

Abstract
Cerebral amyloid angiopathy (CAA), characterized by cerebral vascular amyloid accumulation, neuroinflammation, microbleeds, and white matter (WM) degeneration, is a common comorbidity in Alzheimer disease and a prominent contributor to vascular cognitive impairment and dementia. WM loss was recently reported in the corpus callosum (CC) in the rTg-DI rat model of CAA. The current study shows that the CC exhibits a much lower CAA burden compared with the adjacent cortex. Sequential Window Acquisition of All Theoretical Mass Spectra tandem mass spectrometry was used to show specific proteomic changes in the CC with emerging WM loss and compare them with the proteome of adjacent cortical tissue in rTg-DI rats. In the CC, annexin A3, heat shock protein β1, and cystatin C were elevated at 4 months (M) before WM loss and at 12M with evident WM loss. Although annexin A3 and cystatin C were also enhanced in the cortex at 12M, annexin A5 and the leukodystrophy-associated astrocyte proteins megalencephalic leukoencephalopathy with subcortical cysts 1 and GlialCAM were distinctly elevated in the CC. Pathway analysis indicated neurodegeneration of axons, reflected by reduced expression of myelin and neurofilament proteins, was common to the CC and cortex; activation of Tgf-β1 and F2/thrombin was restricted to the CC. This study provides new insights into the proteomic changes that accompany WM loss in the CC of rTg-DI rats.
Cerebral amyloid angiopathy (CAA), a disease characterized by accumulation of fibrillar amyloid β-protein (Aβ) in blood vessels of the brain, is prevalent in the elderly population and is a common comorbidity of Alzheimer disease.1, 2, 3, 4 There are two major forms of CAA: CAA type 1, in which amyloid accumulates along cerebral microvessels and capillaries, and CAA type 2, in which the Aβ deposition occurs within the walls of small arteries and arterioles.5 CAA can promote degeneration of mural and endothelial cells of the vessel wall, leading to cerebral microbleeds and larger hemorrhages, as well as thrombotic occlusions resulting in ischemic lesions, which collectively can cause white matter (WM) damage and vascular cognitive impairment and dementia.1,2,6, 7, 8 In addition to the apparent loss of myelin proteins, other changes that occur in the proteome of the WM in conjunction with CAA are not well understood and may provide further insight into the pathogenesis of this region.
Prior studies focused on the generation and characterization of a transgenic rat model of CAA type 1 known as rTg-DI.9 The rTg-DI rats produce human Aβ harboring the Dutch (E22Q) and Iowa (D23N) familial CAA mutations in the brain and develop early-onset accumulation of cerebral microvascular amyloid in the absence of parenchymal plaque amyloid deposits. The progressive accumulation of microvascular CAA in this model promotes robust perivascular neuroinflammation, degeneration of pericytes, thalamic cerebral microbleeds and occluded microvessels, and behavioral impairments reminiscent of “cognitive slowing.”9, 10, 11, 12 Recently, rTg-DI rats were shown to present with diffuse WM loss emerging after 6 months (M) and progressing to up to 12M.13 The WM loss was primarily observed in the corpus callosum (CC) and in the globus pallidus and thalamus, as detected by magnetic resonance imaging. Despite the clear loss of WM, an understanding of how cerebral microvascular amyloid accumulation in aging rTg-DI rats affects the CC is unclear.
Liquid chromatography coupled with tandem mass spectrometry (MS/MS) has provided a means for reliable and sensitive high-throughput proteomic investigation.14,15 Data-independent acquisition approaches, such as Sequential Window Acquisition of All Theoretical Mass Spectra (SWATH-MS), which perform quantitative analysis of all detected protein fragments and characterize thousands of proteins within a single sample, are becoming the standard for cohort proteomics.14,16,17 Recently, brain regional proteomic alterations were reported in the cortex, hippocampus, and thalamus of 12M rTg-DI rats using laser microdissection and SWATH-MS.12 Specific proteomic changes with regionally distinct pathologies were used to describe important mechanisms implicated in the development of the differential pathologic phenotypes.
The current study uses laser microdissection and SWATH-MS to investigate the role of progressive CAA type 1 in rTg-DI rats in changing the protein profile of the CC that exhibits prominent WM loss. These proteomic changes were compared with recent findings in the cortex of 12M rTg-DI rats.12 Following the proteomics, bioinformatics analysis was conducted using Ingenuity Pathway Analysis (IPA) (Qiagen Inc., Redwood City, CA) and brain tissue immunolabeling to connect the proteomic alterations with specific pathways and cellular responses implicated in the progressive WM loss.
Materials and Methods
Animals
The rTg-DI transgenic rat model of CAA was generated as previously described9 in which low levels of human Swedish/Dutch/Iowa mutant Aβ protein precursor are expressed in neurons under the control of the Thy1.2 promoter and maintained on a Sprague-Dawley background. In this model, the accumulation of microvascular amyloid begins at approximately 3M and progresses with advancing age. In the current study, three male and three female heterozygous rTg-DI rats and nontransgenic, littermate wild-type (WT) rats were collected at 4M, and three male and two female heterozygous rTg-DI rats and two male and three female WT rats were collected at 12M. The study was neither randomized nor blinded.
All rats were housed in a controlled room (22 ± 2°C and 40% to 60% humidity) on a standard 12-hour light cycle. Rat chow and water were available ad libitum. All work with animals was approved by the University of Rhode Island and Yale University Institutional Animal Care and Use Committees, in accordance with the United States Public Health Service’s Policy on Humane Care and Use of Laboratory Animals, and in compliance with the Animal Research: Reporting of In Vivo Experiments guidelines.
Magnetic Resonance Imaging of WM Loss
The composite data presented were previously acquired from Lee et al.13 Briefly, WT and rTg-DI rats (n = 11 per group) anesthetized with dexmedetomidine and low-dose isoflurane underwent proton density–weighted imaging and were analyzed by using voxel-based morphometry in studying whole-brain-wide WM morphometry as previously described.13,18,19 Spatial registration parameters between tissue probability maps and individual scans were calculated by using the DARTEL algorithm (Diffeomorphic Anatomical Registration Through Exponentiated Lie Algebra) and subsequently smoothed by a 0.6-mm Gaussian smoothing kernel. The resulting smoothed and normalized WM density images were population averaged for the WT and rTg-DI cohorts and overlaid on the corresponding proton density–weighted anatomical template using PMOD version 3.807 software (PMOD Technologies GmbH, Zurich, Switzerland). Two-dimensional displays at the level of the CC and striatum were chosen for the comparisons of WM volume across ages and strains. Additional details are provided in Lee et al.13
Brain Tissue Collection and Preparation
At the designated ages, anesthetized rats were transcardially perfused with phosphate-buffered saline. The rat brain was surgically removed and bisected in the mid-sagittal plane. One hemisphere was fixed in 4% paraformaldehyde for immunohistochemical analysis, and the other hemisphere was snap-frozen in optimal cutting temperature medium (OCT 4585, Fisher Healthcare, Waltham, MA) directly.
Protein Digest of Collected CC Tissue
The CC region from brain sections of WT and rTg-DI rats was collected by using laser capture microdissection. Briefly, 20 sagittal sections (25 μmol/L thick) of fresh frozen 4M and 12M rat brain hemispheres were prepared on laser microdissection framed membrane slides (Leica Microsystems, Buffalo Grove, IL), two sections per slide. The CC from each tissue section was collected by using laser capture microdissection and pooled for each rat. Tissue lysis and sample preparation for MS analysis were performed as previously described.12 Briefly, tissue was lysed in radioimmunoprecipitation assay buffer via sonication. Protein was denatured by adding 25 μL dithiothreitol (100 mmol/L) and incubation with shaking (300 rpm) at 95°C for 15 minutes, and samples were alkylated with 25 μL iodoacetamide (200 mmol/L) and incubated in the dark for 30 minutes at 20°C. Protein was then precipitated and concentrated via chloroform-methanol-water (1:2:1) precipitation and washed with methanol. Samples were resuspended in sodium deoxycholate (3% w/v in 50 mmol/L ammonium bicarbonate). Proteins were digested with tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-treated trypsin at a concentration of 1:20 trypsin:protein (Sciex, Framingham, MA) in a barocycler (Pressure Biosciences Inc., Easton, MA) as previously described.12 Deoxycholate was precipitated by the addition of formic acid (5% v/v, in 50% v/v acetonitrile in water) for a final concentration of 0.5% v/v, followed by centrifugation (15,300 × g, 5 minutes, at room temperature), and the supernatant was collected for liquid chromatography–MS/MS analysis.
Analysis by Liquid Chromatography Quadrupole Time-of-Flight/MS
Proteomic experiments were performed on a SCIEX 5600 TripleTOF mass spectrometer in positive ion mode using a DuoSpray ion source (AB Sciex, Concord, ON, Canada), coupled to an Acquity UPLC H-Class system (Waters Corp., Milford, MA) for chromatographic separation as previously described.12 Peptide separation was achieved by using an Acquity UPLC Peptide BEH C18 column (2.1 × 150 mm, 300 Å, 1.7 μm) preceded by an Acquity VanGuard pre-column (2.1 × 5 mm, 300 Å, 1.7 μm) according to the previously described method.12 Trypsin-digested β-galactosidase was injected every four samples to monitor time-of-flight mass calibration. Data were acquired by using Analyst TF 1.7.1 software (AB Sciex) in data-independent acquisition mode. For all data-independent acquisition experiments, MS settings were exactly as previously described.12
Data Processing
Proteins were identified and quantified by using Spectronaut (release version 13.919110643655, 2019; Biognosys, Schlieren, Switzerland), referencing our previously formed spectral library.12 All Spectronaut settings were factory default, except that “used Biognosys' iRT kit” and “PTM localization” were deselected. After Spectronaut analysis, two samples (one 12M WT and one 12M rTg-DI) were removed from subsequent analysis because the number of proteins identified in those samples was substantially lower (<60% of all identified proteins). Raw protein intensities from Spectronaut were then converted to molar concentrations (picomoles per milligram total protein) using the total protein approach.20,21 For protein quantities of zero in individual samples, we imputed a baseline concentration (0.03 pmol/mg tissue) determined as previously described.12 Differentially expressed proteins were defined as ≥50% increase or ≥30% decrease, and statistical significance (P < 0.05) was determined via the t-test in Excel (Microsoft Corporation, Redmond, WA).
