

Abstract
Leptin is an adipokine with roles in food intake and energy metabolism through its actions on neurons in the hypothalamus. The role of leptin in obesity and cardiovascular disorders is well documented. However, its influence on liver conditions such as cholestasis is poorly understood. The effects of exogenous leptin and leptin-neutralizing antibody on biliary hyperplasia, hepatic fibrosis, and inflammation in the multidrug resistance protein 2 knockout (Mdr2KO) mouse model of cholestasis were assessed by quantifying markers specific for cholangiocytes, activated hepatic stellate cells (HSCs), and cytokines. Serum and hepatic leptin were increased in Mdr2KO mice compared with FVB/NJ (FVBN) controls, and exogenous leptin enhanced biliary hyperplasia and liver fibrosis in Mdr2KO and FVBN mice. Leptin administration increased hepatic expression of C-C motif chemokine ligand 2 and IL-6 in Mdr2KO mice. In contrast, leptin-neutralizing antibody reduced intrahepatic bile duct mass and decreased HSC activation in Mdr2KO mice compared with FVBN controls. Sex-related differences were noted, with female Mdr2KO mice having more leptin than males. In cholangiocytes and LX2 cells in vitro, leptin increased phosphorylated Akt and stimulated cell proliferation. Leptin receptor siRNA and inhibitors of Akt phosphorylation impaired leptin-induced cell proliferation and proinflammatory cytokines. The current data suggest that leptin is abnormally increased in cholestatic mice, and excess leptin increases ductular reaction, hepatic fibrosis, and inflammation via leptin receptor–mediated phosphorylation of Akt in cholangiocytes and HSCs.
Leptin, a product of LEP gene, is a 16-kDa peptide produced primarily in white adipose tissue and acts through receptor isoforms that are products of OBR gene belonging to the class 1 cytokine receptor family.1 Leptin is a pleiotropic molecule acting as a hormone and as a cytokine to modulate a large spectrum of physiological processes. Thus, leptin functions as a hormone with role in regulation of food intake,2 energy expenditure,3 and body weight,4,5 and as a proinflammatory cytokine influencing immune cell functions6 involved in innate and adaptive immunity.7
Although the roles of leptin in obesity,8 diabetes,9 and cancer10 have been extensively investigated, the influence of leptin on cholestasis and liver fibrosis is controversial and still not fully understood.11 An increasing number of studies report roles for leptin in modulation of liver functions in the context of metabolic syndrome and insulin resistance.12 Furthermore, leptin resistance, which is detected as elevated serum leptin, is a contributing factor to insulin resistance in cirrhotic patients.12 Interestingly, unlike nonalcoholic fatty liver disease or nonalcoholic steatohepatitis, liver fibrosis and cirrhosis caused by cholestasis or viral infections are not associated with steatosis or gain of body weight, even though serum leptin is elevated in these liver injuries.13
The role of leptin in cholestatic conditions, such as primary biliary cholangitis, is also debated. Primary biliary cholangitis was associated with increased leptin levels in the serum in several studies.14, 15, 16 However, other studies concluded that serum leptin levels are low in primary biliary cholangitis patients, and leptin does not cause anorexia in liver disease.17 Interestingly, a study on liver cholestasis in dogs pointed out that elevated serum leptin was associated with cholestatic disease in pituitary-dependent hyperadrenocorticism.18 Reports on animal models of cholestasis induced by bile duct ligation suggest that serum leptin is significantly increased early after surgery, followed by normalization and even decrease in long-term cholestasis.19
An imbalance between the orexigenic gastric peptide ghrelin and leptin as its physiological counterpart was reported in primary biliary cholangitis patients having elevated serum levels of leptin and reduced ghrelin compared with healthy controls.20 Previous studies demonstrated that ghrelin treatment attenuated ductular reaction and hepatic fibrosis.21 The current study assessed the effects of leptin in the multidrug resistance protein 2 knockout (Mdr2KO) mouse model of cholestasis.
Materials and Methods
Chemicals, Kits, Antibodies, and Tissue Culture Media
All chemicals were purchased from Millipore-Sigma (Burlington, MA), unless otherwise stated, and were of the highest grade available. Leptin mouse enzyme-linked immunosorbent assay (ELISA) kit was purchased from Abcam (Cambridge, MA). RNeasy kit for isolation of RNA from cells and tissue was from Qiagen (Frederik, MD). Leptin receptor (LepR) siRNA was from OriGene (Rockville, MD); recombinant leptin and signaling inhibitors wortmannin (WMN) and FPA124 were from Tocris (Minneapolis, MN); leptin-neutralizing antibody (Leptin-Ab) was purchased from R&D Systems (Minneapolis, MN). Mouse transforming growth factor-β1 (TGF-β1) was purchased from R&D Systems. Hematoxylin and VectaStain kits for immunohistochemistry (IHC) staining were from Vector Laboratories (Burlingame, CA). For IHC and immunofluorescence (IF) assays, the following antibodies from Abcam were used: leptin, LepR, cytokeratin (CK)-19, CK7, CK8, desmin, α-smooth muscle actin (α-SMA), phosphorylated Akt (p-Akt), and CD11b. Culture media, including Dulbecco’s modified Eagle’s medium, minimal essential medium, and the supplements (ie, fetal bovine serum and penicillin/streptomycin), were from Gibco BRL, purchased through Thermo Fisher Scientific (Waltham, MA).
Animal Experiments
FVB/NJ (FVBN) and Mdr2KO mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and maintained in a temperature-controlled environment at 20°C to 22°C with a 12:12 hours light-dark cycle, having free access to food and drinking water. All animal procedures were performed in accord with the guidelines of University of Texas at Austin (Austin, TX) Institutional Animal Care and Use Committee, with approved protocols. In experiments designed to measure the effect of recombinant leptin and Leptin-Ab on liver fibrosis in Mdr2KO mice, we used 2-month–old, male and female Mdr2KO mice, as well as age-matched FVBN mice as negative controls. Six mice in each group of sex/type (FVBN or Mdr2KO) were used. In parallel, an equal number of 2-month–old, male and female FVBN and Mdr2KO mice were treated with vehicle (saline) only. Leptin was administered via Alzet osmotic minipumps (Cupertino, CA) implanted intraperitoneally at a rate of 100 μg/kg per day (mice weights being 25 to 30 g), as described.22 Leptin-Ab was performed by retro-orbital injections of 2 μg/day for 14 days, after which the mice were euthanized for blood and liver tissue collection. Leptin-Ab inhibits binding to its receptor, negating its downstream cellular effects, such as cell proliferation, and it has been used before, as previously described.23
Serum Biomarker Analysis
Liver enzymes aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were assayed using Catalyst One Analyzer from IDEXX Laboratories (Westbrook, ME).
Assessment of mRNA Expression
Assessment of gene expression at the mRNA level in liver tissue or cultured cells was performed by real-time quantitative PCR for leptin, LepR, CK19, proliferating cell nuclear antigen (PCNA), desmin, α-SMA, collagen type 1A1 (Col1A1), matrix metalloproteinase 2, tissue inhibitor of metalloproteinase 1, TGF-β1 and TGF-β2 and their receptor TGFβR2, and cytokines C-C motif chemokine ligand 2 (CCL2), IL-1β, and IL-6. Fold changes in gene expression were normalized to the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase. Total RNA was isolated by using RNeasy kit, followed by cDNA synthesis with iScript kit from Bio-Rad Life Sciences (Hercules, CA), and real-time quantitative PCR using iTaq Universal SYBR-Green Supermix from the same company. RT2 qPCR Primer Assays and primers for all our real-time quantitative PCR assays were purchased from Qiagen. The AriaMx Real-Time PCR system from Agilent Technologies (Santa Clara, CA) was used for running real-time quantitative PCR. The data were analyzed as previously described.24
Assessment of Liver Histology, Biliary Hyperplasia, and Liver Fibrosis in Mdr2KO and FVBN Mice
Hematoxylin and eosin staining was performed on sections (4 μm thick) of paraffin-embedded livers, as previously described.25 Biliary hyperplasia was assessed by measuring the intrahepatic biliary duct mass by IHC for CK19. Hepatic fibrosis markers, such as α-SMA and desmin, were assayed by IHC of liver tissue from mice treated with leptin or Leptin-Ab. Thus, for IHC, liver tissue sections (4 μm thick) were immunolabeled with primary antibodies specific to proteins of interest, and then processed for staining with VectaStain kits. The IHC slides were scanned with a Leica Aperio AT2 scanner, followed by screenshots and image analysis with using ImageJ version 1.52a software from the NIH (Bethesda, MD; http://imageJ.nih.gov/ij, last accessed October 20, 2021). The percentage areas of stained pixels were calculated and compared for significant differences. Liver samples were also assayed by using Sirius Red–specific staining of collagens I and III, which are increased in hepatic fibrosis, with the kit from IHC World (Ellicott City, MD).
Assessment of Leptin and CCL2 Concentrations Using ELISA Assays
Leptin and CCL2 were assessed in serum and/or liver samples of Mdr2KO and FVBN mice by using ELISA kits purchased from Abcam (Cambridge, MA) and R&D Systems (Minneapolis, MN), respectively. We followed the protocols indicated by the manufacturers.
