

Summary
The airway mucosal surfaces are constantly exposed to inhaled particles that can be potentially toxic, infectious or allergenic and should elicit inflammatory changes. The proximal and distal air spaces, however, are normally infection and inflammation free due to a specialized interplay between cellular and molecular components of the pulmonary innate immune system. Surfactant protein D (SP-D) is an epithelial-cell-derived immune modulator that belongs to the small family of structurally related Ca2+-dependent C-type collagen-like lectins. While collectins can be detected in mucosal surfaces of various organs, SP-A and SP-D (the ‘lung collectins’) are constitutively expressed in the lung at high concentrations. Both proteins are considered important players of the pulmonary immune responses. Under normal conditions however, SP-A−/− mice display no pathological features in the lung. SP-D−/− mice, on the other hand, show chronic inflammatory alterations indicating a special importance of this molecule in regulating immune homeostasis and the function of the innate immune cells. Recent studies in our laboratory and others implied significant associations between changes in SP-D levels and the presence of airway inflammation both in animal models and patients raising a potential usefulness of this molecule as a disease biomarker. Research on wild-type and mutant recombinant molecules in vivo and in vitro showed that SP-D binds carbohydrates, lipids and nucleic acids with a broad spectrum specificity and initiates phagocytosis of inhaled pathogens as well as apoptotic cells. Investigations on gene-deficient and conditional over expressor mice in addition, provided evidence that SP-D directly modulates macrophage and dendritic cell function as well as T cell-dependent inflammatory events. Thus, SP-D has a unique, dual functional capacity to induce pathogen elimination on the one hand and control of pro-inflammatory mechanisms on the other, suggesting a potential suitability for therapeutic prevention and treatment of chronic airway inflammation without compromising the host defence function of the airways. This paper will review recent findings on the mechanisms of immune-protective function of SP-D in the lung.
Introduction
The pulmonary immune system is faced with the dual task of elimination of inhaled pathogens and maintenance of an inflammation-free mucosal environment. Although physical barriers filter out most of the inhaled material, a large amount of small particles (<10 μm in diameter) still reaches the distal air spaces where they encounter components of the innate immune system: alveolar macrophages, dendritic cells and the lung collectins including surfactant protein D (SP-D). Originally identified as a surfactant-associated protein [1], SP-D is expressed not only in the alveolar epithelial cells but also in the epithelium of proximal airways. Although it is constantly released into the airways and alveolar spaces, de novo SP-D expression markedly increases in response to acute lung injury and inflammation. Previous work from our laboratory and others showed that SP-D increased in a time-dependent fashion, coincident with the resolution of the inflammatory response several days after a single allergen challenge [2, 3] or ozone (O3) exposure [4] in mice. This increased expression was mediated by pro-inflammatory cytokines (IL-4/IL-13 and IL-6, respectively), and was preceded by mRNA transcription [3, 5]. Although a number of transcription factors were implicated in the rapid induction of SP-D mRNA, the exact pathways that can lead to an enhanced release of this lung collectin still need to be discovered.
A large body of evidence suggests that SP-D expression in the airways is essential for restoring immune homeostasis following the acute inflammatory response [2, 3, 6–8]. How this molecule works exactly is not well understood but the the C-terminal lectin ‘head’ is implicated in the direct suppression of immune and inflammatory cell activation. On the other hand, the N-terminal collagen tail of SP-D is thought to facilitate the clearance (phagocytosis) of pathogens and apoptotic cells (similar to the triple-helical collagen portion of other molecules with related basic structures such as SP-A, mannose-binding lectin and C1q). Identification of the membrane structures that can mediate these different functions is the current subject of intense investigations by a number of laboratories. The picture is complicated by the fact that SP-D undergoes post-translational modifications in response to oxidative changes in the lung chemical environment, which can profoundly alter the molecule’s complex oligomeric structure as well as it’s function.
Most of our current mechanistic information on the immunomodulatory role of SP-D are derived from studies conducted in mice. These studies, however, have shown not only relevance to human disease but also a potential for therapeutic manipulation of the pulmonary immune system. Models of low or no SP-D expression in different mouse strains and gene-manipulated mice mimic patients who have low SP-D levels in the lung due to genetic reasons or as a consequence of chronic inflammation. For example, SP-D-deficient mice display chronic airway inflammation and a constitutive activation of macrophages and dendritic cells in the proximal and distal air spaces with an increased presence of apoptotic cells. We recently described that current smoker COPD patients had markedly diminished SP-D levels in the lung and lower SP-D was associated with a more severe disease [9]. We also showed that bronchoalveolar macrophages from COPD patients with decreased SP-D had an inability to clear apoptotic cells [10]. Further, changes in serum SP-D levels in healthy volunteers were associated with changes in sputum dendritic cell phenotype in response to O3 inhalation [11]. Locally administered SP-D has been protective in mouse models of airway inflammation and it is possible that it will work in patients too. SP-D has a high degree of evolutionary conservation and showed biological cross-reactivity among different classes of vertebrates including humans [12–15]. SP-D is not only an anti-inflammatory agent but also an important opsonin that binds a wide range of common respiratory pathogens. Thus, unlike the traditional immunosuppressive therapies, an SP-D-based anti-inflammatory approach may have the advantage of a preserved host defence function.
The surfactant protein D structure and function: general considerations
SP-D was described in 1988 as a surfactant-associated collagenous glycoprotein with primary sequence similarities to SP-A, by the laboratory of E. Crouch [1]. Pulmonary surfactant, a phospholipid material that lines the air–liquid interface in the distal air spaces (discovered by Clements and colleagues in the early 1960s [16, 17]), contains four unique proteins: SP-A, SP-B, SP-C and SP-D. The small, hydrophobic molecules of SP-B and SP-C are essential in the structural organization of surfactant and the maintenance of low surface tension. On the other hand, SP-A and SP-D are large, hydrophilic molecules with host defence and immune regulatory functions. By the mid 1980s, cDNA and genomic sequencing revealed that, unlike SP-B and SP-C, lung collectins contain collagenous sequences [18–21] and belong to a separate class of molecules with similar collagen-like lectin structures.
The gene for SP-D is clustered together with the SP-A1, SP-A2 and mannose-binding lectin (MBL) genes on the long arm of chromosome 10q22–23 [22] and is located proximally to the centromere at about 80–100 kb from the SP-A2 gene. The collagen-like sequences are encoded by several short exons [19, 23–25]. The overall length of the collagen domain in each specific collectin is determined by the number of tandem exon duplications. The intron–exon organization (one intron is inserted between two exon duplicates) resembles the genes of non-fibrillar collagens [23] suggesting an evolutionary relationship. The neck domain and the carbohydrate recognition domain (CRD) are each encoded by single discrete exons. The similar structure of the collectin genes indicates that they evolved by duplication from a common ancestral gene, which was probably formed by exon shuffling between genes encoding non-fibrillar collagens and a primordial lectin [26].
While the first collagenous lectin, bovine conglutinin was discovered over a hundred years ago, the significance of carbohydrate recognition, a common essential function of all collectins, was only recognized in the 1950s (reviewed by Gupta and Surolia [27]). The first direct demonstration of lectin-mediated binding of SP-D to Gram-negative bacteria and the resultant bacterial aggregation was published in 1992 by Kuan and colleagues, suggesting a possible role for this molecule in pulmonary host defence[28]. The capability to differentiate between various carbohydrate moieties on pathogen surfaces is today considered an important functional feature of collectins.
In 1989, Tenner and colleagues showed that the triple-helical collagen structure of the first component of complement C1q [29] was similar to that of the collectins and that it could mediate phagocytosis through the C1q-receptor (C1qR) by monocytes/macrophages [30]. The C1qR was later shown to be identical to cell surface calreticulin [31–33]. Although binding to calreticulin does not explain all the biological activities of SP-D, these discoveries prompted a large array of studies on the capability of lung collectins to directly modulate innate immune cell function. Today, we have compelling evidence that SP-D is a potent cellular modulator of immune responses in the lung (reviewed by Wright and colleagues [34–37]). The most recent piece of this evidence comes from studies on genetically modified mice [38–43]. Even though there were discrepancies related to the different background strains or the methods used for genetic manipulations, the collectin gene inactivation generally increased susceptibility to infectious agents, enhanced acute inflammatory response to pathogen exposure and vulnerability to chronic inflammation [26, 38, 40, 42–57]. It is important to note that under normal, non-inflammatory conditions only the SP-D−/− mice but not the SP-A−/− mice display an abnormal immune activation, highlighting the importance of SP-D vs. SP-A in an essential regulation of the pulmonary immune system.
Role of surfactant protein D in allergen and pathogen uptake by phagocytes
The consequence of pathogen binding and aggregation is a more efficient phagocytic elimination. In addition to direct opsonization, collectin-enhanced allergenic particle and pathogen uptake by phagocytes may occur through the regulation of cell-surface pattern recognition receptor expression [34]. Pattern recognition receptors play a major part in host defence and control tissue homeostasis within multicellular hosts [58]. Interactions between collectins and these receptors are significant during both infectious and allergen-induced challenges in the lung. For instance, enhanced mannose receptor expression was shown on human monocyte-derived macrophages involving both the attached sugars and the collagen-like collectin domain. In contrast, alveolar macrophages from collectin-deficient mice had reduced mannose receptor expression when compared with wild-type mice [59]. Such a positive regulation between collectins and pattern recognition receptors was also suggested in another study, using a model of allergic sensitization: Lack of SP-D in gene-deficient mice resulted in a failure to increase TLR4 expression during allergic inflammation in comparison with wild-type mice [60].
Surfactant protein D pattern recognition: carbohydrate and lipid binding
A large body of evidence suggests that CRD-dependent attachment is important between collectins and inhaled particles (reviewed by Hakansson and Reid [61]) and that there is an essential role for the collagen-like domain and the degree of oligomerization for pathogen aggregation and subsequent phagocytosis [62]. SP-D recognizes a wide variety of pathogen surfaces by a broad specificity that is achieved by a very open binding pocket in the CRD. Pattern recognition by the collectins entails the ability to bind lipopolysaccharide, lipoteichoic acid and mannan over the less suitable terminal monosaccharides and macropatterns of mammalian glycoconjugates. Distinguishing between self and non-self can be also achieved through the recognition of a specific orientation of hydroxyl groups of hexoses. These are predominantly found in mannose, fucose, N-acetyl-D-glucosamine and glucose [63], which are often located in repeating structures on the surface of microbes as a result of glycosylation enzymes that differ from higher organisms. Further, organization of the individual low-affinity sugar-binding sites into trimeric arrays in the collectin CRD allows multivalent ligands to have a matching ligand topology (bridging a distance of 45–53Å), which is important to achieve high-affinity binding [26, 33, 35].
In addition to the common carbohydrate-binding activities the collectins have, SP-D is characterized by a relative monosaccharide preference [64]. For instance, while both SP-A and SP-D could recognize the carbohydrate structure of Aspergillus fumigatus [65–67], the interaction with SP-A is inhibited with mannose but with SP-D it can be blocked with maltose [68]. The order of carbohydrate preference for human SP-D in solid-phase competition assays using maltosyl-bovine serum albumin as the ligand, is approximately: maltose (inositol)>glucose, mannose, fucose>galactose, lactose, glucosamine>N-acetylglucosamine (reviewed by Crouch [69]). Saccharide specificity may be determined by the trimeric neck+CRD domains, given that bacterially expressed trimeric CRDs show the same saccharide-inhibition profile as natural human SP-D [70].
Binding of SP-D to lipid moieties may also have important functional consequences. Interactions with surfactant lipids is required to optimally orient the CRDs in relation to the air/lipid or lipid/hypophase (the aqueous lining of the pulmonary epithelium) interface to facilitate the uptake of foreign materials embedded in the surfactant layer of the proximal and distal air spaces. Exposed hydrophobic sites could also mediate interactions with hydrophobic domains displayed on various microorganisms, and perhaps stabilize relatively weak lectin-dependent interactions [69]. The major surfactant-associated ligand of SP-D is phosphatidylinositol (PI) but it also binds to glucosylceramide. The physiological significance of this binding is supported by a compelling demonstration that SP-D−/− mice have striking pathological alterations in the lung including increased surfactant DPPC pool size and abnormalities in the morphology and function of type II alveolar epithelial cells and alveolar macrophages and consequentially in lung structure [26, 38]. Because it is unlikely that SP-D has a primary role in normal surfactant metabolism, these changes could be secondary to an enhanced inflammatory state of the SP-D−/− lung.
