

Abstract
Background
The primary manifestation of coronavirus disease 2019 (COVID‐19) is respiratory insufficiency that can also be related to diffuse pulmonary microthrombosis and thromboembolic events, such as pulmonary embolism, deep vein thrombosis, or arterial thrombosis. People with COVID‐19 who develop thromboembolism have a worse prognosis.
Anticoagulants such as heparinoids (heparins or pentasaccharides), vitamin K antagonists and direct anticoagulants are used for the prevention and treatment of venous or arterial thromboembolism. Besides their anticoagulant properties, heparinoids have an additional anti‐inflammatory potential. However, the benefit of anticoagulants for people with COVID‐19 is still under debate.
Objectives
To assess the benefits and harms of anticoagulants versus active comparator, placebo or no intervention in people hospitalised with COVID‐19.
Search methods
We searched the CENTRAL, MEDLINE, Embase, LILACS and IBECS databases, the Cochrane COVID‐19 Study Register and medRxiv preprint database from their inception to 14 April 2021. We also checked the reference lists of any relevant systematic reviews identified, and contacted specialists in the field for additional references to trials.
Selection criteria
Eligible studies were randomised controlled trials (RCTs), quasi‐RCTs, cluster‐RCTs and cohort studies that compared prophylactic anticoagulants versus active comparator, placebo or no intervention for the management of people hospitalised with COVID‐19. We excluded studies without a comparator group and with a retrospective design (all previously included studies) as we were able to include better study designs. Primary outcomes were all‐cause mortality and necessity for additional respiratory support. Secondary outcomes were mortality related to COVID‐19, deep vein thrombosis, pulmonary embolism, major bleeding, adverse events, length of hospital stay and quality of life.
Data collection and analysis
We used standard Cochrane methodological procedures. We used Cochrane RoB 1 to assess the risk of bias for RCTs, ROBINS‐I to assess risk of bias for non‐randomised studies (NRS) and GRADE to assess the certainty of evidence. We meta‐analysed data when appropriate.
Main results
We included seven studies (16,185 participants) with participants hospitalised with COVID‐19, in either intensive care units, hospital wards or emergency departments. Studies were from Brazil (2), Iran (1), Italy (1), and the USA (1), and two involved more than country. The mean age of participants was 55 to 68 years and the follow‐up period ranged from 15 to 90 days. The studies assessed the effects of heparinoids, direct anticoagulants or vitamin K antagonists, and reported sparse data or did not report some of our outcomes of interest: necessity for additional respiratory support, mortality related to COVID‐19, and quality of life.
Higher‐dose versus lower‐dose anticoagulants (4 RCTs, 4647 participants)
Higher‐dose anticoagulants result in little or no difference in all‐cause mortality (risk ratio (RR) 1.03, 95% CI 0.92 to 1.16, 4489 participants; 4 RCTs) and increase minor bleeding (RR 3.28, 95% CI 1.75 to 6.14, 1196 participants; 3 RCTs) compared to lower‐dose anticoagulants up to 30 days (high‐certainty evidence). Higher‐dose anticoagulants probably reduce pulmonary embolism (RR 0.46, 95% CI 0.31 to 0.70, 4360 participants; 4 RCTs), and slightly increase major bleeding (RR 1.78, 95% CI 1.13 to 2.80, 4400 participants; 4 RCTs) compared to lower‐dose anticoagulants up to 30 days (moderate‐certainty evidence). Higher‐dose anticoagulants may result in little or no difference in deep vein thrombosis (RR 1.08, 95% CI 0.57 to 2.03, 3422 participants; 4 RCTs), stroke (RR 0.91, 95% CI 0.40 to 2.03, 4349 participants; 3 RCTs), major adverse limb events (RR 0.33, 95% CI 0.01 to 7.99, 1176 participants; 2 RCTs), myocardial infarction (RR 0.86, 95% CI 0.48 to 1.55, 4349 participants; 3 RCTs), atrial fibrillation (RR 0.35, 95% CI 0.07 to 1.70, 562 participants; 1 study), or thrombocytopenia (RR 0.94, 95% CI 0.71 to 1.24, 2789 participants; 2 RCTs) compared to lower‐dose anticoagulants up to 30 days (low‐certainty evidence). It is unclear whether higher‐dose anticoagulants have any effect on necessity for additional respiratory support, mortality related to COVID‐19, and quality of life (very low‐certainty evidence or no data).
Anticoagulants versus no treatment (3 prospective NRS, 11,538 participants)
Anticoagulants may reduce all‐cause mortality but the evidence is very uncertain due to two study results being at critical and serious risk of bias (RR 0.64, 95% CI 0.55 to 0.74, 8395 participants; 3 NRS; very low‐certainty evidence). It is uncertain if anticoagulants have any effect on necessity for additional respiratory support, mortality related to COVID‐19, deep vein thrombosis, pulmonary embolism, major bleeding, stroke, myocardial infarction and quality of life (very low‐certainty evidence or no data).
Ongoing studies
We found 62 ongoing studies in hospital settings (60 RCTs, 35,470 participants; 2 prospective NRS, 120 participants) in 20 different countries. Thirty‐five ongoing studies plan to report mortality and 26 plan to report necessity for additional respiratory support. We expect 58 studies to be completed in December 2021, and four in July 2022. From 60 RCTs, 28 are comparing different doses of anticoagulants, 24 are comparing anticoagulants versus no anticoagulants, seven are comparing different types of anticoagulants, and one did not report detail of the comparator group.
Authors' conclusions
When compared to a lower‐dose regimen, higher‐dose anticoagulants result in little to no difference in all‐cause mortality and increase minor bleeding in people hospitalised with COVID‐19 up to 30 days. Higher‐dose anticoagulants possibly reduce pulmonary embolism, slightly increase major bleeding, may result in little to no difference in hospitalisation time, and may result in little to no difference in deep vein thrombosis, stroke, major adverse limb events, myocardial infarction, atrial fibrillation, or thrombocytopenia.
Compared with no treatment, anticoagulants may reduce all‐cause mortality but the evidence comes from non‐randomised studies and is very uncertain. It is unclear whether anticoagulants have any effect on the remaining outcomes compared to no anticoagulants (very low‐certainty evidence or no data).
Although we are very confident that new RCTs will not change the effects of different doses of anticoagulants on mortality and minor bleeding, high‐quality RCTs are still needed, mainly for the other primary outcome (necessity for additional respiratory support), the comparison with no anticoagulation, when comparing the types of anticoagulants and giving anticoagulants for a prolonged period of time.
Keywords: Aged, Humans, Middle Aged, Anticoagulants, Anticoagulants/adverse effects, COVID-19, COVID-19/complications, Heparin, Heparin/adverse effects, SARS-CoV-2, Thromboembolism
Plain language summary
Do blood thinners prevent people who are hospitalised with COVID‐19 from developing blood clots?
Key messages
‐ High‐dose blood thinners result in little or no difference in death rate and increase minor bleeding compared to low‐dose blood thinners for people hospitalised with COVID‐19. Giving blood thinners compared to not giving blood thinners might reduce the death rate.
‐ It is very likely that new studies will not change the evidence about the effects of different doses of blood thinners on death rate and minor bleeding. High‐quality studies are still needed to analyse the need for additional respiratory support, giving blood thinners compared to no blood thinners, comparing different blood thinners, and giving blood thinners for extended periods.
What is COVID‐19?
COVID‐19 typically affects the lungs and airways; however, in addition to respiratory problems, about 16% of people hospitalised with COVID‐19 experience problems with their blood vessels, leading to blood clots forming in the arteries, veins and lungs. Nearly half of all people with severe COVID‐19 in intensive care units develop clots in their veins or arteries.
What are blood thinners?
Blood thinners are medicines that prevent harmful blood clots from forming (deep vein thrombosis). However, they can cause unwanted effects such as bleeding. Some guidelines recommend giving blood thinners when people are first admitted to hospital with COVID‐19 to prevent blood clots from developing, rather than waiting to see whether blood clots develop and then treating them with blood thinners.
What did we want to find out?
We wanted to know whether giving blood thinners to people hospitalised with COVID‐19 as a preventive measure reduced the number of deaths compared to people who received no treatment or those who received a placebo treatment (an identical‐seeming treatment but with no active ingredient). We also wanted to determine whether these individuals needed less support with breathing, whether they still developed harmful blood clots, whether they experienced bleeding and whether they experienced any other unwanted events.
What did we do?
We searched for studies that assessed blood thinners given to people hospitalised with COVID‐19 to prevent blood clots. Studies could be of any design as long as they compared a blood thinner with another blood thinner, no treatment or a placebo. Studies could take place anywhere in the world and participants could be any age as long as they were in hospital with confirmed COVID‐19 disease. We pooled the results when appropriate.
What did we find?
We included seven studies with 16,185 people hospitalised with COVID‐19 in either intensive care units, hospital wards or emergency departments. Studies were from Brazil (2), Iran (1), Italy (1), and the USA (1), and two involved more than country. People in the studies were aged from 55 to 68 years on average. Studies lasted from 15 to 90 days and provided evidence on deaths, bleeding, blood clotting, length of hospital stay and unwanted effects. There was little or no evidence on need for respiratory support (help with breathing), deaths related to COVID‐19, and quality of life.
Higher‐dose of blood thinners compared with lower‐dose (4 studies, 4647 people) In people who received higher compared to lower doses of blood thinners there was little to no difference in death rate. However, people on higher doses were more likely to experience minor bleeding compared to in those on lower doses. People who received higher doses of blood thinners likely had reduced pulmonary embolism (blood clot in the lung or blood vessel leading to the lung), slightly increased major (more severe) bleeding, and probably had little to no difference in time spent in hospital compared to those who received the lower doses of blood thinners. In people who received higher doses of blood thinners, there was little to no difference in the rate of deep vein thrombosis, and other unwanted events compared to those who received the lower dose of blood thinners.
Blood thinners compared with no treatment (3 studies, 11,538 people) People who received blood thinners had a reduced death rate compared to those who did not receive blood thinners, but the evidence is very uncertain.
What are the limitations of the evidence?
We are very confident that higher doses of blood thinners do not change the risk of death but do increase the risk of bleeding in people hospitalised with COVID‐19.
Although our confidence in the evidence is very limited, people who receive blood thinners may have a lower death rate compared to those who did not receive any blood thinners.
What happens next?
Our searches found 62 ongoing studies with 35,470 people. We plan to add the results of these studies to our review when they are published.
How up to date is this evidence?
The evidence is up to date to 14 April 2021.
Summary of findings
Summary of findings 1. Higher‐dose anticoagulants compared to lower‐dose anticoagulants for people hospitalised with COVID‐19.
| Higher‐dose anticoagulants compared to lower‐dose anticoagulants for people hospitalised with COVID‐19 | ||||||
| Patient or population: people hospitalised with COVID‐19 Setting: hospital Intervention: higher‐dose anticoagulants (LMWH, UFH or rivaroxaban) Comparison: lower‐dose anticoagulants (LMWH or UFH) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with lower‐dose anticoagulants (short‐term outcomes) | Risk with higher‐dose anticoagulants | |||||
|
All‐cause mortality Follow‐up: from 28‐30 days |
Study population | RR 1.03 (0.92 to 1.16) | 4489 (4 RCTs) | ⊕⊕⊕⊕ Higha | Higher‐dose anticoagulants results in little to no difference in all‐cause mortality | |
| 191 per 1000 | 196 per 1000 (175 to 221) | |||||
|
Necessity for additional respiratory support Follow‐up: from 28‐30 days |
Study population | RR 0.54 (0.12 to 2.47) | 3407 (3 RCTs) | ⊕⊝⊝⊝ Very lowb,c,d | The evidence is very uncertain about the effect of higher‐dose anticoagulants on necessity for additional respiratory support. | |
| 117 per 1000 | 63 per 1000 (14 to 289) | |||||
| Mortality related to COVID‐19 | No studies measured this outcome | |||||
|
Deep vein thrombosis Follow‐up: from 28‐30 days |
Study population | RR 1.08 (0.57 to 2.03) | 3422 (4 RCTs) | ⊕⊕⊝⊝ Lowd | Higher‐dose anticoagulants may result in little to no difference in DVT | |
| 11 per 1000 | 12 per 1000 (6 to 22) | |||||
|
Pulmonary embolism Follow‐up: from 28‐30 days |
Study population | RR 0.46 (0.31 to 0.70) | 4360 (4 RCTs) | ⊕⊕⊕⊝ Moderateb | Higher‐dose anticoagulants likely reduce PE | |
| 33 per 1000 | 15 per 1000 (10 to 23) | |||||
|
Major bleeding Follow‐up: from 28‐30 days |
Study population | RR 1.78 (1.13 to 2.80) | 4400 (4 RCTs) | ⊕⊕⊕⊝ Moderateb | Higher‐dose anticoagulants likely increase major bleeding slightly | |
| 14 per 1000 | 24 per 1000 (15 to 38) | |||||
|
Adverse events (minor bleeding) Follow‐up: from 28‐30 days |
Study population | RR
3.28 (1.75 to 6.14) |
1196 (3 RCTs) | ⊕⊕⊕⊕ High | Higher‐dose anticoagulants increase adverse events (minor bleeding) | |
| 20 per 1000 | 47 per 1000 (18 to 121) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; COVID‐19: coronavirus disease 2019; DVT: deep vein thrombosis; LMWH: low‐molecular‐weight heparin; PE: pulmonary embolism; RCT: randomised controlled trial; RR: risk ratio; UFH: unfractionated heparin | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aThe largest study in the analysis was at high risk of bias in almost all domains; however, we did not downgrade for study limitations as removing this study in the sensitivity analysis did not change the pooled estimate. bDowngraded one level due to study limitations. One randomised controlled trial provided high risk of bias in almost all domains leading to a different pooled estimate after sensitivity analysis. cDowngraded one level due to inconsistency. We identified substantial unexplained heterogeneity (I² = 60%). dDowngraded two levels due to imprecision. Confidence interval of the absolute difference comprises both important clinical benefit and important clinical harm.
Summary of findings 2. Anticoagulants compared to no treatment for people hospitalised with COVID‐19.
| Anticoagulants compared to no treatment for people hospitalised with COVID‐19 | ||||||
| Patient or population: people hospitalised with COVID‐19 Setting: hospital Intervention: anticoagulants (LMWH, UFH, fondaparinux, DOACs or VKA) Comparison: no treatment (no anticoagulants) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with no treatment | Risk with anticoagulants | |||||
|
All‐cause mortality Follow‐up: from 15‐30 days |
Study population | RR 0.64 (0.55 to 0.74) | 8395 (3 observational studies) | ⊕⊝⊝⊝ Very lowa,b | Anticoagulants may reduce all‐cause mortality but the evidence is very uncertain due to two study results being at critical and serious risk of bias. The numerical results are very unreliable for outcomes where critical risk of bias is an issue | |
| 307 per 1000 | 196 per 1000 (169 to 227) | |||||
| Necessity for additional respiratory support | No studies measured this outcome | |||||
| Mortality related to COVID‐19 | No studies measured this outcome | |||||
|
Deep vein thrombosis Follow‐up: up to 15 days |
Study population | RR 5.67 (1.30 to 24.70) | 1403 (1 observational study) | ⊕⊝⊝⊝ Very lowc,d | It is uncertain if anticoagulants have any effect on DVT. The numerical results are very unreliable for outcomes where critical risk of bias is an issue. | |
| 3 per 1000 | 19 per 1000 (4 to 82) |
|||||
|
Pulmonary embolism Follow‐up: up to 15 days |
Study population | RR 24.19 (3.31 to 176.53) | 1403 (1 observational study) | ⊕⊝⊝⊝ Very lowc,d | It is uncertain if anticoagulants have any effect on PE. The numerical results are very unreliable for outcomes where critical risk of bias is an issue. | |
| 2 per 1000 | 40 per 1000 (5 to 292) |
|||||
|
Major bleeding Follow‐up: from 15‐26 days |
Study population | RR 1.19 (0.66 to 2.12) | 7218 (2 observational studies) | ⊕⊝⊝⊝ Very lowb,c,e | It is uncertain if anticoagulants have any effect on major bleeding. The numerical results are very unreliable for outcomes where critical risk of bias is an issue. | |
| 19 per 1000 | 23 per 1000 (13 to 41) | |||||
| Adverse events (minor bleeding) | No studies measured this outcome | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; COVID‐19: coronavirus disease 2019; DOACs: direct oral anticoagulants; DVT: deep vein thrombosis; LMWH: low‐molecular‐weight heparin; PE: pulmonary embolism; RR: risk ratio; UFH: unfractionated heparin; VKA: vitamin K antagonist | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded two levels due to study limitations. Overall critical/serious risk of bias in two studies, especially related to confounding. bDowngraded one level due to inconsistency. We found moderate unexplained heterogeneity (I² = 30% to 60%). cDowngraded one level due to study limitations. Overall critical risk of bias, especially related to confounding. dDowngraded two levels due to imprecision. Fewer than 300 events were included in the analysis and very large confidence interval. eDowngraded one level due to imprecision. Confidence interval of the absolute difference comprises both unimportant clinical harm and important clinical harm.
Background
See Table 3 for a glossary of terms.
1. Glossary of terms.
| Term | Definition |
| Anticoagulants | Drugs that suppress, delay or prevent blood clots |
| Antiplatelet agents | Drugs that prevent blood clots by inhibiting platelet function |
| Arterial thrombosis | An interruption of blood flow to an organ or body part due to a blood clot blocking the flow of blood |
| Body mass index (BMI) | Body mass divided by the square of the body height, universally expressed in units of kg/m² |
| Catheters | Medical devices (tubes) that can be inserted in the body for a broad range of functions, such as to treat diseases, to perform a surgical procedure, and to provide medicine, fluids and food |
| COVID‐19 | An infectious disease caused by SARS‐CoV‐2 virus |
| Deep vein thrombosis (DVT) | Coagulation or clotting of the blood in a deep vein, that is, far beneath the surface of the skin |
| Disseminated intravascular coagulopathy | A severe condition in which blood clots form throughout the body, blocking small blood vessels and that may lead to organ failure. As clotting factors and platelets are used up, bleeding may occur, throughout the body (e.g. in the urine, in the stool, or bleeding into the skin) |
| Duplex ultrasound | Non‐invasive evaluation of blood flow through the arteries and veins by ultrasound devices |
| Heparin (also known as unfractionated heparin (UFH)) | A drug used to prevent blood clotting (anticoagulant, blood thinner) |
| Hypercoagulability | An abnormality of blood coagulation that increases the risk of blood clot formation in blood vessels (thrombosis) |
| Low‐molecular‐weight heparin | A drug used to prevent blood clotting (anticoagulant) |
| Obesity | Amount of body fat beyond healthy conditions (BMI > 30 kg/m²) |
| Placebo | Substance or treatment with no active effect, like a sugar pill |
| Platelet | Colourless blood cells that help blood to clot by clumping together |
| Pulmonary embolism (PE) | Blood clot in the lung or blood vessel leading to the lung. The clot originates in a vein (e.g. deep vein thrombosis) and travels to the lung |
| Quasi‐randomised controlled trial (quasi‐RCT) | A study in which participants are divided by date of birth or by hospital register number, i.e. not truly randomly divided into separate groups to compare different treatments |
| Randomised controlled trial (RCT) | A study in which participants are divided randomly into separate groups to compare different treatments |
| Respiratory failure | An abnormality that results from inadequate gas exchange by the respiratory system |
| SARS‐CoV‐2 | The virus (coronavirus 2) that causes COVID‐19 |
| Thrombosis | Local coagulation of blood (clot) in a part of the circulatory system |
| Vascular | Relating to blood vessels (arteries and veins) |
| Venous | Relating to a vein |
| Venous thromboembolism (VTE) | A condition that involves a blood clot that forms in a vein and may migrate to another location (e.g. the lung) |
Description of the condition
The novel coronavirus disease strain, coronavirus disease 2019 (COVID‐19), is caused by severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2). COVID‐19 emerged in Wuhan, China and rapidly spread worldwide (Lai 2020). SARS‐CoV‐2 is a highly transmissible virus, and up to 16% of people hospitalised may develop a severe form of the disease (Giannis 2020). Pulmonary effects are typical, but due to high inflammation, hypoxia, immobilisation and diffuse intravascular coagulation, COVID‐19 may predispose patients to both arterial and venous thromboembolism (Ackermann 2020; COVIDSurg 2021; Dolhnikoff 2020; Fox 2020; Long 2020). Venous and arterial thromboembolic complications affect 16% of people hospitalised with COVID‐19 and 31% to 49% of people with COVID‐19 in intensive care units (ICUs), with 90% of such cases being venous thromboembolism (Bilaloglu 2020; Klok 2020a; Klok 2020b). Viral infections induce an imbalance between anticoagulant and procoagulant mechanisms and raise the systemic inflammatory response. Indeed, people with COVID‐19 commonly present with both elevated D‐dimer (fibrin degradation product) and reductions of factors related to clot formation (Giannis 2020). Excessive activation of the coagulation cascade and platelets could explain these haematological findings (Giannis 2020). Coagulopathy and vascular endothelial dysfunction have been proposed as complications of COVID‐19. Emerging data support the hypothesis that asymptomatic individuals with COVID‐19 are at risk of developing pathological thrombosis. The association between large‐vessel stroke and COVID‐19 in young asymptomatic individuals requires further investigation (Oxley 2020); however, Li 2020 found the incidence of stroke among people hospitalised with COVID‐19 to be approximately 5% in a retrospective cohort. Activation of the coagulation system seems to be important in the development of acute respiratory distress syndrome, one of the most typical complications of COVID‐19 infection and it could be related to pulmonary microthrombosis (Ackermann 2020; Dolhnikoff 2020; Fox 2020; Marini 2020).
Description of the intervention
Anticoagulants are pharmacological interventions used in reducing hypercoagulability (Amaral 2020; Dias 2021). The decision to use thromboprophylaxis or not depends on the risk stratification of each patient (NHS 2020).
Anticoagulants are medications used in the prevention and treatment of venous or arterial thromboembolic events (Amaral 2020; Biagioni 2020; Clezar 2020; Dias 2021). When used for a prophylactic purpose, the dose of anticoagulants is usually half or significantly lower than that given for therapeutic purposes (Alquwaizani 2013). Even so, adverse events such as bleeding may occur and can have a significant impact on patient care (Amaral 2020; AVF 2020; Biagioni 2020; Clezar 2020).
How the intervention might work
D‐dimers are a reflection of the pathophysiology in COVID‐19, which is highly associated with increased mortality in people with COVID‐19 infection (Becker 2020). The elevated D‐dimer levels seen are most likely a reflection of the overall clot burden and critically ill people with COVID‐19 have lower levels of fibrinolytic system activation than the reference population (Panigada 2020). Tang 2020 reported decreased mortality after the use of heparin in people with COVID‐19 (40.0% versus 64.2%, P = 0.029). Long 2020 reported that anticoagulation (mainly low‐molecular‐weight heparin), may reduce mortality in people with severe COVID‐19 infection or those with higher levels of D‐dimer (e.g. greater than six times the upper limit).
Some authors had also correlated this effect with the anti‐inflammatory effect of heparinoids, for instance, binding and neutralising a wide variety of mediators released from inflammatory cells, reducing IL‐6 and as potent inhibitors of the complement system, which may have effects on the clinical evolution of people with COVID‐19 (Liu 2019; Shi 2020; Tang 2020; Young 2008). It can attenuate ongoing tissue damage (Liu 2019; Young 2008). Practical guidelines and specialist consensus are addressing the management of thromboprophylaxis and anticoagulation in people with COVID‐19 infection (Bikdeli 2020; NHS 2020; Obe 2020; Ramacciotti 2020). However, the effects of anticoagulants on people with COVID‐19 is still under debate (Sobreira 2020).
Objectives
To assess the benefits and harms of anticoagulants versus active comparator, placebo or no intervention in people hospitalised with COVID‐19.
Methods
Criteria for considering studies for this review
Types of studies
The protocol for this review was prospectively registered with the Open Science Framework on 7 August 2020 (Flumignan 2020a), and a previous version of this review was published on 02 October 2020 (Flumignan 2020b), and disseminated, including a short version published in another international journal (Flumignan 2021).
We considered parallel or cluster‐randomised controlled trials (RCTs), quasi‐RCTs, and cohort studies. Non‐randomised studies (NRS), such as cohort studies, may be useful for rare adverse events and clinical decisions if there is a lack of controlled studies. Related NRS can be developed faster than RCTs and may represent the only available evidence to guide decision making in this setting. To ensure that we captured all relevant study types, we considered a broad range of empirical studies of any size that provided a quantitative measure of impact (Reeves 2021). We did not consider studies without a comparator group or any retrospective NRS because we identified prospective NRS (better study design). We performed meta‐analyses for all of the included studies (RCTs or NRS) with available data to follow Chapter 24 of the Cochrane Handbook for Systematic Reviews of Interventions (Reeves 2021). When at least 400 participants were included from RCTs, we no longer considered NRS for inclusion. We considered all other types of studies irrelevant for this review. Please find further explanations in Appendix 1.