Assessment of Leptin and LepR Expression in Different Types of Liver Cells by IHC and Confocal Microscopy
The expression of leptin and LepR in cholangiocytes, hepatic stellate cells (HSCs), and hepatocytes was determined by IHC using VectaStain kits, according to manufacturer's instructions. The staining was observed in different types of liver cells, based on specific shapes of the cells. The colocalization of LepR with specific markers of hepatic cells (ie, CK7 for cholangiocytes, desmin for HSCs, and CK8 for hepatocytes) was achieved by confocal microscopy. Frozen liver tissue embedded in OCT medium was sectioned at 8 μm using Leica Cryostat CM1850 (Leica Biosystems, Buffalo Grove, IL). The sections were labeled with LepR- and cell type–specific marker antibodies from Abcam, followed by secondary antibodies conjugated to Alexa Fluor 488 or Cy3, purchased from Invitrogen (Waltham, MA). The colocalization of LepR with cell markers was observed with confocal laser capture microscopy from Leica Microsystems Inc. (Buffalo Grove, IL), followed by quantitative PCR.
Assessment of Signaling Pathways Initiated by Leptin in Cholangiocytes and HSCs in Culture
In vitro experiments were run with mouse pooled cholangiocytes (a kind gift from Dr. Yoshiyuki Ueno, Yamagata University, Yamagata, Japan)26 and human LX-2 cells purchased from ATCC (Manassas, VA). The cells were grown according to the instructions from ATCC. In LepR knockdown experiments, the expression of LepR mRNA was silenced in cholangiocytes by transfecting the cells with mouse LepR-specific siRNA from OriGene, using Lipofectamine 2000 Reagent from Thermo Fisher Scientific (Waltham, MA), according to manufacturer's instructions. Cholangiocytes or LX-2 cells were treated with leptin, WMN, or FPA124 for 24 hours, followed by cell harvesting in lysis buffer for either real-time quantitative PCR or ELISA assays. To assess the effect of cholangiocyte-conditioned medium on LX-2 activation, cholangiocytes were treated with leptin, in the absence or presence of WMN or FPA124 for 24 hours, and the conditioned media from cholangiocytes were then used to treat LX-2 for subsequent 24 hours, followed by harvesting the cells for specific assays. Cell proliferation was tested using MTS kit from Abcam. Activation of LX-2 cells was tested by measuring changes in α-SMA and Col1A1 expression by real-time quantitative PCR. For IF, cholangiocytes and LX-2 cells grown on coverslips inside 6-well plates were fixed with 4% paraformaldehyde and blocked with 4% bovine serum albumin in phosphate-buffered saline supplemented with Tween 20, followed by incubation with 2 to 5 μg/mL primary antibody overnight at 4°C, and subsequent labeling with fluorescent dye–conjugated secondary antibody incubation for 1 hour at room temperature. After washings, coverslips were mounted in Prolong Gold Antifade Mounting media with DAPI from Thermo Fisher Scientific. Phosphorylation of Akt in response to leptin treatment was measured in cells in culture as well as in livers of FVBN and Mdr2KO mice, using FastScan Phospho-Akt (Ser473) ELISA kit from Cell Signaling Technology (Danvers, MA). p-Akt was also assessed by IF and confocal microscopy of cells in vitro and frozen sections of livers from FVBN and Mdr2KO mice treated with vehicle or leptin.
To test the expression of LepR in activated compared with quiescent HSC cells, LX-2 cells were treated with 1 ng/mL recombinant TGF-β1 or vehicle for 24 hours, followed by the assessment of LepR at mRNA level by real-time quantitative PCR and at protein level by IF and confocal microscopy.
Assessment of LepR Expression in Specific Cell Types of the Liver by Laser Capture Microdissection
Frozen sections (8 μm thick) of livers from FVBN and Mdr2KO mice were processed for IF labeling for CK8, CK7, and desmin, followed by laser capture microdissection and collection of hepatocytes, cholangiocytes, and HSCs, respectively. The cells were then subjected to RNA isolation using Arcturus PicoPure Frozen RNA isolation kit from Thermo Fisher Scientific. The expression of LepR versus glyceraldehyde-3-phosphate dehydrogenase was further determined by real-time quantitative PCR.
Statistical Analysis
Quantifications of real-time quantitative PCR, ELISA, and image analyses were used to calculate the average and SEM of three replicates for each group of tested animals. The number of animals (N) used for each treatment or as controls was 6, and is specified in Results for each experiment. The statistical difference was calculated between two groups by using the t-test, and was considered significant when P < 0.05. When multiple groups of animals were compared, two-way analysis of variance, followed by an appropriate post hoc test with GraphPad Prism 9.2.0 software (San Diego, CA), was used.
Results
Leptin Levels in Serum and Liver Are Elevated in Mdr2KO Mice Compared with those in FVBN Controls
The expression of leptin and LepR was assessed in FVBN and Mdr2KO mice treated with vehicle. Circulating leptin was significantly elevated in Mdr2KO mice compared with FVBN controls treated with vehicle (Figure 1A). Further assessment of serum leptin in mice treated with leptin or Leptin-Ab confirmed a significant increase in leptin levels in Mdr2KO mice that received leptin and a significant reduction in circulating leptin in Mdr2KO mice that received Leptin-Ab (Figure 1A).
Figure 1.

Leptin and leptin receptor (LepR) are up-regulated in multidrug resistance protein 2 knockout (Mdr2KO) mice. A: Serum leptin levels in FVB/NJ (FVBN) and Mdr2KO mice when treated with vehicle, leptin, or a leptin-neutralizing antibody (Leptin-Ab). Leptin in serum from male and female FVBN and Mdr2KO mice was assayed using enzyme-linked immunosorbent assay kit. B: Leptin mRNA was assessed using real-time quantitative PCR test of liver from Mdr2KO and FVBN mice. C: Leptin immunohistochemistry (IHC), representative images. D: Quantification of liver leptin by image analysis. E: LepR mRNA expression as tested by real-time quantitative PCR. F: Representative images of LepR IHC in liver sections from FVBN and Mdr2KO mice. G: Quantification of liver LepR in FVBN and Mdr2KO mice, by image analysis. H: Laser capture microdissection (LCM) data from liver samples of Mdr2KO and FVBN mice treated with vehicle. Frozen sections of liver were processed for LCM, as described under Materials and Methods, so that specific types of cells [ie, hepatocytes (Hs), cholangiocytes (Cs), and hepatic stellate cells (HSCs)] were obtained. Real-time quantitative PCR was then performed for LepR. Arrows point to hepatocytes, cholangiocytes, or stellate cells (Ss). N = 6 (A, B, D, E, G, and H). ∗P < 0.05 for Mdr2KO versus FVBN mice; †P < 0.05 for leptin or Leptin-Ab versus vehicle. Scale bars = 100 μm (C and F). Original magnifications, ×10 (C and F, top panels); ×20 (C and F, bottom panels).
To test whether leptin could be produced in the liver, real-time quantitative PCR for leptin was performed. It not only demonstrated that leptin mRNA was detected in the livers of FVBN and Mdr2KO mice, but also that leptin mRNA was increased up to threefold in Mdr2KO mice compared with that in FVBN mice (Figure 1B). Immunostaining of leptin in liver sections indicated that leptin in Mdr2KO mice was increased compared with FVBN controls (Figure 1, C and D). LepR was expressed in livers of all tested mice, being significantly more abundant in Mdr2KO than in control mice at mRNA (Figure 1E) and protein levels (Figure 1, F and G). Interestingly, in contrast to a solid, uniform, and low-level distribution of leptin and LepR within all cell types of the liver in FVBN mice, in Mdr2KO mice, both leptin and LepR were concentrated in certain cells that, based on their morphology, were identified as cholangiocytes, HSCs, and hepatocytes (Figure 1, C and F). Further analysis using laser capture microdissection confirmed that LepR mRNA was expressed in these types of hepatic cells, being more abundant in hepatocytes, cholangiocytes, and HSCs obtained from livers of Mdr2KO mice compared with cells from FVBN control mice (Figure 1H).
Colocalization of LepR with markers of hepatocytes, cholangiocytes, and HSCs was also tested by confocal microscopy of liver tissue from Mdr2KO and FVBN mice (Figure 2). LepR colocalized with the hepatocyte marker CK8, the cholangiocyte marker CK7, and the HSC marker desmin. LepR expression was increased in Mdr2KO mice compared with FVBN controls.
Figure 2.

Colocalization of leptin receptor (LepR) with markers of hepatocytes, cholangiocytes, and hepatic stellate cells (HSCs) in livers of multidrug resistance protein 2 knockout (Mdr2KO) mice compared with FVB/NJ (FVBN) control mice. Expression of LepR specifically in hepatocytes, cholangiocytes, and HSCs was investigated by dual-fluorescence immunolabeling of liver sections with LepR antibody and an additional antibody against cytokeratin (CK) 8 marker of hepatocytes (A), CK7 marker of cholangiocytes (B), or desmin marker of HSCs (C). Red is the code color for LepR, and green is the code color for the cell markers. The yellow arrowheads point to areas of colocalization. Scale bars = 75 μm (A–C).