Immunohistochemical Analysis
Sections were cut from frozen paraformaldehyde-fixed brain hemispheres in the sagittal plane at 20 μm thickness using a cryostat (Leica Microsystems) and placed on slides. Antigen retrieval was conducted via 5 minutes’ incubation with proteinase K (0.2 mg/mL) at 22°C. Tissue sections were then blocked in SuperBlock blocking buffer (37518; Thermo Fisher Scientific, Waltham, MA) containing 0.3% Triton X-100 at room temperature for 30 minutes. They were then incubated with individual primary antibodies at the following dilutions overnight: rabbit polyclonal antibody to collagen IV to identify cerebral blood vessels (1:250, SD2365885; Invitrogen); goat polyclonal antibodies to glial fibrillary acidic protein (GFAP; 1:250, ab53554, Abcam) or ionized calcium-binding adapter molecule 1 (Iba-1; 1:250, NB100-1028, Novus) were used to identify astrocytes and microglia, respectively; and rabbit polyclonal antibodies to annexin 5 (1:250, PA578784, Invitrogen), annexin A3 (Anxa3; 1:250, PA5082483, Invitrogen), megalencephalic leukoencephalopathy with subcortical cysts 1 (MLC1; 1:250, PA541042, Invitrogen), and GlialCAM (1:250, PA599999, Invitrogen). The primary antibodies were detected with Alexa Fluor 594– or 488–conjugated secondary antibodies (1:1000). Deposits of fibrillar vascular amyloid were detected by staining with either thioflavin S (123H0598, Sigma Aldrich, St. Louis, MO) or Amylo-Glo (TR-300-AG, Biosensis Inc., Thebarton, SA, Australia) as described by the manufacturer. Myelin was stained and visualized by using the Black Gold II Myelin Staining Kit according to the manufacturer’s protocol (AG105; MilliporeSigma, Burlington, MA). Immunohistologic images were captured on a Keyence BZ-X710 fluorescence microscope and analyzed with BZ-X Analyzer software version 1.3.1.1 (Keyence, Itasca, IL).
Data Availability
Raw MS data can be found in the MassIVE repository (project ID# MSV000088077; password: rtgdi2021cc).
Results
Age-Dependent WM Loss in the CC of rTg-DI Rats
rTg-DI rats were recently shown to exhibit significant WM degeneration compared with WT controls as they age from 6M to approximately 12M with advancing microvascular CAA burden.13 A composite image of this analysis of the WM loss is shown in Figure 1. In the WT cohort, WM volume increased in the CC (as well as in the septum) from 3M to 12M secondary to normal brain maturation and growth. In contrast, in the rTg-DI cohort, the normal growth trajectory of WM volume from 3M to 12M at the level of the CC was not evident due to WM loss secondary to the progression of extensive CAA pathologies. The subsequent studies were designed to further investigate the loss of WM in the CC of aging rTg-DI rats.
Figure 1.

Significant white matter (WM) loss in the corpus callosum (CC) is evident in aged rTg-DI rats. Color-coded population-averaged WM volumetric maps at the level of the CC from a cohort of WT rats scanned at 3 months (3M) (A) and again at approximately 12 months (12M) (B) of age. Corresponding color-coded WM volumetric maps from a cohort of rTg-DI rats scanned at similar 3M (C) and approximately 12M (D) time points overlaid on the anatomical template. The color-coded map represents WM volume in each voxel, and blue and red colors indicate high and low WM volume, respectively. In the WT cohort, WM volume increased in the CC (as well as in the septum) from 3M to 12M secondary to normal brain maturation and growth. In contrast, in the rTg-DI cohort, the normal growth trajectory of WM volume from 3M to 12M, at the level of the CC, is not evident due to WM loss secondary to cerebral amyloid angiopathy pathology. The arrows in D identify the CC exhibiting WM loss. n = 11.
rTg-DI Rats Do Not Exhibit Robust Microvascular Amyloid Accumulation in the CC
Because the magnetic resonance imaging studies presented in Figure 1 indicate the prominent loss of WM in the CC of rTg-DI rats, the presence of CAA was first evaluated in this region and the adjacent cortical area. Prior studies showed that rTg-DI rats develop cerebral microvascular fibrillar amyloid in regions including the cortex, hippocampus, and thalamus, starting at 3M, and sharply increase with age.9,10 At 4M, very low levels of microvascular CAA (percent capillary area coverage) were present in both the cortex and CC but were still significantly higher in the cortex (Figure 2, A, C, and E). However, as rTg-DI rats aged to 12M, the levels of vascular amyloid rose in the cortex to nearly 30% (Figure 2, B and F). In contrast, the adjacent CC region continued to exhibit scant amyloid capillary coverage (Figure 2, D and F). These findings indicate that the CC region develops little CAA accumulation compared with the adjacent cortical regions.
Figure 2.
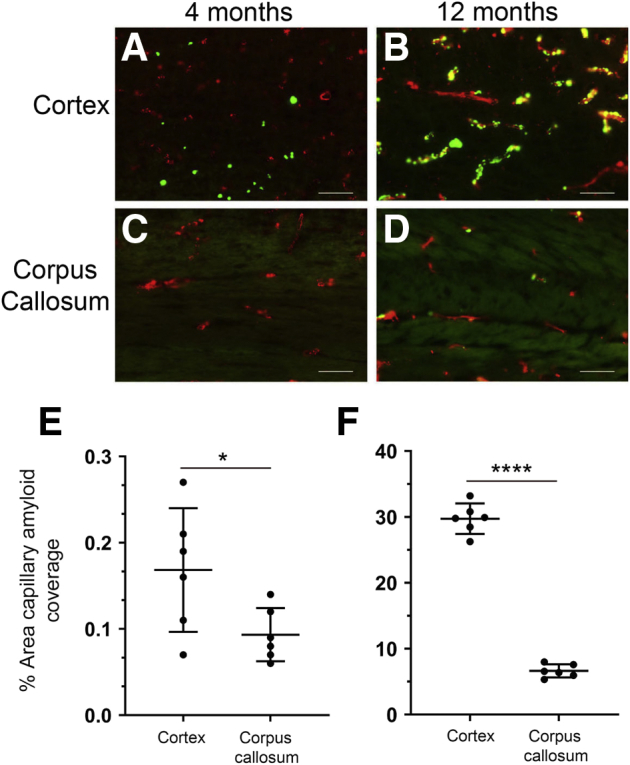
Progressive accumulation of cerebral microvascular amyloid in the cortex and corpus callosum (CC) of rTg-DI rat brain. Brain sections from rTg-DI rats at 4 months (4M) to 12 months (12M) were immunolabeled with rabbit polyclonal antibody to collagen IV to specifically detect cerebral microvessels (red) and stained with thioflavin S to identify fibrillar amyloid (green). The cerebral microvascular fibrillar amyloid deposits in the cortex and CC at 4M (A and C, respectively) and 12M (B and D). Quantitation of cerebral microvascular amyloid load in the cortex and CC of rTg-DI rats at 4M (E) and 12M (F). Data points show the results from each rat and the group mean ± SD of n = 6 rTg-DI rats at each age. ∗P < 0.05; ∗∗∗∗P < 0.0001. Scale bars = 50 μm (A–D).
Proteomic profiling was performed to further understand the basis for the pronounced WM degeneration in the CC of aging rTg-DI rats. Brain tissue sections from rTg-DI and WT rats at 4M to 12M were stained for myelin, and the CC was collected by using laser capture microdissection. Supplemental Figure S1 displays representative images of brain sections that were stained for myelin to identify the CC region that was subsequently microdissected and collected from each rat. Similar to the magnetic resonance images shown in Figure 1, the CC regions of 4M WT and rTg-DI rats were comparable (Supplemental Figure S1, A and C, respectively), whereas at 12M, there was a notable reduction in this region in the rTg-DI rats compared with WT rats (Supplemental Figure S1, B and D) as previously reported.13
Enhanced Proteins in 12M and 4M rTg-DI Rat CC
Proteomic analysis of the laser capture microdissection collected CC sections was performed with a data-independent acquisition liquid chromatography–MS/MS approach as described recently12 and in the Materials and Methods. Average molar concentrations from the rTg-DI and WT rats were compared at 4M and 12M according to the “total protein approach.”20 Because multiple testing–corrected false discovery rates are often too restrictive in small n proteomic studies,22,23 uncorrected P values were used for differentially expressed proteins and effect threshold cutoffs were used to manage false discovery rates as previously described.12 Nineteen and 73 significantly enhanced proteins (≥50% increase over WT; P < 0.05) were identified in the 4M and 12M rTg-DI CC, respectively. These proteins are listed in Supplemental Tables S1 and S2. A Venn diagram comparison of the enhanced proteins at each age is displayed in Figure 3A. Of these, only six proteins, cystatin C, heat shock protein β1 (HspB1), Anxa3, proteosome activator complex subunit 1, clusterin, and amyloid precursor protein, were enhanced both at 4M and 12M.
Figure 3.

