

Abstract
Integrative Conjugative Elements (ICEs) are replicons that can insert and excise from chromosomal locations in a site-specific manner, can conjugate across strains, and which often carry a variety of genes useful for bacterial growth and survival under specific conditions. Although ICEs have been identified and vetted within certain clades of the agricultural pathogen Pseudomonas syringae, the impact of ICE carriage and transfer across the entire P. syringae species complex remains underexplored. Here we identify and vet an ICE (PmaICE-DQ) from P. syringae pv. maculicola ES4326, a strain commonly used for laboratory virulence experiments, demonstrate that this element can excise and conjugate across strains, and highlight that this element contains loci encoding multiple type III effector proteins. Moreover, genome context suggests that another ICE (PmaICE-AOAB) is highly similar in comparison with and found immediately adjacent to PmaICE-DQ within the chromosome of strain ES4326, and also contains multiple type III effectors. Lastly, we present passage data from in planta experiments that suggests that genomic plasticity associated with ICEs may enable strains to more rapidly lose type III effectors that trigger R-gene mediated resistance in comparison to strains where nearly isogenic effectors are not present in active ICEs. Taken together, our study sheds light on a set of ICE elements from P. syringae pv. maculicola ES4326 and suggests how genomic context may lead to different evolutionary dynamics for shared virulence genes between strains.
Keywords: Pseudomonas syringae, integrative conjugative element (ICE), type III effector, phytopathogen, Nicotiana benthamiana
Introduction
Genome fluidity is crucial for survival of bacterial phytopathogens, as selection pressures on the presence and function of specific virulence genes can dramatically change from host to host (Dillon et al., 2019a; de Vries et al., 2020). Much research characterizing gene composition of bacterial pathogens focuses on presence and absence of specific virulence genes, and therefore often extrapolates from lists of loci to predict virulence and evolutionary potential (Baltrus et al., 2011; Dillon et al., 2019b). However, even if virulence genes are conserved across strains of a particular pathogen, differences in genomic flexibility for shared virulence genes could potentiate different evolutionary outcomes between strains under conditions of strong selection. Here we characterize one instance where such differences in genomic context and fluidity for a set of type III effectors affects evolutionary potential for Pseudomonas syringae, and we speculate about the ability of such systems to shift evolutionary dynamics for phytopathogens moving forward.
Integrative Conjugative Elements (ICEs) are mobile replicons that blend characteristics of both plasmids and prophage and are well known for their ability to harbor antibiotic resistance genes and other niche-association traits across bacteria (Johnson and Grossman, 2015). Like conjugative plasmids, ICEs contain all genes and pathways required for conjugation between bacterial cells. Like prophage, ICEs contain site specific recombinases that enable recombination into chromosomal locations and repress the genes responsible for conjugation and replication while in this quiescent state. Most importantly, ICEs also contain cargo regions that can house genes and pathways that directly contribute to dramatic changes in bacterial phenotypes (Lovell et al., 2009; Johnson and Grossman, 2015). Not only can these elements modify phenotypes in their current host cells, but the ability of ICEs to transfer throughout populations and across communities through horizontal gene transfer means that cargo genes present on ICEs can rapidly proliferate across strains if they are beneficial. The rise of sequencing technologies enabling closed bacterial genomes has reinforced the importance of mobile elements in genomes and increased awareness of the prevalence of ICEs throughout bacteria.
The phytopathogen Pseudomonas syringae (sensu lato) is a recognized agricultural pest for many crops throughout the world, with numerous strains well established as laboratory systems for understanding virulence of plant pathogens in vitro and in planta (Baltrus et al., 2017). The presence of a type III secretion system is critical for virulence in planta for many strains of this pathogen, and this system is used to translocate upwards of 40 effectors proteins per strain from the bacterial cytoplasm into plant cells (Dillon et al., 2019a; Laflamme et al., 2020). Once inside the plant cells, effector proteins can disrupt plant immune responses in a variety of ways to promote bacterial growth and infection. The presence of effector proteins can also be monitored by plant immune responses through the action of R-genes, with recognition of effector protein functions leading to an overarching immune reaction termed effector triggered immunity (ETI) (Collmer et al., 2000; Jones and Dangl, 2006; Jayaraman et al., 2021). Triggering of the ETI response can quickly shut down nascent infections in resistant plant cultivars and has thus formed the basis of future plans to engineer durable crop resistance to infection through genetic modification and selective breeding (Dong and Ronald, 2019; Laflamme et al., 2020). Thus, type III effectors sit at an evolutionary inflection point where they can be highly beneficial for bacterial growth in some host backgrounds and highly detrimental in others.
Throughout this manuscript, we use the phrase “genome context” as a catch-all term to represent both the placement and relative location of specific genes within a genome compared to orthologs and homologs of these specific genes. We intend this as an open-ended term to describe how sequence characteristics (gene order, location, etc.) can affect parameters that contribute to evolutionary potential. For instance, imagine two antibiotic resistance genes that are sequence identical and found within a single genome but where one copy is present on a chromosome and stably maintained whereas the other copy is found in a region that is much more easily lost (e.g., on a small mobilizable plasmid). While these two genes may play similar if not redundant phenotypic roles for cells containing this particular version of the genome, differences between copies in rates of loss and horizontal transfer could significantly influence the evolutionary potential of each copy across the population and microbial community by altering population genetic parameters like population size and deletion rates.
The presence of virulence genes, including type III effectors, on plasmids and consequent movement across strains can dramatically alter trajectories of virulence evolution for P. syringae on different host species (Cazorla et al., 2002; Schierstaedt et al., 2019). Plasmids can be lost or modified if effector proteins are recognized by a potential host, which could enable infections to proceed at a population level despite the ETI response (Grant et al., 2006; Bardaji et al., 2019). Plasmids are not the only element within P. syringae genomes displaying plasticity, though. For instance, the effector AvrPphB (aka HopAR1) triggers HR in bean plants that contain the R-gene RPS5 (Qi et al., 2014). AvrPphB is found on an genomic island in certain strains of P. syringae pathovar phaseolicola, and under selective pressure from ETI responses, bacteria with the island excised from the chromosome and maintained independently as an episome rapidly dominate the population (Godfrey et al., 2011; Neale et al., 2018). Excision of the genomic island containing AvrPphB prevents ETI because this Avr gene is downregulated under episomal replication, but enables this gene to be maintained within a population for infection of host plants where it may be beneficial. ICE elements have also been identified and characterized in P. syringae pathovar actinidae strains where their presence/absence is one of the most glaring differences between closely related strains and where they contribute to large scale differences in strain metabolism and resistance to antibacterial compounds (Colombi et al., 2017; McCann et al., 2017; Poulter et al., 2018).
Here we describe how genomic context in a locus encoding an effector protein, HopQ, differentially affects evolutionary flexibility for strains containing this effector. Recognition of HopQ by the plant R-gene Roq1 triggers ETI in the host plant Nicotiana benthamiana, and thus limits growth of strain P. syringae pv. tomato DC3000 (Pto) in this host (Wei et al., 2007; Schultink et al., 2017). We characterize the genomic context for hopQ and other linked virulence genes in both PtoDC3000 and another pathogen, Pseudomonas syringae pv. maculicola ES4326 (PmaES4326). We demonstrate that this and other effectors are differentially present in an active ICE element in PmaES4326 but not in PtoDC3000, and confirm that recognition of HopQ also limits growth of PmaES4326 in N. benthamiana. These differences are somewhat surprising because at a broad scale these regions appear syntenic. We further show how this differential genomic context allows for differential genomic plasticity for hopQ between these two strains, by demonstrating that passage of strains in Nicotiana benthamiana leads hopQ to be more readily lost in PmaES4326 than in PtoDC3000. Thus, we directly demonstrate how genomic context for homologous virulence genes in two closely related pathogens can contribute to different evolutionary trajectories for these strains.
Materials And Methods
Bacterial Strains and Culturing Conditions
E. coli was routinely cultured at 37°C in LB (Lysogeny broth; per 1 L = 10 g tryptone, 5 g yeast extract, 10 g NaCl, pH 7.5 + with 15 g agar for solidified media). P. syringae was routinely cultured at 25°C or 28°C in either LM (LB modified; per 1 L = 10.0 g tryptone, 6.0 g yeast extract, 2.0 g K2HPO4⋅3H2O, 0.6 g NaCl, 0.4 g MgSO4⋅7H2O, with 18 g agar for solidified media), KB (King’s B; per 1 L = 20.0 g proteose peptone 3, 0.4 g MgSO4⋅7H2O, glycerol 10 mL, 2.0 g K2HPO4⋅3H2O, with 18 g agar for solidified media) or KBC (KB amended with boric acid 1.5 g/L and cephalexin 80 mg/L). Liquid cultures were incubated with shaking at 200 rpm. Where appropriate, media was augmented at final concentration with rifampicin (Rf) 40–60 μg/mL, gentamicin (Gm) 10 μg/mL, kanamycin (Km) 50 μg/mL, spectinomycin (Sp) 50 μg/mL, and diaminopimelic acid (DAP) 200 μg/mL for liquid media, 400 μg/mL for solid media for the growth of DAP-auxotrophic E. coli strains.
Genome Sequencing and Assembly
For each strain with a genome sequence reported herein, a frozen stock was streaked to single colonies on King’s B (KB) agar plates, at which point a single colony was picked to 2 mL KB liquid and grown overnight on a shaker at 27°C. Genomic DNA was extracted from these overnight cultures using a Promega (Madison, WI) Wizard kit and including the optional RNAse step. Each strain was sequenced using multiple technologies, and in each case independent DNA isolations were used to prepare libraries for different sequencing platforms. Illumina sequencing for strains was performed by MiGS (Pittsburgh, PA) using their standard workflow. As described in Baym et al. (2015), this workflow uses an Illumina tagmentation kit for library generation, followed by sequencing on a NextSeq 550 instrument with 150-bp paired-end reads. Trimmomatic was used for adaptor trimming (Bolger et al., 2014). For nanopore sequencing for strain PmaES4326-D, a library was prepared from unsheared DNA using the Rapid sequencing kit (SQK-RAD004) and sequenced on a Flongle flowcell. For nanopore sequencing for strains PmaES4326-C, PmaES4326ΔDQ3, and PmaES4326-C-LA-P5-20-1, each library was prepared using unsheared DNA as an input to the LSK109 ligation sequencing kit and was sequenced on R9.4 MinION flowcells. Nucleotide bases were called during sequencing using Guppy v3.2.6 in Fast-Mode. All genomes were assembled using Unicycler v.0.4.8 (Wick et al., 2017). The public facing genomes for PmaES4326-D and PmaES4326-C were annotated using PGAP (Tatusova et al., 2016). Genomes for PmaES4326ΔDQ3, and PmaES4326-C-LA-P5-20-1 found in Figshare doi: 10.6084/m9.figshare.17064080 were annotated using Prokka v 1.14.6 (Seemann, 2014). We used breseq v. 0.35.7 (Deatherage and Barrick, 2014) to identify evolutionary changes that occurred in the passage strain PmaES4326 LA-P5-20-1 (hereafter PmaES4326 LAP5-20). Default parameters were used for all software unless otherwise specified.