In order to minimise selection bias for NRS, we planned to include only studies that used statistical adjustment for baseline factors using multivariate analyses for at least these confounding factors: participants already using anticoagulants (e.g. atrial fibrillation), participants who underwent surgery during the hospitalisation, active cancer treatment, concomitant antiplatelet use and history of venous thromboembolism. We only considered studies with a minimum duration of two weeks.
Types of participants
We included all participants eligible for anticoagulation, both male and female of all ages, hospitalised with the diagnosis of COVID‐19. Any hospitalised participants with confirmed COVID‐19 infection were eligible, independent of the disease severity (e.g. patients hospitalised in ICUs or wards). We also considered participants with a previous history of venous thromboembolism for inclusion in this review. However, participants with COVID‐19 treated outside of hospital, that is, those who were not hospitalised, were not eligible for our review.
In future updates of this review, if we find studies with mixed populations, that is, hospitalised and non‐hospitalised participants, and only a subset of the participants meet our inclusion criteria, we will attempt to obtain data for the subgroup of interest from the study authors in order to include the study. For studies with mixed populations for which we cannot get data for the subgroup of interest but at least 50% of the study population are of interest, we will include all participants in our analysis. Moreover, we will explore the effect of this decision in a sensitivity analysis. We will exclude studies in which less than 50% of the population are of interest and the subgroup of interest data are not available.
Types of interventions
We considered the following pharmacological interventions.
Heparinoids, that is, both unfractionated heparin and low‐molecular‐weight heparin, and pentasaccharides (synthetic and selective anticoagulant drugs similar to low‐molecular‐weight heparin)
Vitamin K antagonists
Direct anticoagulants, both factor Xa inhibitors and direct thrombin inhibitors, that is, direct oral anticoagulants and non‐oral direct anticoagulants (e.g. bivalirudin)
We considered studies that compared different formulations, doses, and schedules of the same intervention (e.g. heparinoids).
Some commonly applicable prophylactic doses of the interventions of interest are low‐molecular‐weight heparin 30 mg twice a day or 40 mg daily, and unfractionated heparin 5000 IU three times a day. However, we considered all doses of anticoagulants when used for primary or secondary prophylaxis of thromboembolism as eligible for our review.
Types of comparisons
We included studies that compared one pharmacological intervention (agent or drug) versus another active comparator, or placebo or no treatment with any combination of interventions, provided that co‐treatments were balanced between the treatment and control arms. We allowed other potential interventions (e.g. antiplatelet agents, elastic stockings, intermittent pneumatic compression) as comparators or additional interventions. We also included studies that compared different doses of drugs. We pooled the studies that addressed the same comparisons.
Anticoagulant versus placebo or no treatment (we planned to pool all anticoagulants together – heparinoids, vitamin K antagonists, direct anticoagulants, etc. – if possible)
Anticoagulant versus a different anticoagulant
Anticoagulant versus a different dose, formulation, or schedule of the same anticoagulant
Anticoagulant versus other pharmacological interventions such as antiplatelet agents
Anticoagulant versus non‐pharmacological interventions
Types of outcome measures
We evaluated core outcomes as pre‐defined by the Core Outcome Measures in Effectiveness Trials Initiative for people with COVID‐19 (COMET 2020). We also considered the outcomes after hospital discharge. We intended to present the outcomes at two different time points following the start of the intervention if data were available: short‐term outcomes (at hospital discharge or before); and long‐term outcomes (after hospital discharge).
Our time point of primary interest is short‐term; we, therefore, intended to produce related summary of findings tables only for this time point, and also planned to report the long‐term outcomes at the longest possible time of follow‐up.
Primary
All‐cause mortality
Necessity for additional respiratory support: oxygen by non‐invasive ventilators or high‐flow intubation and mechanical ventilation or extracorporeal membrane oxygenation.
Secondary
Mortality related to COVID‐19
Deep vein thrombosis, symptomatic or asymptomatic, first episode or recurrent confirmed by ultrasonography or angiography (e.g. by computed tomography (CT), magnetic resonance imaging (MRI) or by digital subtraction) from any site (e.g. lower limbs, upper limbs, abdominal)
Pulmonary embolism (symptomatic or asymptomatic, first episode or recurrent, fatal or non‐fatal): a diagnosis had to be confirmed by angiography (e.g. by CT, MRI or digital subtraction) and ventilation‐perfusion scan, or both. We also considered post mortem examination as an objective confirmation of deep vein thrombosis and pulmonary embolism.
Major bleeding: defined by a haemoglobin concentration decrease of 2 g/dL or more, a retroperitoneal or intracranial bleed, a transfusion of two or more units of blood, or fatal haemorrhagic events, as defined by International Society on Thrombosis and Haemostasis (Schulman 2010)
Adverse events. We will consider all possible adverse events separately, as individual outcomes, such as minor bleeding, gastrointestinal adverse effects (e.g. nausea, vomiting, diarrhoea, abdominal pain), allergic reactions, renal failure and amputations.
Hospitalisation time in days
Quality of life: participant's subjective perception of improvement (yes or no) as reported by the study authors or using any validated scoring system such as the Short Form‐36 Health Survey (SF‐36) (Ware 1992)
We planned to include studies in the review irrespective of whether measured outcome data were reported in a ‘usable’ way.
Search methods for identification of studies
An information specialist (LLA) designed and conducted all searches on 20 June 2020, which were informed and verified by a content expert (RLGF) and independently peer reviewed. The search was updated on 14 April 2021.
Electronic searches
We identified eligible study references through systematic searches of the following bibliographic databases.
Cochrane Central Register of Controlled Trials (CENTRAL; 2020, Issue 6) in the Cochrane Library (searched 20 June 2020)
MEDLINE PubMed (1946 to 20 June 2020)
Embase.com Elsevier (1974 to 20 June 2020)
LILACS Virtual Health Library (Latin American and Caribbean Health Sciences Literature database; 1982 to 20 June 2020)
IBECS Virtual Health Library (Indice Bibliográfico Español de Ciencias de la Salud; 2015 to 20 June 2020)
We adapted the preliminary search strategy for MEDLINE PubMed for use in the other databases. We did not apply any RCT filters for any databases; we selected the study design manually because we also considered NRS for inclusion in this review. See Flumignan 2020b for search strategies conducted in June 2020.
For this update, we subsequently conducted systematic update searches of the following databases for relevant trials without language, publication year or publication status restrictions on 14 April 2021:
Cochrane Central Register of Controlled Trials (CENTRAL; 2021, Issue 3) in the Cochrane Library (searched from 20 June 2020 to 14 April 2021; Appendix 2)
MEDLINE PubMed (searched from 20 June 2020 to 14 April 2021; Appendix 3)
Embase.com Elsevier (searched from 1 January 2020 to 14 April 2021; Appendix 4)
LILACS Virtual Health Library (searched from 1 January 2020 to 14 April 2021; Appendix 5)
IBECS Virtual Health Library (searched from 1 January 2020 to 14 April 2021; Appendix 5)
We searched all databases from their inception to the present, and we did not restrict the language of publication or publication status. We considered the adverse effects described in the included studies only. All relevant MeSH and Emtree index terms for COVID‐19 and SARS‐CoV‐2 will be integrated into electronic search strategies in future updates.
Searching other resources
We also conducted a search of the Cochrane COVID-19 Study Register (Appendix 6), a specialised register containing both trial registry records, journal articles and preprints, and medRxiv (Appendix 7), a preprint server, for ongoing or unpublished studies (both searched 20 June 2020). The Cochrane COVID-19 Study Register is a specialised register built within the Cochrane Register of Studies (CRS) and is maintained by Cochrane Information Specialists. The register contains study reports from several sources, including:
daily searches of ClinicalTrials.gov
weekly searches of PubMed
weekly searches of Embase.com
weekly searches of the WHO International Clinical Trials Registry Platform (ICTRP)
monthly searches of the Cochrane Central Register of Controlled Trials (CENTRAL)
Complete data sources and search methods for the register are available at community.cochrane.org/about-covid-19-study-register.
For this update, we subsequently performed update searches of the following on 14 April 2021:
Cochrane COVID-19 Study Register (Appendix 6);
medRxiv (Appendix 7).
We checked the reference lists of all included studies and any relevant systematic reviews identified for additional references to studies. We examined any relevant retraction statements and errata for included studies. We contacted the authors of the included studies for any possible unpublished data. Furthermore, we contacted field specialists to enquire about relevant ongoing or unpublished studies.
Data collection and analysis
Inclusion of non‐English language studies
We considered abstracts and full texts in all languages for inclusion. All potentially eligible non‐English language abstracts progressed to full‐text review, with methods translated for eligibility, and full text translated for data extraction.
Selection of studies
Two review authors (JDST, LCUN) independently screened titles and abstracts of all the potential studies we identified as a result of the search and coded them as 'retrieve' (eligible or potentially eligible/unclear) or 'do not retrieve', using the Covidence tool. If there were any disagreements, we asked a third review author to arbitrate (RLGF). We retrieved the full‐text study reports/publications, and two review authors (JDST, LCUN) independently screened the full text and identified studies for inclusion, while identifying and recording reasons for the exclusion of ineligible studies. We resolved any disagreement through discussion or, if required, we consulted a third person (RLGF). We identified and excluded duplicates and collated multiple reports of the same study so that each study, rather than each report, is the unit of interest in the review. We recorded the selection process in sufficient detail to complete a PRISMA flow diagram (Page 2021), and Characteristics of excluded studies table. We considered studies reported as full text, those published as abstract only, and unpublished data. We considered abstracts and conference proceedings if they were eligible and had usable data.
Data extraction and management
We managed and synthesised the available data using Review Manager 5 (Review Manager 2020). If there was a conflict between data reported across multiple sources for a single study (e.g. between a published article and a trial registry record), we planned to use the article published for numerical analysis, and we planned to report the differences and consider it on the certainty of evidence (GRADE approach; Schünemann 2013).
We used a data collection form, which we piloted on at least one study in the review, for study characteristics and outcome data. One review author (RLGF) extracted study characteristics from the included studies. We planned to extract the following study characteristics.
Methods: study design, total duration of the study, number of study centres and location, study setting, and date of the study
Participants: comorbidities, ventilation support, pregnancy, number randomised, number lost to follow‐up/withdrawn, number analysed, number of interest, mean age, age range, gender, the severity of the condition, inclusion criteria, and exclusion criteria
Interventions: intervention and comparison characteristics (e.g. manufacture, dosage, additional procedures, method of administration), concomitant medications, and excluded medications
Outcomes: primary and secondary outcomes specified and collected (e.g. how outcomes are measured), and time points reported. For NRS: confounding factors controlled for each relevant analysis presented
Notes: funding for the trial, and notable conflicts of interest of study authors
One review author (RLGF) extracted outcome data from included studies independently, which were verified by the other two review authors (CM, BT). We planned to resolve disagreements by discussion. One review author (RLGF) transferred data into Review Manager 5 (Review Manager 2020). We double‐checked that data were entered correctly by comparing the data presented in the systematic review with the data extraction form. Two review authors (CM, BT) spot‐checked study characteristics for accuracy against the study report.
Assessment of risk of bias in included studies
For data from RCTs we used RoB 1 to analyse the risk of bias in the underlying study results (Higgins 2017). For data from prospective NRS, we used the Risk Of Bias in Non‐randomised Studies of Interventions (ROBINS‐I) tool, version of 2016 (Sterne 2016). We also planned to use ROBINS‐I to assess the risk of bias in quasi‐RCTs or retrospective NRS.
Randomised controlled trials
We planned for one review author (RLGF) to assess the risk of bias for each study, and another review author (LCUN) to check all judgements, using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions for RCTs (RoB 1) (Higgins 2017). We planned to resolve any disagreements by consensus or by involving other review authors (CM, BT). For RCTs, we planned to assess the risk of bias according to the following domains.
Random sequence generation
Allocation concealment
Blinding of participants and personnel
Blinding of outcome assessment
Incomplete outcome data
Selective outcome reporting
Other bias
In cluster‐randomised trials, we planned to consider particular biases as recommended by section 8.15.1.1 of the Cochrane Handbook for Systematic Reviews of Interventions:
recruitment bias;
baseline imbalance;
loss of clusters;
incorrect analysis; and
comparability with individually randomised trials (Higgins 2017).
We planned to grade each potential source of bias as high, low or unclear and provide a quote from the study report together with a justification for our judgement in the risk of bias table. We planned to summarise the risk of bias judgements across different studies for each of the domains listed. Where information on the risk of bias relates to unpublished data or correspondence with a study author, we planned to note this in the risk of bias table.
When considering treatment effects, we planned to take into account the risk of bias for the studies that contributed to that outcome.
We planned to base the overall bias judgement of included RCTs on the following three domains of RoB 1:
adequate sequence generation;
blinding of outcome assessors; and
selective outcome reporting.
An RCT at low risk on all of these domains we planned to label as a low‐risk study. An RCT at high risk on one of these domains we planned to label as a high‐risk study. If there was no clear information on the risk of bias for one or more key domains, but the RCT was not at high risk for any domain, we planned to indicate that the risk of bias in the study was unclear.
Non‐randomised studies
Using the ROBINS‐I tool, version of 2016, we planned to assess the risk of bias of quasi‐RCTs and NRS based on the following seven domains (Sterne 2016).
Bias due to confounding
Bias in selection of participants into the study
Bias in classification of interventions
Bias due to deviations from the intended intervention
Bias due to missing data
Bias in measurement of outcomes
Bias in selection of the reported result
We planned to use our risk of bias judgements for quasi‐RCTs and NRS to label all outcomes at all time points, for each comparison, on these domains as 'critical risk', 'serious risk', 'moderate risk', 'low risk', or 'no information'. We planned to judge the overall risk of bias (across domains) as the worst judgment across all the domains. We were interested in the effect of assignment (intention to treat (ITT)) and ROBINS‐I was used to assess all outcomes at all time points.
We considered the following confounders for the assessment of ROBINS‐I domain on 'confounding' and used the Robvis tool to create the risk of bias graphs for NRS (McGuinness 2020).
Participants already using anticoagulants (e.g. atrial fibrillation)
Participants who underwent surgery during hospitalisation
Active cancer treatment
Concomitant antiplatelet use
History of venous thromboembolism
Measures of treatment effect
Please refer to Appendix 1 for information regarding how we had planned to measure the treatment effects of RCTs, quasi‐RCTs and NRS.
Unit of analysis issues
We included RCTs (for one comparison) and NRS (for another comparison) and performed meta‐analysis when appropriate.
Please refer to Appendix 1 for information regarding how we had planned to combine studies with multiple treatment groups.
Dealing with missing data
We planned to contact investigators or study sponsors in order to verify key study characteristics and obtain missing numerical outcome data where possible (e.g. when a study is identified as abstract only). Where possible, we planned to use the Review Manager 5 calculator to calculate missing standard deviations using other data from the study, such as confidence intervals. Where this was not possible, and the missing data were thought to introduce serious bias, we planned to explore the impact of including such studies in the overall assessment of results by a sensitivity analysis. For all outcomes, we planned to follow ITT principles to the highest degree possible: that is, we planned to analyse participants in their randomised group regardless of what intervention they received. We planned to use available case data for the denominator if ITT data were not available. We estimated the mean difference (MD) using the method reported by Wan 2014 to convert median and interquartile range (IQR) into MD and confidence intervals (CI). When it was not possible, we narratively described skewed data reported as medians and IQRs.
Dealing with sparse data
We planned to adjust comparisons (e.g. grouping broader categories of participants (all ages), grouping broader variations of intervention (all types of anticoagulants) accordingly, regardless of sparse data.
Assessment of heterogeneity
We included RCTs (for one comparison) and NRS (for another comparison) and performed meta‐analysis when appropriate.
Please refer to Appendix 1 for information regarding how we had planned to assess heterogeneity.
Assessment of reporting biases
If we were able to pool more than 10 studies, we planned to create and examine a funnel plot to explore possible small‐study biases for the primary outcomes.
Data synthesis
We planned to use a fixed‐effect model for meta‐analysis when included studies were homogeneous (considering population, interventions, comparators and outcomes characteristics). We planned to use a random‐effects model if we identified at least substantial heterogeneity, or if significant clinical differences regarding participants and interventions existed among included studies.
Please refer to Appendix 1 for information regarding how we had planned to synthesise data from RCTs, quasi‐RCTs and NRS. We meta‐analysed data from RCTs (one comparison) and from NRS (another comparison) when appropriate. We also reported the outcome data of each included study narratively and using tables.
Synthesis without meta‐analysis
We planned to synthesise the data using Review Manager 5 (Review Manager 2020). We planned to report data narratively if it was not appropriate to combine it in a meta‐analysis, and planned to undertake meta‐analyses only where this was meaningful, that is, if the treatments, participants and underlying clinical question were similar enough for pooling to make sense.
We aimed to analyse data from NRS separately in a spreadsheet with the exposure of the sample number and the quantitative and qualitative variables relevant to the review, and we also meta‐analysed data from NRS.
We intended to describe skewed data reported as medians and IQRs narratively.
If a meta‐analysis was not possible, we planned to explore the possibilities above to show data of all relevant outcomes considered in this review. Where there was substantial clinical, methodological, or statistical heterogeneity across studies that prevented the pooling of data, we aimed to use a narrative approach to data synthesis. We planned to describe skewed data reported as medians and IQRs narratively.
Subgroup analysis and investigation of heterogeneity
We planned to explore the following subgroups related to participants or interventions if heterogeneity was substantial.
Different doses of drugs
Duration of prophylaxis (e.g. until 30 days after the start of intervention or more)
Age (e.g. children (up to 18 years), adults (18 years to 64 years) and seniors (65 years and over))
Gender
Comorbidities
Illness severity
-
Type of ventilator support:
oxygen by non‐invasive ventilators or high flow
intubation and mechanical ventilation
extracorporeal membrane oxygenation
Sensitivity analysis
We planned to carry out the following sensitivity analyses to test whether critical methodological factors or decisions have affected the main result. We planned to group according to study design (RCTs or cluster‐RCTs, quasi‐RCTs, NRS).
Only including studies with a low risk of bias, as previously specified ('Assessment of risk of bias in included studies').
We planned to examine both the fixed‐effect model and random‐effects model meta‐analyses, and we planned to explore the differences between the two estimates.
We planned to explore the decision to include all participants when at least 50% were of interest in a study with a mixed population.
We planned to explore the impact of missing data. If we identified studies with missing data that were unobtainable, we planned to repeat analyses excluding these studies to determine their impact on the primary analyses.
We also planned to carry out sensitivity analyses considering cluster‐RCTs and investigate the effect of variation in the intracluster correlation coefficient (ICC), as well as planning to acknowledge heterogeneity in the randomisation unit and perform a sensitivity analysis to investigate the effects of this randomisation unit. We aimed to present these results and compare them with the overall findings. We planned to justify any post hoc sensitivity analyses that arose during the review process in the final report.
Summary of findings and assessment of the certainty of the evidence
We created a summary of findings table for both short‐term and long‐term time points using the following outcomes.
All‐cause mortality
Necessity for additional respiratory support
Mortality related to COVID‐19
Deep vein thrombosis
Pulmonary embolism
Major bleeding
Minor bleeding
We used the five GRADE considerations (study limitations; consistency of effect; imprecision; indirectness; and publication bias) to assess the certainty of a body of evidence as it relates to the studies that contributed data to the analyses for the prespecified outcomes. We used methods and recommendations described in Chapter 14 of the Cochrane Handbook for Systematic Reviews of Interventions (Schünemann 2021), using GRADEpro GDT software. We made a separate summary of findings table for each of the following comparisons with available data.
Anticoagulants (higher dose) versus anticoagulants (lower dose)
Anticoagulant (all types) versus no treatment
We justified all decisions to downgrade the certainty of evidence using footnotes, and made comments to aid the reader's understanding of the review where necessary.
Two review authors (RLGF, VTC) made judgements about the certainty of the evidence, with disagreements resolved by discussion or by involving a third review author (LCUN). We justified, documented and incorporated judgements into reporting of results for each outcome. We extracted study data, formatted our comparisons in data tables and prepared a summary of findings table with meta‐analysis before writing the results and conclusions of our review.
Results
Results of the search
For this update, we identified 7322 new records in addition to the 1148 potentially relevant records from the first version (altogether 8470 records). After removing duplicates, we screened 7329 records based on their titles and abstracts, and excluded 7072 records that did not meet the prespecified inclusion criteria. We selected 257 records for full‐text reading. We excluded 129 studies after a full‐text analysis as we considered them not relevant and we excluded 59 studies for at least one reason (see Characteristics of excluded studies). Sixty‐two studies are ongoing (see Characteristics of ongoing studies).
For this review, we found seven studies with available data for inclusion; four RCTs (Lemos 2020; Lopes 2021; Sadeghipour 2021; Zarychanski 2021), and three NRS (Albani 2020; Rentsch 2020; Santoro 2020). See Figure 1 for the study flow diagram (Page 2021). As there is now evidence available from RCTs, and prospective NRS, we excluded the studies analysed in the previous version of this review because they were all retrospective NRS (Ayerbe 2020; Liu 2020; Paranjpe 2020; Russo 2020; Shi 2020; Tang 2020; Trinh 2020) (Flumignan 2020b).
1.
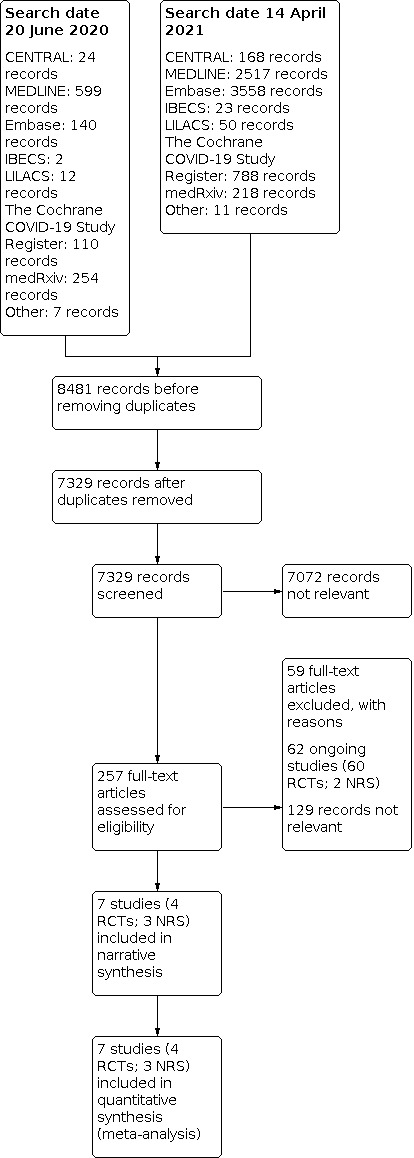
Study flow diagram RCT: randomised controlled trial; NRS: non‐randomised study
Included studies
See Table 4 for the summarised characteristics of included studies.
2. Summary of characteristics of included studies.
| Study (design) | Country | Participant age (mean ± SD) | Setting | Intervention type (dose) | Comparator | All‐cause mortality | Necessity for additional respiratory support | Follow‐up time (mean days) | Total participants allocated | Intervention group participants (anticoagulant) |
|
Albani 2020 (Prospective cohort) |
Italy | 68.66 ± 12.62 (experimental), 70.6 ± 15.01 (comparator) | Hospitala | Enoxaparin (40‐80 mg once daily, duration 3‐9 days) | NA | In‐hospital mortality: aOR 0.53 (95% CI 0.10 to 0.70), in favour of intervention group | NR | Until death or hospital discharge (time in days NR) | 1403 | 799 |
|
Lemos 2020 (RCT) |
