

Abstract

A copper-catalyzed enantioselective cyclopropanation involving trifluorodiazoethane in the presence of alkenyl boronates has been developed. This transformation enables the preparation of 2-substituted-3-(trifluoromethyl)cyclopropylboronates with high levels of stereocontrol. The products are valuable synthetic intermediates by transformation of the boronate group. This methodology can be applied to the synthesis of novel trifluoromethylated analogues of trans-2-arylcyclopropylamines, which are prevalent motifs in biologically active compounds.
Cyclopropanes are widespread carbocycles in bioactive natural and synthetic compounds.1 It is currently a standard fragment in drug discovery, which allows one to modulate properties such as lipophilicity, metabolic stability, pKa or binding, among others.2 Nowadays, it is present in numerous drugs, for example Ticagrelor,3 which is active against cardiovascular diseases, or Tezacaftor,4 which is used to treat cystic fibrosis.
Numerous methods have been described for the synthesis of substituted cyclopropanes.5 Among all the different possibilities, the preparation of cyclopropanes with fluorinated groups, in particular trifluoromethyl, is of special interest.6 This functional group is present in a vast number of therapeutic compounds.7 However, the enantioselective procedures for the preparation of trifluoromethylcyclopropanes are scarce in the literature.8 All the existing protocols, which are summarized in Scheme 1a, led to cyclopropanes with an unsubstituted carbon on the three-membered ring. For this reason, there is still a need to develop efficient enantioselective methodologies to prepare all-carbon-substituted trifluoromethylcyclopropanes.
Scheme 1. Previous Synthesis of Trifluoromethylcyclopropanes and Trifluoromethyl-Cyclopropylboronates.

On the other hand, the synthesis of versatile cyclopropanes, such as cyclopropylboronates, has also attracted the interest of the synthetic community.9 A boronate group can be easily transformed into a wide range of different functional groups.10 This allows the generation of compound libraries from a common structure. In this area, several strategies have been recently developed to prepare optically active cyclopropylboronates, including cyclopropanation of alkenyl boronates with diazo compounds,11 borylative cyclization of allylic carbonates, phosphonates,12 or epoxides,13 hydroboration of cyclopropenes,14 zinco-cyclopropanation of allylic alcohols15 and C–H borylation.16
In this context, we focused our attention on the enantioselective preparation of cyclopropanes that include simultaneously a trifluoromethyl group and a pinacol boronate as substituents. These versatile compounds would give access to a wide range of trifluoromethyl–cyclopropane derivatives. In the literature, there are only three examples of these types of compounds, all of them have been obtained as racemates from monosubstituted vinyl boron derivatives (see Scheme 1b).17
Herein, we report the enantioselective cyclopropanation of trans-alkenyl boronates with trifluorodiazoethane catalyzed by a copper(I)-bisoxazoline complex to obtain versatile 2-substituted-3-(trifluoromethyl)cyclopropylboronates. It is worth mentioning that the reactivity between alkenyl boroxines and trifluorodiazoethane has been recently reported to prepare α-trifluoromethyl allylboronic acids,18 by formation of highly electrophilic BINOL boronate derivatives in a metal-free procedure.
The cyclopropanation was initially studied with (E)-styryl pinacolboronate (1a) as a model substrate. We commenced using Cu(I)-tBuBOX (5 mol %) as a catalyst formed in situ in DCE. Initial experiments showed that alkenyl boronate was not fully consumed with 2 equiv of diazo added over the course of 2 h (Table 1, entry 1). This point was crucial from a practical point of view, as cyclopropane 2a was not easily separable from the starting material by column chromatography. Further increases in the amount of the diazo compound (4 equiv) combined with a longer reaction time (6 h) raised the conversion to 90% (Table 1, entries 2–4). The relative configuration of cyclopropane 2a was determined by 1H NMR experiments (see the Supporting Information).
Table 1. Optimization of the Reaction Conditionsa.

