transcription and viral replication while evading the host innate immune response remain unclear. Growing evidence suggests an important role for the RLR in initiating a type I IFN response in human endothelial cells [31, 58]. Recent work from our lab demonstrates that ISG expression in HUVEC following HTNV infection was completely ablated in RIG-I and MDA5 double knockout cells. Interestingly, ISG expression was delayed in RIG-I−/− cells and unaffected in MDA5−/− cells, suggesting temporally specific yet redundant roles for each of the RLR in response to HTNV infection. However, the mechanism and viral motifs recognized by the RLR are still unknown. Interestingly, the long non-coding RNA NEAT1 was implicated in activating RIG-I during HTNV infection[58] and may play a role in innate immune signaling even if the viral RNA is not recognized directly by the RLR. Due to the limited nature and disparate results f\of these in vitro studies, further research should be pursued to better define the roles of each of these PRR for innate immune signaling and to determine the downstream consequences of specific transcription factor activation on antiviral functions and immune disease.
Viral antagonism of antiviral responses
Recently, investigations into differences between pathogenic and non-pathogenic orthohantavirus species has led to observations that ISG upregulation may be delayed in infections with pathogenic hantaviruses when compared to infections with non-disease-causing species, such as Prospect Hill virus (PHV) or Tula virus (TULV)[36, 43, 46, 59]. While the mechanisms are not clear, infection of human hepatocytes with the non-pathogenic PHV led to increase MxA expression and earlier activation of antiviral expression when compared with HTNV infection[40]. Likewise, this increased innate immune signaling led to decreased vial protein detection in these cells, suggesting that innate immune activation restricted viral replication. Similar studies performed in primary human endothelial cells demonstrated a reduction in IFN-beta production following ANDV infection compared to that produced in PHV infections[60]. Surprisingly, although Spiropoulou and colleagues observed decreased phosphorylation of STAT1/2 in ANDV infections compared to PHV, this effect was likely a result of reduced IFN-beta levels because both viruses were able to inhibit STAT1/2 activation in IFN-treated Vero cells. This robust and effective response for viral clearance has been postulated to explain the lack of observed human infections or disease with the putative non-pathogenic viruses. While in contrast, viral antagonism of host antiviral responses may explain the ability of other orthohantaviruses to establish productive infections.
Due to the strong pressure placed on viral replication by the RLR pathway and NF-kB/IRF3 activation, all viruses have evolved at least one method to antagonize these signaling cascades. Hantaviruses are no exception, although the mechanisms may differ between species and between pathogenic and non-pathogenic viruses. Using a molecular approach, Mackow and colleagues determined that the nucleoprotein of ANDV can modulate the innate immune signaling cascade by direct interaction with TBK-1 to inhibit phosphorylation of IRF3[61, 62]. Follow-up work identified a unique serine residue at position 386 in the nucleoprotein of ANDV that allows for phosphorylation to occur on the viral protein and inhibition of IRF3 phosphorylation when cells were stimulated by RLR signaling[62]. Addition of this residue to the nucleoprotein of Maporal virus, a related non-pathogenic orthohantavirus, conveyed inhibitory activity for IRF3 phosphorylation. Pan and colleagues identified a similar role for the nucleoprotein of HTNV in disrupting polyl:C-induced IFNβ and ISG expression in A549 cells[51]. Future work is needed to determine whether the mechanisms inhibiting antiviral signaling through nucleoprotein activity are similar between Old World and New World orthohantaviruses.
Researchers have also identified role for the viral glycoproteins (Gn and Gc) in host innate immune antagonism. A significant amount of work has been compiled from the Mackow lab investigating the role for the cytoplasmic tail of the N-terminal glycoprotein (Gn) in disrupting immune signaling cascades. The glycoproteins of orthohantaviruses are transmembrane proteins embedded in endoplasmic reticulum and Golgi membranes with the Gn containing an 142-nucleotide cytoplasmic tail (GnT)[63]. Years of work from the Mackow group has determined that New World (NY-1V, ANDV) and Old World (TULV) orthohantaviruses encode an antagonist of type I IFN in their GnT[47, 48, 64, 65]. It was first identified that the NY-1V Gn could modulate type I IFN responses during infection in HUVEC through targeted disruption of TRAF3-TBK1 binding and activation [64, 65]. Subsequent studies identified differences between the low-pathogenic Old World TULV and the non-pathogenic New World PHV in their ability to modulate antiviral signaling downstream of RIG-I[66]. Ultimately, more studies added support to the hypothesis that pathogenic, but not most non-pathogenic, orthohantaviruses can interfere with innate immune activation downstream of RLR activity[48]. The previously discussed report by Spiropoulou et al. identified the glycoproteins of both PHV and ANDV as responsible for inhibition of STAT1/2 activation in response to type I IFN signaling [60]. Similarly, HTNV replication was resistant to post-infection treatment with type I, II, and III IFNs and prevented significant STAT1 phosphorylation, suggesting a common mechanism of interference [67]. Inhibition of downstream IFN signaling prevents the amplification of innate immune activation and production of numerous ISGs, giving these viruses greater opportunities for replication and spread. Orthohantaviruses therefore have evolved many potential mechanisms for counteracting the RLR pathway and type I IFN signaling in their mammalian hosts, a key factor to enhance their ability to survive, replicate, and infect new hosts.
While most other members of the order Bunyavirales encode an overlapping reading frame for a non-structural protein (NSs) in their small genome segment, only orthohantaviruses endemic to Arvicolinae and Sigmodontinae rodents have this gene[68]. The bunyavirus NSs typically have some role in antiviral antagonism and this seems to be the case also for those encoded by orthohantaviruses[69]. Jääyäskelyäinen and colleagues described inhibitory activity for PUUV and TULV NSs against IFNβ, NF-κB, and IRF3 promoter activation in COS-7 cells following overexpression[70]. This group later described accumulation of the NSs protein from TULV in perinuclear space during infection of HUVEC, suggesting a possible mechanism for transcriptional inhibition[71 ]. Work by Vera-Otarola and colleagues identified an interaction between ANDV NSs and MAVS that inhibits downstream activation of NF-κB, IRF3, and IRFβ promoters[72]. Interestingly, the authors found that ANDV NSs interactions with MAVS did not disrupt MAVS-TBK1 interactions, but did result in decreased ubiquitination of MAVS, potentially altering MAVS aggregation or signaling in still unknown ways. Dissecting the similarities and differences between the functions of NSs in other bunyaviruses to those of orthohantaviruses may also provide insights into the evolutionary history of NSs and the relatedness of rodentia-borne orthohantaviruses with their viral cousins.
Antiviral response does not always involve the transcription of ISGs and antiviral effector molecules. Cell responses such as host translational shutoff and apoptosis limit viral spread through uniform alteration of cellular functions that are also required for viral replication. Wang and Mir noted a lack of translational inhibition in ANDV-infected human cells and determined that the nucleoprotein effectively disrupts protein kinase R (PKR) dimerization and subsequent inhibition through phosphorylation of EIF2α, an essential subunit of the host translation initiation complex[73]. They found this to be true even though PKR is an ISG and was transcriptionally upregulated during ANDV infection. Similarly, Christ and colleagues investigated whether hantavirus infection could inhibit the formation of cellular stress granules as part of its interference with PKR-dependent translational inhibition[74]. They found that the pathogenic orthohantaviruses PUUV and ANDV efficiently inhibited stress granule formation and translational repression in a PKR-dependent manner.
Another stress response driven by viral infection is apoptosis. Through the mechanism of programmed cell death, virus-infected cells can limit further tissue spread and curb the infection. In 2013, it was reported that ANDV and HTNV could inhibit NK cell-mediated killing of infected endothelial cells through both caspase-3 and granzyme B inhibition[75]. Subsequent studies demonstrated that apoptosis inhibition was not unique to ANDV and HTNV, but may be a conserved mechanism of inhibition of immune clearance[76]. Specific induction of BCL-2 to prevent mitochondrial membrane loss and downregulation of the cellular death receptor 5 were recently implicated as mechanisms to prevent chemically-induced and TRAIL-mediated NK cell killing of orthohantavirus infected endothelial cells[77, 78]. In contrast, last year Chen and colleagues proposed that HTNV infection of HUVEC induces an upregulation of TNF-related apoptosis-inducing ligand (TRAIL) and sensitizes infected cells to apoptosis[79]. They further observed that administration of recombinant TRAIL to suckling mice post-infection significantly reduced viral load and resulted in less mortality, suggesting that TRAIL-mediated apoptosis could be protective to the host in HTNV infection. Importantly, in vitro infections of endothelial cells with orthohantaviruses consistently lack observable cell death, providing stronger support for the direct mechanisms of inhibition, rather than induction, of apoptosis [52]. Follow-up studies will be required to dissect these differing conclusions and determine whether study system or design may explain the seemingly contradictory results.