Brazil | 55 ± 10 (experimental), 58 ± 16 (comparator) | Hospitala | Therapeutic anticoagulation: heparin (SC enoxaparin, adjusted dose by age and CrCl (maximum dose allowed 140 mg twice daily) | Prophylactic anticoagulation: SC UFH 5000 IU three times/day (if weight < 120 kg) and 7500 IU 3 times/day (if weight > 120 kg) or enoxaparin 40 mg once daily (if weight < 120 kg) and 40 mg twice daily (if weight > 120 kg) according to the doctor's judgment | RR 0.33 (95% CI 0.04 to 2.69) | NR | 28 | 20 | 10 |
|
Lopes 2021 (RCT) |
Brazil | 56.7 ± 14.1 (experimental), 56.5 ± 14.5 (comparator) | Hospitala | Therapeutic anticoagulation: stable participants = rivaroxaban 20 mg once daily; unstable participants = enoxaparin 1 mg/kg twice daily. Followed by rivaroxaban for 30 days, irrespective of the duration of hospitalisation |
Prophylactic anticoagulation: enoxaparin 40 mg once daily | RR 1.49 (95% CI 0.90 to 2.46) | RR 0.16 (95% CI 0.02 to 1.35) | 30 | 615 | 310 |
|
Rentsch 2020 (Prospective cohort) |
USA | 67.03 ± 12.31 (experimental), 67.83 ± 13.74 (comparator) | Hospitala |
|
NA | Inpatient mortality: aHR 0.69 (95% CI 0.61 to 0.77) 30‐day mortality: aHR 0.73 (95% CI 0.66 to 0.81) |
NR | 30 | 4297 | 3627 |
|
Sadeghipour 2021 (RCT) |
Iran | 61.23 ± 14.68 (experimental), 59.66 ± 17.88 (comparator) | Hospitala | Higher‐dose anticoagulation: enoxaparin 1 mg/kg once daily, modified according to body weight and CrCl | Lower‐dose anticoagulation: enoxaparin 40 mg once daily, modified according to body weight and CrCl | Short‐term time point: RR 1.05 (95% CI 0.87 to 1.28) Long‐term time point: RR 1.07 (95% CI 0.89 to 1.29) |
Short‐term time point: no events in both groups Long‐term time point: no events in both groups |
90 | 562 | 276 |
|
Santoro 2020 (Prospective cohort) |
Spain, Italy, Ecuador, Cuba, Germany, China, Canada, Serbia, USA, Chile, and Colombia | 66 ± 15 (experimental), 63 ± 27 (comparator) | Hospitala | Anticoagulant (oral, SC, or IV):
|
NA | RR 0.91 (95% CI 0.89 to 0.93), in all participants (N = 3089) RR 0.58 (95% CI 0.49 to 0.67), in those non‐anticoagulated before admission (N = 2695) RR 0.50 (95% CI 0.37 to 0.70), in those undergoing invasive ventilation (N = 391) RR 0.72 (95% CI 0.51 to 1.01), in those undergoing non‐invasive ventilation (N = 583) |
NR | 15 | 5838 | 2601 |
|
Zarychanski 2021 (RCT) |
UK, USA, Canada, Brazil, Ireland, Netherlands, Australia, Nepal, Saudi Arabia, and Mexico | Critically ill: 60.2 ± 13.1 (experimental), 61.6 ± 12.5 (comparator) Moderate‐severity illness: 59.0 ± 14.1 (experimental), 58.8 ± 13.9 (comparator) |
Hospitala | Therapeutic anticoagulation: LMWH or UFH according to local protocols used for the treatment of acute VTE for up to 14 days or until recovery (defined as hospital discharge, or liberation from supplemental oxygen for ≥ 24 h) | Prophylactic anticoagulation: LMWH or UFH according to local practice or with guidance from the trial protocol on maximum dosing, which included either standard low‐dose thromboprophylaxis or enhanced intermediate dose thromboprophylaxis | Short‐term time point: moderate‐severity RR 0.89 (95% CI 0.67 to 1.19), critically ill RR 1.03 (95% CI 0.88 to 1.21) Long‐term time point: NR |
Short‐term time point: moderate‐severity RR 0.89 (95% CI 0.74 to 1.08), critically ill: NR Long‐term time point: NR |
90 | 3450 | 1780 |
| Total | Australia: 1 Brazil: 3 Canada: 2 Chile: 1 China: 1 Colombia: 1 Cuba: 1 Ecuador: 1 Germany: 1 Iran: 1 Ireland: 1 Italy: 2 Mexico: 1 Nepal: 1 Netherlands: 1 Saudi Arabia: 1 Serbia:1 Spain: 1 UK: 1 USA: 3 | 55 to 68.66 (mean, 7 studies) | ‐ | ‐ | ‐ | 7 studies considered mortality | 4 studies considered additional respiratory support | 15 to 90 (7 studies) | 16,185 | 9403 |
| aHR: adjusted hazard ratio; aOR: adjusted odds ratio; twice daily: twice a day; CI: confidence interval; CrCl: creatinine clearance; DOACs: direct oral anticoagulants; GFR: glomerular filtration rate;HR: hazard ratio; ICU: intensive care units; IU: international unit;LMWH: low‐molecular‐weight heparin; NA: no anticoagulation; NR: not reported; NRS: non‐randomised study;OR: odds ratio; RCT: randomised controlled trial; RR: risk ratio; SC: subcutaneous; SD: standard deviation; SIC: sepsis‐induced coagulopathy; TID: three times a day; UFH: unfractionated heparin; VKA: vitamin K antagonist | ||||||||||
aHospital: includes intensive care unit, hospital wards or emergency department. bAnticoagulation used twice daily if glomerular filtration rate (GFR) was > 30 mL/min, or once daily if GFR was 30 mL/min or less.
We included seven studies describing 16,185 participants in this review, of whom at least 9403 received anticoagulants (Albani 2020; Lemos 2020; Lopes 2021; Rentsch 2020; Sadeghipour 2021; Santoro 2020; Zarychanski 2021). From the seven included studies, four were RCTs (Lemos 2020; Lopes 2021; Sadeghipour 2021; Zarychanski 2021) and the other three NRS of interventions (Albani 2020; Rentsch 2020; Santoro 2020), with a comparator group. Of the seven included studies, two originated from Brazil (Lemos 2020; Lopes 2021), one from Iran (Sadeghipour 2021), one from Italy (Albani 2020), and one from the USA (Rentsch 2020), while two involved several countries (Santoro 2020; Zarychanski 2021).
All included RCTs compared different doses of anticoagulant (lower versus higher) (Lemos 2020; Lopes 2021; Sadeghipour 2021; Zarychanski 2021), and all included NRS compared anticoagulation versus no anticoagulation (Albani 2020; Rentsch 2020; Santoro 2020). All included studies reported a follow‐up period that varied from 15 to 90 days. All included studies considered participants from all settings (ICU, hospital wards and emergency departments), but only Zarychanski 2021 reported data separately by disease severity (critically ill and moderate severity). All included studies reported data regarding the age of participants; the mean age varied from 55 to 68.66 years. All studies reported data on mortality, and four reported data for the necessity for additional respiratory support (Lemos 2020; Lopes 2021; Sadeghipour 2021; Zarychanski 2021).
All studies described the type and dose of anticoagulation. Four studies used only heparin in the intervention group (Albani 2020; Lemos 2020; Sadeghipour 2021; Zarychanski 2021), and the other three analysed data from heparin, vitamin K antagonist or direct anticoagulants. Only Sadeghipour 2021 compared higher‐dose anticoagulation (enoxaparin 1 mg/kg once daily, modified according to body weight and creatinine clearance) versus lower‐dose anticoagulation (enoxaparin 40 mg once daily, modified according to body weight and creatinine clearance) without a therapeutic dose. The other three RCTs compared therapeutic (higher) dose anticoagulation versus prophylactic (lower) dose anticoagulation.
Please refer to the Characteristics of included studies for detailed information.
Excluded studies
We excluded 59 studies for at least one reason (Characteristics of excluded studies). Four studies had ineligible interventions because they evaluated aspirin (NCT04365309), anticoagulants for arterial line heparinisation (Maurer 2020), or anti‐inflammatory drugs (EUCTR2020‐001748‐24‐SE; Mareev 2020), and there was no difference between the intervention groups regarding anticoagulants. Ten studies evaluated ineligible participants (CTRI/2021/01/030373; Kukin 2020; NCT04483830; NCT04492254; NCT04504032; NCT04516941; NCT04662684; NCT04673214; NCT04715295; NCT04757857), and all other excluded studies had an ineligible study design for one of the following reasons:
retrospective cases series without a consistent comparator group;
prospective cohort study without a comparator group (single‐arm study);
prospective cohort study without an intervention purpose;
prospective before‐after cohort study without a parallel comparator group;
editorial articles;
retrospective NRS (new registers and previously included studies from the first version of this review (Flumignan 2020b));
prospective cohort study without a parallel comparator group of intervention.
Ongoing studies
Sixty‐two ongoing studies met our inclusion criteria. They plan to evaluate 35,470 participants (120 participants from two NRS and 35,350 participants from 60 RCTs). From 60 RCTs, 28 are comparing different doses of anticoagulants, 24 are comparing anticoagulants versus no anticoagulants, seven are comparing different types of anticoagulants, and one did not report detail of the comparator group (Wilkinson 2020). We tried to contact the study authors and also searched by study registration number and title of the study on all databases of interest for this review. However, there are no additional data for all these ongoing studies. See the Characteristics of ongoing studies table for further details.
Six of the ongoing studies plan to include 1000 participants or more in RCTs (CTRI/2020/11/029345; EUCTR2020‐001708‐41‐IT; NCT04333407; NCT04366960; NCT04512079; Wilkinson 2020).
CTRI/2020/11/029345 plans to compare prophylactic enoxaparin, full‐dose enoxaparin and apixaban versus no anticoagulant in 3600 participants to assess the composite of all‐cause mortality, intubation requiring mechanical ventilation, systemic thromboembolism or ischaemic stroke.
EUCTR2020‐001708‐41‐IT plans to compare 40 mg enoxaparin once daily versus twice daily in 2000 participants to assess the incidence of venous thromboembolism.
NCT04333407 plans to compare aspirin, clopidogrel, rivaroxaban, atorvastatin, and omeprazole with no treatment in 3170 participants to assess mortality at 30 days.
NCT04366960 plans to compare 40 mg subcutaneous enoxaparin twice daily versus 40 mg subcutaneous enoxaparin once daily to assess venous thromboembolism in 2712 participants. NCT04512079 plans to compare apixaban versus prophylactic enoxaparin and full‐dose enoxaparin in 3600 participants to assess overt bleeding plus haemoglobin drop, cardiac tamponade, bleeding requiring surgical intervention for control, bleeding requiring vasoactive agents, or intracranial haemorrhage (time to event).
Wilkinson 2020 plans to compare several possible interventions (without details about type or dose of anticoagulants) in 1800 participants to assess time to clinical improvement.
See Table 5 for a summary of the characteristics of ongoing studies.
3. Summary of characteristics of ongoing studies.
| Study | Country | Design | Experimental intervention | Comparator intervention | Primary outcomes | Estimated number of participants | Estimated primary completion date |
| ACTRN12620000517976 | Australia | RCT | Nebulised heparin (UFH) | Standard care (without anticoagulants) | Time to separation from invasive ventilation | 172 | 25 July 2021 |
| Busani 2020 | Italy | RCT | Enoxaparin | UFH | All‐cause mortality at day 28, defined as the comparison of proportions of patients' deaths for any cause at day 28 from randomisation | 210 | 6 May 2021 |
| Chambers 2020 | USA | RCT | Intermediate‐dose enoxaparin | Standard prophylactic dose enoxaparin | Risk of all‐cause mortality (time frame: 30 days post‐intervention) | 170 | 16 April 2021 |
| ChiCTR2000030700 | China | RCT | Enoxaparin | Standard care (without anticoagulants) | Time to virus eradication | 60 | 30 September 2020 |
| ChiCTR2000030701 | China | RCT | Enoxaparin | Standard care (without anticoagulants) | Time to virus eradication | 60 | 30 September 2020 |
| ChiCTR2000030946 | China | Prospective cohort | LMWH | Mechanical prevention | Biochemical indicators | 120 | 24 April 2020 |
| CTRI/2020/06/026220 | India | RCT | Nafamostat (synthetic serine proteinase inhibitor) | Standard care (without anticoagulants) | Proportion of patients showing clinical improvement | 40 | 27 January 2021 |
| CTRI/2020/08/027033 | India | RCT | Enoxaparin | Standard care (without anticoagulants) | Reduction in clinical symptoms and RT‐PCR test negative | 100 | 27 January 2021 |
| CTRI/2020/11/029175 | India | RCT | Nebulised heparin | Standard care (without anticoagulants) | Time to separation from mechanical ventilation (duration of mechanical ventilation) up to day 28 | 58 | 27 January 2021 |
| CTRI/2020/11/029345 | India | RCT | Higher‐dose enoxaparin | Lower‐dose enoxaparin; apixaban | Time to first event rate within 30 days of randomisation of the composite of all‐cause mortality, intubation requiring mechanical ventilation, systemic thromboembolism (including PE) confirmed by imaging or requiring surgical intervention or ischaemic stroke confirmed by imaging | 3600 | 27 January 2021 |
| EUCTR2020‐001302‐30‐AT | Austria | RCT | Rivaroxaban | Standard care (without anticoagulants) | Time to sustained improvement of one category from admission | 500 | 11 January 2021 |
| EUCTR2020‐001708‐41‐IT | Italy | RCT | Higher‐dose enoxaparin | Lower‐dose enoxaparin | Incidence of VTE (a composite of incident asymptomatic and symptomatic proximal DVT diagnosed by serial compression ultrasonography, and symptomatic PE diagnosed by CT scan), in patients with SARS‐CoV‐2 infection | 2000 | 30 October 2020 |
| EUCTR2020‐001709‐21‐FR | France | RCT | Higher‐dose LMWH | Lower‐dose LMWH | VTE (causing death or not) | 230 | 11 May 2020 |
| EUCTR2020‐001891‐14‐ES | Spain | RCT | Enoxaparin | Standard care (without anticoagulants) | Need for oxygen therapy escalation due to oxygen saturation (Sat O2) = 92% with FiO2 = 0.5 and respiratory rate = 30 (IROX index = SatO2/FiO2)/FR < 5.5) or invasive mechanical ventilation or mortality during admission | 140 | 16 November 2020 |
| EUCTR2020‐002234‐32‐IT | Switzerland | RCT | Higher‐dose edoxaban | Lower‐dose edoxaban | Major vascular thrombotic events at 25 (+/‐3) days defined as a composite of:
|
420 | 11 January 2021 |
| EUCTR2020‐002504‐39‐DE | Germany | RCT | Edoxaban | Fondaparinux | Composite of all‐cause mortality and/or VTE and/or arterial thromboembolism within 42 days | 172 | 5 January 2021 |
| EUCTR2020‐003349‐12‐IE | Ireland | RCT | Heparin | Standard care (without anticoagulants) | D‐dimer profile, with data collected on days 1, 3, 5 and 10 | 40 | 19 October 2020 |
| Goldin 2020 | USA | RCT | Higher‐dose LMWH | Lower‐dose LMWH | Composite outcome of arterial thromboembolic events, venous thromboembolic events and all‐cause mortality at day 30 ± 2 days (time frame: day 30 ± 2 days). Risk of arterial thromboembolic events (including myocardial infarction, stroke, systemic embolism), VTE (including symptomatic DVT of the upper or lower extremity, asymptomatic proximal DVT of the lower extremity, non‐fatal PE), and all‐cause mortality at day 30 ± 2 days | 308 | 26 April 2021 |
| IRCT20200515047456N1 | Iran | RCT | UFH | Standard care (without anticoagulants) | Decrease D‐dimer level Improve compliance Improve of oxygenation Improve SOFA score |
15 | 13 July 2020 |
| ISRCTN14212905 | UK | RCT | Nafamostat (synthetic serine proteinase inhibitor) | Standard care (without anticoagulants) | Safety of candidate agents as add‐on therapy to standard care in patients with COVID‐19 measured at 30, 60 and 90 days post‐treatment | 100 | 3 August 2020 |
| Kharma 2020 | Qatar | RCT | Bivalirudin (DOAC) | LMWH or UFH | PaO2/FiO2 ratio (time frame: 3 days of intervention) | 100 | 24 June 2020 |
| Lasky 2021 | USA | RCT | Dociparstat (heparinoid) | Placebo | Proportion of participants who are alive and free of invasive mechanical ventilation | 525 | 17 February 2021 |
| Lins 2020 | Brazil | RCT | UFH | Standard care (without UFH) | The percentage of clotted dialysers within 72 h in each of the studied groups | 90 | 27 July 2020 |
| Marietta 2020 | Italy | RCT | Higher‐dose LMWH | Lower‐dose LMWH | Clinical worsening (includes death and necessity for additional respiratory support) | 300 | June 2021 |
| NCT04333407 | UK | RCT | Rivaroxaban | Standard care (without anticoagulants) | All‐cause mortality at 30 days after admission | 3170 | 30 March 2021 |
| NCT04344756 | France | RCT | Higher‐dose LMWH or UFH | Lower‐dose LMWH or UFH | Survival without ventilation | 808 | 31 July 2020 |
| NCT04345848 | Switzerland | RCT | Higher‐dose LMWH or UFH | Lower‐dose LMWH or UFH | Composite outcome of arterial or venous thrombosis, disseminated intravascular coagulation and all‐cause mortality | 200 | 30 November 2020 |
| NCT04352400 | Italy | RCT | Nafamostat (synthetic serine proteinase inhibitor) | Placebo | Time to clinical improvement | 256 | December 2021 |
| NCT04366960 | Italy | RCT | Higher‐dose enoxaparin | Lower‐dose enoxaparin | Incidence of VTE detected by imaging | 2712 | August 2020 |
| NCT04367831 | USA | RCT | Higher‐dose enoxaparin | Lower‐dose enoxaparin | Total number of patients with clinically relevant venous or arterial thrombotic events in ICU | 100 | November 2020 |
| NCT04373707 | France | RCT | Higher‐dose enoxaparin | Lower‐dose enoxaparin | VTE | 602 | September 2020 |
| NCT04377997 | USA | RCT | Higher‐dose LMWH or UFH | Lower‐dose LMWH or UFH | Risk of composite efficacy endpoint of death, cardiac arrest, symptomatic DVT, PE, arterial thromboembolism, myocardial infarction, or haemodynamic shock Risk of major bleeding event according to the ISTH definition |
300 | 1 January 2021 |
| NCT04397510 | USA | RCT | Nebulised heparin | Placebo | Mean daily PaO2/FiO2 | 50 | 31 December 2020 |
| NCT04406389 | USA | RCT | Higher‐dose heparinoid or fondaparinux | Lower‐dose heparinoid or fondaparinux | 30‐day mortality | 186 | December 2021 |
| NCT04409834 | USA | RCT | Higher‐dose heparinoid plus antiplatelet agent | Lower‐dose heparinoid without antiplatelet agent | Venous or arterial thrombotic events | 750 | May 2021 |
| NCT04416048 | Germany | RCT | Higher‐dose DOAC (rivaroxaban) | Lower‐dose heparinoid | Composite endpoint of VTE (DVT and/or fatal or non‐fatal PE), arterial thromboembolism, new myocardial infarction, non‐haemorrhagic stroke, all‐cause mortality or progression to intubation and invasive ventilation (time frame: 35 days post‐randomisation) | 400 | 30 May 2021 |
| NCT04420299 | Spain | RCT | Higher‐dose heparin | Lower‐dose heparin | Combined worsening variable. Presence of any of the following will be considered worsening
|
120 | 31 March 2021 |
| NCT04444700 | Brazil | RCT | Higher‐dose enoxaparin | Lower‐dose enoxaparin | Composite outcome of ICU admission (yes/no), non‐invasive positive pressure ventilation (yes/no), invasive mechanical ventilation (yes/no), or all‐cause death (yes/no) up to 28 days | 462 | 31 December 2020 |
| NCT04485429 | Brazil | RCT | Higher‐dose heparin | Lower‐dose heparin | Rate of invasive mechanical ventilation | 268 | 31 December 2020 |
| NCT04508439 | Mexico | RCT | Higher‐dose enoxaparin | Lower‐dose enoxaparin | Ventilatory support time Thrombotic complications Length of hospital stay Mortality rate |
130 | 30 December 2020 |
| NCT04511923 | Ireland | RCT | Nebulised heparin | Standard care (without anticoagulants) | D‐dimer profile up to day 10 Frequency of severe adverse outcomes up to day 60 |
40 | January 2022 |
| NCT04512079 | USA | RCT | Apixaban (DOAC) | Lower‐dose enoxaparin; higher‐dose enoxaparin | Time to first event (time frame: 30 days) Number of in‐hospital rate of BARC 3 or 5 (time frame: 30 days) Number of in‐hospital rate of BARC 3 or 5 bleeding (binary). BARC Type 3: a. Overt bleeding plus haemoglobin drop of 3 to < 5 g/dL (provided haemoglobin drop is related to bleed); transfusion with overt bleeding b. Overt bleeding plus haemoglobin drop < 5 g/dL (provided haemoglobin drop is related to bleed); cardiac tamponade; bleeding requiring surgical intervention for control; bleeding requiring IV vasoactive agents c. Intracranial haemorrhage confirmed by autopsy, imaging, or lumbar puncture; intraocular bleed compromising vision |
3600 | March 2022 |
| NCT04530578 | Argentina | RCT | Nebulised heparin | Enoxaparin | Percentage of patients requiring mechanical ventilation (time frame: 15 days) | 200 | 1 June 2021 |
| NCT04542408 | Germany | RCT | Higher‐dose LMWH | Lower‐dose LMWH | Combined endpoint: all‐cause mortality and/or VTE and/or arterial thromboembolism (time frame: 42 days)
|
172 | 30 September 2021 |
| NCT04545541 | USA | RCT | Nebulised heparin | Placebo | Alive and Ventilator‐Free Score (time frame: day 28) | 300 | June 2022 |
| NCT04584580 | Egypt | RCT | Higher‐dose LMWH | Lower‐dose LMWH | Mortality (time frame: until patient is discharged or up to 4 weeks whichever comes first) Occurrence of venous and/or arterial thrombosis (time frame: until patient is discharged or up to 4 weeks whichever comes first) |
50 | 31 December 2020 |
| NCT04600141 | Brazil | RCT | Higher‐dose LMWH or UFH | Lower‐dose LMWH or UFH | Proportion of patients with clinical improvement (time frame: 30 days)
|
308 | 31 December 2020 |
| NCT04604327 | Spain | RCT | Higher‐dose LMWH | Lower‐dose LMWH | Clinical deterioration (time frame: 10 days) Combined outcome that includes number of patients who suffer any of the following: death, ICU admission, mechanical ventilatory support, progression to moderate or severe ARDS (according to Berlin criteria) or arterial or venous thrombosis |
164 | 31 July 2021 |
| NCT04623177 | Spain | Prospective cohort | Higher‐dose LMWH | Lower‐dose LMWH; no anticoagulation | ICU mortality rate (time frame: from admission to ICU discharge, an average of 1 month) | 950 | 30 September 2020 |
| NCT04640181 | USA | RCT | Rivaroxaban at low, intermediate or therapeutic dose | Enoxaparin at low, intermediate or therapeutic dose | Death or 30‐day all‐cause mortality (time frame: 30 days) Mechanical ventilation, intubation (time frame: 30 days) Transfer to an ICU setting (time frame: 30 days) |
150 | 31 July 2021 |
| NCT04646655 | Italy | RCT | Higher‐dose enoxaparin | Lower‐dose enoxaparin | Mortality rate (time frame: 30 days from enrolment ) Progression of respiratory failure (time frame: 30 days from enrolment) Progression of respiratory failure (time frame: 30 days from enrolment) Progression of respiratory failure (time frame: 30 days from enrolment) Number of major bleeding episodes (time frame: up to 6 months from randomisation) |
300 | 31 July 2021 |
| NCT04655586 | USA | RCT | Higher‐dose heparin | Lower‐dose heparin | Change in D‐dimer level from baseline to day 8, or day of discharge if prior to day 8 Number of major or non‐major clinically relevant bleeding events within 8 days of randomisation Time to recovery within 30 days of randomisation |
100 | 31 May 2021 |
| NCT04723563 | USA | RCT | Nebulised heparin | Placebo | Need for mechanical ventilation at day 28 | 50 | 29 May 2021 |
| NCT04730856 | Spain | RCT | Higher‐dose heparin | Lower‐dose heparin | Reduction of suspicion of systemic thrombotic symptomatic events (time frame: 30 days) Use of mechanical ventilation (time frame: 30 days) Progression on the WHO Progression Scale during follow‐up (time frame: 30 days) Overall survival at 30 days (time frame: 30 days) Length of hospital stay (days) (time frame: 30 days) Length of ICU stay (days) (time frame: 30 days) |
600 | 31 July 2021 |
| NCT04743011 | Brazil | RCT | Nebulised heparin | Placebo | Change in aPTT > 1.5 (time frame: immediately or up to 8 days after starting treatment) Viral load in nasal swab RT‐PCR (time frame: immediately or up to 8 days after starting treatment) |
50 | 31 December 2021 |
| NCT04745442 | Spain | RCT | Heparin | No anticoagulant | Combined variable: mortality or worsening rate with need for non‐invasive mechanical ventilation or with need for invasive mechanical ventilation (time frame: at day 31 after randomisation or hospital discharge (whichever occurs first) | 48 | 15 January 2021 |
| PACTR202007606032743 | Egypt | RCT | Nebulised heparin | No anticoagulant | The average daily ratio of partial pressure of oxygen to FiO2 (PaO2/FiO2) while the patient is on room air for 7 days | 100 | 22 February 2021 |
| RBR‐7y8j2bs | Brazil | RCT | Nebulised heparin | Placebo | Efficacy: relative to the proposed treatment, through the analysis of the viral load of the SARS‐CoV‐2 virus in the participants treated by the sequential evaluation of the viral load in RT‐PCR of nasal swab. Safety: related to the use of inhalational high‐molecular‐weight heparin in patients with SARS‐CoV‐2 through the assessment of haemorrhagic events of any nature, alteration of the coagulogram that indicates an increase in aPTT > 1.5 and HIT |
40 | 11 October 2021 |
| Sholzberg 2021a | Canada | RCT | Higher‐dose heparinoids | Lower‐dose heparinoids | Composite outcome of ICU admission (yes/no), non‐invasive positive pressure ventilation (yes/no), invasive mechanical ventilation (yes/no), or all‐cause death (yes/no) up to 28 days | 462 | April 2022 |
| Vanassche 2020 | Belgium | RCT | LMWH | DOAC plus aprotinin | The overall objective of the study is to evaluate the clinical efficacy and safety of different investigational therapeutics relative to standard care in patients hospitalised with COVID‐19 | 210 | 18 August 2020 |
| Van Haren 2020 | Argentina | RCT | Nebulised heparin | No anticoagulant | Intubation rate (time frame: day 28) Proportion of patients requiring invasive mechanical ventilation |
712 | 1 June 2021 |
| Wilkinson 2020 | UK | RCT | Anticoagulants (no details) | NA | Time to clinical improvement of at least 2 points (from randomisation) on a 9‐point category ordinal scale, live discharge from the hospital, or considered fit for discharge (a score of 0, 1, or 2 on the ordinal scale), whichever comes first, by day 29 (this will also define the 'responder' for the response rate analyses) | 1800 | 04 September 2021 |
| Total number of studies | Argentina: 2 Australia: 1 Austria: 1 Belgium: 1 Brazil: 6 Canada: 1 China: 3 Egypt: 2 France: 3 Germany: 3 India: 4 Iran: 1 Ireland: 2 Italy: 6 Mexico: 1 Qatar: 1 Spain: 6 Switzerland: 2 UK: 3 USA: 13 |
Prospective cohort: 2 RCT: 60 |
35 studies considered mortality 26 studies considered additional respiratory support |
35,470 participants (120 from NRS; 35,350 from RCTs) | 58 studies to December 2021 Four studies to July 2022 |
||
| aPTT: activated partial thromboplastin time; ARDS: acute respiratory distress syndrome; BARC: Bleeding Academic Research Consortium; CNS: central nervous system; DOACs: direct oral anticoagulants; DVT: deep vein thrombosis; ECMO: extracorporeal membrane oxygenation; FiO2: fraction of inspired oxygen; HIT: heparin‐induced thrombocytopenia; ICU: intensive care unit; ISTH: International Society on Thrombosis and Haemostasis; LMWH: low‐molecular‐weight heparin; NA: not available; NRS: non‐randomised studies; PaO2: arterial oxygen pressure;PE: pulmonary embolism; RCT: randomised controlled trial; RT‐PCR: reverse transcription polymerase chain reaction; SOFA: sequential organ failure assessment; UFH: unfractionated heparin; VKA: vitamin K antagonist; VTE: venous thromboembolism; WHO: World Health Organization | |||||||
Risk of bias in included studies
Risk of bias in randomised controlled trials
Overall judgement
We assessed the risk of bias at the study level using RoB 1 for RCTs (Higgins 2017). The specific judgements ('high risk', 'low risk' or 'unclear risk') by available studies are presented in Figure 2 and Figure 3, and the support for judgement is explained in the related risk of bias tables (Characteristics of included studies). Lopes 2021 and Sadeghipour 2021 had a low overall risk of bias. We judged the other two RCTs at a high overall risk of bias because of 'blinding of outcomes assessment' domain issues (Lemos 2020; Zarychanski 2021), and 'selective reporting' domain issues (Zarychanski 2021).
2.

