

Abstract
Background
Breast cancer (BC) is the leading malignancy among women worldwide.
Aim
This work aimed to present a comprehensively bioinformatic analysis of gene expression profiles and to identify the hub genes during BC tumorigenesis, providing potential biomarkers and targets for the diagnosis and therapy of BC.
Materials & Methods
In this study, multiple public databases, bioinformatics approaches, and online analytical tools were employed and the real‐time reverse transcription polymerase chain reaction was implemented.
Results
First, we identified 10, 107, and 3869 differentially expressed genes (DEGs) from three gene expression datasets (GSE9574, GSE15852, and GSE42568, covering normal, para‐cancerous, and BC samples, respectively), and investigated different biological functions and pathways involved. Then, we screened out 8, 16, and 29 module genes from these DEGs, respectively. Next, 10 candidate genes were determined through expression and survival analyses. We noted that seven candidate genes JUN, FOS, FOSB, EGR1, ZFP36, CFD, and PPARG were downregulated in BC compared to normal tissues and lower expressed in aggressive types of BC (basal, HER2+, and luminal B), TP53 mutation group, younger patients, higher stage BC, and lymph node metastasis BC, while CD27, PSMB9, and SELL were upregulated. The present study discovered that the expression levels of these candidate genes were correlated with the infiltration of immune cells (CD8+ T cell, macrophage, natural killer [NK] cell, and cancer‐associated fibroblast) in BC, as well as biomarkers of immune cells and immune checkpoints. We also revealed that promoter methylation, amplification, and deep deletion might contribute to the abnormal expressions of candidate genes. Moreover, we illustrated downstream‐targeted genes of JUN, FOS, FOSB, EGR1, and ZFP36 and demonstrated that these targeted genes were involved in “positive regulation of cell death”, “pathways in cancer”, “PI3K‐Akt signaling pathway”, and so on.
Discussion & Conclusion
We presented differential gene expression profiles among normal, para‐cancerous, and BC tissues and further identified candidate genes that might contribute to tumorigenesis and progression of BC, as potential diagnostic and prognostic targets for BC patients.
Keywords: bioinformatics, breast cancer, diagnosis, oncogenesis, prognosis
In this work, we focused on the transcriptional aberrations during the formation of breast cancer (BC) and explored the molecular mechanisms that might be relevant using bioinformatic approaches, and found the initial expression alterations of BC might occur to transcription factors. Our findings provide novel insights in gene expression during BC tumorigenesis and promising biomarkers for early diagnosis, prevention of metastasis, and targeted therapies in BC.

1. INTRODUCTION
Breast cancer (BC) is currently the most commonly diagnosed cancer according to Global Cancer Statistics 2020 covering 36 cancers in 185 countries, the new cases of which were up to 2,261,419, and the new deaths were 684,996. 1 Women are the main sufferers with the high incidence and mortality, obviously increasing female extra cancer burden. According to the morphological, molecular, and genetic heterogeneities, 2 BC is divided into four major subtypes with the expression of markers (estrogen receptor, ER; progesterone receptor, PR; human epidermal growth factor receptor 2, HER2): luminal A (ER+ and/or PR+, HER2−, Ki‐67−), luminal B (ER+ and/or PR+, HER2+, Ki‐67+), HER2+ (ER− and PR−, HER2+), and basal or triple‐negative (ER−, PR− and HER2−). 3 Numerous studies proved that therapeutic approaches and patients' outcomes of BC varied largely based on cancer stages and subtypes at diagnosis, as well as ages, tumor grades, and lymphovascular status. 4 The 5‐year overall survival (OS) rates for stage I and II patients were over 90%, receiving surgery with or without adjuvant radiation and chemotherapy. However, it was less than 30% for stage IV patients treated with palliative/noncurative‐intent therapy or radiation/chemotherapy alone. Compared with ER+/PR+ and HER2+ BC patients benefiting from endocrine and targeted therapies with chemotherapy, triple‐negative, or basal BC patients have poorer prognoses due to limited treatment options. 4 , 5 , 6 In the last decades, PD‐L1 inhibition has been proved to ameliorate the progression‐free survival (PFS) of triple‐negative BC (TNBC) patients. 7 However, the mechanisms of carcinogenesis and progression of BC are still ambiguous, which need to be further elucidated. More effectively therapeutic targets are as well essential to be explored.
In the last two decades, high‐throughput technologies and next‐generation sequencing 8 have developed rapidly, as well as bioinformatics, 9 which make it possible to deeply explore the etiology, pathogenic mechanism, progress, treatment of diseases at the genetic level. Moreover, public databases, data processing software, and online tools 10 , 11 make it convenient for researchers to brainstorm all over the world. Nevertheless, the common practices are to identify differentially expressed genes (DEGs) between normal and aberrant tissues as potential biomarkers for diagnosis, prognosis, or therapeutic targets of diseases with bioinformatic approaches. 12 , 13 , 14 , 15 , 16 , 17 In this study, we also investigated the differences between para‐cancerous (histologically normal) tissues from BC patients and normal tissues from cancer‐free patients in addition to the well‐studied changes between histologically normal and cancer tissues, attempting to reveal the main disturbances and any subtle clues from normal breast tissue to BC based on differential gene expression profiles among them. And further to identify candidate genes that contribute to tumorigenesis and progression of BC, and are associated with the prognosis of BC patients as potential diagnostic and therapeutic targets.
First, we screened and selected three gene expression datasets GSE9574 (containing normal and para‐cancerous samples), GSE15852 (containing para‐cancerous and BC samples), and GSE42568 (containing normal and BC samples) from Gene Expression Omnibus (GEO) database, and identified DEGs from them, respectively. Then, the corresponding functions and pathways enriched by these DEGs were explored using the Gene Ontology (GO) function and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses. The corresponding module genes were screened out from these DEGs based on networks and module analysis. Next, the candidate genes were determined through expression validations of these module genes with The Cancer Genome Atlas (TCGA) and survival analysis. Subsequently, we evaluated expression levels of these candidate genes based on clinical features and TP53 mutation status, and also investigated the correlations between their expression and immune cell infiltration in BC, as well as biomarkers of immune cells and immune checkpoints. Moreover, we estimated genetic alteration and methylation of candidate genes and explored targeted genes of transcription factors (TFs) in them. More convincingly, we further assessed candidate gene expression levels in clinical samples. In summary, our work implemented a comprehensive bioinformatic analysis on BC, showing different transcriptional variations in para‐cancerous and BC tissues, and enriching our understanding of the mechanisms of BC tumorigenesis, invasion, relapse, progression, and prognosis. Our results provided promisingly diagnostic and therapeutic targets for BC patients.
2. MATERIALS AND METHODS
2.1. Raw data acquisition
Three gene expression datasets (GSE9574, GSE15852, and GSE42568) of BC were screened and selected from GEO (https://www.ncbi.nlm.nih.gov/geo/), 18 including raw CEL files and their corresponding platform annotation files (i.e., GPL files). In dataset GSE9574 based on GPL96 platforms ([HG‐U133A] Affymetrix Human Genome U133A Array), 29 samples were obtained from histologically normal breast epithelium, of which 14 samples were from epithelium adjacent to a breast tumor, and 15 samples were from patients undergoing reduction mammoplasty without apparent BC. 19 In dataset GSE15852 also based on GPL96 Platforms, 86 sets of gene expression data were obtained from 43 pairs of tumors and adjacent‐tumor tissues. 20 The GSE42568 dataset was based on GPL570 Platforms ([HG‐U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array) and contained 104 primary BC and 17 normal breast biopsies gene chips. 21
2.2. Raw data quality control
The normalized unscaled standard error (NUSE) plots 22 were selected to test the consistency of the data, and RNA degradation plots were to estimate the RNA degradation of the microarray gene chips with affyPLM and affy packages in R (version 4.0.3). For box charts of NUSE, the median standard error across gene chips close to 1 and not higher than 1.05 indicated good consistency. The chips not meeting the criteria were omitted to assure the data quality. As for RNA degradation plots, in a general way, much higher slope and poor uniformity with other chips, especially the latter, indicated that their RNA degradation was outrageous and substantially impacts data quality. 22
2.3. Raw data preprocessing
The log scale robust multi‐array analysis 23 was applied to transform probe intensity data of gene chips stored in CEL files into expression values, including three main steps: background correction, normalization, and log2 transformation of PM values, through rma function of R package affy. Probe IDs were then turned into gene symbols by R with the expression files of probe set and their relevant GPL files. The mean of their expression values was used to avoid more than one probe corresponding to one gene. Finally, K Nearest Neighbors algorithm 24 was used to estimate and impute missing value of gene expression matrix carried out by R package impute after the rows without gene symbols were deleted.
2.4. DEG identification
The limma 25 package of R was used to identify DEGs from processed data according to the cutoff criterion of adjusted p value <0.05 and log2(fold change) > 1 or log2(fold change) < −1. To visualize the discrepancy, we invoked ggpubr package based on ggplot2 and ggthemes package of R to draw volcano plots. Furthermore, the Venn diagram was portrayed using an online tool (http://bioinformatics.psb.ugent.be/webtools/Venn/) to observe the relationships among three DEGs groups clearly. Meanwhile, the concrete overlapping and nonoverlapping genes among them were also obtained.
2.5. GO function and KEGG pathway enrichment analyses of DEGs
The GO function 26 , 27 and KEGG pathway 28 enrichment analyses of DEGs were accomplished using R packages org. Hs.eg.db, AnnotationDbi, and clusterProfiler with adjusted p value cutoff = 0.05 and merged terms of biological process in GO by the cutoff = 0.7. Furthermore, bubble diagrams were drawn using R packages to make the results more intuitionistic.
2.6. Network construction and module analysis
The STRING online database (version 11, http://string‐db.org/) 29 was used to construct protein‐protein interaction (PPI) networks of DEGs identified from GSE9574 and GSE15852 datasets with minimum required interaction score: medium confidence (0.400) and applied TSV files containing the detail information of the networks. The weighted gene co‐expression network analysis (WGCNA) 30 was performed to obtain the detail information of the gene co‐expression network for DEGs from GSE42568 dataset through the corresponding R package WGCNA 31 (version 1.70‐3) with signed R 2 > 0.85. Cytoscape (version 3.8.2) 32 was then employed to reproduce PPI networks via importing the TSV files and building a gene co‐expression network. Subsequently, plug‐in MCODE 33 of Cytoscape was used to detect the densely connected regions (i.e., modules) of networks, the criteria of which were a degree cutoff = 2, MCODE scores ≥ 4, Max. Depth = 100, k‐score = 2, and node score cutoff = 0.2. The online tool Metascape (https://metascape.org/gp/index.html#/main/step1) 34 was applied to conduct functional enrichment analysis of module DEGs, and the R package clusterProfiler (version 3.16.1) was applied to explore cellular component terms of GO and KEGG pathways enriched by these module genes with adjusted p value cutoff = 0.05.
2.7. TF analysis
Transcription factor information was downloaded from Transcriptional Regulatory Relationships Unraveled by Sentence‐based Text mining 35 (TRRUST; www.grnpedia.org/trrust ). The online tool Draw Venn Diagram was applied to identify TFs from module genes screened out from the three datasets.