Significantly enhanced and reduced proteins in the rTg-DI corpus callosum (CC) at 12 months (12M) and 4 months (4M). A: Venn diagram comparing significantly enhanced proteins by ≥50% of the wild-type concentration in the CC of rTg-DI rats at 12M and 4M. B: Venn diagram comparing significantly reduced proteins by ≥30% of the wild-type concentration in the CC of rTg-DI rats at 12M and 4M. C: Heat map depicting relative expression of most enhanced proteins at 12M only (top), at both 12M and 4M (middle), and at 4M only (bottom), along with the corresponding expression in the other age group, with red indicating enhanced expression, green representing reduced expression, and color intensity correlating with degree of change. D: Heat map depicting relative expression of most reduced proteins arranged as in C with color shading as in C. n = 5, 6 (A and B). P < 0.05 (A and B). Anxa3, annexin A3; ApoE, apolipoprotein E; App, amyloid precursor protein; Clu, clusterin; Cp, ceruloplasmin; Cst3, cystatin C; GFAP, glial fibrillary acidic protein; HTRA1, high-temperature requirement A serine peptidase 1; HSPB1, heat shock protein β1; Krt8, keratin 8; Mag, myelin-associated glycoprotein; Mbp, myelin basic protein; MLC1, megalencephalic leukoencephalopathy with subcortical cysts 1; Mog, myelin-oligodendrocyte glycoprotein; Nefh, neurofilament heavy chain; Nefl, neurofilament light polypeptide; Nefm, neurofilament medium polypeptide; Plp1, proteolipid protein; Psme1, proteosome activator complex subunit 1; S100a4, S100 calcium binding protein A4; S100A13, S100 calcium binding protein A13; Slc14a1, solute carrier family 14 member 1.
Relative expression of these proteins is depicted in the heat map in Figure 3C. Sixty-seven proteins were enhanced only at 12M and not at 4M. Relative expression of the 25 displaying the greatest percent increase is depicted by the heat map in Figure 3C, along with corresponding relative expression of those proteins in the 4M rTg-DI CC, with red shading indicating enhanced expression, green indicating decreased expression, and color intensity indicating degree of change in expression. Notable among these proteins are S100 calcium binding protein A4, high-temperature requirement A serine peptidase 1 (HTRA1), GFAP, MLC1, and ceruloplasmin, as enhanced expression of these proteins was recently reported in other brain regions of 12M rTg-DI rats.12 Thus, despite a relatively low CAA burden, the rTg-DI CC displays enhancement of multiple proteins in common with other rTg-DI brain regions with a much higher CAA burden. Thirteen proteins were elevated in the 4M rTg-DI CC that were not at 12M, and relative expression of these proteins is similarly depicted in Figure 3C. Of these proteins, only thymopoietin was previously reported as enhanced in other brain regions of rTg-DI rats. However, the six proteins commonly enhanced in the rTg-DI CC at 4M and 12M were among those reported as enhanced in other regions of rTg-DI rats. Therefore, before emergence of robust CAA pathology in other brain regions, many of the rTg-DI specifically enhanced proteins are already enhanced at 4M in the CC. Interestingly, despite the lack of robust CAA in the rTg-DI CC (Figure 2), significant increases in GFAP, a common astrocyte marker, were observed in both the 12M and 4M rTg-DI CC.24, 25, 26
Decreased Proteins in 12M and 4M rTg-DI Rat CC
Fourteen and 10 significantly decreased proteins (≥30% decrease, WT expression ≥2 pmol/mg; P < 0.05) were identified in the 12M and 4M rTg-DI CC, respectively, and are listed in Supplemental Tables S3 and S4. A Venn diagram comparing the significantly down-regulated proteins at each age is depicted in Figure 3B, revealing no proteins shared between the two age groups. Expression of these proteins relative to WT are depicted in the heat map in Figure 3D, alongside corresponding relative expression from the other rTg-DI CC age group. Of particular interest in this study are the significant reductions in the myelin related proteins and the neurofilament proteins. For example, myelin basic protein (Mbp) was reduced by 30% (one-tailed t-test, P = 0.033), consistent with recent reported findings via quantitative immunoblotting.13 Dysregulation of neurofilament proteins is a hallmark of neurodegenerative diseases,27 whereas myelin-associated glycoprotein (Mag), Mbp, and myelin proteolipid protein (Plp1) are all important for axonal myelination, and reductions in their expression could lead to demyelination and disrupted axonal integrity.28, 29, 30 These results are consistent with the previously reported age-dependent WM degeneration,13 and shown in Figure 1, as these dramatic decreases in protein expression are only seen in the 12M rTg-DI CC.
Differentially Expressed CC Proteins Compared with Those in Cortex
Regional pathologic characteristics and proteomic changes in the cortex, hippocampus, and thalamus of rTg-DI rats were reported recently.12 Although neuroinflammation was present in each region, CAA load in the cortex was moderate compared with higher levels of CAA present in the other two regions. Also, pathway analysis revealed that neurodegeneration of axons was implicated both in the cortex and hippocampus of rTg-DI rats.12 The dramatic loss of WM (Figure 1) and the differential expression of proteins associated with neurodegeneration despite minimal CAA load in rTg-DI CC could indicate an impact of the pathology of neighboring regions on the CC. For example, Wallerian degeneration, or the progressive degeneration of distal portions of an injured axon,31 could account for degeneration of axons in the CC of nerves originating and damaged in other brain regions. Therefore, the enhanced proteins in the CC were compared with those in the cortex of 12M rTg-DI rats, as these neighboring regions share characteristics of neurodegeneration, whereas the higher CAA load is restricted to the cortex. Of the 73 significantly enhanced proteins in the CC, 18 were also elevated in the cortex (Figure 4A). Fifty-five proteins were uniquely elevated in the CC, whereas 42 proteins were elevated in the cortex, but not in the CC.
Figure 4.

Comparison of significantly enhanced and reduced proteins in the rTg-DI corpus callosum (CC) and cortex at 12 months (12M). A: Venn diagram comparing significantly enhanced proteins by ≥50% of the wild-type concentration in the CC and cortex of 12M rTg-DI rats. B: Venn diagram comparing significantly reduced proteins by ≥30% of the wild-type concentration in the CC and cortex of 12M rTg-DI rats. C: Heat map depicting relative expression of most enhanced proteins in the CC only (top), both CC and cortex (middle), and cortex only (bottom), along with the corresponding expression in the other region, with red indicating enhanced expression, green representing reduced expression, and color intensity representing the degree of expression change. D: Heat map depicting relative expression of most reduced proteins arranged as in C with color shading as in C. n = 5, 6 (A and B). P < 0.05 (A and B). Anxa11, annexin A11; other abbreviations are as indicated in Figure 3.
These proteins were ranked according to increased expression over WT, and the relative expression for the top 25 proteins uniquely enhanced in the CC and in the cortex, along with all proteins commonly enhanced in those regions, is depicted in a heat map in Figure 4C. Of the commonly elevated proteins, GFAP, S100 calcium binding protein A4, apolipoprotein E, keratin 8, and amyloid precursor protein were more robustly increased in the cortex and could reflect responses to the higher CAA burden in this region. However, in the CC, S100 calcium binding protein A13, solute carrier family 14 member 1, and ceruloplasmin were more strongly elevated than in the cortex and could reflect responses unique to this region. Also of note is the dramatic increase in HspB1 observed in the CC. Significantly increased HspB1 expression was recently reported in the hippocampus and thalamus but not in the cortex of rTg-DI rats.12 Here in the CC, HspB1 represents a >500% increase over WT expression. In addition, the common enhancement of HTRA1 in the CC and cortex is intriguing. Proteomic analysis in human CAA cases has also revealed elevated expression of HTRA1.22 Furthermore, mutations in the HTRA1 gene and loss of HTRA1 function cause cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy, an inherited non-amyloidal cerebral small vessel disease also associated with intracerebral hemorrhage and vascular cognitive impairment and dementia.32, 33, 34, 35 Therefore, the increases in HTRA1 observed here are noteworthy, as is the potential impact on CAA pathologies in the rTg-DI model.
The two regions displayed less commonality in proteins with significantly reduced expression. Six proteins were reduced in both regions (Figure 4B). A heat map comparing the relative expression of proteins specifically decreased in the CC or the cortex, and those common to both regions, is displayed in Figure 4D. Commonly decreased proteins included Mbp, Plp1, Mag, neurofilament light polypeptide, neurofilament medium polypeptide, and neurofilament heavy chain. As discussed in Decreased Proteins in 12M and 4M rTg-DI Rat CC, down-regulation of these proteins can be an indication of neurodegeneration, demyelination, and damaged axonal integrity. In addition, myelin-oligodendrocyte glycoprotein was decreased in the cortex by 41%, and, although it did not meet the effect cutoff, it was reduced 23% in the CC. Myelin-oligodendrocyte glycoprotein is also important for myelination and axonal integrity,36 and thus its reduced expression has implications similar to the other commonly down-regulated proteins.
IPA Upstream Regulators and Causal Networks
Bioinformatics analysis was performed with IPA to provide disease function and pathway context to the proteomic changes. Differentially expressed proteins from the rTg-DI CC were analyzed both individually and comparatively with those from the cortex. Proteins with ≥50% increases or ≥30% decreases over WT expression were included. Based on directional differential expression of downstream target molecules, IPA predicts activation (z score > 2) or inhibition (z score less than −2) states of upstream regulators.37 Comparative analysis of the CC and cortex indicated the anti-inflammatory transforming growth factor-β1 (TGF-β1) as activated (z score = 2.065) in the CC but not in the cortex. However, it is worth noting that although they did not meet the threshold for predicted activation (z score > 2), many of the affiliated proteins are indeed significantly enhanced in the cortex. A heat map with the relative changes in marker expression in each region is depicted in Figure 5A, with increased expression indicated in red, decreased expression indicated in green, and color intensity based on degree of change from the WT expression.
Figure 5.