Leptin Exacerbates Liver Histopathology and Biliary Hyperplasia in Cholestatic Mice
The hepatic transaminases ALT and AST were elevated in male and female Mdr2KO mice compared with those in the FVBN counterparts (Figure 3, A and B). Upon leptin administration, ALT and AST levels were significantly increased in Mdr2KO and in some of the FVBN mice compared with vehicle-treated mice (Figure 3, A and B).
Figure 3.

Exogenous leptin increases circulating transaminases, biliary hyperplasia, and liver cell proliferation in multidrug resistance protein 2 knockout (Mdr2KO) mice. A and B: Alanine aminotransferase (ALT; A) and aspartate aminotransferase (AST; B) transaminases were assessed in serum from male and female Mdr2KO and FVB/NJ (FVBN) control mice treated with vehicle or leptin, as described under Materials and Methods. C: Representative images of hematoxylin and eosin–stained liver sections of male and female Mdr2KO and FVBN mice treated with vehicle or leptin. D and E: Expression of cytokeratin (CK) 19 (D) and proliferating cell nuclear antigen (PCNA; E) mRNA in mice treated with leptin versus vehicle was assessed. F and G: Images of immunohistochemical (IHC) staining for CK19 (F) were quantified by image analysis (G), as described in Materials and Methods. F: Arrows point to cholangiocytes (Cs). H and I: Images of PCNA IHC (H) were also quantified (I). H: Arrows point to hepatocytes (Hs), cholangiocytes, or stellate cells (Ss). N = 6 (A, B, D, E, G, and I). ∗P < 0.05 for leptin versus vehicle; †P < 0.05 for Mdr2KO versus FVBN. Scale bars = 100 μm (C, F, and H).
Liver samples from mice administered recombinant leptin or vehicle were analyzed with regard to histopathology and expression of biomarkers for bile ducts. The hematoxylin and eosin staining suggested that leptin increased the number of bile ducts and level of fibrosis in male and female Mdr2KO mice (Figure 3C). The mRNA expression of CK19 and PCNA was increased in leptin-treated Mdr2KO mice compared with that in counterparts treated with vehicle (Figure 3, D and E). PCNA mRNA was enhanced more than CK19 mRNA in Mdr2KO mice. Interestingly, PCNA mRNA, unlike CK19 mRNA, was increased also in FVBN mice when treated with leptin, suggesting that other cell types in addition to cholangiocytes, such as HSCs and/or macrophages, could be stimulated to proliferate (Figure 3E). At protein level, CK19, as assessed by IHC, was increased by leptin treatment in Mdr2KO mice (Figure 3, F and G). PCNA IHC revealed a robust increase of liver cell proliferation in Mdr2KO mice treated with leptin compared with that in vehicle-treated Mdr2KO mice (Figure 3, H and I). Moreover, an increase in PCNA was found also in leptin-treated FVBN mice compared with vehicle-treated FVBN mice. In addition to high-magnification images of PCNA IHC in Figure 3H, images at lower magnification are presented in Supplemental Figure S1, showing large areas of liver tissue with PCNA-expressing cells.
Leptin Aggravates Hepatic Fibrosis and Inflammation in Cholestatic Mice
The effects of leptin on biomarkers of liver fibrosis were measured in Mdr2KO and FVBN mice. At mRNA level, biomarkers, including desmin, α-SMA, Col1A1, matrix metalloproteinase 2, tissue inhibitor of metalloproteinase 1, and fibrogenic gene TGF-β1, were highly increased on leptin administration in Mdr2KO mice compared with those in vehicle-treated Mdr2KO counterparts (Figure 4). The up-regulation of Col1A1, matrix metalloproteinase 2, and tissue inhibitor of metalloproteinase 1 was remarkable (ie, 20- to 30-fold higher in Mdr2KO mice with leptin versus 2- to 4-fold enhancement in Mdr2KO mice with vehicle). Most of the fibrosis biomarkers were up-regulated in FVBN mice when treated with leptin. These data suggest that leptin strongly stimulates up-regulation of genes involved in fibrogenesis in cholestatic and control mice.
Figure 4.

Exogenous leptin increases mRNA expression of gene markers of fibrosis in multidrug resistance protein 2 knockout (Mdr2KO) mice. mRNA expression of a series of genes was assessed in livers of male and female Mdr2KO and FVB/NJ (FVBN) mice that had been treated with leptin or vehicle. Gene markers of fibrosis included desmin (A), α-smooth muscle actin (α-SMA; B), collagen type 1A1 (Col1A1; C), matrix metalloproteinase 2 (MMP2; D), tissue inhibitor of metalloproteinase 1 (TIMP1; E), and transforming growth factor (TGF)-β1 (F). N = 6 (A–F). ∗P < 0.05 for leptin versus vehicle; †P < 0.05 for Mdr2KO versus FVBN mice.
The expression of desmin, α-SMA, and collagens type I and III was quantified at the protein level using IHC and Sirius Red staining (Figure 5). All these fibrosis biomarkers were increased in Mdr2KO and FVBN mice treated with leptin compared with vehicle, except for desmin, which was up-regulated in Mdr2KO groups only.
Figure 5.

Leptin enhances fibrosis markers at protein level in multidrug resistance protein 2 knockout (Mdr2KO) mice. A–F: Immunohistochemical (IHC) images of liver fibrosis markers and the results of their quantification by image analysis are shown for desmin (A and B), α-smooth muscle actin (α-SMA; C and D), and Sirius Red staining for collagen type I and III (E and F), in liver sections from Mdr2KO and FVB/NJ (FVBN) mice treated with vehicle or leptin. A and C: Arrows point to desmin and α-SMA, respectively. N = 6 (B, D, and F). ∗P < 0.05 for leptin versus vehicle; †P < 0.05 for Mdr2KO versus FVBN mice. Scale bars = 100 μm (A, C, and E).
The expression of inflammation genes known to have roles in hepatic fibrogenesis was tested (Figure 6, A–C). Leptin induced up-regulation of IL-1β, IL-6, and CCL2 mRNA in Mdr2KO but not in FVBN mice, with CCL2 being the most up-regulated, followed by IL-6, whereas IL-1β was less affected. Moreover, serum CCL2 was increased by leptin treatment in Mdr2KO mice (Figure 6D). Assessment of CD11b, a marker of monocyte-derived macrophages that are recruited from blood circulation into the liver in response to increased chemotactic cytokines, such as CCL2, demonstrated enhanced accumulation of this type of macrophages in liver tissue of leptin-treated Mdr2KO mice compared with vehicle-treated counterparts (Figure 6, E and F).
Figure 6.

Leptin increases inflammation of the liver in multidrug resistance protein 2 knockout (Mdr2KO) mice. A–C: Gene expression of proinflammatory cytokines IL-1β (A), IL-6 (B), and C-C motif chemokine ligand 2 (CCL2; C) was assessed in liver samples from Mdr2KO and FVB/NJ (FVBN) mice treated with vehicle or leptin. D: CCL2 level was assayed in serum samples of mice treated with vehicle or leptin. E: The amount of CD11b+ macrophages was quantified by image analysis of images obtained by immunohistochemistry (IHC) for CD11b in liver samples from Mdr2KO and FVBN mice treated with leptin versus vehicle. F: Representative images of IHC for CD11b. F: Arrows point to CD11b+ cells. N = 6 (A–E). ∗P < 0.05 for leptin versus vehicle; †P < 0.05 for Mdr2KO versus FVBN mice. Scale bars = 100 μm (F).
Leptin-Ab Attenuates Serum Biochemistry and Biliary Hyperplasia in Mdr2KO Mice
An alternative strategy to assess the influence of leptin on liver ductular reaction, hepatic fibrosis, and inflammation was to treat FVBN and Mdr2KO mice with Leptin-Ab and measure serum transaminases, liver histopathology, intrahepatic biliary duct mass, and PCNA (Figure 7). The levels of ALT and AST were significantly reduced in Mdr2KO mice (Figure 7, A and B). The histology of livers was improved by Leptin-Ab in both male and female Mdr2KO mice (Figure 7C). The intrahepatic biliary duct mass was significantly lowered by Leptin-Ab treatment of Mdr2KO mice, as demonstrated by CK19 expression at mRNA and protein levels (Figure 7, D–F). A robust effect of Leptin-Ab was found on PCNA, in livers of Mdr2KO mice, compared with Mdr2KO mice treated with vehicle only (Figure 7, G–I), suggesting a lower level of cell proliferation as a result of Leptin-Ab treatment. The reduction of cells positively stained for PCNA by IHC in livers of Mdr2KO mice as a result of treatment with Leptin-Ab can be seen at high magnification (Figure 7H), and at low magnification for the assessment of larger areas of liver tissue (Supplemental Figure S2).
Figure 7.