2.8. Expression validation and survival analysis
All identified module genes were validated with UALCAN 36 (http://ualcan.path.uab.edu/) among a total of 114 normal and 1097 primary breast tumor samples based on TCGA, 37 with a threshold of p < 0.05. For validated genes, the Breast Cancer Gene‐Expression Miner v4.7 38 (bc‐GenExMiner v4.7; http://bcgenex.ico.unicancer.fr/BC‐GEM/GEM‐Accueil.php?js=1) were used for survival analysis based on DNA microarrays (n = 11,359) in “PROGNOSIS” of “ANALYSIS Module”. Clearly, the OS for all module genes was primarily performed among all BC patients and then for genes whose expression levels were significantly associated with OS, and the disease‐free survival (DFS) and distant metastasis‐free survival (DMFS) analysis were performed. Next, we downloaded TCGA dataset of BC, consisting of gene expression RNAseq data (n = 1218), clinical phenotype data (n = 1247), curated survival data (n = 1236), and somatic mutation data (MC3 gene‐level non‐silent mutation, n = 791) from UCSC Xena web (https://xenabrowser.net/DATAPAGES/) for the univariate and multivariate OS analysis of genes associated with OS above using Cox Proportional Hazards model carried out by R package survival. Briefly, we first applied separate univariate Cox regressions to assess the statistical significance for each gene, BC patient stage, age, ER/PR/HER2 status, molecular subtype, tumor/node/metastasis (TNM) status, and TP53 mutation status in relation to OS, then performed multivariate Cox regression analysis for each significant gene combining with all the variables. Expression analysis based on BC patients’ clinical phenotype and TP53 status.
Subsequently, the bc‐GenExMiner v4.7 resource 39 was applied to explore candidate gene expression levels with DNA microarrays data (n = 11,359) in the “EXPRESSION” of “ANALYSIS Module” based on intrinsic molecular subtypes (PAM50 subtypes), patient's ages, nodal metastasis statuses, and TP53 mutation statuses. Then, the UALCAN web resource was employed to investigate candidate gene expression levels among 1,097 BC samples of TCGA data based on individual cancer stages with the “Expression Link” of “TCGA analysis module”.
2.9. Immune infiltration analysis
The “gene module” of “Immune Association” in Tumor Immune Estimation Resource, version 2.0 40 (TIMER2.0, http://timer.cistrome.org/) 40 was used to estimate the correlations between the expression levels of candidate genes and the infiltration of CD8+ T cells, macrophages, NK cells, and cancer‐associated fibroblasts (CAFs) among four major subtypes of BC using all algorithms provided, like EPIC, TIMER, CIBERSORT, CIBERSORT‐ABS, QUANTISEQ, XCELL, and MCPCOUNTER algorithm with Spearman's rank correlation coefficient and statistical significance: p < 0.05. Moreover, for CD8+ T cells and macrophages, we further employed the Breast Cancer Gene‐Expression Miner v4.7 tool to estimate the correlations between the expression of candidate genes and the biomarkers of these two immune cells, as well as six immune checkpoints (PDCD1, CD274, CTLA4, TIGIT, LAG3, and BTLA).
2.10. Promoter methylation, genetic alteration, and TF‐targeted genes analysis
Promoter methylation analysis was implemented using UALCAN web based on TCGA data containing 793 BC and 97 normal samples. The genetic alteration analysis of candidate genes was carried out in cBioPortal 41 web (https://www.cbioportal.org/) based on 8196 BC samples. For TF candidate genes, we applied TRRUST web to predict the potential downstream‐targeted genes and visualized with Cytoscape software. Meanwhile, the functional annotation of them was investigated with Metascape.
2.11. BC sample collection, mRNA extraction, and reverse transcription polymerase chain reaction
Four BC samples (1 luminal A, 2 luminal B, and 1 HER2+ subtypes) and paired normal breast samples, as well as five samples of benign breast disease, were obtained from patients at Zhongnan Hospital of Wuhan University with informed consent. The Ethics Committee of Zhongnan Hospital of Wuhan University approved the use of these samples for total RNA isolation and reverse transcription polymerase chain reaction (RT‐PCR) analysis. The tissues were instantly frozen by liquid nitrogen and then stored at −80°C until used. Total RNA was isolated from the tissues and then converted to complementary DNA (cDNA) with the HiScript II Q RT SuperMix (Vazyme) according to the manufacturer's instruction. The real‐time RT‐PCR was implemented using the Universal SYBR Green Fast qPCR Mix in triplicates. The 2−ΔΔCT method was used to normalize each gene with GAPDH as internal control. The primer sequences for RT‐PCR used in this study were designed and selected from Primer designing tool (http://www.ncbi.nlm.nih.gov/tools/primer‐blast/) and listed in Table S1.
2.12. Gene set enrichment analysis
Gene set enrichment analysis (GSEA) 42 was used to assess pathway variations in these three datasets via dividing para‐cancerous and BC samples into high/low‐expression groups according to the median expression value of FOS, respectively, and with the cutoff: false discovery rate (FDR) q < 0.25.
3. RESULTS
3.1. Raw data quality control
Based on the box charts of NUSE, there was no aberrant chip in the GSE9574 dataset, 1 aberrant chip from para‐cancer tissues in GSE15852, and 13 aberrant chips in GSE42568, consisting of three normal breast tissue and 10 BC chips. After excluding the abnormal samples, the median standard error of all gene chips shown in the NUSE boxplots (Figure S1A–C) was close to 1 and no more than 1.05, indicating a good agreement among the rest samples. With RNA degradation plots, one chip from tumor paired normal tissues in GSE15852 was inconsistent with others and had a lower slope compared to the other chips. RNA degradation of the remaining gene chips was considered to be acceptable (Figure S1D–F). Therefore, we guaranteed the quality of raw data and made them more rigorous.
3.2. Raw data standardization and probe set annotation
Raw data were processed with the rma algorithm of R package affy and interpreted probe set expression to gene expression profile. As a result, a total of 12,413 genes were detected in GSE9574 and GSE15852 based on GPL96, while 20,460 genes in GSE42568 based on GPL570.
3.3. DEG identification
In GSE9574 dataset, only 10 DEGs were extracted, and all of them were downregulated in the para‐cancerous compared with breast normal tissues (Figure 1A). In GSE15852 dataset, 14 upregulated and 93 downregulated genes in BC compared with para‐cancerous tissues were identified (Figure 1B), while 2936 upregulated and 933 downregulated genes in BC compared with normal tissues were obtained in GSE42568 (Figure 1C). In addition, through further comparison among these DEGs, we found that two downregulated DEGs (FOS and FOSB) were overlapped within all the three datasets. In addition, there were other three common downregulated DEGs (ZFP36, EGR1, and JUN) between GSE9574 and GSE42568 datasets, and 95 shared DEGs between GSE15852 and GSE42568 datasets, of which 13 were upregulated and 82 downregulated (Figure 1D; Table 1).
FIGURE 1.
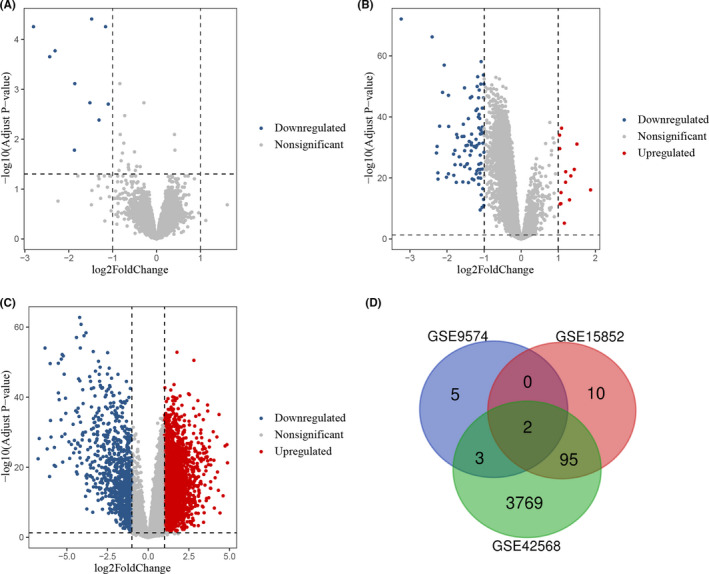
Differentially expressed gene (DEG) identification for each GEO dataset. (A–C) Volcano plots of all genes detected by probes in GSE9574, GSE15852, and GSE42568 datasets, according to the criterion: |log2FC| > 1 and adjusted p < 0.05. The red dots represent upregulated DEGs, the gray dots represent genes without significant difference between groups, and the blue dots represent downregulated DEGs. (D) The Venn diagram of DEGs identified from the three datasets
TABLE 1.
Overlapping differentially expressed genes in each dataset
| Datasets | Upregulated genes | Downregulated genes |
|---|---|---|
| GSE9574&GSE15852&GSE42568 | FOS FOSB | |
| GSE9574&GSE42568 | ZFP36 EGR1 JUN | |
| GSE15852&GSE42568 | DSP GATA3 CD24 KRT18 BUB1B KRT19 EFNA1 SLC38A1 S100A14 SPP1 CD9 TFF3 EPCAM | ESYT1 DPT CHRDL1 PRKAR2B ITIH5 ECM2 ITGB1BP1 LEP SERPINF1 ECH1 RBP4 S100A4 PYGL EPB41L2 GHR GNAI1 GBE1 LGALS1 MAOA ALDH2 IGFBP6 PPAP2A PALMD AKAP12 CD36 ASS1 CAV1 EGFL6 ACSL1 PPARG PC PDZD2 ADIRF HBB GNG11 FHL1 SRPX COX7A1 C2CD2 FTL NEK7 PLSCR4 ABCA8 AOC3 ADH1B AKR1C1 PEMT MMD LPL PTPRM VIM FABP4 ACACB TNMD PPP1R1A ADCK3 PCK1 RGCC CIDEC RRAS HSD11B1 PFKFB3 AKR1C3 ITGA7 SPARC ECHDC3 F3 G0S2 PCOLCE2 ADAMTS5 RETSAT WASF3 GYG2 FOXO1 CDH5 CFD TMEM100 FAM13A EFEMP1 PLIN1 COPG2IT1 ADIPOQ |
3.4. GO terms and pathways enriched by DEGs
To further explore the mechanism of the occurrence and development of BC, we conducted GO function and KEGG pathway annotation for the DEGs extracted from these three datasets. We discovered that 112 main biological processes (BP) were significantly affected in para‐cancerous compared to normal tissues with adjusted p < 0.05, of which the top three were cellular response to extracellular stimulus, cellular response to external stimulus, and response to starvation. Compared to para‐cancerous tissues, 175 main BP were affected in BC tissues, of which the top three were response to acid chemical, lipid localization, and lipid transport. Compared to normal tissues, 164 main BP were influenced in BC tissues, the top three of which were organelle fission, leukocyte migration, and extracellular matrix organization (Figure 2A). Most DEGs identified from GSE9574 or their corresponding proteins were elements of transcription regulator complex, RNA polymerase II transcription regulator complex, and mRNA cap‐binding complex (top three of 10 in total). While the majority of DEGs from GSE15852 were mainly constituents of collagen‐containing extracellular matrix, lipid droplet, platelet alpha granule, and platelet alpha granule membrane, and most DEGs from GSE42568 were main contributors of collagen‐containing extracellular matrix, cell–cell junction, and apical part of cell (Figure 2B). Meanwhile, these DEGs from these datasets also had diversely main molecular functions (Figure 2C).
FIGURE 2.