Ingenuity Pathway Analysis (IPA) identified upstream regulators, causal networks, and disease functions. A: Heat map depicting the differentially expressed proteins (≥50% increase or ≥30% decrease) in the corpus callosum (CC) and cortex of rTg-DI rats at 12 months (12M) associated with the upstream regulator Tgf-β1. B: Heat map depicting the differentially expressed proteins in the CC and cortex of 12M rTg-DI rats associated with the F2/thrombin causal network. C: Heat map depicting the differentially expressed proteins in the CC and cortex of 12M rTg-DI rats associated with the disease function Neurodegeneration of Axons. Red indicates increased, green indicates decreased, and white indicates not differentially expressed proteins; color intensity correlates with degree of change. P < 0.05 (A). Anxa5, annexin A5; other abbreviations are as indicated in Figure 3.
TGF-β1 mRNA expression was recently shown to be significantly enhanced in 12M rTg-DI rats and IPA analysis of brain regional proteomics identified TGF-β1 as an activated upstream regulator in the hippocampus and thalamus of 12M rTg-DI rats,10,12 consistent with the current findings. Notable among these proteins is HspB1. HspB1 can reportedly inhibit induced endothelial to mesenchymal transition, and preserve endothelial cell integrity.38 Treatment with TGF-β1 has been reported to enhance HspB1 mRNA expression in astrocytes in vitro.39 Furthermore, astrocytes release HspB1 when treated with Aβ in vitro.40 Thus, TGF-β1–regulated HspB1 may be an important component of the astrocyte-mediated inflammatory response in the CC, while also serving protective functions against degeneration of the BBB.
A “causal network analysis” was conducted within IPA that connects differentially expressed downstream target proteins via direct and indirect relationships to predict activation states of causative networks.37 It revealed significant activation (z score = 4.158) of the thrombin (F2) network in the CC but not the cortex (Figure 5B). Twenty-eight downstream target molecules in the F2 causal network were differentially expressed in the CC, only seven of which were altered in the cortex. IPA activates the F2 network specifically in the thalamus of 12M rTg-DI rats, a region that uniquely exhibits numerous thrombotic events, including small vessel occlusions and microbleeds.12 None of these events has been observed in the CC of rTg-DI rats (data not shown). However, thrombin has also been reported to induce neurodegeneration after activation of astrocytes, microglia, and neuroinflammatory responses.41, 42, 43, 44, 45 Thus, thrombin signaling could mediate activation of microglia and astrocytes in the rTg-DI CC and induce neuroinflammatory responses that contribute to the degeneration of WM observed in this region.
Downstream Effects Analysis of IPA
IPA also identifies specific processes and functions causally affected by differentially expressed proteins via downstream effects analysis. IPA provides activation scores for diseases and functions when causal reference information is available.37 Comparative downstream effects analysis revealed that neurodegeneration of axons was enhanced in both the CC and cortex, with activation z scores of 2.217 and 2.783, respectively. It is important to note that although IPA did not include Mbp in the network, it was appropriate to add it in the analysis, as loss of Mbp reportedly can lead to axon demyelination. Additionally, Mbp expression is reduced in the CC of rTg-DI rats exhibiting dramatic WM loss.13,28 Relative expression compared with WT in the CC and cortex was mostly similar for the WM-associated proteins Plp1, Mag, and Mbp (Figure 5C). The considerable losses in Plp1, Mbp, and Mag, all with reported roles in myelination and axon integrity, could therefore contribute to the dramatic WM loss characterized in the rTg-DI CC.
Glial Cell Densities in the Cortex and CC
In rTg-DI rats, the presence of cerebral microvascular amyloid drives strong neuroinflammation, as evidenced by a marked elevation of reactive perivascular glial cells.10,12 Many of the differentially expressed proteins in this study can be expressed in both astrocytes and microglia. Therefore, the impact of astrocyte and microglia numbers on the proteomic changes observed in the CC was investigated. In the cortex or CC of 12M WT rats, there was a normal distribution of astrocytes (Figure 6, A and B). In contrast, in the presence of abundant cortical CAA in the rTg-DI rats, there was a large increase in perivascular activated astrocytes (Figure 6, C and E). However, although the CC of rTg-DI rats lacks appreciable microvascular CAA (Figure 2), there was a highly significant increase (P < 0.001) in astrocyte numbers in this region (Figure 6, D and E).
Figure 6.
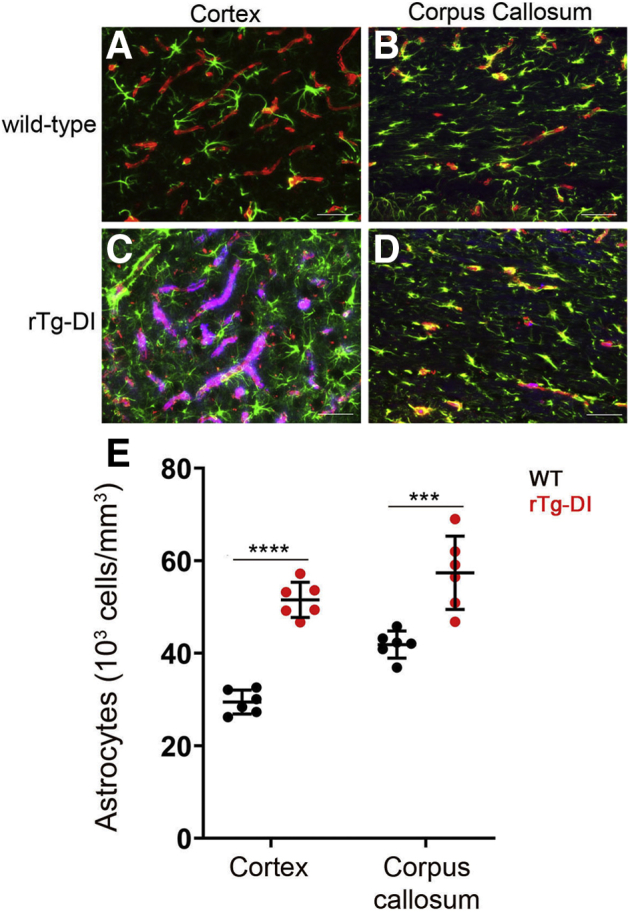
Increased astrocytes in the cortex and corpus callosum (CC) of rTg-DI rats. Brain sections from wild-type (WT) rats (A and B) and rTg-DI rats (C and D) at 12 months were labeled with Amylo-Glo to detect microvascular fibrillar amyloid (blue), goat polyclonal antibody to collagen IV to detect cerebral microvessels (red), and rabbit polyclonal antibody to glial fibrillary acidic protein to detect astrocytes (green). E: Quantitation of astrocyte numbers from rTg-DI rats (red circles) and WT rats (black circles) at 12 months in the cortex and CC. Data points show the results from each rat and the group mean ± SD of n = 6 rTg-DI rats and n = 6 WT rats. Compared with WT rats, the astrocyte numbers were significantly elevated in the cortex and CC of the rTg-DI rats. ∗∗∗P < 0.001; ∗∗∗∗P < 0.0001. Scale bars = 50 μm (A–D).
Similar observations were made in the evaluation of activated microglia. In the presence of cortical microvascular CAA, there was a striking increase in the number and activation of perivascular microglia (Figure 7, A–E). Again, in the CC of rTg-DI rats, with small amounts of microvascular amyloid, there was a strong elevation of microglia (Figure 7, D and E). Together, these findings indicate that even with minimal presence of microvascular amyloid, there are robust perivascular glial cell responses that accompany the WM degeneration which occurs in the CC.
Figure 7.
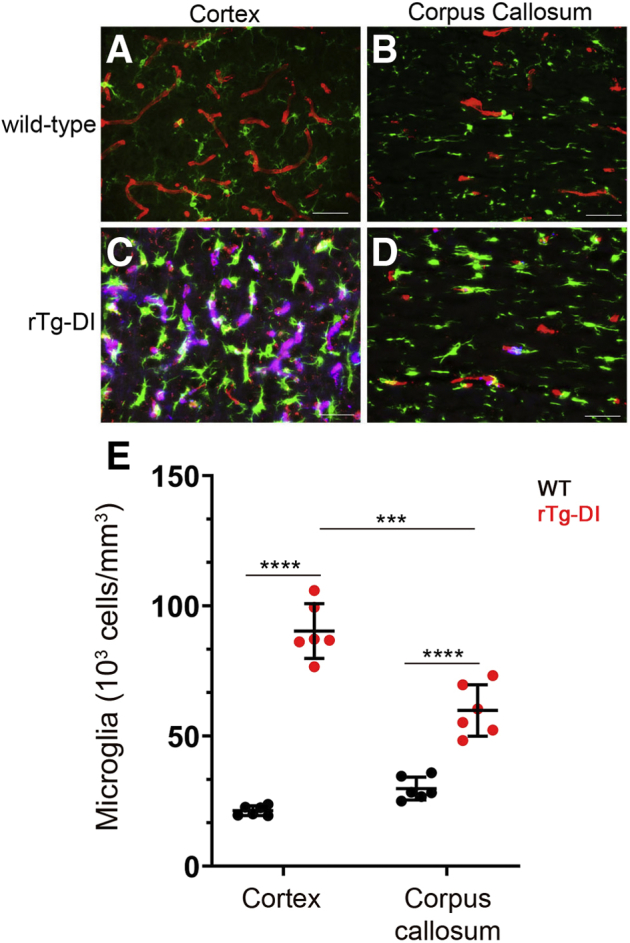
Increased microglia in the cortex and corpus callosum (CC) of rTg-DI rats. Brain sections from wild-type (WT) rats (A and B) and rTg-DI rats (C and D) at 12 months were labeled with Amylo-Glo to detect microvascular fibrillar amyloid (blue), rabbit polyclonal antibody to collagen IV to detect cerebral microvessels (red), and goat polyclonal antibody ionized calcium-binding adapter molecule 1 to identify microglia (green). E: Quantitation at 12 months of microglia in rTg-DI rats (red circles) and WT rats (black circles) in the cortex and CC. Data points show the results from each rat and the group mean ± SD of n = 6 rTg-DI rats and n = 6 WT rats. Compared with WT rats, the microglia were significantly elevated in the cortex and CC of rTg-DI rats. Microglia were elevated more in the cortex compared with the CC of rTg-DI rats. ∗∗∗P < 0.001; ∗∗∗∗P < 0.0001. Scale bars = 50 μm (A–D).