Binding to nucleic acids and removal of apoptotic cells
SP-D binds nucleic acids from a variety of origins, including components of disrupted cells (cell debris), liberated and cell surface DNA, thereby contributing to general clearance activities. SP-D effectively binds and aggregates alveolar macrophage DNA, and it enhances the uptake of DNA by human monocytic cells. Binding to cell-surface DNA might be one mechanism by which collectins mediate enhanced phagocytosis of apoptotic cells. Indeed, SP-D is capable of recognizing and binding to apoptotic cells and facilitates their removal by macrophages [71–75]. Collectins may recognize damaged cells via either their CRD [76], collagen region [72] or independently from those [77].
Several receptors were proposed as candidates to mediate apoptotic cell removal by collectins. Among these, currently the calreticulin/CD91 complex appears to be the most accepted [74]. The role of the collagen domain-mediated calreticulin/CD91 binding, however, was contradicted by an in vivo study in which the treatment of SP-D−/− mice with a C-terminal mutant SP-D molecule (that does not contain the collagen domain) restored the macrophage defect and reduced the number of apoptotic cells in the lung [55].
Aggregation and phagocytosis of inhaled pathogens and allergens
Critical evidence for the significance of collectin-mediated phagocytosis of inhaled pathogens were provided by studies on bacterial infection. Collectin gene-targeted mice [47] are highly susceptible to group B Streptoccus [78, 79], Haemophilus influenzae [44] and Pseudomonas aeruginosa infections [80]. Collectin-enhanced phagocytosis and aggregation before uptake was also important for Escherichia coli [28], Streptococcus pneumoniae, Staphylococcus aureus [81] and Cryptococcus neoformans [82]. Phagocytosis through direct SP-D binding was shown for P. aeruginosa [83], H. influenzae [44] and the unencapsulated variants of Klebsiella pneumoniae [84]. It is important that SP-D is also involved in neutralization and clearance of inhaled endotoxin [85]. SP-D prefers rough LPS to the smooth LPS [86] and unlike SP-A, SP-D does not recognize lipid A as a ligand [87]. In addition, the LPS component of the pathogens has been identified as a major ligand for SP-D on E. coli [28], K. pneumoniae and P. aeruginosa [70]. The binding occurs via the CRD, and accordingly is Ca dependent.
Fungal pathogens and allergens frequently gain access to the host via the respiratory tract. Under normal conditions, fungal infections of the lung are very rare, although fungi like C. neoformans and A. fumigatus are commonly found in the environment and are continuously inhaled. This suggests that efficient defence mechanisms must exist to protect against these pathogens. The acidic capsule of the fungi is the most important in their pathogenesis as it inhibits phagocytosis and is poorly immunogenic, and thus it enables the infectious agents to persist for extended periods in the host [88, 89]. Both rat and human SP-D can bind to Pneumocystis carinii via its CRD [90] and mediate its aggregation in vitro [91]. Unlike SP-A, SP-D enhances the agglutination of unencapsulated C. neoformans [82]. The clinical importance of these observations is illustrated by the fact that a heightened susceptibility of children with cystic fibrosis to a range of infectious and allergenic pathogens is linked with extremely low levels of SP-A and SP-D in the BAL fluid [92].
Depending on the host’s immunological and genetic status, different types of A. fumigatus-associated respiratory diseases have been recognized, the most common of which is allergic bronchopulmonary aspergillosis (ABPA). When the lung of an immunocompromised subject is exposed to A. fumigatus, a systemic infection, invasive aspergillosis ensues. In an immunocompetent subject, however, it often results in IgE-mediated asthma. A number of studies using murine models of ABPA or allergic airway sensitization have emphasized the importance of lung collectins in protection against A. fumigatus-induced pathologies. Treatment with recombinant collectins not only protected against mortality but also inhibited the Ig, eosinophil and Th2 cytokine responses in murine models [3, 93–97].
In addition to fungi, both lung collectins were shown to bind inhaled allergenic particles of a wide range of sources. For example, pollen starch granules extracted from Dactylis glomerata (Cocksfoot grass) were aggregated by the CRD of SP-D [98, 99] and this interaction inhibited the release of β-hexosaminidase by peritoneal mast cells. SP-A also readily bound water-extractable particles of various pollen grains including pollen from Populus nigra italica (Lombardy poplar), Poa pratensis (Kentucky blue grass), Secale cerale (Cultivated rye) and Ambrosira elatior (Short ragweed) [100]. SP-D and SP-A were also shown to bind to whole mite extracts (Dermatophagoides pteronyssimus) and the purified allergen Der p 1 in a carbohydrate-specific and calcium-dependent manner [101, 102]. Lung collectins, in fact, inhibited mite-extract-specific IgE binding and histamine release in vitro [103] indicating a protective importance of calcium and sugar-dependent allergen binding by collectins [104].
Binding to cell surface receptors: the importance of a dual biological activity
Binding through the collagen tail
The first indications of collectin binding to immune cells came from studies in 1989 investigating the C1qR function on alveolar macrophage [29]. The details of cell function modulation by the lung collectins are still not well understood. Because these proteins are ‘sticky’ and strongly bind carbohydrate, lipid and protein ligands, promiscuous binding partners and several putative receptors have been identified. The amino-terminal collagen tail of SP-D may serve as a ligand for the collectin receptor [100, 105]. This receptor, originally termed cC1qR, was later shown to be identical to calreticulin. As the endoplasmic reticulum (ER) membrane fuses with the plasma membrane during phagocytosis, calreticulin (which is a cytoplasmic molecule with an ER retention sequence) is expressed on the cell surface during this process. Calreticulin does not have a transmembrane domain but appears to mediate signalling through the LDL-receptor-related protein, CD91 [32] which is found on macrophages [35, 106, 107]. As calreticulin, CD91, SP-A and SP-D can all bind multiple ligands in a promiscuous manner, the importance and specificity of this receptor–ligand mechanism need further investigations. Although the calreticulin/CD91 complex explains some of the biological actions mediated by SP-D, it is still unclear whether ER recruitment and calreticulin binding are necessary and/or sufficient for phagocytosis [32, 74, 105, 108, 109]. Through collagen receptor binding, collectins were shown to up-regulate pattern recognition receptor expression [59], stimulate cytokine [110–115] and reactive molecular species production [116, 117], induce chemotaxis [118–120] and influence intracellular signalling pathways [109, 119, 121] in innate immune cells.
The collagenous tail may also act as a ligand for receptors other than calreticulin. A 340-kDa glycoprotein (gp-340) was originally identified as a pulmonary SP-D-binding protein [122] but later it was shown to also bind SP-A [123] in the presence of calcium but independent of the CRD region. This protein has both a soluble and a membrane-associated form and functions as an opsonin receptor for various pathogens [124–126]. Gp-340, however, has a questionable significance as its presence on the membrane surface was not necessary to mediate collectin function (chemotaxis) on macrophages [123].
Binding through the carbohydrate recognition domain
CRD-specific interactions with lung cells may be mediated through a number of receptors. Apart from pathogen binding, the CRD region has been involved in modulating functions of various cell types of the immune system. The ability of SP-D to exert chemotactic activity on neutrophils and monocytes for instance was suggested to depend on its lectin activity [127]. In fact, the functionally active part of the lectin head of the lung collectins in immunosuppression appeared to be the CRD [52, 55, 81, 128], which was specifically inhibited by treatment with sugars (glucose) and calcium chelation [103].
Through the CRD, SP-D can also recognize, bind and modulate function of CD14 [129, 130], TLR2 [131, 132] and TLR4 [121, 132]. SP-D bound both TLR2 and TLR4 in a concentration- and calcium-dependent manner. Epitope mapping with recombinant proteins consisting of the CRD or the neck domain plus CRD, indicated that human SP-D binds the extracellular domains of TLR2 and TLR4 through its CRD by a mechanism different from its binding to PI and LPS [132].
Dual collectin activity on innate immune cells
Early indications of dual collectin activity on macrophages came from studies on SP-A and mycobacterial infections: SP-A enhanced phagocytosis of Mycobacterium tuberculosis and Mycobacterium bovis BCG by alveolar macrophages [133, 134] and augmented the production of IL-6 in the presence of infection. In the absence of infection however, SP-A inhibited IL-6 [135]. These studies suggested that the nature of collectin-mediated immune functions may depend on whether an immune intervention is needed following infection or allergen exposure or whether an immunosuppressive/anti-inflammatory action would be necessary to prevent chronic tissue damage during inflammatory changes. SP-A (but not C1q and type IV collagen) was also shown to inhibit Candida albicans-induced cytokine production by macrophages [136] independent of Candida-SP-A binding or C1qR occupancy. These data confirmed that collectin inhibition of innate immune cells occurs by a direct binding to the cell through the lectin head and it is not related to the collagen receptor binding.
Gardai and colleagues provided in vitro as well as in vivo evidence that SP-A and SP-D (but not MBL or C1q) binding to signal inhibitory regulatory protein (SIRP-α) on macrophages through their lectin head inhibited LPS- and H2O2-stimulated cytokine production [109] as well as phagocytosis [137]. SIRP-α is a type I transmembrane receptor of the Ig superfamily expressed on dendritic cells as well as other leucocytes of the myelogranulocytic lineage (monocytes, macrophages and neutrophils). The SIRP-α intracellular domain contains two ITIM as well as two immunoreceptor tyrosine-based switch-like motifs, which recruit Src homology region 2 domain-containing phosphatase (SHP)-1 and -2, thus inhibiting intracellular signalling. SIRP-α is important in regulating a number of dendritic cell functions and is particularly involved in dendritic cell maturation and migration [138].
According to the ‘head or tail hypothesis’, (Fig. 1) SP-A and SP-D interacting with pathogens via their lectin head, and presenting their collagenous tail on phagocytes to calreticulin/CD91 induce phagocytosis and a pro-inflammatory response [109]. In subsequent studies, the Gardai group demonstrated that SP-A or SP-D binding to SIRP-α on residential alveolar macrophages via the CRD suppressed phagocytic function of these cells through SHP-1 signalling [137]. Thus, SP-D is capable of eliciting differential and indeed opposing functions depending on the binding orientation [109]; CRD binding by the native molecule mediates inhibitory signals while collagen binding induces cell activation. While this hypothesis is very elegant and could explain some of the confusions that surround the role of lung collectins in innate immune regulation, a number of questions remain unanswered. For example, in case of SP-D, given the cruciform dodecameric structure, it is still unclear, how the collagen tail may bind calreticulin when pathogens are present. It is also difficult to understand why there are striking differences between the pulmonary phenotype of the SP-A−/− and SP-D −/− mice if SP-A and SP-D operated the same way. And finally, what is the relevance of all the in vitro and animal findings to how the human pulmonary immune system functions?
Fig. 1.