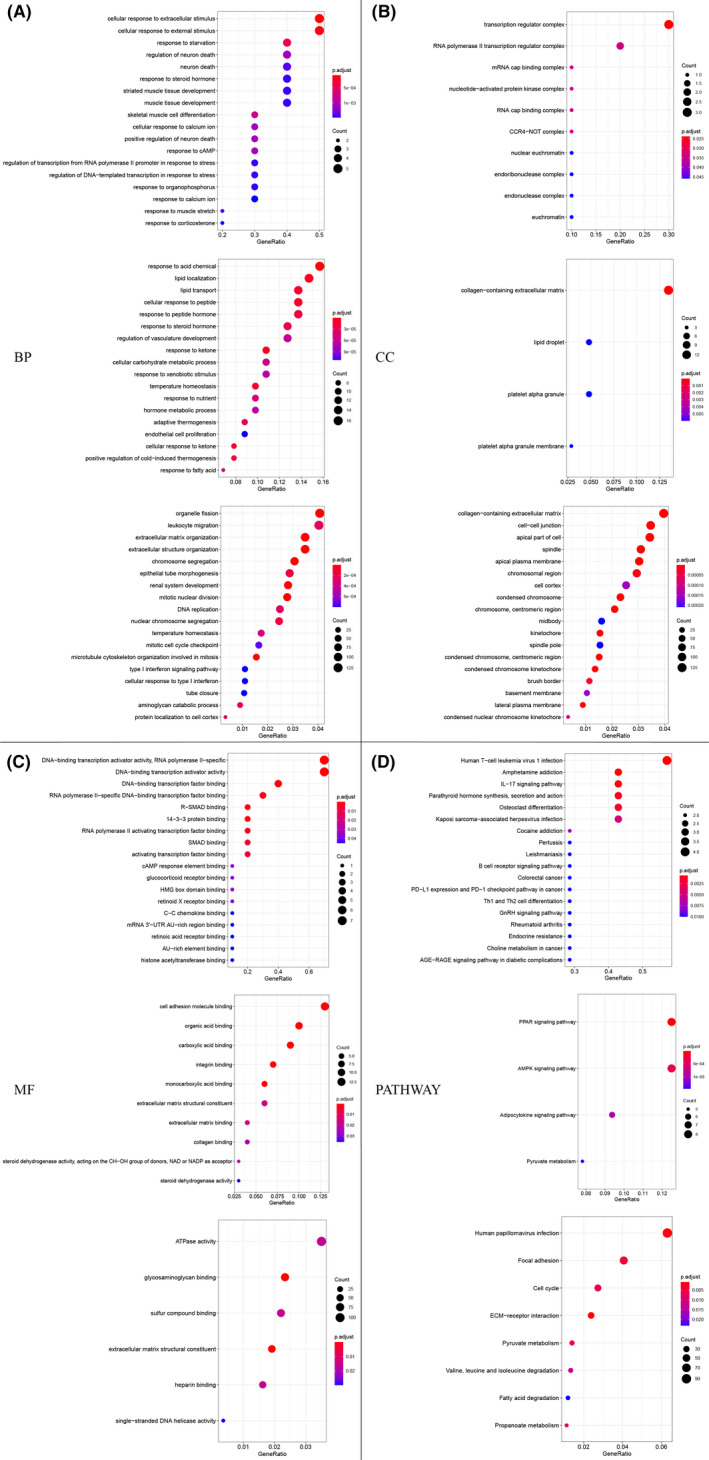
Gene Ontology (GO) function and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses of DEGs. (A) The top 18 biological process terms of GO enriched by most differentially expressed genes (DEGs) in GSE9574, GSE15852, and GSE42568 datasets, respectively. (B) The major cellular components terms of GO enriched by DEGs in GSE9574, GSE15852, and GSE42568. (C) The main molecular functions of DEGs in GSE9574, GSE15852, and GSE42568. (D) The significantly enriched KEGG pathways of DEGs in GSE9574, GSE15852, and GSE42568. BP, biological processes; CC, cellular components; MF, molecular functions
With KEGG pathway analysis, we found 40 pathways significantly gathered by DEGs of GSE9574, the top three of which were human T‐cell leukemia virus 1 infection, amphetamine addition, and IL‐17 signaling pathway. The DEGs of GSE15852 were mainly gathered in the peroxisome proliferator‐activated receptors (PPAR) signaling pathway, AMPK signaling pathway, adipocytokine signaling pathway, and pyruvate metabolism pathway. The eight pathways significantly enriched by DEGs of GSE42568 were human papillomavirus infection, focal adhesion, cell cycle, ECM‐receptor interaction, pyruvate metabolism, valine, leucine and isoleucine degradation, fatty acid degradation, and propanoate metabolism (Figure 2D).
3.5. Network construction and module gene identification
To estimate the relationships or interactions among products encoded by DEGs and further screen out candidate genes, we constructed PPI networks by DEGs identified from GSE9574 and GSE15852. A network containing nine nodes and 30 edges were created with DEGs of GSE9574 (Figure 3A), from which a module was recognized with MCODE scores = 7.429 including eight nodes (i.e., eight downregulated DEGs, NR4A2, FOSB, immediate early response 2 [IER2], ZFP36, FOS, EGR1, ATF3, and JUN) and 26 edges (Figure 3B). Furthermore, multiple function enrichment analysis was performed on module genes using Metascape resource, which mainly gathered in PID‐AP1 pathway, HTLV‐1 infection, and response to extracellular stimulus (Figure 3C; Table S2). In addition, FOS, NR4A2, and JUN are members of the transcription regulator complex. JUN also participates in transcription repression, and ZFP36 was reported to compose mRNA cap‐binding complex (Figure 3D). JUN and FOS were involved in lots of pathways, including MAPK signaling pathway, PD‐L1 expression and PD‐1 checkpoint pathway in BC, colorectal cancer, and even coronavirus disease––COVID‐19 pathway (Figure 3E).
FIGURE 3.
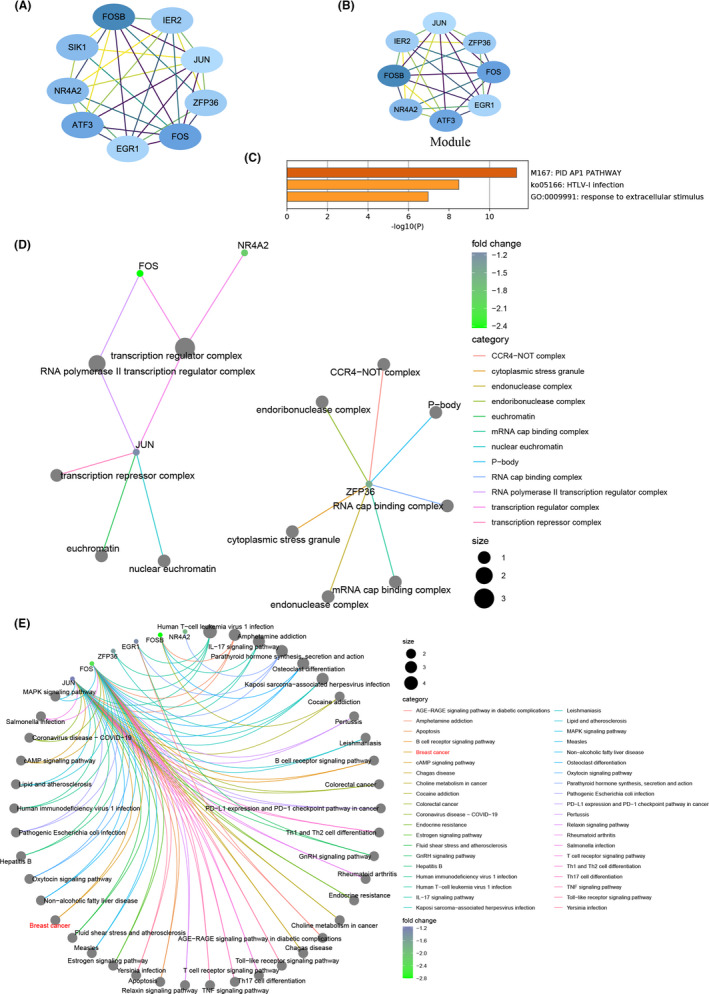
PPI network and module analysis of differentially expressed genes (DEGs) identified from GSE9574 dataset. (A) PPI network constructed by DEGs from GSE9574 dataset. Each node represents a gene. The blue nodes denote the downregulated genes in adjacent‐tumor tissues compared with normal tissues. The darker the nodes, the more differentially expressed the genes between groups. The degrees between them indicate their interactions. (B) A module identified from PPI network. (C) Multiple functions and pathways enriched by module genes. (D) cellular components (CC) terms of Gene Ontology enriched by module genes. (E) Kyoto Encyclopedia of Genes and Genomes pathways enriched by module genes
From the network structured by 74 nodes and 194 edges with DEGs of the GSE15852 dataset (Figure 4A), three modules were identified, of which the first two were selected to further analysis with MCODE scores = 9.400 and 4.000. One was formed by 11 nodes (i.e., 11 downregulated DEGs, ACSL1, PCK1, CIDEC, RBP4, CFD, LEP, LPL, FABP4, PLIN1, ADIPOQ, and PPARG) and 47 edges (Figure 4B). The other was composed of five nodes and eight edges, and all DEGs (KRT18, CD24, KRT19, EPCAM, and GATA3) were upregulated (Figure 4C). These module genes were mainly enriched in adipogenesis pathway, response to acid chemical term of GO, lipid localization of GO (Figure 4D; Table S3). Moreover, we also found that PLIN1, FABP4, and CIDEC were located in lipid droplet (Figure 4E) and PPARG, PLIN1, ADIPOQ, LPL, FABP4, ACSL1, and PCK1 were involved in the PPAR signaling pathway.
FIGURE 4.
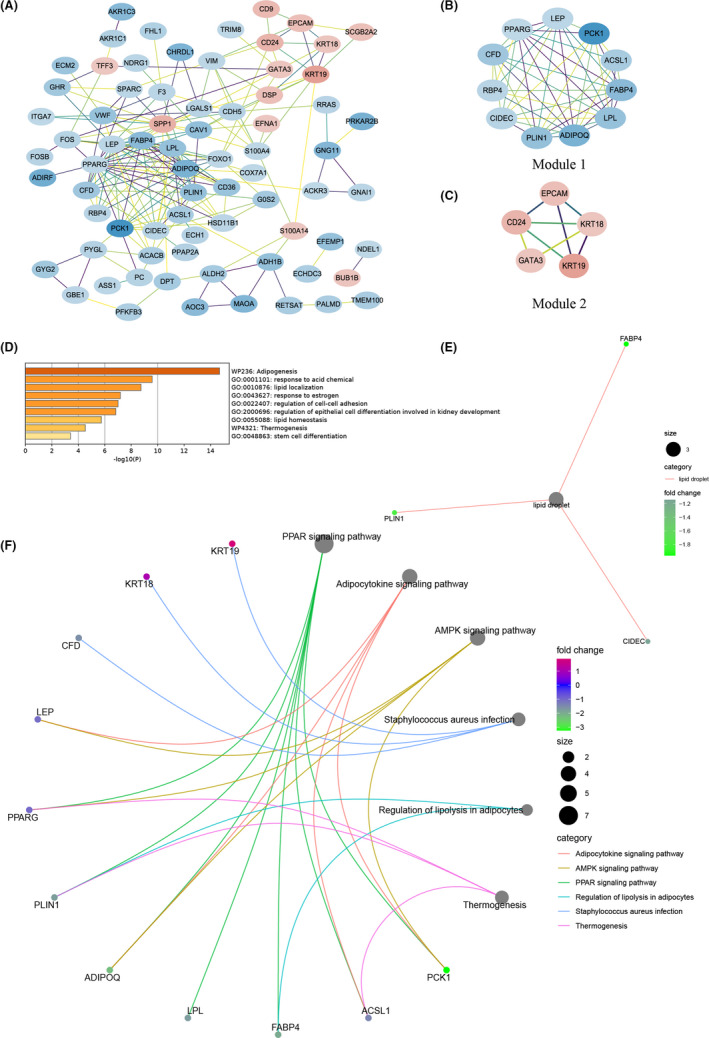
PPI network and module analysis of differentially expressed genes (DEGs) identified from GSE15852 dataset. (A) PPI network constructed by DEGs from GSE15852 dataset. Each node represents a gene. The blue nodes represent the downregulated genes, and the red nodes represent the upregulated genes in BC tissues compared with adjacent‐tumor tissues. The darker the nodes, the more differentially expressed the genes between groups. The degrees between them indicate their interactions. (B, C) Two modules identified from PPI network. (D) Multiple functions and pathways enriched by module genes. (E) Cellular components terms of Gene Ontology enriched by module genes. (F) Kyoto Encyclopedia of Genes and Genomes pathways enriched by module genes
As for the DEGs of the GSE42568 dataset, the number of them was too large to create a PPI network using STRING. Therefore, we performed WGCNA to generate a gene co‐expression network with power = 6, minimum size = 4, and threshold = 0.4 (Figure S2), and subsequently to mine module genes via applying plug‐in MCODE in Cytoscape. In consequence, five modules including 29 genes were sorted out with MCODE scores = 5.333, 5.000, 5.000, 4.500, and 4.000, respectively (Figure 5A–F). Involved genes were mainly aggregated in “immunoregulatory interactions between a Lymphoid and a non‐Lymphoid cell” of “Reactome Gene Sets”, “lymphocyte activation” of “GO BP”, and “pathogenesis of SARS‐CoV‐2 mediated by nsp9‐nsp10 complex” of “WikiPathways” (Figure 5G; Table S4). In addition, HLA‐B, HLA‐C, and HLA‐F contributed to multiple cellular components, like MHC protein complex, ER to Golgi transport vesicle membrane, early endosome, and others. Meanwhile, CCR7, CD27, CD2, CD3G, and CD8A were constituents of external side of plasma membrane, as well as HLA‐B, HLA‐C, and HLA‐F (Figure 5H). Moreover, CD247, CD3G, HLA‐B, HLA‐C, and HLA‐F took part in various pathways, including Th1 and Th2 cell differentiation, PD‐L1 expression and PD‐1 checkpoint pathway in cancer, Th17 cell differentiation, antigen processing presentation, allograft rejection, and cell adhesion molecular (Figure 5I).