Leptin-neutralizing antibody (Leptin-Ab) attenuates serum transaminases and biliary hyperplasia in multidrug resistance protein 2 knockout (Mdr2KO) mice. A–C: Serum transaminases alanine aminotransferase (ALT; A) and aspartate aminotransferase (AST; B) and hematoxylin and eosin staining (C) of liver sections were assessed for Mdr2KO and FVB/NJ (FVBN) mice treated with vehicle or Leptin-Ab. D–F: Hepatic expression of cytokeratin (CK) 19 mRNA (D) and protein (images in E, quantifications in F) was assessed. E: All arrows point to cholangiocytes (Cs). G–I: Cell proliferation marker, proliferating cell nuclear antigen (PCNA), was assessed at mRNA (G) and protein level, and immunohistochemical (IHC) images (H) and quantification (I) are shown. H: Arrows point to different types of cells expressing PCNA. N = 6 (A, B, D, F, G, and I). ∗P < 0.05 for leptin versus vehicle; †P < 0.05 for Mdr2KO versus FVBN mice. Scale bars = 100 μm (C, E, and H). H, hepatocyte; S, hepatic stellate cell.
Leptin-Ab Attenuates Liver LepR Expression, Fibrosis, and Inflammation in Mdr2KO Mice
The influence of Leptin-Ab treatment on the expression level of LepR in Mdr2KO mice was assessed using IHC (Figure 8A). Leptin-Ab significantly reduced LepR in Mdr2KO mice, suggesting a leptin-dependent regulation of LepR expression in the liver.
Figure 8.

Leptin-neutralizing antibody (Leptin-Ab) reduces liver fibrosis in multidrug resistance protein 2 knockout (Mdr2KO) mice. A: Immunohistochemistry (IHC) of leptin receptor (LepR) in livers of Mdr2KO and FVB/NJ (FVBN) mice treated with vehicle or Leptin-Ab. Desmin, α-smooth muscle actin (α-SMA), and collagen were assessed to measure hepatic fibrosis in male and female Mdr2KO and FVBN mice treated with vehicle or Leptin-Ab. A: Arrows point to cells expressing LepR. B–D: Desmin: mRNA (B), IHC images (C), and quantifications from image analysis (D). E–G: α-SMA: mRNA (E), IHC images (F), and quantification (G). C and F: Arrows indicate stellate cells expressing desmin and α-SMA, respectively. H–J: Collagens: collagen type 1A1 (Col1A1) mRNA (H), Sirius Red staining images (I), and quantification (J). N = 6 (B, D, E, G, H, and J). ∗P < 0.05 for leptin-Ab versus vehicle; †P < 0.05 for Mdr2KO versus FVBN mice. Scale bars = 100 μm (A, C, F, and I). C, cholangiocyte; H, hepatocyte; S, hepatic stellate cell.
Analysis of desmin expression in liver samples from Mdr2KO and FVBN mice treated with Leptin-Ab versus vehicle indicated a strong decrease in Mdr2KO mice, whereas no effects were seen in FVBN mice (Figure 8, B–D). α-SMA expression at the mRNA and protein levels was also diminished by the Leptin-Ab treatment compared with vehicle-treated Mdr2KO mice (Figure 8, E–G). Col1A1 mRNA was reduced by Leptin-Ab treatment in Mdr2KO mice (Figure 8H). Sirius Red staining of collagen in liver sections was significantly decreased in Leptin-Ab–treated Mdr2KO mice compared with vehicle-treated Mdr2KO counterparts (Figure 8, I and J).
The fibrogenic gene TGF-β1 as well as genes involved in cholestasis-related inflammation, such as IL-1β, IL-6, and CCL2, were tested for mRNA expression and were found to be significantly reduced on treatment with Leptin-Ab in Mdr2KO mice (Figure 9, A–D). IL-1β and IL-6, which were increased in Mdr2KO mice compared with controls, were down-regulated by Leptin-Ab to the level of expression in FVBN control mice (Figure 9, B and C). However, CCL2, which had high expression in the liver of Mdr2KO mice of approximately 20-fold in excess of normal level, was significantly reduced by Leptin-Ab but did not reach the levels of FVBN mice (Figure 9D). Serum CCL2 was also reduced in Mdr2KO mice treated with Leptin-Ab compared with vehicle (Figure 9E). This was associated with a significant reduction in CD11b+ macrophages as a result of treatment of Mdr2KO mice with Leptin-Ab (Figure 9, F and G).
Figure 9.

Leptin-neutralizing antibody (Leptin-Ab) reduces fibrogenic and proinflammatory genes in multidrug resistance protein 2 knockout (Mdr2KO) mice. A–D: The mRNA expression of transforming growth factor (TGF)-β1 fibrogenic gene (A) and of IL-1β (B), IL-6 (C), and C-C motif chemokine ligand 2 (CCL2) proinflammatory genes (D) was assessed in male and female Mdr2KO and FVB/NJ (FVBN) mice. E: CCL2 was assessed in serum of Mdr2KO and FVBN mice treated with vehicle or Leptin-Ab. F: Representative images of immunohistochemistry (IHC) for CD11b+ macrophages in liver sections. G: Image analysis results of CD11b IHC in liver samples. Arrows in IHC images point to cells expressing the protein of interest. N = 6 (A–E and G). ∗P < 0.05 for leptin-Ab versus vehicle; †P < 0.05 for Mdr2KO versus FVBN mice. Scale bars = 100 μm (F).
In summary, the treatment of Mdr2KO mice with Leptin-Ab resulted in considerable attenuation of liver fibrosis and inflammation markers while having no negative effects on FVBN mice.
Leptin Stimulates Proliferation and Activation of Cholangiocytes and HSCs in Vitro via LepR, phosphoinositide 3 (PI3) Kinase, and Akt Phosphorylation
Mouse cholangiocytes were used in vitro to study possible signaling pathways underlying the proliferative effects of leptin on bile ducts in Mdr2KO mice in vivo. Initial experiments checked whether LepR was expressed in our mouse cholangiocyte cell line and the receptor was detected by IF and confocal microscopy (Figure 10A). On treatment of cholangiocytes with leptin in the absence or presence of PI3 kinase inhibitor, WMN, and Akt phosphorylation inhibitor, FPA124, the cells displayed increased proliferation with leptin only in the absence of inhibitors (Figure 10B). An ELISA test of p-Akt in cholangiocytes over time indicated that Akt phosphorylation increased within the first 30 minutes after leptin treatment (Figure 10C). This was confirmed by immunostaining of cholangiocytes for p-Akt at 30 minutes after leptin treatment in the absence of inhibitors (Figure 10D).
Figure 10.

Leptin increases proliferation of cholangiocytes (Chols) via Akt phosphorylation. A: Leptin receptor (LepR) was detected in mouse cholangiocytes using immunofluorescence. B: Changes in cell proliferation rate were measured using MTS kit on cholangiocytes treated with vehicle (Veh) or leptin in the absence or presence of phosphoinositide 3 (PI3) kinase inhibitor wortmannin (WMN) or Akt phosphorylation inhibitor FPA124 (FPA). C: Akt phosphorylation time course was determined using enzyme-linked immunosorbent assay. D: Phosphorylated Akt (p-Akt) at 30 minutes after leptin versus vehicle treatment of cholangiocytes was detected by immunolabeling and confocal microscopy. E: LepR was silenced using three different sequences of siRNAs [sequence 1 (Seq1), sequence 2 (Seq2), and sequence 3 (Seq3) versus negative control seq (Neg Ctr)], followed by real-time quantitative PCR assay of genes of interest, including LepR, to confirm the knockdown, leptin, proliferating cell nuclear antigen (PCNA; marker of cell proliferation), cytokeratin (CK) 19 (housekeeping gene of cholangiocytes), and IL-1β and C-C motif chemokine ligand 2 (CCL2) cytokines. F: LX-2 cells were assessed for proliferation after being treated with conditioned medium from cholangiocytes in which LepR was silenced with siRNA or negative control siRNA (NCsiRNA), followed by treatment with vehicle or leptin. N = 3 (B, C, E, and F). ∗P < 0.05 for vehicle versus any other treatment; †P < 0.05 for time 0 versus any other time point; ‡P < 0.05 for Neg Ctr versus Seq1, Seq2, and Seq3; §P < 0.05 for vehicle versus any other treatment. Scale bars: 100 μm (A); 50 μm (D). GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
The expression of LepR in cholangiocytes was silenced using siRNA, and the expression of PCNA, IL-1β, and CCL2 was assessed. LepR knockdown resulted in significant reduction in leptin mRNA without affecting the housekeeping gene CK19 (Figure 10E). The mRNA expression of IL-1β and CCL2 cytokines was also reduced following LepR knockdown.
The effect of cholangiocyte-conditioned culture medium on LX-2 stellate cell proliferation was studied with cholangiocytes grown in the absence or presence of leptin (Figure 10F). Thus, cholangiocytes with wild-type genotype expressing LepR, as well as cholangiocytes with knocked-down LepR, were treated with vehicle or leptin, and their culture media were used for stimulating LX-2 cells to grow. The medium from wild-type cholangiocytes increased proliferation of LX-2 cells, whereas medium conditioned from LepR-knockdown cholangiocytes did not stimulate LX-2 proliferation.