Integrative Conjugative Element Identification
Potential ICE elements were identified by manual searches of gene annotations for loci that could code for proteins critical for processing of conjugative elements. These include a site specific recombinase, a TraG-like NTPase, an ATPase, and numerous proteins involved in creation of the conjugation pilus. To be present within an ICE, these elements must all be present in a contiguous segment of the genome which is bordered by a potential attachment site (often tRNA loci).
DNA Manipulation
Plasmid DNA was routinely purified using the GeneJet plasmid miniprep kit (Thermo Fisher Scientific). PCR was conducted with Phusion HiFi polymerase (Thermo Fisher Scientific). PCR/reaction cleanup and gel extraction were conducted with the Monarch PCR and DNA cleanup kit and Monarch DNA gel extraction kits (NEB). E. coli transformation was conducted by either by preparing competent cells using the Mix and Go! E. coli transformation kit (Zymo Research) or standard electro-transformation protocols. PmaES4326 strains were transformed with pCPP5372hopQ (pHopQ) (Wei et al., 2018) plasmid DNA via electro-transformation after washing and concentration of overnight liquid cultures with 300 mM sucrose (Choi et al., 2006). Restriction enzymes, T4 ligase, and Gibson Assembly Mastermix were purchased from NEB. Oligonucleotide primers were synthesized by IDT. Commercial molecular biology reagents were used in accordance with their manufacturer’s recommendations.
To create the site-specific Tn7 3xmCherry labeling vector pURR25DK-3xmcherry, pURR25 (Teal et al., 2006) was first digested with PstI and recircularized with T4 ligase to remove the nptII KmR marker gene creating pURR25DK. To replace the gfp gene in pURR25DK, the 3xmCherry cassette was PCR amplified from pGGC026 (pGGC026 was a gift from Jan Lohmann (Addgene plasmid # 488311; RRID:Addgene_48831) using primers bko374 (5′ACATCTAGAATTAAAGAGGAGAAATTAAGCATGGTG AGCAAGGGCGAGGAGGATAACATG 3′) and bko375 (5′CAGGAGTCCAAGCTCAGCTAATTAAGCTTACTTGTAC AACTCATCCATACCACCTGTTGA 3′) to introduce 30 bp 5′ overlaps corresponding to the gfp flanking regions. Gibson assembly was used to join the 3xmCherry PCR amplicon with pURR25DK backbone digested with BseRI and partially digested with HindIII.
Bacterial Conjugation and Creation of Mutant Strains
All genetic manipulations of PmaES4326 were conducted with the PmaES4326-C strain. Conjugations were performed by mixing 15 μL of fivefold concentrated, washed, overnight LM liquid cultures of each parent strain and co-culturing at 28°C overnight on sterile nitrocellulose membranes on either LM or LM + DAP plates (for conjugations with DAP-auxotrophic E. coli donor strains). Tn7 transposition conjugations always included the E. coli RHO3 pTNS3 Tn7 transposase helper strain as a third parent. For all conjugation experiments cultures of each parent strain were included separately as controls. PmaES4326 merodiploid exconjugants of pCPP5729 (pK18msGmΔhopQ1) were recovered on LM Km (Kvitko et al., 2009). Resolved Pma ES4326 pCPP5729 merodiploids were recovered via counter-selection on LM Rf + 10% sucrose and sucrose resistant clones were screened for kanamycin sensitivity by patch plating indicating the loss of the pK18ms plasmid backbone (Kvitko and Collmer, 2011). Derivative attTn7-3xmCherry transposant strains were recovered on LM Sp and pink clones were selected after 4 days incubation at 4°C and restreaked to isolation. To test the native mobility of ICE-DQ, conjugation was conducted as described above with the PmaES4326 pCPP5729 merodiploid as the donor parent and attTn7-3xmCherry derivatives of PtoDC3000 and PmaES4326 ΔICE-DQ strains (Pma DQ3 and Pma LAP5-20) as recipients. ICE-DQ exconjugants were recovered on LM Rf Sp Km. For all conjugation experiments cultures of each parent strain were included separately as controls. Conjugation frequency was calculated as the number of SpRKmR colonies recovered per recipient CFU as determined by dilution plating.
Nicotiana benthamiana Growth, Inoculation and Bacterial Passage Assays
Nicotiana benthamiana, WT LAB accession, and roq1-1 (Qi et al., 2018) were grown in a Conviron Adaptis growth chamber with 12 h light (125 μmol/m2/s) at 26°C and 12 h dark at 23°C. Plants were used at 5-7 weeks post germination. To prepare inoculum, P. syringae cultures were recovered from fresh KB plate cultures, resuspended in 0.25 mM MgCl2, standardized to OD600 0.2, and serially diluted 10,000X to ∼3 × 104 CFU/mL. Cell suspensions were infiltrated with a blunt syringe into either the 3rd, 4th, or 5th leaves. Infiltrated spots were allowed to dry fully and then plants were covered with a humidity dome and kept at 100% humidity for 6–8 days to allow symptoms to develop. At end point, leaves were photographed to document symptoms and four 4 mm diameter leaf punches (∼0.5 cm2 total) were collected with a 4 mm diameter biopsy punch from each infiltrated area. Discs were macerated in 0.1 mL of 0.25 mM MgCl2 using an Analytik Jena SpeedMill Plus homogenizer and the bacterial CFU/cm2 leaf tissue was determined by serial dilution spot plating from 10 μL volumes on LM Rf.
For P. syringae passaging in N. benthamiana, single colonies of Pto DC3000 WT and PmaES4326 were inoculated into LM Rf liquid cultures. Samples of the initial cultures (P0) were cryo-preserved at −80°C in 15% final strength sterile glycerol. The liquid cultures were diluted 1000X in 0.25 mM MgCl2 to approximately 5 × 105 CFU/mL prior to syringe infiltration into three leaf areas establishing three lineages (A, B, C) each for PtoDC3000 and PmaES4326. Tissue samples were collected 6–7 days post inoculation and processed as described above. Bacteria cultured from the 10–1 tissue macerate dilutions of each lineage were directly scraped from the dilution plate with an inoculation loop and suspended in 1 mL 0.25 mM MgCl2. These suspensions were then sub-cultured 5 μL into 5 mL LM Rf and diluted as described above to create inoculum for the next passage. This passaging scheme was repeated five times and samples of both the post-passage recovered bacteria (P1) and the corresponding sub-cultured bacteria (P1c) were cryo-preserved for each lineage and each passage. To screen for changes in N. benthamiana disease compatibility with passaged strains in a medium-throughput format, bacteria from the P5 cryo-preserved samples were streaked to isolation on KBC plates and isolated “P5” colonies were cultured in 200 μL of LM Rf in sterile 96 well microtiter plates along with Pto DC3000, Pto DC3000ΔhopQ, Pma ES4326 and PmaES4326ΔDQ3 control strains. Cultures were serially diluted 5000X in 0.25 mM MgCl2 to ∼3 × 104 CFU/mL and inoculated into N. benthamiana leaves as described. Inoculum concentration was verified via serial dilution spot plating. Inoculated areas were monitored visually for qualitative changes to disease symptoms compared to their respective WT and targeted hopQ deletion control strains over the course to 6–8 days. The CFU/cm2 leaf tissue bacterial load of select clones was determined for each strain as described above. Isolated colonies of strains that displayed bacterial load and symptoms comparable to their respective ΔhopQ backgrounds were subcultured from the dilution spot count plates and retained.
We estimate that Ne of these passage populations to be on the order of 2.5 × 104 over the course of five passages. This number was calculated by taking the inoculum of each passage (5 × 105 CFU/mL in 50 μL) and estimating that approximately 10 μL were inoculated per cm2 for a total of 2.5 × 104 for the area inoculated (∼5 cm2). We further estimate that this is the smallest bottleneck over the course of the passages, and that populations undergo ∼10 generations of division, as these populations grow from 104 to 107 cm2 in planta). Given these assumptions, Ne can be estimated by taking the harmonic mean of fluctuating population sizes (Sjödin et al., 2005) and will be highly skewed by smaller numbers. Since 1 cm2 is harvested at the end of each cycle, a conservative estimate for Ne over five passages is therefore approximately 2.5 × 104 CFU.
Results
Complete Genome Sequences for Multiple Isolates of PmaES4326
We previously reported a draft genome sequence for an isolate of PmaES4326 acquired from the lab of Jeff Dangl (Baltrus et al., 2011), and our first goal was to generate a complete genome sequence for this strain (referred to herein as PmaES4326-D, Table 1). We used MiGS (Pittsburgh, PA) to generate Illumina reads for PmaES4326-D, and their workflow generated a total of 3,284,990 paired reads and 418 Mb (∼64× coverage) of sequence. We also independently isolated genomic DNA and sequenced using an Oxford Nanopore MinION to generate 32,291 reads for a total of 465 Mb (∼71× coverage) of sequence with a read N50 of 30,656 bp. Assembly of these reads resulted in a complete circular chromosome and four separate plasmids. Notably, our isolate of PmaES4326-D contains three previously reported plasmids (pPma4326A, pPma4326B, pPma4326E) but also appears to have lost two different plasmids (pPma4326C and pPma4326D) first reported as present in this strain by Stavrinides and Guttman (2004). The assembly for this strain appears to contain an additional ∼350 kb plasmid that was not reported by Stavrinides and Guttman (2004) and which we name pPma4326F following earlier naming conventions.
TABLE 1.