RoB 1.0 assessments for randomised controlled trials
3.

RoB 1.0 graph: assessments for randomised controlled trials presented as percentages across studies.
Allocation (selection bias)
All four studies had a low risk of bias for random sequence generation and for allocation concealment (Lemos 2020; Lopes 2021; Sadeghipour 2021; Zarychanski 2021).
Blinding (performance bias and detection bias)
Although anticoagulation is a pharmacological intervention that allows the blinding of participants and personnel, all included studies had a high risk of bias (Lemos 2020; Lopes 2021; Sadeghipour 2021; Zarychanski 2021).
We assessed two studies to be at low risk of bias for blinding of outcome assessment (Lopes 2021; Sadeghipour 2021), and two at high risk of bias for this domain (Lemos 2020; Zarychanski 2021).
Incomplete outcome data (attrition bias)
Two studies had a high risk of bias for incomplete outcome reporting (Lemos 2020; Zarychanski 2021). Conversely, Lopes 2021 and Sadeghipour 2021 had a low risk of bias.
Selective reporting (reporting bias)
Zarychanski 2021 was at high risk of bias for selective reporting and none was at unclear risk of bias for this domain. All other included studies (3/4) had a low risk of bias for this domain (Lemos 2020; Lopes 2021; Sadeghipour 2021).
Other potential sources of bias
Zarychanski 2021 was at high risk of bias for other potential sources of bias and all other studies were at low risk of bias for this domain (Lemos 2020; Lopes 2021; Sadeghipour 2021).
Although the study authors declare that they harmonised their protocols into a "prospectively multiplatform uniformisation", Zarychanski 2021 combined the results from three different trials registries, with different 'centres' of randomisation and documentation. There is a possibility of additional heterogeneity in overall results when combining these three trials as a unique trial:
There was an imbalance of losses to follow‐up (moderate‐severity: experimental = 19 losses (1.5%), comparator = 7 losses (0.6%)).
There was a factorial randomisation for antiplatelet agent intervention in one of the considered trials (REMAP‐CAP).
There was a change in the primary outcome specified in the registered protocols compared to the unique reported primary outcome.
We contacted the study authors requesting the data separately, without success. Therefore, we considered Zarychanski 2021 data as a unique study.
Risk of bias in non‐randomised controlled trials
Overall judgement
We assessed the risk of bias at the results level using ROBINS‐I tool for all NRS (Sterne 2016). The specific judgements ('critical risk', 'serious risk', 'moderate risk', 'low risk', or 'no information') by available outcomes are presented in Figure 4, Figure 5, Figure 6, Figure 7, Figure 8, Figure 9 and Figure 10. The support for ROBINS‐I judgement is explained in the related risk of bias tables (Table 6; Table 7; Table 8; Table 9; Table 10; Table 11; Table 12). We will provide detailed risk of bias assessment data on request. The overall risk of bias for all‐cause mortality, deep vein thrombosis, pulmonary embolism, major bleeding, adverse events (stroke and myocardial infarction) in the comparison 'anticoagulants (all types) versus no treatment' was critical. The overall risk of bias for hospitalisation was serious for the same comparison.
4.
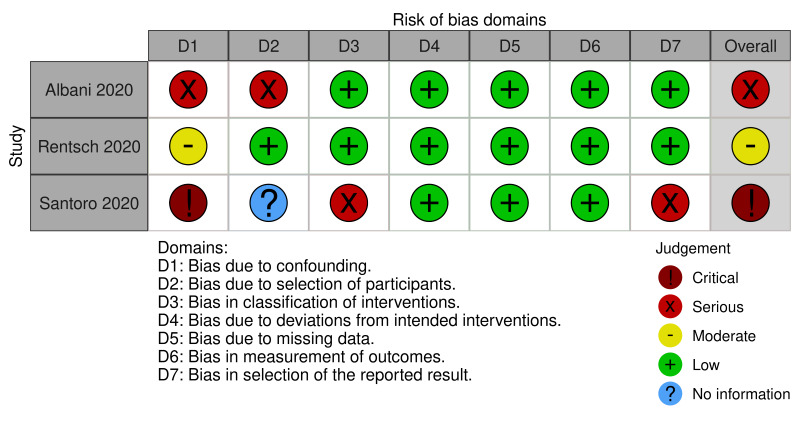
ROBINS‐I assessments: anticoagulants (all types) versus no treatment for people hospitalised with COVID‐19 (all‐cause mortality)
5.
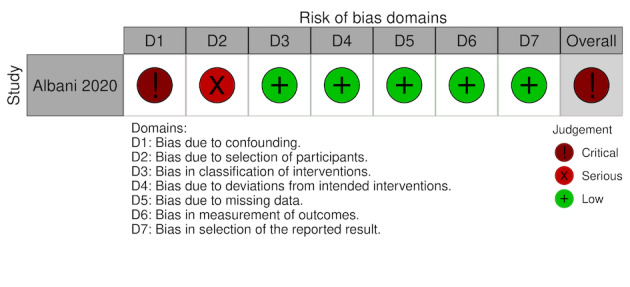
ROBINS‐I assessments: anticoagulants (all types) versus no treatment for people hospitalised with COVID‐19 (deep vein thrombosis)
6.
ROBINS‐I assessments: anticoagulants (all types) versus no treatment for people hospitalised with COVID‐19 (pulmonary embolism)
7.

ROBINS‐I assessments: anticoagulants (all types) versus no treatment for people hospitalised with COVID‐19 (major bleeding)
8.
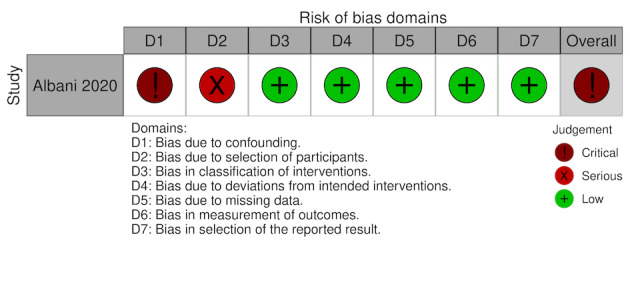
ROBINS‐I assessments: anticoagulants (all types) versus no treatment for people hospitalised with COVID‐19 (adverse events: stroke)
9.
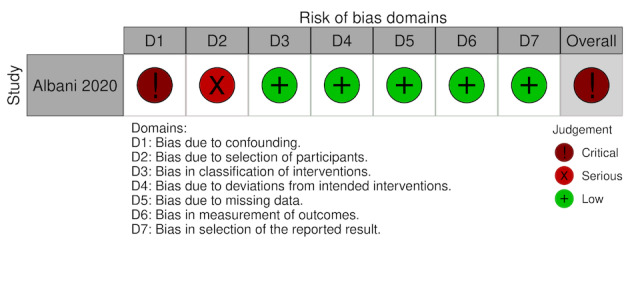
ROBINS‐I assessments: anticoagulants (all types) versus no treatment for people hospitalised with COVID‐19 (adverse events: myocardial infarction)
10.

ROBINS‐I assessments: anticoagulants (all types) versus no treatment for people hospitalised with COVID‐19 (hospitalisation)
4. ROBINS‐I assessments: anticoagulants (all types) versus no treatment for people hospitalised with COVID‐19 (all‐cause mortality).
| Study | Bias due to confounding | Bias in selection of participants into the study | Bias in classification of interventions | Bias due to deviations from the intended intervention | Bias due to missing data | Bias in measurement of outcomes | Bias in selection of the reported result | Overall risk of bias |
| Albani 2020 | Serious risk | Serious risk | Low risk | Low risk | Low risk | Low risk | Low risk | Serious risk |
| Judgement | One or more prognostic variables are likely to be unbalanced between the compared groups. To minimise the impact of the absence of randomisation, an adjusted analysis with propensity scores was performed considering age, sex, disease severity, admission to ICU and COVID‐19 treatment. However, the essential confounding factors: 'participants already using anticoagulants', 'participants who underwent surgery during the hospitalisation', 'active cancer treatment', 'concomitant antiplatelet use' and 'history of venous thromboembolism' were not considered. | Participants included in both groups were selected from a single hospital, and the first dose of anticoagulant was administered between 0 and 3 days after hospital admission. The start of follow‐up and start of intervention possibly did not coincide for most participants, and adjustment techniques to correct the presence of selection bias were not used. It is not clear how prevalent use of anticoagulation was handled. | The intervention groups were clearly defined and recorded at the start of the intervention. Intervention status was probably not affected by knowledge of the outcome or the risk of the outcome. | No deviations from the intended intervention were reported in the study, and if any deviation occurred from usual practice, it was unlikely to impact on the outcome. | There were missing outcome data for 27 participants (1.9% of the total) and balanced between the groups. These missing data possibly could not cause an important impact on the estimate. | It is unlikely that the outcome assessment (objective outcome) was influenced by the knowledge of the intervention received by the study participants. | The study protocol was not identified but all reported results corresponded to the intended outcome. | The study has some important problems. |
| Rentsch 2020 | Moderate risk | Low risk | Low risk | Low risk | Low risk | Low risk | Low risk | Moderate risk |
| Judgement | One or more prognostic variables are likely to be unbalanced between the compared groups. Essential characteristics, such as participants who underwent surgery during the hospitalisation, and history of venous thromboembolism, were not considered. However, an appropriate analysis method to control for measured confounders was used (inverse probability of treatment weighting), and all the important confounding domains for this study were probably controlled. | Participants included in both groups were selected from a nationwide cohort of patients receiving care in the Department of Veterans Affairs in the USA, and selection may have not been related to intervention and outcome. The start of follow‐up and start of intervention coincided for most participants (the first 24 h of hospitalisation). | The intervention groups were clearly defined and recorded at the start of the intervention. Intervention status was probably not affected by knowledge of the outcome or the risk of the outcome. | No deviations from the intended intervention were reported in the study, and if any deviation occurred from usual practice, it was unlikely to impact on the outcome. | No missing data were reported for the outcome. | It is unlikely that the outcome assessment (objective outcome) was influenced by the knowledge of the intervention received by the study participants. | The study protocol was not identified but all reported results corresponded to the intended outcome. | The study is sound for a non‐randomised
study with regard to this domain but cannot be considered comparable to a well‐ performed randomised trial. |
| Santoro 2020 | Critical risk | No information | Serious risk | Low risk | Low risk | Low risk | Serious risk | Critical risk |
| Judgement | One or more prognostic variables are likely to be unbalanced between the compared groups. Essential characteristics, such as participants who underwent surgery during the hospitalisation, and antiplatelet use were not considered. The Cox's multivariable regression analysis was performed to define independent risk factors for the mortality outcome, but only for participants with respiratory failure. |
Insufficient information to judge. There was insufficient information if the start of follow‐up and the start of intervention coincided for most participants. | The intervention groups were not clearly defined and recorded at the start of the intervention. Information about frequency and dose was not provided. | No deviations from the intended intervention were reported in the study, and if any deviation occurred from usual practice, it was unlikely to impact on the outcome. | No missing data were reported for the outcome. | It is unlikely that the outcome assessment (objective outcome) was influenced by the knowledge of the intervention received by the study participants. | The study protocol was available, but it is not possible to exclude bias in selection of reported effect estimate, based on the results, from multiple outcome measurements within the outcome domain (mortality), and multiple effect estimates for different subgroups were provided, omitting varying proportions of the original cohort. |
The study is too problematic to provide useful evidence. |
| COVID‐19: coronavirus disease 2019; ICU: intensive care unit | ||||||||
5. ROBINS‐I assessments: anticoagulants (all types) versus no treatment for people hospitalised with COVID‐19 (deep vein thrombosis).
| Study | Bias due to confounding | Bias in selection of participants into the study | Bias in classification of interventions | Bias due to deviations from the intended intervention | Bias due to missing data | Bias in measurement of outcomes | Bias in selection of the reported result | Overall risk of bias |
| Albani 2020 | Critical risk | Serious risk | Low risk | Low risk | Low risk | Low risk | Low risk | Critical risk |
| Judgement | One or more prognostic variables are likely to be unbalanced between the compared groups. Essential characteristics, such as participants who underwent surgery during the hospitalisation, concomitant antiplatelet use, and history of venous thromboembolism, were not considered. The outcome was reported without any adjustment. | Participants included in both groups were selected from a single hospital, and the first dose of anticoagulant was administered between 0 and 3 days after hospital admission. The start of follow‐up and start of intervention possibly did not coincide for most participants, and adjustment techniques to correct the presence of selection bias were not used. | The intervention groups were clearly defined and recorded at the start of the intervention. Intervention status was probably not affected by knowledge of the outcome or the risk of the outcome. | No deviations from the intended intervention were reported in the study, and if any deviation occurred from usual practice, it was unlikely to impact on the outcome. | There were missing outcome data for 27 participants (1.9% of the total), balanced between the groups. These missing data would probably not have an important impact on the estimate. | It is unlikely that the outcome assessment (objective outcome) was influenced by the knowledge of the intervention received by the study participants. | The study protocol was not identified but all reported results corresponded to the intended outcome. | The study is too problematic to provide useful evidence. |
6. ROBINS‐I assessments: anticoagulants (all types) versus no treatment for people hospitalised with COVID‐19 (pulmonary embolism).
| Study | Bias due to confounding | Bias in selection of participants into the study | Bias in classification of interventions | Bias due to deviations from the intended intervention | Bias due to missing data | Bias in measurement of outcomes | Bias in selection of the reported result | Overall risk of bias |
| Albani 2020 | Critical risk | Serious risk | Low risk | Low risk | Low risk | Low risk | Low risk | Critical risk |
| Judgement | One or more prognostic variables are likely to be unbalanced between the compared groups. Essential characteristics, such as participants who underwent surgery during the hospitalisation, concomitant antiplatelet use, and history of venous thromboembolism, were not considered. The outcome was reported without any adjustment. | Participants included in both groups were selected from a single hospital, and the first dose of anticoagulant was administered between 0 and 3 days after hospital admission. The start of follow‐up and start of intervention possibly did not coincide for most participants, and adjustment techniques to correct the presence of selection bias were not used. | The intervention groups were clearly defined and recorded at the start of the intervention. Intervention status was probably not affected by knowledge of the outcome or the risk of the outcome. | No deviations from the intended intervention were reported in the study, and if any deviation occurred from usual practice, it was unlikely to impact on the outcome. | There were missing outcome data for 27 participants (1.9% of the total), balanced between the groups. These missing data would probably not have an important impact on the estimate. | It is unlikely that the outcome assessment (objective outcome) was influenced by the knowledge of the intervention received by the study participants. | The study protocol was not identified but all reported results corresponded to the intended outcome. | The study is too problematic to provide useful evidence. |
7. ROBINS‐I assessments: anticoagulants (all types) versus no treatment for people hospitalised with COVID‐19 (major bleeding).
| Study | Bias due to confounding | Bias in selection of participants into the study | Bias in classification of interventions | Bias due to deviations from the intended intervention | Bias due to missing data | Bias in measurement of outcomes | Bias in selection of the reported result | Overall risk of bias |
| Albani 2020 | Critical risk | Serious risk | Low risk | Low risk | Low risk | Low risk | Low risk | Critical risk |
| Judgement | One or more prognostic variables are likely to be unbalanced between the compared groups. Essential characteristics, such as participants who underwent surgery during the hospitalisation, concomitant antiplatelet use, and history of venous thromboembolism, were not considered. The outcome was reported without any adjustment. | Participants included in both groups were selected from a single hospital, and the first dose of anticoagulant was administered between 0 and 3 days after hospital admission. The start of follow‐up and start of intervention possibly did not coincide for most participants, and adjustment techniques to correct the presence of selection bias were not used. | The intervention groups were clearly defined and recorded at the start of the intervention. Intervention status was probably not affected by knowledge of the outcome or the risk of the outcome. | No deviations from the intended intervention were reported in the study, and if any deviation occurred from usual practice, it was unlikely to impact on the outcome. | There were missing outcome data for 27 participants (1.9% of the total) and balanced between the groups. These missing data would probably not have an important impact on the estimate. | It is unlikely that the outcome assessment (objective outcome) was influenced by the knowledge of the intervention received by the study participants. | The study protocol was not identified but all reported results corresponded to the intended outcome. | The study is too problematic to provide useful evidence. |
| Santoro 2020 | Critical risk | No information | Serious risk | Low risk | Low risk | Low risk | Low risk | Critical risk |
| Judgement | One or more prognostic variables are likely to be unbalanced between the compared groups. Essential characteristics, such as participants who underwent surgery during the hospitalisation, and antiplatelet use were not considered. The Cox's multivariable regression analysis was performed to define independent risk factors only for the mortality outcome. |
Insufficient information to judge. There was insufficient information if the start of follow‐up and the start of intervention coincided for most participants. | The intervention groups were not clearly defined and recorded at the start of the intervention. Information about frequency and dose was not provided. | No deviations from the intended intervention were reported in the study, and if any deviation occurred from usual practice, it was unlikely to impact on the outcome. | No missing data were reported for the outcome. | It is unlikely that the outcome assessment (objective outcome) was influenced by the knowledge of the intervention received by the study participants. | The study protocol was available, and the reported results corresponded to the intended outcome. | The study is too problematic to provide useful evidence. |
8. ROBINS‐I assessments: anticoagulants (all types) versus no treatment for people hospitalised with COVID‐19 (adverse events (stroke)).
| Study | Bias due to confounding | Bias in selection of participants into the study | Bias in classification of interventions | Bias due to deviations from the intended intervention | Bias due to missing data | Bias in measurement of outcomes | Bias in selection of the reported result | Overall risk of bias |
| Albani 2020 | Critical risk | Serious risk | Low risk | Low risk | Low risk | Low risk | Low risk | Critical risk |
| Judgement | One or more prognostic variables are likely to be unbalanced between the compared groups. Essential characteristics, such as participants who underwent surgery during the hospitalisation, concomitant antiplatelet use, and history of venous thromboembolism, were not considered. The outcome was reported without any adjustment. | Participants included in both groups were selected from a single hospital, and the first dose of anticoagulant was administered between 0 and 3 days after hospital admission. The start of follow‐up and start of intervention possibly did not coincide for most participants, and adjustment techniques to correct the presence of selection bias were not used. | The intervention groups were clearly defined and recorded at the start of the intervention. Intervention status was probably not affected by knowledge of the outcome or the risk of the outcome. | No deviations from the intended intervention were reported in the study, and if any deviation occurred from usual practice, it was unlikely to impact on the outcome. | There were missing outcome data for 27 participants (1.9% of the total), balanced between the groups. These missing data would probably not have an important impact on the estimate. | It is unlikely that the outcome assessment (objective outcome) was influenced by the knowledge of the intervention received by the study participants. | The study protocol was not identified but all reported results corresponded to the intended outcome. | The study is too problematic to provide useful evidence. |
9. ROBINS‐I assessments: anticoagulants (all types) versus no treatment for people hospitalised with COVID‐19 (adverse events (myocardial infarction)).
| Study | Bias due to confounding | Bias in selection of participants into the study | Bias in classification of interventions | Bias due to deviations from the intended intervention | Bias due to missing data | Bias in measurement of outcomes | Bias in selection of the reported result | Overall risk of bias |
| Albani 2020 | Critical risk | Serious risk | Low risk | Low risk | Low risk | Low risk | Low risk | Critical risk |
| Judgement | One or more prognostic variables are likely to be unbalanced between the compared groups. Essential characteristics, such as participants who underwent surgery during the hospitalisation, concomitant antiplatelet use, and history of venous thromboembolism, were not considered. The outcome was reported without any adjustment. | Participants included in both groups were selected from a single hospital, and the first dose of anticoagulant was administered between 0 and 3 days after hospital admission. The start of follow‐up and start of intervention possibly did not coincide for most participants, and adjustment techniques to correct the presence of selection bias were not used. | The intervention groups were clearly defined and recorded at the start of the intervention. Intervention status was probably not affected by knowledge of the outcome or the risk of the outcome. | No deviations from the intended intervention were reported in the study, and if any deviation occurred from usual practice, it was unlikely to impact on the outcome. | There were missing outcome data for 27 participants (1.9% of the total), balanced between the groups. These missing data would probably not have an important impact on the estimate. | It is unlikely that the outcome assessment (objective outcome) was influenced by the knowledge of the intervention received by the study participants. | The study protocol was not identified but all reported results corresponded to the intended outcome. | The study is too problematic to provide useful evidence. |
10. ROBINS‐I assessments: anticoagulants (all types) versus no treatment for people hospitalised with COVID‐19 (hospitalisation).
| Study | Bias due to confounding | Bias in selection of participants into the study | Bias in classification of interventions | Bias due to deviations from the intended intervention | Bias due to missing data | Bias in measurement of outcomes | Bias in selection of the reported result | Overall risk of bias |
| Albani 2020 | Serious risk | Serious risk | Low risk | Low risk | Low risk | Low risk | Low risk | Serious risk |
| Judgement | One or more prognostic variables are likely to be unbalanced between the compared groups. Essential characteristics, such as participants who underwent surgery during the hospitalisation, concomitant antiplatelet use, and history of venous thromboembolism, were not considered. | Participants included in both groups were selected from a single hospital, and the first dose of anticoagulant was administered between 0 and 3 days after hospital admission. The start of follow‐up and start of intervention possibly did not coincide for most participants, and adjustment techniques to correct the presence of selection bias were not used. | The intervention groups were clearly defined and recorded at the start of the intervention. Intervention status was probably not affected by knowledge of the outcome or the risk of the outcome. | No deviations from the intended intervention were reported in the study, and if any deviation occurred from usual practice, it was unlikely to impact on the outcome. | There were missing outcome data for 27 participants (1.9% of the total) and balanced between the groups. These missing data would probably not have an important impact on the estimate. | It is unlikely that the outcome assessment (objective outcome) was influenced by the knowledge of the intervention received by the study participants. | The study protocol was not identified but all reported results corresponded to the intended outcome. | The study has some important problems. |
All‐cause mortality
'Three studies reported mortality for the comparison 'anticoagulants (all types) versus no treatment'. We rated Albani 2020 as serious risk due to confounding and selection of participants. We rated Albani 2020 as low risk for all other domains. We rated Rentsch 2020 as moderate risk for confounding and low risk for all other domains. We rated Santoro 2020 as a critical risk due to confounding, serious risk due to problems with the 'classification of interventions' and 'selection of reported results' items. There was no information about bias due to the selection of participants in Santoro 2020, and all other domains were at low risk. See Figure 4 and Table 6.
Deep vein thrombosis
Albani 2020 reported deep vein thrombosis for the comparison 'anticoagulants (all types) versus no treatment'. We rated Albani 2020 as a critical risk for confounding, serious risk for the selection of participants and low risk for all other domains. See Figure 5 and Table 7.
Pulmonary embolism
Albani 2020 reported pulmonary embolism for the comparison 'anticoagulants (all types) versus no treatment'. We rated Albani 2020 as a critical risk for confounding, serious risk for the selection of participants and low risk for all other domains. See Figure 6 and Table 8.
Major bleeding
Albani 2020 and Santoro 2020 reported major bleeding for the comparison 'anticoagulants (all types) versus no treatment'. We rated both studies as a critical risk of confounding, Albani 2020 as serious risk for the selection of participants and Santoro 2020 as a serious risk for the classification of interventions. Albani 2020 did not report information on the selection of participants and we rated all other domains as low risk for both studies. See Figure 7 and Table 9.
Adverse events (stroke)
Albani 2020 reported stroke for the comparison 'anticoagulants (all types) versus no treatment'. We rated Albani 2020 as a critical risk to confounding, serious risk to the selection of participants and low risk to all other domains. See Figure 8 and Table 10.
Adverse events (myocardial infarction)
Albani 2020 reported myocardial infarction for the comparison 'anticoagulants (all types) versus no treatment'. We rated Albani 2020 as a critical risk for confounding, serious risk for the selection of participants and low risk for all other domains. See Figure 9 and Table 11.
Hospitalisation
Albani 2020 reported hospitalisation for the comparison 'anticoagulants (all types) versus no treatment'. We rated Albani 2020 as serious risk for confounding and for the selection of participants, and as low risk for all other domains. See Figure 10 and Table 12.
Effects of interventions
We included three NRS of interventions for the comparison 'anticoagulants (all types) versus no treatment' (short‐term time point) and four RCTs for the comparison 'higher‐dose anticoagulants versus lower‐dose anticoagulants' (short‐term and long‐term time points) and performed quantitative data analysis (meta‐analysis) when appropriate. We did not perform any funnel plot analysis because there is no comparison with 10 or more studies in this review.
1. Higher‐dose anticoagulants versus lower‐dose anticoagulants (short term)
Four RCTs (Lemos 2020; Lopes 2021; Sadeghipour 2021; Zarychanski 2021), compared heparins (unfractionated heparin) or low‐molecular‐weight heparin) or direct oral anticoagulants (rivaroxaban) in higher doses (2376 participants) versus heparins (unfractionated heparin or low‐molecular‐weight heparin) or direct oral anticoagulants (rivaroxaban) in lower doses (2271 participants). Only Zarychanski 2021 reported outcomes data separately by moderate‐severity and critically ill disease, and we, therefore, included this study in both subgroups. More than 82% of participants in Lopes 2021 had moderate‐severity disease; therefore, we included them in the moderate‐severity subgroup. Lemos 2020 and Sadeghipour 2021 included only participants under invasive ventilatory support or in ICU; therefore, we analysed their data in the critically ill subgroup. See Table 1.
Primary outcomes
All‐cause mortality
All studies reported all‐cause mortality with a follow‐up of up to 30 days. Higher‐dose anticoagulants result in little to no difference in all‐cause mortality compared to lower‐dose anticoagulants for up to 30 days (RR 1.03, 95% CI 0.92 to 1.16; I² = 5%; 4 studies, 4489 participants; high‐certainty evidence; Analysis 1.1). The test for subgroup differences suggested that the severity of the condition has no modifying effect on the all‐cause mortality (Chi² = 0.07, df = 1 (P = 0.80), I² = 0%; Analysis 1.1).
1.1. Analysis.