| entry | ligand | diazo (equiv) | t (h) | conv (%) | dr | er |
|---|---|---|---|---|---|---|
| 1 | L1 | 2 | 2 | 72 | 92:8 | – |
| 2 | L1 | 2 | 6 | 58 | 92:8 | – |
| 3 | L1 | 4 | 2 | 89 | 92:8 | – |
| 4 | L1 | 4 | 6 | 90 | 92:8 | 95:5 |
| 5 | L2 | 4 | 6 | 72 | 79:21 | 88:12 |
| 6 | L3 | 4 | 6 | 87 | 94:6 | 95:5 |
| 7 | L3 | 4b | 6 | 100 | 94:6 | 95:5 |
| 8 | L3 | 2b | 6 | 100 (69)c | 94:6 | 95:5 |
Reaction conditions: 1 (0.4 mmol), [Cu(NCMe)4]PF6 (0.02 mmol, 5 mol %), L (0.02 mmol, 5 mol %), DCE (1 mL), inert atmosphere, trifluorodiazoethane (0.5 M DCE, 2–4 equiv) 6 h slow addition. Conversion measured by 1H NMR. Diastereomeric ratio (dr) determined by 19F NMR analysis of the crude reaction mixture. Enantiomeric ratio (er) determined by HPLC analysis of the isolated product.
Trifluorodiazoethane (1.06 M DCE).
Isolated yield.
Gratifyingly, good results of diastereo- and enantiocontrol were obtained under these catalytic conditions (92:8 dr, 95:5 er). We examined different organic solvents such as THF or toluene (see SI). Toluene significantly reduced reactivity and diastereoselectivity, and THF led to no conversion of the olefin. Subsequently we investigated different com-mercially available BOX ligands. Whereas the iPrBOX (L2) ligand decreased the conversion and stereocontrol of the reaction, PhBOX (L3) slightly improved the diastereoselec-tivity (entries 5-6). At this stage, concentration of trifluorodiazoethane was increased from ca. 0.5 to 1 M, con-ducting to complete conversion (entry 7). Furthermore, the amount of diazo compound could be reduced to 2 equiva-lents (entry 8).
Under the optimized conditions, using 5 mol % of [Cu(NCMe)4]PF6 and tBuBOX as the catalyst and 2 equiv of trifluorodiazoethane added during 6 h, 69% of cyclopropylboronate 2a was isolated, with high level of stereocontrol (94:6 dr, 95:5 er).
With the optimized conditions in hand, the scope of the cyclopropanation was examined (Scheme 2). The procedure was successful with a variety of (E)-alkenyl boronates, considering electron-withdrawing and electron-donating groups (alkyl, halogens, trifluoromethyl, ether and ester substituents) at different positions in the aromatic substituent of the olefin. Moderate to good yields were obtained for the entire series (40%–77%) and high stereoselectivity was also achieved, in terms of diastereoselectivity (up to 95:5) and enantioselectivity (up to 97:3). Notably, both parameters increase as the electron density of the aromatic ring decreases. A similar result was obtained with an electron-rich heterocycle such as thiophene (2l), with moderate enantioselectivity (90:10 er). Furthermore, an aliphatic-substituted cyclopropane (2m) was also accessible with moderate yield and levels of enantioinduction. In several substrates, an increase of the equivalents of trifluorodiazoethane was necessary to achieve complete conversion, whereas the reaction was suppressed in the presence of functional groups such as nitrile or nitro. The absolute configuration of the stereogenic centers of the cyclopropane were determined by single-crystal X-ray diffraction (XRD) analysis of p-bromo and p-methoxy derivatives 2i and 2l (Scheme 2).19
Scheme 2. Substrate Scope of Copper-Catalyzed Cyclopropanation of Alkenyl Boronates.