Finally, autophagy is yet another mechanism for viral sequestration and clearance[80, 81]. Autophagy is the normal cellular process of recycling materials for re-use facilitating protein and organelle turnover. In some cases, as reported for SNV, autophagic cycling can help promote viral replication[82]. In their study, Hussein and colleagues observed Gn degradation by host cell autophagic machinery and that Gn expression alone was able to induce autophagy in HeLa cells[82]. Through siRNA knockdowns of autophagy genes during SNV infection, they found that viral replication is enhanced by autophagy and autophagic clearance of Gn in cells. Follow up work identified a mechanism by which co-expression of the SNV nucleoprotein helped to stabilize Gn for viral assembly[83]. It has been recently proposed that viral control of autophagic flux could be a mechanism to remove antiviral effector molecules from the cytosol during initial viral invasion. Wang and colleagues find similar involvement of N and Gn in induction of autophagy in HTNV infection in HUVEC[84]. They propose that Gn-induced mitophagy acts to inhibit type I IFN signaling by degrading MAVS present on mitochondrial-associated membranes. Subsequently, they found that N competes for Gn binding to the autophagy regulator LC3B to inhibit autophagic cycling later in infection and to promote virion assembly with Gn. Thus, inhibition of cellular stress responses are a conserved mechanism for orthohantaviruses to evade cellular restriction to establish productive infection.
Human Clinical and Genetic Association Studies:
Although infection with pathogenic New World orthohantaviruses can be severe and fatal, it remains relatively rare compared to other respiratory infections such as influenza or rhinoviruses. Thus, genome-wide association studies have also been rare and somewhat limited. However, an excellent study from Estevez Ribeiro and colleagues recently found links between genetic variation in complement factor H related genes 1 and 3 and severe HPS associated with ANDV infection in Chile[85]. They also found a significant association in patients with mild disease for a 1.81kb deletion in the SIRPB1 gene, which encodes for a protein involved in ITAM signaling and neutrophil transendothelial migration. Studying more common, but milder infections such as HFRS and nephropathia epidemica, associated with PUUV infection, have also implicated innate immunity in playing a protective role. Exome sequencing of seven PUUV patients suffering from encephalitis identified heterozygosity for a TLR3 p.L742F variant in two of seven patients examined[86]. Functional analysis with immortalized fibroblasts from the two heterozygous patients found reduced responses to poly(I:C) stimulation. Similar associations with the TLR3 locus have been made for patients suffering Herpes simplex virus and varicella zoster virus associated encephalitis[87, 88]. Not only do these studies identify potential risk factors for severe disease, but insights into the genetic components driving mild versus severe pathogenesis allows the field to further dissect the mechanisms and course of hantavirus disease.
Insights from animal models:
The effort to understand orthohantavirus infections and the role of the innate immune response in pathogenesis requires the continued development of tractable animal models. As mentioned previously, immunocompetent laboratory mice and other non-reservoir rodent hosts largely clear an orthohantavirus infection with no overt signs of disease. Notably, while research has demonstrated that Mus muculus support some level of viral replication when infected with Old World orthohantaviruses, there is a distinct lack of data to suggest that New World orthohantaviruses are also supported in this model. Early studies with Old World orthohantaviruses HTNV and SEOV reported that adult wild type mice (C57B6 strains) were resistant to infection and disease, with little to no viral RNA or infectious virus recovered from tissues[89–91]. More recently, several groups have reported detecting viral RNA and even infectious virus from WT mice within 14 days post-infection, indicating that WT adult mice likely support limited viral replication but manage to clear the infection without the onset of disease symptoms[31, 92, 93]. Suckling and newborn mice are susceptible to infection but succumb to neurologic disease and paralysis within days[41, 94–98]. Despite the limitations in recapitulating human disease, experiments with newborn mouse models revealed early on that splenic cell transfer from immunized adults protected newborns from lethal challenge with HTNV[99]. Further, researchers demonstrated that innate immune priming with muramyldipepetide (MDP), a component of bacterial cell walls, prior to infection with 4 times the lethal dose protected newborn mice from mortality[100]. These early studies revealed that immune-mediated protection could be achieved and that innate immune priming is sufficient to prevent disease in this model. Infections in immunocompromised adult mice, either the SCID model, lacking both B and T cells as well as other immune deficiencies, or “nude” mice lacking normal thymic development and absent T cell populations, have resulted in productive infections but disease symptoms remain absent or non-representative of human illness[101, 102]. Recently, it was reported that neutrophil depletion in the SCID mouse model of HTNV infection greatly reduced pulmonary vascular permeability and lung disease, lending greater evidence to the hypothesis that disease is immune-mediated, specifically through proinflammatory neutrophil activity[103].
More targeted approaches to investigate the role of type I IFN in replication and pathology in the mouse model has yielded somewhat unexpected results. While intraperitoneal (i.p.) infection of ifnar−/− mice with HTNV yielded detectable viral loads at later times post-infection compared to infections in WT mice, viral RNA was undetectable by day 14 with still no signs of disease or mortality[31]. Infection in mice lacking the RLR signaling adapter MAVS resulted in an insignificant impact on viral replication, dissemination or persistence. Another recent publication describes intranasal infection of ifnar−/− mice (A129) resulting in viral detection as late as 14 days post-challenge, suggesting the intranasal route may yield a more sustained infection[93]. No viral RNA was detected on day 14 when animals were infected via i.p. or intramuscularly. Still, no animals showed any signs of disease. It should be noted that an earlier study did report significant mortality in WT and ifnar−/− laboratory mice following HTNV infection[92]. The authors reported that i.p. infection of adult, wild type C57BL/6, SJL/J, BALB/c, and AKR/J mice with 105 pfu HTNV resulted in 100% mortality in all strains except AKR/J, which saw 80% mortality. In this system, an LD50 study reported 75% mortality in C57BL/6 at doses as low as 102 pfu. Because this work is in contrast with other reports, it will be essential to determine whether differences in animal housing, genetics, or viral propagation can explain the disparate results.
Importantly, repeated passaging HTNV in mice has just recently allowed for the recovery of a mouse-adapted pathogenic strain of HTNV in wild type adult mice[104]. Future research into the functional differences between the passaged virus and its progenitor virus will reveal essential factors for viral replication and novel mechanisms of immune-meditated disease. This model will also serve the essential function of drug screening and vaccine efficacy verification.
Humanized mice have been investigated for use as models to recapitulate human disease and their use in VFH research is nicely summarized in a recent review by Schönrich and Raftery[105]. Kobak and colleagues examined HTNV-infected NOD. Cg-Prkdcscid IL2rgtm1wjl ISzJ mice for viral replication and pathology[106]. While pathology has been observed and in a manner that nicely recapitulates human depletion of platelets, dissecting the role of innate immunity in driving disease has yet to be explored in this model[105, 106].
The Syrian hamster model of lethal disease for ANDV infection is currently the only small animal model that accurately recapitulates human HPS. This model provides a platform to test therapeutics and vaccines in an immunocompetent adult host[107]. While Old World orthohantaviruses cause no apparent disease in hamsters, even at doses upwards of 104 ffu, intramuscular ANDV infection with as little as 20 ffu drove up to 75% mortality. Because not all NW orthohantaviruses infect healthy, adult Syrian hamsters with pathogenic effect, there is likely a unique interaction between certain orthohantaviruses and these hosts for pathogenesis[108–113]. Further supporting a genetic determinant to disease, an ANDV strain-specific pathogenic effect in the Syrian hamster model was presented earlier this year[114].Dissecting these interactions that lead to divergent disease outcomes will highlight conserved mechanisms for hantavirus disease. Similar to human infections, respiratory distress preceded mortality by about 24 hours and histology revealed intra-alveolar edema and significant lung infiltration of inflammatory macrophages[107]. Given the reproducibility of human pathology for HPS in this model, studies over the subsequent 20 years have offered insight into early infection dynamics and immune-mediated disease. In efforts to understand the mechanisms of disease and protection in this model, researchers have performed targeted depletion of immune system components and investigated their role in pathogenesis. With both general and targeted T cell depletion had no significant effect on survival, viral replication, or disease course[115, 116], further studies focused on depletion of inflammatory alveolar macrophages[117]. While neutrophil recruitment was increased early in infection, it was significantly decreased at later time points in disease in hamsters for which alveolar macrophages were depleted using chlodronate-encapsidated liposomes. Importantly, however, depletion of alveolar macrophages had no effect on viral load, survival or tissue damage.
Using this model to better understand immune correlates of susceptibility, immunosuppression using cyclophosphomide (suppresses B and T cell function, causes apoptosis in neutrophils, macs and DC) and dexamethasone (inhibits NF-kB activation and lymphocyte proliferation) was induced prior to SNV infection[118]. In healthy adult hamsters, SNV establishes a limited asymptomatic infection, while dual treatment with cyclophosphomide and dexamethasone resulted in significantly increased viral titers and induced vascular leakage in the lungs. Curiously, dexamethasone treatment alone had no effect on pathogenesis or SNV detection, suggesting that immune damage and viral control is not dependent on NF-κB in this model[118]. This work was subsequently repeated using other NW orthohantaviruses yielding identical results[119]. Thus, while this model is invaluable for testing antivirals and vaccine candidates, we still have much to learn about virus-host interactions that either drive or protect against hantavirus disease. Understanding the potential genetic factors that make these rodents susceptible to so many NW orthohantaviruses where other rodents remain resistant may also provide new clues for antiviral resistance.