Comparison 1: Higher‐dose anticoagulants versus lower‐dose anticoagulants (short term), Outcome 1: All‐cause mortality
The sensitivity analysis including only trials at low risk of bias (RR 1.16, 95% CI 0.86 to 1.57; Analysis 1.2) did not substantially change the effect estimate.
1.2. Analysis.

Comparison 1: Higher‐dose anticoagulants versus lower‐dose anticoagulants (short term), Outcome 2: All‐cause mortality ‐ trials at low risk of bias
Necessity for additional respiratory support
Three studies reported the necessity for additional respiratory support with a follow‐up for up to 30 days (Lopes 2021; Sadeghipour 2021; Zarychanski 2021). Lemos 2020 did not report the necessity for additional respiratory support and Zarychanski 2021 reported this outcome only for moderate‐severity participants. Lopes 2021 and Sadeghipour 2021 reported these outcomes for all participants. The evidence is very uncertain about the effect of higher‐dose anticoagulants on necessity for additional respiratory support compared to lower‐dose anticoagulants up to 30 days (RR 0.54, 95% CI 0.12 to 2.47; I² = 60%; 3 studies, 3407 participants; very low‐certainty evidence; Analysis 1.3). The test for subgroup differences was not applicable because the effect in Sadeghipour 2021 was not estimable (no events).
1.3. Analysis.

Comparison 1: Higher‐dose anticoagulants versus lower‐dose anticoagulants (short term), Outcome 3: Necessity for additional respiratory support
The sensitivity analysis including only trials at low risk of bias (RR 0.16, 95% CI 0.02 to 1.35; Analysis 1.4) substantially changed the effect estimate.
1.4. Analysis.

Comparison 1: Higher‐dose anticoagulants versus lower‐dose anticoagulants (short term), Outcome 4: Necessity for additional respiratory support ‐ trials at low risk of bias
Secondary outcomes
Mortality related to COVID‐19
There were no available data for this outcome.
Deep vein thrombosis
Although Zarychanski 2021 reported this outcome only for moderate severity participants, all studies reported deep vein thrombosis with a follow‐up of up to 30 days. Higher‐dose anticoagulants may result in little to no difference in deep vein thrombosis compared to lower‐dose anticoagulants up to 30 days (RR 1.08, 95% CI 0.57 to 2.03; I² = 0%; 4 studies, 3422 participants; low‐certainty evidence; Analysis 1.5). The test for subgroup differences suggested that the severity of the condition has no modifying effect on deep vein thrombosis (Chi² = 0.82, df = 1 (P = 0.36), I² = 0%; Analysis 1.5).
1.5. Analysis.

Comparison 1: Higher‐dose anticoagulants versus lower‐dose anticoagulants (short term), Outcome 5: Deep vein thrombosis
The sensitivity analysis including only trials at low risk of bias (RR 1.21, 95% CI 0.53 to 2.79; Analysis 1.6) did not change the effect estimate substantially.
1.6. Analysis.

Comparison 1: Higher‐dose anticoagulants versus lower‐dose anticoagulants (short term), Outcome 6: Deep vein thrombosis ‐ trials at low risk of bias
Pulmonary embolism
All studies reported pulmonary embolism with a follow‐up of up to 30 days. Higher‐dose anticoagulants may reduce pulmonary embolism compared to lower‐dose anticoagulants for up to 30 days (RR 0.46, 95% CI 0.31 to 0.70; I² = 0%; 4 studies, 4360 participants; moderate‐certainty evidence; Analysis 1.7). The test for subgroup differences suggested that the severity of the condition has no modifying effect on pulmonary embolism (Chi² = 0.08, df = 1 (P = 0.78), I² = 0%; Analysis 1.7).
1.7. Analysis.

Comparison 1: Higher‐dose anticoagulants versus lower‐dose anticoagulants (short term), Outcome 7: Pulmonary embolism
The sensitivity analysis including only trials at low risk of bias (RR 0.50, 95% CI 0.23 to 1.10; Analysis 1.8) changed the effect estimate substantially.
1.8. Analysis.

Comparison 1: Higher‐dose anticoagulants versus lower‐dose anticoagulants (short term), Outcome 8: Pulmonary embolism ‐ trial at low risk of bias
Major bleeding
All studies reported major bleeding with a follow‐up of up to 30 days. Higher‐dose anticoagulants likely increase major bleeding slightly compared to lower‐dose anticoagulants up to 30 days (RR 1.78, 95% CI 1.13 to 2.80; I² = 0%; 4 studies, 4400 participants; moderate‐certainty evidence; Analysis 1.9). The test for subgroup differences suggested that the severity of the condition has no modifying effect on major bleeding (Chi² = 1.03, df = 1 (P = 0.31), I² = 2.8%; Analysis 1.9).
1.9. Analysis.

Comparison 1: Higher‐dose anticoagulants versus lower‐dose anticoagulants (short term), Outcome 9: Major bleeding
The sensitivity analysis including only trials at low risk of bias (RR 2.13, 95% CI 0.92 to 4.90; Analysis 1.10) substantially changed the effect estimate.
1.10. Analysis.

Comparison 1: Higher‐dose anticoagulants versus lower‐dose anticoagulants (short term), Outcome 10: Major bleeding ‐ trials at low risk of bias
Adverse events (minor bleeding, gastrointestinal adverse effects (e.g. nausea, vomiting, diarrhoea, abdominal pain), allergic reactions, renal failure and amputations)
Minor bleeding
Three studies reported minor bleeding with a follow‐up of up to 30 days (Lemos 2020; Lopes 2021; Sadeghipour 2021). Higher‐dose anticoagulants increase adverse events (minor bleeding) compared to lower‐dose anticoagulants up to 30 days (RR 3.28, 95% CI 1.75 to 6.14; I² = 0%; 3 studies, 1196 participants; high‐certainty evidence; Analysis 1.11). The test for subgroup differences suggested that the severity of the condition has no modifying effect on minor bleeding (Chi² = 1.50, df = 1 (P = 0.22), I² = 33.5%; Analysis 1.11).
1.11. Analysis.

Comparison 1: Higher‐dose anticoagulants versus lower‐dose anticoagulants (short term), Outcome 11: Adverse events (minor bleeding)
The sensitivity analysis including only trials at low risk of bias (RR 3.67, 95% CI 1.82 to 7.40; Analysis 1.12) did not change the effect estimate substantially.
1.12. Analysis.

Comparison 1: Higher‐dose anticoagulants versus lower‐dose anticoagulants (short term), Outcome 12: Adverse events (minor bleeding) ‐ trials at low risk of bias
Stroke
Three studies reported stroke with a follow‐up of up to 30 days (Lopes 2021; Sadeghipour 2021; Zarychanski 2021). Higher‐dose anticoagulants may result in little to no difference in adverse events (stroke) compared to lower‐dose anticoagulants for up to 30 days (RR 0.91, 95% CI 0.40 to 2.03; I² = 0%; 3 studies, 4349 participants; low‐certainty evidence; Analysis 1.13). We downgraded two levels due to imprecision (CI of the absolute difference comprises both important clinical benefit and important clinical harm). The test for subgroup differences suggested that the severity of the condition has no modifying effect on stroke (Chi² = 0.00, df = 1 (P = 0.97), I² = 0%; Analysis 1.13).
1.13. Analysis.

Comparison 1: Higher‐dose anticoagulants versus lower‐dose anticoagulants (short term), Outcome 13: Adverse events (stroke)
The sensitivity analysis including only trials at low risk of bias (RR 1.62, 95% CI 0.20 to 13.13; Analysis 1.14) did not substantially change the effect estimate.
1.14. Analysis.

Comparison 1: Higher‐dose anticoagulants versus lower‐dose anticoagulants (short term), Outcome 14: Adverse events (stroke) ‐ trials at low risk of bias
Major adverse limb events
Two studies reported major adverse limb events with a follow‐up of up to 30 days (Lopes 2021; Sadeghipour 2021). Higher‐dose anticoagulants may result in little to no difference in major adverse limb events compared to lower‐dose anticoagulants for up to 30 days (RR 0.33, 95% CI 0.01 to 7.99; I² not applicable; 2 studies, 1176 participants; low‐certainty evidence; Analysis 1.15). We downgraded two levels due to imprecision (CI of the absolute difference comprises both important clinical benefit and important clinical harm). The test for subgroup differences was not applicable because Sadeghipour 2021 reported no events in both groups.
1.15. Analysis.

Comparison 1: Higher‐dose anticoagulants versus lower‐dose anticoagulants (short term), Outcome 15: Adverse events (major adverse limb event)
We judged both studies as low risk of bias and, therefore, no sensitivity analysis was applicable.
Myocardial infarction
Three studies reported myocardial infarction with a follow‐up of up to 30 days (Lopes 2021; Sadeghipour 2021; Zarychanski 2021). Higher‐dose anticoagulants may result in little to no difference in myocardial infarction compared to lower‐dose anticoagulants for up to 30 days (RR 0.86, 95% CI 0.48 to 1.55; I² = 0%; 3 studies, 4349 participants; low‐certainty evidence; Analysis 1.16). We downgraded two levels due to imprecision (CI of the absolute difference comprises both important clinical benefit and important clinical harm). The test for subgroup differences suggested that the severity of the condition has no modifying effect on stroke (Chi² = 0.08, df = 1 (P = 0.78), I² = 0%; Analysis 1.16).
1.16. Analysis.

Comparison 1: Higher‐dose anticoagulants versus lower‐dose anticoagulants (short term), Outcome 16: Adverse events (myocardial infarction)
The sensitivity analysis including only trials at low risk of bias (RR 0.91, 95% CI 0.44 to 1.91; Analysis 1.17) did not substantially change the effect estimate.
1.17. Analysis.

Comparison 1: Higher‐dose anticoagulants versus lower‐dose anticoagulants (short term), Outcome 17: Adverse events (myocardial infarction) ‐ trials at low risk of bias
Atrial fibrillation
Sadeghipour 2021 reported atrial fibrillation with a follow‐up of up to 30 days. Higher‐dose anticoagulants may result in little to no difference in atrial fibrillation compared to lower‐dose anticoagulants for up to 30 days (RR 0.35, 95% CI 0.07 to 1.70, I² not applicable; 1 study, 562 participants; low‐certainty evidence; Analysis 1.18). We downgraded two levels due to imprecision (CI of the absolute difference comprises both important clinical benefit and important clinical harm).
1.18. Analysis.

Comparison 1: Higher‐dose anticoagulants versus lower‐dose anticoagulants (short term), Outcome 18: Adverse events (atrial fibrillation)
Thrombocytopenia
Two studies reported thrombocytopenia with a follow‐up of up to 30 days (Sadeghipour 2021; Zarychanski 2021). Higher‐dose anticoagulants may result in little to no difference in thrombocytopenia compared to lower‐dose anticoagulants for up to 30 days (RR 0.94, 95% CI 0.71 to 1.24; I² not applicable; 2 studies, 2789 participants; low‐certainty evidence; Analysis 1.19). We downgraded two levels due to imprecision (CI of the absolute difference comprises both important clinical benefit and important clinical harm). The test for subgroup differences and the sensitivity analysis were not applicable because Zarychanski 2021 reported no events in both groups.
1.19. Analysis.

Comparison 1: Higher‐dose anticoagulants versus lower‐dose anticoagulants (short term), Outcome 19: Adverse events (thrombocytopenia)
Hospitalisation time in days
Two studies reported the hospitalisation time in days with a follow‐up of up to 30 days for moderate‐severity (Lopes 2021), and critically ill (Lemos 2020), participants. Higher‐dose anticoagulants probably result in little to no difference in hospitalisation time compared to lower‐dose anticoagulants up to 30 days (MD 0.28 days, 95% CI −0.87 to 1.44; I² = 0%; 2 studies, 634 participants; moderate‐certainty evidence; Analysis 1.20). The test for subgroup differences (Chi² = 0.06, df = 1 (P = 0.81), I² = 0%; Analysis 1.20) did not change the effect estimate.
1.20. Analysis.

Comparison 1: Higher‐dose anticoagulants versus lower‐dose anticoagulants (short term), Outcome 20: Hospitalisation time
The sensitivity analysis including only trials at low risk of bias (MD 0.30, −0.86 to 1.46; Analysis 1.21) did not change the effect estimate substantially.
1.21. Analysis.

Comparison 1: Higher‐dose anticoagulants versus lower‐dose anticoagulants (short term), Outcome 21: Hospitalisation time ‐ trials at low risk of bias
Quality of life
There were no available data for this outcome.
2. Higher‐dose anticoagulants versus lower‐dose anticoagulants (long term)
Sadeghipour 2021 compared enoxaparin (low‐molecular‐weight heparin) in higher doses (299 participants) versus enoxaparin in lower doses (299 participants) for participants in ICU and reported data at the follow‐up of up to 90 days (long term).
Primary outcomes
All‐cause mortality
Sadeghipour 2021 reported all‐cause mortality with a follow‐up of up to 90 days. Higher‐dose anticoagulants may result in little to no difference in all‐cause mortality compared to lower‐dose anticoagulants up to 90 days (RR 1.07, 95% CI 0.89 to 1.28; I² not applicable; 1 study, 590 participants; moderate‐certainty evidence; Analysis 2.1). We downgraded the evidence one level due to imprecision (fewer than 300 events were included in the analysis).
2.1. Analysis.

Comparison 2: Higher‐dose anticoagulants versus lower‐dose anticoagulants (long term), Outcome 1: All‐cause mortality
Necessity for additional respiratory support
Sadeghipour 2021 reported the necessity for additional respiratory support with a follow‐up of up to 90 days. The evidence is not estimable about the effect of higher‐dose anticoagulants on necessity for additional respiratory support compared to lower‐dose anticoagulants up to 90 days (no events in both groups; I² not applicable; 1 study, 590 participants; low‐certainty evidence; Analysis 2.2). We downgraded the evidence two levels due to imprecision (no events).
2.2. Analysis.

Comparison 2: Higher‐dose anticoagulants versus lower‐dose anticoagulants (long term), Outcome 2: Necessity for additional respiratory support
Secondary outcomes
Mortality related to COVID‐19
There were no available data for this outcome.
Deep vein thrombosis
Sadeghipour 2021 reported deep vein thrombosis with a follow‐up of up to 90 days. Higher‐dose anticoagulants may result in little to no difference in deep vein thrombosis compared to lower‐dose anticoagulants up to 90 days (RR 1.39, 95% CI 0.45 to 4.33; I² not applicable; 1 study, 590 participants; low‐certainty evidence; Analysis 2.3). We downgraded the evidence two levels due to imprecision (CI of the absolute difference comprises both important clinical benefit and harm, and fewer than 300 events were included in the analysis).
2.3. Analysis.

Comparison 2: Higher‐dose anticoagulants versus lower‐dose anticoagulants (long term), Outcome 3: Deep vein thrombosis
Pulmonary embolism
Sadeghipour 2021 reported pulmonary embolism with a follow‐up of up to 90 days. Higher‐dose anticoagulants may reduce pulmonary embolism compared to lower‐dose anticoagulants up to 90 days (RR 0.40, 95% CI 0.08 to 2.03; I² not applicable; 1 study, 590 participants; low‐certainty evidence; Analysis 2.4). We downgraded the evidence two levels due to imprecision (CI of the absolute difference comprises both important clinical benefit and harm, and fewer than 300 events were included in the analysis).
2.4. Analysis.

Comparison 2: Higher‐dose anticoagulants versus lower‐dose anticoagulants (long term), Outcome 4: Pulmonary embolism
Major bleeding
Sadeghipour 2021 reported major bleeding with a follow‐up of up to 90 days. Higher‐dose anticoagulants may result in little to no difference in major bleeding compared to lower‐dose anticoagulants up to 90 days (RR 1.74, 95% CI 0.51 to 5.87; I² not applicable; 1 study, 590 participants; low‐certainty evidence; Analysis 2.5). We downgraded the evidence two levels due to imprecision (CI of the absolute difference comprises both important clinical benefit and harm, and fewer than 300 events were included in the analysis).
2.5. Analysis.

Comparison 2: Higher‐dose anticoagulants versus lower‐dose anticoagulants (long term), Outcome 5: Major bleeding
Adverse events (minor bleeding, gastrointestinal adverse effects (e.g. nausea, vomiting, diarrhoea, abdominal pain), allergic reactions, renal failure and amputations)
Minor bleeding
Sadeghipour 2021 reported minor bleeding with a follow‐up of up to 90 days. Higher‐dose anticoagulants may increase minor bleeding compared to lower‐dose anticoagulants up to 90 days (RR 2.32, 95% CI 0.90 to 5.95; I² not applicable; 1 study, 590 participants; low‐certainty evidence; Analysis 2.6). We downgraded the evidence two levels due to imprecision (CI of the absolute difference comprises both important clinical benefit and harm, and fewer than 300 events were included in the analysis).
2.6. Analysis.

Comparison 2: Higher‐dose anticoagulants versus lower‐dose anticoagulants (long term), Outcome 6: Adverse events (minor bleeding)
Stroke
Sadeghipour 2021 reported stroke with a follow‐up of up to 90 days. Higher‐dose anticoagulants may result in no difference in stroke compared to lower‐dose anticoagulants up to 90 days (RR 0.99, 95% CI 0.06 to 15.80; I² not applicable; 1 study, 590 participants; low‐certainty evidence; Analysis 2.7). We downgraded the evidence two levels due to imprecision (CI of the absolute difference comprises both important clinical benefit and harm, and fewer than 300 events were included in the analysis).
2.7. Analysis.

Comparison 2: Higher‐dose anticoagulants versus lower‐dose anticoagulants (long term), Outcome 7: Adverse events (stroke)
Acute peripheral arterial thrombosis
Sadeghipour 2021 reported acute peripheral arterial thrombosis with a follow‐up of up to 90 days. The evidence is not estimable about the effect of higher‐dose anticoagulants on acute peripheral arterial thrombosis compared to lower‐dose anticoagulants up to 90 days (no events in both groups; I² not applicable; 1 study, 590 participants; low‐certainty evidence; Analysis 2.8). We downgraded the evidence two levels due to imprecision (no events).
2.8. Analysis.