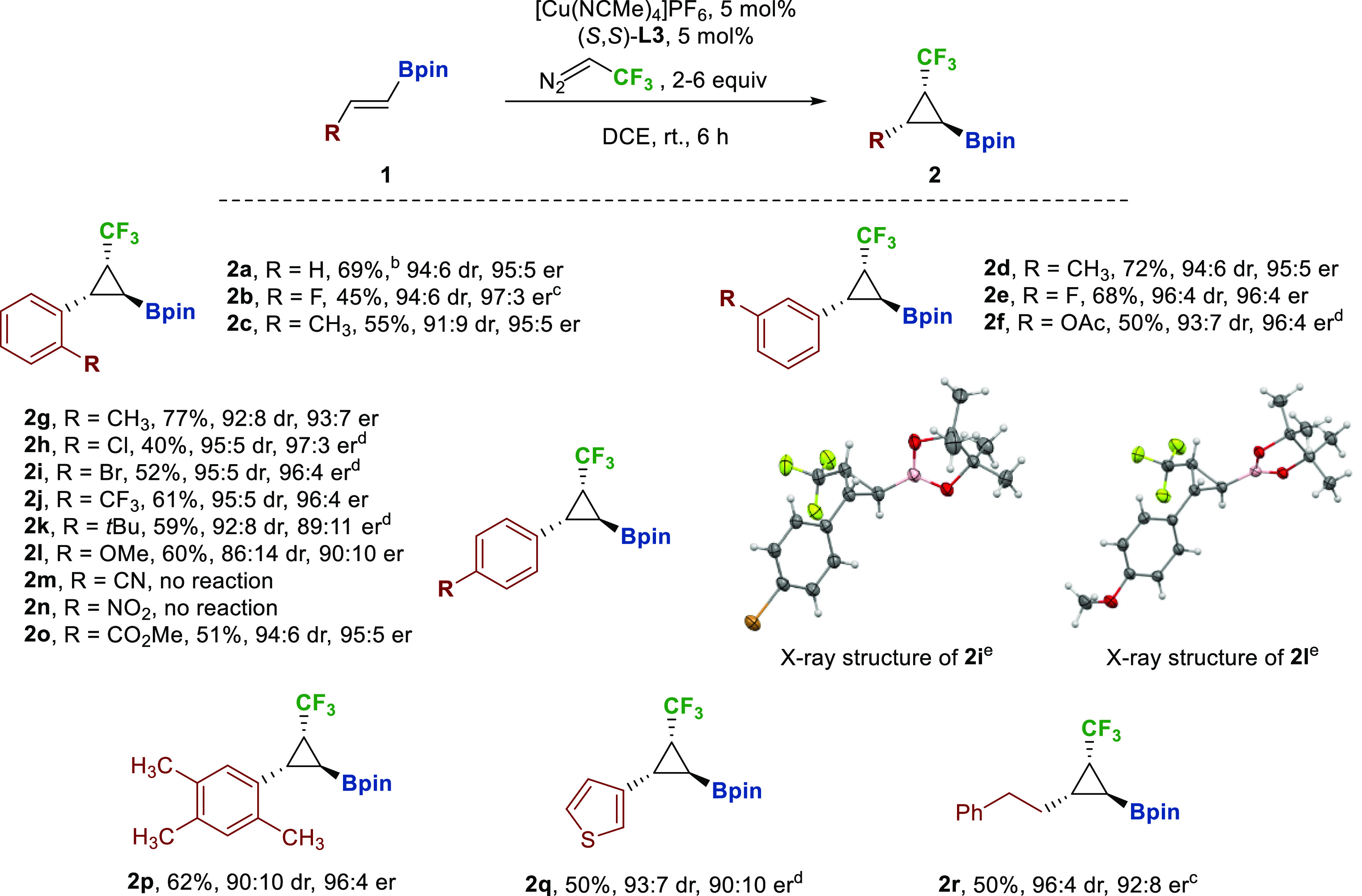
Reaction conditions: 1 (0.61 mmol), [Cu(NCMe)4]PF6 (0.03 mmol, 5 mol %), (S,S)-L3 (0.03 mmol, 5 mol %), DCE (1.5 mL), inert atmosphere trifluorodiazoethane in DCE (2 equiv), 6 h slow addition. Isolated yields.
76% at 1.25 mmol scale.
Trifluorodiazoethane (6 equiv).
Trifluorodiazoethane (4 equiv).
Thermal ellipsoids are drawn at the 50% probability level.
As mentioned above, cyclopropylboronates are versatile intermediates in organic synthesis by the transformation of the C–B bond. To highlight the synthetic utility of the new compounds, we performed several transformations of the pinacol boronate group, following reported methodologies (Scheme 3). Boronic acid 3 was smoothly obtained by treatment with methylboronic acid.20 Standard conditions of Suzuki–Miyaura cross-coupling led to 3-trifluoromethyl-1,2-diarylsubstituted cyclopropane 4 in good yield. Furthermore, oxidation of the boronate group could be achieved under basic conditions to get alcohol 5.10 Finally, amination of the cyclopropylboronate was accomplished by using BCl3 and BnN3 to get the benzylamine derivative in good yield (6).21 The latter transformations gave access to substituted trans-2-trifluoromethylcyclopropan-1-amine and trans-2-trifluoromethylcyclopropanol, rarely described in the literature in an enantioselective manner.22
Scheme 3. Transformations of Cyclopropylboronate Ester.