Reservoir innate immunity:
Studies of innate immunity to orthohantavirus infection in their natural reservoir rodent hosts have been limited primarily to in vivo studies due to few tractable tools and incomplete host genome annotations. In recent years, the need to develop in vitro model tools has been recognized and thoroughly reviewed[120]. Nonetheless, the field has learned a considerable amount from these in vivo studies regarding the development of immune tolerance and the innate immune signaling response early in infection. A central theme of many of these studies is the relationship between the reservoir and their respective orthohantavirus species that leads to a tolerance-like phenotype versus the viral clearance seen in non-reservoir infections. Important questions regarding the role of innate immunity in viral persistence and the generality of immune mediated tolerance for other pathogens in reservoir hosts still remain. Many of these questions, lessons learned, and limitations to reservoir studies are well-described in a recent review[121]. Here, we will review those studies focused primarily on innate immune responses.
PUUV in Myodes glareolus
A model system to investigate innate reservoir host responses to orthohantavirus infection was developed a decade ago for Myodes glareolus (bank vole) infections with Puumala virus (PUUV)[122]. While in vivo studies for viral dynamics and pathology had been previously executed, the presentation of a cell culture system to investigate virus-host interactions at the cellular level was critical. Using embryonic fibroblasts, Stoltz and colleagues found that reservoir cells infected with PUUV did not upregulate mRNA for known antiviral genes Mx and IFNβ, even though increased transcription of those same genes occurred in response to infection with other viral agents[122]. These results suggest that either PUUV is able to evade recognition by reservoir host cells or that this virus has evolved mechanisms to inhibit antiviral signaling in reservoir infection. More recently, Strandin and colleagues isolated PUUV from a wild, infected bank vole on a novel bank vole renal epithelial cell line and investigated its phenotypic properties including pathogenesis and persistence[123]. Here, the authors note that persistence and antiviral responses induced by virus stocks passaged in reservoir hosts (in vitro cell culture or in vivo lung homogenates) differed from Vero-passaged viral stocks. This issue has also been raised for other orthohantaviruses[124]. Unfortunately, while impressive tools are being developed with improved technology, the bank vole genome has yet to be fully sequenced and annotated, limiting the impact of this novel in vitro system to study virus-host interaction in depth, at least for the time being.
In vivo studies of bank vole immune dynamics following infection with PUUV have been many, with overall consistent findings. A detailed summary of these studies was recently published by Madrieres et al.[121] Recent genetic surveys investigating tolerance in reservoirs hosts have identified key innate immune signaling genes whose expression profiles and mutations have been hypothesized to alter the immune response to orthohantaviruses and determine the tolerance phenotype. Comparisons between bank vole populations with endemic PUUV infections and those with low PUUV prevalence identified variations in promoter sequences for innate immune effector genes and altered TNFα expression[125]. In populations with increased PUUV prevalence, TNFα expression was significantly lower and single-nucleotide polymorphisms in the promoter sequences were identified that increase basal TNFα gene expression in animals from non-endemic areas. Genetic variation in MHC class II gene expression was also noted to be linked to increased susceptibility to PUUV infection in the bank vole, but importantly it was later found that this genetic variability has been lost in some laboratory colony populations of bank voles, indicating the importance of studying wild, genetically diverse reservoir populations[126, 127]. Future studies that combine tractable cell culture models with observations in wild reservoir populations will be necessary to determine the molecular basis for reservoir tolerance and disease mitigation, with lessons for therapeutic development to treat human pathologies.
SNV in Peromyscus maniculatus
The HPS-causing Sin Nombre virus has been studied in vivo and in vitro to better understand the immune dynamics of reservoir infections. Like many other reservoirs for orthohantaviruses, the North American deer mouse (Peromyscus maniculatus) is not a model organism, with genetic and molecular tools for study still limited. To address this hurdle, technically challenging efforts to clone predicted innate immune genes and develop in vitro methods for dissecting host responses have made a significant impact on what we understand about SNV interactions with its reservoir host[128, 129]. Early investigations into the immune response of the SNV reservoir revealed transient immune activation with elevated levels of chemokines and cytokines (such as CCL2, CCL5, and GM-CSF) early in infection, but then declining rapidly, concomitant with signatures of regulatory T cell upregulation[130, 131]. As noted by Shountz et al., many immune genes were transcriptionally unchanged during acute infection, and most of those that were upregulated, returned to baseline by day 2 post-infection[130]. Viral recrudescence has been proposed for persistently infected deer mice, with waning immunity through stress and possibly metabolic fluctuations being a prime suspect to explain cycles of viremia and control[15, 132–134]. Ex vivo isolation of bone marrow-derived antigen presenting cells (APC) from the deer mouse holds potential for in vitro studies of reservoir APC interactions with SNV[135]. To date, however, these cells have been used to facilitate propagation of reservoir T cells in vitro for in depth investigation into adaptive immune responses and antibody development. While significant progress has been made in understanding adaptive immune responses to SNV by its reservoir host, there remains a gap in understanding of the molecular interactions occurring within infected endothelial cells and potentially program systemic responses leading to persistence.
P. maniculatus has also been developed as a model to study non-reservoir rodent host responses. Important contrasts have been made between host responses to SNV and ANDV or Maporal virus (MAPV), for which the deer mouse does not serve as a common reservoir host. Studies characterizing the course of infection and clearance of ANDV in experimental infections of deer mice observed detectable levels of viral RNA for 4-6 weeks post-infection with humoral immune response detected at 14 days post-infection [136]. Animals infected with ANDV produced measurable neutralizing antibodies, mounted a CD8+ T cell response, and showed no visible signs of disease. Importantly, no viral RNA was detected by 56 days post-infection suggested full clearance of the infection following a robust and effective immune response. Follow-up studies have investigated leukocyte-specific responses of the deer mouse infected with ANDV and direct comparisons of experimental infections with ANDV or SNV [137, 138]. Surprisingly, infection of the deer mouse with MAPV, a NW orthohantavirus not associated with human disease, resulted in mild pathology [139]. Due to the lower risk of human pathogenesis, this model may represent a potential animal host to study NW orthohantavirus disease at biosafety level 3. One of the more important conclusions from these studies as a whole is that, although immune suppression is often observed in reservoir infections, this immune tolerance or suppression is virus species specific and not a general feature of how these animals respond to all infections. This further supports the hypothesis that specific virus-host interactions have evolved in reservoir hosts to determine disease outcomes in orthohantavirus infections.
SEOV in Rattus norvegicus
SEOV was originally identified as a Hantaan-like virus in urban Rattus novegicus populations in Seoul, Korea[140]. It was later determined that SEOV and HTNV were separate viral species with different reservoirs. Once identified, SEOV isolates were collected around the world in brown, urban rat populations, making SEOV the most widespread orthohantavirus and posing a risk to people worldwide. Due to the tractability and development of the rat as a laboratory model organism, more comprehensive and exploratory work has been performed in vivo with this reservoir host-virus system.
It was first reported that SEOV does not transmit vertically from infected rat dam to progeny but that newborn rats were susceptible to persistent viral infection, the first indication that maternal antibody responses are elicited upon SEOV infection and can protect newborn rats from infection[141–143]. Bone marrow-derived dendritic cells and macrophages from rats were interrogated for alteration in response to SEOV infection[144]. Stimulation of APCs prior to infection drove increased expression of activation markers and proinflammatory cytokines. However, SEOV-infected cells displayed a reduction in activation and innate immune signaling. These results are consistent with those described for PUUV infection in bank vole reservoir cells, suggesting a conserved strategy of host innate immune antagonism to increase replication and reduce disease.
Male rats are reported to produce higher antibody titers, higher Th1 responses and shed virus longer than female rats[145]. Subsequent investigations by Klein and colleagues revealed important immunological differences between male and female rats in SEOV infection and transmission[50, 146–151]. Hannah et al. observed that female rats exhibited elevated induction of pathogen recognition receptors and other ISGs upon SEOV infection when compared to male rats[151]. Putative androgen and estrogen response elements were identified in the promoter regions of several differentially regulated ISGs and were hypothesized to mediate these observed differences. Regulatory T cells were identified to be important to establish persistence in reservoir rats infected with SEOV[152]. In support of these findings, Easterbrook and Klein subsequently found that male rats induced a significantly higher regulatory T cell response than female rats and that this differences was driven primarily by the expression of corticosteroids[147, 152]. As the field learns more about sex-dependent differences in innate immunity, the rat reservoir model will certainly become increasingly useful to dissect the immune correlates of protection and susceptibility to hantavirus disease.