Comparison 2: Higher‐dose anticoagulants versus lower‐dose anticoagulants (long term), Outcome 8: Adverse events (acute peripheral arterial thrombosis)
Myocardial infarction
Sadeghipour 2021 reported myocardial infarction with a follow‐up of up to 90 days. The evidence is not estimable about the effect of higher‐dose anticoagulants on myocardial infarction compared to lower‐dose anticoagulants up to 90 days (no events in both groups; I² not applicable; 1 study, 590 participants; low‐certainty evidence; Analysis 2.9). We downgraded the evidence two levels due to imprecision (no events).
2.9. Analysis.

Comparison 2: Higher‐dose anticoagulants versus lower‐dose anticoagulants (long term), Outcome 9: Adverse events (myocardial infarction)
Atrial fibrillation
Sadeghipour 2021 reported atrial fibrillation with a follow‐up of up to 90 days. Higher‐dose anticoagulants may result in little to no difference in atrial fibrillation compared to lower‐dose anticoagulants up to 90 days (RR 0.50, 95% CI 0.13 to 1.97; I² not applicable; 1 study, 590 participants; low‐certainty evidence; Analysis 2.10). We downgraded two levels due to imprecision (CI of the absolute difference comprises both important clinical benefit and important clinical harm).
2.10. Analysis.

Comparison 2: Higher‐dose anticoagulants versus lower‐dose anticoagulants (long term), Outcome 10: Adverse events (atrial fibrillation)
Thrombocytopenia
Sadeghipour 2021 reported thrombocytopenia with a follow‐up of up to 90 days. Higher‐dose anticoagulants may result in little to no difference in adverse events (thrombocytopenia) compared to lower‐dose anticoagulants up to 90 days (RR12.91, 95% CI 0.73 to 228.18; I² not applicable; 1 study, 590 participants; low‐certainty evidence; Analysis 2.11). We downgraded two levels due to imprecision (CI of the absolute difference comprises both important clinical benefit and important clinical harm).
2.11. Analysis.

Comparison 2: Higher‐dose anticoagulants versus lower‐dose anticoagulants (long term), Outcome 11: Adverse events (thrombocytopenia)
Hospitalisation time in days
There were no available data for this outcome.
Quality of life
There were no available data for this outcome.
3. Anticoagulants (all types) versus no treatment
Three studies compared enoxaparin (low‐molecular‐weight heparin) (Albani 2020), heparinoids (unfractionated heparin, low‐molecular‐weight heparin or fondaparinux), direct anticoagulants or vitamin K antagonists (Rentsch 2020; Santoro 2020) (7027 participants) to no treatment (4511 participants). Albani 2020 and Rentsch 2020 compared "prophylactic anticoagulation" (including oral, subcutaneous, or intravenous forms) to no treatment. Santoro 2020 compared "prophylactic anticoagulation" in 83% of cases, while 15% received a full dose of low‐molecular‐weight heparin, 1% oral anticoagulation with AVK, and 1% direct anticoagulants (including oral, subcutaneous, or intravenous forms) no treatment. See Table 2.
The Cochrane Handbook for Systematic Reviews of Interventions states that studies judged to be at critical risk of bias should be excluded from the meta‐analysis (Reeves 2021). However, given the small number of studies, there is a balance between loss of information and excluding unreliable information. Therefore, we retained all studies in the analyses, but we also stated the critical risk with the related evidence.
Primary outcomes
All‐cause mortality
Albani 2020 reported all‐cause mortality (follow‐up of up to 15 days) as the proportion of participants and as odds ratio (OR) after adjusting for some covariates (e.g. age, sex, disease severity, admission to ICU and COVID‐19 treatment). They found 200 (25%) deaths in the intervention group and 154 (25.5%) deaths in the comparator group (adjusted OR 0.53, 95% CI 0.40 to 0.70; 1403 participants), in favour of the intervention group after all adjustments (serious risk of bias).
Rentsch 2020 reported all‐cause mortality (follow‐up of up to 30 days) as the proportion of participants and as hazard ratio (HR) after adjusting for some covariates (inverse probability of treatment weighting). They found 418 (11.5%) deaths in the intervention group and 92 (13.7%) deaths in the comparator group (adjusted HR 0.69, 95% CI 0.61 to 0.77; 4297 participants), in favour of the intervention group after all adjustments (moderate risk of bias).
Santoro 2020 reported all‐cause mortality (follow‐up of up to 26 days) as the proportion of participants and as RR after the Cox's multivariable regression analysis only for participants with respiratory failure (2859 participants, 49%). They found 467 (32%) deaths in the intervention group and 588 (42%) deaths in the comparator group (adjusted RR 0.58, 95% CI 0.49 to 0.67; 2859 participants), in favour of the intervention group after all adjustments (critical risk of bias).
We combined these results in a meta‐analysis of adjusted values (Analysis 3.1). Anticoagulants may reduce all‐cause mortality but the evidence is very uncertain due to two study results being at critical and serious risk of bias (RR 0.64, 95% CI 0.55 to 0.74; I² = 53%; 3 NRS, 8395 participants; very low‐certainty evidence; Analysis 3.1). It was not possible to test for subgroup differences and carry out sensitivity analysis.
3.1. Analysis.

Comparison 3: Anticoagulants versus no treatment (short term), Outcome 1: All‐cause mortality
Necessity for additional respiratory support
There were no available data for this outcome.
Secondary outcomes
Mortality related to COVID‐19
There were no available data for this outcome.
Deep vein thrombosis
Albani 2020 reported deep vein thrombosis (follow‐up of up to 15 days) as the proportion of participants but without any adjustment for covariates (e.g. age, sex, disease severity, admission to ICU and COVID‐19 treatment). They found 15 (1.87%) deep vein thromboses in the intervention group and 2 (0.33%) in the comparator group (critical risk of bias). The evidence on DVTs is uncertain (RR 5.67, 95% CI 1.30 to 24.70; I² not applicable; 1 NRS, 1403 participants, very low‐certainty evidence; Analysis 3.2).
3.2. Analysis.

Comparison 3: Anticoagulants versus no treatment (short term), Outcome 2: Deep vein thrombosis
Pulmonary embolism
Albani 2020 reported pulmonary embolism (follow‐up of up to 15 days) as the proportion of participants but without any adjustment for covariates (e.g. age, sex, disease severity, admission to ICU and COVID‐19 treatment). They found 32 (4%) pulmonary embolism in the intervention group and 1 (0.1%) pulmonary embolism in the comparator group (critical risk of bias). The evidence on pulmonary embolism is uncertain (RR 24.19, 95% CI 3.31 to 176.53; I² not applicable; 1 NRS, 1403 participants; very low‐certainty evidence; Analysis 3.3).
3.3. Analysis.

Comparison 3: Anticoagulants versus no treatment (short term), Outcome 3: Pulmonary embolism
Major bleeding
Albani 2020 reported major bleeding (follow‐up of up to 15 days) as the proportion of participants but without any adjustment for covariates (e.g. age, sex, disease severity, admission to ICU and COVID‐19 treatment). They found 16 (2%) major bleeding in the intervention group and 15 (2.4%) major bleeding in the comparator group (critical risk of bias).
Santoro 2020 reported major bleeding (follow‐up of up to 26 days) as the proportion of participants but without any adjustment for covariates. The Cox's multivariable regression analysis was performed to define independent risk factors only for the mortality outcome. They found 70 (2.7%) major bleeding in the intervention group and 58 (1.8%) major bleeding in the comparator group (critical risk of bias).
The evidence on major bleeding is uncertain (RR 1.19, 95% CI 0.66 to 2.12; I² = 58%; 2 NRS, 7218 participants; very low‐certainty evidence; Analysis 3.4). It was not possible to test for subgroup differences and carry out sensitivity analysis.
3.4. Analysis.

Comparison 3: Anticoagulants versus no treatment (short term), Outcome 4: Major bleeding
Adverse events (minor bleeding, gastrointestinal adverse effects (e.g. nausea, vomiting, diarrhoea, abdominal pain), allergic reactions, renal failure and amputations)
Stroke
Albani 2020 reported stroke (follow‐up of up to 15 days) as the proportion of participants but without any adjustment for covariates (e.g. age, sex, disease severity, admission to ICU and COVID‐19 treatment). They found 6 (0.7%) stroke events in the intervention group and 4 (0.6%) in the comparator group (critical risk of bias). The evidence on stroke is uncertain (RR 1.13, 95% CI 0.32 to 4.0; I² not applicable; 1 NRS, 1403 participants; very low‐certainty evidence; Analysis 3.5). We downgraded one level due to study limitations (overall critical risk of bias, especially related to confounding) and two levels due to imprecision (fewer than 300 events were included in the analysis and very large CI).
3.5. Analysis.

Comparison 3: Anticoagulants versus no treatment (short term), Outcome 5: Adverse events (stroke)
Myocardial infarction
Albani 2020 reported myocardial infarction (follow‐up of up to 15 days) as the proportion of participants but without any adjustment for covariates (e.g. age, sex, disease severity, admission to ICU and COVID‐19 treatment). They found 10 (1.2%) myocardial infarction events in the intervention group and no events in the comparator group (critical risk of bias). The evidence on myocardial infarction is uncertain (RR 15.88, 95% CI 0.93 to 270.48; I² not applicable; 1 NRS, 1403 participants; very low‐certainty evidence; Analysis 3.6). We downgraded one level due to study limitations (overall critical risk of bias, especially related to confounding) and two levels due to imprecision (fewer than 300 events were included in the analysis and very large CI).
3.6. Analysis.

Comparison 3: Anticoagulants versus no treatment (short term), Outcome 6: Adverse events (myocardial infarction)
Hospitalisation time in days
Albani 2020 reported hospitalisation time in days (follow‐up of up to 15 days) but without any adjustment for covariates (e.g. age, sex, disease severity, admission to ICU and COVID‐19 treatment). Anticoagulants may increase hospitalisation time compared to no anticoagulation (MD 5.00, 95% CI 4.47 to 5.53; I² not applicable; 1 NRS, 1376 participants; moderate‐certainty evidence; Analysis 3.7). We downgraded one level due to study limitations (overall serious risk of bias, especially related to confounding).
3.7. Analysis.