Validation of Proteomic Analysis and Regional Marker Distribution
To gain further insight into the cellular responses associated with these proteomic changes, regional immunolabeling was conducted for selected proteins of interest. Anxa3 and specific expression in microglia is enhanced in various brain regions of 12M rTg-DI rats.12 Here, despite minimal CAA burden (Figure 2) and lower microglia density (Figure 7) in the CC compared with the cortex, proteomic results indicated higher total protein concentration (Supplemental Tables S1 and S3) and greater increase in Anxa3 (Figure 4C) in the CC than in the cortex. Interestingly, Anxa3 displayed strong co-localization with the microglia marker Iba1 in both the cortex and CC (Figure 8, A–C), indicating its expression in microglia. After normalizing the total protein concentration (picomoles per milligram total protein) by the microglia density (103 cells/mm3), Anxa3 levels per microglia number in the CC were nearly double that in the cortex. This finding suggests that the more robust increase in Anxa3 in the CC is not merely a function of microgliosis in the presence of vascular amyloid but likely a specific microglial response to WM degeneration.
Figure 8.

Increased immunolabeling for glial markers in the corpus callosum (CC) of rTg-DI rats. Brain sections at 12 months from the current cohort of rTg-DI rats were immunolabeled with antibodies to annexin A3 (Anxa3) (A–C), annexin A5 (Anxa5) (D–F), megalencephalic leukoencephalopathy with subcortical cysts 1 (MLC1) (G–I), or GlialCAM (J–L) (red) and ionized calcium-binding adapter molecule 1 (Iba-1) (C, green), or glial fibrillary acidic protein (GFAP) (F, I, and L, green). Representative images show increased Anxa3 labeling in microglia in the cortex and CC and increased Anxa5, MLC1, and GlialCAM labeling of astrocytes only in the CC of rTg-DI rats. Scale bars = 50 μm.
Annexin A5 (Anxa5), whose expression was recently reported in WM-associated and -activated microglia using single-cell RNA-sequencing,46 was up-regulated in the CC (Supplemental Table S1) but not in the cortex of rTg-DI rats. Anxa5 has also been reported to prevent atherothrombosis and reverse Aβ-induced toxicity in choroid plexus cell cultures.47,48 Based on the reported expression and function of Anxa5, its unique elevation in the CC of rTg-DI rats, and the suggested activation of thrombin in the CC by IPA, the regional distribution of Anxa5 via immunolabeling (Figure 8, D–F) was investigated further. Consistent with the proteomic results, Anxa5 displayed specific enhancement in the CC of rTg-DI rats but not in the cortex (Figure 8, D and E), while double-labeling revealed its expression in astrocytes rather than microglia (Figure 8F).
IPA indication of the F2 network and diffuse WM loss in the thalamus of 12M rTg-DI rats,12,13 both share characteristics in the CC, but in no other investigated regions in the rTg-DI rats. However, the CC does not share extensive CAA load (Figure 2), nor occluded small vessels/microbleeds with the thalamus. It is possible that based on the reported functions of Anxa5, in the context of atherothrombosis and Aβ toxicity, it harbors protective effects in the CC.
Another protein of interest is MLC1, which displayed dramatic increases (>400% over WT) in the rTg-DI CC and only modest changes (<50% over WT) in the cortex (Figure 4B). Missense mutations in the MLC1 gene lead to the development of MLC, a leukodystrophy characterized by myelin vacuolation, brain edema, and macrocephaly.49 Although these phenotypic changes are different from the diffuse WM loss observed in rTg-DI rats, the distinct association of MLC1 with leukodystrophy prompted further investigation of the regional distribution of MLC1. Consistent with the SWATH-MS analysis, immunolabeling revealed extensive up-regulation of MLC1 in the CC, but not in the cortex of rTg-DI rats (Figure 8, G and H). MLC1 is reportedly expressed specifically in astrocytes,49, 50, 51 and double-immunolabeling with GFAP confirmed strong astrocytic localization in the CC of rTg-DI rats (Figure 8I). MLC can also be caused by missense mutations in GlialCAM, a cell adhesion molecule expressed primarily in glial cells but also found in hepatocytes.51 Furthermore, GlialCAM and MLC1 bind one another and co-localize in the astrocytic end feet surrounding cerebral blood vessels and astrocyte–astrocyte contacts.51, 52, 53 Interestingly, proteomic analysis revealed an approximately 40% increase in GlialCAM expression in the CC of rTg-DI rats, with no change in the cortex (data not shown). Although this increase was below the 50% threshold cutoff, because of the increase in GlialCAM protein expression and close association with MLC1, its regional distribution was investigated further. Immunolabeling revealed dramatic enhancements of GlialCAM in the CC of rTg-DI rats but not in the cortex, which was co-localized with astrocytic endfeet (Figure 8, J–L).
Discussion
CAA is a prevalent feature in most patients with Alzheimer disease, a common cause of cerebral small vessel disease in the elderly, and a prominent cause of vascular cognitive impairment and dementia.1, 2, 3, 4,6,7 Furthermore, the association between CAA and significant diffuse WM loss and WM hypersensitivities has been well documented.13,54,55 Although the link between the accumulation of vascular amyloid in the brain and resultant neurodegenerative pathologies remains poorly understood, proteomic analysis can provide further mechanistic insight.
Proteomic approaches in human CAA cases have been reported.22,56,57 However, a challenge in investigating human CAA cases is that they originate from diverse backgrounds, including numerous environmental, lifestyle, and genetic differences, thus potentially confounding CAA-specific investigation. Preclinical rodent models provide an advantageous avenue for more uniform population studies, as genetic backgrounds and environmental conditions can be tightly maintained. The rTg-DI model is particularly beneficial because it develops consistent and progressive microvascular CAA, occluded microvessels, and microbleeds.9 In addition, rTg-DI rats develop significant perivascular neuroinflammation, disruption of axonal integrity, diffuse WM loss, and behavioral deficits.9, 10, 11,13 Recently, regional proteomic changes in the cortex, hippocampus, and thalamus of rTg-DI rats were shown to exhibit both similar and distinct differentially expressed proteins reflecting the common and distinguishing pathologic features observed in each region.12 Therefore, this approach of regional isolation and SWATH-MS proteomics is an effective method for reporting on specific regional proteomic alterations associated with common and distinct pathologic features of CAA type 1.
The aim of the current study was to identify proteomic changes within the CC of rTg-DI rats that could provide mechanistic insight into the WM loss associated with CAA type 1. Moreover, by isolating and analyzing CC from 4M to 12M rTg-DI rats, progressive proteomic changes in animals at early stages of CAA accumulation with no WM loss (4M) were compared to those with extensive CAA, vascular pathologies, and diffuse WM loss (12M).
Several of the decreased proteins identified at 12M in rTg-DI CC, including Plp1, Mag, Mbp, neurofilament light polypeptide, neurofilament medium polypeptide, and neurofilament heavy chain, are important for myelination of axons, axonal integrity, and WM homeostasis. Notably, all six of these proteins were also decreased in the cortex (Figure 4D), and neurodegeneration of axons was indicated by IPA analysis in both regions (Figure 5C). Although the observed fold decrease of these proteins was similar between the two regions, the concentrations of these proteins ranged from 2.5 to 20× higher in the CC (Supplemental Table S4). Thus, in terms of total protein, these decreased levels represent much more dramatic changes in the CC versus the cortex, which is consistent with the significant diffuse WM loss observed in the CC (Figure 1).13
Activation of thrombin in the CC, indicated by IPA (Figure 5B), could also be a contributing factor to demyelination/neurodegeneration of axons. Although no overt signs of microbleeds or vessel occlusions were observed in the CC (data not shown) thrombin can promote neuroinflammation, astrocytosis, and microgliosis.41, 42, 43, 44, 45 Recently, it was reported that proteolysis of neurofascin 155, an adhesion protein responsible for the attachment of myelin to axons, is thrombin-dependent and occurs through direct binding of thrombin.58,59 In addition, myelin structure/stability can be maintained by paranodal astrocytes through the release of thrombin inhibitors, and blockade of these inhibitors results in demyelination.58 Furthermore, deletion of the thrombin receptor protease activated receptor 1 in mice results in enhanced myelination and increased numbers of oligodendrocytes, and blockade of protease activated receptor 1 can promote myelin regeneration in demyelinated lesions.60, 61, 62 Thus, thrombin could directly contribute to the neurodegeneration, demyelination, and diffuse WM loss observed in the CC and thalamus of rTg-DI rats.13
The common enhancement of HTRA1 in the cortex and CC is also an important observation. Enhancement of HTRA1 has been reported in human CAA cases,22 whereas mutations in the HTRA1 gene and loss of HTRA1 function reported causes cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy.32, 33, 34, 35 Thus, HTRA1 is implicated in both amyloidal and non-amyloidal forms of cerebral small vessel disease associated with leukodystrophies. IPA analysis linked HTRA1 to both the TGF-β1 and thrombin networks (Figure 5, A and B). HTRA1 has been reported to regulate TGF-β1 signaling, and enhanced HTRA1 mRNA expression can be induced by TGF-β1.35,63,64 Furthermore, dysregulation of TGF-β1 signaling as a result of dysfunctional HTRA1 has been suggested as an underlying mechanism of cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy pathologies.35 IPA’s indicated association of HTRA1 to thrombin is indirect and based on reported links between thrombin and TGF-β1. Thrombin can alter TGF-β1 signaling in epithelial cells, whereas thrombin injections in the cerebrospinal fluid induced TGF-β1 pathway-dependent inflammatory responses in rats.65,66 Hence, HTRA1 may be an important factor in CAA pathology, possibly related to thrombin and TGF-β1 signaling, although its exact role will require further investigation.