FIGURE 5.
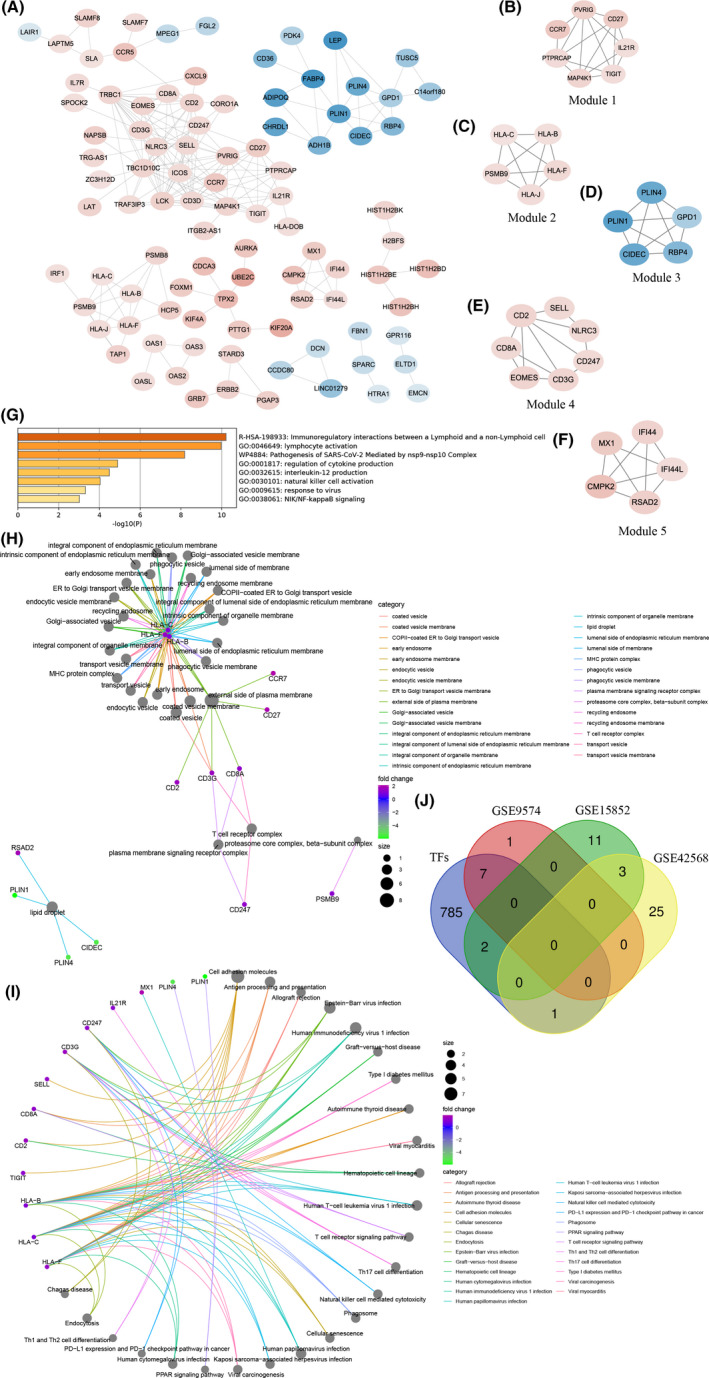
Gene co‐expression network and module analysis of differentially expressed genes (DEGs) identified from GSE42568 dataset and transcription factor screening. (A) Gene co‐expression network formed by DEGs from GSE42568 dataset. Each node represents a gene. The blue nodes represent the downregulated genes, and the red nodes represent the upregulated genes in BC tissues compared with adjacent‐tumor tissues. The darker the nodes, the more differentially expressed the genes between groups. The degrees between them indicate their co‐expressive relationships. (B‐F) Five modules identified from the co‐expression network. (G) Multiple functions and pathways enriched by module genes. (H) Cellular components terms of Gene Ontology enriched by module genes. (I) Kyoto Encyclopedia of Genes and Genomes pathways enriched by module genes. (J) Venn diagram of transcription factors download from TRRUST database and module genes extracted from GSE9574, GSE15852, and GSE42568 datasets
Furthermore, through TRRUST database we found that seven genes (JUN, FOS, FOSB, ATF3, EGR1, NR4A2, and ZFP36) identified from GSE9574, two genes (GATA3 and PPARG) from GSE15852, and one gene EOMES from GSE42568 were TFs (Figure 5J).
3.6. Module gene expression validation and prognostic values for BC patients
A total of 50 unique module genes in PPI and gene co‐expression networks were identified from three GEO datasets covering breast tissues in different statuses. To further confirm our findings, UALCAN web was applied to investigate the distinctions of their transcriptional levels between BC and normal samples based on TCGA. The expressions of all module genes were significantly discrepant by comparing normal samples with tumor, including three genes (ATF3, IER2, and NNR4A2) downregulated in para‐cancerous compared to normal tissues, which were also low expressed in BC compared to normal samples. Whereas, we noted that five genes (HLA‐J, NLRC3, CD247, CD3G, and CD8A) turned to be downregulated in BC compared with normal tissues, which were opposite to the result of GSE42568 of the GEO database (Table S5). Thus, they were excluded from the following analysis.
Subsequently, the OS, DMFS, and RFS were analyzed using the bc‐GenExMiner v4.7 resource based on all DNA microarray data. For module genes from GSE9574, five genes JUN, FOS, FOSB, EGR1, and ZFP36 were positively associated with OS and DFS, among which four genes (FOS, FOSB, EGR1, and ZFP36) were also positively associated with DMFS with statistical significance p < 0.05. For module genes from GSE15852, 13 genes were associated with OS, of which the high expressions of CFD, LEP, LPL, FABP4, PLIN1, ADIPOQ, PPARG, and GATA3 predicted better prognosis, while ACSL1, CD24, KRT18, KRT19, and EPCAM were reverse. Among them, eight and 10 genes were significantly correlated with DMFS and DFS, respectively. For module genes from GSE42568, except for the overlapping gene PLIN1, 11 genes were related to OS, of which PVRIG, CD27, GPD1, PLIN4, and SELL were positively associated with OS, while PSMB9, CMPK2, RSAD2, MX1, IFI44, and IFI44L were reverse. Among them, nine and 11 genes were also significantly correlated with DMFS and DFS, respectively. (Figure 6; Table S5).
FIGURE 6.
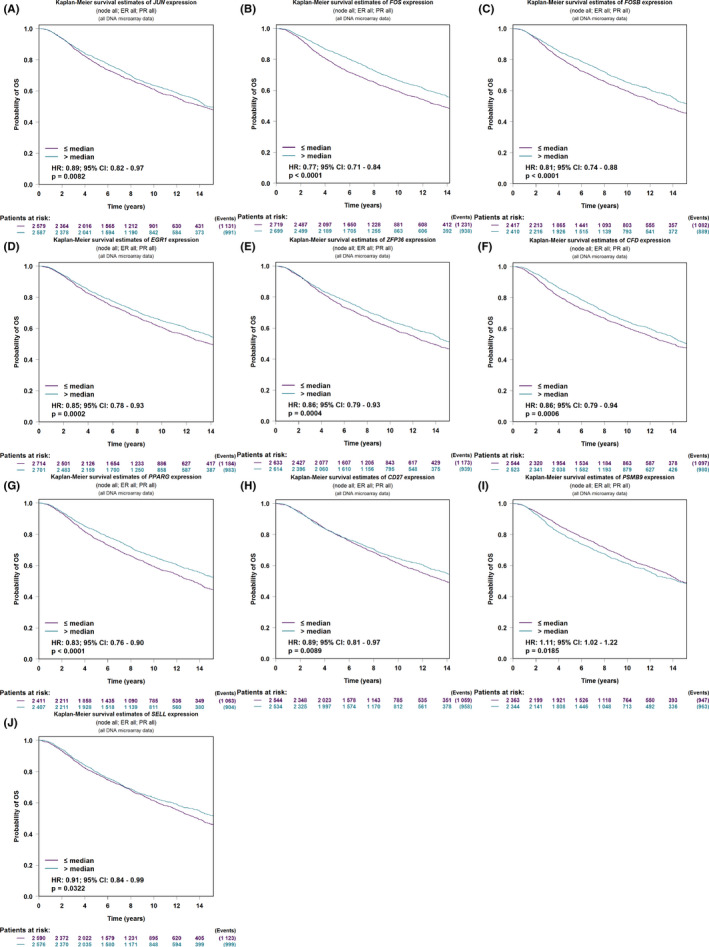
Associations of candidate genes with the overall survival (OS) in breast cancer (BC) patients. (A–E) Five candidate genes JUN, FOS, FOSB, EGR1, and ZFP36 from the GSE9574 dataset significantly associated with the OS in BC patients. (E–J) Five candidate genes CFD, PPARG, CD27, PSMB9, and SELL were significantly associated with the OS evaluated by multivariate Cox survival analysis in BC patients
Moreover, we further predicted the prognostic values of these genes associated with OS in BC patients with different stages, ages, molecular subtypes, ER/PR/HER2 status, TNM status, and TP53 mutation status, in addition to their expression levels using Cox Proportional‐Hazards model based on TCGA cohorts. As a result, a total of 789 BC patients were enrolled, and the clinical characteristics and TP53 status are listed in Table S6. According to the univariate Cox regression analysis, we found that stage, age, tumor status, node status, metastasis status, and the expression levels of CD24 were significantly negatively correlated to OS as risk factors, while the expression levels of JUN, FOS, FOSB, ZFP36, CFD, LEP, PLIN1, ADIPOQ, PPARG, PVRIG, CD27, PSMB9, GPD1, PLIN4, and SELL were significantly positively correlated to OS as protective factors (Table S7). Then, according to multivariate analysis, only CFD (HR = 0.84, p = 0.0327), PPARG (HR = 0.80, p = 0.0478), CD27 (HR = 0.75, p = 0.0001), PSMB9 (HR = 0.73, p = 0.00299), and SELL (HR = 0.77, p = 0.0062) were still significantly positively associated with OS as potentially independent prognostic factors in BC patients (Table S8).