Sequencing of PmaES4326 strains.
| Strain | Reads (Illumina/Nanopore) | Total bp sequenced Mb (Illumina/Nanopore) | Read N50 Nanopore bp | Total sequencing depth (Illumina/Nanopore) | SRA accession (Illumina/Nanopore) |
| PmaES4326-D | 3,284,990/32,291 | 418.372/465.212 | 30,656 | 64×/71× | SRR1598872 SRR15988724 |
| PmaES4326-C | 3,757,284/55,845 | 485.469/700.483 | 23,669 | 74×/107× | SRR15988571/ SRR15988568 |
| PmaES4326ΔDQ3 | 2,479,212/15,275 | 331.276/178.118 | 31,491 | 51×/27× | SRR15988570/ SRR15988567 |
| PmaES4326-C-LA-P5-20-1 | 1,372,720/9,934 | 189.971/146.897 | 31,020 | 29×/22× | SRR17005742/ SRR15988569 |
Given the absence of two plasmids from PmaES4326-D, we sought to generate a genome assembly from a different lab isolate, acquired from the lab of Alan Collmer referred to here as PmaES4326-C (Table 1). We used MiGS (Pittsburgh, PA) to generate Illumina reads for the Collmer lab version of PmaES4326, and their workflow generated a total of 3,757,284 paired reads and 485 Mb (∼74× coverage) of sequence. We also independently isolated genomic DNA and sequenced using an Oxford Nanopore MinION to generate 55,845 reads for a total of 700 Mb (∼107× coverage) of sequence with a read N50 of 24,669 bp. Hybrid assembly of all read types resulted in a single chromosome that appears nearly complete but was not circularized by Unicycler. However, this assembly does contain all predicted circular plasmids as well as pPmaES4326F.
Sequencing and assembly characteristics for all strains can be found in Tables 1, 2, respectfully.
TABLE 2.
Assembly of PmaES4326 strains.
| Strain | Complete genome | Number of contigs in assembly | Replicons present | Assembly accession |
| PmaES4326-D | Yes | 5 | Chromosome (circular), pPmaES4326ABEF | GCA_000145845.2 |
| PmaES4326-C | Yes | 7 | Chromosome (circular), pPmaES4326ABCDEF | GCA_020309905.1 |
| PmaES4326ΔDQ3 | Yes | 6 | Chromosome (circular), pPmaES4326ABCF | doi: 10.6084/m9.figshare.17064080 |
| PmaES4326-C-LA-P5-20-1 | No | 7 | Chromosome (not circular), pPmaES4326ABCDEF | doi: 10.6084/m9.figshare.17064080 |
An Integrative Conjugative Element Hotspot in PmaES4326
Genomic inspection of the region containing type III effectors hopQ and hopD in strains PmaES4326-C and PmaES4326-D indicate that this area is a potential hotspot for genomic plasticity. Specifically, in a region bordered by clpB and the type III effector hopR, gene content characterization strongly suggests the presence of two independent integrative conjugative elements (ICEs, Figure 1). Both ICEs are approximately 70 kb in length, and are found adjacent to tRNA loci encoding a proline anticodon. Moreover, roughly 70% of each element is composed of sequences with > 95% nucleotide similarity and which encode many of the structural genes predicted to be involved in ICE proliferation and integration. These conserved genes include predicted integrases/recombinases, pilus proteins and ATPases, regulator proteins, a topoisomerase (topB) and helicase, chromosome partitioning proteins (parB), DNA coupling proteins (traD), an NTPase (traG), and numerous loci annotated as “integrative conjugative element proteins.” We have included annotations for all genes within these two ICEs in Table 3.
FIGURE 1.
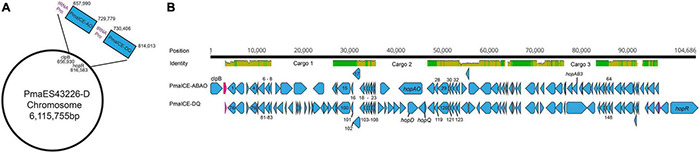
Comparison of PmaICE-ABAO and PmaICE-DQ. (A) A representation of the circular chromosome of strain PmaES4326-D, highlighting the region where both ICE elements are found as well as the tRNA-Pro loci that border these ICE elements. (B) We aligned nucleotide sequences from the pair of potential ICEs found within the clpB-hopR region in strain PmaES4326 against each other, and visualize the results here. The top line of the figure displays overall nucleotide identity between these two ICEs, with green bars representing conserved nucleotides and yellow representing slight to modest divergence. If no colored bars on this top line, the sequences are completely divergent, and we highlight the three potential cargo regions that differ between these ICE elements. Predicted loci for each ICE are shown on the lines immediately below the nucleotide identity comparison. PmaICE-ABAQ is positioned immediately adjacent to clpB in the PmaES4326 genome while PmaICE-DQ is positioned immediately adjacent to hopR in the same genome. Each predicted locus is represented in the figure in blue, while predicted tRNA loci are represented in magenta. We labeled all identified type III effector loci within these regions as well as clpB. All loci that are implicated in ICE function, by annotation, have been labeled with numbers that correspond to their annotations within Table 3.
TABLE 3.
Gene annotations in PmaES4326 ICEs.
| Number | Name | Type | Minimum | Maximum | Length | Direction |
| tRNA-Asn | tRNA | 2,862 | 2,937 | 76 | Forward | |
| tRNA-Pro | tRNA | 2,967 | 3,043 | 77 | Forward | |
| tRNA-Lys | tRNA | 3,049 | 3,124 | 76 | Forward | |
| tRNA-Pro | tRNA | 3,220 | 3,296 | 77 | Forward | |
| 1 | Tyrosine-type recombinase/integrase | CDS | 3,625 | 5,064 | 1,440 | Reverse |
| 2 | DNA-binding domain-containing protein | CDS | 5,061 | 6,992 | 1,932 | Reverse |
| 3 | DUF3742 family protein | CDS | 7,604 | 8,005 | 402 | Forward |
| 4 | traG conjugal transfer protein | CDS | 8,047 | 9,585 | 1,539 | Reverse |
| 5 | Hypothetical protein | CDS | 9,582 | 9,950 | 369 | Reverse |
| 6 | Integrating conjugative element protein | CDS | 9,953 | 11,323 | 1,371 | Reverse |
| 7 | TIGR03756 family integrating conjugative elementprotein | CDS | 11,344 | 12,282 | 939 | Reverse |
| 8 | TIGR03757 family integrating conjugative elementprotein | CDS | 12,279 | 12,755 | 477 | Reverse |
| 9 | Hypothetical protein | CDS | 13,053 | 13,241 | 189 | Forward |
| 10 | Hypothetical protein | CDS | 13,259 | 13,798 | 540 | Forward |
| 11 | Hypothetical protein | CDS | 13,839 | 14,393 | 555 | Forward |
| 12 | Hypothetical protein | CDS | 14,752 | 15,297 | 546 | Reverse |
| 13 | Thioredoxin domain-containing protein | CDS | 15,609 | 16,307 | 699 | Reverse |
| 14 | Hypothetical protein | CDS | 16,304 | 16,603 | 300 | Reverse |
| 15 | Conjugative transfer ATPase | CDS | 16,600 | 19,347 | 2,748 | Reverse |
| 16 | TIGR03751 family conjugal transfer lipoprotein | CDS | 19,557 | 20,012 | 456 | Reverse |
| 17 | TIGR03752 family integrating conjugative elementprotein | CDS | 19,990 | 21,486 | 1,497 | Reverse |
| 18 | TIGR03749 family integrating conjugative elementprotein | CDS | 21,476 | 22,399 | 924 | Reverse |
| 19 | TIGR03746 family integrating conjugative elementprotein | CDS | 22,396 | 23,064 | 669 | Reverse |
| 20 | TIGR03750 family conjugal transfer protein | CDS | 23,061 | 23,453 | 393 | Reverse |
| 21 | TIGR03745 family integrating conjugative elementmembrane protein | CDS | 23,469 | 23,837 | 369 | Reverse |
| 22 | TIGR03758 family integrating conjugative elementprotein | CDS | 23,857 | 24,096 | 240 | Reverse |
| 23 | Conjugal transfer protein | CDS | 24,093 | 24,458 | 366 | Reverse |
| 24 | DUF4177 domain-containing protein | CDS | 24,538 | 24,870 | 333 | Reverse |
| 25 | Hypothetical protein | CDS | 25,041 | 25,472 | 432 | Forward |
| 26 | hopAO type III effector | CDS | 25,861 | 26,877 | 1,017 | Reverse |
| 27 | UvrD-helicase domain-containing protein | CDS | 27,062 | 28,519 | 1,458 | Reverse |
| 28 | TIGR03747 family integrating conjugative elementmembrane protein | CDS | 28,529 | 29,278 | 750 | Reverse |
| 29 | traD | CDS | 29,318 | 31,477 | 2,160 | Reverse |
| 30 | Integrating conjugative element protein | CDS | 31,489 | 31,992 | 504 | Reverse |
| 31 | Transglycosylase SLT domain-containing protein | CDS | 31,989 | 32,546 | 558 | Reverse |
| 32 | TIGR03759 family integrating conjugative element protein | CDS | 32,531 | 33,277 | 747 | Reverse |
| 33 | Hypothetical protein | CDS | 33,286 | 33,990 | 705 | Reverse |
| 34 | dcm | CDS | 34,335 | 35,390 | 1,056 | Forward |
| 35 | vsr | CDS | 35,329 | 35,766 | 438 | Reverse |
| 36 | DNA mismatch repair protein | CDS | 35,850 | 37,871 | 2,022 | Forward |
| 37 | DEAD/DEAH box helicase family protein | CDS | 37,973 | 40,231 | 2,259 | Reverse |
| 38 | Class I SAM-dependent methyltransferase | CDS | 40,335 | 41,786 | 1,452 | Reverse |
| 39 | Hypothetical protein | CDS | 41,816 | 42,415 | 600 | Reverse |
| 40 | DUF3275 family protein | CDS | 42,536 | 43,147 | 612 | Reverse |
| 41 | Hypothetical protein | CDS | 43,248 | 43,610 | 363 | Reverse |
| 42 | Hypothetical protein | CDS | 43,672 | 43,986 | 315 | Reverse |
| 43 | DUF3577 domain-containing protein | CDS | 44,290 | 44,859 | 570 | Reverse |
| 44 | Hypothetical protein | CDS | 44,879 | 45,403 | 525 | Forward |
| 45 | Hypothetical protein | CDS | 45,480 | 45,848 | 369 | Reverse |
| 46 | Regulator | CDS | 46,056 | 46,589 | 534 | Reverse |
| 47 | topB | CDS | 47,650 | 49,671 | 2,022 | Reverse |
| 48 | Hypothetical protein | CDS | 49,794 | 50,363 | 570 | Reverse |
| 49 | Hypothetical protein | CDS | 50,657 | 51,094 | 438 | Reverse |
| 50 | ATP-dependent helicase | CDS | 51,329 | 53,281 | 1,953 | Reverse |
| 51 | Hypothetical protein | CDS | 53,284 | 54,849 | 1,566 | Reverse |
| 52 | CrpP family protein | CDS | 55,287 | 55,466 | 180 | Reverse |
| 53 | Hypothetical protein | CDS | 55,471 | 55,644 | 174 | Reverse |
| 54 | AAA family ATPase | CDS | 55,991 | 56,941 | 951 | Reverse |
| 55 | hopAB type III effector | CDS | 56,958 | 58,115 | 1,158 | Reverse |