Cholangiocytes were shown to express LepR. This is essential in mediating an autocrine effect of leptin to maintain leptin expression in cholangiocytes along with cell proliferation and activation by up-regulation of proinflammatory cytokines IL-1β and CCL2 and an exocrine effect of leptin to induce proliferation of LX-2 cells.
LX-2 cells were also shown to express LepR (Figure 11A). The involvement of Akt phosphorylation in leptin-induced signaling has been demonstrated in LX-2 cells by a p-Akt time course assay, which indicated a twofold increase in p-Akt following leptin stimulation (Figure 11B). Akt phosphorylation was less intense (ie, twofold increase in p-Akt in LX-2 cells compared with fivefold in cholangiocytes), and short-lived, reaching a maximum at 15 minutes in LX-2 cells compared with 30 minutes in cholangiocytes (Figure 11B versus Figure 10C). The expression of several LX-2 cell fibrogenic genes, such as TGF-β1 and TGF-β2, as well as genes of HSC activation, including Col1A1 and α-SMA, were assessed in LX-2 cells treated with vehicle, leptin, or leptin plus PI3 kinase inhibitor WMN or Akt phosphorylation, FPA124 (Figure 11C). Leptin stimulated all tested genes but less so in the presence of WMN or FPA124. The expression of CCL2 and IL-6 in LX2 cells stimulated with leptin was also tested, demonstrating that leptin-induced expression of these genes was impaired by FPA124 but not WMN (Figure 11D). These results indicate that leptin has a large spectrum of effects on gene expression in HSCs and induces HSC activation. Some of these effects are mediated via PI3 kinase and p-Akt, whereas others are mediated via p-Akt and signaling molecules other than PI3 kinase.
Figure 11.

Leptin induces LX-2 cell proliferation and activation via Akt phosphorylation. A: Leptin receptor (LepR) was detected in LX-2 cells by immunostaining and confocal microscopy. B: Phosphorylated Akt (p-Akt) quantification assayed by enzyme-linked immunosorbent assay (ELISA) in LX-2 cells treated with leptin versus vehicle over a time course up to 2 hours. C: mRNA expression of genes associated with fibrosis in LX-2 cells treated with vehicle or leptin in the absence or presence of phosphoinositide 3 (PI3) kinase inhibitor wortmannin (WMN) or Akt phosphorylation inhibitor FPA124 (FPA). D: Same as in C, but mRNA was assayed for inflammatory genes C-C motif chemokine ligand 2 (CCL2) and IL-6. E: Cell proliferation assay of LX-2 cells treated with vehicle or leptin in the absence or presence of WMN and FPA124. N = 3 (B–E). ∗P < 0.05 for time 0 versus any other time point; †P < 0.05 for leptin versus vehicle; ‡P < 0.05 for leptin plus WMN versus vehicle; §P < 0.05 for leptin plus FPA124 versus vehicle; ¶P < 0.05 for vehicle versus any other treatment. Scale bars = 100 μm (A). Col1A1, collagen type 1A1; α-SMA, α-smooth muscle actin; FN1, fibronectin 1; TGF-β1, transforming growth factor-β1.
The effect of leptin on LX-2 cell proliferation was also tested (Figure 11E). Leptin increased cell proliferation only in the absence of PI3 kinase and p-Akt inhibitors, suggesting that LepR in LX-2 cells triggers proliferation via the same pathways as in cholangiocytes.
Phosphorylation of Akt due to activation of PI3 kinase in both cholangiocytes and LX-2 cells was confirmed by quantification of p-Akt using ELISA in cells in vitro, treated with leptin in the absence or presence of WMN (Supplemental Figure S3). p-Akt was also assessed in liver tissue from Mdr2KO and FVBN mice that had been treated with vehicle or leptin (Supplemental Figure S4), and the data from ELISA and IF assays indicated a significant increase of p-Akt in leptin-treated mice compared with vehicle counterparts.
To explore the expression of LepR in activated versus quiescent HSCs, LepR mRNA and protein were assesed in LX-2 cells when treated with TGF-β1 compared with vehicle (Supplemental Figure S5). Both real-time quantitative PCR and IF data demonstrated that LepR was expressed in LX-2 cells treated with TGF-β1 or vehicle. Activation of HSCs did not cause changes in LepR expression while significantly up-regulating α-SMA.
Discussion
The present study investigated the role of leptin, a known proinflammatory adipokine, on hepatic inflammation, fibrosis, and biliary hyperplasia in the Mdr2KO mouse model, based on publications reporting on the association of leptin with conditions of chronic inflammation of the liver,27 hepatic fibrosis,28 and cell proliferation in liver cancer.29
The study also investigated the role of leptin in biliary hyperplasia and hepatic fibrosis in a rodent model of chronic cholestasis. The data on the effects of leptin in cholestasis-induced liver injury in Mdr2KO mice suggest that excess leptin stimulated the expression of fibrogenic genes, such as TGF-β1, and resulted in increased HSC number and activation as well as extracellular matrix proteins, thus aggravating liver fibrogenesis. These data are consistent with reports suggesting a possible role for leptin in epithelial-mesenchymal transition in cancer,30 and reports from clinical research studies in which serum leptin was elevated in patients with advanced hepatic fibrosis. Thus, studies on circulating leptin levels and the stage of hepatic fibrosis due to chronic hepatitis B and C concluded that increased leptin was a negative prognostic factor in both conditions.31,32 In the current experiments, when using the alternative strategy of reducing the systemic leptin level by using Leptin-Ab in Mdr2KO mice, the liver fibrosis markers were significantly attenuated, indicating again that leptin has functions in stimulating and maintaining fibrogenesis in cholestasis-induced liver injury. Interestingly, in search for drugs to alleviate liver fibrosis, it has been discovered that peroxisome proliferator-activated receptor-α/γ agonists are effective through regulating leptin.33 Such studies suggest that the antifibrotic effect is explained by the suppression of leptin in the liver, followed by down-regulation of fibrogenic genes, including TGF-β and platelet-derived growth factor-β.33
Several recent publications reported that, in liver diseases, HSCs express LepR and are influenced by leptin via miRNAs34,35 or by altering DNA methylation.36 The current experiments noted a large increase in HSC markers in Mdr2KO mice on leptin treatment, and an opposite effect when Leptin-Ab was used. LX-2 cells in vitro were used to demonstrate that leptin induced cell proliferation and activation via PI3 kinase and Akt phosphorylation. The results of the current study are thus consistent with other published reports, indicating that leptin via LepR is able to affect various cellular processes conducive to HSC proliferation and activation.
The influence of excessive leptin on hepatic inflammatory markers in Mdr2KO mice addressed in this study suggested that exogenous leptin significantly increased the expression of proinflammatory cytokines characteristic for cholestasis (ie, IL-1β, IL-6, and CCL2). Contrary to leptin treatment, Leptin-Ab administered to Mdr2KO mice caused a remarkable reduction in these proinflammatory cytokines. The findings are in line with data from clinical studies in patients experiencing liver inflammation attributable to various causes. For example, it was reported that serum leptin could be used as a biomarker for inflammatory activity in autoimmune hepatitis to monitor the response to steroid treatments.37,38
Numerous publications point to a possible association of leptin with cell proliferation, especially in various types of cancer, including liver malignancy.39, 40, 41 There is a higher frequency of cancers in obese compared with normal weight population.42, 43, 44 In particular, clinical studies on breast cancer found a positive correlation between obesity and elevated serum leptin with this type of cancer.45,46 Even though there are some inconsistencies and contradictory results regarding liver-related cancers such as hepatocellular carcinoma, it is generally agreed that leptin has a role in the development of hepatocellular carcinoma.40,47,48 Because leptin is elevated in hepatic cholestasis,49 and there is extensive ductular reaction and cholangiocyte proliferation associated with cholestatic injuries in patients as well as in animal models, the effects of leptin or Leptin-Ab were investigated on cholangiocyte and HSC proliferation. The in vivo data demonstrate that elevated leptin had a stimulatory effect on intrahepatic biliary duct mass growth as well as desmin and α-SMA–expressing HSCs in Mdr2KO mice, whereas Leptin-Ab inhibited proliferation of these cells. Furthermore, in vitro experiments with cholangiocytes and LX-2 cells indicated that leptin acted via its LepR and induced cell proliferation by signaling through PI3 kinase and Akt phosphorylation. In in vitro experiments, mouse cholangiocytes expressed leptin and LepR mRNAs. Moreover, knockdown of LepR reduced the expression of PCNA marker of cell proliferation.