Challenges and future goals:
In this author’s opinion, there are two central challenges facing the orthohantavirus field in pursuit of understanding host innate immunity and pathogenesis outlined here:
Need for reverse genetics system for orthohantaviruses: In order to fully dissect the molecular interactions that determine host recognition and immune antagonism, a tractable system to introduce targeted mutations into the viral genome is required. An easily manipulated, full infectious clone encoded within plasmid DNA would be a revolutionary tool for reductionist molecular biology studying orthohantavirus-host interactions. Strong progress towards this end has been made, yet the orthohantavirus field is still far behind other members of the Bunyaviridae family in this regard [153–155]. Most notable are the minigenome systems published for HTNV and ANDV. Typically, minigenome systems entail plasmid DNA encoding the 5’ and 3’ untranslated regions (UTR) of the viral genome, with the protein-coding regions replaced with a reporter such as GFP or luciferase. These minigenomes do not produce infectious virus, but allow for the study of UTR regulation of viral RNA translation and replication [156–158]. The minigenome system also has the added value that aspects of the viral life-cycle for high-containment pathogens can be studied at biosafety level 2. As early as 2003, Flick et al. developed the first minigenome rescue system for any orthohantavirus[156]. Their CAT and pol I driven HTNV minigenome system was the first to demonstrate the feasibility of this approach for an orthohantavirus. However, use of this system did not become widespread, suggesting consistency or tractability may have been suboptimal. Nearly a decade later, Brown et al developed a minigenome system for ANDV to study replication and host factors required for infection[159]. Unfortunately, the authors note issues with reproducibility in virus rescue and unstable expression of the polymerase as major hurdles to tractability. Recently, the issue of poor polymerase expression in mammalian cells has been overcome with a single amino acid substitution near the N-terminus of the protein, with no detectable effect on specific polymerase activity[160]. One potentially significant hurdle to the development of a tractable, full infectious clone, reverse genetics system for orthohantaviruses may be specific host requirements for replication that exist only in the reservoir and not in common cell lines used for laboratory studies, such as Vero E6, BHK, or HEK293 cells. Thus, the development of tractable cell culture tools for reservoir hosts may aid in this effort. The impact and usefulness of a reverse genetics system for the study on innate immunity in viral infection cannot be understated and has been evident in other fields of virology (eg. influenza A virus, togaviruses, and poxviruses) [161–164]. Targeted disruption of putative protein interactions between orthohantaviruses and their hosts and investigations into the consequences for viral replication and disease would not only elucidate novel interactions, but also provide a mechanistic basis for disease and persistence.
Shortage of small animal models to study human disease and reservoir dynamics: The need for further development of animal models to study human pathogenesis as well as reservoir dynamics is well recognized by the community and has been the subject of several commentaries and reviews in recent years[121]. While the ability to genetically manipulate the Syrian hamster models has improved considerably over the years, this immunocompetent model only truly recapitulates human disease when infected with one of the dozens of pathogenic orthohantaviruses. Thus, the need for further development of models to investigate the causative factors in hantavirus disease remains. Resources such as the Collaborative Cross mouse model[165–167], a large panel of multi-parental recombinant inbred mouse lines designed to reproduce the genetic complexity of the human genome, should be harnessed to identify genetic determinants of resistance observed for previously-investigated “wild-type” mouse lines. Such studies may reveal essential host factors required for viral replication, and may also uncover specific innate immune factors and pathways that limit infection and prevent disease. Reservoir models themselves are scarce, with the most common being those discussed in the previous section. Further investment is needed to develop reservoir colonies, in addition to a greater set of genetic and immunologic tools to dissect host responses in these infections. Full genome sequencing and annotation for reservoir hosts of pathogenic orthohantaviruses should be prioritized. In addition to animal model development for in vivo research, cell culture models for reservoir hosts is also severely limited. As mentioned above, the development of the bank vole embryonic fibroblast in vitro culture system could provide important insights into the host innate response and viral replication kinetics in reservoirs[168]. Use of primary endothelial cells derived from rats also represent an untapped resource to investigate host factors and host responses to orthohantavirus infections. The need still exists for cell culture systems to study NW orthohantavirus-reservoir interactions, however. Such systems, combined with the creation of novel molecular tools, can be used to interrogate innate immunity in the context of reservoir and non-reservoir hosts.
Conclusion:
Over the 45 years since the first isolation of an orthohantavirus, the field has learned much about innate host responses to viral infection and the role of the immune response in mediating tissue damage and disease. Unsurprisingly, type I IFN and ISGs are involved in the acute antiviral response in both reservoir and non-reservoir hosts. However, the molecular mechanisms by which orthohantaviruses are recognized and restricted by the host innate immune response remains incompletely understood for most rodent-borne pathogenic orthohantaviruses. Further, the signaling cascades that drive the infection outcome towards either asymptomatic, persistent infection or proinflammatory vascular disease are largely unknown. Continued research, and development of essential tools, will provide boundless opportunity for molecular virology research and novel targets to modulate hosts responses to mitigate human hantavirus disease.
Research Highlights:
Orthohantaviruses trigger type I IFN responses in reservoir and human hosts
Strength/timing of innate immune activation may determine pathogenicity in humans
Molecular mechanisms that determine viral recognition are poorly defined
Innate immune interactions in reservoir hosts is largely unknown
Development of reverse genetics system and novel reservoir models needed
Acknowledgements:
This work has been supported by NIH grant 5K22AI141680-02. Figures were made using Biorender software.
Abbreviations:
- HTNV
Hantaan virus
- HFRS
hemorrhagic fever with renal syndrome
- HPS
hantavirus pulmonary syndrome
- NW
New World
- OW
Old World
- ANDV
Andes virus
- SNV
Sin Nombre virus
- PHV
Prospect Hill virus
- SEOV
Seoul virus
- PUUV
Pumaala virus
- TULV
Tula virus
- RLR
RIG-I-like receptors
- IFN
interferon
- TLR
Toll-like receptor
- IRF
interferon regulatory factor
- ISG
interferon stimulated gene
- Jak
JNK-associated kinase
- STAT
signal transducer and activator of transcription
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CRediT Author statement
Alison Kell: conceptualization, visualization, Writing-Original draft preparation, Writing – reviewing and editing
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References:
- [1].(CDC) CfDC. Management of patients with suspected viral hemorrhagic fever. MMWR Suppl. 1988;37:1–16. [PubMed] [Google Scholar]
- [2].EARLE DP. Analysis of sequential physiologic derangements in epidemic hemorrhagic fever; with a commentary on management. Am J Med. 1954;16:690–709. [DOI] [PubMed] [Google Scholar]
- [3].SHEEDY JA, FROEB HF, BATSON HA, CONLEY CC, MURPHY JP, HUNTER RB, et al. The clinical course of epidemic hemorrhagic fever. Am J Med. 1954;16:619–28. [DOI] [PubMed] [Google Scholar]
- [4].Henttonen H, Buchy P, Suputtamongkol Y, Jittapalapong S, Herbreteau V, Laakkonen J, et al. Recent discoveries of new hantaviruses widen their range and question their origins. Ann N Y Acad Sci. 2008;1149:84–9. [DOI] [PubMed] [Google Scholar]
- [5].Klempa B, Koivogui L, Sylla O, Koulemou K, Auste B, Krüger DH, et al. Serological evidence of human hantavirus infections in Guinea, West Africa. J Infect Dis. 2010;201:1031–4. [DOI] [PubMed] [Google Scholar]
- [6].Klempa B, Fichet-Calvet E, Lecompte E, Auste B, Aniskin V, Meisel H, et al. Hantavirus in African wood mouse, Guinea. Emerg Infect Dis. 2006;12:838–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Green W, Feddersen R, Yousef O, Behr M, Smith K, Nestler J, et al. Tissue distribution of hantavirus antigen in naturally infected humans and deer mice. J Infect Dis. 1998;177:1696–700. [DOI] [PubMed] [Google Scholar]