| 56 | IS91 family transposase | CDS | 58,481 | 59,101 | 621 | Reverse |
| 57 | RulB protein | CDS | 59,132 | 59,200 | 69 | Reverse |
| 58 | IS481 family transposase | CDS | 59,217 | 60,878 | 1,662 | Reverse |
| 59 | Recombinase family protein | CDS | 60,859 | 61,434 | 576 | Reverse |
| 60 | Hypothetical protein | CDS | 61,751 | 62,224 | 474 | Reverse |
| 61 | Single-stranded DNA-binding protein | CDS | 62,425 | 62,886 | 462 | Reverse |
| 62 | Phage regulatory protein | CDS | 62,898 | 63,730 | 833 | Reverse |
| 63 | DUF3158 family protein | CDS | 63,736 | 64,257 | 522 | Reverse |
| 64 | TIGR03761 family integrating conjugative elementprotein | CDS | 64,294 | 65,073 | 780 | Reverse |
| 65 | Hypothetical protein | CDS | 65,066 | 65,662 | 597 | Reverse |
| 66 | Hypothetical protein | CDS | 65,992 | 67,353 | 1,362 | Reverse |
| 67 | DUF2857 family protein | CDS | 67,350 | 68,087 | 738 | Reverse |
| 68 | parBchromosome partitioning protein | CDS | 68,124 | 69,884 | 1,761 | Reverse |
| 69 | Arc family DNA-binding protein | CDS | 69,887 | 70,204 | 318 | Reverse |
| 70 | Hypothetical protein | CDS | 70,382 | 70,786 | 405 | Reverse |
| 71 | Hypothetical protein | CDS | 70,788 | 71,117 | 330 | Reverse |
| 72 | Hypothetical protein | CDS | 71,437 | 72,123 | 687 | Reverse |
| 73 | Hypothetical protein | CDS | 72,563 | 73,153 | 591 | Reverse |
| 74 | dnaB | CDS | 73,150 | 74,496 | 1,347 | Reverse |
| 75 | AAA family ATPase | CDS | 74,554 | 75,414 | 861 | Reverse |
| tRNA-Pro | tRNA | 75,651 | 75,727 | 77 | Forward | |
| 76 | Tyrosine-type recombinase/integrase | CDS | 76,041 | 77,495 | 1,455 | Reverse |
| 77 | DNA-binding domain-containing protein | CDS | 77,492 | 79,423 | 1,932 | Reverse |
| 78 | DUF3742 family protein | CDS | 80,037 | 80,438 | 402 | Forward |
| 79 | traG conjugal transfer protein | CDS | 80,499 | 82,031 | 1,533 | Reverse |
| 80 | Hypothetical protein | CDS | 82,028 | 82,402 | 375 | Reverse |
| 81 | Integrating conjugative element protein | CDS | 82,405 | 83,772 | 1,368 | Reverse |
| 82 | TIGR03756 family integrating conjugative elementprotein | CDS | 83,793 | 84,731 | 939 | Reverse |
| 83 | TIGR03757 family integrating conjugative elementprotein | CDS | 84,728 | 85,159 | 432 | Reverse |
| 84 | DUF1440 domain-containing protein | CDS | 85,713 | 86,201 | 489 | Reverse |
| 85 | Lytic murein transglycosylase | CDS | 86,453 | 87,694 | 1,242 | Reverse |
| 86 | Hypothetical protein | CDS | 88,186 | 88,395 | 210 | Reverse |
| 87 | IS5 family transposase | CDS | 88,841 | 89,818 | 978 | Reverse |
| 88 | Carbon storage regulator | CDS | 90,586 | 90,810 | 225 | Forward |
| 89 | TraR/DksA family transcriptional regulator | CDS | 90,846 | 91,262 | 417 | Forward |
| 90 | Hypothetical protein | CDS | 91,297 | 91,518 | 222 | Forward |
| 91 | GNAT family N-acetyltransferase | CDS | 92,339 | 92,830 | 492 | Forward |
| 92 | AAA family ATPase | CDS | 93,208 | 93,489 | 282 | Forward |
| 93 | tnpB | CDS | 93,794 | 94,150 | 357 | Forward |
| 94 | IS66 family transposase | CDS | 94,167 | 95,699 | 1,533 | Forward |
| 95 | IS5 family transposase | CDS | 95,741 | 96,382 | 642 | Reverse |
| 96 | AAA family ATPase | CDS | 96,453 | 97,271 | 819 | Forward |
| 97 | (p)ppGpp synthetase | CDS | 97,274 | 98,092 | 819 | Forward |
| 98 | Thioredoxin domain-containing protein | CDS | 98,266 | 98,964 | 699 | Reverse |
| 99 | Hypothetical protein | CDS | 98,961 | 99,260 | 300 | Reverse |
| 100 | Conjugative transfer ATPase | CDS | 99,257 | 102,004 | 2,748 | Reverse |
| 101 | TIGR03751 family conjugal transfer lipoprotein | CDS | 102,214 | 102,669 | 456 | Reverse |
| 102 | TIGR03752 family integrating conjugative elementprotein | CDS | 102,647 | 104,143 | 1,497 | Reverse |
| 103 | TIGR03749 family integrating conjugative elementprotein | CDS | 104,133 | 105,056 | 924 | Reverse |
| 104 | TIGR03746 family integrating conjugative elementprotein | CDS | 105,053 | 105,721 | 669 | Reverse |
| 105 | TIGR03750 family conjugal transfer protein | CDS | 105,718 | 106,110 | 393 | Reverse |
| 106 | TIGR03745 family integrating conjugative elementmembrane protein | CDS | 106,126 | 106,494 | 369 | Reverse |
| 107 | TIGR03758 family integrating conjugative elementprotein | CDS | 106,514 | 106,753 | 240 | Reverse |
| 108 | Conjugal transfer protein | CDS | 106,750 | 107,115 | 366 | Reverse |
| 109 | DUF4177 domain-containing protein | CDS | 107,195 | 107,525 | 331 | Reverse |
| 110 | MFS transporter | CDS | 108,102 | 109,286 | 1,185 | Forward |
| 111 | Transposase | CDS | 109,422 | 109,937 | 516 | Forward |
| 112 | Amidinotransferase | CDS | 110,235 | 111,335 | 1,101 | Forward |
| 113 | Serine kinase - nikkomycin | CDS | 111,422 | 112,675 | 1,254 | Forward |
| 114 | Serine kinase | CDS | 112,728 | 113,972 | 1,245 | Forward |
| 115 | hopDtype III effector protein | CDS | 114,438 | 116,555 | 2,118 | Forward |
| 116 | hopQtype III effector protein | CDS | 116,671 | 118,014 | 1,344 | Reverse |
| 117 | Phosphoribulokinase | CDS | 118,048 | 118,365 | 318 | Forward |
| 118 | UvrD-helicase domain-containing protein | CDS | 118,603 | 120,060 | 1,458 | Reverse |
| 119 | TIGR03747 family integrating conjugative elementmembrane protein | CDS | 120,070 | 120,819 | 750 | Reverse |
| 120 | traD | CDS | 120,859 | 123,018 | 2,160 | Reverse |
| 121 | Integrating conjugative element protein | CDS | 123,030 | 123,536 | 507 | Reverse |
| 122 | Tansglycosylase SLT domain-containing protein | CDS | 123,533 | 124,090 | 558 | Reverse |
| 123 | TIGR03759 family integrating conjugative elementprotein | CDS | 124,075 | 124,821 | 747 | Reverse |
| 124 | Hypothetical protein | CDS | 124,830 | 125,534 | 705 | Reverse |
| 125 | IS5 family transposase | CDS | 125,711 | 126,561 | 851 | Reverse |
| 126 | Hypothetical protein | CDS | 126,700 | 127,581 | 882 | Forward |
| 127 | Hypothetical protein | CDS | 127,745 | 128,107 | 363 | Reverse |
| 128 | DEAD/DEAH box helicase family protein | CDS | 128,202 | 130,460 | 2,259 | Reverse |
| 129 | Class I SAM-dependent methyltransferase | CDS | 130,564 | 132,015 | 1,452 | Reverse |
| 130 | Hypothetical protein | CDS | 132,045 | 132,644 | 600 | Reverse |
| 131 | DUF3275 family protein | CDS | 132,770 | 133,381 | 612 | Reverse |
| 132 | Hypothetical protein | CDS | 133,482 | 133,844 | 363 | Reverse |
| 133 | DUF3577 domain-containing protein | CDS | 134,172 | 134,726 | 555 | Reverse |
| 134 | Hypothetical protein | CDS | 134,746 | 135,270 | 525 | Forward |
| 135 | Hypothetical protein | CDS | 135,347 | 135,715 | 369 | Reverse |
| 136 | Regulator | CDS | 135,923 | 136,456 | 534 | Reverse |
| 137 | topB | CDS | 137,517 | 139,538 | 2,022 | Reverse |
| 138 | Hypothetical protein | CDS | 139,661 | 140,230 | 570 | Reverse |
| 139 | Hypothetical protein | CDS | 140,774 | 141,457 | 684 | Forward |
| 140 | ATP-dependent helicase | CDS | 141,779 | 143,731 | 1,953 | Reverse |
| 141 | Hypothetical protein | CDS | 143,734 | 145,259 | 1,526 | Reverse |
| 142 | CrpP family protein | CDS | 145,697 | 145,897 | 201 | Reverse |
| 143 | IS3 family transposase | CDS | 146,020 | 147,260 | 1,241 | Forward |
| 144 | Hypothetical protein | CDS | 147,274 | 147,519 | 246 | Reverse |
| 145 | Single-stranded DNA-binding protein | CDS | 147,725 | 148,186 | 462 | Reverse |
| 146 | Phage regulatory protein | CDS | 148,198 | 149,031 | 834 | Reverse |
| 147 | DUF3158 family protein | CDS | 149,037 | 149,600 | 564 | Reverse |
| 148 | TIGR03761 family integrating conjugative elementprotein | CDS | 149,597 | 150,376 | 780 | Reverse |
| 149 | Hypothetical protein | CDS | 150,369 | 150,918 | 550 | Reverse |
| 150 | Hypothetical protein | CDS | 151,362 | 152,723 | 1,362 | Reverse |
| 151 | DUF2857 family protein | CDS | 152,720 | 153,457 | 738 | Reverse |
| 152 | parBchromosome partitioning protein | CDS | 153,494 | 155,254 | 1,761 | Reverse |
| 153 | Arc family DNA-binding protein | CDS | 155,257 | 155,574 | 318 | Reverse |
| 154 | Hypothetical protein | CDS | 155,510 | 155,755 | 246 | Reverse |
| 155 | Hypothetical protein | CDS | 155,752 | 156,156 | 405 | Reverse |
| 156 | Hypothetical protein | CDS | 156,158 | 156,487 | 330 | Reverse |
| 157 | Hypothetical protein | CDS | 156,797 | 157,387 | 591 | Reverse |
| 158 | dnaB | CDS | 15,7384 | 158,730 | 1,347 | Reverse |
| 159 | AAA family ATPase | CDS | 158,788 | 159,648 | 861 | Reverse |
| tRNA-Pro | tRNA | 159,887 | 159,963 | 77 | Forward |
Despite relatively high sequence similarity across these two closely related ICEs positioned successively in the genome of PmaES4326, gene comparisons suggest the presence of three distinct cargo regions that clearly differentiate these elements (Figure 1). The first ICE contains type III effector proteins hopAO and hopAB3-1 (aka hopPmaL) in two different predicted cargo regions, and so we name this region ICE-ABAO. The second ICE contains loci for type III effectors hopD and hopQ as well as the lytic transglycosylase hopAJ1, and so we name this ICE ICE-DQ. Cargo region one contains four predicted ORFs in PmaECE-ABAO and fourteen predicted ORFs in ICE-DQ. Cargo region two contains two predicted ORFs in ICE-ABAO and seven predicted ORFs in ICE-DQ, including the effectors hopAO, hopD, and hopQ. Cargo region three contains six predicted ORFs in ICE-ABAO and two in ICE-DQ. Aside from the type III effectors, many of loci potentially encoded by these cargo regions are annotated as hypothetical proteins or as parts of IS elements and transposons.