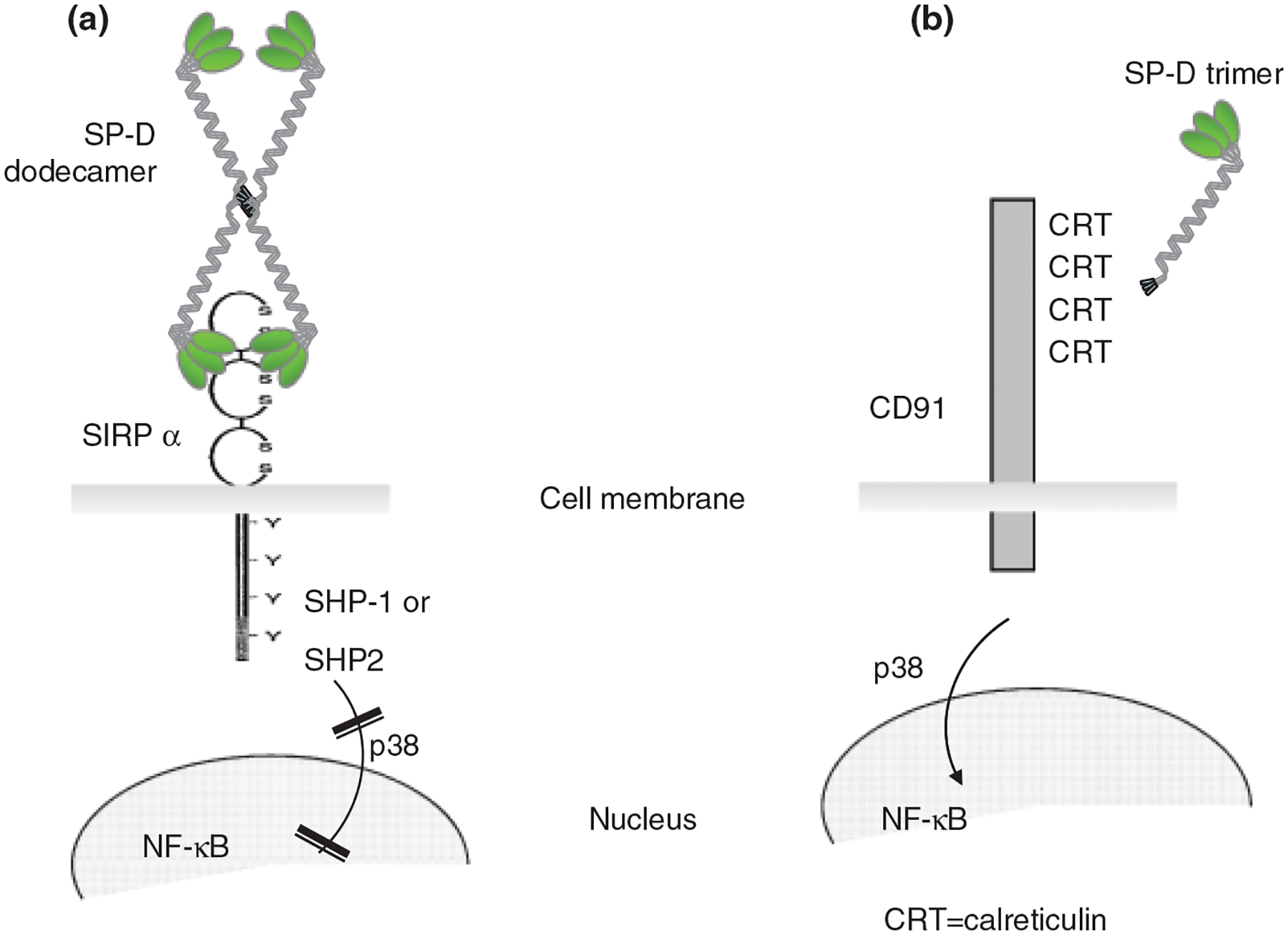
The ‘Head or Tail Hypothesis’ (adapted from Gardai et al. [109]): SP-D is capable of differential binding through either the CRD or the collagen domain to cell membrane receptors and eliciting respective anti- or pro-inflammatory, signalling pathways. (A) SP-D is assembled as a lectin head containing the carbohydrate recognition domain (CRD), a neck region and a collagenous tail. SP-D (a 43 kDa monomer) under normal baseline conditions forms a higher order quaternary structure (usually a dodecamer) assembled from four homotrimers with the N-terminal collagen tail of the molecule hidden in the centre of the oligomeric structures bound together by cysteine residues. The C-terminal CRD binding to signal inhibitory regulatory protein (SIRP-α) inhibits cytokine production [109]. SIRP-α is expressed on dendritic cells and macrophages. It contains two ITIM as well as two immunoreceptor tyrosine-based switch-like motifs, which recruit Src homology region 2 domain-containing phosphatase (SHP)-1 and -2, thus inhibiting intracellular signalling [138]. (B) When the collagen domain of SP-D is exposed in lower oligomers (trimers), it can initiate binding to the collagen-receptor calreticulin/CD91. Ligation of this receptor complex induces pro-inflammatory cellular functions mediated by the activation of p38 and NF-κB signalling molecules. Oxidative changes under pro-inflammatory circumstances can alter the structure of the native SP-D [119, 120, 143] leading to an impaired inhibitory function of the CRD [143] or de-oligomerization of the SP-D molecule from dodecameric to trimeric forms exposing the otherwise hidden N-terminal collagen domain [119].
Susceptibility to oxidative damage
One possible way to make the SP-D structure available for the reversible ‘head or tail’ orientation is by its susceptibility to oxidative post-translational modifications. The SP-D monomer forms a higher order quaternary structure (the normal, native SP-D is a dodecamer assembled from four identical homotrimers) [139]. Because of its long collagen tail, SP-D is capable of forming combined structures of these multimers, the so-called ‘fuzzy balls’ with a total mass of several million Da and a size of about 100 nm [140]. Under baseline conditions, the N-terminal collagen tail of the molecule is well hidden in the centre of the oligomeric structures bound together by cysteine residues. Unmasking the collagen domain of SP-D in lower oligomers (trimers) may initiate or amplify proinflammatory cellular changes that can play a significant modulatory role during airway inflammation. When exposed, the collagen tail of SP-D can serve as a ligand for the collagen-receptor calreticulin/CD91 [32, 74, 141, 142]. Ligation of this receptor complex induces pro-inflammatory cellular functions mediated by the activation of p38 and NF-κB signalling molecules [109, 119]. Through the collagen receptor, collectins up-regulate pattern recognition receptor expression, stimulate cytokine and reactive molecular species production, induce chemotaxis and influence intracellular signalling pathways in innate immune cells [109, 119, 120].
Under pro-inflammatory circumstances, oxidative changes can alter the structure of the native SP-D [119, 120, 143]. Nitration of the molecule, for example, resulted in an impaired CRD function [143]. Nitrosylation, on the other hand, induced de-oligomerization of the SP-D molecule from dodecameric to trimeric forms exposing the otherwise hidden N-terminal collagen domain [119]. We have recently showed that O3 inhalation induced trimeric SP-D formation in the airways of mice [120, 144]. In two recent publications [119, 120], bleomycin-induced acute lung injury resulted in oxidative damage to the SP-D molecular structure with a loss of the dodecameric form. Investigations on the structural integrity of SP-D in the BAL fluid using native gel electrophoresis showed the appearance of SP-D trimers, in addition to dodecamers in the O3-exposed mice [120]. Based on these results, we propose that inflammation induces conformational changes in SP-D and the appearance of trimers. The trimeric form may function differently as it has an exposed collagen tail (as well as a C-terminal head). Because the collagen tail can serve as a ligand for collectin receptors such as the calreticulin/CD91 [74] and engagement of this receptor may induce pro-inflammatory signals in dendritic cells or macrophages, it is possible that there are domain-specific changes in the SP-D molecule induced by oxidative damage. Therefore, loss of SP-D immunoprotection and activation of innate immune cells during allergic airway inflammation, could be due to impaired CRD function and/or exposure of the collagen domain with a calreticulin/CD91-binding ability. Indeed, in a model of O3-induced exacerbation of allergic airway changes, we recently found that the appearance of abnormal, lower molecular weight forms of SP-D was associated with a heightened activation of alveolar macrophages and dendritic cells in the airways [144]. The pathogenic importance of oxidative modifications in the SP-D molecule is however still hypothetical and will require further investigations.
Collectin regulation of macrophage function
Pathogen recognition and binding by SP-D lead to aggregation, phagocytosis and a regulated release of inflammatory mediators. The importance of this mechanism is supported by evidence obtained using gene-targeted mice in models of infection and allergic sensitization as these animals show an impaired clearance of a number of bacteria, virus and fungal pathogens. A large body of studies also strongly suggests that direct binding of both collectins to phagocytes in the presence and absence of pathogens may result in different outcomes. However, under normal conditions, the lung epithelium must remain infection and inflammation free. That the lung collectins serve an anti-inflammatory function has been clearly demonstrated more than a decade ago. Both SP-D and SP-A inhibited TNF-α and other pro-inflammatory cytokines and chemokines by LPS-stimulated human alveolar macrophages in vitro [136, 145]. The immune regulatory effects of the lung collectins were later extensively studied in SP-A−/−, SP-D−/− and double (SP-A/SP-D) knockout mice. Interestingly, while the SP-A−/− mice appear normal under baseline conditions, SP-D−/− and double knockout mice show serious signs of constitutive activation of the immune system indicating that SP-D has an essential immunoprotective role. That this immunoprotection parallels pathogen elimination is demonstrated by the fact that in spite of an exaggerated inflammatory response, upon injury or infection mice lacking SP-D have an impaired host defence capability [38, 43, 44, 49, 146, 147]. Cells of both the innate and adaptive immune system in SP-D−/− mice display multiple abnormalities including altered morphology, constitutive activation with a heightened release of cytoplasmic enzymes, cytokines, chemokines and oxidants. The ensuing disease conditions of SP-D−/− mice include an accelerated development of emphysema, susceptibility to various inflammatory stimuli and allergic sensitization and infections.
Indeed, a hallmark of the SP-D−/− lung is the presence of enlarged, foamy macrophages with an increased proinflammatory mediator expression and high levels of MMP-9 and MMP-12 [43]. That SP-D protects against pro-inflammatory mediator release including the ones that favour the development of a Th2-type inflammation was more recently confirmed in a model of allergic sensitization. Culture of alveolar macrophages from oval-bumin-sensitized C57BL/6 mice together with SP-D and allergen resulted in an increased production of the immunosuppressive IL-10 as well as IL-12, and IFN-γ cytokines unfavourable for development of Th2-type changes [148]. SP-D also directly inhibited Th2 cytokine release in allergen-stimulated splenic mononuclear cells in vitro, derived from Balb/c mice sensitized with A. fumigatus [3].
In studies performed in the early 90s, SP-D and SP-A were shown to enhance the amount of oxygen radicals produced by alveolar macrophages and neutrophils [117] and two independent studies showed that SP-A stimulated the release of reactive oxygen metabolites [116, 149]. These reported effects, however, were not confirmed in later studies using SP-D knock-out mice, which had increased oxidant production [44, 147]. In support of the protective function of SP-D against free radical release, a recent report on allergic sensitization in mice showed that recombinant SP-D significantly inhibited allergen and LPS-induced NO production by isolated alveolar macrophages [150]. Interestingly, lung collectins had additional protective effects as they appeared to be potent endogenous inhibitors of lipid peroxidation and oxidative cellular injury [151].
Alveolar macrophages need to home to the lung in order to perform their immune surveillance function without the presence of inflammation. Lung collectins have significant chemotactic activities on alveolar macrophages. Both SP-A- and SP-D-stimulated chemotaxis in a concentration-dependent manner [152] and induced directional actin polymerization in alveolar macrophages in vitro [123]. In this latter study, it was also demonstrated that this effect is cell and collectin specific, a SP-A did not stimulate the chemotaxis of monocytes [118], while in another study SP-D attracted these cells [127]. Binding of collectins to macrophages probably occurred independently from the C1qR, because C1q did not show the same chemotactic effect on macrophages.
Taken together, these works indicate that SP-D recognizes and binds pathogens that leads to aggregation, phagocytosis and the regulated release of inflammatory mediators. The dual role of SP-D in immunoprotection and pathogen elimination is demonstrated in gene-deficient mice. In spite of an exaggerated inflammatory response, these animals show an impaired clearance of a number of bacteria, virus and fungal pathogens.
Effects of surfactant protein D on dendritic cells
Dendritic cell maturation
While the role of collectins in regulating alveolar macrophages has been extensively studied, the literature on how these innate immune molecules affect the function of dendritic cells is scant. Studies describing the effects of lung collectins on dendritic cell function indicated a regulatory effect in antigen presentation and T cell stimulation [153–155] but there is virtually no information on the effects of SP-A or SP-D on differentiation or migration of the pulmonary dendritic cell subpopulations. Our studies suggested that lack of SP-D in the lung of SP-D−/− mice resulted in a dendritic cell population with a pro-inflammatory, myeloid phenotype and constitutive TNF-α expression. In contrast, the administration of SP-D in bone marrow dendritic cell cultures suppressed the activation marker and TNF-α expression [156] indicating that native SP-D inhibits the maturation and activation of the pro-inflammatory dendritic cells.