3.7. Expression pattern of candidate genes based on clinical features and association with immune microenvironment in BC
Through expression validation and survival analysis, 10 candidate genes (JUN, FOS, FOSB, EGR1, ZFFP36, PPARG, CFD, CD27, PSMB9, and SELL) were determined. To dissect their variations and roles in BC, we further explored their expression levels based on clinical features, and whether their aberrations were correlated with BC immune microenvironment (CD8+ T cell, macrophage, NK cell, and CAF) in four main subtypes of BC. As a result, seven candidate genes (JUN, FOS, FOSB, EGR1, ZFP36, CFD, and PPARG) were significantly upregulated in luminal A type of BC and normal breast‐like samples compared with luminal B, HER2+, and TNBC, while CD27, PSMB9, and SELL were opposite (Figure 7), as well as in TP53‐normal relative to TP53‐mutant group (Figure S3). The expressions levels of FOS, FOSB, EGR1, JUN, ZFP36, PPARG, and CFD were lower as the stage got worse, whereas the expression of PSMB9 was higher (Figure S4). Meanwhile, JUN, FOS, EGR1, ZFP36, CFD, and PPARG were lower expressed in patients aged 21–40 compared to 40–70 and 70–97, while CD27, PSMB9, and SELL were higher expressed (Figure S5). In addition, FOS and FOSB were downregulated when lymph node metastasis was present, while CD27 and SELL were upregulated (Figure S6).
FIGURE 7.
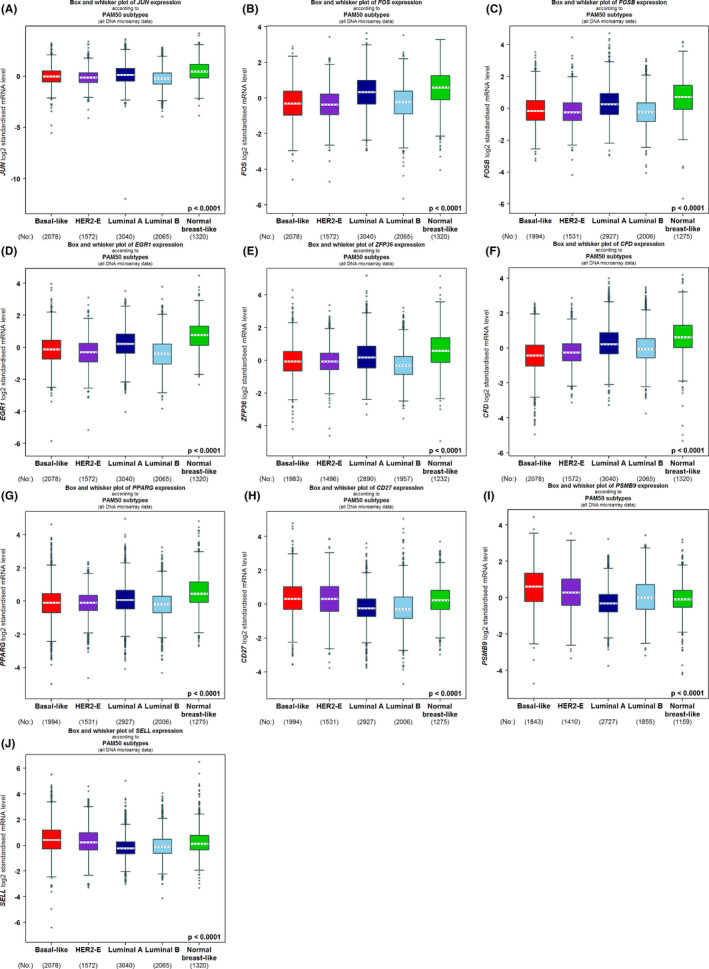
Expression levels of candidate genes based on major subclasses of breast cancer (BC). (A–E) Five candidate genes JUN, FOS, FOSB, EGR1, and ZFP36 from GSE9574 dataset (F–G) Two candidate genes CFD and PPARG from GSE15852 dataset (H–J) Three candidate genes CD27, PSMB9, and SELL from GSE42568 dataset
Next, we estimated the correlations of candidate gene expression levels with infiltration levels of four immune cells (CD8+ T cell, macrophage, NK cell, and CAF) in four subtypes of BC, respectively. Multiple algorithm results displayed that JUN was negatively correlated with CD8+ T cell infiltration in basal BC, while positively with CAF infiltration in basal and luminal A type of BC (Figure 8A,D). FOS, FOSB, EGR1, ZFP36, CFD, and PPARG were positively correlated with M2 macrophage and CAF infiltration in basal BC, while they were positively correlated with CD8+ T cells, activated NK cells, and CAFs, and negatively correlated with resting NK cells in luminal A type of BC (Figure 8A,D). Of interest, the correlations of CD27, PSMB9, and SELL expression with immune cell infiltration were almost the same among four subtypes of BC (Figure 8). Additionally, we noted that CFD and PPARG were positively correlated with CD8+ T cells, macrophages, activated NK cells, and CAFs in HER2+ type of BC, but only with macrophages in luminal B type of BC (Figure 8B,C). FOS, FOSB, EGR1, and ZFP36 were positively correlated with CAFs in luminal B type of BC (Figure 8C). Furthermore, CD27, PSMB9, and SELL were positively correlated with the biomarkers of CD8+ T cells and macrophages, as well as immune checkpoints, such as CTLA4, TIGIT, and BTLA (Figure S7).
FIGURE 8.

Correlations between candidate gene expression and immune cell infiltration (CD8+ T cell, macrophage, natural killer cell, and cancer‐associated fibroblast) in four major types of breast cancer (BC). (A) In basal BC. (B) In HER2+ BC. (C) In luminal A type of BC. (D) In luminal B type of BC. The color of the rectangle indicates the corresponding degree of correlation. The number in the rectangle represents the corresponding Spearman correlation coefficient and those with p < 0.05 are presented
3.8. Promoter methylation, genetic alteration, and downstream‐targeted genes of candidate genes
Then, we explored the mechanism of candidate gene expression changes from DNA promoter methylation status and genetic alteration. The promoter methylation levels of three genes PPARG, CFD, and SELL were significantly higher in BC than normal samples (Figure S8A–C), and the main genetic alterations of these candidate genes in BC were amplification and deep deletion. Nine genes existed amplification with the highest alteration frequency, especially SELL with almost 14% amplification, while CFD existed more deep deletion.
After TF identification, we noted that seven out of eight module genes from GSE9574 were TFs, and through survival analysis, five of them (JUN, FOS, FOSB, EGR1, and ZFP36) were associated with the OS of BC patients. Therefore, we explored their downstream‐targeted genes (Figure 9A) and investigated functions and pathways enriched by these targeted genes (Figure 9B), including “positive regulation of cell death”, “pathways in cancer”, and “PI3K‐Akt signaling pathway”. Among these targeted genes, we noted TP53 regulated by JUN, and PPARG regulated by EGR1 were included (Figure 9A). Thereby, we further estimated expression correlations of candidate genes (Figure 9C) and found that the Pearson correlation coefficient between EGR1 and PPARG was 0.37 with p < 0.05 based on the TCGA cohort. Therefore, we further validated their correlation using the bc‐GenExMiner v4.7 resource with r = 0.24 and p < 0.0001 (Figure 9D).
FIGURE 9.
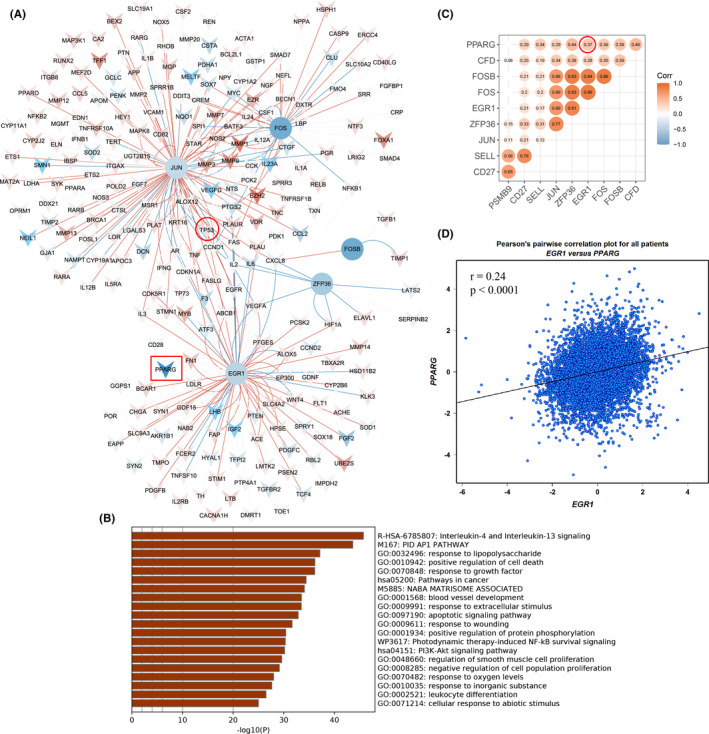
Downstream‐targeted genes of transcription factors, function enrichment, and correlation analysis. (A) Downstream‐targeted genes of JUN, FOS, FOSB, EGR1, and ZFP36. The circles represent transcription factors and the V shapes represent targeted genes, those with red indicate the corresponding genes are upregulated in BC compared with normal samples, while those with blue indicate the corresponding genes are downregulated. The red connectors present activation, while the blue connectors present suppression. (B) Main functions enriched by downstream‐targeted genes of JUN, FOS, FOSB, EGR1, and ZFP36. (C) Expression correlations of candidate genes. The number in the circle represents the corresponding Pearson correlation coefficient and those with p < 0.05 are presented. (D) Expression correlation of EGR1 with PPARG
3.9. Expression validation of candidate genes among BC samples by RT‐PCR
To validate our findings in this study, we collected four BC samples (one luminal A, two luminal B, and one HER2+ subtype) and four paired normal breast samples, and five samples of benign breast disease. JUN, FOS, FOSB, EGR1, ZFP36, and PPARG were detectable in luminal A, luminal B, and HER2+ BC patients. All of them were downregulated in HER2+ and at least one luminal B patients by comparing cancer tissue to the corresponding normal tissue, except that FOSB was only downregulated in HER2+, and that PPARG was downregulated in luminal A and one luminal B patients (Figure S9A–F without p‐value). For CFD, CD27, PSMB9, and SELL, whose expression data were only available in HER2+ BC, and the results were consistent with those predicted by microarray data (Figure S9G without p‐value). Next, we compared expression levels of candidate genes in BC and non‐BC tissues obtained from patients with benign breast disease. All downregulated genes were lower expressed in cancer tissues from the HER2+ BC patient compared to non‐BC tissues, and all upregulated genes were obviously higher in cancer tissues from one luminal B BC patient (Figure S10).
3.10. Pathway variations correlated with the expression of FOS
FOS was identified as a DEG in all the three datasets and positively associated with the OS, DMFS, and PFS in BC patients, as well as immune cell infiltration. FOS might play vital roles in neoplasia and progression in BC as a TF and immune‐associated gene. Thereby, we performed GSEA based on the expression values of FOS and discovered that nine pathways (containing “Leishmania infection”, “Fc gamma r mediated phagocytosis”, and “Antigen processing and presentation”) were positively correlated with the expression of FOS and one pathway (“Linoleic acid metabolism”) was negatively correlated in para‐cancerous samples of GSE9574 with the cutoff: FDR q < 0.25 (Figure 10A,B; Table S9). Diversely, in BC samples of GSE15852, two pathways (“Focal adhesion” and “Retinol metabolism”) were positively correlated with the expression of FOS (Figure 10C; Table S9), while in BC samples obtained from GSE42568, 14 pathways were positively correlated with the expression of FOS, of which the top three were “Focal adhesion”, “Adherens junction”, and “Colorectal cancer” according to normalized enrichment score (Figure 10D–F; Table S9).
FIGURE 10.