Reaction conditions: (a) MeB(OH)2 (5 equiv), TFA (5%)/DCM, 8 h, 72%. (b) 4-iodoanisole (1.5 equiv), Pd2(dba)3·CHCl3 (10 mol %), PPh3 (1 equiv), Ag2O (1.5 equiv), THF, 70 °C, 24 h, 45%. (c) 3 M NaOH 30% H2O2 THF, 30 min, 68%. (d) BCl3 (5.0 equiv, CH2Cl2, 25 °C, 1.5 h), then BnN3 (3.0 equiv, CH2Cl2, from 0 to 25 °C, 2 h), 51%.
Then, we focused our interest in amine derivative 6, as a trifluoromethylated analogue of trans-2-arylcyclopropylamines. This scaffold is common to numerous biological active compounds23 and is present in drugs such as Tranylcypromine (an antidepressant), Ticagrelor (a platelet aggregation inhibitor), or candidates under clinical trials for the treatment of cancer and neurodegenerative diseases.23,24 Because of the implication of F atoms in the properties of bioactive compounds,25 we targeted the enantioselective synthesis of a CF3 analogue of a lysine-specific demethylase 1 (LSD1) inhibitor (Scheme 4). The amination of cyclopropylboronate 2a with 3-(azidomethyl)-2-methoxypyridine (7) allowed us to obtain the trifluoromethyl analogue 8 of LSD1 inhibitor in a good yield.
Scheme 4. Preparation of a Trifluoromethyl Analogue of LSD1 Inhibitor.

Reaction conditions: (a) BCl3 (5.0 equiv, CH2Cl2, 25 °C, 1.5 h), then 7 (3.0 equiv, CH2Cl2, from 0 to 25 °C, 4 h), 55%.
In summary, we have developed a catalytic approach for the preparation of enantiomerically enriched 2-substituted-3-(trifluoromethyl)cyclopropylboronates by cyclopropanation of (E)-alkenyl boronates with trifluorodiazoethane. This methodology is general for a variety of substrates, using commercially available copper catalyst and ligand. Valuable synthetic intermediates can be obtained by the functionalization of the C–B bond. This route provides straightforward access to enantioenriched 2-aryl-3-(trifluoromethyl)cyclopropylamines, a relevant scaffold in medicinal chemistry.
Acknowledgments
We gratefully acknowledge MICINN (PID2019-105007GA-I00, CTQ2017-85263-R), Instituto de Salud Carlos III (FEDER funds, ISCIII RETIC REDINREN RD16/0009/0015), Comunidad de Madrid and Universidad de Alcalá (CM/JIN/2019-025, CCG19/CC-038) for financial support. We thank Dr. C. Golz, University of Göttingen, for the X-ray analyses of compounds 2i and 2l. We also thank Dr. Pedro Pérez and Dr. Ana Caballero, Universidad de Huelva, for fruitful discussions and Pilar Franco (Chiral Technologies) for HPLC advice. J.A. thanks MEFP for a predoctoral contract.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.orglett.1c02420.
Experimental procedures; characterization data; 1H, 13C, 11B and 19F NMR spectral data; HPLC; mass spectrometry data of new compounds and X-ray crystallographic data for 2i and 2l (PDF)
Accession Codes
CCDC 2079480 and 2079481 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: + 44 1223 336033.
The authors declare no competing financial interest.