Comparison 3: Anticoagulants versus no treatment (short term), Outcome 7: Hospitalisation time
Quality of life
There were no available data for this outcome.
Discussion
This review aimed to assess the effects of anticoagulants versus active comparator, placebo or no intervention on mortality and need for additional respiratory support for people hospitalised with COVID‐19.
Summary of main results
We found no quasi‐RCTs with available data assessing the effects of anticoagulants compared to active comparator, placebo or no intervention on mortality and the need for additional respiratory support for people hospitalised with COVID‐19. Since we found better study designs for the comparisons of interest, we excluded all retrospective studies from this review.
We found four RCTs that compared higher versus lower doses of anticoagulants (unfractionated heparin, low‐molecular‐weight heparin, or direct anticoagulants (rivaroxaban)) in 4647 participants hospitalised with COVID‐19 (Table 4). Higher‐dose anticoagulants result in little to no difference in all‐cause mortality and increase minor bleeding compared to lower‐dose anticoagulants for up to 30 days. Higher‐dose anticoagulants probably reduce pulmonary embolism, slightly increase major bleeding, and result in little to no difference in hospitalisation time. They may result in little to no difference in deep vein thrombosis and in adverse events (stroke, major adverse limb event, myocardial infarction, atrial fibrillation, or thrombocytopenia). We are uncertain about the effects on necessity for additional respiratory support because the certainty of evidence is very low. There were no data regarding mortality related to COVID‐19, and quality of life. See Table 1.
One included RCT, which compared higher versus lower doses of anticoagulants (low‐molecular‐weight heparin) in 590 participants hospitalised with COVID‐19 also provided data for the long‐term time point (after hospital discharge) of up to 90 days after intervention (Table 4). Higher‐dose anticoagulants probably result in little to no difference in all‐cause mortality, deep vein thrombosis, and major bleeding, and may reduce pulmonary embolism, increase adverse events (minor bleeding), and result in little to no difference in adverse events (stroke, atrial fibrillation, and thrombocytopenia) compared to lower‐dose anticoagulants for up to 90 days. The evidence is not estimable about the effect of higher‐dose anticoagulants on necessity for additional respiratory support, adverse events (acute peripheral arterial thrombosis, and myocardial infarction) compared to lower‐dose anticoagulants up to 90 days because there were no events. There were no data regarding mortality related to COVID‐19 and quality of life.
We also found three prospective NRS, which compared anticoagulants (heparinoids (unfractionated heparin, low‐molecular‐weight heparin or fondaparinux), direct anticoagulants or vitamin K antagonists) versus no anticoagulants in 11,538 participants hospitalised with COVID‐19 (Table 4). Anticoagulants may reduce all‐cause mortality but the evidence is very uncertain due to two study results being at critical and serious risk of bias. Anticoagulants may increase hospitalisation time compared to no anticoagulants for up to 30 days. We are uncertain about the effects on deep vein thrombosis, pulmonary embolism, major bleeding, adverse events (stroke, and myocardial infarction) because the certainty of evidence is very low. There were no data regarding need for additional respiratory support, mortality related to COVID‐19 or quality of life. See Table 2.
We found 62 ongoing studies (from Argentina: 2, Australia: 1, Austria: 1, Belgium: 1, Brazil: 6, Canada: 1, China: 3, Egypt: 2, France: 3, Germany: 3, India: 4, Iran: 1, Ireland: 2, Italy: 6, Mexico: 1, Qatar: 1, Spain: 6, Switzerland: 2, the UK: 3, and the USA: 13) that plan to evaluate 35,470 participants in this setting, of whom 35,350 individuals are from 60 RCTs, and 120 are from two prospective NRS. Thirty‐five ongoing studies plan to report data for mortality. Twenty‐six ongoing studies plan to report data on the need for additional respiratory support. Fifty‐eight ongoing studies are expected to be completed in December 2021, and four in July 2022. Six of these ongoing studies plan to include 1000 participants or more. See Table 5.
One of the studies plans to compare prophylactic enoxaparin, full‐dose enoxaparin and apixaban versus no anticoagulant in 3600 participants to assess the composite of all‐cause mortality, intubation requiring mechanical ventilation, systemic thromboembolism or ischaemic stroke. One study plans to compare higher‐dose enoxaparin versus lower‐dose enoxaparin in 2000 participants to assess the incidence of venous thromboembolism, while another plans to compare aspirin, clopidogrel, rivaroxaban, atorvastatin, and omeprazole with no treatment in 3170 participants to assess mortality at 30 days. One study plans to compare higher‐dose enoxaparin versus lower‐dose enoxaparin to assess venous thromboembolism in 2712 participants, another plans to compare apixaban versus prophylactic enoxaparin and full‐dose enoxaparin in 3600 participants to assess overt bleeding plus haemoglobin drop, cardiac tamponade, bleeding requiring surgical intervention for control, bleeding requiring vasoactive agents, or intracranial haemorrhage, and another study plans to compare several possible interventions (without details about type or dose of anticoagulants) in 1800 participants to assess time to clinical improvement.
Overall completeness and applicability of the evidence
While all of the studies reported our primary outcome of all‐cause mortality, we found sparse data relating to the need for additional respiratory support and hospitalisation time. It is also noteworthy that none of the studies measured our secondary outcomes such as mortality related to COVID‐19 and quality of life. Furthermore, there are neither data comparing different types of anticoagulants or anticoagulants versus non‐pharmacological interventions, nor data from more than 30 days after the intervention.
There was moderate heterogeneity in the methods of the included studies and many did not provide complete and clear information about their data. This hindered the qualitative analyses and the assessment of the risk of bias of many outcomes in some studies.
The number of studies for each of the possible comparisons was small, ranging from three to four studies. However, the included studies had relatively large primary sample sizes (six studies with 562 or more participants), except for only one study that evaluated 20 participants. The largest study involved 5838 participants, 2601 of whom were treated with anticoagulation in a non‐randomised design but did not provide data regarding one of our primary outcomes (necessity for additional respiratory support).
There was considerable variation in the use of the same intervention (e.g. dosages, type, method of application). The variation in assessment for the confounding factor in NRS also impaired the results.
It is noteworthy that the studies included in this review were conducted in 21 different countries, most of which (52%) were high‐income countries. Social and cultural aspects of the evaluated interventions can also interfere with their acceptability and effectiveness for the treatment of people hospitalised with COVID‐19. Therefore, the external validity of the overall evidence presented in this review should be considered with caution.
We acknowledge that designing and conducting an appropriate study with available data for this topic is difficult. The new approach regarding prophylactic anticoagulants for people hospitalised with COVID‐19 has been used to provide high levels of anticoagulants for these people, although there is now available evidence based on RCTs against their use. This reinforces the importance of this review and serves as an incentive for further investigation.
Certainty of the evidence
We found four RCTs with data for one comparison ('higher‐dose anticoagulants versus lower‐dose anticoagulants') at two different time points and three prospective NRS with data for another comparison ('anticoagulants versus no treatment') at a short‐term time point for this review; we also excluded all retrospective studies.
The overall risk of bias was low for two and high for two RCTs included in the comparison 'higher‐dose anticoagulants versus lower‐dose anticoagulants'. We judged the bias domains due to random sequence generation, allocation concealment, blinding of participants and personnel, blinding of outcome assessment, incomplete outcome data, selective outcome reporting, and other biases from low to high. Although it did not change the effect estimate of all‐cause mortality when excluded in a sensibility analysis, there was a high risk of bias for one large RCT (the only study reporting some of the outcomes of interest). Despite the increasing number of studies on prophylactic anticoagulants for people hospitalised with COVID‐19 in the past months, the overall risk of bias for all‐cause mortality, deep vein thrombosis, pulmonary embolism, major bleeding and adverse events (stroke and myocardial infarction) in the comparison 'anticoagulants versus no treatment' was critical and for hospitalisation was serious in the same comparison. We judged the bias domains due to confounding, selection of participants into the study, classification of interventions, deviations from the intended intervention, measurement of outcomes, and selection of the reported results from low to critical risk of bias.
The certainty of evidence is high to very low. We downgraded the certainty of evidence due to the risk of bias, particularly with regard to detection, performance and attrition in two RCTs and also to selection and other bias in one of them. Although three RCTs (3407 participants) assessed the necessity for additional respiratory support, there is considerable uncertainty about this primary outcome (very low‐certainty evidence). In the NRS, we downgraded the certainty of evidence due to the risk of bias, particularly with regard to the overall critical/serious risk of bias across studies, especially related to confounding or selection bias. We downgraded the certainty of evidence due to study limitations (risk of bias), inconsistency (unexplained heterogeneity) and imprecision (few events and large CI). We decided to pool data, even in NRS, due to the clinically relevant question related to mortality, but the judgements of critical risk of bias mean that these data are particularly unreliable.
Potential biases in the review process
We performed a comprehensive search of the literature and performed study selection according to the Cochrane Handbook for Systematic Reviews of Interventions (Lefebvre 2021). We believe that we identified all of the relevant studies that met our inclusion criteria. However, the possibility remains that we may have missed some studies, particularly in the grey literature. Although we considered 'COVID‐19' and 'SARS‐CoV‐2' as 'Supplementary Concept' or 'free terms' in our search strategies, they were included as 'index terms' in 2021 for databases such as MEDLINE. Therefore, in the future versions of this review, we plan to include these relevant terms also as 'index terms' in our search strategies. We adhered to the inclusion and exclusion criteria prespecified in the protocol in order to limit subjectivity (Flumignan 2020a). We made efforts to obtain additional relevant data from study authors but were unable to do so for all of the included studies. If we can source supplementary data, we will consider them in future updates. Two review authors selected studies in duplicate, independently, to reduce the potential bias of the review process. One review author extracted data and assessed the risk of bias of the included studies while another checked the data extraction and 'Risk of bias' judgements, to accelerate the process and also to reduce the potential bias of the review process. Additional analysis (subgroups and sensitivity analysis) was performed as planned in our protocol, but the conclusions were based on the primary analysis (Flumignan 2020a). We assumed the pragmatic decision to include NRS at critical and serious risk of bias in meta‐analysis due to the relevance of the clinical question. It is perhaps reasonable to have included these in analyses given the small number of studies, but we note that this was a decision taken in the review process. We ensure that any such syntheses were presented throughout the review with unequivocal warnings about the risk of bias and note that the findings cannot rely upon this very low‐certainty evidence.
The synthesis of evidence is a field in constant transformation. Therefore, the Cochrane Reviews are periodically updated, mainly the rapid reviews. During the final process of this review, we identified at least three other trials (898 participants together) that seem to reach our inclusion criteria (Perepu 2021; Sholzberg 2021b; Spyropoulos 2021). We will consider these trials in the next update of this review, but we did not assess them for this rapid review updating.
Agreements and disagreements with other studies or reviews
Since the publication of the latest version of this review (Flumignan 2020b), a number of systematic reviews have addressed the role of anticoagulants in people with COVID‐19.
Abdel‐Maboud 2021 searched MEDLINE, Scopus, Cochrane Library, Science direct, OVID, medRxiv, bioRxiv, and Web of Science without language limits on 2 July 2020. They did not specify the inclusion criteria for study design and limited their search to eight keywords related to intervention and population for all databases and only for registers from December 2019. Abdel‐Maboud 2021 included only NRS, most retrospective cohorts or consecutive series, did not assess the risk of bias or the certainty of evidence and concluded that "current evidence is not sufficient to support the role of prophylactic heparin in reducing mortality among COVID‐19 patients."
Hasan 2020 searched PubMed, Google Scholar, medRxiv and SSRN (preprint server) up to 25 June 2020. They did not specify the inclusion criteria for study design and limited their search to some keywords related to heparin (without other anticoagulant terms) and population and only for data from 2020. Hasan 2020 combined 12 prospective and retrospective cohorts with cross‐sectional studies but did not assess the risk of bias or the certainty of evidence and concluded that prophylactic anticoagulants in higher doses may fail less than those in lower doses for people with COVID‐19 admitted to ICU."
Kamel 2021 searched Google Scholar, PubMed, Scopus, the Cochrane Library and Clinical Trials.gov up to 5 July 2020. They included case‐control and cohort studies and limited their search to English‐language studies. Kamel 2021 used an obsolete risk of bias tool (The Modified Newcastle–Ottawa Scoring System), did not assess the certainty of evidence and concluded that anticoagulants may reduce mortality in people with COVID‐19 and that higher‐dose anticoagulants might offer an advantage over lower‐dose anticoagulants in this setting.
Lazaridis 2021 searched PubMed, Ovid, Google Scholar, MEDLINE and Embase databases from December 2019 to 30 May 2020 with limited terms. They considered only randomised clinical trials, quasi‐experimental studies, case reports and case series for inclusion. Lazaridis 2021 combined four retrospective NRS without an assessment with a validated risk of bias and certainty of evidence tool and concluded that anticoagulants may reduce the mortality in severely ill people with COVID‐19.
Matli 2021 searched Ovid MEDLINE, Web of Science, PubMed and Google Scholar from March 2020 to January 2021 with limited terms related to anticoagulants and antiplatelet agents. They included only English‐language published studies and combined 12 NRS without any risk of bias or certainty of evidence assessment. Matli 2021 concluded that anticoagulants reduced mortality and reduced thromboembolic events in people hospitalised with COVID‐19, but there is a paucity of data on antiplatelet use in combination with anticoagulants in this setting.
McBane 2020 searched MEDLINE and Embase from November 2019 to May 2020. They did not specify the inclusion criteria for study design and the limits regarding study language but limited their search to studies with 100 participants or more. McBane 2020 used an obsolete risk of bias tool (The Modified Newcastle–Ottawa Scoring System), did not assess the certainty of evidence and combined 27 NRS in meta‐analyses to include in their recommendations: 1) lower‐dose anticoagulants for all people hospitalised with COVID‐19, 2) a baseline screening venous ultrasound of lower limbs upon admission in ICU, and 3) extending anticoagulation prophylaxis to 35–45 days post‐hospital discharge to reduce venous thromboembolism while it can increase bleeding, even under low‐quality available evidence.
Moonla 2021 searched PubMed, Embase, and the Cochrane Library from the inception of COVID‐19 (specific date not provided) to 22 October 2020. They included only studies reporting mortality and anticoagulant use in people hospitalised with COVID‐19 without limit regarding the study design, but they limited their inclusion to studies of 10 participants or more. Moonla 2021 reported only one of our included studies' results (Lemos 2020), used an obsolete risk of bias tool (The Modified Newcastle–Ottawa Scoring System) for NRS, and did not assess the certainty of evidence. They combined 17 studies into meta‐analyses and concluded that lower‐dose anticoagulants were associated with lower in‐hospital mortality without excess bleeding compared to no anticoagulation and that the higher‐dose anticoagulation revealed no survival benefit but a three‐fold increase in major bleeding.
Parisi 2021 searched MEDLINE, Embase, PubMed, Web of Science, CENTRAL, medRxiv, and Preprints.org on 8 January 2021. They reported following the Cochrane Handbook for Systematic Reviews of Interventions but did not provide a full search strategy and did not describe the date and language limits. Parisi 2021 considered two of our included studies (Albani 2020; Rentsch 2020), used an obsolete risk of bias tool (The Modified Newcastle–Ottawa Scoring System) for NRS, and did not assess the certainty of evidence. They combined 29 NRS in meta‐analyses, including one study with a mixed population (hospitalised and non‐hospitalised people), and concluded that both higher‐ and lower‐dose anticoagulant regimens are associated with better survival in people with COVID‐19, particularly the severely ill. However, Parisi 2021 added that in non‐critically ill individuals with COVID‐19, the lower‐dose anticoagulant is probably preferred due to the higher risk of bleeding at higher doses.
Patell 2021 searched MEDLINE, Embase, and Cochrane CENTRAL from inception to 29 August 2020. They considered RCTs, retrospective and prospective NRS, or case series of adults hospitalised with COVID‐19 for inclusion and limited their search to studies in English and with 10 or more participants. Patell 2021 used the validated methodological index for non‐randomised studies (MINORS) to assess the risk of bias in the included studies and did not assess the certainty of evidence. They combined 35 NRS of hospitalised people with COVID‐19 in meta‐analyses and concluded that hospitalised patients with COVID‐19 treated with lower‐dose anticoagulants have a decreased rate of thrombosis compared with those receiving no anticoagulant, while higher‐dose anticoagulant regimens were not associated with decreased in‐hospital thrombotic events compared with lower‐dose anticoagulants.
Talasaz 2021 systematically searched ClinicalTrials.gov and the World Health Organization (WHO) International Clinical Trials Registry Platform for ongoing RCTs regarding antithrombotic drugs for people hospitalised and non‐hospitalised with COVID‐19 and reported the results in a narrative review. They reported the results of only one of our included studies (Lemos 2020), and concluded that the "optimal thromboprophylaxis has not been established for patients with this disease".
In order to prevent thrombosis, some clinicians use higher‐dose anticoagulants rather than standard prophylactic (lower) dosing for inpatients with COVID‐19 (AVF 2020; Bikdeli 2020; Obe 2020). However, this practice is not supported by robust evidence. Although some practical guidelines address the management of prophylactic anticoagulation in people with COVID‐19, some of these recommendations are based on non‐COVID‐19 populations or low‐quality COVID‐19‐related evidence (AVF 2020; Bikdeli 2020; NHS 2020; Obe 2020; Ramacciotti 2020). Cuker 2021 searched Cochrane COVID‐19 study register, Embase, Epistemonikos COVID‐19 Evidence, MEDLINE, and WHO Global Research Database in August 2020 without time or language limitations to perform a living guideline under a GRADE approach. Cuker 2021 found very low‐certainty evidence, based mainly on an RCT that we also included in this review (Zarychanski 2021), and made two conditional recommendations in favour of lower‐dose anticoagulation over higher‐dose (intermediate or therapeutic‐intensity) anticoagulation for critical or acute illness patients with COVID‐19 who do not have confirmed or suspected venous thromboembolism.
Our review seems to be more comprehensive than the previous reviews identified here, which used limited search strategies, imposed language or date limits, searched overlapping databases (e.g. SCOPUS, Pubmed, MEDLINE and Web of Science in the same review) or searched a limited number of databases (e.g. ClinicalTrials.gov and the WHO International Clinical Trials Registry Platform only). None of the identified systematic reviews used the GRADE approach (excepting a guideline under GRADE approach (Cuker 2021)) to assess the certainty of evidence. Although some previous reviews identified the potential of anticoagulants in lower doses and no difference with higher doses, the evidence found is conflicting. Since we identified high‐certainty evidence, our conclusions are more decisive for clinical practice.
Authors' conclusions
Implications for practice
Higher‐dose anticoagulants result in little to no difference in all‐cause mortality and increase minor bleeding compared to lower‐dose anticoagulants for people hospitalised with COVID‐19 for up to 30 days. Higher‐dose anticoagulants are likely to reduce pulmonary embolism, slightly increase major bleeding, probably result in little to no difference in hospitalisation time, may result in little to no difference in deep vein thrombosis and in stroke, major adverse limb events, myocardial infarction, atrial fibrillation, or thrombocytopenia. We are uncertain about the effects on necessity for additional respiratory support, mortality related to COVID‐19, and quality of life because the certainty of evidence is very low or there were no data.
Higher‐dose anticoagulants may result in little to no difference in all‐cause mortality, deep vein thrombosis and major bleeding, may reduce pulmonary embolism and increase minor bleeding, and may result in little to no difference in stroke, atrial fibrillation, and thrombocytopenia compared to lower‐dose anticoagulants for up to 90 days. There is a lack of evidence about the effect of higher‐dose anticoagulants on the need for additional respiratory support, mortality related to COVID‐19, acute peripheral arterial thrombosis, myocardial infarction and quality of life compared to lower‐dose anticoagulants for up to 90 days.
Anticoagulants may reduce all‐cause mortality compared to no anticoagulants, but the evidence is very uncertain. We are uncertain about the effects on the need for additional respiratory support, mortality related to COVID‐19, deep vein thrombosis, pulmonary embolism, major bleeding, stroke, myocardial infarction, and quality of life because the certainty of evidence is very low or there were no data.
Implications for research
Although we are very confident that new RCTs will not change the conclusion when comparing anticoagulant doses, high‐quality RCTs that compare anticoagulants for people hospitalised with COVID‐19 are still needed, mainly for the other primary outcome (necessity for additional respiratory support), and the comparison with no anticoagulation. There is further lack of evidence when comparing the types of anticoagulants and the effects of giving anticoagulants for a prolonged period of time (e.g. after hospital discharge).
Since there are 62 ongoing studies (60 RCTs) that plan to evaluate 35,470 participants in this setting, robust evidence may be available soon. Fifty‐eight ongoing studies are expected to be completed in December 2021, and four in July 2022. Six of these plan to include 1000 participants or more, with two studies aiming for 3600 and 3170 participants, respectively, which should be compared to different anticoagulant regimens or to no anticoagulation. There is still a need for RCTs with high methodological quality, that is, adequate reporting of randomisation, allocation concealment and blinding, to assess the effects on this population prospectively in an unconfounded randomised study of anticoagulants for people hospitalised with COVID‐19.
The most notable outcomes to be measured are death and the need for additional respiratory support. Other important issues to be considered are deep vein thrombosis, pulmonary embolism, major bleeding, adverse events, hospitalisation time, and quality of life.
What's new
| Date | Event | Description |
|---|---|---|
| 13 January 2022 | New search has been performed | Search updated to 14 April 2021; new studies incorporated |
| 14 April 2021 | New citation required and conclusions have changed | Updated search. Conclusions changed |
History
Review first published: Issue 10, 2020
| Date | Event | Description |
|---|---|---|
| 2 October 2020 | New citation required but conclusions have not changed | First version published |
Acknowledgements
This review was published in collaboration with the Cochrane Editorial and Methods Department. We particularly thank Sarah Hodgkinson and Liz Bickerdike (Associate Editors), Clare Dooley (Managing Editor), Denise Mitchell (Copy Editor), Theresa Moore (Cochrane Methods Support Unit), Robin Featherstone (Information Specialist) and Leticia Rodrigues (Cochrane Editorial and Methods Department) for their methodological and editorial support. Many thanks to Teo Aminah Wasteneys Quay (Managing Editor, Cochrane Emergency and Critical Care), Harald Harkner (Co‐ordinating Editor, Cochrane Emergency and Critical Care) and Mike Brown (Network Senior Editor, Cochrane Acute and Emergency Care) for their support and contributions at various stages of the editorial process. Thanks to Analysis of Review Group Output (ARGO) for their comments on the Abstract and Plain language summary.
Thanks to Marcelly S Cossi (State University of Rio Grande do Norte, Natal, Brazil), Larissa Souza (Hospital Central Coronel Pedro Germano, Natal, Brazil), Maria ICD Fernandes and Isabelle KF Costa (Federal University of Rio Grande do Norte, Natal, Brazil) for their contributions to the first version of this review.
Thanks to the Division of Vascular and Endovascular Surgery, Federal University of Sao Paulo, Brazil, and Cochrane Brazil for their continuous support of this review.
The following people conducted the editorial process for this review update.
Sign‐off Editor (final editorial decision): Harald Harkner (Co‐ordinating Editor, Cochrane Emergency and Critical Care)
Managing Editor (selected peer reviewers, collated peer‐reviewer comments, provided editorial guidance to authors, edited the article): Anne‐Marie Stephani, Cochrane Central Editorial Service
Editorial Assistant (conducted editorial policy checks and supported editorial team): Leticia Rodrigues, Cochrane Central Editorial Service
Copy Editor (copy‐editing and production): Denise Mitchell, Senior Copy Editor, Cochrane Central Executive
Peer‐reviewers (provided comments and recommended an editorial decision): Vicky Mai (University of British Columbia, Canada), Roberto Pola (Tufts University School of Medicine, USA) (clinical/content review), Stella Maria O'Brien (consumer review), Liz Bickerdike and Rachel Richardson, Cochrane Editorial and Methods Department (methods review), Robin Featherstone, Cochrane Editorial and Methods Department (search review)
Appendices
Appendix 1. Planned methodology for randomised controlled trials (RCTs) and non‐randomised studies (NRS) of interventions
Types of studies
We planned to use the Cochrane Handbook for Systematic Reviews of Interventions to guide the whole of this review process (Higgins 2020a). To assess the effects of prophylactic anticoagulants for people hospitalised with COVID‐19 we had planned to include randomised controlled trials (RCTs) only, as such studies if performed appropriately, currently give the best evidence for experimental therapies in highly controlled therapeutic settings.
In case of insufficient evidence (very low‐certainty evidence or no evidence) available from RCTs to answer this review's questions, we had planned to include prospective controlled non‐randomised studies (NRS) of interventions, including quasi‐randomised controlled trials (e.g. assignment to treatment by alternation, medical register or by date of birth).
In case of insufficient evidence (very low‐certainty evidence or no evidence) available from RCTs, quasi‐RCTs, and prospective NRS, we planned to include retrospective observational studies with a control group.
As there was evidence from RCTs, and prospective NRS, we no longer included retrospective NRS and followed the methodology as specified in the protocol (Flumignan 2020a).
Measures of treatment effect