- [8].Hepojoki J, Vaheri A, Strandin T. The fundamental role of endothelial cells in hantavirus pathogenesis. Front Microbiol. 2014;5:727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Vaheri A, Strandin T, Hepojoki J, Sironen T, Henttonen H, Makela S, et al. Uncovering the mysteries of hantavirus infections. Nat Rev Microbiol. 2013;11:539–50. [DOI] [PubMed] [Google Scholar]
- [10].Spiropoulou CF, Srikiatkhachorn A. The role of endothelial activation in dengue hemorrhagic fever and hantavirus pulmonary syndrome. Virulence. 2013;4:525–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Muyangwa M, Martynova EV, Khaiboullina SF, Morzunov SP, Rizvanov AA. Hantaviral Proteins: Structure, Functions, and Role in Hantavirus Infection. Front Microbiol. 2015;6:1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Klingstrom J, Smed-Sorensen A, Maleki KT, Sola-Riera C, Ahlm C, Bjorkstrom NK, et al. Innate and adaptive immune responses against human Puumala virus infection: immunopathogenesis and suggestions for novel treatment strategies for severe hantavirus-associated syndromes. J Intern Med. 2019;285:510–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Schönrich G, Raftery MJ. Dendritic Cells (DCs) as “Fire Accelerants” of Hantaviral Pathogenesis. Viruses. 2019; 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Noack D, Goeijenbier M, Reusken CBEM, Koopmans MPG, Rockx BHG. Orthohantavirus Pathogenesis and Cell Tropism. Front Cell Infect Microbiol. 2020; 10:399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Warner BM, Stein DR, Griffin BD, Tierney K, Leung A, Sloan A, et al. Development and Characterization of a Sin Nombre Virus Transmission Model in. Viruses. 2019; 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Botten J, Mirowsky K, Ye C, Gottlieb K, Saavedra M, Ponce L, et al. Shedding and intracage transmission of Sin Nombre hantavirus in the deer mouse (Peromyscus maniculatus) model. J Virol. 2002;76:7587–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Maas M, van Heteren M, de Vries A, Kuiken T, Hoornweg T, Veldhuis Kroeze E, et al. Seoul Virus Tropism and Pathology in Naturally Infected Feeder Rats. Viruses. 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Schountz T, Prescott J. Hantavirus immunology of rodent reservoirs: current status and future directions. Viruses. 2014;6:1317–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Holmes EC, Zhang YZ. The evolution and emergence of hantaviruses. Curr Opin Virol. 2015;10:27–33. [DOI] [PubMed] [Google Scholar]
- [20].Zhang YZ, Holmes EC. What is the time-scale of hantavirus evolution? Infect Genet Evol. 2014;25:144–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Plyusnin A, Sironen T. Evolution of hantaviruses: co-speciation with reservoir hosts for more than 100 MYR. Virus Res. 2014;187:22–6. [DOI] [PubMed] [Google Scholar]
- [22].Rivera PC, González-Ittig RE, Gardenal CN. Preferential host switching and its relation with Hantavirus diversification in South America. J Gen Virol. 2015;96:2531–42. [DOI] [PubMed] [Google Scholar]
- [23].Nemirov K, Henttonen H, Vaheri A, Plyusnin A. Phylogenetic evidence for host switching in the evolution of hantaviruses carried by Apodemus mice. Virus Res. 2002;90:207–15. [DOI] [PubMed] [Google Scholar]
- [24].Schoggins JW. Interferon-Stimulated Genes: What Do They All Do? Annu Rev Virol. 2019;6:567–84. [DOI] [PubMed] [Google Scholar]
- [25].Hemann EA, Gale M, Savan R. Interferon Lambda Genetics and Biology in Regulation of Viral Control. Front Immunol. 2017;8:1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chan YK, Gack MU. RIG-I-like receptor regulation in virus infection and immunity. Curr Opin Virol. 2015;12:7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gack MU. What viruses can teach us about the human immune system. PLoS Pathog. 2017;13:e1006364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Czerkies M, Korwek Z, Prus W, Kochańczyk M, Jaruszewicz-Błońska J, Tudelska K, et al. Cell fate in antiviral response arises in the crosstalk of IRF, NF-κB and JAK/STAT pathways. Nat Commun. 2018;9:493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Iwanaszko M, Kimmel M. NF-κB and IRF pathways: cross-regulation on target genes promoter level. BMC Genomics. 2015; 16:307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Weber M, Weber F. Segmented negative-strand RNA viruses and RIG-I: divide (your genome) and rule. Curr Opin Microbiol. 2014;20:96–102. [DOI] [PubMed] [Google Scholar]
- [31].Kell AM, Hemann EA, Turnbull JB, Gale M. RIG-I-like receptor activation drives type I IFN and antiviral signaling to limit Hantaan orthohantavirus replication. PLoS Pathog. 2020;16:e1008483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ye W, Xu Y, Wang Y, Dong Y, Xi Q, Cao M, et al. Hantaan virus can infect human keratinocytes and activate an interferon response through the nuclear translocation of IRF-3. Infect Genet Evol. 2015;29:146–55. [DOI] [PubMed] [Google Scholar]
- [33].Nam JH, Hwang KA, Yu CH, Kang TH, Shin JY, Choi WY, et al. Expression of interferon inducible genes following Hantaan virus infection as a mechanism of resistance in A549 cells. Virus Genes. 2003;26:31–8. [DOI] [PubMed] [Google Scholar]
- [34].Prescott JB, Hall PR, Bondu-Hawkins VS, Ye C, Hjelle B. Early innate immune responses to Sin Nombre hantavirus occur independently of IFN regulatory factor 3, characterized pattern recognition receptors, and viral entry. J Immunol. 2007;179:1796–802. [DOI] [PubMed] [Google Scholar]
- [35].Popugaeva E, Witkowski PT, Schlegel M, Ulrich RG, Auste B, Rang A, et al. Dobrava-Belgrade hantavirus from Germany shows receptor usage and innate immunity induction consistent with the pathogenicity of the virus in humans. PLoS One. 2012;7:e35587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bourquain D, Bodenstein C, Schürer S, Schaade L. Puumala and Tula Virus Differ in Replication Kinetics and Innate Immune Stimulation in Human Endothelial Cells and Macrophages. Viruses. 2019; 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gallo G, Caignard G, Badonnel K, Chevreux G, Terrier S, Szemiel A, et al. Interactions of Viral Proteins from Pathogenic and Low or Non-Pathogenic Orthohantaviruses with Human Type I Interferon Signaling. Viruses. 2021; 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kim IW, Hwang JY, Kim SK, Kim JK, Park HS. Interferon-stimulated genes response in endothelial cells following Hantaan virus infection. J Korean Med Sci. 2007;22:987–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Shim SH, Park MS, Moon S, Park KS, Song JW, Song KJ, et al. Comparison of innate immune responses to pathogenic and putative non-pathogenic hantaviruses in vitro. Virus Res. 2011;160:367–73. [DOI] [PubMed] [Google Scholar]
- [40].Handke W, Oelschlegel R, Franke R, Kruger DH, Rang A. Hantaan virus triggers TLR3-dependent innate immune responses. J Immunol. 2009;182:2849–58. [DOI] [PubMed] [Google Scholar]
- [41].Shin OS, Song GS, Kumar M, Yanagihara R, Lee HW, Song JW. Hantaviruses induce antiviral and pro-inflammatory innate immune responses in astrocytic cells and the brain. Viral Immunol. 2014;27:256–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ye W, Lei Y, Yu M, Xu Y, Cao M, Yu L, et al. NLRP3 inflammasome is responsible for Hantavirus inducing interleukin-1β in THP-1 cells. Int J Mol Med. 2015;35:1633–40. [DOI] [PubMed] [Google Scholar]
- [43].Geimonen E, Neff S, Raymond T, Kocer SS, Gavrilovskaya IN, Mackow ER. Pathogenic and nonpathogenic hantaviruses differentially regulate endothelial cell responses. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:13837–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Yu HT, Jiang H, Zhang Y, Nan XP, Li Y, Wang W, et al. Hantaan virus triggers TLR4-dependent innate immune responses. Viral Immunol. 2012;25:387–93. [DOI] [PubMed] [Google Scholar]
- [45].Jiang H, Wang PZ, Zhang Y, Xu Z, Sun L, Wang LM, et al. Hantaan virus induces toll-like receptor 4 expression, leading to enhanced production of beta interferon, interleukin-6 and tumor necrosis factor-alpha. Virology. 2008;380:52–9. [DOI] [PubMed] [Google Scholar]
- [46].Kraus AA, Raftery MJ, Giese T, Ulrich R, Zawatzky R, Hippenstiel S, et al. Differential antiviral response of endothelial cells after infection with pathogenic and nonpathogenic hantaviruses. Journal of virology. 2004;78:6143–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Matthys V, Gorbunova EE, Gavrilovskaya IN, Pepini T, Mackow ER. The C-terminal 42 residues of the Tula virus Gn protein regulate interferon induction. Journal of virology. 2011;85:4752–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Matthys VS, Cimica V, Dalrymple NA, Glennon NB, Bianco C, Mackow ER. Hantavirus GnT elements mediate TRAF3 binding and inhibit RIG-I/TBK1-directed beta interferon transcription by blocking IRF3 phosphorylation. Journal of virology. 2014;88:2246–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Mackow ER, Dalrymple NA, Cimica V, Matthys V, Gorbunova E, Gavrilovskaya I. Hantavirus interferon regulation and virulence determinants. Virus Res. 2014;187:65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Klein SL, Cernetich A, Hilmer S, Hoffman EP, Scott AL, Glass GE. Differential expression of immunoregulatory genes in male and female Norway rats following infection with Seoul virus. J Med Virol. 2004;74:180–90. [DOI] [PubMed] [Google Scholar]