Anxa3 was identified as a novel marker for microglia and was implicated in proliferation and migration.67 We recently reported enhanced microglial expression of Anxa3 in several brain regions of 12M rTg-DI rats.12 Here, despite the significantly lower number of microglia in the CC compared with the cortex (Figure 7), Anxa3 expression/microglia number in the CC was double compared with that in the cortex. Furthermore, Anxa3 was elevated in the CC of 4M rTg-DI rats, when CAA burden is minimal (Figure 2). Therefore, Anxa3 may indicate a distinct population of activated microglia in the CC compared with the cortex, with specific roles in areas of WM degeneration that is not affected by CAA burden.
Anxa5 was specifically elevated in the CC of 12M rTg-DI rats and not in the cortex (Supplemental Table S2 and Figure 8E). Up-regulation of Anxa5 has been reported in the choroid plexus of patients with Alzheimer disease,68 can prevent Aβ aggregation,69,70 and has been implicated as an inflammatory marker in Aβ-treated microglia.71 In the rTg-DI CC, however, Anxa5 expression was observed primarily in astrocytes (Figure 8E). There was no increased Anxa5 expression observed in the cortex, and we did not find Anxa5 up-regulation in other rTg-DI rat brain regions.12 Based on its reported direct interactions with Aβ, its specific up-regulation in the rTg-DI CC, and its localization in astrocytes, it is possible that Anxa5 represents a component of a WM-specific astrocyte response to degeneration and CAA pathology. Alternatively, Anxa5 has been reported as a marker of Wallerian degeneration,31 and its presence in the CC could indicate occurrence of this process.
The up-regulation of MLC1 and GlialCAM and their relevance to leukodystrophies is also of keen interest. Mutations in the MLC1 and GlialCAM genes are uniquely linked to the incidence of MLC, a leukodystrophy characterized by WM vacuolization, swelling, and brain edema.49 MLC1 and GlialCAM bind one another, and GlialCAM serves as a chaperone for membrane localization of MLC1 in astrocytic endfeet contacting blood vessels and other glial cells.49,51,52,72 Furthermore, MLC1 and GlialCAM maintain volume-regulated anion currents through interaction with the chloride channel ClC-2.72 However, previous studies have solely focused on effects of MLC1 and GlialCAM knockout or missense mutations, and conditions of increased expression have not been reported. Based on their prominent role in other leukodystrophies, it is possible that MLC1 and GlialCAM up-regulation is linked to the diffuse WM loss and myelin degeneration; however, this relationship in the rTg-DI CC requires further investigation.
Limitations of the Study
First, the current study focused on proteomic changes that occurred in the CC region of the rTg-DI rat model of microvascular CAA. Because the CC exhibited a combination of WM loss and low levels of CAA, the reported proteomic changes in this specific region could result from a combination of these localized pathologic events. Second, because larger vessel CAA was largely absent in the rTg-DI rats, it is unclear if this form of vascular amyloid may promote different proteomic changes. Third, due to the large number of proteins changed in the CC, only a subset was confirmed by immunohistochemical analysis. Fourth, this study is limited in its ability to report sex-specific proteomic changes due to the small number of female and male animals used. Although no significant differences in protein expression were observed between male and female rTg-DI rats, and their microvascular amyloid loads remained consistent, determination of definitive sex-specific changes in the rTg-DI model will require larger studies with a greater number of male and female rats.
Conclusions
The current study reveals novel information on proteomic changes that uniquely occur in the CC of rTg-DI rats with emerging WM degeneration. These findings provide new information on potential pathogenic mechanisms that occur in the CC subsequent to the onset and progression of robust microvascular CAA in adjacent regions of the brain and warrant further systematic investigation in future studies.
Acknowledgments
We thank Dr. Xiaoyue Zhu and Ms. Judianne Davis for technical assistance.
Footnotes
Supported by NIH research grant RO1NS094201 (W.E.V.N.) and RF-AG053991 (H.B., W.E.V.N.).
Disclosures: None declared.
Current address of B.B., Merck & Co., Boston, MA.
Supplemental material for this article can be found at http://doi.org/10.1016/j.ajpath.2021.11.010.
Author Contributions
J.M.S., F.X., H.L., and B.B. performed experiments; J.M.S. drafted the manuscript; W.E.V.N. conceptualized and supervised the study; and W.E.V.N. and H.B acquired funding, and reviewed and edited the manuscript.
Supplemental Data
Supplemental Figure S1.

Collection of corpus callosum (CC) from young and older wild-type (WT) and rTg-DI rats for proteomic analysis. The CC of each rat from 4 months to 12 months (12M) in WT and rTg-DI rats was collected from fresh frozen brain sections using laser capture microdissection. Here, representative fresh frozen brain sections from 4 months to 12M WT rats (A and B, respectively) and 12M rTg-DI rats (C and D) were stained for myelin. The CC regions that were collected are outlined in yellow. n = 5, 6 per age. Scale bar = 1 mm.
References
- 1.Attems J., Jellinger K., Thal D.R., Van Nostrand W. Review: sporadic cerebral amyloid angiopathy. Neuropathol Appl Neurobiol. 2011;37:75–93. doi: 10.1111/j.1365-2990.2010.01137.x. [DOI] [PubMed] [Google Scholar]
- 2.Rensink A.A.M., de Waal R.M.W., Kremer B., Verbeek M.M. Pathogenesis of cerebral amyloid angiopathy. Brain Res Rev. 2003;43:207–223. doi: 10.1016/j.brainresrev.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 3.Arvanitakis Z., Leurgans S.E., Wang Z., Wilson R.S., Bennett D.A., Schneider J.A. Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann Neurol. 2011;69:320–327. doi: 10.1002/ana.22112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Viswanathan A., Greenberg S.M. Cerebral amyloid angiopathy in the elderly. Ann Neurol. 2011;70:871–880. doi: 10.1002/ana.22516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thal D.R., Ghebremedhin E., Rüb U., Yamaguchi H., Del Tredici K., Braak H. Two types of sporadic cerebral amyloid angiopathy. J Neuropathol Exp Neurol. 2002;61:282–293. doi: 10.1093/jnen/61.3.282. [DOI] [PubMed] [Google Scholar]
- 6.Boyle P.A., Yu L., Nag S., Leurgans S., Wilson R.S., Bennett D.A., Schneider J.A. Cerebral amyloid angiopathy and cognitive outcomes in community-based older persons. Neurology. 2015;85:1930–1936. doi: 10.1212/WNL.0000000000002175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greenberg S.M., Vernooij M.W., Cordonnier C., Viswanathan A., Al-Shahi Salman R., Warach S., Launer L.J., Van Buchem M.A., Breteler M.M., Microbleed Study Group Cerebral microbleeds: a guide to detection and interpretation. Lancet Neurol. 2009;8:165–174. doi: 10.1016/S1474-4422(09)70013-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reijmer Y.D., van Veluw S.J., Greenberg S.M. Ischemic brain injury in cerebral amyloid angiopathy. J Cereb Blood Flow Metab. 2016;36:40–54. doi: 10.1038/jcbfm.2015.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davis J., Xu F., Hatfield J., Lee H., Hoos M.D., Popescu D., Crooks E., Kim R., Smith S.O., Robinson J.K., Benveniste H., Van Nostrand W.E. A novel transgenic rat model of robust cerebral microvascular amyloid with prominent vasculopathy. Am J Pathol. 2018;188:2877–2889. doi: 10.1016/j.ajpath.2018.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu X., Hatfield J., Sullivan J.K., Xu F., Van Nostrand W.E. Robust neuroinflammation and perivascular pathology in rTg-DI rats, a novel model of microvascular cerebral amyloid angiopathy. J Neuroinflammation. 2020;17:78. doi: 10.1186/s12974-020-01755-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Popescu D.L., Van Nostrand W.E., Robinson J.K. Longitudinal cognitive decline in a novel rodent model of cerebral amyloid angiopathy type-1. Int J Mol Sci. 2020;21:2348. doi: 10.3390/ijms21072348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schrader J.M., Xu F., Nostrand W.E.V. Distinct brain regional proteome changes in the rTg-DI rat model of cerebral amyloid angiopathy. J Neurochem. 2021;159:273–291. doi: 10.1111/jnc.15463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee H., Xu F., Liu X., Koundal S., Zhu X., Davis J., Yanez D., Schrader J., Stanisavljevic A., Rothman D.L., Wardlaw J., Nostrand W.E.V., Benveniste H. Diffuse white matter loss in a transgenic rat model of cerebral amyloid angiopathy. J Cereb Blood Flow Metab. 2020;41:1103–1118. doi: 10.1177/0271678X20944226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ludwig C., Gillet L., Rosenberger G., Amon S., Collins B.C., Aebersold R. Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial. Mol Syst Biol. 2018;14:e8126. doi: 10.15252/msb.20178126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aebersold R., Mann M. Mass-spectrometric exploration of proteome structure and function. Nature. 2016;537:347–355. doi: 10.1038/nature19949. [DOI] [PubMed] [Google Scholar]