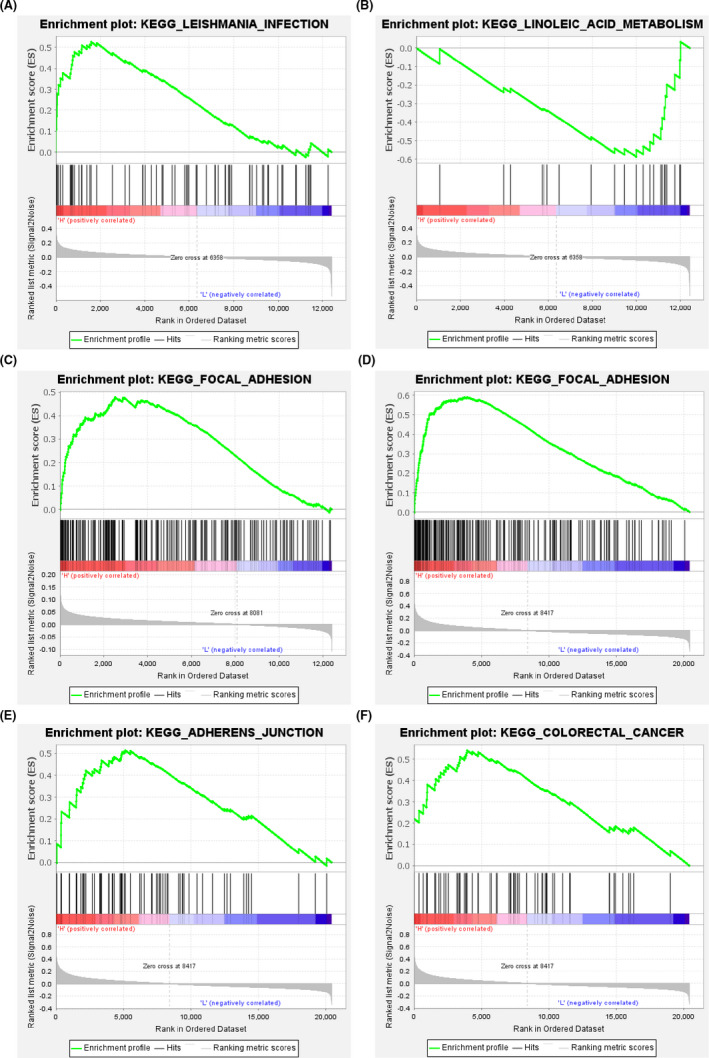
Gene set enrichment analysis based on the expression levels of FOS and the gene sets of KEGG pathways. (A) The top pathway positively correlated with the expression levels of FOS according to NES and false discovery rate (FDR) q < 0.25 based on the para‐cancerous samples in GSE9574 dataset. (B) The pathway negatively correlated with the expression levels of FOS according to FDR q < 0.25 based on the para‐cancerous samples in GSE9574 dataset. (C) The pathway positively correlated with the expression levels of FOS according to NES and FDR q < 0.25 based on the BC samples in GSE15852 dataset. (D–F) The top three pathways positively correlated with the expression levels of FOS according to NES and FDR q < 0.25 based on the BC samples in GSE42568 dataset
4. DISCUSSION
BC is the most common carcinoma in women worldwide with the leading cause of cancer death. 1 With the quick development of medical care, the mortality of BC has sharply decreased and approximately 70%–80% BC can be curable in recent years. 4 However, a complete cure for BC patients is not yet possible with currently available therapies due to the ambiguous mechanism of tumorigenesis and tumor progression in BC. Consequently, it is necessary and a trend to dissect mechanisms of tumorigenesis and progression of BC more explicitly at the level of genes. In the present work, we focused on the transcriptional aberrations from normal tissues to BC.
First, we revealed different gene expression profiles among normal, para‐cancerous, and BC tissues, and initially identified 10 downregulated genes in para‐cancerous compared with normal samples, 14 upregulated and 93 downregulated genes in BC compared with para‐cancerous samples, and 2936 upregulated and 933 downregulated genes in BC compared with normal samples. Since para‐cancerous tissue is the intermediate stage from normal to BC. If the para‐cancerous tissue is not removed, cancer definitely will return. 43 Thus, the disturbance in para‐cancerous tissue could help us unmask tumor formation. Following the thought, we further implemented function and pathway annotation analysis for these genes and found they participated in various BP, located in different cellular components, possessed diverse molecular functions, and were involved in different pathways, suggesting that in the process of tumor development, different stages had different main functional changes caused by these DEGs. Thereby, we next constructed PPI and gene co‐expression networks to show interrelationships among these DEGs, and further identify key genes (module genes). As a result, for the GSE9574 dataset, eight module genes were stood out and mainly involved in the AP1 pathway, HTLV‐1 infection, and response to extracellular stimulus. For GSE15852, 16 module genes were screened out and mainly involved in adipogenesis, response to acid chemical, regulation of cell–cell adhesion, etc. For GSE42568, 29 module genes were identified and mainly involved in immunoregulatory interactions between a lymphoid and a non‐lymphoid cell, NK cell activation, and NF‐kappa B signaling. These module genes might play crucial roles during tumorigenesis and progression of BC, through biological function and pathway perturbances caused by them.
TFs regulate gene expression and their regulatory networks are dynamic. 44 In the study combined with the existed database of TFs, we found that seven module genes (JUN, FOS, FOSB, ATF3, EGR1, NR4A2, and ZFP36) identified from GSE9574 were TFs, while only two module genes (GATA3 and PPARG) from GSE15852 and one module gene EOMES from GSE42568 were TFs, indicating that JUN, FOS, FOSB, ATF3, EGR1, NR4A2, and ZFP36 might be responsible for tumor initiation even early before pathological diagnosis, and existed tumors might first control the expression of TFs and then influence the expression of their downstream target genes to create a suitable environment for survival and expansion.
Subsequently, with validations of the expression levels of these module genes and survival analysis, we further screened out 10 candidate genes for the following analysis. Five candidate genes (JUN, FOS, FOSB, EGR1, and ZFP36) were differentially expressed in para‐cancerous compared with normal tissues, and significantly associated with the OS of BC patients. The other five candidate genes (CFD, PPARG, CD27, PSMB9, and SELL) could be independent prognostic factors in BC. Among them, seven genes (JUN, FOS, FOSB, EGR1, ZFP36, CFD, and PPARG) were downregulated in BC compared to normal tissues, and in aggressive BC (basal, HER2+, and luminal B), TP53 mutation group, younger patients, higher stages, and lymph node metastasis, while CD27, PSMB9, and SELL were upregulated, suggesting that these candidate genes could be indicators of aggressive types of BC, like HER2+ and TNBC, and might be responsible for the characteristics of invasive BC.
Prior studies noted that the immune system modulated the development and progression of BC, and in turn, cancer influenced the immune microenvironment. 45 Previous studies discovered that immunity at early stages of cancer showed anticancer profiles, while as the tumor progressed, it could transfer to the tumor‐protective profile. 46 Moreover, immune cell infiltrations, especially lymphocytes, were reported to be positively correlated with patients' outcomes at early stages of HER2+ BC and TNBC. 4 , 47 , 48 On the contrast, the infiltration of CAFs and M2 macrophages may contribute to cancer progression. 49 , 50 The present study discovered that the expression levels of candidate genes were correlated with the infiltration of immune cells (CD8+ T cell, macrophage, NK cell, and CAF) in four subtypes of BC, especially in luminal A type of BC. We noted that correlations of three upregulated genes (CD27, PSMB9, and SELL) with immune cell infiltrations were almost the same in four subtypes of BC, while seven downregulated genes were not. Moreover, CD27, PSMB9, and SELL were also highly correlated with biomarkers of CD8+ T cells and macrophages, as well as immune checkpoints. These findings suggested that these candidate genes might be involved in tumor immuno‐editing and promisingly therapeutic targets for advanced BC.
Abnormal gene expression is regulated by genetic and epigenetic mechanisms in cancer. 51 , 52 Here, we further estimated promoter methylation and genetic alteration of candidate genes and found that the promoters of PPARG, CFD, and SELL were hypermethylated, while almost all of them existed amplification with highest frequency except for CFD with the high deep deletion. These results might partly explain the expression aberrations of candidate genes. Moreover, JUN, FOS, FOSB, EGR1, and ZFP36 were differentially expressed in para‐cancerous compared with normal tissue and associated with OS of BC patients, which may participate in BC tumorigenesis. As all of them were TFs, we further explored downstream‐ targeted genes of them and found that JUN‐regulated TP53 expression o, while EGR1‐regulated PPARG expression. These targeted genes were mainly involved in “positive regulation of cell death”, “pathways in cancer”, and “PI3K‐Akt signaling pathway”. These findings further revealed that these genes might be important for us to understand BC initiation. Moreover, we observed that the expression of FOS (one of the candidate genes and differentially expressed among breast tissues of three distinct statuses) was associated with different pathway variations in para‐cancerous and BC tissues through GSEA, indicating its various functions in BC.
In addition, for gene perturbations in para‐cancerous tissues, the original study identified 105 DEGs in histologically normal tissues near breast tumors compared to the normal tissues using Cyber‐T‐test and concluded that the characteristics of para‐cancerous tissues were more like BC tissues. 19 Another study elucidated that expression profiles varied with distance from the tumor in eight BC patients. 53 In our study, using R package limma with statistical significance: adjusted p < 0.05 and |log2 fold change| > 1, 10 DEGs were identified, and all of them were downregulated in tumor adjacent compared with the normal tissues, of which eight DEGs (JUN, FOSB, FOS, ATF3, EGR1, IER2, ZFP36, and NR4A2) were overlapped with the original study mentioned above. 19 Whereas another study didn't discern differences between these two states in breast tissues, which also applied limma package of R, but with the significance of false discovery rate adjusted p ≤ 0.01 and fold change ≥ 2.0, 54 and this may partly explain the inconsistency. In addition, though the original study 19 found no gene expression differences by unsupervised hierarchical clustering or principal component analysis of all genes, we were not sure if the aforesaid reason was included because the concrete cutoff of significance did not elucidate. Therefore, the gene expression perturbations in para‐cancerous tissues are hard to be determined, which might depend on how far away from the tumor to get the tissue, analytical methods used, and a certain number of samples. For DEGs between BC and para‐cancerous or normal tissues, we will not discuss them here, since they have been extensively studied.
In fact, JUN, FOS, FOSB, and ATF3 are components of TF activator protein‐1 (AP‐1), a type of dimeric TF that has been widely studied and is formed by JUN, FOS, ATF, and MAF protein families, which regulates a variety of functions, such as cell proliferation, differentiation, transformation, apoptosis, survival, 55 , 56 migration, and oncogenesis in the response to extracellular and external stimuli such as cytokines, growth factors, stress signals, bacterial and viral infections, and oncoproteins. 57 , 58 Moreover, previous studies concluded that AP‐1 was mainly associated with autoimmune disorders, carcinomas, and hematological malignancies. 58 Initially, proteins of the AP‐1 family, JUN, FOS, and ATF were considered to be oncogenic, whereafter JUNB and FOS were reported to suppress tumorigenesis. 59 JUN was then illustrated to suppress tumorigenesis. 60 On the other hand, some studies indicated that AP‐1 might play important roles in tumor cell invasion 61 , 62 and metastasis. 63 These studies as well confirmed different effects of FOS on the tumor.