Supplementary Material
References
- For selected reviews, see:; a Ebner C.; Carreira E. M. Cyclopropanation Strategies in Recent Total Syntheses. Chem. Rev. 2017, 117, 11651–11679. 10.1021/acs.chemrev.6b00798. [DOI] [PubMed] [Google Scholar]; b Časar Z. Synthetic Approaches to Contemporary Drugs That Contain the Cyclopropyl Moiety. Synthesis 2020, 52, 1315–1345. 10.1055/s-0039-1690058. [DOI] [Google Scholar]
- a Talele T. T. The “Cyclopropyl Fragment” Is a Versatile Player That Frequently Appears in Preclinical/Clinical Drug Molecules. J. Med. Chem. 2016, 59, 8712–8756. 10.1021/acs.jmedchem.6b00472. [DOI] [PubMed] [Google Scholar]; b Bauer M. R.; Di Fruscia P.; Lucas S. C. C.; Michaelides I. N.; Nelson J. E.; Storer R. I.; Whitehurst B. C. Put a Ring on It: Application of Small Aliphatic Rings in Medicinal Chemistry. RSC Med. Chem. 2021, 12, 448–471. 10.1039/D0MD00370K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijeyeratne Y. D.; Joshi R.; Heptinstall S. Ticagrelor: A P2Y12 Antagonist for Use in Acute Coronary Syndromes. Expert Rev. Clin. Pharmacol. 2012, 5, 257–269. 10.1586/ecp.12.17. [DOI] [PubMed] [Google Scholar]
- Hughes D. L. Patent Review of Synthetic Routes and Crystalline Forms of the CFTR-Modulator Drugs Ivacaftor, Lumacaftor, Tezacaftor, and Elexacaftor. Org. Process Res. Dev. 2019, 23, 2302–2322. 10.1021/acs.oprd.9b00326. [DOI] [Google Scholar]
- Pellissier H., Lattanzi A.; Dalpozzo R.. Asymmetric Cyclopropanation. In Asymmetric Synthesis of Three-Membered Rings; John Wiley & Sons, Ltd., 2017; pp 1–204. [Google Scholar]
- For a selected reviews, see:; a Decaens J.; Couve-Bonnaire S.; Charette A. B.; Poisson T.; Jubault P. Synthesis of Fluoro-, Monofluoromethyl-, Difluoromethyl-, and Trifluoromethyl-Substituted Three-Membered Rings. Chem. - Eur. J. 2021, 27, 2935–2962. 10.1002/chem.202003822. [DOI] [PubMed] [Google Scholar]; For recent examples:; b Cyr P.; Flynn-Robitaille J.; Boissarie P.; Marinier A. Mild and Diazo-Free Synthesis of Trifluoromethyl-Cyclopropanes Using Sulfonium Ylides. Org. Lett. 2019, 21, 2265–2268. 10.1021/acs.orglett.9b00557. [DOI] [PubMed] [Google Scholar]; c Chen G.-S.; Yan X.-X.; Chen S.-J.; Mao X.-Y.; Li Z.-D.; Liu Y.-L. Diastereoselective Synthesis of 1,3-Diyne-Tethered Trifluoromethylcyclopropanes through a Sulfur Ylide Mediated Cyclopropanation/DBU-Mediated Epimerization Sequence. J. Org. Chem. 2020, 85, 6252–6260. 10.1021/acs.joc.0c00162. [DOI] [PubMed] [Google Scholar]; d Chernykh A. V.; Olifir O. S.; Kuchkovska Y. O.; Volochnyuk D. M.; Yarmolchuk V. S.; Grygorenko O. O. Fluoroalkyl-Substituted Cyclopropane Derivatives: Synthesis and Physicochemical Properties. J. Org. Chem. 2020, 85, 12692–12702. 10.1021/acs.joc.0c01848. [DOI] [PubMed] [Google Scholar]
- Meanwell N. A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. 10.1021/acs.jmedchem.7b01788. [DOI] [PubMed] [Google Scholar]
- a Le Maux P.; Juillard S.; Simonneaux G. Asymmetric Synthesis of Trifluoromethylphenyl Cyclopropanes Catalyzed by Chiral Metalloporphyrins. Synthesis 2006, 2006, 1701–1704. 10.1055/s-2006-926451. [DOI] [Google Scholar]; b Denton J. R.; Sukumaran D.; Davies H. M. L. Enantioselective Synthesis of Trifluoromethyl-Substituted Cyclopropanes. Org. Lett. 2007, 9, 2625–2628. 10.1021/ol070714f. [DOI] [PubMed] [Google Scholar]; c Morandi B.; Mariampillai B.; Carreira E. M. Enantioselective Cobalt-Catalyzed Preparation of Trifluoromethyl-Substituted Cyclopropanes. Angew. Chem., Int. Ed. 2011, 50, 1101–1104. 