Differential Contexts for the Genomic Island Containing hopD and hopQ Across PmaES4326 and PtoDC3000
The type III effectors HopD and HopQ are nearly sequence identical in two phytopathogens commonly used as laboratory models for studying P. syringae virulence, PtoDC3000 and PmaES4326 (Baltrus et al., 2011). Furthermore, these effectors are found in roughly the same genomic locations in the two strains, bordered on one side by clpB and on the other by the type III effector hopR (Figures 1, 2). Broad comparisons between PtoDC3000 and PmaES4326 further suggest that these effectors form a genomic island along with a third effector (hopR) across these two strains (Figure 2), an island which has been referred to as effector cluster IV in in the PtoDC3000 genome (Kvitko et al., 2009). Moreover, the size of the region between clpB and hopR in PtoDC3000 is roughly 70 kb, which closely approximates the size of each ICE in PmaES4326. While this region within PtoDC3000 does contain hints that it previously housed a functional ICE, there are other sections that are quite divergent from that in PmaES4326 (Figure 2). Specifically, the predicted recombinase in PtoDC3000 is truncated due to a frameshift mutation and multiple other loci that encode functions important for ICE conjugation and transfer are disrupted by IS elements (Figure 2). Lastly, we note that conservation of nucleotide and protein sequences between PmaICEs coupled with nucleotide diversity in PtoDC3000 at this genomic location renders accurate evaluation and comparison of evolutionary histories between PtoDC3000 and the ICEs of PmaES4326 challenging. However, the most parsimonious explanation does appear that an ICE element housed both hopQ and hopD within an ancestor of strain PtoDC3000 and that this ICE was locked in place through both IS element insertions and loss of recombinase activity.
FIGURE 2.
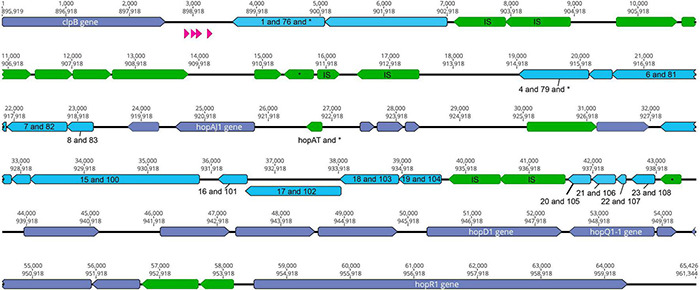
The clpB-hopR Region in PtoDC3000. Here we visualize the region between clpB and hopR from the PtoDC3000 genome, and compare this region to both predicted ICE elements from PmaES4326. Each predicted locus is colored as purple (present in one of the PmaES4326 ICE elements), blue (present in both PmaES4326 ICE elements as per nucleotide identity), and green (only present in PtoDC3000). Predicted tRNA loci are shown in magenta. We also show two nucleotide positions within this region: top number is from the start of clpB and the bottom number is from the start of the genome (from dnaA). We label all identified type III effector loci and clpB, as well as all annotated IS elements (with an “IS”) and genes that have predicted frameshifts (with an “*”). All genes potentially implicated in ICE function have been labeled with numbers corresponding to potential orthologs from strain PmaES4326 ICEs and listed in Table 3.
An Evictable Integrative Conjugative Element Containing HopQ and HopD in PmaES4326
ICEs are categorized by their ability to cleanly excise from the genome, and we confirmed the prediction that the type three effectors hopQ and hopD are contained in an excisable region in PmaES4326 in two distinct ways. As a first step, we characterized the size of the region excised during intentional creation of a hopQ- mutant. To do this, we generated a merodiploid strain in which plasmid pCPP5729 was recombined into a region adjacent to hopQ, and then selected for resolution of the merodiploid through plating on sucrose. Presence of sacB in plasmid pCPP5729 renders the merodiploid sensitive to killing by sucrose. Notably, this merodiploid also contains regions sequence identical to those surrounding hopQ in PmaES4326 and so we originally expected that the hopQ gene could be locally deleted through RecA-dependent homologous recombination. We isolated sucrose resistant isolates after plating this merodiploid, and identified strains that were neither WT reverants nor clean hopQ deletions by PCR genotyping using previously validated genotyping primers. Interestingly, while no clean hopQ deletion mutants were identified after sucrose counter-selection of merodiploid strains, WT revertants were. We then performed whole genome sequencing to confirm whether the ICE-DQ genomic region was lost under these selective conditions. We refer to this strain recovered by in vitro selection hereafter as Pma DQ3. As one can see in Figure 3, hopQ and the surrounding regions that are predicted to be part of ICE-DQ have been lost in the targeted deletion strain Pma DQ3, confirming that this region can be cleanly and completely excised from the chromosome in a manner consistent with the action of site-specific recombinases contained by ICEs. Searches of both the raw Illumina and Nanopore reads for remnants of hopQ or hopD yielded no hits despite extensive depth (∼51× for Illumina reads and ∼27× for Nanopore reads), which suggests that ICE-DQ was fully lost and not retained as an episome in Pma DQ3 (data not shown).
FIGURE 3.

Identical deletions of PmaICE-DQ following laboratory selection and passage under selective conditions in planta. We compared genomic regions bordering ICE-DQ in a strain where the region containing hopQ was intentionally deleted (Pma DQ3) as well as a strain that arose after five passages in planta in N. benthamiana (Pma LAP5-20). Genomic segments from each of the three strains are arranged vertically, with each line labeled by strain name and genes within each strain outlined in white boxes underneath the alignment. Each contiguous segment is oriented so that clpB is found on the left side of the segment and so that hopR is found on the right side. Sections within the alignment colored red are those that are aligned into contiguous blocks by Mauve and are shared by all three strains. A white section within the wild type PmaES4326 alignment represents a region (PmaICE-DQ) that is not present in the other two strains. Mauve alignments of these regions demonstrate that identical evictions of ICE-DQ occurred in each strain.
Loss of PmaICE-DQ Enables Virulence of PmaES436 on Nicotiana benthamiana
The presence of hopQ in Pma would be expected to elicit the ETI response in Nicotiana benthamiana accessions containing the R-gene Roq1, and a lack of compatible infection and disease. Given this information, we tested whether Pma DQ3 would gain compatibility with N. benthamiana. The Pma DQ3 strain did in fact gain disease compatibility with N. benthamiana in a manner similar to the gain-of-compatibility phenotypes previously observed for PtoDC3000 ΔhopQ1-1 mutants and Xanthomonas euvesicatoria 85–10 ΔxopQ mutants (Wei et al., 2007; Adlung et al., 2016). Furthermore, N. benthamiana incompatibility could be restored in PmaICEΔDQ3 by hopQ complementation in trans and PmaES4326 incompatibility was not observed in roq1-1 CRISPR-edited N. benthamiana as shown in Figure 4 (Qi et al., 2018).
FIGURE 4.