An important implication of both phagocytosis and the direct binding of collectins to antigen-presenting cells is the ensuing regulation of adaptive immune response. Studies on this function showed that SP-A inhibited while SP-D augmented antigen presentation by bone marrow-derived (myeloid) dendritic cells [153, 154]. SP-D, however, inhibited antigen presentation when dendritic cells were isolated from the lung [155]. Under baseline conditions, dendritic cells reside in the lung tissue in an immature state. These resting cells are scattered throughout in the bronchial and alveolar wall capturing inhaled pathogens but unable to present them to T cells. In response to ‘danger’ signals such as pathogens, proinflammatory cytokines, or necrotic cells, dendritic cells start to mature and switch from an antigen-capturing to an antigen-presenting and a T cell-stimulating, pro-inflammatory state [157]. Although all mature dendritic cells share a common ability to process and present antigen to naïve T cells for the initiation of an immune response, they differ in origin, migratory patterns, localization and cytokine production [158]. Plasmocytoid dendritic cells make up the majority in the lung under baseline conditions [159] and these cells were shown to be tolerogenic in allergen-induced inflammation. On the other hand, myeloid dendritic cells migrate rapidly to the lung during a Th2-type inflammation and exert potent Th2 cell activation [160–164].
Dendritic cells in the BAL of SP-D−/− mice had a myeloid (bone marrow derived) phenotype and constitutive TNF-α expression. In bone marrow-derived dendritic cell cultures, an administration of SP-D suppressed both the activation marker and autocrine TNF-α expression. Thus, SP-D can alter the differentiation and inhibit activation of immature dendritic cells through the inhibition of TNF-α [156]. Interestingly, dendritic cells in the lung of SP-D−/− mice were unable to migrate to the regional lymph nodes and accumulated instead in the airway submucosal tissue (S. Kierstein and A. Haczku, unpublished observation), possibly due to a markedly increased expression of thymus and activation-regulated chemokine (TARC) [3].
Dendritic cell migration
Dendritic cell migration is essential for both the onset and resolution of the inflammatory airway changes. There are a number of potential chemokine receptors that can mediate epithelium-directed or lymph-node-directed migration. For example, Ccr2, Ccr4 and Ccr5 could mediate pro-inflammatory migration of immune cells into the airway epithelium. Lymph node homing of dendritic cells and lymphocytes can be facilitated by Cxcr4 and Ccr7. Nevertheless, in our model of A. fumigatus-induced allergic airway inflammation, the Ccl17-Ccr4 and Ccl19/21-Ccr7 ligand–receptor pairs were significantly up-regulated at the mRNA level in a time-dependent manner indicating the importance of these genes in immune cell migration in response to allergen inhalation. The expression of the Ccr4 ligand Ccl17 was selectively induced while the expression of Ccr7 was selectively abolished in vivo in SP-D−/− mice [165, 166]. The significance of this effect of SP-D on the Ccr7 expression is that this receptor was shown to be specific not only for lymph node homing but also for immune tolerance [167]. Ccr4 is considered to be specific for Th2 cells and activated dendritic cells; therefore, this receptor and its ligand Ccl17 (TARC) are very relevant to studies on the allergic airway response.
Ccl17 is produced by epithelial cells, macrophages and dendritic cells and is responsible for attracting Th2 lymphocytes and activated myeloid dendritic cells to the airway submucosal tissue through its receptor, Ccr4, which is expressed specifically on Th2 cells and activated dendritic cells [164]. Our studies showed that myeloid dendritic cells accumulated in the airways of SP-D−/− mice [156] paralleled by the elevated expression of TARC [6] indicating that the presence of SP-D maybe important in suppressing the release of this pro-inflammatory mediator and a consequent epithelial migration of dendritic cells. Activated myeloid dendritic cells themselves release large amounts of TARC, creating a ‘vicious’ Th2-type inflammatory circle, which explains the susceptibility of the SPD−/− mice to allergic sensitization [6]. Because TNF-α is a potent activator of TARC release, it is possible that native SP-D or the CRD prevents epithelial migration of myeloid dendritic cells by inhibiting the TNF-α/TARC pro-inflammatory axis.
Following the uptake of inhaled pathogens, immature dendritic cells mature and start migrating towards the mediastinal lymph nodes where they exert their antigen-presenting and T cell-stimulating function. Lymph-node-directed migration also serves the purpose of confining the immune response to the lymphoid area leaving the airways pathogen and inflammation free. This process is driven by Ccl19 or Ccl21, both of which act on the chemokine receptor Ccr7. The Ccr7 expression on dendritic cells was shown to be important not only in lymph node homing but also in protecting from autoimmune pathologies, atherosclerosis and allergic airway sensitization. We have found that the Ccr7 gene was highly expressed in the lung of wild-type mice 48 h after allergen inhalation, coincident with the inflammatory resolution [166]. Expression of Ccl19 and Ccr7 in wild-type mice paralleled the appearance of labelled dendritic cells in the thoracic lymph nodes. SP-D−/− mice, however, were not able to induce Ccr7 expression and had virtually no CFSE-labelled cells in the thoracic lymph nodes after allergen challenge [166]. These intriguing data suggested that the presence of SP-D is necessary to suppress TNF-α/TARC-mediated epithelial migration and to promote lymph node-directed migration (Fig. 2). These speculations, however, will need to be substantiated and the significance of SP-D in dendritic cell migrations should be clarified.
Fig. 2.
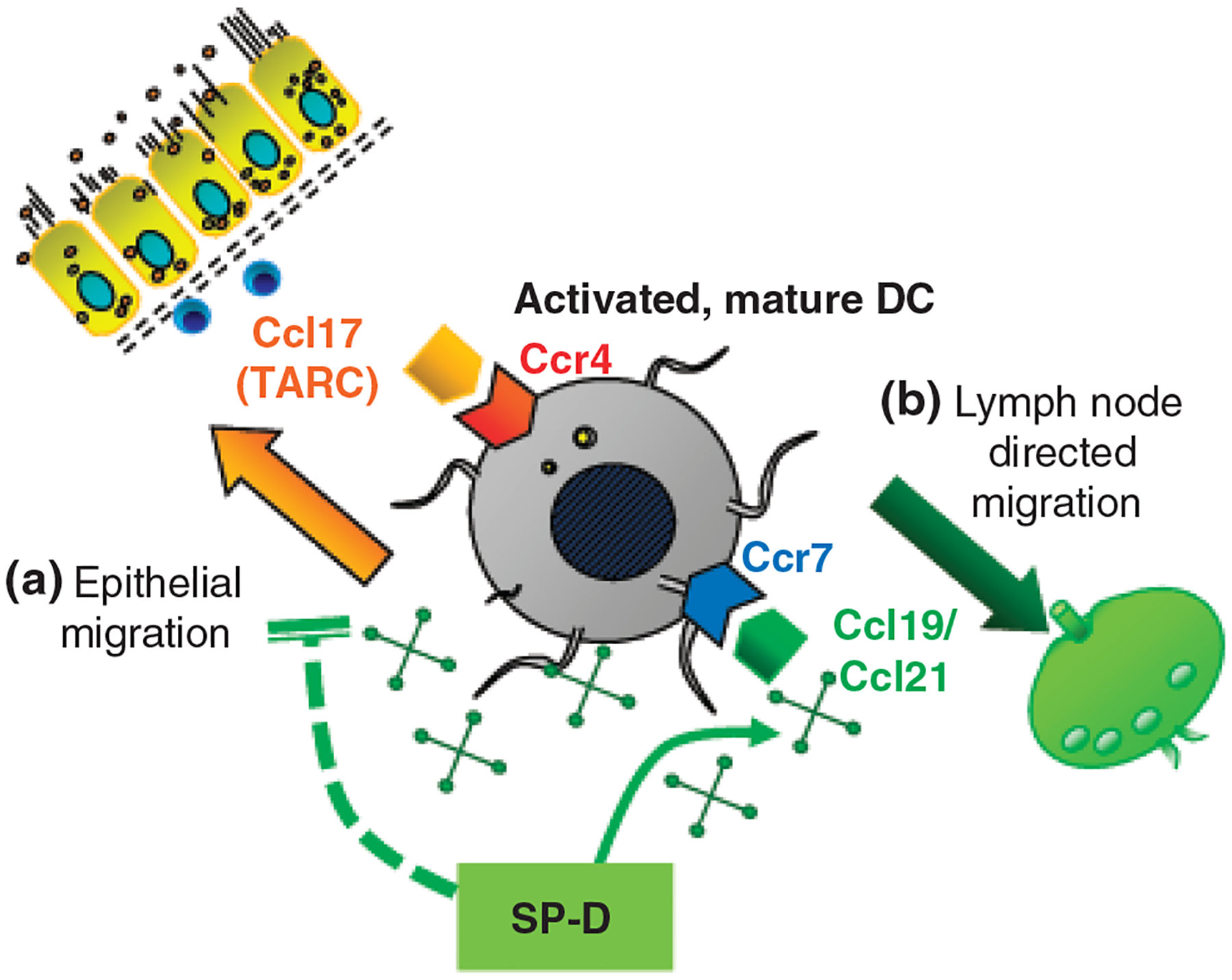
Hypothetic role of SP-D in dendritic cell migration. In Aspergillus fumigatus-induced allergic airway inflammation, the Ccl17-Ccr4 and Ccl19/21-Ccr7 ligand–receptor pairs were significantly up-regulated at the mRNA level in a time-dependent manner in mice [4, 165, 166]. (a) SP-D prevents the epithelial migration of myeloid dendritic cells by inhibiting autocrine Ccl17 release. Ccl17 (thymus and activation-regulated chemokine, TARC) is responsible for attracting Th2 lymphocytes and activated myeloid dendritic cells to the airway submucosal tissue through its receptor, Ccr4 which is expressed specifically on Th2 cells and activated dendritic cells. (b) SP-D promotes the uptake of inhaled pathogens and the migration towards the mediastinal lymph nodes. In addition to the initiation of naïve T cells, lymph-node-directed migration serves the purpose of confining the immune response to the lymphoid area leaving the airways pathogen and inflammation free. This process is driven by Ccl19 or Ccl21, both of which act on the chemokine receptor Ccr7.
Effects of surfactant protein D on the adaptive immune response
Pulmonary lymphocytes are hyporesponsive to a variety of antigenic stimuli when compared with peripheral blood leucocytes. Early studies provided evidence that the alveolar lining fluid contributes to the induction and maintenance of such cellular hyporesponsiveness in vivo and in vitro [168–170]. These observations were more recently confirmed by studies on gene-deficient mice. While no significant alterations have been observed in SP-A-deficient mice, in SP-D−/− animals an increased percentage of both CD4+ and CD8+ T cells were found together with an increased expression of the activation markers CD69 and CD25 in the BAL [3, 41]. That the presence of SP-D can directly suppress T cell function was demonstrated in vitro on isolated lymphocytes in a number of studies [3, 15, 171, 172]. What are the possible mechanisms of T cell SP-D interactions? Binding and sequestering of the antigens may be one possibility to interfere with T cell activation. However, as SP-D was also capable of inhibiting mitogen-induced T cells [3, 14], blocking of antigen-T cell receptor interaction may not be the major pathway for the inhibition of lymphocyte function. Interestingly, SP-A was shown to inhibit T cell proliferation and decrease IL-2 production [173] via its collagen-like domain through surfactant protein receptor 210 (SPR-210; named after its 210 kDa size) [174]. However, no such receptor has been identified for SP-D. Further, SP-D was implicated in T cell inhibition in an IL-2-independent manner but cell viability was not affected by this collectin [175]. The SP-D effects on mitogen- and Der p-stimulated lymphocytes were attenuated when cells were derived from asthmatic children with acute attacks [103]. These results indicated that T cells that are already activated may be resistant to the inhibitory effects of SP-D.
The in vivo consequences of SP-D interaction with T cell-dependent inflammatory events were demonstrated in various models of allergic airway sensitization. Treatment of allergen-sensitized mice with recombinant or purified SP-D was effective in reducing the eosinophilic inflammation and specific IgE production [93–96, 150, 176]. Furthermore, airway hyperresponsiveness was also inhibited and there was a shift from a Th2 cytokine pattern towards a Th1 response ex vivo [93, 94] and in vitro [3, 148]. SP-A- and SP-D-deficient mice exhibited intrinsic hypereosinophilia [57], increased IL-13 [3, 57, 60] and TARC [3, 177] levels in the BAL indicating a diminished control of inflammation in these animals. The currently studied SP-D−/− mice are either of a mixed background or have a C57BL/6 genotype. An important question would be whether the lack of SP-D would result in more prominent effects in Balb/c mice, which are intrinsically more susceptible to allergic sensitization than C57BL/6 mice. The relevance of this question to disease is that individuals who are genetically predisposed to allergic sensitization of the airways may be protected by sufficient levels of SP-D. However, if susceptibility to allergy is paralleled with low SP-D production (due to genetic or acquired factors), a more severe disease outcome may ensue.