Interestingly, the results on the effects of leptin in the liver pathology in Mdr2KO mice suggested sex-specific differences. Thus, serum leptin level was higher in female versus male Mdr2KO mice. In the liver, there was no difference in leptin concentration between the two sexes; however, LepR was expressed more in male than female Mdr2KO mice. Exogenous leptin increased ALT and AST levels in male but less so in female Mdr2KO mice. The regulation of proinflammatory cytokines by leptin was sex-specific, being altered differently in male versus female Mdr2KO mice. IL-1β was up-regulated in males but not females, whereas IL-6 and CCL2 were up-regulated in both male and female groups, and more in females compared with male Mdr2KO mice. Leptin had a stronger fibrogenic effect in female compared with male Mdr2KO mice. Leptin had a remarkable effect on HSCs in the in vivo experiments. In Mdr2KO mice, leptin induced up-regulation of desmin and α-SMA more in females than in males. These data suggest that even though female Mdr2KO mice exhibited more sensitivity to leptin in regard to cholangiocyte and HSC proliferation and activation, the hepatic functional injury, reflected by transaminase activity in the serum, was less when compared with male Mdr2KO mice. These results indicate that at least in this model, females tolerate the leptin effects better than males, having better protection mechanisms to prevent hepatocyte injury.
A recent publication on dysfunctions of leptin in cholelithiasis shows that leptin stimulates the production of total bile acids by more than twofold in L-02 cells, a human hepatocyte cell line that is used for studies of liver functions in vitro.50 These results are consistent with our data that demonstrate a negative effect of leptin on cholestasis-induced liver fibrosis in Mdr2KO mice.
In conclusion, the current data indicate that leptin stimulates ductular reaction and liver fibrosis in Mdr2KO mice as well as in cells in vitro.
Acknowledgments
We thank Dr. Yoshiyuki Ueno (Yamagata University, Yamagata, Japan) for providing the mouse pooled cholangiocytes used in this study.
Footnotes
Supported by NIH R01 awards DK082435 and DK112803 and a VA Merit awardBX002638 from the US Department of Veterans Affairs Biomedical Laboratory Research and Development Service (S.D.). This work was completed with support from the Veterans Health Administration and with resources and the use of facilities at the Central Texas Veterans Health Care System (Temple, TX).
Disclosures: None declared.
The contents of this manuscript do not represent the views of the US Department of Veterans Affairs or the US Government.
Supplemental material for this article can be found at http://doi.org/10.1016/j.ajpath.2021.11.008.
Author Contributions
A.D.P., S.G., E.W., S.Y.A., M.M., G.F., N.S., M.S., C.T., T.A., Y.N., L.W., M.T., C.S.C., and J.V. performed experiments and analyzed data; S.G. and E.W. performed animal experiments and collected tissue samples; S.D. formulated study; S.D. and A.D.P. wrote the manuscript; A.P., S.G., G.F., E.W., S.Y.A., M.M., G.F., N.S., M.S., C.T., T.A., Y.N., L.W., M.T., C.S.C., J.V., and S.D. critically edited and approved the final manuscript.
Supplemental Data
Supplemental Figure S1.
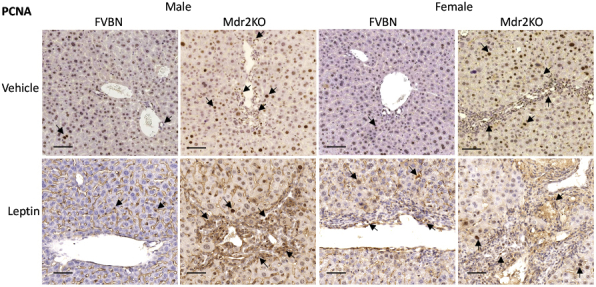
Low-magnification images of immunohistochemistry for proliferating cell nuclear antigen (PCNA) in liver samples from multidrug resistance protein 2 knockout (Mdr2KO) and FVB/NJ (FVBN) mice treated with vehicle or leptin. Arrows point to representative cells in which nuclei are stained for PCNA. Scale bars = 100 μm.
Supplemental Figure S2.
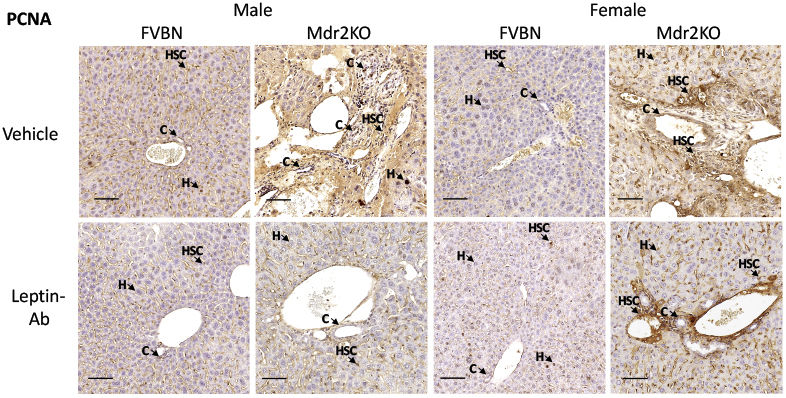
Low-magnification images of immunohistochemistry for proliferating cell nuclear antigen (PCNA) in liver samples from multidrug resistance protein 2 knockout (Mdr2KO) and FVB/NJ (FVBN) mice treated with vehicle and leptin-neutralizing antibody (Leptin-Ab). Arrows point to representative cells in which nuclei are stained for PCNA. Scale bars = 100 μm. C, cholangiocyte; H, hepatocyte; HSC, hepatic stellate cell.
PI3 kinase inhibitor wortmannin (WMN) prevents phosphorylation of Akt in cholangiocytes and LX-2 cells in vitro, when treated with leptin. Based on time-course results shown in Figures 10 and 11, phosphorylated Akt (p-Akt) was assayed by enzyme-linked immunosorbent assay at 30 minutes for cholangiocytes and at 15 minutes for LX-2 cells when treated with vehicle or leptin in the absence or presence of WMN. A: Results for mouse cholangiocytes. B: Results for LX-2 cells. N = 3 (A and B). ∗P < 0.05 versus vehicle; †P < 0.05 versus leptin.
Supplemental Figure S4.
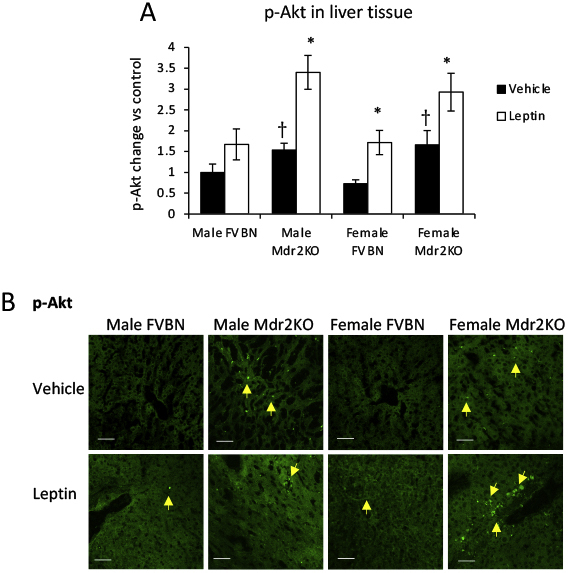
Akt phosphorylation is enhanced in livers from multidrug resistance protein 2 knockout (Mdr2KO) mice compared with FVB/NJ (FVBN) control mice. A: Enzyme-linked immunosorbent assay of phosphorylated Akt (p-Akt) in liver samples of male and female FVBN and Mdr2KO mice that had been treated with vehicle or leptin. B: Immunofluorescence labeling and confocal microscopy images of p-Akt in liver sections of FVBN and Mdr2KO mice treated with vehicle or leptin. Arrows in immunohistochemical images point to cells expressing the protein of interest. N = 6 (A). ∗P < 0.05 for leptin versus vehicle; †P < 0.05 for Mdr2KO versus FVBN mice. Scale bars = 100 μm (B).
Supplemental Figure S5.
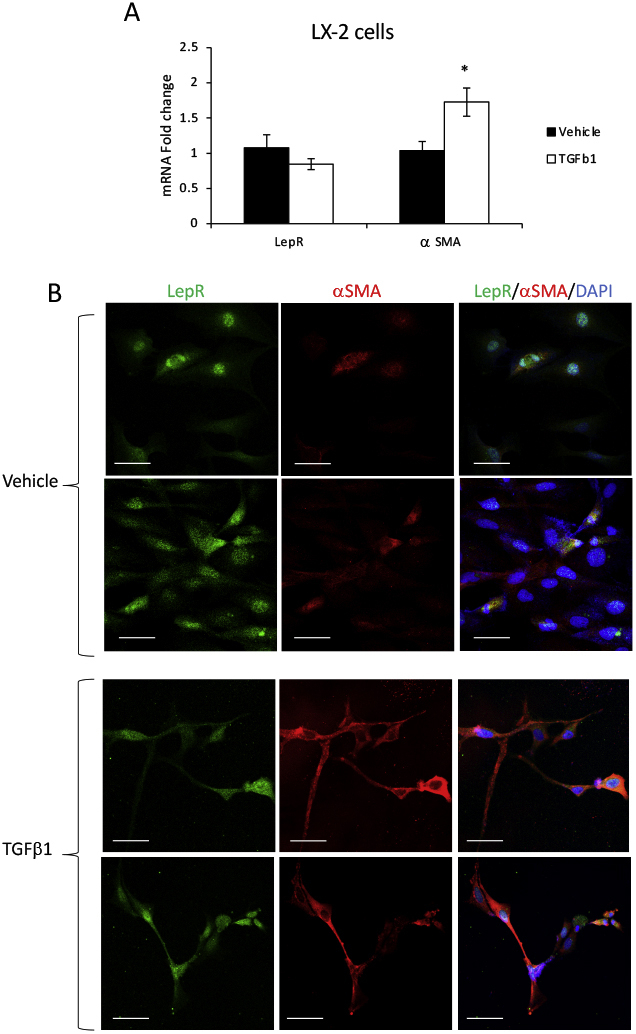
Leptin receptor (LepR) is equally expressed in quiescent and activated hepatic stellate cells (HSCs). LX-2 cells were treated with either vehicle or transforming growth factor (TGF)-β1, as described under Materials and Methods. α-Smooth muscle actin (α-SMA) was used as a marker of HSC activation, and LepR was assessed for expression at mRNA and protein levels. A: Real-time quantitative PCR results. B: Confocal microscopy images of α-SMA (red) and LepR (green) in LX-2 cells when quiescent (vehicle treatment) or activated (TGF-β1 treatment). N = 3 (A). ∗P < 0.05 for TGF-β1 versus vehicle. Scale bars = 50 μm (B).