- 16.López-Sánchez L.M., Jiménez-Izquierdo R., Peñarando J., Mena R., Guil-Luna S., Toledano M., Conde F., Villar C., Díaz C., Ortea I., De la Haba-Rodríguez J.R., Aranda E., Rodríguez-Ariza A. SWATH-based proteomics reveals processes associated with immune evasion and metastasis in poor prognosis colorectal tumours. J Cell Mol Med. 2019;23:8219–8232. doi: 10.1111/jcmm.14693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gillet L.C., Navarro P., Tate S., Röst H., Selevsek N., Reiter L., Bonner R., Aebersold R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics. 2012;11 doi: 10.1074/mcp.O111.016717. O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koundal S., Liu X., Sanggaard S., Mortensen K., Wardlaw J., Nedergaard M., Benveniste H., Lee H. Brain morphometry and longitudinal relaxation time of spontaneously hypertensive rats (SHRs) in early and intermediate stages of hypertension investigated by 3D VFA-SPGR MRI. Neuroscience. 2019;404:14–26. doi: 10.1016/j.neuroscience.2019.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Valdés-Hernández P.A., Sumiyoshi A., Nonaka H., Haga R., Aubert-Vásquez E., Ogawa T., Iturria-Medina Y., Riera J.J., Kawashima R. An in vivo MRI template set for morphometry, tissue segmentation, and fMRI localization in rats. Front Neuroinform. 2011;5:26. doi: 10.3389/fninf.2011.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wiśniewski J.R., Rakus D. Multi-enzyme digestion FASP and the ‘Total Protein Approach’-based absolute quantification of the Escherichia coli proteome. J Proteomics. 2014;109:322–331. doi: 10.1016/j.jprot.2014.07.012. [DOI] [PubMed] [Google Scholar]
- 21.Wiśniewski J.R., Ostasiewicz P., Duś K., Zielińska D.F., Gnad F., Mann M. Extensive quantitative remodeling of the proteome between normal colon tissue and adenocarcinoma. Mol Syst Biol. 2012;8:611. doi: 10.1038/msb.2012.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hondius D.C., Eigenhuis K.N., Morrema T.H.J., van der Schors R.C., van Nierop P., Bugiani M., Li K.W., Hoozemans J.J.M., Smit A.B., Rozemuller A.J.M. Proteomics analysis identifies new markers associated with capillary cerebral amyloid angiopathy in Alzheimer’s disease. Acta Neuropathol Commun. 2018;6:46. doi: 10.1186/s40478-018-0540-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pascovici D., Handler D.C.L., Wu J.X., Haynes P.A. Multiple testing corrections in quantitative proteomics: a useful but blunt tool. Proteomics. 2016;16:2448–2453. doi: 10.1002/pmic.201600044. [DOI] [PubMed] [Google Scholar]
- 24.Eng L.F., Vanderhaeghen J.J., Bignami A., Gerstl B. An acidic protein isolated from fibrous astrocytes. Brain Res. 1971;28:351–354. doi: 10.1016/0006-8993(71)90668-8. [DOI] [PubMed] [Google Scholar]
- 25.Eng L.F., Ghirnikar R.S., Lee Y.L. Glial fibrillary acidic protein: GFAP-thirty-one years (1969-2000) Neurochem Res. 2000;25:1439–1451. doi: 10.1023/a:1007677003387. [DOI] [PubMed] [Google Scholar]
- 26.Guttenplan K.A., Liddelow S.A. Astrocytes and microglia: models and tools. J Exp Med. 2019;216:71–83. doi: 10.1084/jem.20180200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Q., Xie F., Alvarado-Diaz A., Smith M.A., Moreira P.I., Zhu X., Perry G. Neurofilamentopathy in neurodegenerative diseases. Open Neurol J. 2011;5:58–62. doi: 10.2174/1874205X01105010058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weil M.-T., Möbius W., Winkler A., Ruhwedel T., Wrzos C., Romanelli E., Bennett J.L., Enz L., Goebels N., Nave K.-A., Kerschensteiner M., Schaeren-Wiemers N., Stadelmann C., Simons M. Loss of myelin basic protein function triggers myelin breakdown in models of demyelinating diseases. Cell Rep. 2016;16:314–322. doi: 10.1016/j.celrep.2016.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gould E.A., Busquet N., Shepherd D., Dietz R.M., Herson P.S., Simoes de Souza F.M., Li A., George N.M., Restrepo D., Macklin W.B. Mild myelin disruption elicits early alteration in behavior and proliferation in the subventricular zone. Elife. 2018;7:e34783. doi: 10.7554/eLife.34783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pronker M.F., Lemstra S., Snijder J., Heck A.J.R., Thies-Weesie D.M.E., Pasterkamp R.J., Janssen B.J.C. Structural basis of myelin-associated glycoprotein adhesion and signalling. Nat Commun. 2016;7:13584. doi: 10.1038/ncomms13584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sievers C., Platt N., Perry V.H., Coleman M.P., Conforti L. Neurites undergoing Wallerian degeneration show an apoptotic-like process with annexin V positive staining and loss of mitochondrial membrane potential. Neurosci Res. 2003;46:161–169. doi: 10.1016/s0168-0102(03)00039-7. [DOI] [PubMed] [Google Scholar]
- 32.Fukutake T. Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL): from discovery to gene identification. J Stroke Cerebrovasc Dis. 2011;20:85–93. doi: 10.1016/j.jstrokecerebrovasdis.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 33.Chen Y., He Z., Meng S., Li L., Yang H., Zhang X. A novel mutation of the high-temperature requirement A serine peptidase 1 (HTRA1) gene in a Chinese family with cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL) J Int Med Res. 2013;41:1445–1455. doi: 10.1177/0300060513480926. [DOI] [PubMed] [Google Scholar]
- 34.Uemura M., Nozaki H., Koyama A., Sakai N., Ando S., Kanazawa M., Kato T., Onodera O. HTRA1 mutations identified in symptomatic carriers have the property of interfering the trimer-dependent activation cascade. [Erratum appeared in Front Neurol 2021, 12:756038.] Front Neurol. 2019;10:693. doi: 10.3389/fneur.2019.00693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beaufort N., Scharrer E., Kremmer E., Lux V., Ehrmann M., Huber R., Houlden H., Werring D., Haffner C., Dichgans M. Cerebral small vessel disease-related protease HTRA1 processes latent TGF-[beta] binding protein 1 and facilitates TGF-[beta] signaling. Proc Natl Acad Sci U S A. 2014;111:16496–16501. doi: 10.1073/pnas.1418087111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peschl P., Bradl M., Höftberger R., Berger T., Reindl M. Myelin oligodendrocyte glycoprotein: deciphering a target in inflammatory demyelinating diseases. Front Immunol. 2017;8:529. doi: 10.3389/fimmu.2017.00529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krämer A., Green J., Pollard J., Jr., Tugendreich S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics. 2014;30:523–530. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Choi S.-H., Nam J.-K., Kim B.-Y., Jang J., Jin Y.-B., Lee H.-J., Park S., Ji Y.H., Cho J., Lee Y.-J. HSPB1 inhibits the endothelial-to-mesenchymal transition to suppress pulmonary fibrosis and lung tumorigenesis. Cancer Res. 2016;76:1019–1030. doi: 10.1158/0008-5472.CAN-15-0952. [DOI] [PubMed] [Google Scholar]
- 39.Hamby M.E., Coppola G., Ao Y., Geschwind D.H., Khakh B.S., Sofroniew M.V. Inflammatory mediators alter the astrocyte transcriptome and calcium signaling elicited by multiple G-protein-coupled receptors. J Neurosci. 2012;32:14489–14510. doi: 10.1523/JNEUROSCI.1256-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nafar F., Williams J.B., Mearow K.M. Astrocytes release HspB1 in response to amyloid-[beta] exposure in vitro. J Alzheimers Dis. 2016;49:251–263. doi: 10.3233/JAD-150317. [DOI] [PubMed] [Google Scholar]
- 41.Lin C.-C., Lee I.-T., Wu W.-B., Liu C.-J., Hsieh H.-L., Hsiao L.-D., Yang C.-C., Yang C.-M. Thrombin mediates migration of rat brain astrocytes via PLC, Ca2+, CaMKII, PKC[alpha], and AP-1-dependent matrix metalloproteinase-9 expression. Mol Neurobiol. 2013;48:616–630. doi: 10.1007/s12035-013-8450-6. [DOI] [PubMed] [Google Scholar]
- 42.Lee D.Y., Park K.W., Jin B.K. Thrombin induces neurodegeneration and microglial activation in the cortex in vivo and in vitro: proteolytic and non-proteolytic actions. Biochem Biophys Res Commun. 2006;346:727–738. doi: 10.1016/j.bbrc.2006.05.174. [DOI] [PubMed] [Google Scholar]
- 43.Huang C., Ma R., Sun S., Wei G., Fang Y., Liu R., Li G. JAK2-STAT3 signaling pathway mediates thrombin-induced proinflammatory actions of microglia in vitro. J Neuroimmunol. 2008;204:118–125. doi: 10.1016/j.jneuroim.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 44.Ye X., Zuo D., Yu L., Zhang L., Tang J., Cui C., Bao L., Zan K., Zhang Z., Yang X., Chen H., Tang H., Zu J., Shi H., Cui G. ROS/TXNIP pathway contributes to thrombin induced NLRP3 inflammasome activation and cell apoptosis in microglia. Biochem Biophys Res Commun. 2017;485:499–505. doi: 10.1016/j.bbrc.2017.02.019. [DOI] [PubMed] [Google Scholar]
- 45.Yin M., Chen Z., Ouyang Y., Zhang H., Wan Z., Wang H., Wu W., Yin X. Thrombin-induced, TNFR-dependent miR-181c downregulation promotes MLL1 and NF-[kappa]B target gene expression in human microglia. J Neuroinflammation. 2017;14:132. doi: 10.1186/s12974-017-0887-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Safaiyan S., Besson-Girard S., Kaya T., Cantuti-Castelvetri L., Liu L., Ji H., Schifferer M., Gouna G., Usifo F., Kannaiyan N., Fitzner D., Xiang X., Rossner M.J., Brendel M., Gokce O., Simons M. White matter aging drives microglial diversity. Neuron. 2021;109:1100–1117.e10. doi: 10.1016/j.neuron.2021.01.027. [DOI] [PubMed] [Google Scholar]