10.1002/anie.201004269. [DOI] [PubMed] [Google Scholar]; d Tinoco A.; Steck V.; Tyagi V.; Fasan R. Highly Diastereo- and Enantioselective Synthesis of Trifluoromethyl-Substituted Cyclopropanes via Myoglobin-Catalyzed Transfer of Trifluoromethylcarbene. J. Am. Chem. Soc. 2017, 139, 5293–5296. 10.1021/jacs.7b00768. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Kotozaki M.; Chanthamath S.; Fujii T.; Shibatomi K.; Iwasa S. Highly Enantioselective Synthesis of Trifluoromethyl Cyclopropanes by Using Ru(II)–Pheox Catalysts. Chem. Commun. 2018, 54, 5110–5113. 10.1039/C8CC02286K. [DOI] [PubMed] [Google Scholar]; f Huang W.-S.; Schlinquer C.; Poisson T.; Pannecoucke X.; Charette A. B.; Jubault P. General Catalytic Enantioselective Access to Monohalomethyl and Trifluoromethyl Cyclopropanes. Chem. - Eur. J. 2018, 24, 10339–10343. 10.1002/chem.201802685. [DOI] [PubMed] [Google Scholar]; g Carminati D. M.; Decaens J.; Couve-Bonnaire S.; Jubault P.; Fasan R. Biocatalytic Strategy for the Highly Stereoselective Synthesis of CHF2-Containing Trisubstituted Cyclopropanes. Angew. Chem., Int. Ed. 2021, 60, 7072–7076. 10.1002/anie.202015895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín-Heras V.; Parra A.; Tortosa M. Cyclopropyl- and Cyclobutylboronates and -Silanes: A Stereoselective Approach. Synthesis 2018, 50, 470–484. 10.1055/s-0036-1589133. [DOI] [Google Scholar]
- Sandford C.; Aggarwal V. K. Stereospecific Functionalizations and Transformations of Secondary and Tertiary Boronic Esters. Chem. Commun. 2017, 53, 5481–5494. 10.1039/C7CC01254C. [DOI] [PubMed] [Google Scholar]
- a Carreras J.; Caballero A.; Pérez P. J. Enantio- and Diastereoselective Cyclopropanation of 1-Alkenylboronates: Synthesis of 1-Boryl-2,3-Disubstituted Cyclopropanes. Angew. Chem., Int. Ed. 2018, 57, 2334–2338. 10.1002/anie.201710415. [DOI] [PubMed] [Google Scholar]; and references cited therein.; b Sun X.; Gu P.; Qin J.; Su Y. Rhodium-Catalysed Diastereo- and Enantio-Selective Cyclopropanation of α-Boryl Styrenes. Chem. Commun. 2020, 56, 12379–12382. 10.1039/D0CC02549F. [DOI] [PubMed] [Google Scholar]
- a Ito H.; Kosaka Y.; Nonoyama K.; Sasaki Y.; Sawamura M. Synthesis of Optically Active Boron–Silicon Bifunctional Cyclopropane Derivatives through Enantioselective Copper(I)-Catalyzed Reaction of Allylic Carbonates with a Diboron Derivative. Angew. Chem., Int. Ed. 2008, 47, 7424–7427. 10.1002/anie.200802342. [DOI] [PubMed] [Google Scholar]; b Zhong C.; Kunii S.; Kosaka Y.; Sawamura M.; Ito H. Enantioselective Synthesis of Trans-Aryl- and -Heteroaryl-Substituted Cyclopropylboronates by Copper(I)-Catalyzed Reactions of Allylic Phosphates with a Diboron Derivative. J. Am. Chem. Soc. 2010, 132, 11440–11442. 10.1021/ja103783p. [DOI] [PubMed] [Google Scholar]
- Amenós L.; Trulli L.; Nóvoa L.; Parra A.; Tortosa M. Stereospecific Synthesis of α-Hydroxy-Cyclopropylboronates from Allylic Epoxides. Angew. Chem., Int. Ed. 2019, 58, 3188–3192. 10.1002/anie.201812836. [DOI] [PubMed] [Google Scholar]
- a Rubina M.; Rubin M.; Gevorgyan V. Catalytic Enantioselective Hydroboration of Cyclopropenes. J. Am. Chem. Soc. 2003, 125, 7198–7199. 10.1021/ja034210y. [DOI] [PubMed] [Google Scholar]; b Tian B.; Liu Q.; Tong X.; Tian P.; Lin G.-Q. Copper(I)-Catalyzed Enantioselective Hydroboration of Cyclopropenes: Facile Synthesis of Optically Active Cyclopropylboronates. Org. Chem. Front. 2014, 1, 1116–1122. 10.1039/C4QO00157E. [DOI] [Google Scholar]; c Parra A.; Amenós L.; Guisán-Ceinos M.; López A.; García Ruano J. L.; Tortosa M. Copper-Catalyzed Diastereo- and Enantioselective Desymmetrization of Cyclopropenes: Synthesis of Cyclopropylboronates. J. Am. Chem. Soc. 2014, 136, 15833–15836. 10.1021/ja510419z. [DOI] [PubMed] [Google Scholar]; and references cited therein.