References
- 1.Almabhouh F.A., Md Mokhtar A.H., Malik I.A., Aziz N., Durairajanayagam D., Singh H.J. Leptin and reproductive dysfunction in obese men. Andrologia. 2020;52:e13433. doi: 10.1111/and.13433. [DOI] [PubMed] [Google Scholar]
- 2.Huo L., Maeng L., Bjorbaek C., Grill H.J. Leptin and the control of food intake: neurons in the nucleus of the solitary tract are activated by both gastric distension and leptin. Endocrinology. 2007;148:2189–2197. doi: 10.1210/en.2006-1572. [DOI] [PubMed] [Google Scholar]
- 3.Boucsein A., Kamstra K., Tups A. Central signalling cross-talk between insulin and leptin in glucose and energy homeostasis. J Neuroendocrinol. 2021;33:e12944. doi: 10.1111/jne.12944. [DOI] [PubMed] [Google Scholar]
- 4.Gasmi A., Noor S., Menzel A., Dosa A., Pivina L., Bjorklund G. Obesity and insulin resistance: associations with chronic inflammation, genetic and epigenetic factors. Curr Med Chem. 2021;28:800–826. doi: 10.2174/0929867327666200824112056. [DOI] [PubMed] [Google Scholar]
- 5.Friedman J.M. Leptin, leptin receptors, and the control of body weight. Nutr Rev. 1998;56:s38–s46. doi: 10.1111/j.1753-4887.1998.tb01685.x. discussion s54-s75. [DOI] [PubMed] [Google Scholar]
- 6.Kiernan K., MacIver N.J. The role of the adipokine leptin in immune cell function in health and disease. Front Immunol. 2020;11:622468. doi: 10.3389/fimmu.2020.622468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.La Cava A., Matarese G. The weight of leptin in immunity. Nat Rev Immunol. 2004;4:371–379. doi: 10.1038/nri1350. [DOI] [PubMed] [Google Scholar]
- 8.Francisco V., Pino J., Campos-Cabaleiro V., Ruiz-Fernandez C., Mera A., Gonzalez-Gay M.A., Gomez R., Gualillo O. Obesity, fat mass and immune system: role for leptin. Front Physiol. 2018;9:640. doi: 10.3389/fphys.2018.00640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moran O., Phillip M. Leptin: obesity, diabetes and other peripheral effects--a review. Pediatr Diabetes. 2003;4:101–109. doi: 10.1034/j.1399-5448.2003.00017.x. [DOI] [PubMed] [Google Scholar]
- 10.Garofalo C., Surmacz E. Leptin and cancer. J Cell Physiol. 2006;207:12–22. doi: 10.1002/jcp.20472. [DOI] [PubMed] [Google Scholar]
- 11.Martinez-Una M., Lopez-Mancheno Y., Dieguez C., Fernandez-Rojo M.A., Novelle M.G. Unraveling the role of leptin in liver function and its relationship with liver diseases. Int J Mol Sci. 2020;21:9368. doi: 10.3390/ijms21249368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kosuta I., Mrzljak A., Kolaric B., Vucic Lovrencic M. Leptin as a key player in insulin resistance of liver cirrhosis? a cross-sectional study in liver transplant candidates. J Clin Med. 2020;9:560. doi: 10.3390/jcm9020560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Testa R., Franceschini R., Giannini E., Cataldi A., Botta F., Fasoli A., Tenerelli P., Rolandi E., Barreca T. Serum leptin levels in patients with viral chronic hepatitis or liver cirrhosis. J Hepatol. 2000;33:33–37. doi: 10.1016/s0168-8278(00)80156-7. [DOI] [PubMed] [Google Scholar]
- 14.Floreani A., Variola A., Niro G., Premoli A., Baldo V., Gambino R., Musso G., Cassader M., Bo S., Ferrara F., Caroli D., Rizzotto E.R., Durazzo M. Plasma adiponectin levels in primary biliary cirrhosis: a novel perspective for link between hypercholesterolemia and protection against atherosclerosis. Am J Gastroenterol. 2008;103:1959–1965. doi: 10.1111/j.1572-0241.2008.01888.x. [DOI] [PubMed] [Google Scholar]
- 15.Voumvouraki A., Koulentaki M., Notas G., Sfakianaki O., Kouroumalis E. Serum surrogate markers of liver fibrosis in primary biliary cirrhosis. Eur J Intern Med. 2011;22:77–83. doi: 10.1016/j.ejim.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 16.Szalay F., Folhoffer A., Horvath A., Csak T., Speer G., Nagy Z., Lakatos P., Horvath C., Habior A., Tornai I., Lakatos P.L. Serum leptin, soluble leptin receptor, free leptin index and bone mineral density in patients with primary biliary cirrhosis. Eur J Gastroenterol Hepatol. 2005;17:923–928. doi: 10.1097/00042737-200509000-00007. [DOI] [PubMed] [Google Scholar]
- 17.Ben-Ari Z., Schafer Z., Sulkes J., Manhaim V., Tur-Kaspa R., Fainaru M. Alterations in serum leptin in chronic liver disease. Dig Dis Sci. 2002;47:183–189. doi: 10.1023/a:1013248427783. [DOI] [PubMed] [Google Scholar]
- 18.Lee S., Kweon O.K., Kim W.H. Relationship of serum leptin concentration with pituitary-dependent hyperadrenocorticism and cholestatic disease in dogs. J Small Anim Pract. 2019;60:601–606. doi: 10.1111/jsap.13044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rioux K.P., Beck P.L., Hoppin A.G., Ezedi I., Kaplan L., Le T., Swain M.G. Differential leptin responses to acute and chronic biliary obstruction in rats. J Hepatol. 2000;33:19–25. doi: 10.1016/s0168-8278(00)80154-3. [DOI] [PubMed] [Google Scholar]
- 20.Breidert M., Zimmermann T.F., Schneider R., Ehninger G., Brabant G. Ghrelin/leptin-imbalance in patients with primary biliary cirrhosis. Exp Clin Endocrinol Diabetes. 2004;112:123–126. doi: 10.1055/s-2004-817819. [DOI] [PubMed] [Google Scholar]
- 21.Petrescu A.D., Grant S., Williams E., Frampton G., Reinhart E.H., Nguyen A., An S., McMillin M., DeMorrow S. Ghrelin reverses ductular reaction and hepatic fibrosis in a rodent model of cholestasis. Sci Rep. 2020;10:16024. doi: 10.1038/s41598-020-72681-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rafael J., Herling A.W. Leptin effect in ob/ob mice under thermoneutral conditions depends not necessarily on central satiation. Am J Physiol Regul Integr Comp Physiol. 2000;278:R790–R795. doi: 10.1152/ajpregu.2000.278.3.R790. [DOI] [PubMed] [Google Scholar]
- 23.Konstantinides S., Schafer K., Neels J.G., Dellas C., Loskutoff D.J. Inhibition of endogenous leptin protects mice from arterial and venous thrombosis. Arterioscler Thromb Vasc Biol. 2004;24:2196–2201. doi: 10.1161/01.ATV.0000146531.79402.9a. [DOI] [PubMed] [Google Scholar]
- 24.McMillin M., Frampton G., Grant S., DeMorrow S. The neuropeptide galanin is up-regulated during cholestasis and contributes to cholangiocyte proliferation. Am J Pathol. 2017;187:819–830. doi: 10.1016/j.ajpath.2016.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McMillin M., Grant S., Frampton G., Petrescu A.D., Williams E., Jefferson B., Thomas A., Brahmaroutu A., DeMorrow S. Elevated circulating TGFbeta1 during acute liver failure activates TGFbetaR2 on cortical neurons and exacerbates neuroinflammation and hepatic encephalopathy in mice. J Neuroinflammation. 2019;16:69. doi: 10.1186/s12974-019-1455-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frampton G., Ueno Y., Quinn M., McMillin M., Pae H.Y., Galindo C., Leyva-Illades D., DeMorrow S. The novel growth factor, progranulin, stimulates mouse cholangiocyte proliferation via sirtuin-1-mediated inactivation of FOXO1. Am J Physiol Gastrointest Liver Physiol. 2012;303:G1202–G1211. doi: 10.1152/ajpgi.00104.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Becerril S., Rodriguez A., Catalan V., Ramirez B., Unamuno X., Gomez-Ambrosi J., Fruhbeck G. iNOS gene ablation prevents liver fibrosis in leptin-deficient ob/ob mice. Genes (Basel) 2019;10:184. doi: 10.3390/genes10030184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mousa N., Abdel-Razik A., Sheta T., Shabana W., Zakaria S., Awad M., Abd Elsalam M., El-Wakeel N., Elkashef W., Effat N., Elgamal A., Deiab A.G., Eldars W. Serum leptin and homeostasis model assessment-IR as novel predictors of early liver fibrosis in chronic hepatitis B virus infection. Br J Biomed Sci. 2018;75:192–196. doi: 10.1080/09674845.2018.1505187. [DOI] [PubMed] [Google Scholar]