Significance
During the inflammatory airway response, SP-D plays immune-protective roles at multiple levels. The mechanistic significance from the onset to resolution of the inflammatory airway response is highlighted by the fact that SP-D is capable of the differential (pro- or anti-inflammatory) regulation of innate immune cell function. The outcome of SP-D-cell interactions may be dictated by the binding orientation (either via the CRD or through the collagen region [33, 109]), receptor signalling and the cytokine/inflammatory mediator milieu of the local environment [178]. The ability to achieve differential effects on the innate immune response by the same molecules carries obvious benefits. Based upon recent human studies on genetic polymorphisms and chronic inflammatory lung disease, it appears that a deficiency in expression and/or function of SP-D is associated with an enhanced susceptibility to infections and inflammation, particularly in severe conditions.
Four polymorphisms have been identified within the SP-D gene, including the non-synonymous Met11Thr, Ala160Thr, Ser270Thr, and a synonymous Ala286Ala mutation. The Met11Thr and Ala160Thr polymorphisms in the amino terminal and collagen domain have a frequency exceeding 20%, whereas the Ser270Thr and Ala286Ala polymorphisms are relatively rare. To date, only polymorphism in amino acid 11 has been associated with disease [179, 180]. Importantly, individuals with the Met11Thr-encoding genotype had significantly lower SP-D serum levels than individuals with the Met11Met genotype [181]. The Met11Thr allele is linked with protection from severe RSV infection in infants, whereas the Met11Met allele is associated with RSV bronchiolitis, and the Thr11-coding allele is associated with a susceptibility to M. tuberculosis infection [180, 182]. Studies have suggested that the Met11Met allele produces SP-D of both low- and high-molecular-weight forms (monomers, trimers and dodecamers) whereas the Thr11 produces mainly low-molecular-weight structures (monomers) [181]. It was also shown that the high-molecular-weight form of SP-D has an increased binding affinity to complex microorganisms while the low-molecular weight SP-D preferentially bound LPS. Further, in a recent study, significantly more atopic black children had the Met allele compared with non-atopic black children, indicating a differential regulation of allergic sensitization [177]. It appears, therefore, that the different alleles of codon 11 can influence SP-D oligomerization, which can result in markedly different SP-D serum levels and function [181].
Nucleotide polymorphisms that do not result in amino acid changes were also identified in the lung collectin genes. These are called ‘silent’ or, if exons are involved, ‘synonymous’ mutations and generally thought to be evolutionarily neutral as these polymorphisms do not alter protein function. However, due to possible codon usage biases, translational stability, splicing, or transcriptional control of expression of the lung collectins may be significantly affected. Indeed, COPD susceptibility was associated with such polymorphisms in the SP-A and SP-D genes [183, 184]. Further studies are needed, however, to understand better the clinical significance of allelic variations in SP-D expression and function and to aid future clinical investigations into the use of collectin preparations for therapeutic manipulations of the pulmonary immune system.
Acknowledgements
Dr Imre Redai (Columbia University) is greatfully acknowledged for his help in the preparation of this manuscript.
This work was supported by the ALA Career Investigator Award, R01AI055593, R01HL076646, RC1ES018505 and the Center of Excellence in Environmental Toxicology, University of Pennsylvania.
References
- 1.Persson A, Rust K, Chang D, Moxley M, Longmore W, Crouch E. CP4: a pneumocyte-derived collagenous surfactant-associated protein. Evidence for heterogeneity of collagenous surfactant proteins. Biochemistry 1988; 27:8576–84. [DOI] [PubMed] [Google Scholar]
- 2.Haczku A, Atochina EN, Tomer Y et al. Aspergillus fumigatus-induced allergic airway inflammation alters surfactant homeostasis and lung function in BALB/c mice. Am J Respir Cell Mol Biol 2001; 25:45–50. [DOI] [PubMed] [Google Scholar]
- 3.Haczku A, Cao Y, Vass G et al. IL-4 and IL-13 form a negative feedback circuit with surfactant protein-D in the allergic airway response. J Immunol 2006; 176:3557–65. [DOI] [PubMed] [Google Scholar]
- 4.Kierstein S, Poulain FR, Cao Y et al. Susceptibility to ozone-induced airway inflammation is associated with decreased levels of surfactant protein D. Respir Res 2006; 7:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cao Y, Tao JQ, Bates SR, Beers MF, Haczku A. IL-4 induces production of the lung collectin surfactant protein-D. J Allergy Clin Immunol 2004; 113:439–44. [DOI] [PubMed] [Google Scholar]
- 6.Haczku A. Role and regulation of lung collectins in allergic airway sensitization. Pharmacol Ther 2006; 110:14–34. [DOI] [PubMed] [Google Scholar]
- 7.Haczku A, Atochina EN, Tomer Y et al. The late asthmatic response is linked with increased surface tension and reduced surfactant protein B in mice. Am J Physiol Lung Cell Mol Physiol 2002; 283:L755–65. [DOI] [PubMed] [Google Scholar]
- 8.Atochina EN, Beers MF, Tomer Y et al. Attenuated allergic airway hyperresponsiveness in C57BL/6 mice is associated with enhanced surfactant protein (SP)-D production following allergic sensitization. Respir Res 2003; 4:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sims MW, Tal-Singer RM, Kierstein S et al. Chronic obstructive pulmonary disease and inhaled steroids alter surfactant protein D (SP-D) levels: a cross-sectional study. Respir Res 2008; 9:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krytska K, Ogden CA, S K et al. Impaired apoptotic cell clearance is associated with lack of surfactant protein D (SP-D) in bronchoalveolar lavage (BAL) of gene deficient mice as well as in current smokers and patients with chronic obstructive pulmonary disease (COPD). Am J Respir Crit Care Med 2008: A345. [Google Scholar]
- 11.Alexis NE, Lay JC, Haczku A et al. Fluticasone propionate protects against ozone-induced airway inflammation and modified immune cell activation markers in healthy volunteers. Environ Health Perspect 2008; 116:799–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sullivan LC, Daniels CB, Phillips ID, Orgeig S, Whitsett JA. Conservation of surfactant protein A: evidence for a single origin for vertebrate pulmonary surfactant. J Mol Evol 1998; 46:131–8. [DOI] [PubMed] [Google Scholar]
- 13.Drickamer K, Dodd RB. C-Type lectin-like domains in caenorhabditis elegans: predictions from the complete genome sequence. Glycobiology 1999; 9:1357–69. [DOI] [PubMed] [Google Scholar]
- 14.Scanlon ST, Milovanova T, Kierstein S et al. Surfactant protein-A inhibits Aspergillus fumigatus-induced allergic T-cell responses. Respir Res 2005; 6:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vass G, Scanlon ST, Beers MF, Haczku A. Surfactant protein (SP)-D suppresses antigenic and mitogenic T cell activation in vitro. J Allergy Clin Immunol 2004; 113:S252. [Google Scholar]
- 16.Klaus MH, Clements JA, Havel RJ. Composition of surface-active material isolated from beef lung. Proc Natl Acad Sci USA 1961; 47:1858–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clements JA. Surfactant in pulmonary disease. N Engl J Med 1965; 272:1336–7. [DOI] [PubMed] [Google Scholar]
- 18.White RT, Damm D, Miller J et al. Isolation and characterization of the human pulmonary surfactant apoprotein gene. Nature 1985; 317:361–3. [DOI] [PubMed] [Google Scholar]
- 19.Benson B, Hawgood S, Schilling J et al. Structure of canine pulmonary surfactant apoprotein: cDNA and complete amino acid sequence. Proc Natl Acad Sci USA 1985; 82:6379–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Floros J, Steinbrink R, Jacobs K et al. Isolation and characterization of cDNA clones for the 35-kDa pulmonary surfactant-associated protein. J Biol Chem 1986; 261:9029–33. [PubMed] [Google Scholar]
- 21.Sano K, Fisher J, Mason RJ et al. Isolation and sequence of a cDNA clone for the rat pulmonary surfactant-associated protein (PSP-A). Biochem Biophys Res Commun 1987; 144:367–74. [DOI] [PubMed] [Google Scholar]
- 22.Holmskov UL. Collectins and collectin receptors in innate immunity. APMIS Suppl 2000; 100:1–59. [PubMed] [Google Scholar]
- 23.Drickamer K, McCreary V. Exon structure of a mannose-binding protein gene reflects its evolutionary relationship to the asia-loglycoprotein receptor and nonfibrillar collagens. J Biol Chem 1987; 262:2582–9. [PubMed] [Google Scholar]
- 24.Lawson PR, Perkins VC, Holmskov U, Reid KB. Genomic organization of the mouse gene for lung surfactant protein D. Am J Respir Cell Mol Biol 1999; 20:953–63. [DOI] [PubMed] [Google Scholar]
- 25.Rust K, Grosso L, Zhang V et al. Human surfactant protein D: SP-D contains a C-type lectin carbohydrate recognition domain. Arch Biochem Biophys 1991; 290:116–26. [DOI] [PubMed] [Google Scholar]
- 26.Hawgood S, Poulain FR. The pulmonary collectins and surfactant metabolism. Annu Rev Physiol 2001; 63:495–519. [DOI] [PubMed] [Google Scholar]
- 27.Gupta G, Surolia A. Collectins: sentinels of innate immunity. Bioessays 2007; 29:452–64. [DOI] [PubMed] [Google Scholar]
- 28.Kuan SF, Rust K, Crouch E. Interactions of surfactant protein D with bacterial lipopolysaccharides. Surfactant protein D is an Escherichia coli-binding protein in bronchoalveolar lavage. J Clin Invest 1992; 90:97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tenner AJ, Robinson SL, Borchelt J, Wright JR. Human pulmonary surfactant protein (SP-A), a protein structurally homologous to C1q, can enhance FcR- and CR1-mediated phagocytosis. J Biol Chem 1989; 264:13923–8. [PubMed] [Google Scholar]
- 30.Malhotra R, Thiel S, Reid KB, Sim RB. Human leukocyte C1q receptor binds other soluble proteins with collagen domains. J Exp Med 1990; 172:955–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stuart GR, Lynch NJ, Day AJ, Schwaeble WJ, Sim RB. The C1q and collectin binding site within C1q receptor (cell surface calreticulin). Immunopharmacology 1997; 38:73–80. [DOI] [PubMed] [Google Scholar]
- 32.Ogden CA, deCathelineau A, Hoffmann PR et al. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J Exp Med 2001; 194:781–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fraser DA, Tenner AJ. Directing an appropriate immune response: the role of defense collagens and other soluble pattern recognition molecules. Curr Drug Targets 2008; 9:113–22. [DOI] [PubMed] [Google Scholar]
- 34.Wright JR. Immunoregulatory functions of surfactant proteins. Nat Rev 2005; 5:58–68. [DOI] [PubMed] [Google Scholar]
- 35.Kishore U, Greenhough TJ, Waters P et al. Surfactant proteins SP-A and SP-D: structure, function and receptors. Mol Immunol 2006; 43:1293–315. [DOI] [PubMed] [Google Scholar]
- 36.Crouch E, Wright JR. Surfactant proteins a and d and pulmonary host defense. Annu Rev Physiol 2001; 63:521–54. [DOI] [PubMed] [Google Scholar]