In BC, previous studies found that FOS and JUN were downregulated in BC compared with adjacent and normal tissues, 64 , 65 but FOSB had no difference. 65 In other studies, FOS and FOSB were found to increase cancer cell invasion, 61 and JUN was involved in cancer cell invasion and metastasis. 66 Recently, researchers have further elucidated the mechanisms of their functions in tumorigenesis, tumor cell invasion, and metastasis. For instance, Xie et al. 67 reported that JUN was upregulated in TNBC than non‐TNBC tumors, and the JUN N‐terminal kinase (JNK)/JUN signaling pathway might regulate cancer stem‐like cell phenotype and tumorigenesis through Notch1 signaling pathway in TNBC, while Anders et al. 68 demonstrated that hyper‐phosphorylation of JUN by JNK inhibited tumor cell migration and invasion. Diann. et al. found that FOS promoted the expression of CRADD and UQCRC2, two apoptosis‐effector genes, and was associated with better survival outcomes. 69 A recent study found that FOSB was significantly decreased in different subtypes of BC samples. Its higher expression was related to better RFS in TNBC and Her2‐ BC based on Oncomine and TCGA datasets, and increased TNBC cell proliferation in the FOSB knockout model conversely indicated its ability of tumor inhibition. 70 Furthermore, studies on drug‐treated TNBC cells showed that FOSB might be a pro‐apoptotic protein and led to BC cell death, 71 whereas the upregulated FOS in response to eribulin treatment in TNBC cells might be responsible for the low drug sensitivity. 72 ATF3 was highly expressed when the organism encountered cancer cells as an adaptive‐response gene, which could induce apoptosis as a tumor suppressor or enhance BC cell tumorigenicity as an oncogene, contributing to epithelial‐to‐mesenchymal transition and metastasis. 73 , 74 A current research showed that ATF3 was downregulated in BC compared with adjacent tissues, and overexpression of which enhanced BC cell apoptosis and suppress cell growth, migration, and invasion. 75
Early growth response 1 (EGR1) was considered as a tumor suppressor regulating the expressions of several cancer suppressor genes, including TGFβ1, IGF‐II, PTEN, fibronectin, p53, and p73, while in prostate cancer as a tumor enhancer. 76 The low expression of EGR1 led to poor prognosis and therapeutic resistance. 77 Human IER2 was upregulated in plenty of primary tumors compared with paired adjacent normal tissues including BC, 78 whereas it was downregulated in hepatocellular carcinoma compared with matched normal tissues, but enhanced hepatocellular carcinoma cell motility and metastasis. 79 ZFP36 is downregulated in the tumor compared with normal samples, and co‐expressed with JUN, JUNB, FOS, and FOSB, downregulation of which was associated with poor outcomes and BC progression. Thereby, ZFP36 was regarded as a tumor suppressor, and several non‐coding RNAs could restrain tumor growth through it. 80 , 81 Nuclear receptor subfamily four group A member 2 (NR4A2) were involved in cell proliferation, apoptosis, inflammation, and cancer, 82 and highly expressed in CD133+ colorectal carcinoma cells, which could be a tumor suppressor or promoter depending on the tumor provenance, 83 and no research linked it to BC yet. However, little emphasis was put on the mechanisms of these low‐expressed genes in para‐cancerous compared with normal tissues. Therefore, our findings provided more evidence to lay a foundation for that.
PPARs, a kind of transcriptional factors belonging to nuclear receptor superfamily, are made up of PPAR‐a, PPAR‐b/d, and PPAR‐g, which are associated with adipocyte differentiation, lipid metabolism, hyperlipidemia, insulin sensitization, cancer, inflammation, and atherosclerosis. 84 According to KEGG pathway enrichment and module function analyses, the PPAR signaling pathway was significantly enriched in our research model. A recent study 12 combining four GEO and TCGA databases of BC also identified PPAR signaling pathway as a main pathway involved in BC, indicating that the PPAR signaling pathway‐regulated BC progression. As Figure 4F showed that seven module genes were gathered in PPAR pathway, namely PCK1, PPARG, PLIN1, ADIPOQ, FABP4, LPL, and ACSL1, of which PPARG showed the highest degrees in the PPI network and the upstream location in this pathway (Figure S11). PPARG constitutes heterodimer with retinoid X receptor and thus conducts transcription of multiple genes, which mainly regulates adipocyte differentiation and is related to cancer, inflammation, and atherosclerosis. 84 In BC, researches indicated that PPARG inhibited tumor progression 85 , 86 , 87 as a promising antineoplastic agent in the clinic. 88
Complement factor D (CFD), also known as ADIPSIN, targeted by PPARG, 89 is secreted from adipocytes. It was reported to be expressed in adipose tissues surrounding a tumor, which could promote BC cell growth and cancer stem cell‐like properties. 90 CD27 is an immune checkpoint gene, 91 whose signal contributes to antitumor immunity in humans. Ongoing clinical trial targeting CD27 has been established. 92 PSMB9, a kind of immunoproteasome gene, consistent with our findings, was reported to be highly expressed in BC and associated with long survival. 93 Selectin L (SELL) was also highly expressed in BC and associated with better outcomes of BC patients according to public data. 94
In summary, our work implemented a comprehensive bioinformatic analysis on BC, showing different transcriptional variations in para‐cancerous tissues and BC. Our findings deepened our understanding of the mechanisms of tumorigenesis, invasion, relapse, progression, and prognosis of BC, and provided promisingly diagnostic and therapeutic targets for BC patients.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTION
Xingxing Dong, Yan Gong, and Gaosong Wu conceived the study; Xingxing Dong, Gaoran Xu, and Zelin Tian delt with the data; Xingxing Dong and Qian Yang performed the visualization; Xingxing Dong wrote the draft; Yalong Yang, Yan Gong, and Gaosong Wu revised the paper. All authors have read and agreed to the published version of the manuscript.
ETHICS STATEMENT
All studies involving human samples have received the patient's informed consent and the approval of the Wuhan University Ethics Committee.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank Jianing Tang for his help at the beginning of this work and Xiaoping Liu for his advice. The authors also appreciate GEO database and the researchers providing the original data.
Dong X, Yang Y, Xu G, et al. The initial expression alterations occurring to transcription factors during the formation of breast cancer: Evidence from bioinformatics. Cancer Med. 2022;11:1371–1395. doi: 10.1002/cam4.4545
Funding information
This research received no external funding.
Contributor Information
Yan Gong, Email: yan.gong@whu.edu.cn.
Gaosong Wu, Email: wugaosongtj@163.com.
DATA AVAILABILITY STATEMENT
The data presented in this study are openly available in GEO database (https://www.ncbi.nlm.nih.gov/geo/), reference number. 18 , 62 , 63
REFERENCES
- 1. Sung H, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209‐249. [DOI] [PubMed] [Google Scholar]
- 2. Aleskandarany MA, Vandenberghe ME, Marchiò C, et al. Tumour heterogeneity of breast cancer: from morphology to personalised medicine. Pathobiology. 2018;85(1–2):23‐34. [DOI] [PubMed] [Google Scholar]
- 3. Dai X, Li T, Bai Z, et al. Breast cancer intrinsic subtype classification, clinical use and future trends. Am J Cancer Res. 2015;5(10):2929‐2943. [PMC free article] [PubMed] [Google Scholar]
- 4. Harbeck N, Penault‐Llorca F, Cortes J, et al. Breast cancer. Nat Rev Dis Primers. 2019;5(1):66. [DOI] [PubMed] [Google Scholar]
- 5. Waks AG, Winer EP. Breast cancer treatment: a review. JAMA. 2019;321(3):288‐300. [DOI] [PubMed] [Google Scholar]
- 6. DeSantis CE, Ma J, Gaudet MM, et al. Breast cancer statistics, 2019. CA Cancer J Clin. 2019;69(6):438‐451. [DOI] [PubMed] [Google Scholar]
- 7. Schmid P, Adams S, Rugo HS, et al. Atezolizumab and Nab‐paclitaxel in advanced triple‐negative breast cancer. N Engl J Med. 2018;379(22):2108‐2121. [DOI] [PubMed] [Google Scholar]
- 8. Zhang H. Overview of sequence data formats. Methods Mol Biol. 2016;1418:3‐17. [DOI] [PubMed] [Google Scholar]
- 9. Gentleman R, Carey VJ, Huber W, et al. Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Springer; 2005. [Google Scholar]
- 10. Zeng Q, et al. Identification of therapeutic targets and prognostic biomarkers among CXC chemokines in the renal cell carcinoma microenvironment. Front Oncol. 2019;9:1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cui X, Zhang X, Liu M, et al. A pan‐cancer analysis of the oncogenic role of staphylococcal nuclease domain‐containing protein 1 (SND1) in human tumors. Genomics. 2020;112(6):3958‐3967. [DOI] [PubMed] [Google Scholar]
- 12. Xu Y‐H, Deng J‐L, Wang L‐P, et al. Identification of candidate genes associated with breast cancer prognosis. DNA Cell Biol. 2020;39(7):1205‐1227. [DOI] [PubMed] [Google Scholar]
- 13. Meng L‐B, Shan M‐J, Qiu Y, et al. TPM2 as a potential predictive biomarker for atherosclerosis. Aging. 2019;11:6960‐6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ge W‐H, Lin Y, Li S, et al. Identification of biomarkers for early diagnosis of acute myocardial infarction. J Cell Biochem. 2018;119(1):650‐658. [DOI] [PubMed] [Google Scholar]
- 15. Li L, Lei Q, Zhang S, et al. Screening and identification of key biomarkers in hepatocellular carcinoma: evidence from bioinformatic analysis. Oncol Rep. 2017;38(5):2607‐2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guo Y, Bao Y, Ma M, et al. Identification of key candidate genes and pathways in colorectal cancer by integrated bioinformatical analysis. Int J Mol Sci. 2017;18(4):722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang J, Wang Y, Kong F, et al. Identification of a six‐gene prognostic signature for oral squamous cell carcinoma. J Cell Physiol. 2020;235(3):3056‐3068. [DOI] [PubMed] [Google Scholar]
- 18. Clough E, Barrett T. The gene expression omnibus database. Methods Mol Biol. 2016;1418:93‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tripathi A, King C, de la Morenas A, et al. Gene expression abnormalities in histologically normal breast epithelium of breast cancer patients. Int J Cancer. 2008;122(7):1557‐1566. [DOI] [PubMed] [Google Scholar]
- 20. Pau Ni IB, Zakaria Z, Muhammad R, et al. Gene expression patterns distinguish breast carcinomas from normal breast tissues: the Malaysian context. Pathol Res Pract. 2010;206(4):223‐228. [DOI] [PubMed] [Google Scholar]
- 21. Clarke C, Madden SF, Doolan P, et al. Correlating transcriptional networks to breast cancer survival: a large‐scale coexpression analysis. Carcinogenesis. 2013;34(10):2300‐2308. [DOI] [PubMed] [Google Scholar]
- 22. Heber S, Sick B. Quality assessment of Affymetrix GeneChip data. OMICS J Integr Biol. 2006;10(3):358‐368. [DOI] [PubMed] [Google Scholar]
- 23. Irizarry RA, et al. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31(4):e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Troyanskaya O, Cantor M, Sherlock G, et al. Missing value estimation methods for DNA microarrays. Bioinformatics. 2001;17(6):520‐525. [DOI] [PubMed] [Google Scholar]
- 25. Smyth GK. Limma: Linear Models for Microarry Data. Springer. 2005;397‐420. [Google Scholar]
- 26. Gene Ontology Consortium . The gene ontology resource 20 years and still GOing strong. Nucleic Acids Res. 2018;47:D330‐D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Consortium , Ashburner M, Ball CA, Blake JA, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kanehisa M, Sato Y, Furumichi M, et al. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019;47(D1):D590‐D595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein‐protein association networks with increased coverage, supporting functional discovery in genome‐wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607‐D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang B, Horvath S. A general framework for weighted gene co‐expression network analysis. Stat Appl Genet Mol Biol. 2005;4(1). [DOI] [PubMed] [Google Scholar]