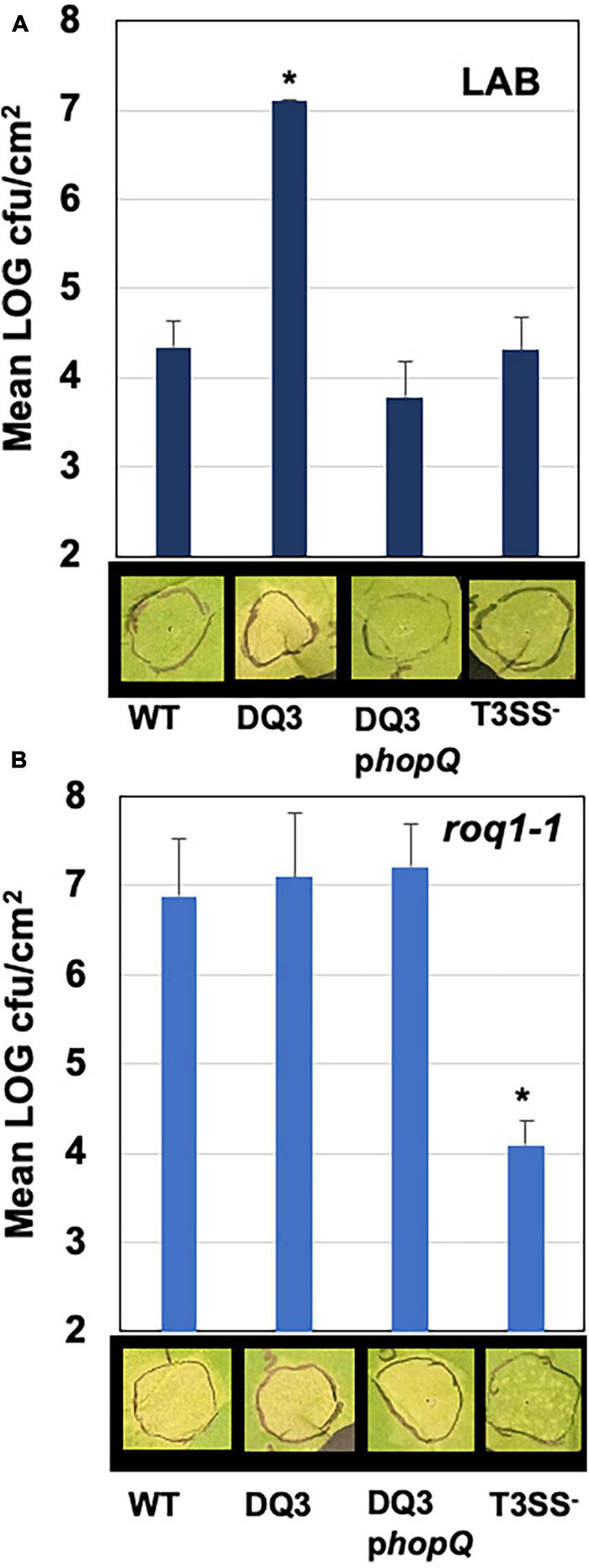
PmaES4326 gains disease compatibility with N. benthamiana in the absence of hopQ/Roq1-mediated ETI. The PmaES4326 WT strain, Pma DQ3 (ΔICE-DQ), DQ3 complemented with pCPP5372hopQ (DQ3pQ), and the non-pathogenic T3SS- strain hrcN:Tn5 were infiltrated into leaves of the N. benthamiana Lab accession (A) or the roq1-1 CRISPR mutant derivative line (B) at 3 × 104 CFU/mL. Eight days post inoculation leaves were photographed to document symptoms and bacterial load from four 4 mm diameter leaf discs from each infiltrated area by dilution plating was determined as CFUs/cm2 leaf tissue. Values are the means and standard deviations of LOG transformed CFUs/cm2 from three inoculated plants. *p < 0.05 compared to PmaES4326 WT strain as determined by unpaired one-tailed t-test.
Loss of PmaICE-DQ Under Selection in planta
Observing that we could readily recover strains containing spontaneous evictions of the ICE-DQ containing a single crossover pK18 merodiploid under the selective pressure of sucrose counter-selection, and that the targeted Pma DQ3 strain gained disease compatibility with N. benthamiana, we were curious whether the selective pressure of ETI would also result in recovery of strains with ICE-DQ evicted. We inoculated N. benthamiana with PmaES4236-C and PtoDC3000 at approximately 5 × 105 CFU/mL establishing three lineages for each strain (Lineage A, LA; Lineage B, LB; Lineage C, LC). Six to seven days post inoculation bacteria from each lineage were recovered and used to create new inoculum to passage the bacteria through N. benthamiana a total of five times. Twenty-two isolated clones of each passage 5 (P5) lineage were screened for altered N. benthamiana disease compatibility phenotypes. For PtoDC3000, none of the 66 P5 clones tested produced disease symptoms consistent with the PtoDC3000ΔhopQ N. benthamiana compatible strain and thus were not examined further. However, for PmaES4326, 7 of 66 (2 Lineage A, 3 Lineage B, 2 Lineage C) P5 clones were able to cause disease symptoms similar to the targeted deletion strain Pma DQ3 (see Figure 5A). For a subset of clones bacterial populations were determined to correlate qualitative differences in symptoms with differences in bacterial load (see Figure 5B). Genome sequencing of PmaES4326 clone (PmaES43226 LAP5-20) confirmed a loss of ICE-DQ in this strain identical to that observed in the Pma DQ3 targeted deletion strain recovered after sucrose counter-selection (Table 3 and Figure 3). We have further queried for additional evolutionary changes in PmaES4326 LAP5-20 by using breseq (Deatherage and Barrick, 2014) to map Illumina reads to the PmaES4326-C genome sequence. While it is possible, but not entirely clear, that a handful of additional changes have occurred in intergenic regions of this genome, there is no compelling data that any changes occurred in known virulence genes (see breseq output file on Figshare doi: 10.6084/m9.figshare.17064080).
FIGURE 5.
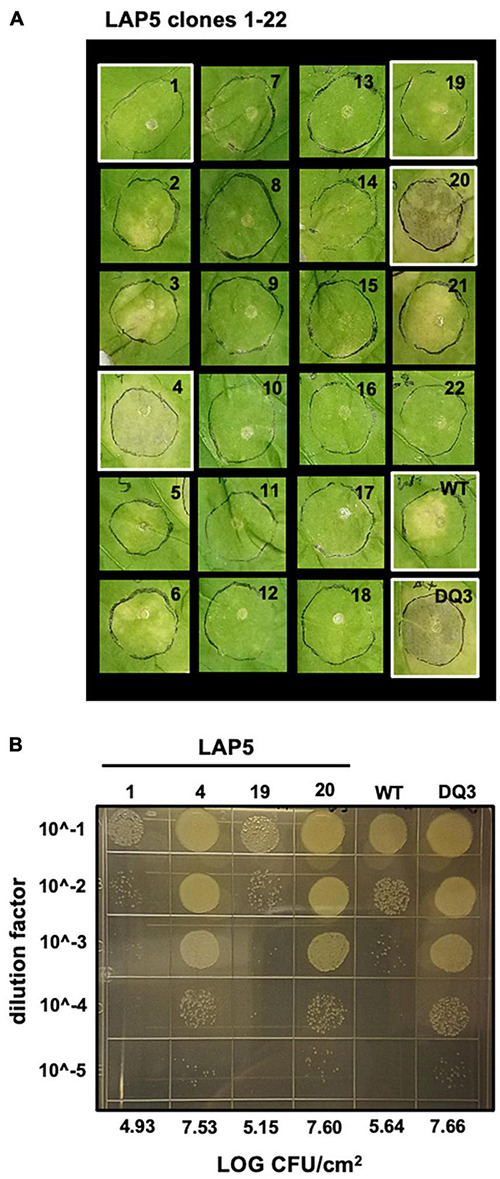
A subset of PmaES4326 isolates passaged through N. benthamiana gain disease compatibility. (A) Two isolates out of 22 PmaES4326 lineage A (LA) passage 5 (P5) isolates produce disease symptoms in the N. benthamiana LAB accession comparable to the targeted Pma DQ3 (ΔICE-DQ) strain. Leaves were photographed eight days post inoculation with 3 × 104 CFU/mL bacterial suspensions. (B) Tenfold dilution series 10 μL spot plate for assessing bacterial load of select strains highlighted in white in panel A.
Transfer of PmaICE-DQ Between Strains
Aside from being able to excise from the genome, ICE elements are also categorized by their ability to transfer across bacterial strains, and we therefore tested whether ICE-DQ could undergo conjugation to a naive strain. To do this, ICE-DQ was marked with kanamycin resistance by generating a single-crossover merodiploid with a pK18ms-derivative plasmid (Pma ICE-DQ:pK18). Three recipient strains, PtoDC3000 and two independent PmaΔICE-DQ strains (the targeted Pma DQ3 strain and the passage derived Pma LAP5-20) were labeled with a Tn7 3xmCherry resistant transposon to confer spectinomycin resistance as well as visually differentiate them from the donor strain. The Pma ICE-DQ:pK18, KmR marked donor strain was co-cultured with each of the three Tn7 3xmCherry SpR recipients. Conjugations between these strains were then plated on kanamycin and spectinomycin selective media (Figure 6A). No kanamycin and spectinomycin resistant mutants or exconjugants were recovered from the PtoDC3000 conjugation or from the single parent control experiments. However, we were able to recover kanamycin and spectinomycin resistant mCherry + exconjugants in PmaES4326ΔICE-DQ strains at rates of 2.50 × 10–7 stdev ± 1.23–7 and 3.05 × 10–7 stdev ± 1.51–7 exconjugants per recipient respectively in Pma DQ3and Pma LAP5-20 recipient strains (Figure 6B) which strongly suggests that the ICE-DQ element is readily transmissible into PmaES4326 strains lacking ICE-DQ.
FIGURE 6.