- 37.van de Wetering JK, van Golde LM, Batenburg JJ. Collectins: players of the innate immune system. Eur J Biochem 2004; 271: 1229–49. [DOI] [PubMed] [Google Scholar]
- 38.Botas C, Poulain F, Akiyama J et al. Altered surfactant homeostasis and alveolar type II cell morphology in mice lacking surfactant protein D. Proc Natl Acad Sci USA 1998; 95: 11869–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elhalwagi BM, Zhang M, Ikegami M et al. Normal surfactant pool sizes and inhibition-resistant surfactant from mice that overexpress surfactant protein A. Am J Respir Cell Mol Biol 1999; 21:380–7. [DOI] [PubMed] [Google Scholar]
- 40.Fisher JH, Sheftelyevich V, Ho YS et al. Pulmonary-specific expression of SP-D corrects pulmonary lipid accumulation in SP-D gene-targeted mice. Am J Physiol Lung Cell Mol Physiol 2000; 278:L365–73. [DOI] [PubMed] [Google Scholar]
- 41.Fisher JH, Larson J, Cool C, Dow SW. Lymphocyte activation in the lungs of SP-D null mice. Am J Respir Cell Mol Biol 2002; 27:24–33. [DOI] [PubMed] [Google Scholar]
- 42.Zhang L, Ikegami M, Dey CR, Korfhagen TR, Whitsett JA. Reversibility of pulmonary abnormalities by conditional replacement of surfactant protein D (SP-D) in vivo. J Biol Chem 2002; 277:38709–13. [DOI] [PubMed] [Google Scholar]
- 43.Hawgood S, Ochs M, Jung A et al. Sequential targeted deficiency of SP-A and -D leads to progressive alveolar lipoproteinosis and emphysema. Am J Physiol Lung Cell Mol Physiol 2002; 283:L1002–10. [DOI] [PubMed] [Google Scholar]
- 44.LeVine AM, Whitsett JA, Gwozdz JA et al. Distinct effects of surfactant protein A or D deficiency during bacterial infection on the lung. J Immunol 2000; 165:3934–40. [DOI] [PubMed] [Google Scholar]
- 45.Vaandrager AB, van Golde LM. Lung surfactant proteins A and D in innate immune defense. Biol Neonate 2000; 77 (Suppl. 1):9–13. [DOI] [PubMed] [Google Scholar]
- 46.LeVine AM, Whitsett JA, Hartshorn KL, Crouch EC, Korfhagen TR. Surfactant protein D enhances clearance of influenza A virus from the lung in vivo. J Immunol 2001; 167:5868–73. [DOI] [PubMed] [Google Scholar]
- 47.LeVine AM, Whitsett JA. Pulmonary collectins and innate host defense of the lung. Microbes Infect 2001; 3:161–6. [DOI] [PubMed] [Google Scholar]
- 48.Ikegami M, Hull WM, Yoshida M, Wert SE, Whitsett JA. SP-D and GM-CSF regulate surfactant homeostasis via distinct mechanisms. Am J Physiol Lung Cell Mol Physiol 2001; 281: L697–703. [DOI] [PubMed] [Google Scholar]
- 49.Hawgood S, Akiyama J, Brown C, Allen L, Li G, Poulain FR. GM-CSF mediates alveolar macrophage proliferation and type II cell hypertrophy in SP-D gene-targeted mice. Am J Physiol Lung Cell Mol Physiol 2001; 280:L1148–56. [DOI] [PubMed] [Google Scholar]
- 50.Zhang L, Ikegami M, Crouch EC, Korfhagen TR, Whitsett JA. Activity of pulmonary surfactant protein-D (SP-D) in vivo is dependent on oligomeric structure. J Biol Chem 2001; 276: 19214–9. [DOI] [PubMed] [Google Scholar]
- 51.Zhang P, McAlinden A, Li S et al. The amino-terminal heptad repeats of the coiled-coil neck domain of pulmonary surfactant protein d are necessary for the assembly of trimeric subunits and dodecamers. J Biol Chem 2001; 276:19862–70. [DOI] [PubMed] [Google Scholar]
- 52.Clark H, Palaniyar N, Strong P, Edmondson J, Hawgood S, Reid KB. Surfactant protein D reduces alveolar macrophage apoptosis in vivo. J Immunol 2002; 169:2892–9. [DOI] [PubMed] [Google Scholar]
- 53.Zhang L, Hartshorn KL, Crouch EC, Ikegami M, Whitsett JA. Complementation of pulmonary abnormalities in SP-D(−/−) mice with an SP-D/conglutinin fusion protein. J Biol Chem 2002; 277:22453–9. [DOI] [PubMed] [Google Scholar]
- 54.Li G, Siddiqui J, Hendry M et al. Surfactant protein-A–deficient mice display an exaggerated early inflammatory response to a beta-resistant strain of influenza A virus. Am J Respir Cell Mol Biol 2002; 26:277–82. [DOI] [PubMed] [Google Scholar]
- 55.Clark H, Palaniyar N, Hawgood S, Reid KB. A recombinant fragment of human surfactant protein D reduces alveolar macrophage apoptosis and pro-inflammatory cytokines in mice developing pulmonary emphysema. Ann NY Acad Sci 2003; 1010:113–6. [DOI] [PubMed] [Google Scholar]
- 56.Collins RA, Ikegami M, Korfhagen TR, Whitsett JA, Sly PD. In vivo measurements of changes in respiratory mechanics with age in mice deficient in surfactant protein D. Pediatr Res 2003; 53:463–7. [DOI] [PubMed] [Google Scholar]
- 57.Madan T, Reid KB, Singh M, Sarma PU, Kishore U. Susceptibility of mice genetically deficient in the surfactant protein (SP)-A or SP-D gene to pulmonary hypersensitivity induced by antigens and allergens of Aspergillus fumigatus. J Immunol 2005; 174:6943–54. [DOI] [PubMed] [Google Scholar]
- 58.Gordon S Pattern recognition receptors: doubling up for the innate immune response. Cell 2002; 111:927–30. [DOI] [PubMed] [Google Scholar]
- 59.Beharka AA, Gaynor CD, Kang BK, Voelker DR, McCormack FX, Schlesinger LS. Pulmonary surfactant protein A up-regulates activity of the mannose receptor, a pattern recognition receptor expressed on human macrophages. J Immunol 2002; 169: 3565–73. [DOI] [PubMed] [Google Scholar]
- 60.Schaub B, Westlake RM, He H et al. Surfactant protein D deficiency influences allergic immune responses. Clin Exp Allergy 2004; 34:1819–26. [DOI] [PubMed] [Google Scholar]
- 61.Hakansson K, Reid KB. Collectin structure: a review. Protein Sci 2000; 9:1607–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ogasawara Y, Voelker DR. The role of the amino-terminal domain and the collagenous region in the structure and the function of rat surfactant protein D. J Biol Chem 1995; 270:19052–8. [DOI] [PubMed] [Google Scholar]
- 63.Kawasaki T, Etoh R, Yamashina I. Isolation and characterization of a mannan-binding protein from rabbit liver. Biochem Biophys Res Commun 1978; 81:1018–24. [DOI] [PubMed] [Google Scholar]
- 64.Kolatkar AR, Weis WI. Structural basis of galactose recognition by C-type animal lectins. J Biol Chem 1996; 271:6679–85. [PubMed] [Google Scholar]
- 65.Allen MJ, Harbeck R, Smith B, Voelker DR, Mason RJ. Binding of rat and human surfactant proteins A and D to Aspergillus fumigatus conidia. Infect Immun 1999; 67:4563–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Allen MJ, Voelker DR, Mason RJ. Interactions of surfactant proteins A and D with Saccharomyces cerevisiae and Aspergillus fumigatus. Infect Immun 2001; 69:2037–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Allen MJ, Laederach A, Reilly PJ, Mason RJ. Polysaccharide recognition by surfactant protein D: novel interactions of a C-type lectin with nonterminal glucosyl residues. Biochemistry 2001; 40:7789–98. [DOI] [PubMed] [Google Scholar]
- 68.Madan T, Eggleton P, Kishore U et al. Binding of pulmonary surfactant proteins A and D to Aspergillus fumigatus conidia enhances phagocytosis and killing by human neutrophils and alveolar macrophages. Infect Immun 1997; 65:3171–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Crouch EC. Collectins and pulmonary host defense. Am J Respir Cell Mol Biol 1998; 19:177–201. [DOI] [PubMed] [Google Scholar]
- 70.Lim BL, Wang JY, Holmskov U, Hoppe HJ, Reid KB. Expression of the carbohydrate recognition domain of lung surfactant protein D and demonstration of its binding to lipopolysaccharides of gram-negative bacteria. Biochem Biophys Res Commun 1994; 202:1674–80. [DOI] [PubMed] [Google Scholar]
- 71.Palaniyar N, Zhang L, Kuzmenko A et al. The role of pulmonary collectin N-terminal domains in surfactant structure, function, and homeostasis in vivo. J Biol Chem 2002; 277:26971–9. [DOI] [PubMed] [Google Scholar]
- 72.Palaniyar N, Clark H, Nadesalingam J, Hawgood S, Reid KB. Surfactant protein D binds genomic DNA and apoptotic cells, and enhances their clearance, in vivo. Ann NY Acad Sci 2003; 1010:471–5. [DOI] [PubMed] [Google Scholar]
- 73.Palaniyar N, Nadesalingam J, Clark H, Shih MJ, Dodds AW, Reid KB. Nucleic acid is a novel ligand for innate immune pattern recognition collectins surfactant proteins A and D and man-nose-binding lectin. J Biol Chem 2004; 279:32728–36. [DOI] [PubMed] [Google Scholar]
- 74.Vandivier RW, Ogden CA, Fadok VA et al. Role of surfactant proteins A, D, and C1q in the clearance of apoptotic cells in vivo and in vitro: calreticulin and CD91 as a common collectin receptor complex. J Immunol 2002; 169:3978–86. [DOI] [PubMed] [Google Scholar]
- 75.Cheng G, Ueda T, Nakajima H et al. Surfactant protein A exhibits inhibitory effect on eosinophils IL-8 production. Biochem Biophys Res Commun 2000; 270:831–5. [DOI] [PubMed] [Google Scholar]
- 76.Nauta AJ, Raaschou-Jensen N, Roos A et al. Mannose-binding lectin engagement with late apoptotic and necrotic cells. Eur J Immunol 2003; 33:2853–63. [DOI] [PubMed] [Google Scholar]
- 77.Schagat TL, Wofford JA, Wright JR. Surfactant protein A enhances alveolar macrophage phagocytosis of apoptotic neutrophils. J Immunol 2001; 166:2727–33. [DOI] [PubMed] [Google Scholar]
- 78.LeVine AM, Bruno MD, Huelsman KM, Ross GF, Whitsett JA, Korfhagen TR. Surfactant protein A-deficient mice are susceptible to group B streptococcal infection. J Immunol 1997; 158:4336–40. [PubMed] [Google Scholar]
- 79.LeVine AM, Kurak KE, Wright JR et al. Surfactant protein-A binds group B streptococcus enhancing phagocytosis and clearance from lungs of surfactant protein-A-deficient mice. Am J Respir Cell Mol Biol 1999; 20:279–86. [DOI] [PubMed] [Google Scholar]
- 80.LeVine AM, Kurak KE, Bruno MD, Stark JM, Whitsett JA, Korfhagen TR. Surfactant protein-A-deficient mice are susceptible to Pseudomonas aeruginosa infection. Am J Respir Cell Mol Biol 1998; 19:700–8. [DOI] [PubMed] [Google Scholar]
- 81.Hartshorn KL, Crouch E, White MR et al. Pulmonary surfactant proteins A and D enhance neutrophil uptake of bacteria. Am J Physiol 1998; 274:L958–69. [DOI] [PubMed] [Google Scholar]
- 82.Schelenz S, Malhotra R, Sim RB, Holmskov U, Bancroft GJ. Binding of host collectins to the pathogenic yeast Cryptococcus neoformans: human surfactant protein D acts as an agglutinin for acapsular yeast cells. Infect Immun 1995; 63:3360–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Restrepo CI, Dong Q, Savov J, Mariencheck WI, Wright JR. Surfactant protein D stimulates phagocytosis of Pseudomonas aeruginosa by alveolar macrophages. Am J Respir Cell Mol Biol 1999; 21:576–85. [DOI] [PubMed] [Google Scholar]
- 84.Ofek I, Mesika A, Kalina M et al. Surfactant protein D enhances phagocytosis and killing of unencapsulated phase variants of Klebsiella pneumoniae. Infect Immun 2001; 69:24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Van Rozendaal BA, Van de Lest CH, Van Eijk M, Van Helden HP, Haagsman HP. Pulmonary surfactant proteins A and D are involved in the early response to intratracheally aerosolized lipopolysaccharide. Biochem Soc Trans 1997; 25:S656. [DOI] [PubMed] [Google Scholar]
- 86.Sahly H, Ofek I, Podschun R et al. Surfactant protein D binds selectively to Klebsiella pneumoniae lipopolysaccharides containing mannose-rich O-antigens. J Immunol 2002; 169:3267–74. [DOI] [PubMed] [Google Scholar]