- 31. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Smoot ME, Ono K, Ruscheinski J, et al. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27(3):431‐432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. 2003;4(2):1471‐2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhou Y, Zhou B, Pache L, et al. Metascape provides a biologist‐oriented resource for the analysis of systems‐level datasets. Nat Commun. 2019;10(1):1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Han H, Cho J‐W, Lee S, et al. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018;46(D1):D380‐D386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chandrashekar DS, Bashel B, Balasubramanya SAH, et al. UALCAN: a portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia. 2017;19(8):649‐658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tomczak K, Czerwinska P, Wiznerowicz M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol (Pozn). 2015;19(1A):A68‐A77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jézéquel P, Campone M, Gouraud W, et al. bc‐GenExMiner: an easy‐to‐use online platform for gene prognostic analyses in breast cancer. Breast Cancer Res Treat. 2012;131(3):765‐775. [DOI] [PubMed] [Google Scholar]
- 39. Jezequel P, Frenel J‐S, Campion L, et al. bc‐GenExMiner 3.0: new mining module computes breast cancer gene expression correlation analyses. Database (Oxford). 2013;2013:bas060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li T, Fu J, Zeng Z, et al. TIMER2.0 for analysis of tumor‐infiltrating immune cells. Nucleic Acids Res. 2020;48(W1):W509‐W514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cerami E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wideexpression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545‐15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pilewskie M, Morrow M. Margins in breast cancer: how much is enough? Cancer. 2018;124(7):1335‐1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lambert SA, Jolma A, Campitelli LF, et al. The human transcription factors. Cell. 2018;172(4):650‐665. [DOI] [PubMed] [Google Scholar]
- 45. Nagarajan D, McArdle SEB. Immune landscape of breast cancers. Biomedicines. 2018;6:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011;331(6024):1565‐1570. [DOI] [PubMed] [Google Scholar]
- 47. Savas P, Salgado R, Denkert C, et al. Clinical relevance of host immunity in breast cancer: from TILs to the clinic. Nat Rev Clin Oncol. 2016;13(4):228‐241. [DOI] [PubMed] [Google Scholar]
- 48. Gao G, Wang Z, Qu X, et al. Prognostic value of tumor‐infiltrating lymphocytes in patients with triple‐negative breast cancer: a systematic review and meta‐analysis. BMC Cancer. 2020;20(1):179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ishii G, Ochiai A, Neri S. Phenotypic and functional heterogeneity of cancer‐associated fibroblast within the tumor microenvironment. Adv Drug Deliv Rev. 2016;99(Pt B):186‐196. [DOI] [PubMed] [Google Scholar]
- 50. Xia Y, Rao L, Yao H, et al. Engineering macrophages for cancer immunotherapy and drug delivery. Adv Mater. 2020;32(40):e2002054. [DOI] [PubMed] [Google Scholar]
- 51. Esteller M. CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene. 2002;21(35):5427‐5440. [DOI] [PubMed] [Google Scholar]
- 52. Kuscu C, Evensen N, Kim D, et al. Transcriptional and epigenetic regulation of KIAA1199 gene expression in human breast cancer. PLoS One. 2012;7(9):e44661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Abdalla M, Tran‐Thanh D, Moreno J, et al. Mapping genomic and transcriptomic alterations spatially in epithelial cells adjacent to human breast carcinoma. Nat Commun. 2017;8(1):1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Finak G, Sadekova S, Pepin F, et al. Gene expression signatures of morphologically normal breast tissue identify basal‐like tumors. Breast Cancer Res. 2006;8(5):R58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hess J, Angel P, Schorpp‐Kistner M. AP‐1 subunits: quarrel and harmony among siblings. J Cell Sci. 2004;117(Pt 25):5965‐5973. [DOI] [PubMed] [Google Scholar]
- 56. Shaulian E, Karin M. AP‐1 as a regulator of cell life and death. Nat Cell Biol. 2002;4:131‐136. [DOI] [PubMed] [Google Scholar]
- 57. Lopez‐Bergami P, Lau E, Ronai Z. Emerging roles of ATF2 and the dynamic AP1 network in cancer. Nat Rev Cancer. 2010;10(1):65‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Trop‐Steinberg S, Azar Y. AP‐1 expression and its clinical relevance in immune disorders and cancer. Am J Med Sci. 2017;353(5):474‐483. [DOI] [PubMed] [Google Scholar]
- 59. Eferl R, Wagner EF. AP‐1: a double‐edged sword in tumorigenesis. Nat Rev Cancer. 2003;3(11):859‐868. [DOI] [PubMed] [Google Scholar]
- 60. Shaulian E. AP‐1–The Jun proteins: oncogenes or tumor suppressors in disguise? Cell Signal. 2010;22(6):894‐899. [DOI] [PubMed] [Google Scholar]
- 61. Ozanne BW, Spence HJ, McGarry LC, et al. Transcription factors control invasion: AP‐1 the first among equals. Oncogene. 2007;26(1):1‐10. [DOI] [PubMed] [Google Scholar]
- 62. Vleugel MM, Greijer AE, Bos R, et al. c‐Jun activation is associated with proliferation and angiogenesis in invasive breast cancer. Hum Pathol. 2006;37(6):668‐674. [DOI] [PubMed] [Google Scholar]
- 63. Zhang Y, et al. Critical role of c‐Jun overexpression in liver metastasis of human breast cancer xenograft model. BMC Cancer. 2007;7:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Smith LM, Birrer MJ, Stampfer MR, Brown PH. Breast cancer cells have lower activity protein 1 transcription factor activity than normal mammary epithelial cells. Can Res. 1997;57:3046‐3054. [PubMed] [Google Scholar]
- 65. Kharman‐Biz A, Gao H, Ghiasvand R, et al. Expression of activator protein‐1 (AP‐1) family members in breast cancer. BMC Cancer. 2013;13:441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhang Y, Pu X, Shi M, et al. c‐Jun, a crucial molecule in metastasis of breast cancer and potential target for biotherapy. Oncol Rep. 2007;18(5):1207‐1212. [PubMed] [Google Scholar]
- 67. Xie X, Kaoud TS, Edupuganti R, et al. c‐Jun N‐terminal kinase promotes stem cell phenotype in triple‐negative breast cancer through upregulation of Notch1 via activation of c‐Jun. Oncogene. 2017;36(18):2599‐2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sundqvist A, et al. JNK‐dependent cJun phosphorylation mitigates TGFbeta‐ and EGF‐induced pre‐malignant breast cancer cell invasion by suppressing AP‐1‐mediated transcriptional responses. Cells. 2019;8(12):1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fisler D, Sikaria D, Yavorski J, et al. Elucidating feed‐forward apoptosis signatures in breast cancer datasets: higher FOS expression associated with a better outcome. Oncol Lett. 2018;16(2):2757‐2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhang R, Li X, Liu Z, et al. EZH2 inhibitors‐mediated epigenetic reactivation of FOSB inhibits triple‐negative breast cancer progress. Cancer Cell Int. 2020;20:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Park JA, Na H‐H, Jin H‐O, Kim K‐C. Increased expression of FosB through reactive oxygen species accumulation functions as pro‐apoptotic protein in piperlongumine treated MCF7 breast cancer cells. Mol Cells. 2019;42(12):884‐892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tanaka S, Ishii T, Sato F, et al. Eribulin mesylate‐induced c‐Fos upregulation enhances cell survival in breast cancer cell lines. Biochem Biophys Res Comm. 2020;526(1):154‐157. [DOI] [PubMed] [Google Scholar]
- 73. Yin X, Wolford CC, Chang Y‐S et al. ATF3, an adaptive‐response gene, enhances TGF beta signaling and cancer‐initiating cell features in breast cancer cells. J Cell Sci. 2010;123(20):3558‐3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wolford CC, McConoughey SJ, Jalgaonkar SP, et al. Transcription factor ATF3 links host adaptive response to breast cancer metastasis. J Clin Invest. 2013;123(7):2893‐2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Li LQ, Sun RM, Jiang GQ. ATF3 demethylation promotes the transcription of ARL4C, which acts as a tumor suppressor in human breast cancer. Onco Targets Ther. 2020;13:3467‐3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Baron V, Adamson ED, Calogero A, Ragona G, Mercola D. The transcription factor Egr1 is a direct regulator of multiple tumor suppressors including TGFβ1, PTEN, p53 and fibronectin: Egr1 is a potential target of gene therapy for prostate cancer. Cancer Gene Ther. 2006;13(2):115‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Shajahan‐Haq AN, Boca SM, Jin LU, et al. EGR1 regulates cellular metabolism and survival in endocrine resistant breast cancer. Oncotarget. 2017;8(57):96865‐96884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Neeb A, Wallbaum S, Novac N, et al. The immediate early gene Ier2 promotes tumor cell motility and metastasis, and predicts poor survival of colorectal cancer patients. Oncogene. 2012;31(33):3796‐3806. [DOI] [PubMed] [Google Scholar]
- 79. Xu X, Zhou W, Chen Y, et al. Immediate early response protein 2 promotes the migration and invasion of hepatocellular carcinoma cells via regulating the activity of Rho GTPases. Neoplasma. 2020;67(3):614‐622. [DOI] [PubMed] [Google Scholar]
- 80. Canzoneri R, Naipauer J, Stedile M, et al. Identification of an AP1‐ZFP36 regulatory network associated with breast cancer prognosis. J Mammary Gland Biol Neoplasia. 2020;25(2):163‐172. [DOI] [PubMed] [Google Scholar]
- 81. Mao Y, Lv M, Cao W, et al. Circular RNA 000554 represses epithelial‐mesenchymal transition in breast cancer by regulating microRNA‐182/ZFP36 axis. FASEB J. 2020;34(9):11405‐11420. [DOI] [PubMed] [Google Scholar]
- 82. Yamamoto S, Komiya T. PS01.32: high throughput screening of small molecule Inhibitors for Nuclear Receptor Subfamily 4 Group A Member 2 (NR4A2) in human cancers. J Thorac Oncol. 2016;11(11):S288‐S289. [Google Scholar]
- 83. Han YF, Cao GW. Role of nuclear receptor NR4A2 in gastrointestinal inflammation and cancers. World J Gastroenterol. 2012;18(47):6865‐6873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kota BP, Huang THW, Roufogalis BD. An overview on biological mechanisms of PPARs. Pharmacol Res. 2005;51(2):85‐94. [DOI] [PubMed] [Google Scholar]
- 85. Yang PB, et al. Blocking PPAR gamma interaction facilitates Nur77 interdiction of fatty acid uptake and suppresses breast cancer progression. Proc Natl Acad Sci USA. 2020;117(44):27412‐27422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Xu YY, et al. PPAR gamma inhibits breast cancer progression by upregulating PTPRF expression. Eur Rev Med Pharmacol Sci. 2019;23(22):9965‐9977. [DOI] [PubMed] [Google Scholar]
- 87. Lightbody ED, et al. PPAR gamma loss increases the metastatic potential of HER2+ breast cancer. Can Res. 2018;78(13). [Google Scholar]
- 88. Augimeri G, et al. The role of PPAR gamma ligands in breast cancer: from basic research to clinical studies. Cancers. 2020;12(9):2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Brown JD, Plutzky J. Peroxisome proliferator‐activated receptors as transcriptional nodal points and therapeutic targets. Circulation. 2007;115(4):518‐533. [DOI] [PubMed] [Google Scholar]
- 90. Goto H, Shimono Y, Funakoshi Y, et al. Adipose‐derived stem cells enhance human breast cancer growth and cancer stem cell‐like properties through adipsin. Oncogene. 2019;38(6):767‐779. [DOI] [PubMed] [Google Scholar]
- 91. Fang J, Chen F, Liu D, et al. Prognostic value of immune checkpoint molecules in breast cancer. Biosci Rep. 2020;40(7):1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Buchan SL, Rogel A, Al‐Shamkhani A. The immunobiology of CD27 and OX40 and their potential as targets for cancer immunotherapy. Blood. 2018;131(1):39‐48. [DOI] [PubMed] [Google Scholar]
- 93. Rouette A, et al. Expression of immunoproteasome genes is regulated by cell‐intrinsic and ‐extrinsic factors in human cancers. Sci Rep. 2016;6:34019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kumari S, Arora M, Singh J, et al. L‐Selectin expression is associated with inflammatory microenvironment and favourable prognosis in breast cancer. 3 Biotech. 2021;11(2):38. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
The data presented in this study are openly available in GEO database (https://www.ncbi.nlm.nih.gov/geo/), reference number. 18 , 62 , 63