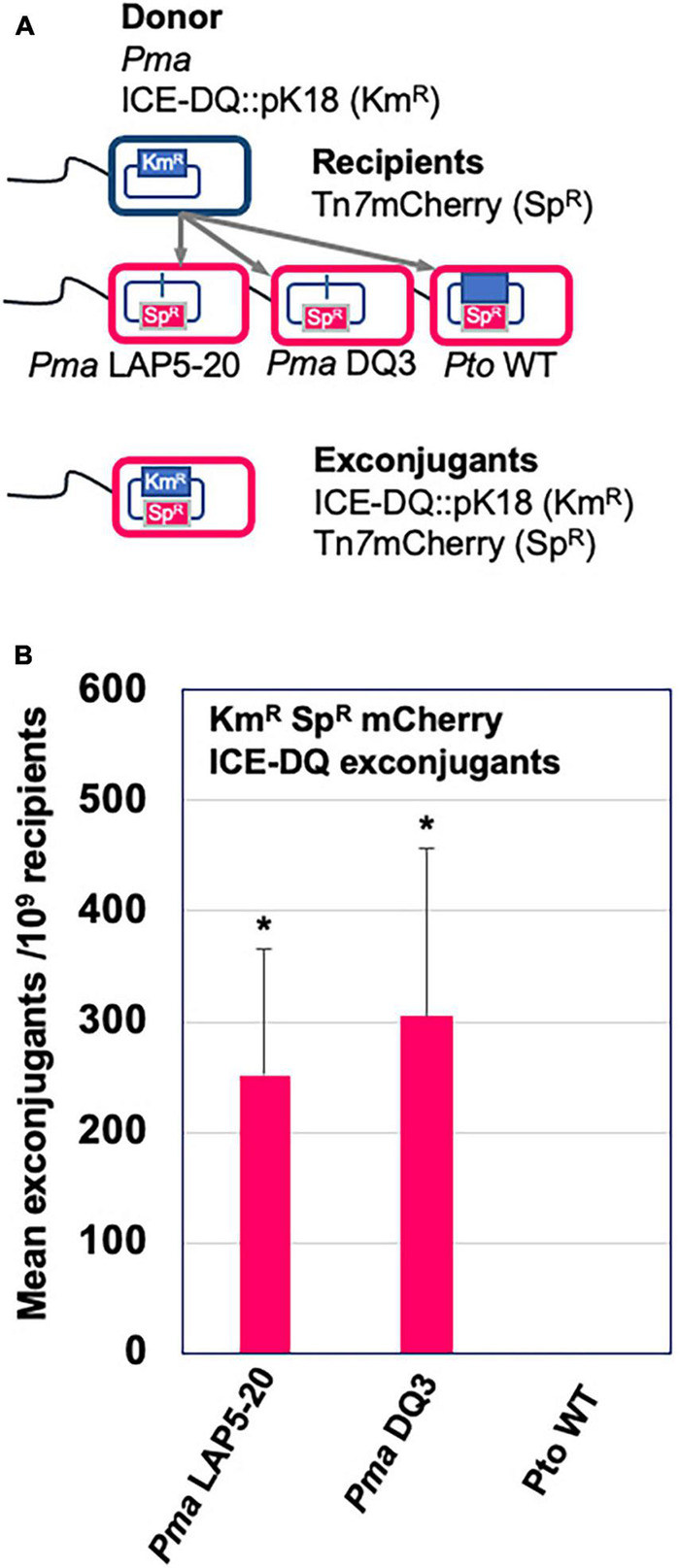
ICE-DQ can be readily transferred into PmaES4326 strains that lack the ICE element. (A) Schematic representation of the conjugation strategy for monitoring ICE-DQ transfer between strains. The ICE-DQ of the donor strain was marked with kanamycin resistance by single crossover integration the pK18ms plasmid (Pma ICE-DQ:pK18). The recipient strains Pma DQ3 (ΔICE-DQ targeted), Pma LAP5-20 (ΔICE-DQ passage-derived) and PtoDC3000 were marked by introduction of a spectinomycin resistance Tn73xmCherry transposon. Donor and recipients were co-cultured on LM media and ICE-DQ:pK18 mCherry expressing ICE-DQ exconjugants were recovered on kanamycin and spectinomycin. (B) ICE-DQ:pK18 exconjugant recovery frequencies/109 recipients were determined based on kanamycin and spectinomycin exconjugants per spectinomycin resistant recipients as determined by dilution plating. Values are means and standard deviations of three biological replicates. *p < 0.05 compared to Pto WT strain as determined by unpaired one-tailed t-test.
Discussion
Integrative Conjugative Elements (ICEs) are important drivers of evolutionary dynamics within and across bacterial populations and communities because of their potential to disseminate genes and pathways through horizontal transfer (Johnson and Grossman, 2015). Genes encoded by ICEs often impart phenotypes critical for survival under specific environmental conditions, including antibiotic resistance loci as well as phage defense systems (Botelho and Schulenburg, 2021; LeGault et al., 2021). In the phytopathogen Pseudomonas syringae, plasticity of ICE elements encoding genes involved in heavy metal resistance, type III effectors, and carbon metabolism has been identified as the main difference between epidemic strains causing disease across kiwi orchards in New Zealand and with previous outbreaks (Colombi et al., 2017; McCann et al., 2017; Poulter et al., 2018). Despite the accumulation of numerous complete genomes sequences for P. syringae and related species and the likelihood of ICEs to impart traits that affect growth in agricultural settings, to date there have been relatively few ICEs identified and vetted for this pathogen.
Placement of genes and pathways in ICEs may be especially important under conditions of fluctuating selection pressures where selection pressures on cargo genes in an ICE switch from positive to negative depending on the environment. One such scenario involves the presence of type three effector genes, which are bacterial proteins delivered from symbionts into eukaryotic host cells and which are critical for pathogenesis of P. syringae strains in planta (Grant et al., 2006). Type three effectors are particularly well studied in phytopathogens such as P. syringae, where they can be of exceptional benefit on some host plants by enabling strains to overcome host immune responses but may also directly trigger immune responses based on the presence of cognate immune receptors on other host genotypes (Dillon et al., 2019a; Laflamme et al., 2020). The presence of type three effectors on ICEs would enable the rapid transmission of these critical virulence genes across strains, while also enabling genes to be rapidly lost from lineages if host genotypes shift such that the effectors are recognized by R gene pathways. Extending this thought, effectors in ICEs that are recognized by hosts are potentially lost more readily through ICE element excision than through mutation (given dedicated excision machinery of the ICEs), which may facilitate adaptation if a strain encounters a resistant host background. While not an extensive experiment, our passage experiment where a subset of PmaES4326 but not PtoDC3000 clones becomes compatible with N. benthamiana does demonstrate that parameter space exists for such a scenario. Moreover, effectors that are inactivated through mutation can only be reactivated by reversion mutations or through horizontal transfer and acquisition from different strains. In contrast, if effectors are found on ICEs and lost through excision, it’s possible that a small resident pool of strains containing these ICEs will remain on other host plants or throughout the environment, facilitating rapid reacquisition of effectors that could be beneficial under different contexts. Lastly, there are a plethora of type III effectors in P. syringae that carry out a variety of functions across different host plants and resistance backgrounds (Dillon et al., 2019a). Some of these effectors can be considered “core” and found in syntenic locations across strains while others are more variably present. It’s possible that presence of effectors in ICEs could itself reflect something about the characteristics of these effectors, in that the proteins found as cargo on ICEs may be subject to higher levels of fluctuating selection across hosts than those found in the core set.
HopQ and HopD are often found together on genomic islands in P. syringae genomes, and have been associated with adaptation to agriculturally important crop plants (Wei et al., 2007; Baltrus et al., 2011; Monteil et al., 2016). Presence of hopQ in an active ICE in strain PmaES4326 sets up an interesting scenario because a nearly identical version of this effector is present in a somewhat syntenic position in a relatively closely related strain PtoDC3000 except that this effector is not part of a functional ICE. Therefore, this genomic context suggests that hopQ would experience more evolutionary flexibility in PmaES4326 than in PtoDC3000 because it can be more easily lost and/or transferred from this strain due to its presence in a region of the chromosome that can excised at a relatively high rate. To test for differences in evolutionary flexibility, we passaged replicate populations of both PmaES4326 and PtoDC3000 in an accession of N. benthamiana which can recognize and mount an immune response to HopQ. As such, wild type versions of PmaES4326 and PtoDC3000 are recognized by this accession and trigger an immune response which prevents disease. However, we found that passage of PmaES4326 from plant to plant resulted in recovery of strains of PmaES4326 that didn’t trigger the immune response. Therefore, presence of this type III effector inside of an ICE enables rapid loss of the recognized effector under conditions of negative selection and there is relatively less flexibility for this effector to be lost in a strain where it is not present in an active ICE.
One other curious feature of the genome of PmaES4326 is that it encodes two distinct but highly similar ICE elements (Figure 1). The configuration is particularly interesting as two distinct ICE elements have integrated successively into the PmaES4326 genome and many of the genes involved in ICE functions are quite conserved between the two. It also appears as though integration of the first element recreated a tRNA with a proline anticodon and that the second ICE element can use this as a further integration point. Indeed, excision of ICE-DQ in both the targeted Pma DQ3 and passage-derived Pma LAP5-20 strains reported here also deletes one of the predicted proline tRNAs. It may therefore be no coincidence that two ICE elements that are highly similar (but which contain different cargo regions) can integrate successively into the genome as it’s straightforward to imagine that highly specific recombinases would have similar target sequences.
Although not the main focus of this manuscript, we also report on plasticity of the PmaES4326 genome writ large across lab derived strains (Table 2). As the number of complete P. syringae genome sequences accumulates, it is becoming more apparent that strain PmaES4326 is notable compared to the rest of the species complex because it contains so many secondary replicons as well as additional elements that contribute to genomic plasticity (Stavrinides and Guttman, 2004; Stavrinides et al., 2012). Many of these plasmids appear to contain genes involved in virulence and so acquisition of these replicons through horizontal gene transfer has likely contributed to the pathogenic ability of this strain across multiple hosts (Stavrinides and Guttman, 2004). However, there is a clear downside to the large plasmid repertoire of PmaES4326 as demonstrated by genome sequences reported here. Although we can’t definitively catalog passage histories, genome sequences from isolates from the Dangl and Collmer labs are likely not diverged by > 10 passages total. In this time, the Dangl isolate (PmaES4326-D) has lost two different plasmids that were originally reported by Stavrinides et al. and which are contained in the Collmer lab isolate (PmaES4326-C). Indeed, genome assemblies for the targeted ICE-DQ excision strain Pma DQ3 suggest that this strain has also lost at least one plasmid in addition to the ICE-DQ after only three additional passages in culture. Lastly, even though this strain was extensively surveyed for plasmids through gel electrophoresis and Sanger sequencing, we report the existence of an additional large plasmid present within all sequenced isolates of this strain.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Author Contributions
DB and BK conceived, conceptualized, analyzed the experiments, and co-wrote the manuscript. QF conducted a subset of experiments described in the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Karl Schreiber and Darrell Desveaux for providing PmaES4326 hrcN:Tn5 as well as Alex Schultink and Brian Staskawicz for providing the roq1-1 N. benthamiana line.
Footnotes
Funding
This work was supported in part by the University of Georgia Office of Research as well as the College of Agricultural and Environmental Sciences’ Research Office.