- 87.Van Iwaarden JF, Pikaar JC, Storm J et al. Binding of surfactant protein A to the lipid A moiety of bacterial lipopolysaccharides. Biochem J 1994; 303 (Part 2):407–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Deitsch KW, Moxon ER, Wellems TE. Shared themes of antigenic variation and virulence in bacterial, protozoal, and fungal infections. Microbiol Mol Biol Rev 1997; 61:281–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.van de Wetering JK, Coenjaerts FE, Vaandrager AB, van Golde LM, Batenburg JJ. Aggregation of cryptococcus neoformans by surfactant protein D is inhibited by its capsular component glucuronoxylomannan. Infect Immun 2004; 72: 145–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vuk-Pavlovic Z, Standing JE, Crouch EC, Limper AH. Carbohydrate recognition domain of surfactant protein D mediates interactions with Pneumocystis carinii glycoprotein A. Am J Respir Cell Mol Biol 2001; 24:475–84. [DOI] [PubMed] [Google Scholar]
- 91.Yong SJ, Vuk-Pavlovic Z, Standing JE, Crouch EC, Limper AH. Surfactant protein D-mediated aggregation of Pneumocystis carinii impairs phagocytosis by alveolar macrophages. Infect Immun 2003; 71:1662–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Postle AD, Mander A, Reid KB et al. Deficient hydrophilic lung surfactant proteins A and D with normal surfactant phospholipid molecular species in cystic fibrosis. Am J Respir Cell Mol Biol 1999; 20:90–8. [DOI] [PubMed] [Google Scholar]
- 93.Madan T, Kishore U, Singh M et al. Surfactant proteins A and D protect mice against pulmonary hypersensitivity induced by Aspergillus fumigatus antigens and allergens. J Clin Invest 2001; 107:467–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Madan T, Kishore U, Singh M et al. Protective role of lung surfactant protein D in a murine model of invasive pulmonary aspergillosis. Infect Immun 2001; 69:2728–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kishor U, Madan T, Sarma PU, Singh M, Urban BC, Reid KB. Protective roles of pulmonary surfactant proteins, SP-A and SP-D, against lung allergy and infection caused by Aspergillus fumigatus. Immunobiology 2002; 205:610–8. [DOI] [PubMed] [Google Scholar]
- 96.Strong P, Reid KB, Clark H. Intranasal delivery of a truncated recombinant human SP-D is effective at down-regulating allergic hypersensitivity in mice sensitized to allergens of Aspergillus fumigatus. Clin Exp Immunol 2002; 130:19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Strong P, Townsend P, Mackay R, Reid KB, Clark HW. A recombinant fragment of human SP-D reduces allergic responses in mice sensitized to house dust mite allergens. Clin Exp Immunol 2003; 134:181–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Malherbe DC, Erpenbeck VJ, Abraham SN, Crouch EC, Hohlfeld JM, Wright JR. Surfactant protein D decreases pollen-induced IgE-dependent mast cell degranulation. Am J Physiol Lung Cell Mol Physiol 2005; 289:L856–66. [DOI] [PubMed] [Google Scholar]
- 99.Erpenbeck VJ, Malherbe DC, Sommer S et al. Surfactant protein D increases phagocytosis and aggregation of pollen-allergen starch granules. Am J Physiol Lung Cell Mol Physiol 2005; 288:L692–8. [DOI] [PubMed] [Google Scholar]
- 100.Malhotra R, Haurum J, Thiel S, Jensenius JC, Sim RB. Pollen grains bind to lung alveolar type II cells (A549) via lung surfactant protein A (SP-A). Biosci Rep 1993; 13:79–90. [DOI] [PubMed] [Google Scholar]
- 101.Wang JY, Kishore U, Lim BL, Strong P, Reid KB. Interaction of human lung surfactant proteins A and D with mite (dermatophagoides pteronyssinus) allergens. Clin Exp Immunol 1996; 106:367–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Deb R, Shakib F, Reid K, Clark H. Major house dust mite allergens dermatophagoides pteronyssinus 1 and dermatophagoides farinae 1 degrade and inactivate lung surfactant proteins A and D. J Biol Chem 2007; 282:36808–19. [DOI] [PubMed] [Google Scholar]
- 103.Wang JY, Shieh CC, You PF, Lei HY, Reid KB. Inhibitory effect of pulmonary surfactant proteins A and D on allergen-induced lymphocyte proliferation and histamine release in children with asthma. Am J Respir Crit Care Med 1998; 158:510–8. [DOI] [PubMed] [Google Scholar]
- 104.Hohlfeld JM, Erpenbeck VJ, Krug N. Surfactant proteins SP-A and SP-D as modulators of the allergic inflammation in asthma. Pathobiology 2002; 70:287–92. [DOI] [PubMed] [Google Scholar]
- 105.McGreal E, Gasque P. Structure-function studies of the receptors for complement C1q. Biochem Soc Trans 2002; 30:1010–4. [DOI] [PubMed] [Google Scholar]
- 106.Henson PM, Bratton DL, Fadok VA. The phosphatidylserine receptor: a crucial molecular switch? Nat Rev Mol Cell Biol 2001; 2:627–33. [DOI] [PubMed] [Google Scholar]
- 107.Kuroki Y, Takahashi M, Nishitani C. Pulmonary collectins in innate immunity of the lung. Cell Microbiol 2007; 9:1871–9. [DOI] [PubMed] [Google Scholar]
- 108.Ackerman AL, Kyritsis C, Tampe R, Cresswell P. Early phagosomes in dendritic cells form a cellular compartment sufficient for cross presentation of exogenous antigens. Proc Natl Acad Sci USA 2003; 100:12889–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gardai SJ, Xiao YQ, Dickinson M et al. By binding SIRPalpha or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cell 2003; 115:13–23. [DOI] [PubMed] [Google Scholar]
- 110.Keisari Y, Wang H, Mesika A et al. Surfactant protein D-coated Klebsiella pneumoniae stimulates cytokine production in mononuclear phagocytes. J Leukoc Biol 2001; 70:135–41. [PubMed] [Google Scholar]
- 111.Weikert LF, Lopez JP, Abdolrasulnia R, Chroneos ZC, Shepherd VL. Surfactant protein A enhances mycobacterial killing by rat macrophages through a nitric oxide-dependent pathway. Am J Physiol Lung Cell Mol Physiol 2000; 279:L216–23. [DOI] [PubMed] [Google Scholar]
- 112.Stamme C, Walsh E, Wright JR. Surfactant protein A differentially regulates IFN-gamma- and LPS-induced nitrite production by rat alveolar macrophages. Am J Respir Cell Mol Biol 2000; 23:772–9. [DOI] [PubMed] [Google Scholar]
- 113.Kremlev SG, Phelps DS. Surfactant protein A stimulation of inflammatory cytokine and immunoglobulin production. Am J Physiol 1994; 267:L712–9. [DOI] [PubMed] [Google Scholar]
- 114.Kremlev SG, Umstead TM, Phelps DS. Surfactant protein A regulates cytokine production in the monocytic cell line THP-1. Am J Physiol 1997; 272:L996–1004. [DOI] [PubMed] [Google Scholar]
- 115.Blau H, Riklis S, Kravtsov V, Kalina M. Secretion of cytokines by rat alveolar epithelial cells: possible regulatory role for SP-A. Am J Physiol 1994; 266:L148–55. [DOI] [PubMed] [Google Scholar]
- 116.van Iwaarden F, Welmers B, Verhoef J, Haagsman HP, van Golde LM. Pulmonary surfactant protein A enhances the host-defense mechanism of rat alveolar macrophages. Am J Respir Cell Mol Biol 1990; 2:91–8. [DOI] [PubMed] [Google Scholar]
- 117.van Iwaarden JF, Shimizu H, Van Golde PH, Voelker DR, Van Golde LM. Rat surfactant protein D enhances the production of oxygen radicals by rat alveolar macrophages. Biochem J 1992; 28 (Part 1):5–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tino MJ, Wright JR. Surfactant proteins A and D specifically stimulate directed actin-based responses in alveolar macrophages. Am J Physiol 1999; 276:L164–74. [DOI] [PubMed] [Google Scholar]
- 119.Guo CJ, Atochina-Vasserman EN, Abramova E et al. S-nitrosylation of surfactant protein-D controls inflammatory function. PLoS biol 2008; 6:e266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Atochina-Vasserman EN, Gow AJ, Abramova H et al. Immune reconstitution during pneumocystis lung infection: disruption of surfactant component expression and function by S-nitrosylation. J Immunol 2009; 182:2277–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Guillot L, Balloy V, McCormack FX, Golenbock DT, Chignard M, Si-Tahar M. Cutting edge: the immunostimulatory activity of the lung surfactant protein-A involves Toll-like receptor 4. J Immunol 2002; 168:5989–92. [DOI] [PubMed] [Google Scholar]
- 122.Holmskov U, Lawson P, Teisner B et al. Isolation and characterization of a new member of the scavenger receptor superfamily, glycoprotein-340 (gp-340), as a lung surfactant protein-D binding molecule. J Biol Chem 1997; 272:13743–9. [DOI] [PubMed] [Google Scholar]
- 123.Tino MJ, Wright JR. Glycoprotein-340 binds surfactant protein-A (SP-A) and stimulates alveolar macrophage migration in an SP-A-independent manner. Am J Respir Cell Mol Biol 1999; 20:759–68. [DOI] [PubMed] [Google Scholar]
- 124.Madsen J, Tornoe I, Nielsen O et al. CRP-ductin, the mouse homologue of gp-340/deleted in malignant brain tumors 1 (DMBT1), binds gram-positive and gram-negative bacteria and interacts with lung surfactant protein D. Eur J Immunol 2003; 33:2327–36. [DOI] [PubMed] [Google Scholar]
- 125.Hartshorn KL, White MR, Mogues T, Ligtenberg T, Crouch E, Holmskov U. Lung and salivary scavenger receptor glycoprotein-340 contribute to the host defense against influenza A viruses. Am J Physiol Lung Cell Mol Physiol 2003; 285: L1066–76. [DOI] [PubMed] [Google Scholar]
- 126.Ligtenberg TJ, Bikker FJ, Groenink J et al. Human salivary agglutinin binds to lung surfactant protein-D and is identical with scavenger receptor protein gp-340. Biochem J 2001; 359:243–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Crouch EC, Persson A, Griffin GL, Chang D, Senior RM. Interactions of pulmonary surfactant protein D (SP-D) with human blood leukocytes. Am J Respir Cell Mol Biol 1995; 12:410–5. [DOI] [PubMed] [Google Scholar]
- 128.Clark H, Reid K. The potential of recombinant surfactant protein D therapy to reduce inflammation in neonatal chronic lung disease, cystic fibrosis, and emphysema. Arch Dis Child 2003; 88:981–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sano H, Chiba H, Iwaki D, Sohma H, Voelker DR, Kuroki Y. Surfactant proteins A and D bind CD14 by different mechanisms. J Biol Chem 2000; 275:22442–51. [DOI] [PubMed] [Google Scholar]
- 130.Chiba H, Sano H, Iwaki D et al. Rat mannose-binding protein a binds CD14. Infect Immun 2001; 69:1587–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Murakami S, Iwaki D, Mitsuzawa H et al. Surfactant protein A inhibits peptidoglycan-induced tumor necrosis factor-alpha secretion in U937 cells and alveolar macrophages by direct interaction with toll-like receptor 2. J Biol Chem 2002; 277: 6830–7. [DOI] [PubMed] [Google Scholar]
- 132.Ohya M, Nishitani C, Sano H et al. Human pulmonary surfactant protein D binds the extracellular domains of toll-like receptors 2 and 4 through the carbohydrate recognition domain by a mechanism different from its binding to phosphatidylinositol and lipopolysaccharide. Biochemistry 2006; 45:8657–64. [DOI] [PubMed] [Google Scholar]
- 133.Gaynor CD, McCormack FX, Voelker DR, McGowan SE, Schlesinger LS. Pulmonary surfactant protein A mediates enhanced phagocytosis of mycobacterium tuberculosis by a direct interaction with human macrophages. J Immunol 1995; 155: 5343–51. [PubMed] [Google Scholar]