- [51].Pan W, Bian G, Wang K, Feng T, Dai J. Effects of Different Doses of Nucleocapsid Protein from Hantaan Virus A9 Strain on Regulation of Interferon Signaling. Viral Immunol. 2015;28:448–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sundstrom JB, McMullan LK, Spiropoulou CF, Hooper WC, Ansari AA, Peters CJ, et al. Hantavirus infection induces the expression of RANTES and IP-10 without causing increased permeability in human lung microvascular endothelial cells. J Virol. 2001;75:6070–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Gavrilovskaya IN, Shepley M, Shaw R, Ginsberg MH, Mackow ER. beta3 Integrins mediate the cellular entry of hantaviruses that cause respiratory failure. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:7074–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Gavrilovskaya IN, Brown EJ, Ginsberg MH, Mackow ER. Cellular entry of hantaviruses which cause hemorrhagic fever with renal syndrome is mediated by beta3 integrins. Journal of virology. 1999;73:3951–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Jangra RK, Herbert AS, Li R, Jae LT, Kleinfelter LM, Slough MM, et al. Protocadherin-1 is essential for cell entry by New World hantaviruses. Nature. 2018;563:559–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Dieterle ME, Solà-Riera C, Ye C, Goodfellow SM, Mittler E, Kasikci E, et al. Genetic depletion studies inform receptor usage by virulent hantaviruses in human endothelial cells. Elife. 2021;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Mittler E, Dieterle ME, Kleinfelter LM, Slough MM, Chandran K, Jangra RK. Hantavirus entry: Perspectives and recent advances. Adv Virus Res. 2019;104:185–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ma H, Han P, Ye W, Chen H, Zheng X, Cheng L, et al. The Long Noncoding RNA NEAT1 Exerts Antihantaviral Effects by Acting as Positive Feedback for RIG-I Signaling. Journal of virology. 2017;91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Khaiboullina SF, Rizvanov AA, Otteson E, Miyazato A, Maciejewski J, St Jeor S. Regulation of cellular gene expression in endothelial cells by sin nombre and prospect hill viruses. Viral Immunol. 2004;17:234–51. [DOI] [PubMed] [Google Scholar]
- [60].Spiropoulou CF, Albariño CG, Ksiazek TG, Rollin PE. Andes and Prospect Hill hantaviruses differ in early induction of interferon although both can downregulate interferon signaling. J Virol. 2007;81:2769–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Cimica V, Dalrymple NA, Roth E, Nasonov A, Mackow ER. An innate immunity-regulating virulence determinant is uniquely encoded by the Andes virus nucleocapsid protein. mBio. 2014;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Simons MJ, Gorbunova EE, Mackow ER. Unique Interferon Pathway Regulation by the Andes Virus Nucleocapsid Protein Is Conferred by Phosphorylation of Serine 386. J Virol. 2019;93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Serris A, Stass R, Bignon EA, Muena NA, Manuguerra JC, Jangra RK, et al. The Hantavirus Surface Glycoprotein Lattice and Its Fusion Control Mechanism. Cell. 2020;183:442–56.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Alff PJ, Gavrilovskaya IN, Gorbunova E, Endriss K, Chong Y, Geimonen E, et al. The pathogenic NY-1 hantavirus G1 cytoplasmic tail inhibits RIG-I- and TBK-1-directed interferon responses. Journal of virology. 2006;80:9676–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Alff PJ, Sen N, Gorbunova E, Gavrilovskaya IN, Mackow ER. The NY-1 hantavirus Gn cytoplasmic tail coprecipitates TRAF3 and inhibits cellular interferon responses by disrupting TBK1-TRAF3 complex formation. Journal of virology. 2008;82:9115–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Matthys V, Gorbunova EE, Gavrilovskaya IN, Pepini T, Mackow ER. The C-terminal 42 residues of the Tula virus Gn protein regulate interferon induction. J Virol. 2011;85:4752–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Stoltz M, Ahlm C, Lundkvist A, Klingstrom J. Lambda interferon (IFN-lambda) in serum is decreased in hantavirus-infected patients, and in vitro-established infection is insensitive to treatment with all IFNs and inhibits IFN-gamma-induced nitric oxide production. Journal of virology. 2007;81:8685–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Plyusnin A Genetics of hantaviruses: implications to taxonomy. Arch Virol. 2002;147:665–82. [DOI] [PubMed] [Google Scholar]
- [69].Hedil M, Kormelink R. Viral RNA Silencing Suppression: The Enigma of Bunyavirus NSs Proteins. Viruses. 2016;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Jaaskelainen KM, Kaukinen P, Minskaya ES, Plyusnina A, Vapalahti O, Elliott RM, et al. Tula and Puumala hantavirus NSs ORFs are functional and the products inhibit activation of the interferon-beta promoter. J Med Virol. 2007;79:1527–36. [DOI] [PubMed] [Google Scholar]
- [71].Virtanen JO, Jääskeläinen KM, Djupsjöbacka J, Vaheri A, Plyusnin A. Tula hantavirus NSs protein accumulates in the perinuclear area in infected and transfected cells. Arch Virol. 2010;155:117–21. [DOI] [PubMed] [Google Scholar]
- [72].Vera-Otarola J, Solis L, Lowy F, Olguín V, Angulo J, Pino K, et al. The Andes Orthohantavirus NSs Protein Antagonizes the Type I Interferon Response by Inhibiting MAVS Signaling. J Virol. 2020;94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Wang Z, Mir MA. Andes virus nucleocapsid protein interrupts protein kinase R dimerization to counteract host interference in viral protein synthesis. J Virol. 2015;89:1628–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Christ W, Tynell J, Klingström J. Puumala and Andes Orthohantaviruses Cause Transient Protein Kinase R-Dependent Formation of Stress Granules. J Virol. 2020;94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Gupta S, Braun M, Tischler ND, Stoltz M, Sundström KB, Björkström NK, et al. Hantavirus-infection confers resistance to cytotoxic lymphocyte-mediated apoptosis. PLoS Pathog. 2013;9:e1003272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Solà-Riera C, Gupta S, Ljunggren HG, Klingström J. Orthohantaviruses belonging to three phylogroups all inhibit apoptosis in infected target cells. Sci Rep. 2019;9:834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Solà-Riera C, García M, Ljunggren HG, Klingström J. Hantavirus inhibits apoptosis by preventing mitochondrial membrane potential loss through up-regulation of the pro-survival factor BCL-2. PLoS Pathog. 2020;16:e1008297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Solà-Riera C, Gupta S, Maleki KT, González-Rodriguez P, Saidi D, Zimmer CL, et al. Hantavirus Inhibits TRAIL-Mediated Killing of Infected Cells by Downregulating Death Receptor 5. Cell Rep. 2019;28:2124–39.e6. [DOI] [PubMed] [Google Scholar]
- [79].Chen QZ, Wang X, Luo F, Li N, Zhu N, Lu S, et al. HTNV Sensitizes Host Toward TRAIL-Mediated Apoptosis-A Pivotal Anti-hantaviral Role of TRAIL. Front Immunol. 2020;11:1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Mao J, Lin E, He L, Yu J, Tan P, Zhou Y. Autophagy and Viral Infection. Adv Exp Med Biol. 2019;1209:55–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Choi Y, Bowman JW, Jung JU. Autophagy during viral infection - a double-edged sword. Nat Rev Microbiol. 2018;16:341–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Hussein IT, Cheng E, Ganaie SS, Werle MJ, Sheema S, Haque A, et al. Autophagic clearance of Sin Nombre hantavirus glycoprotein Gn promotes virus replication in cells. J Virol. 2012;86:7520–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Ganaie SS, Mir MA. The role of viral genomic RNA and nucleocapsid protein in the autophagic clearance of hantavirus glycoprotein Gn. Virus Res. 2014;187:72–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Wang K, Ma H, Liu H, Ye W, Li Z, Cheng L, et al. The Glycoprotein and Nucleocapsid Protein of Hantaviruses Manipulate Autophagy Flux to Restrain Host Innate Immune Responses. Cell Rep. 2019;27:2075–91.e5. [DOI] [PubMed] [Google Scholar]
- [85].Ribeiro GE, Leon LE, Perez R, Cuiza A, Vial PA, Ferres M, et al. Deletions in Genes Participating in Innate Immune Response Modify the Clinical Course of Andes Orthohantavirus Infection. Viruses. 2019; 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Partanen T, Chen J, Lehtonen J, Kuismin O, Rusanen H, Vapalahti O, et al. Heterozygous TLR3 Mutation in Patients with Hantavirus Encephalitis. J Clin Immunol. 2020;40:1156–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007;317:1522–7. [DOI] [PubMed] [Google Scholar]
- [88].Sironi M, Peri AM, Cagliani R, Forni D, Riva S, Biasin M, et al. TLR3 Mutations in Adult Patients With Herpes Simplex Virus and Varicella-Zoster Virus Encephalitis. J Infect Dis. 2017;215:1430–4. [DOI] [PubMed] [Google Scholar]
- [89].Kariwa H, Kamimura M, Arikawa J, Yoshimatsu K, Takashima I, Hashimoto N. Characterization of the mode of Hantaan virus infection in adult mice using a nested reverse transcriptase polymerase chain reaction: transient virus replication in adult mice. Microbiol Immunol. 1995;39:35–41. [DOI] [PubMed] [Google Scholar]