References
- Adlung N., Prochaska H., Thieme S., Banik A., Blüher D., John P., et al. (2016). Non-host resistance induced by the Xanthomonas effector XopQ Is Widespread within the Genus Nicotiana and functionally depends on EDS1. Front. Plant Sci. 7:1796. 10.3389/fpls.2016.01796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltrus D. A., McCann H. C., Guttman D. S. (2017). Evolution, genomics and epidemiology of Pseudomonas syringae: challenges in bacterial molecular plant pathology. Mol. Plant Pathol. 18 152–168. 10.1111/mpp.12506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltrus D. A., Nishimura M. T., Romanchuk A., Chang J. H., Mukhtar M. S., Cherkis K., et al. (2011). Dynamic evolution of pathogenicity revealed by sequencing and comparative genomics of 19 Pseudomonas syringae isolates. PLoS Pathog. 7:e1002132. 10.1371/journal.ppat.1002132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardaji L., Añorga M., Echeverría M., Ramos C., Murillo J. (2019). The toxic guardians — multiple toxin-antitoxin systems provide stability, avoid deletions and maintain virulence genes of Pseudomonas syringae virulence plasmids. Mob. DNA 10:7. 10.1186/s13100-019-0149-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baym M., Kryazhimskiy S., Lieberman T. D., Chung H., Desai M. M., Kishony R. (2015). Inexpensive multiplexed library preparation for megabase-sized genomes. PLoS One 10:e0128036. 10.1371/journal.pone.0128036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger A. M., Lohse M., Usadel B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botelho J., Schulenburg H. (2021). The role of integrative and conjugative elements in antibiotic resistance evolution. Trends Microbiol. 29 8–18. 10.1016/j.tim.2020.05.011 [DOI] [PubMed] [Google Scholar]
- Cazorla F. M., Arrebola E., Sesma A., Pérez-García A., Codina J. C., Murillo J., et al. (2002). Copper resistance in Pseudomonas syringae strains isolated from mango is encoded mainly by plasmids. Phytopathology 92 909–916. 10.1094/PHYTO.2002.92.8.909 [DOI] [PubMed] [Google Scholar]
- Choi K. H., Kumar A., Schweizer H. P. (2006). A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J. Microbiol. Methods 64 391–397. 10.1016/j.mimet.2005.06.001 [DOI] [PubMed] [Google Scholar]
- Collmer A., Badel J. L., Charkowski A. O., Deng W.-L., Fouts D. E., Ramos A. R., et al. (2000). Pseudomonas syringae Hrp type III secretion system and effector proteins. Proc. Natl. Acad. Sci. U.S.A. 97 8770–8777. 10.1073/pnas.97.16.8770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombi E., Straub C., Künzel S., Templeton M. D., McCann H. C., Rainey P. B. (2017). Evolution of copper resistance in the kiwifruit pathogen Pseudomonas syringae pv. actinidiae through acquisition of integrative conjugative elements and plasmids. Environ. Microbiol. 19 819–832. 10.1111/1462-2920.13662 [DOI] [PubMed] [Google Scholar]
- de Vries S., Stukenbrock E. H., Rose L. E. (2020). Rapid evolution in plant–microbe interactions – an evolutionary genomics perspective. New Phytol. 226 1256–1262. 10.1111/nph.16458 [DOI] [PubMed] [Google Scholar]
- Deatherage D. E., Barrick J. E. (2014). Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using Breseq. Methods Mol. Biol. 1151 165–188. 10.1007/978-1-4939-0554-6_12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon M. M., Almeida R. N. D., Laflamme B., Martel A., Weir B. S., Desveaux D., et al. (2019a). Molecular evolution of Pseudomonas syringae Type III secreted effector proteins. Front. Plant Sci. 10:418. 10.3389/fpls.2019.00418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon M. M., Thakur S., Almeida R. N. D., Wang P. W., Weir B. S., Guttman D. S. (2019b). Recombination of ecologically and evolutionarily significant loci maintains genetic cohesion in the Pseudomonas syringae species complex. Genome Biol. 20 1–28. 10.1186/s13059-018-1606-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong O. X., Ronald P. C. (2019). Genetic engineering for disease resistance in plants: recent progress and future perspectives. Plant Physiol. 180 26–38. 10.1104/pp.18.01224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey S. A. C., Lovell H. C., Mansfield J. W., Corry D. S., Jackson R. W., Arnold D. L. (2011). The stealth episome: suppression of gene expression on the excised genomic island PPHGI-1 from Pseudomonas syringae pv. phaseolicola. PLoS Pathog. 7:e1002010. 10.1371/journal.ppat.1002010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant S. R., Fisher E. J., Chang J. H., Mole B. M., Dangl J. L. (2006). Subterfuge and manipulation: type III effector proteins of phytopathogenic bacteria. Annu. Rev. Microbiol. 60 425–449. 10.1146/annurev.micro.60.080805.142251 [DOI] [PubMed] [Google Scholar]
- Jayaraman J., Chatterjee A., Hunter S., Chen R., Stroud E. A., Saei H., et al. (2021). Rapid methodologies for assessing Pseudomonas syringae pv. actinidiae colonization and effector-mediated hypersensitive response in Kiwifruit. Mol. Plant Microbe Interact. 34 880–890. 10.1094/MPMI-02-21-0043-R [DOI] [PubMed] [Google Scholar]
- Johnson C. M., Grossman A. D. (2015). Integrative and conjugative elements (ICEs): what they do and how they work. Annu. Rev. Genet. 49 577–601. 10.1146/annurev-genet-112414-055018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones J. D. G., Dangl J. L. (2006). The plant immune system. Nature 444 323–329. [DOI] [PubMed] [Google Scholar]
- Kvitko B. H., Collmer A. (2011). Construction of Pseudomonas syringae pv. tomato DC3000 mutant and polymutant strains. Methods Mol. Biol. 712 109–128. 10.1007/978-1-61737-998-7_10 [DOI] [PubMed] [Google Scholar]
- Kvitko B. H., Park D. H., Velásquez A. C., Wei C.-F., Russell A. B., Martin G. B., et al. (2009). Deletions in the Repertoire of Pseudomonas syringae pv. tomato DC3000 Type III secretion effector genes reveal functional overlap among effectors. PLoS Pathog. 5:e1000388. 10.1371/journal.ppat.1000388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laflamme B., Dillon M. M., Martel A., Almeida R. N. D., Desveaux D., Guttman D. S. (2020). The pan-genome effector-triggered immunity landscape of a host-pathogen interaction. Science 367 763–768. 10.1126/science.aax4079 [DOI] [PubMed] [Google Scholar]
- LeGault K. N., Hays S. G., Angermeyer A., McKitterick A. C., Johura F. T., Sultana M., et al. (2021). Temporal shifts in antibiotic resistance elements govern phage-pathogen conflicts. Science 373 eabg2166. 10.1126/science.abg2166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell H. C., Mansfield J. W., Godfrey S. A. C., Jackson R. W., Hancock J. T., Arnold D. L. (2009). Bacterial evolution by genomic Island transfer occurs via DNA transformation in planta. Curr. Biol. 19 1586–1590. 10.1016/j.cub.2009.08.018 [DOI] [PubMed] [Google Scholar]
- McCann H. C., Li L., Liu Y., Li D., Pan H., Zhong C., et al. (2017). Origin and evolution of the Kiwifruit canker pandemic. Genome Biol. Evol. 9:932. 10.1093/gbe/evx055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteil C. L., Yahara K., Studholme D. J., Mageiros L., Méric G., Swingle B., et al. (2016). Population-genomic insights into emergence, crop adaptation and dissemination of Pseudomonas syringae pathogens. Microb. Genom. 2:e000089. 10.1099/mgen.0.000089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale H. C., Jackson R. W., Preston G. M., Arnold D. L. (2018). Supercoiling of an excised genomic island represses effector gene expression to prevent activation of host resistance. Mol. Microbiol. 110 444–454. 10.1111/mmi.14111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulter R. T. M., Ho J., Handley T., Taiaroa G., Butler M. I. (2018). Comparison between complete genomes of an isolate of Pseudomonas syringae pv. actinidiae from Japan and a New Zealand isolate of the pandemic lineage. Sci. Rep. 8:10915. 10.1038/s41598-018-29261-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi D., Dubiella U., Kim S. H., Sloss D. I., Dowen R. H., Dixon J. E., et al. (2014). Recognition of the protein kinase AVRPPHB SUSCEPTIBLE1 by the disease resistance protein RESISTANCE TO PSEUDOMONAS SYRINGAE5 is dependent on s-acylation and an exposed loop in AVRPPHB SUSCEPTIBLE1. Plant Physiol. 164 340–351. 10.1104/pp.113.227686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi T., Seong K., Thomazella D. P. T., Kim J. R., Pham J., Seo E., et al. (2018). NRG1 functions downstream of EDS1 to regulate TIR-NLR-mediated plant immunity in Nicotiana benthamiana. Proc. Natl. Acad. Sci. U.S.A. 115 E10979–E10987. 10.1073/pnas.1814856115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schierstaedt J., Bziuk N., Kuzmanović N., Blau K., Smalla K., Jechalke S. (2019). Role of plasmids in plant-bacteria interactions. Curr. Issues Mol. Biol. 30 17–38. 10.21775/cimb.030.017 [DOI] [PubMed] [Google Scholar]
- Schultink A., Qi T., Lee A., Steinbrenner A. D., Staskawicz B. (2017). Roq1 mediates recognition of the Xanthomonas and Pseudomonas effector proteins XopQ and HopQ1. Plant J. 92 787–795. 10.1111/tpj.13715 [DOI] [PubMed] [Google Scholar]
- Seemann T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30 2068–2069. 10.1093/bioinformatics/btu153 [DOI] [PubMed] [Google Scholar]
- Sjödin P., Kaj I., Krone S., Lascoux M., Nordborg M. (2005). On the meaning and existence of an effective population size. Genetics 169 1061–1070. 10.1534/genetics.104.026799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavrinides J., Guttman D. S. (2004). Nucleotide sequence and evolution of the five-plasmid complement of the phytopathogen Pseudomonas syringae pv. maculicola ES4326. J. Bacteriol. 186 5101–5115. 10.1128/JB.186.15.5101-5115.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavrinides J., Kirzinger M. W. B., Beasley F. C., Guttman D. S. (2012). E622, a miniature, virulence-associated mobile element. J. Bacteriol. 194:509. 10.1128/JB.06211-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusova T., DiCuccio M., Badretdin A., Chetvernin V., Nawrocki E. P., Zaslavsky L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44 6614–6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teal T. K., Lies D. P., Wold B. J., Newman D. K. (2006). Spatiometabolic Stratification of Shewanella oneidensis biofilms. Appl. Environ. Microbiol. 72:7324. 10.1128/aem.01163-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C. F., Kvitko B. H., Shimizu R., Crabill E., Alfano J. R., Lin N. C., et al. (2007). A Pseudomonas syringae pv. tomato DC3000 mutant lacking the type III effector HopQ1-1 is able to cause disease in the model plant Nicotiana benthamiana. Plant J. 51 32–46. 10.1111/j.1365-313X.2007.03126.x [DOI] [PubMed] [Google Scholar]
- Wei H. L., Zhang W., Collmer A. (2018). Modular study of the Type III effector repertoire in Pseudomonas Syringae Pv. Tomato DC3000 reveals a matrix of effector interplay in pathogenesis. Cell Rep. 23 1630–1638. 10.1016/j.celrep.2018.04.037 [DOI] [PubMed] [Google Scholar]
- Wick R. R., Judd L. M., Gorrie C. L., Holt K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. 10.1371/journal.pcbi.1005595 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.