- 134.Weikert LF, Edwards K, Chroneos ZC, Hager C, Hoffman L, Shepherd VL. SP-A enhances uptake of bacillus calmetteguerin by macrophages through a specific SP-A receptor. Am J Physiol 1997; 272:L989–95. [DOI] [PubMed] [Google Scholar]
- 135.Gold JA, Hoshino Y, Tanaka N et al. Surfactant protein A modulates the inflammatory response in macrophages during tuberculosis. Infect Immun 2004; 72:645–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rosseau S, Hammerl P, Maus U et al. Surfactant protein A down-regulates proinflammatory cytokine production evoked by Candida albicans in human alveolar macrophages and monocytes. J Immunol 1999; 163:4495–502. [PubMed] [Google Scholar]
- 137.Janssen WJ, McPhillips KA, Dickinson MG et al. Surfactant proteins A and D suppress alveolar macrophage phagocytosis via interaction with SIRP alpha. Am J Respir Crit Care Med 2008; 178:158–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Matozaki T, Murata Y, Okazawa H, Ohnishi H. Functions and molecular mechanisms of the CD47-SIRPalpha signalling pathway. Trends Cell Biol 2009; 19:72–80. [DOI] [PubMed] [Google Scholar]
- 139.Holmskov U, Thiel S, Jensenius JC. Collections and ficolins: humoral lectins of the innate immune defense. Annu Rev Immunol 2003; 21:547–78. [DOI] [PubMed] [Google Scholar]
- 140.Crouch E, Persson A, Chang D, Heuser J. Molecular structure of pulmonary surfactant protein D (SP-D). J Biol Chem 1994; 269:17311–9. [PubMed] [Google Scholar]
- 141.Vandivier RW, Fadok VA, Ogden CA et al. Impaired clearance of apoptotic cells from cystic fibrosis airways. Chest 2002; 121:89S. [DOI] [PubMed] [Google Scholar]
- 142.Malhotra R, Haurum J, Thiel S, Sim RB. Interaction of C1q receptor with lung surfactant protein A. Eur J Immunol 1992; 22:1437–45. [DOI] [PubMed] [Google Scholar]
- 143.Matalon S, Shrestha K, Kirk M et al. Belaaouaj A, crouch EC, modification of surfactant protein D by reactive oxygen-nitrogen intermediates is accompanied by loss of aggregating activity, in vitro and in vivo. Faseb J 2009; 23:1415–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Koziol-White C, Forbes LR, Ducka B et al. Ozone-induced exacerbation Of allergic airway inflammation is associated with altered surfactant protein-D (SP-D) structure and activation of macrophages and dendritic cells of the airways. Am J Respir Crit Care Med 2010, in press. [Google Scholar]
- 145.McIntosh JC, Mervin-Blake S, Conner E, Wright JR. Surfactant protein A protects growing cells and reduces TNF-alpha activity from LPS-stimulated macrophages. Am J Physiol 1996; 271:L310–9. [DOI] [PubMed] [Google Scholar]
- 146.Yoshida M, Whitsett JA. Alveolar macrophages and emphysema in surfactant protein-D-deficient mice. Respirology 2006; 11 (Suppl.):S37–40. [DOI] [PubMed] [Google Scholar]
- 147.Atochina EN, Gow AJ, Beck JM et al. Delayed clearance of pneumocystis carinii infection, increased inflammation, and altered nitric oxide metabolism in lungs of surfactant protein-D knockout mice. J Infect Dis 2004; 189:1528–39. [DOI] [PubMed] [Google Scholar]
- 148.Takeda K, Miyahara N, Rha YH et al. Surfactant protein D regulates airway function and allergic inflammation through modulation of macrophage function. Am J Respir Crit Care Med 2003; 168:783–9. [DOI] [PubMed] [Google Scholar]
- 149.Weissbach S, Neuendank A, Pettersson M, Schaberg T, Pison U. Surfactant protein A modulates release of reactive oxygen species from alveolar macrophages. Am J Physiol 1994; 267: L660–6. [DOI] [PubMed] [Google Scholar]
- 150.Liu CF, Chen YL, Shieh CC, Yu CK, Reid KB, Wang JY. Therapeutic effect of surfactant protein D in allergic inflammation of mite-sensitized mice. Clin Exp Allergy 2005; 35:515–21. [DOI] [PubMed] [Google Scholar]
- 151.Bridges JP, Davis HW, Damodarasamy M et al. Pulmonary surfactant proteins A and D are potent endogenous inhibitors of lipid peroxidation and oxidative cellular injury. J Biol Chem 2000; 275:38848–55. [DOI] [PubMed] [Google Scholar]
- 152.Wright JR, Youmans DC. Pulmonary surfactant protein A stimulates chemotaxis of alveolar macrophage. Am J Physiol 1993; 264:L338–44. [DOI] [PubMed] [Google Scholar]
- 153.Brinker KG, Garner H, Wright JR. Surfactant protein A modulates the differentiation of murine bone marrow-derived dendritic cells. Am J Physiol Lung Cell Mol Physiol 2003; 284:L232–41. [DOI] [PubMed] [Google Scholar]
- 154.Brinker KG, Martin E, Borron P et al. Surfactant protein D enhances bacterial antigen presentation by bone marrow-derived dendritic cells. Am J Physiol Lung Cell Mol Physiol 2001; 281:L1453–63. [DOI] [PubMed] [Google Scholar]
- 155.Hansen S, Lo B, Evans K, Neophytou P, Holmskov U, Wright JR. Surfactant protein D augments bacterial association but attenuates major histocompatibility complex class II presentation of bacterial antigens. Am J Respir Cell Mol Biol 2007; 36:94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hortobagyi L, Kierstein S, Krytska K et al. Surfactant protein D inhibits TNF-alpha production by macrophages and dendritic cells in mice. J Allergy Clin Immunol 2008; 122:521–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Gallucci S, Matzinger P. Danger signals: SOS to the immune system. Curr Opin Immunol 2001; 13:114–9. [DOI] [PubMed] [Google Scholar]
- 158.Wu L, Liu YJ. Development of dendritic-cell lineages. Immunity 2007; 26:741–50. [DOI] [PubMed] [Google Scholar]
- 159.Grayson MH, Ramos MS, Rohlfing MM et al. Controls for lung dendritic cell maturation and migration during respiratory viral infection. J Immunol 2007; 179:1438–48. [DOI] [PubMed] [Google Scholar]
- 160.Lambrecht BN, De Veerman M, Coyle AJ, Gutierrez-Ramos JC, Thielemans K, Pauwels RA. Myeloid dendritic cells induce Th2 responses to inhaled antigen, leading to eosinophilic airway inflammation. J Clin Invest 2000; 106:551–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kohl K, Schnautz S, Pesch M et al. Subpopulations of human dendritic cells display a distinct phenotype and bind differentially to proteins of the extracellular matrix. Eur J Cell Biol 2007; 86:719–30. [DOI] [PubMed] [Google Scholar]
- 162.Kohl J, Wills-Karp M. Complement regulates inhalation tolerance at the dendritic cell/T cell interface. Mol Immunol 2007; 44:44–56. [DOI] [PubMed] [Google Scholar]
- 163.Hammad H, Lambrecht BN. Recent progress in the biology of airway dendritic cells and implications for understanding the regulation of asthmatic inflammation. J Allergy Clin Immunol 2006; 118:331–6. [DOI] [PubMed] [Google Scholar]
- 164.Hammad H, Lambrecht BN. Lung dendritic cell migration. Adv Immunol 2007; 93:265–78. [DOI] [PubMed] [Google Scholar]
- 165.Kierstein S, Hortobagyi L, Krytska K, Haczku A. Regulation of dendritic cell function by surfactant Protein D. J Allergy Clin Immunol 2007; 122:521–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kierstein S, Hortobagyi L, Yang X, Vass G, Haczku A . Local gene expression profiling suggests altered dendritic cell (DC) function in surfactant protein-D (SP-D) deficient mice. Keysone Symposia; Allergy, Allergic Inflamm Asthma Abstract Book, 2006, D7, 57. [Google Scholar]
- 167.Forster R, Davalos-Misslitz AC, Rot A. CCR7 and its ligands: balancing immunity and tolerance. Nat Rev 2008; 8:362–71. [DOI] [PubMed] [Google Scholar]
- 168.Wilsher ML, Hughes DA, Haslam PL. Immunoregulatory properties of pulmonary surfactant: effect of lung lining fluid on proliferation of human blood lymphocytes. Thorax 1988; 43:354–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Wilsher ML, Parker DJ, Haslam PL. Immunosuppression by pulmonary surfactant: mechanisms of action. Thorax 1990; 45:3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wilsher ML, Hughes DA, Haslam PL. Immunomodulatory effects of pulmonary surfactant on natural killer cell and antibody-dependent cytotoxicity. Clin Exp Immunol 1988; 74: 465–70. [PMC free article] [PubMed] [Google Scholar]
- 171.Borron PJ, Crouch EC, Lewis JF, Wright JR, Possmayer F, Fraher LJ. Recombinant rat surfactant-associated protein D inhibits human T lymphocyte proliferation and IL-2 production. J Immunol 1998; 161:4599–03. [PubMed] [Google Scholar]
- 172.Haczku A Protective role of the lung collectins surfactant protein A and surfactant protein D in airway inflammation. J Allergy Clin Immunol 2008; 122:861–79; quiz 880–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Borron P, Veldhuizen RA, Lewis JF et al. Surfactant associated protein-A inhibits human lymphocyte proliferation and IL-2 production. Am J Respir Cell Mol Biol 1996; 15:115–21. [DOI] [PubMed] [Google Scholar]
- 174.Borron P, McCormack FX, Elhalwagi BM et al. Surfactant protein A inhibits T cell proliferation via its collagen-like tail and a 210-kDa receptor. Am J Physiol 1998; 275:L679–86. [DOI] [PubMed] [Google Scholar]
- 175.Borron PJ, Mostaghel EA, Doyle C, Walsh ES, McHeyzer-Williams MG, Wright JR. Pulmonary surfactant proteins A and D directly suppress CD3+/CD4+ cell function: evidence for two shared mechanisms. J Immunol 2002; 169:5844–50. [DOI] [PubMed] [Google Scholar]
- 176.Singh M, Madan T, Waters P, Parida SK, Sarma PU, Kishore U. Protective effects of a recombinant fragment of human surfactant protein D in a murine model of pulmonary hypersensitivity induced by dust mite allergens. Immunol Lett 2003; 86:299–307. [DOI] [PubMed] [Google Scholar]
- 177.Brandt EB, Mingler MK, Stevenson MD et al. Surfactant protein D alters allergic lung responses in mice and human subjects. J Allergy Clin Immunol 2008; 121:1140–7, e1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Reidy MF, Wright JR. Surfactant protein A enhances apoptotic cell uptake and TGF-beta1 release by inflammatory alveolar macrophages. Am J Physiol Lung Cell Mol Physiol 2003; 285: L854–61. [DOI] [PubMed] [Google Scholar]
- 179.DiAngelo S, Lin Z, Wang G et al. Novel, non-radioactive, simple and multiplex PCR-cRFLP methods for genotyping human SP-A and SP-D marker alleles. Dis Markers 1999; 15: 269–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lahti M, Lofgren J, Marttila R et al. Surfactant protein D gene polymorphism associated with severe respiratory syncytial virus infection. Pediatr Res 2002; 51:696–9. [DOI] [PubMed] [Google Scholar]
- 181.Leth-Larsen R, Garred P, Jensenius H et al. A common polymorphism in the SFTPD gene influences assembly, function, and concentration of surfactant protein D. J Immunol 2005; 174:1532–8. [DOI] [PubMed] [Google Scholar]
- 182.Floros J, Lin HM, Garcia A et al. Surfactant protein genetic marker alleles identify a subgroup of tuberculosis in a Mexican population. J Infect Dis 2000; 182:1473–8. [DOI] [PubMed] [Google Scholar]
- 183.Guo X, Lin HM, Lin Z et al. Polymorphisms of surfactant protein gene A, B, D, and of SP-B-linked microsatellite markers in COPD of a Mexican population. Chest 2000; 117:249S–50S. [DOI] [PubMed] [Google Scholar]
- 184.Guo X, Lin HM, Lin Z et al. Surfactant protein gene A, B, and D marker alleles in chronic obstructive pulmonary disease of a Mexican population. Eur Respir J 2001; 18:482–90. [DOI] [PubMed] [Google Scholar]