- [90].Taruishi M, Yoshimatsu K, Hatsuse R, Okumura M, Nakamura I, Arikawa J. Lack of vertical transmission of Hantaan virus from persistently infected dam to progeny in laboratory mice. Arch Virol. 2008;153:1605–9. [DOI] [PubMed] [Google Scholar]
- [91].Yoo YC, Yoshimatsu K, Yoshida R, Tamura M, Azuma I, Arikawa J. Comparison of virulence between Seoul virus strain SR-11 and Hantaan virus strain 76-118 of hantaviruses in newborn mice. Microbiol Immunol. 1993;37:557–62. [DOI] [PubMed] [Google Scholar]
- [92].Wichmann D, Grone HJ, Frese M, Pavlovic J, Anheier B, Haller O, et al. Hantaan virus infection causes an acute neurological disease that is fatal in adult laboratory mice. Journal of virology. 2002;76:8890–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Dowall SD, Graham VA, Aram M, Findlay-Wilson S, Salguero FJ, Emery K, et al. Hantavirus infection in type I interferon receptor-deficient (A129) mice. J Gen Virol. 2020;101:1047–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Ebihara H, Yoshimatsu K, Ogino M, Araki K, Ami Y, Kariwa H, et al. Pathogenicity of Hantaan virus in newborn mice: genetic reassortant study demonstrating that a single amino acid change in glycoprotein G1 is related to virulence. Journal of virology. 2000;74:9245–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Kim GR, McKee KT, Jr. Pathogenesis of Hantaan virus infection in suckling mice: clinical, virologic, and serologic observations. Am J Trop Med Hyg. 1985;34:388–95. [DOI] [PubMed] [Google Scholar]
- [96].Lokugamage K, Kariwa H, Lokugamage N, Iwasa M, Hagiya T, Araki K, et al. Comparison of virulence of various hantaviruses related to hemorrhagic fever with renal syndrome in newborn mouse model. Jpn J Vet Res. 2004;51:143–9. [PubMed] [Google Scholar]
- [97].Tamura M, Asada H, Kondo K, Takahashi M, Yamanishi K. Effects of human and murine interferons against hemorrhagic fever with renal syndrome (HFRS) virus (Hantaan virus). Antiviral Res. 1987;8:171–8. [DOI] [PubMed] [Google Scholar]
- [98].Klingström J, Hardestam J, Lundkvist A. Dobrava, but not Saaremaa, hantavirus is lethal and induces nitric oxide production in suckling mice. Microbes Infect. 2006;8:728–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Nakamura T, Yanagihara R, Gibbs CJ, Gajdusek DC. Immune spleen cell-mediated protection against fatal Hantaan virus infection in infant mice. J Infect Dis. 1985;151:691–7. [DOI] [PubMed] [Google Scholar]
- [100].Yoo YC, Yoshimatsu K, Hatsuse R, Tamura M, Yoshida R, Tono-oka S, et al. Effect of MDP-Lys(L18), a derivative of MDP, on enhancing host resistance against Hantaan virus infection in newborn mice. Vaccine. 1995;13:1300–5. [DOI] [PubMed] [Google Scholar]
- [101].Yoshimatsu K, Arikawa J, Ohbora S, Itakura C. Hantavirus infection in SCID mice. J Vet Med Sci. 1997;59:863–8. [DOI] [PubMed] [Google Scholar]
- [102].Koma T, Yoshimatsu K, Nagata N, Sato Y, Shimizu K, Yasuda SP, et al. Neutrophil depletion suppresses pulmonary vascular hyperpermeability and occurrence of pulmonary edema caused by hantavirus infection in C.B-17 SCID mice. Journal of virology. 2014;88:7178–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Koma T, Yoshimatsu K, Nagata N, Sato Y, Shimizu K, Yasuda SP, et al. Neutrophil depletion suppresses pulmonary vascular hyperpermeability and occurrence of pulmonary edema caused by hantavirus infection in C.B-17 SCID mice. J Virol. 2014;88:7178–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Shimizu K, Koma T, Yoshimatsu K, Tsuda Y, Isegawa Y, Arikawa J. Appearance of renal hemorrhage in adult mice after inoculation of patient-derived hantavirus. Virol J. 2017;14:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Schonrich G, Raftery MJ. Exploring the Immunopathogenesis of Viral Hemorrhagic Fever in Mice with a Humanized Immune System. Front Immunol. 2017;8:1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Kobak L, Raftery MJ, Voigt S, Kuhl AA, Kilic E, Kurth A, et al. Hantavirus-induced pathogenesis in mice with a humanized immune system. J Gen Virol. 2015;96:1258–63. [DOI] [PubMed] [Google Scholar]
- [107].Hooper JW, Larsen T, Custer DM, Schmaljohn CS. A lethal disease model for hantavirus pulmonary syndrome. Virology. 2001;289:6–14. [DOI] [PubMed] [Google Scholar]
- [108].Campen MJ, Milazzo ML, Fulhorst CF, Obot Akata CJ, Koster F. Characterization of shock in a hamster model of hantavirus infection. Virology. 2006;356:45–9. [DOI] [PubMed] [Google Scholar]
- [109].Milazzo ML, Eyzaguirre EJ, Fulhorst CF. Pneumonitis in Syrian golden hamsters (Mesocricetus auratus) infected with Rio Mamoré virus (family Bunyaviridae, genus Hantavirus). Virus Res. 2014;191:39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Sanada T, Kariwa H, Nagata N, Tanikawa Y, Seto T, Yoshimatsu K, et al. Puumala virus infection in Syrian hamsters (Mesocricetus auratus) resembling hantavirus infection in natural rodent hosts. Virus Res. 2011;160:108–19. [DOI] [PubMed] [Google Scholar]
- [111].Milazzo ML, Eyzaguirre EJ, Molina CP, Fulhorst CF. Maporal viral infection in the Syrian golden hamster: a model of hantavirus pulmonary syndrome. J Infect Dis. 2002;186:1390–5. [DOI] [PubMed] [Google Scholar]
- [112].Gu SH, Kim YS, Baek LJ, Kurata T, Yanagihara R, Song JW. Lethal disease in infant and juvenile Syrian hamsters experimentally infected with Imjin virus, a newfound crocidurine shrew-borne hantavirus. Infect Genet Evol. 2015;36:231–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Hardcastle K, Scott D, Safronetz D, Brining DL, Ebihara H, Feldmann H, et al. Laguna Negra Virus Infection Causes Hantavirus Pulmonary Syndrome in Turkish Hamsters (Mesocricetus brandti). Vet Pathol. 2016;53:182–9. [DOI] [PubMed] [Google Scholar]
- [114].Warner BM, Sloan A, Deschambault Y, Dowhanik S, Tierney K, Audet J, et al. Differential pathogenesis between Andes virus strains CHI-7913 and Chile-9717869in Syrian Hamsters. J Virol. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Hammerbeck CD, Hooper JW. T cells are not required for pathogenesis in the Syrian hamster model of hantavirus pulmonary syndrome. Journal of virology. 2011;85:9929–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Prescott J, Safronetz D, Haddock E, Robertson S, Scott D, Feldmann H. The adaptive immune response does not influence hantavirus disease or persistence in the Syrian hamster. Immunology. 2013;140:168–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Hammerbeck CD, Brocato RL, Bell TM, Schellhase CW, Mraz SR, Queen LA, et al. Depletion of Alveolar Macrophages Does Not Prevent Hantavirus Disease Pathogenesis in Golden Syrian Hamsters. Journal of virology. 2016;90:6200–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Brocato RL, Hammerbeck CD, Bell TM, Wells JB, Queen LA, Hooper JW. A lethal disease model for hantavirus pulmonary syndrome in immunosuppressed Syrian hamsters infected with Sin Nombre virus. J Virol. 2014;88:811–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Vergote V, Laenen L, Vanmechelen B, Van Ranst M, Verbeken E, Hooper JW, et al. A lethal disease model for New World hantaviruses using immunosuppressed Syrian hamsters. PLoS Negl Trop Dis. 2017; 11:e0006042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Eckerle I, Lenk M, Ulrich RG. More novel hantaviruses and diversifying reservoir hosts--time for development of reservoir-derived cell culture models? Viruses. 2014;6:951–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Madrières S, Castel G, Murri S, Vulin J, Marianneau P, Charbonnel N. The Needs for Developing Experiments on Reservoirs in Hantavirus Research: Accomplishments, Challenges and Promises for the Future. Viruses. 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Stoltz M, Sundström KB, Hidmark Å, Tolf C, Vene S, Ahlm C, et al. A model system for in vitro studies of bank vole borne viruses. PLoS One. 2011;6:e28992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Strandin T, Smura T, Ahola P, Aaltonen K, Sironen T, Hepojoki J, et al. Orthohantavirus Isolated in Reservoir Host Cells Displays Minimal Genetic Changes and Retains Wild-Type Infection Properties. Viruses. 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Prescott J, Feldmann H, Safronetz D. Amending Koch’s postulates for viral disease: When “growth in pure culture” leads to a loss of virulence. Antiviral Res. 2017;137:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Guivier E, Galan M, Salvador AR, Xuéreb A, Chaval Y, Olsson GE, et al. Tnf-α expression and promoter sequences reflect the balance of tolerance/resistance to Puumala hantavirus infection in European bank vole populations. Infect Genet Evol. 2010;10:1208–17. [DOI] [PubMed] [Google Scholar]
- [126].Deter J, Bryja J, Chaval Y, Galan M, Henttonen H, Laakkonen J, et al. Association between the DQA MHC class II gene and Puumala virus infection in Myodes glareolus, the bank vole. Infect Genet Evol. 2008;8:450–8. [DOI] [PubMed] [Google Scholar]
- [127].Guivier E, Galan M, Malé PJ, Kallio ER, Voutilainen L, Henttonen H, et al. Associations between MHC genes and Puumala virus infection in Myodes glareolus are detected in wild populations, but not from experimental infection data. J Gen Virol. 2010;91:2507–12. [DOI] [PubMed] [Google Scholar]