- 29.Fava G., Alpini G., Rychlicki C., Saccomanno S., DeMorrow S., Trozzi L., Candelaresi C., Venter J., Di Sario A., Marzioni M., Bearzi I., Glaser S., Alvaro D., Marucci L., Francis H., Svegliati-Baroni G., Benedetti A. Leptin enhances cholangiocarcinoma cell growth. Cancer Res. 2008;68:6752–6761. doi: 10.1158/0008-5472.CAN-07-6682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olea-Flores M., Juarez-Cruz J.C., Zuniga-Eulogio M.D., Acosta E., Garcia-Rodriguez E., Zacapala-Gomez A.E., Mendoza-Catalan M.A., Ortiz-Ortiz J., Ortuno-Pineda C., Navarro-Tito N. New actors driving the epithelial-mesenchymal transition in cancer: the role of leptin. Biomolecules. 2020;10:1676. doi: 10.3390/biom10121676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Manolakopoulos S., Bethanis S., Liapi C., Stripeli F., Sklavos P., Margeli A., Christidou A., Katsanika A., Vogiatzakis E., Tzourmakliotis D., Theocharis S. An assessment of serum leptin levels in patients with chronic viral hepatitis: a prospective study. BMC Gastroenterol. 2007;7:17. doi: 10.1186/1471-230X-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crespo J., Rivero M., Fabrega E., Cayon A., Amado J.A., Garcia-Unzeta M.T., Pons-Romero F. Plasma leptin and TNF-alpha levels in chronic hepatitis C patients and their relationship to hepatic fibrosis. Dig Dis Sci. 2002;47:1604–1610. doi: 10.1023/a:1015835606718. [DOI] [PubMed] [Google Scholar]
- 33.Makled M.N., Sharawy M.H., El-Awady M.S. The dual PPAR-alpha/gamma agonist saroglitazar ameliorates thioacetamide-induced liver fibrosis in rats through regulating leptin. Naunyn Schmiedebergs Arch Pharmacol. 2019;392:1569–1576. doi: 10.1007/s00210-019-01703-5. [DOI] [PubMed] [Google Scholar]
- 34.Li Z., Ji L., Su S., Zhu X., Cheng F., Jia X., Zhou Q., Zhou Y. Leptin up-regulates microRNA-27a/b-3p level in hepatic stellate cells. Exp Cell Res. 2018;366:63–70. doi: 10.1016/j.yexcr.2018.03.015. [DOI] [PubMed] [Google Scholar]
- 35.Cao Q., Zhu X., Zhai X., Ji L., Cheng F., Zhu Y., Yu P., Zhou Y. Leptin suppresses microRNA-122 promoter activity by phosphorylation of foxO1 in hepatic stellate cell contributing to leptin promotion of mouse liver fibrosis. Toxicol Appl Pharmacol. 2018;339:143–150. doi: 10.1016/j.taap.2017.12.007. [DOI] [PubMed] [Google Scholar]
- 36.Cheng F., Su S., Zhu X., Jia X., Tian H., Zhai X., Guan W., Zhou Y. Leptin promotes methionine adenosyltransferase 2A expression in hepatic stellate cells by the downregulation of E2F-4 via the beta-catenin pathway. FASEB J. 2020;34:5578–5589. doi: 10.1096/fj.201903021RR. [DOI] [PubMed] [Google Scholar]
- 37.Lal D., Thakur M., Bihari C. Serum leptin serves as an inflammatory activity marker and predicts steroid response in autoimmune hepatitis. J Clin Exp Hepatol. 2020;10:574–580. doi: 10.1016/j.jceh.2020.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perez-Perez A., Vilarino-Garcia T., Fernandez-Riejos P., Martin-Gonzalez J., Segura-Egea J.J., Sanchez-Margalet V. Role of leptin as a link between metabolism and the immune system. Cytokine Growth Factor Rev. 2017;35:71–84. doi: 10.1016/j.cytogfr.2017.03.001. [DOI] [PubMed] [Google Scholar]
- 39.Zou H., Liu Y., Wei D., Wang T., Wang K., Huang S., Liu L., Li Y., Ge J., Li X., Zhu H., Wang L., Zhao S., Zhang X., Wang L. Leptin promotes proliferation and metastasis of human gallbladder cancer through OB-Rb leptin receptor. Int J Oncol. 2016;49:197–206. doi: 10.3892/ijo.2016.3530. [DOI] [PubMed] [Google Scholar]
- 40.Wang Y.Y., Lin S.Y. Leptin in relation to hepatocellular carcinoma in patients with liver cirrhosis. Horm Res. 2003;60:185–190. doi: 10.1159/000073231. [DOI] [PubMed] [Google Scholar]
- 41.Lopez I., Pineda C., Raya A.I., Rodriguez-Ortiz M.E., Diaz-Tocados J.M., Rios R., Rodriguez J.M., Aguilera-Tejero E., Almaden Y. Leptin directly stimulates parathyroid hormone secretion. Endocrine. 2017;56:675–678. doi: 10.1007/s12020-016-1207-z. [DOI] [PubMed] [Google Scholar]
- 42.Atoum M.F., Alzoughool F., Al-Hourani H. Linkage between obesity leptin and breast cancer. Breast Cancer (Auckl) 2020;14 doi: 10.1177/1178223419898458. 1178223419898458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gallagher E.J., LeRoith D. Obesity and diabetes: the increased risk of cancer and cancer-related mortality. Physiol Rev. 2015;95:727–748. doi: 10.1152/physrev.00030.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gravena A.A.F., Romeiro Lopes T.C., Demitto M.O., Borghesan D.H.P., Dell' Agnolo C.M., Brischiliari S.C.R., Carvalho M.D.B., Pelloso S.M. The obesity and the risk of breast cancer among pre and postmenopausal women. Asian Pac J Cancer Prev. 2018;19:2429–2436. doi: 10.22034/APJCP.2018.19.9.2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ando S., Gelsomino L., Panza S., Giordano C., Bonofiglio D., Barone I., Catalano S. Obesity, leptin and breast cancer: epidemiological evidence and proposed mechanisms. Cancers (Basel) 2019;11:62. doi: 10.3390/cancers11010062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Surmacz E. Obesity hormone leptin: a new target in breast cancer? Breast Cancer Res. 2007;9:301. doi: 10.1186/bcr1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Andrighetto L.V., Poziomyck A.K. Serum leptin levels and hepatocellular carcinoma: review article. Arq Bras Cir Dig. 2016;29:276–278. doi: 10.1590/0102-6720201600040015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ataseven H., Bahcecioglu I.H., Kuzu N., Yalniz M., Celebi S., Erensoy A., Ustundag B. The levels of ghrelin, leptin, TNF-alpha, and IL-6 in liver cirrhosis and hepatocellular carcinoma due to HBV and HDV infection. Mediators Inflamm. 2006;2006:78380. doi: 10.1155/MI/2006/78380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sarac S., Atamer A., Atamer Y., Can A.S., Bilici A., Tacyildiz I., Kocyigit Y., Yenice N. Leptin levels and lipoprotein profiles in patients with cholelithiasis. J Int Med Res. 2015;43:385–392. doi: 10.1177/0300060514561134. [DOI] [PubMed] [Google Scholar]
- 50.Wen J., Jiang Y., Lei Z., He J., Ye M., Fu W. Leptin influence cholelithiasis formation by regulating bile acid metabolism. Turk J Gastroenterol. 2021;32:97–105. doi: 10.5152/tjg.2020.19594. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PI3 kinase inhibitor wortmannin (WMN) prevents phosphorylation of Akt in cholangiocytes and LX-2 cells in vitro, when treated with leptin. Based on time-course results shown in Figures 10 and 11, phosphorylated Akt (p-Akt) was assayed by enzyme-linked immunosorbent assay at 30 minutes for cholangiocytes and at 15 minutes for LX-2 cells when treated with vehicle or leptin in the absence or presence of WMN. A: Results for mouse cholangiocytes. B: Results for LX-2 cells. N = 3 (A and B). ∗P < 0.05 versus vehicle; †P < 0.05 versus leptin.