- Zimmer L. E.; Charette A. B. Enantioselective Synthesis of 1,2,3-Trisubstituted Cyclopropanes Using gem-Dizinc Reagents. J. Am. Chem. Soc. 2009, 131, 15624–15626. 10.1021/ja906033g. [DOI] [PubMed] [Google Scholar]
- Shi Y.; Gao Q.; Xu S. Chiral Bidentate Boryl Ligand Enabled Iridium-Catalyzed Enantioselective C(sp3)–H Borylation of Cyclopropanes. J. Am. Chem. Soc. 2019, 141, 10599–10604. 10.1021/jacs.9b04549. [DOI] [PubMed] [Google Scholar]
- a Duncton M. A. J.; Ayala L.; Kaub C.; Janagani S.; Edwards W. T.; Orike N.; Ramamoorthy K.; Kincaid J.; Kelly M. G. Dibutyl 2-(Trifluoromethyl)Cyclopropylboronate as a Useful (Trifluoromethyl)Cyclopropyl Donor: Application to Antagonists of TRPV1. Tetrahedron Lett. 2010, 51, 1009–1011. 10.1016/j.tetlet.2009.12.073. [DOI] [Google Scholar]; b Duncton M. A. J.; Singh R. Synthesis of Trans-2-(Trifluoromethyl)Cyclopropanes via Suzuki Reactions with an N-Methyliminodiacetic Acid Boronate. Org. Lett. 2013, 15, 4284–4287. 10.1021/ol401636d. [DOI] [PubMed] [Google Scholar]; c Hryschuk O. V.; Yurov Y.; Tymtsunik A. V.; Kovtunenko V. O.; Komarov I. V.; Grygorenko O. O. Multigram Synthesis and C–C/C–N Couplings of Functionalized 1,2-Disubstituted Cyclopropyltrifluoroborates. Adv. Synth. Catal. 2019, 361, 5428–5439. 10.1002/adsc.201900879. [DOI] [Google Scholar]; d A chiral cyclopropyl PIDA boronate has been described in a patent: Duncton M.; Burke M. D. Cyclopropyl PIDA Boronate. U.S. Patent No. US20130331585 A1, 2013.
- Jonker S. J. T.; Jayarajan R.; Kireilis T.; Deliaval M.; Eriksson L.; Szabó K. J. Organocatalytic Synthesis of α-Trifluoromethyl Allylboronic Acids by Enantioselective 1,2-Borotropic Migration. J. Am. Chem. Soc. 2020, 142, 21254–21259. 10.1021/jacs.0c09923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CCDC 2079481 (2i) and CCDC 2079480 (2l) contain the supplementary crystallographic data. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
- Hinkes S. P. A.; Klein C. D. P. Virtues of Volatility: A Facile Transesterification Approach to Boronic Acids. Org. Lett. 2019, 21, 3048–3052. 10.1021/acs.orglett.9b00584. [DOI] [PubMed] [Google Scholar]
- Hupe E.; Marek I.; Knochel P. Diastereoselective Reduction of Alkenylboronic Esters as a New Method for Controlling the Stereochemistry of up to Three Adjacent Centers in Cyclic and Acyclic Molecules. Org. Lett. 2002, 4, 2861–2863. 10.1021/ol0262486. [DOI] [PubMed] [Google Scholar]
- See reference (8e). For some racemic examples, see:; a Mykhailiuk P. K.; Afonin S.; Ulrich A. S.; Komarov I. V. A Convenient Route to Trifluoromethyl-Substituted Cyclopropane Derivatives. Synthesis 2008, 2008, 1757–1760. 10.1055/s-2008-1067041. [DOI] [Google Scholar]; b Yarmolchuk V.; Bezdudny A.; Tolmacheva N.; Lukin O.; Boyko A.; Chekotylo A.; Tolmachev A.; Mykhailiuk P. An Efficient and Safe Method for the Multigram Synthesis of Trans-2-(Trifluoromethyl)Cyclopropylamine. Synthesis 2012, 44, 1152–1154. 10.1055/s-0031-1289711. [DOI] [Google Scholar]; and references cited therein.