- 47.Cederholm A., Frostegård J. Annexin A5 as a novel player in prevention of atherothrombosis in SLE and in the general population. Ann N Y Acad Sci. 2007;1108:96–103. doi: 10.1196/annals.1422.011. [DOI] [PubMed] [Google Scholar]
- 48.Bartolome F., Krzyzanowska A., de la Cueva M., Pascual C., Antequera D., Spuch C., Villarejo-Galende A., Rabano A., Fortea J., Alcolea D., Lleo A., Ferrer I., Hardy J., Abramov A.Y., Carro E. Annexin A5 prevents amyloid-β-induced toxicity in choroid plexus: implication for Alzheimer’s disease. Sci Rep. 2020;10:9391. doi: 10.1038/s41598-020-66177-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hwang J., Vu H.M., Kim M.-S., Lim H.-H. Plasma membrane localization of MLC1 regulates cellular morphology and motility. Mol Brain. 2019;12:116. doi: 10.1186/s13041-019-0540-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dubey M., Bugiani M., Ridder M.C., Postma N.L., Brouwers E., Polder E., Jacobs J.G., Baayen J.C., Klooster J., Kamermans M., Aardse R., de Kock C.P.J., Dekker M.P., van Weering J.R.T., Heine V.M., Abbink T.E.M., Scheper G.C., Boor I., Lodder J.C., Mansvelder H.D., van der Knaap M.S. Mice with megalencephalic leukoencephalopathy with cysts: a developmental angle. Ann Neurol. 2015;77:114–131. doi: 10.1002/ana.24307. [DOI] [PubMed] [Google Scholar]
- 51.Hoegg-Beiler M.B., Sirisi S., Orozco I.J., Ferrer I., Hohensee S., Auberson M., Gödde K., Vilches C., de Heredia M.L., Nunes V., Estévez R., Jentsch T.J. Disrupting MLC1 and GlialCAM and ClC-2 interactions in leukodystrophy entails glial chloride channel dysfunction. Nat Commun. 2014;5:3475. doi: 10.1038/ncomms4475. [DOI] [PubMed] [Google Scholar]
- 52.López-Hernández T., Ridder M.C., Montolio M., Capdevila-Nortes X., Polder E., Sirisi S., Duarri A., Schulte U., Fakler B., Nunes V., Scheper G.C., Martínez A., Estévez R., van der Knaap M.S. Mutant GlialCAM causes megalencephalic leukoencephalopathy with subcortical cysts, benign familial macrocephaly, and macrocephaly with retardation and autism. Am J Hum Genet. 2011;88:422–432. doi: 10.1016/j.ajhg.2011.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.López-Hernández T., Sirisi S., Capdevila-Nortes X., Montolio M., Fernández-Dueñas V., Scheper G.C., van der Knaap M.S., Casquero P., Ciruela F., Ferrer I., Nunes V., Estévez R. Molecular mechanisms of MLC1 and GLIALCAM mutations in megalencephalic leukoencephalopathy with subcortical cysts. Hum Mol Genet. 2011;20:3266–3277. doi: 10.1093/hmg/ddr238. [DOI] [PubMed] [Google Scholar]
- 54.Reijmer Y.D., Fotiadis P., Charidimou A., van Veluw S.J., Xiong L., Riley G.A., Martinez-Ramirez S., Schwab K., Viswanathan A., Gurol M.E., Greenberg S.M. Relationship between white matter connectivity loss and cortical thinning in cerebral amyloid angiopathy. Hum Brain Mapp. 2017;38:3723–3731. doi: 10.1002/hbm.23629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fotiadis P., van Rooden S., van der Grond J., Schultz A., Martinez-Ramirez S., Auriel E., Reijmer Y., van Opstal A.M., Ayres A., Schwab K.M., Alzheimer’s Disease Neuroimaging Initiative. Hedden T., Rosand J., Viswanathan A., Wermer M., Terwindt G., Sperling R.A., Polimeni J.R., Johnson K.A., van Buchem M.A., Greenberg S.M., Gurol M.E. Cortical atrophy in hereditary and sporadic forms of cerebral amyloid angiopathy: an observational case-control study. Lancet Neurol. 2016;15:811–819. doi: 10.1016/S1474-4422(16)30030-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Manousopoulou A., Gatherer M., Smith C., Nicoll J.A.R., Woelk C.H., Johnson M., Kalaria R., Attems J., Garbis S.D., Carare R.O. Systems proteomic analysis reveals that clusterin and tissue inhibitor of metalloproteinases 3 increase in leptomeningeal arteries affected by cerebral amyloid angiopathy. Neuropathol Appl Neurobiol. 2017;43:492–504. doi: 10.1111/nan.12342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Manousopoulou A., Yuen H.M., Sharp M.M., Saito S., Aldea R., Mazer N., Garbis S.D., Carare R.O. Quantitative proteomic profiling of white matter in cases of cerebral amyloid angiopathy reveals upregulation of extracellular matrix proteins and clusterin. Free Neuropathol. 2020;1:28. doi: 10.17879/freeneuropathology-2020-2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dutta D.J., Woo D.H., Lee P.R., Pajevic S., Bukalo O., Huffman W.C., Wake H., Basser P.J., SheikhBahaei S., Lazarevic V., Smith J.C., Fields R.D. Regulation of myelin structure and conduction velocity by perinodal astrocytes. Proc Natl Acad Sci U S A. 2018;115:11832–11837. doi: 10.1073/pnas.1811013115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dutta D.J., Fields R.D. Deletion of the thrombin proteolytic site in neurofascin 155 causes disruption of nodal and paranodal organization. Front Cell Neurosci. 2021;15:576609. doi: 10.3389/fncel.2021.576609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Choi C.-I., Yoon H., Drucker K.L., Langley M.R., Kleppe L., Scarisbrick I.A. The thrombin receptor restricts subventricular zone neural stem cell expansion and differentiation. Sci Rep. 2018;8:9360. doi: 10.1038/s41598-018-27613-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yoon H., Radulovic M., Drucker K.L., Wu J., Scarisbrick I.A. The thrombin receptor is a critical extracellular switch controlling myelination. Glia. 2015;63:846–859. doi: 10.1002/glia.22788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yoon H., Choi C.-I., Triplet E.M., Langley M.R., Kleppe L.S., Kim H.N., Simon W.L., Scarisbrick I.A. Blocking the thrombin receptor promotes repair of demyelinated lesions in the adult brain. J Neurosci. 2020;40:1483–1500. doi: 10.1523/JNEUROSCI.2029-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Friedrich U., Datta S., Schubert T., Plössl K., Schneider M., Grassmann F., Fuchshofer R., Tiefenbach K.-J., Längst G., Weber B.H.F. Synonymous variants in HTRA1 implicated in AMD susceptibility impair its capacity to regulate TGF-[beta] signaling. Hum Mol Genet. 2015;24:6361–6373. doi: 10.1093/hmg/ddv346. [DOI] [PubMed] [Google Scholar]
- 64.Xu L., Golshirazian I., Asbury B.J., Li Y. Induction of high temperature requirement A1, a serine protease, by TGF-beta1 in articular chondrocytes of mouse models of OA. Histol Histopathol. 2014;29:609–618. doi: 10.14670/HH-29.10.609. [DOI] [PubMed] [Google Scholar]
- 65.Bae J.-S., Kim I.-S., Rezaie A.R. Thrombin down-regulates the TGF-beta-mediated synthesis of collagen and fibronectin by human proximal tubule epithelial cells through the EPCR-dependent activation of PAR-1. J Cell Physiol. 2010;225:233–239. doi: 10.1002/jcp.22249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li T., Zhang P., Yuan B., Zhao D., Chen Y., Zhang X. Thrombin-induced TGF-β1 pathway: a cause of communicating hydrocephalus post subarachnoid hemorrhage. Int J Mol Med. 2013;31:660–666. doi: 10.3892/ijmm.2013.1253. [DOI] [PubMed] [Google Scholar]
- 67.Zhang Z., Li Z., Ma Z., Deng M., Xing M., Wu J., Jiang S., Wang Q., Guo Q., Zou W. Annexin A3 as a marker protein for microglia in the central nervous system of rats. Neural Plast. 2021;2021:5575090. doi: 10.1155/2021/5575090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Krzyzanowska A., García-Consuegra I., Pascual C., Antequera D., Ferrer I., Carro E. Expression of regulatory proteins in choroid plexus changes in early stages of Alzheimer disease. J Neuropathol Exp Neurol. 2015;74:359–369. doi: 10.1097/NEN.0000000000000181. [DOI] [PubMed] [Google Scholar]
- 69.Lee G., Pollard H.B., Arispe N. Annexin 5 and apolipoprotein E2 protect against Alzheimer's amyloid-β-peptide cytotoxicity by competitive inhibition at a common phosphatidylserine interaction site. Peptides. 2002;23:1249–1263. doi: 10.1016/s0196-9781(02)00060-8. [DOI] [PubMed] [Google Scholar]
- 70.Hung L.W., Ciccotosto G.D., Giannakis E., Tew D.J., Perez K., Masters C.L., Cappai R., Wade J.D., Barnham K.J. Amyloid-beta peptide (Abeta) neurotoxicity is modulated by the rate of peptide aggregation: Abeta dimers and trimers correlate with neurotoxicity. J Neurosci. 2008;28:11950–11958. doi: 10.1523/JNEUROSCI.3916-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Correani V., Di Francesco L., Mignogna G., Fabrizi C., Leone S., Giorgi A., Passeri A., Casata R., Fumagalli L., Maras B., Schininà M.E. Plasma membrane protein profiling in beta-amyloid-treated microglia cell line. Proteomics. 2017;17 doi: 10.1002/pmic.201600439. [DOI] [PubMed] [Google Scholar]
- 72.Capdevila-Nortes X., López-Hernández T., Apaja P.M., López de Heredia M., Sirisi S., Callejo G., Arnedo T., Nunes V., Lukacs G.L., Gasull X., Estévez R. Insights into MLC pathogenesis: GlialCAM is an MLC1 chaperone required for proper activation of volume-regulated anion currents. Hum Mol Genet. 2013;22:4405–4416. doi: 10.1093/hmg/ddt290. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw MS data can be found in the MassIVE repository (project ID# MSV000088077; password: rtgdi2021cc).