- [128].Schountz T, Green R, Davenport B, Buniger A, Richens T, Root JJ, et al. Cloning and characterization of deer mouse (Peromyscus maniculatus) cytokine and chemokine cDNAs. BMC Immunol. 2004;5:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Herbst MM, Prescott J, Palmer AD, Schountz T. Sequence and expression analysis of deer mouse interferon-gamma, interleukin-10, tumor necrosis factor, and lymphotoxin-alpha. Cytokine. 2002;17:203–13. [DOI] [PubMed] [Google Scholar]
- [130].Schountz T, Acuna-Retamar M, Feinstein S, Prescott J, Torres-Perez F, Podell B, et al. Kinetics of immune responses in deer mice experimentally infected with Sin Nombre virus. Journal of virology. 2012;86:10015–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Schountz T, Prescott J, Cogswell AC, Oko L, Mirowsky-Garcia K, Galvez AP, et al. Regulatory T cell-like responses in deer mice persistently infected with Sin Nombre virus. Proc Natl Acad Sci U S A. 2007;104:15496–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Pearce-Duvet JM, St Jeor SC, Boone JD, Dearing MD. Changes in sin nombre virus antibody prevalence in deer mice across seasons: the interaction between habitat, sex, and infection in deer mice. J Wildl Dis. 2006;42:819–24. [DOI] [PubMed] [Google Scholar]
- [133].Safronetz D, Drebot MA, Artsob H, Cote T, Makowski K, Lindsay LR. Sin Nombre virus shedding patterns in naturally infected deer mice (Peromyscus maniculatus) in relation to duration of infection. Vector Borne Zoonotic Dis. 2008;8:97–100. [DOI] [PubMed] [Google Scholar]
- [134].Lehmer EM, Jones JD, Bego MG, Varner JM, Jeor SS, Clay CA, et al. Long-term patterns of immune investment by wild deer mice infected with Sin Nombre virus. Physiol Biochem Zool. 2010;83:847–57. [DOI] [PubMed] [Google Scholar]
- [135].Davenport BJ, Willis DG, Prescott J, Farrell RM, Coons TA, Schountz T. Generation of competent bone marrow-derived antigen presenting cells from the deer mouse (Peromyscus maniculatus). BMC Immunol. 2004;5:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Spengler JR, Haddock E, Gardner D, Hjelle B, Feldmann H, Prescott J. Experimental Andes virus infection in deer mice: characteristics of infection and clearance in a heterologous rodent host. PLoS One. 2013;8:e55310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Schountz T, Quackenbush S, Rovnak J, Haddock E, Black WCt, Feldmann H, et al. Differential lymphocyte and antibody responses in deer mice infected with Sin Nombre hantavirus or Andes hantavirus. Journal of virology. 2014;88:8319–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Schountz T, Shaw TI, Glenn TC, Feldmann H, Prescott J. Expression profiling of lymph node cells from deer mice infected with Andes virus. BMC Immunol. 2013;14:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].McGuire A, Miedema K, Fauver JR, Rico A, Aboellail T, Quackenbush SL, et al. Maporal Hantavirus Causes Mild Pathology in Deer Mice (Peromyscus maniculatus). Viruses. 2016;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Lee HW, Baek LJ, Johnson KM. Isolation of Hantaan virus, the etiologic agent of Korean hemorrhagic fever, from wild urban rats. J Infect Dis. 1982;146:638–44. [DOI] [PubMed] [Google Scholar]
- [141].Morita C, Morikawa S, Sugiyama K, Komatsu T, Ueno H, Kitamura T. Inability of a strain of Seoul virus to transmit itself vertically in rats. Jpn J Med Sci Biol. 1993;46:215–9. [DOI] [PubMed] [Google Scholar]
- [142].Dohmae K, Nishimune Y. Protection against hantavirus infection by dam’s immunity transferred vertically to neonates. Arch Virol. 1995;140:165–72. [DOI] [PubMed] [Google Scholar]
- [143].Kariwa H, Kimura M, Yoshizumi S, Arikawa J, Yoshimatsu K, Takashima I, et al. Modes of Seoul virus infections: persistency in newborn rats and transiency in adult rats. Arch Virol. 1996;141:2327–38. [DOI] [PubMed] [Google Scholar]
- [144].Au RY, Jedlicka AE, Li W, Pekosz A, Klein SL. Seoul virus suppresses NF-kappaB-mediated inflammatory responses of antigen presenting cells from Norway rats. Virology. 2010;400:115–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Klein SL, Bird BH, Glass GE. Sex differences in immune responses and viral shedding following Seoul virus infection in Norway rats. Am J Trop Med Hyg. 2001;65:57–63. [DOI] [PubMed] [Google Scholar]
- [146].Klein SL, Marson AL, Scott AL, Ketner G, Glass GE. Neonatal sex steroids affect responses to Seoul virus infection in male but not female Norway rats. Brain Behav Immun. 2002;16:736–46. [DOI] [PubMed] [Google Scholar]
- [147].Easterbrook JD, Klein SL. Corticosteroids modulate Seoul virus infection, regulatory T-cell responses and matrix metalloprotease 9 expression in male, but not female, Norway rats. J Gen Virol. 2008;89:2723–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Easterbrook JD, Kaplan JB, Glass GE, Pletnikov MV, Klein SL. Elevated testosterone and reduced 5-HIAA concentrations are associated with wounding and hantavirus infection in male Norway rats. Horm Behav. 2007;52:474–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Klein S Host factors mediating sex differences in viral infection. Gend Med. 2005;2:197–207. [DOI] [PubMed] [Google Scholar]
- [150].Easterbrook JD, Klein SL. Seoul virus enhances regulatory and reduces proinflammatory responses in male Norway rats. J Med Virol. 2008;80:1308–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Hannah MF, Bajic VB, Klein SL. Sex differences in the recognition of and innate antiviral responses to Seoul virus in Norway rats. Brain Behav Immun. 2008;22:503–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Easterbrook JD, Zink MC, Klein SL. Regulatory T cells enhance persistence of the zoonotic pathogen Seoul virus in its reservoir host. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:15502–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Xu M, Wang B, Deng F, Wang H, Wang M, Hu Z, et al. Establishment of a Reverse Genetic System of Severe Fever with Thrombocytopenia Syndrome Virus Based on a C4 Strain. Virol Sin. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Takenaka-Uema A, Sugiura K, Bangphoomi N, Shioda C, Uchida K, Kato K, et al. Development of an improved reverse genetics system for Akabane bunyavirus. J Virol Methods. 2016;232:16–20. [DOI] [PubMed] [Google Scholar]
- [155].Bouloy M, Flick R. Reverse genetics technology for Rift Valley fever virus: current and future applications for the development of therapeutics and vaccines. Antiviral Res. 2009;84:101–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Flick K, Hooper JW, Schmaljohn CS, Pettersson RF, Feldmann H, Flick R. Rescue of Hantaan virus minigenomes. Virology. 2003;306:219–24. [DOI] [PubMed] [Google Scholar]
- [157].Nelson EV, Pacheco JR, Hume AJ, Cressey TN, Deflubé LR, Ruedas JB, et al. An RNA polymerase II-driven Ebola virus minigenome system as an advanced tool for antiviral drug screening. Antiviral Res. 2017;146:21–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Groseth A, Feldmann H, Theriault S, Mehmetoglu G, Flick R. RNA polymerase I-driven minigenome system for Ebola viruses. J Virol. 2005;79:4425–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Brown KS, Ebihara H, Feldmann H. Development of a minigenome system for Andes virus, a New World hantavirus. Arch Virol. 2012;157:2227–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Rothenberger S, Torriani G, Johansson MU, Kunz S, Engler O. Conserved Endonuclease Function of Hantavirus L Polymerase. Viruses. 2016;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Choi WS, Jeong JH, Lloren KKS, Ahn SJ, Antigua KJC, Kim YI, et al. Development of a rapid, simple and efficient one-pot cloning method for a reverse genetics system of broad subtypes of influenza A virus. Sci Rep. 2019;9:8318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Anchisi S, GonÇalves AR, Mazel-Sanchez B, Cordey S, Schmolke M. Influenza A Virus Genetic Tools: From Clinical Sample to Molecular Clone. Methods Mol Biol. 2018;1836:33–58. [DOI] [PubMed] [Google Scholar]
- [163].Phuektes P, Chu JJ. Reverse Genetics Approaches for Chikungunya Virus. Methods Mol Biol. 2016;1426:283–95. [DOI] [PubMed] [Google Scholar]
- [164].Warren RD, Cotter CA, Moss B. Reverse genetics analysis of poxvirus intermediate transcription factors. J Virol. 2012;86:9514–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Maurizio PL, Ferris MT. The Collaborative Cross Resource for Systems Genetics Research of Infectious Diseases. Methods Mol Biol. 2017;1488:579–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Abu Toamih Atamni H, Nashef A, Iraqi FA. The Collaborative Cross mouse model for dissecting genetic susceptibility to infectious diseases. Mamm Genome. 2018;29:471–87. [DOI] [PubMed] [Google Scholar]
- [167].Noll KE, Ferris MT, Heise MT. The Collaborative Cross: A Systems Genetics Resource for Studying Host-Pathogen Interactions. Cell Host Microbe. 2019;25:484–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Stoltz M, Sundstrom KB, Hidmark A, Tolf C, Vene S, Ahlm C, et al. A model system for in vitro studies of bank vole borne viruses. PloS one. 2011;6:e28992. [DOI] [PMC free article] [PubMed] [Google Scholar]