- a Miyamura S.; Itami K.; Yamaguchi J. Syntheses of Biologically Active 2-Arylcyclopropylamines. Synthesis 2017, 49, 1131–1149. 10.1055/s-0036-1588390. [DOI] [Google Scholar]; For recent synthetic examples, see:; b Miyamura S.; Araki M.; Suzuki T.; Yamaguchi J.; Itami K. Stereodivergent Synthesis of Arylcyclopropylamines by Sequential C-H Borylation and Suzuki–Miyaura Coupling. Angew. Chem., Int. Ed. 2015, 54, 846–851. 10.1002/anie.201409186. [DOI] [PubMed] [Google Scholar]; c Li Z.; Zhang M.; Zhang Y.; Liu S.; Zhao J.; Zhang Q. Multicomponent Cyclopropane Synthesis Enabled by Cu-Catalyzed Cyclopropene Carbometalation with Organoboron Reagent: Enantioselective Modular Access to Polysubstituted 2-Arylcyclopropylamines. Org. Lett. 2019, 21, 5432–5437. 10.1021/acs.orglett.9b01650. [DOI] [PubMed] [Google Scholar]
- a Hitchin J. R.; Blagg J.; Burke R.; Burns S.; Cockerill M. J.; Fairweather E. E.; Hutton C.; Jordan A. M.; McAndrew C.; Mirza A.; Mould D.; Thomson G. J.; Waddell I.; Ogilvie D. J. Development and Evaluation of Selective, Reversible LSD1 Inhibitors Derived from Fragments. MedChemComm 2013, 4, 1513–1522. 10.1039/c3md00226h. [DOI] [Google Scholar]; b Schulz-Fincke J.; Hau M.; Barth J.; Robaa D.; Willmann D.; Kürner A.; Haas J.; Greve G.; Haydn T.; Fulda S.; Lübbert M.; Lüdeke S.; Berg T.; Sippl W.; Schüle R.; Jung M. Structure-Activity Studies on N-Substituted Tranylcypromine Derivatives Lead to Selective Inhibitors of Lysine Specific Demethylase 1 (LSD1) and Potent Inducers of Leukemic Cell Differentiation. Eur. J. Med. Chem. 2018, 144, 52–67. 10.1016/j.ejmech.2017.12.001. [DOI] [PubMed] [Google Scholar]; c Herrlinger E.-M.; Hau M.; Redhaber D. M.; Greve G.; Willmann D.; Steimle S.; Müller M.; Lübbert M.; Miething C. C.; Schüle R.; Jung M. Nitroreductase-Mediated Release of Inhibitors of Lysine-Specific Demethylase 1 (LSD1) from Prodrugs in Transfected Acute Myeloid Leukaemia Cells. ChemBioChem 2020, 21, 2329–2347. 10.1002/cbic.202000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monofluorinated tranylcypromine analogues have been previously studied:Borrello M. T.; Schinor B.; Bartels K.; Benelkebir H.; Pereira S.; Al-Jamal W. T.; Douglas L.; Duriez P. J.; Packham G.; Haufe G.; Ganesan A. Fluorinated Tranylcypromine Analogues as Inhibitors of Lysine-Specific Demethylase 1 (LSD1, KDM1A). Bioorg. Med. Chem. Lett. 2017, 27, 2099–2101. 10.1016/j.bmcl.2017.03.081. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.