Dichotomous data
For dichotomous variables, we planned to calculate the risk ratio (RR) and 95% confidence intervals (CIs).
Continuous data
For continuous data, we planned to calculate mean differences (MD) and 95% CIs between treatment groups where studies reported the same outcomes. Where similar outcomes are reported on different scales, we planned to calculate the standardised mean difference (SMD) and 95% CI. To interpret SMD, we planned to use the following thresholds.
SMD less than 0.2 = trivial or no effect
SMD equal to or greater than 0.2 and less than 0.5 = small effect
SMD equal to or greater than 0.5 and less than 0.8 = medium effect
SMD equal to or greater than 0.8 = large effect
Unit of analysis issues
We planned to seek advice from a statistician (VTC) to address issues relating to double‐counting, correlation or unit of analysis posed by the following.
Cluster‐RCTs
Episodes of disease
Multi‐arm studies
We planned for individuals to be our unit of analysis. If studies included multi‐arm interventions, we planned to consider only the arms relevant to the scope of our review.
Cluster‐randomised trials
We planned to include cluster‐randomised trials in the analyses along with individually randomised RCTs. We planned to adjust their sample sizes using the methods described in Section 23.1.5 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2020b), using an estimate of the intracluster correlation coefficient (ICC) derived from the trial (if possible), from a similar trial, or a study of a similar population. If we used ICCs from other sources, we planned to report this and conduct sensitivity analyses to investigate the effect of variation in the ICC. If we identified both cluster‐randomised trials and individually randomised trials, we planned to synthesise the relevant information. We planned to consider it reasonable to combine the results from both types of studies if there was little heterogeneity between the study designs, and we considered the interaction between the effect of the intervention and the choice of randomisation unit to be unlikely. We also planned to acknowledge heterogeneity in the randomisation unit and perform a sensitivity analysis to investigate the effects of the randomisation unit.
Assessment of heterogeneity
We planned to inspect forest plots visually to consider the direction and magnitude of effects and the degree of overlap between confidence intervals. We planned to use the I² statistic (Higgins 2003), to measure heterogeneity among the studies in each analysis, but acknowledge that there is substantial uncertainty in the value of the I² statistic when there is only a small number of studies: we therefore also planned to consider the P value from the Chi² test. If we identified substantial heterogeneity, we planned to report it and explore possible causes by prespecified subgroup analysis.
As strict thresholds for interpretation of the I² statistic are not recommended, we intended to follow the rough guide to interpretation in the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2021).
0% to 40%: might not be important
30% to 60%: may represent moderate heterogeneity
50% to 90%: may represent substantial heterogeneity
75% to 100%: considerable heterogeneity
When the I² statistic lies in an area of overlap between two categories (e.g. between 50% and 60%), we planned to consider differences in participants and interventions among the studies contributing data to the analysis (Deeks 2021).
Data synthesis
In preparation for synthesis (either meta‐analyses or synthesis without meta‐analysis), we planned to assess how much data are available for each of our comparisons by the following.
Table to compare PICO elements/study design features
Conversion of numerical data for meta‐analysis
Forest plots
Qualitative synthesis
Synthesis without meta‐analysis
Appendix 2. Cochrane Central Register of Controlled Trials (CENTRAL; via the Cochrane Library) search strategy
#1(2019 novel coronavirus infection) or (COVID‐19 pandemic) or (coronavirus disease‐19) or (COVID19) or (2019 novel coronavirus disease) or (coronavirus disease 2019) or COVID‐19
#2MeSH descriptor: [Severe Acute Respiratory Syndrome] explode all trees
#3(Wuhan coronavirus) or (Wuhan seafood market pneumonia virus) or (COVID19 virus) or (COVID‐19 virus) or (coronavirus disease 2019 virus) or (SARS‐CoV‐2) or (SARS2) or (2019 novel coronavirus)
#4MeSH descriptor: [Coronavirus] explode all trees
#5Coronavirus* or Deltacoronavirus* or Deltacoronavirus*
#6#1 OR #2 OR #3 OR #4 OR #5
#7MeSH descriptor: [Antithrombins] explode all trees
#8(Direct Thrombin Inhibitor*) or (Direct Antithrombin*) or (thrombin inhibitor)
#9MeSH descriptor: [Coumarins] explode all trees
#10Coumarin* or (Benzopyran 2 ones) or (Coumarin Derivative*)
#11MeSH descriptor: [Dabigatran] explode all trees
#12Pradaxa or (Dabigatran Etexilate) or (Dabigatran Etexilate Mesylate)
#13MeSH descriptor: [Anticoagulants] explode all trees #14(Anticoagulation Agent*) or (Anticoagulant Drug*) or Anticoagulant* or (Indirect Thrombin Inhibitor*) #15MeSH descriptor: [Heparin] explode all trees #16(Unfractionated Heparin) or (Heparinic Acid) or Liquaemin or (Sodium Heparin) or alpha‐Heparin or (alpha Heparin) or UFH or heparin* #17MeSH descriptor: [Fondaparinux] explode all trees #18(Fondaparinux Sodium) or Quixidar or Arixtra #19MeSH descriptor: [Hirudin Therapy] explode all trees #20Leeching or Hirudin* #21MeSH descriptor: [Phenindione] explode all trees #22Phenylindanedione or Phenyline or Pindione or Fenilin or Dindevan #23MeSH descriptor: [Polysaccharides] explode all trees #24Glycans #25MeSH descriptor: [Rivaroxaban] explode all trees #26Xarelto or Rivaroxaban #27MeSH descriptor: [Warfarin] explode all trees #28Apo‐Warfarin or Aldocumar or Gen‐Warfarin or Warfant or Coumadin* or Marevan or Tedicumar or warfarin* #29MeSH descriptor: [Factor Xa Inhibitors] explode all trees #30(factor Xa inhibitor*) #31MeSH descriptor: [Enoxaparin] explode all trees #32Enoxaparin* or Lovenox or Clexane #33reviparin* or Clivarine or reviparin‐sodium or (reviparin sodium) or Clivarin #34MeSH descriptor: [Dalteparin] explode all trees #35Tedelparin or (Dalteparin Sodium) or Fragmin or Fragmine #36danaproid or Orgaran or Lomoparan or (danaparoid sodium) or (danaproid sodium) or danaparoid* or DOAC or embolex or Liquemine or (oral anticoagulants) or Pentasaccharide* or (vitamin k antagonist) or Savaysa or (edoxaban tosylate) or edoxaban or xi‐melagatran or Exanta #37MeSH descriptor: [Phenprocoumon] explode all trees #38Phenylpropylhydroxycumarinum or Phenprocoumalol or Phenprocoumarol or Phenprogramma or Marcoumar or Marcumar or Falithrom or Liquamar or Oligosaccharides or (idraparinux sodium) #39MeSH descriptor: [Tinzaparin] explode all trees #40(Tinzaparin Sodium) or Innohep #41MeSH descriptor: [Heparin, Low‐Molecular‐Weight] explode all trees #42(Heparin Low Molecular Weight) or LMWH or (Low‐Molecular‐Weight Heparin) or parnaparin or Azetidines or Benzylamines #43MeSH descriptor: [Nadroparin] explode all trees #44Nadroparin* or Fraxiparin*#45MeSH descriptor: [Acenocoumarol] explode all trees #46Nicoumalone or Acenocoumarin or Sinthrome or Synthrom or Syncoumar or Syncumar or Sinkumar or Sintrom or Mini‐Sintrom or (Mini Sintrom) or MiniSintrom or Lactones or Pyridines #47#7 OR #8 OR #9 OR #10 OR #11 OR #12 OR #13 OR #14 OR #15 OR #16 OR #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR #23 OR #24 OR #25 OR #26 OR #27 OR #28 OR #29 OR #30 OR #31 OR #32 OR #33 OR #34 OR #35 OR #36 OR #37 OR #38 OR #39 OR #40 OR #41 OR #42 OR #43 OR #44 OR #45 OR #46 #48#6 AND #47 #49#48 AND trials #50#49 AND Filter: Custom date range 20/06/2020 to 14/04/2021
Appendix 3. MEDLINE (PubMed) search strategy
1 "COVID‐19" [Supplementary Concept] or (2019 novel coronavirus infection) or (2019‐nCoV infection) or (COVID‐19 pandemic) or (coronavirus disease‐19) or (2019‐nCoV disease) or (COVID19) or (2019 novel coronavirus disease) or (coronavirus disease 2019) or COVID‐19
2 "severe acute respiratory syndrome coronavirus 2" [Supplementary Concept] or (Wuhan coronavirus) or (Wuhan seafood market pneumonia virus) or (COVID19 virus) or (COVID‐19 virus) or (coronavirus disease 2019 virus) or (SARS‐CoV‐2) or (SARS2) or (2019‐nCoV) or (2019 novel coronavirus)
3 "Coronavirus"[Mesh] or Coronavirus* or Deltacoronavirus*
4 #1 OR #2 OR #3
5 "Antithrombins"[Mesh] or (Direct Thrombin Inhibitor*) or (Direct Antithrombin*) or (thrombin inhibitor)
6 "Coumarins"[Mesh] or Coumarin* or (1,2‐Benzopyrone Derivative*) or (1,2 Benzopyrone Derivative*) or Benzopyran‐2‐ones or (Benzopyran 2 ones) or (Coumarin Derivative*) or (1,2‐Benzopyrones) or (1,2 Benzopyrones) or (1,2‐Benzo‐Pyrones) or (1,2 Benzo Pyrones)
7 "Dabigatran"[Mesh] or Pradaxa or Dabigatran*
8 "Anticoagulants"[Mesh] or Anticoagulant* or (Indirect Thrombin Inhibitor*)
9 "Heparin"[Mesh] or (Unfractionated Heparin) or (Heparinic Acid) or Liquaemin or (Sodium Heparin) or alpha‐Heparin or (alpha Heparin) or UFH or heparin*
10 "Fondaparinux"[Mesh] or (Fondaparinux Sodium) or Quixidar or Arixtra
11 "Hirudin Therapy"[Mesh] or Leeching or Hirudin*
12 "Phenindione"[Mesh] or Phenylindanedione or Phenyline or Pindione or Fenilin or Dindevan
13 "Polysaccharides"[Mesh] or Glycans
14 "Rivaroxaban"[Mesh] or Xarelto or Rivaroxaban
15 "Warfarin"[Mesh] or Apo‐Warfarin or Aldocumar or Gen‐Warfarin or Warfant or Coumadin* or Marevan or Tedicumar or warfarin*
16 "Factor Xa Inhibitors" [Pharmacological Action] or (factor Xa inhibitor*)
17 "Enoxaparin"[Mesh] or Enoxaparin* or Lovenox or Clexane
18 "reviparin" [Supplementary Concept] or reviparin* or Clivarine or reviparin‐sodium or (reviparin sodium) or Clivarin
19 "Dalteparin"[Mesh] or Tedelparin or (Dalteparin Sodium) or Fragmin*
20 "danaparoid" [Supplementary Concept] or danaproid* or Orgaran or Lomoparan or danaparoid*
21 DOAC or embolex or Liquemine or (oral anticoagulants) or Pentasaccharide* or (vitamin k antagonist)
22 "edoxaban" [Supplementary Concept] or Savaysa or (edoxaban tosylate) or edoxaban
23 "ximelagatran" [Supplementary Concept] or xi‐melagatran or Exanta
24 "Phenprocoumon"[Mesh] or Phenylpropylhydroxycumarinum or Phenprocoumalol or Phenprocoumarol or Phenprogramma or Marcoumar or Marcumar or Falithrom or Liquamar
25 "idrabiotaparinux" [Supplementary Concept] or (Biotin/analogs and derivatives) or Oligosaccharides
26 "idraparinux" [Supplementary Concept] or (idraparinux sodium)
27 "Tinzaparin"[Mesh] or (Tinzaparin Sodium) or Innohep
28 "Heparin, Low‐Molecular‐Weight"[Mesh] or (Heparin Low Molecular Weight) or LMWH or (Low‐Molecular‐Weight Heparin) or parnaparin
29 "melagatran" [Supplementary Concept] or Azetidines or Benzylamines
30 "Nadroparin"[Mesh] or Nadroparin* or Fraxiparin or Fraxiparine
31 "Acenocoumarol"[Mesh] or Nicoumalone or Acenocoumarin or Sinthrome or Synthrom or Syncoumar or Syncumar or Sinkumar or Sintrom or Mini‐Sintrom or (Mini Sintrom) or MiniSintrom
32 "vorapaxar" [Supplementary Concept] or Lactones or Pyridines
33 #5 OR #6 OR #7 OR #8 OR #9 OR #10 OR #11 OR #12 OR #13 OR #14 OR #15 OR #16 OR #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR #23 OR #24 OR #25 OR #26 OR #27 OR #28 OR #29 OR #30 OR #31 OR #32
34 #4 AND #33
35 #34 AND Filters: from 2020/6/20 ‐ 2021/4/14
Appendix 4. Embase.com (Elsevier) search strategy
1 ('coronavirus disease 2019'/exp or (2019 novel coronavirus infection) or (COVID‐19 pandemic) or (coronavirus disease‐19) or (COVID19) or (2019 novel coronavirus disease) or (coronavirus disease 2019) or COVID‐19 OR 'Severe acute respiratory syndrome coronavirus 2'/exp OR (Wuhan coronavirus) or (Wuhan seafood market pneumonia virus) or (COVID19 virus) or (COVID‐19 virus) or (coronavirus disease 2019 virus) or (SARS‐CoV‐2) or (SARS2) or (2019 novel coronavirus) OR 'Coronavirus infection'/exp OR Coronavirus* or Deltacoronavirus* or Deltacoronavirus*) AND ('antithrombin'/exp OR (Direct Thrombin Inhibitor*) or (Direct Antithrombin*) or (thrombin inhibitor) OR 'coumarin derivative'/exp OR Coumarin* or (Benzopyran 2 ones) or (Coumarin Derivative*) OR 'dabigatran'/exp OR Pradaxa or (Dabigatran Etexilate) or (Dabigatran Etexilate Mesylate) OR 'anticoagulant agent'/exp OR (Anticoagulation Agent*) or (Anticoagulant Drug*) or Anticoagulant* or (Indirect Thrombin Inhibitor*) OR 'heparin derivative'/exp OR (Unfractionated Heparin) or (Heparinic Acid) or Liquaemin or (Sodium Heparin) or alpha‐Heparin or (alpha Heparin) or UFH or heparin* OR 'fondaparinux'/exp OR (Fondaparinux Sodium) or Quixidar or Arixtra OR 'anticoagulant therapy'/exp OR Hirudins or Leeching or Hirudin* OR 'phenindione'/exp OR Phenylindanedione or Phenyline or Pindione or Fenilin or Dindevan OR 'polysaccharide'/exp OR Glycans OR 'rivaroxaban'/exp OR Xarelto or Rivaroxaban OR 'warfarin'/exp OR Apo‐Warfarin or Aldocumar or Gen‐Warfarin or Warfant or Coumadin* or Marevan or Tedicumar or warfarin* OR 'blood clotting factor 10a inhibitor'/exp OR (factor Xa inhibitor*) OR 'enoxaparin'/exp OR Enoxaparin* or Lovenox or Clexane OR reviparin* or Clivarine or reviparin‐sodium or (reviparin sodium) or Clivarin OR 'dalteparin'/exp OR Tedelparin or (Dalteparin Sodium) or Fragmin* OR danaproid or Orgaran or Lomoparan or danaparoid* or DOAC or embolex or Liquemine or (oral anticoagulants) or Pentasaccharide* or (vitamin k antagonist) or Savaysa or (edoxaban tosylate) or edoxaban or xi‐melagatran or Exanta OR 'phenprocoumon h 3'/exp OR Phenylpropylhydroxycumarinum or Phenprocoumalol or Phenprocoumarol or Phenprogramma or Marcoumar or Marcumar or Falithrom or Liquamar or Oligosaccharides or (idraparinux sodium) OR 'tinzaparin'/exp OR (Tinzaparin Sodium) OR 'low molecular weight heparin'/exp OR (Heparin Low Molecular Weight) or LMWH or (Low‐Molecular‐Weight Heparin) or parnaparin or Azetidines or Benzylamines OR 'nadroparin'/exp OR Nadroparin* or Fraxiparin or Fraxiparine OR 'acenocoumarol'/exp OR Nicoumalone or Acenocoumarin or Sinthrome or Synthrom or Syncoumar or Syncumar or Sinkumar or Sintrom or Mini‐Sintrom or (Mini Sintrom) or MiniSintrom or Lactones or Pyridines)
2 #1 AND [embase]/lim NOT ([embase]/lim AND [medline]/lim)
3 #2 AND (2020:py OR 2021:py)
Appendix 5. LILACS and IBECS (Virtual Health Library) search strategy
tw:((tw:(mh: "Coronavirus Infections" OR mh: "Infecciones por Coronavirus" OR mh: "Infecções por Coronavirus" OR covid‐19 OR (coronavirus infection*) OR mers OR (middle east respiratory syndrome) OR (novel coronavirus pneumonia) OR (wuhan seafood market pneumonia) OR (brote por el nuevo coronavirus 2019) OR (brote por el coronavirus de wuhan) OR (epidemia de neumonía por coronavirus de wuhan) OR (síndrome respiratório de oriente medio) OR (síndrome respiratorio de oriente medio por coronavirus) OR (epidemia de pneumonia por coronavirus de wuhan) OR (epidemia de pneumonia por coronavírus de wuhan) OR (epidemia de pneumonia por coronavírus de wuhan de 2019‐2020) OR mh: betacoronavirus OR (2019 new coronavirus) OR (2019 novel coronavirus) OR betacoronavirus* OR sars‐cov‐2 OR (severe acute respiratory syndrome coronavirus 2) OR (wuhan coronavirus) OR (wuhan seafood market pneumonia virus) OR (coronavirus de wuhan) OR (coronavirus del síndrome respiratorio agudo grave 2) OR (nuevo coronavirus 2019) OR (virus de la neumonía del mercado de pescado y marisco de wuhan) OR (wuhan coronavirus) OR (coronavírus da síndrome respiratória aguda grave 2) OR (coronavírus de wuhan) OR (vírus de pneumonia no mercado de frutos do mar de wuhan) OR mh: coronavirus OR (coronavirus* rabbit) OR coronavirus* OR deltacoronavirus* OR (coronavirus del conejo) OR (coronavirus do coelho))) AND (tw:(tw:((tw:(mh: antithrombins OR mh: antitrombinas OR (direct antithrombins) OR (direct thrombin inhibitors) OR (antitrombinas directas) OR (antitrombinas diretas) OR d27.505.519.389.745.800.449 OR d27.505.954.502.119.500)) OR (tw:(mh: coumarins OR mh: cumarinas OR mh: cumarínicos OR (coumarin derivative*) OR coumarin* OR cumarina* OR d03.383.663.283.446 OR d03.633.100.150.446)) OR (tw:(mh: dabigatran OR mh: dabigatrán OR mh: dabigatrana OR (dabigatran* etexilat*) OR (dabigatran etexilate mesylate) OR pradaxa OR (etexilato de dabigatrana) OR d03.383.725.192 OR d03.633.100.103.280)) OR (tw:(mh: anticoagulants OR mh: anticoagulantes OR (agent* anticoagulant*) OR anticoagulant* OR (anticoagulant drug*) OR (anticoagulation agents) OR (indirect thrombin inhibitor*) OR (agentes anticoagulantes) OR (agentes de anticoagulación) OR anticoagulante*)) OR (tw:(mh: heparin OR mh: heparina OR (heparin sodium) OR (heparin unfractionated) OR (heparinic acid) OR liquaemin OR (alpha heparin) OR alpha‐heparin OR alfa‐heparina OR (ácido heparínico) OR (heparina alfa) OR heparina‐alfa)) OR (tw:(mh: fondaparinux OR arixtra OR (fondaparinux sodium) OR quixidar OR (fondaparinux sódico))) OR (tw:(mh: "Hirudin Therapy" OR mh: "Terapia con Hirudina" OR mh: "Terapia com Hirudina")) OR (tw:(mh: phenindione OR mh: fenindiona OR dindevan OR fenilin OR phenylindanedione OR phenyline OR pindione OR d02.455.426.559.847.486.487.750 OR d04.615.486.487.750)) OR (tw:(mh: polysaccharides OR mh: polisacáridos OR mh: polissacarídeos OR glycans OR glican*)) OR (tw:(mh: rivaroxaban OR mh: rivaroxabán OR mh: rivaroxabana OR xarelto OR d02.886.778.727 OR d03.383.533.640.713 OR d03.383.903.727)) OR (tw:(mh: warfarin OR mh: warfarina OR mh: varfarina OR aldocumar OR apo‐warfarin OR coumadin OR coumadine OR gen‐warfarin OR marevan OR tedicumar OR warfant OR (warfarin potassium) OR (warfarin sodium) OR d03.383.663.283.446.520.914 OR d03.633.100.150.446.520.914)) OR (tw:(mh: "Factor Xa Inhibitors" OR mh: "Inhibidores del Factor Xa" OR mh: "Inibidores do Fator Xa" OR (anticoagulant* direct‐acting oral) OR (direct acting oral anticoagulant*) OR (direct factor xa inhibitor*) OR d27.505.519.389.745.800.449.500 OR d27.505.954.502.119.500.500 OR (anticoagulantes orales de acción directa) OR (inhibidor del factor xa) OR (inhibidores directos del factor xa) OR (anticoagulantes orais de ação direta) OR (inibidor do fator xa) OR (inibidores diretos do fator xa))) OR (tw:(mh: enoxaparin OR mh: enoxaparin* OR clexane OR lovenox)) OR (tw:(mh: dalteparin OR mh: dalteparina OR (dalteparin sodium) OR fragmin* OR tedelparin*)) OR (tw:(doac OR embolex OR liquemine OR (oral anticoagulants) OR pentasaccharide* OR (vitamin k antagonist) OR savaysa OR (edoxaban tosylate) OR edoxaban OR xi‐melagatran OR exanta OR danaproid* OR orgaran OR lomoparan OR danaparoid* OR reviparin* OR clivarine OR reviparin‐sodium OR (reviparin sodium) OR clivarin OR azetidines OR benzylamines OR lactones OR pyridines)) OR (tw:(mh: phenprocoumon OR mh: fenprocumón OR mh: femprocumona OR falithrom OR liquamar OR marcoumar OR marcumar OR phenprocoumalol OR phenprocoumarol OR phenprogramma OR phenylpropylhydroxycumarinum OR d03.383.663.283.446.520.750 OR d03.633.100.150.446.520.750 OR fenilpropilhidroxicumarina OR fenprocumalol OR fenprocumarol OR femprocumalol OR femprocumarol OR fenilpropilidroxicumarina OR (feno procumarol) OR fenoprocumalol OR fenoprocumona)) OR (tw:(mh: tinzaparin OR mh: tinzaparina OR innohep OR (tinzaparin sodium) OR (tinzaparina sódica))) OR (tw:(mh: "Heparin, Low‐Molecular‐Weight" OR mh: "Heparina de Bajo‐Peso‐Molecular" OR mh: "Heparina de Baixo Peso Molecular" OR (heparin low molecular weight) OR lmwh OR (low molecular weight heparin) OR (low‐molecular‐weight heparin) OR hbpm)) OR (tw:(mh: nadroparin OR mh: nadroparina OR (calcium nadroparin) OR fraxiparin* OR nadroparin*)) OR (tw:(mh: acenocoumarol OR mh: acenocumarol OR acenocoumarin OR (mini sintrom) OR mini‐sintrom OR minisintrom OR nicoumalone OR sinkumar OR sinthrome OR sintrom* OR syncoumar OR syncumar OR synthrom OR d03.383.663.283.446.520.079 OR d03.633.100.150.446.520.079 OR acenocumarina OR nicumalon*)))))) AND ( db:("LILACS" OR "IBECS")) AND (year_cluster:[2020 TO 2021])
Appendix 6. Cochrane COVID‐19 Study Register search strategy
"Anticoagulant* or Heparin* or Rivaroxaban or Warfarin or Enoxaparin or DOAC or LMWH" AND Filter created: 20 Jun '20 ‐ 14 Apr '21
Appendix 7. medRxiv search strategy
"Anticoagulant OR anticoagulants OR Heparin OR Rivaroxaban OR Warfarin OR Enoxaparin OR DOAC OR LMWH" (match whole all) and posted between "20 Jun, 2020 and 14 Apr, 2021"
Data and analyses
Comparison 1. Higher‐dose anticoagulants versus lower‐dose anticoagulants (short term).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 All‐cause mortality | 4 | 4489 | Risk Ratio (IV, Random, 95% CI) | 1.03 [0.92, 1.16] |
| 1.1.1 Moderate severity | 2 | 2833 | Risk Ratio (IV, Random, 95% CI) | 1.11 [0.68, 1.81] |
| 1.1.2 Critical ill | 3 | 1656 | Risk Ratio (IV, Random, 95% CI) | 1.04 [0.91, 1.17] |
| 1.2 All‐cause mortality ‐ trials at low risk of bias | 2 | 1176 | Risk Ratio (IV, Random, 95% CI) | 1.16 [0.86, 1.57] |
| 1.2.1 Moderate severity | 1 | 614 | Risk Ratio (IV, Random, 95% CI) | 1.49 [0.90, 2.46] |
| 1.2.2 Critically ill | 1 | 562 | Risk Ratio (IV, Random, 95% CI) | 1.05 [0.87, 1.28] |
| 1.3 Necessity for additional respiratory support | 3 | 3407 | Risk Ratio (IV, Random, 95% CI) | 0.54 [0.12, 2.47] |
| 1.3.1 Moderate severity | 2 | 2845 | Risk Ratio (IV, Random, 95% CI) | 0.54 [0.12, 2.47] |
| 1.3.2 Critically ill | 1 | 562 | Risk Ratio (IV, Random, 95% CI) | Not estimable |
| 1.4 Necessity for additional respiratory support ‐ trials at low risk of bias | 2 | 1176 | Risk Ratio (IV, Random, 95% CI) | 0.16 [0.02, 1.35] |
| 1.4.1 Moderate severity | 1 | 614 | Risk Ratio (IV, Random, 95% CI) | 0.16 [0.02, 1.35] |
| 1.4.2 Critically ill | 1 | 562 | Risk Ratio (IV, Random, 95% CI) | Not estimable |
| 1.5 Deep vein thrombosis | 4 | 3422 | Risk Ratio (IV, Random, 95% CI) | 1.08 [0.57, 2.03] |
| 1.5.1 Moderate severity | 2 | 2840 | Risk Ratio (IV, Random, 95% CI) | 0.85 [0.38, 1.92] |
| 1.5.2 Critically ill | 2 | 582 | Risk Ratio (IV, Random, 95% CI) | 1.55 [0.56, 4.26] |
| 1.6 Deep vein thrombosis ‐ trials at low risk of bias | 2 | 1176 | Risk Ratio (IV, Random, 95% CI) | 1.21 [0.53, 2.79] |
| 1.6.1 Moderate severity | 1 | 614 | Risk Ratio (IV, Random, 95% CI) | 0.98 [0.29, 3.35] |
| 1.6.2 Critically ill | 1 | 562 | Risk Ratio (IV, Random, 95% CI) | 1.45 [0.47, 4.52] |
| 1.7 Pulmonary embolism | 4 | 4360 | Risk Ratio (IV, Random, 95% CI) | 0.46 [0.31, 0.70] |
| 1.7.1 Moderate severity | 2 | 2840 | Risk Ratio (IV, Random, 95% CI) | 0.49 [0.27, 0.88] |
| 1.7.2 Critically ill | 3 | 1520 | Risk Ratio (IV, Random, 95% CI) | 0.44 [0.25, 0.78] |
| 1.8 Pulmonary embolism ‐ trial at low risk of bias | 2 | 1176 | Risk Ratio (IV, Random, 95% CI) | 0.50 [0.23, 1.10] |
| 1.8.1 Moderate severity | 1 | 614 | Risk Ratio (IV, Random, 95% CI) | 0.53 [0.21, 1.31] |
| 1.8.2 Critically ill | 1 | 562 | Risk Ratio (IV, Random, 95% CI) | 0.41 [0.08, 2.12] |
| 1.9 Major bleeding | 4 | 4400 | Risk Ratio (IV, Random, 95% CI) | 1.78 [1.13, 2.80] |
| 1.9.1 Moderate severity | 2 | 2841 | Risk Ratio (IV, Random, 95% CI) | 2.25 [1.19, 4.27] |
| 1.9.2 Critically ill | 3 | 1559 | Risk Ratio (IV, Random, 95% CI) | 1.41 [0.75, 2.67] |
| 1.10 Major bleeding ‐ trials at low risk of bias | 2 | 1176 | Risk Ratio (IV, Random, 95% CI) | 2.13 [0.92, 4.90] |
| 1.10.1 Moderate severity | 1 | 614 | Risk Ratio (IV, Random, 95% CI) | 2.45 [0.78, 7.73] |
| 1.10.2 Critically ill | 1 | 562 | Risk Ratio (IV, Random, 95% CI) | 1.81 [0.54, 6.13] |
| 1.11 Adverse events (minor bleeding) | 3 | 1196 | Risk Ratio (IV, Random, 95% CI) | 3.28 [1.75, 6.14] |
| 1.11.1 Moderate severity | 1 | 614 | Risk Ratio (IV, Random, 95% CI) | 5.10 [1.98, 13.11] |
| 1.11.2 Critically ill | 2 | 582 | Risk Ratio (IV, Random, 95% CI) | 2.31 [1.00, 5.36] |
| 1.12 Adverse events (minor bleeding) ‐ trials at low risk of bias | 2 | 1176 | Risk Ratio (IV, Random, 95% CI) | 3.67 [1.82, 7.40] |
| 1.12.1 Moderate severity | 1 | 614 | Risk Ratio (IV, Random, 95% CI) | 5.10 [1.98, 13.11] |
| 1.12.2 Critical ill | 1 | 562 | Risk Ratio (IV, Random, 95% CI) | 2.49 [0.89, 6.97] |
| 1.13 Adverse events (stroke) | 3 | 4349 | Risk Ratio (IV, Random, 95% CI) | 0.91 [0.40, 2.03] |
| 1.13.1 Moderate severity | 2 | 2840 | Risk Ratio (IV, Random, 95% CI) | 0.88 [0.13, 5.97] |
| 1.13.2 Critical ill | 2 | 1509 | Risk Ratio (IV, Random, 95% CI) | 0.91 [0.37, 2.23] |
| 1.14 Adverse events (stroke) ‐ trials at low risk of bias | 2 | 1176 | Risk Ratio (IV, Random, 95% CI) | 1.62 [0.20, 13.13] |
| 1.14.1 Moderate severity | 1 | 614 | Risk Ratio (IV, Random, 95% CI) | 2.94 [0.12, 71.94] |
| 1.14.2 Critical ill | 1 | 562 | Risk Ratio (IV, Random, 95% CI) | 1.04 [0.07, 16.49] |
| 1.15 Adverse events (major adverse limb event) | 2 | 1176 | Risk Ratio (IV, Random, 95% CI) | 0.33 [0.01, 7.99] |
| 1.15.1 Moderate severity | 1 | 614 | Risk Ratio (IV, Random, 95% CI) | 0.33 [0.01, 7.99] |
| 1.15.2 Critically ill | 1 | 562 | Risk Ratio (IV, Random, 95% CI) | Not estimable |
| 1.16 Adverse events (myocardial infarction) | 3 | 4349 | Risk Ratio (IV, Random, 95% CI) | 0.86 [0.48, 1.55] |
| 1.16.1 Moderate severity | 2 | 2840 | Risk Ratio (IV, Random, 95% CI) | 0.91 [0.45, 1.85] |
| 1.16.2 Critically ill | 2 | 1509 | Risk Ratio (IV, Random, 95% CI) | 0.76 [0.27, 2.17] |
| 1.17 Adverse events (myocardial infarction) ‐ trials at low risk of bias | 2 | 1176 | Risk Ratio (IV, Random, 95% CI) | 0.91 [0.44, 1.91] |
| 1.17.1 Moderate severity | 1 | 614 | Risk Ratio (IV, Random, 95% CI) | 0.91 [0.44, 1.91] |
| 1.17.2 Critically ill | 1 | 562 | Risk Ratio (IV, Random, 95% CI) | Not estimable |
| 1.18 Adverse events (atrial fibrillation) | 1 | 562 | Risk Ratio (IV, Random, 95% CI) | 0.35 [0.07, 1.70] |
| 1.19 Adverse events (thrombocytopenia) | 2 | 2789 | Risk Ratio (IV, Random, 95% CI) | 0.94 [0.71, 1.24] |
| 1.19.1 Moderate severity | 1 | 2227 | Risk Ratio (IV, Random, 95% CI) | Not estimable |
| 1.19.2 Critically ill | 1 | 562 | Risk Ratio (IV, Random, 95% CI) | 0.94 [0.71, 1.24] |
| 1.20 Hospitalisation time | 2 | 634 | Mean Difference (IV, Random, 95% CI) | 0.28 [‐0.87, 1.44] |
| 1.20.1 Moderate severity | 1 | 614 | Mean Difference (IV, Random, 95% CI) | 0.30 [‐0.86, 1.46] |
| 1.20.2 Critically ill | 1 | 20 | Mean Difference (IV, Random, 95% CI) | ‐1.00 [‐11.58, 9.58] |
| 1.21 Hospitalisation time ‐ trials at low risk of bias | 1 | 614 | Mean Difference (IV, Random, 95% CI) | 0.30 [‐0.86, 1.46] |
Comparison 2. Higher‐dose anticoagulants versus lower‐dose anticoagulants (long term).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 All‐cause mortality | 1 | 590 | Risk Ratio (IV, Random, 95% CI) | 1.07 [0.89, 1.28] |
| 2.2 Necessity for additional respiratory support | 1 | 590 | Risk Ratio (IV, Random, 95% CI) | Not estimable |
| 2.3 Deep vein thrombosis | 1 | 590 | Risk Ratio (IV, Random, 95% CI) | 1.39 [0.45, 4.33] |
| 2.4 Pulmonary embolism | 1 | 590 | Risk Ratio (IV, Random, 95% CI) | 0.40 [0.08, 2.03] |
| 2.5 Major bleeding | 1 | 590 | Risk Ratio (IV, Random, 95% CI) | 1.74 [0.51, 5.87] |
| 2.6 Adverse events (minor bleeding) | 1 | 590 | Risk Ratio (IV, Random, 95% CI) | 2.32 [0.90, 5.95] |
| 2.7 Adverse events (stroke) | 1 | 590 | Risk Ratio (IV, Random, 95% CI) | 0.99 [0.06, 15.80] |
| 2.8 Adverse events (acute peripheral arterial thrombosis) | 1 | 590 | Risk Ratio (IV, Random, 95% CI) | Not estimable |
| 2.9 Adverse events (myocardial infarction) | 1 | 590 | Risk Ratio (IV, Random, 95% CI) | Not estimable |
| 2.10 Adverse events (atrial fibrillation) | 1 | 590 | Risk Ratio (IV, Random, 95% CI) | 0.50 [0.13, 1.97] |
| 2.11 Adverse events (thrombocytopenia) | 1 | 590 | Risk Ratio (IV, Random, 95% CI) | 12.91 [0.73, 228.18] |
Comparison 3. Anticoagulants versus no treatment (short term).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 3.1 All‐cause mortality | 3 | Risk Ratio (IV, Random, 95% CI) | 0.64 [0.55, 0.74] | |
| 3.2 Deep vein thrombosis | 1 | 1403 | Risk Ratio (IV, Random, 95% CI) | 5.67 [1.30, 24.70] |
| 3.3 Pulmonary embolism | 1 | 1403 | Risk Ratio (IV, Random, 95% CI) | 24.19 [3.31, 176.53] |
| 3.4 Major bleeding | 2 | 7218 | Risk Ratio (IV, Random, 95% CI) | 1.19 [0.66, 2.12] |
| 3.5 Adverse events (stroke) | 1 | 1403 | Risk Ratio (IV, Random, 95% CI) | 1.13 [0.32, 4.00] |
| 3.6 Adverse events (myocardial infarction) | 1 | 1403 | Risk Ratio (IV, Random, 95% CI) | 15.88 [0.93, 270.48] |
| 3.7 Hospitalisation time | 1 | 1376 | Mean Difference (IV, Random, 95% CI) | 5.00 [4.47, 5.53] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Albani 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
Inclusion criteria
Exclusion criteria
|
|
| Interventions |
|
|
| Outcomes | Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
Lemos 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
Inclusion criteria
Exclusion criteria
|
|
| Interventions |
|
|
| Outcomes | Primary (specified)
Primary (collected)
Secondary (specified)
Secondary (collected)
Time points reported: at 0, 4, 7, 14 and 28 days after the start of the intervention |
|
| Notes |
|
|
| Item | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Yes | Quote "We used blocked randomisation, and the participants were randomised in a 1:1 ratio within two blocks of ten patients each." |
| Allocation concealment (selection bias) | Yes | Quote " The patients were assigned to each treatment by drawing the sequential numbering of opaque envelopes containing the treatment allocation." |
| Blinding of participants and personnel (performance bias) | No | Quote "In this randomised, controlled, open‐label..." |
| Blinding of outcome assessment (detection bias) | No | Quote "In this randomised, controlled, open‐label..." |
| Incomplete outcome data (attrition bias) | No | Quote "The third arm of this study with therapeutic intravenous unfractionated heparin (UFH) was abandoned due to difficulties in adjusting the activated partial thromboplastin time (aPTT) during the pandemic." |
| Selective reporting (reporting bias) | Yes | All prespecified outcomes were reported |
| Other bias | Yes | We do not suspect any other bias related to this study. |
Lopes 2021.
| Study characteristics | ||
| Methods |
|
|
| Participants |
Inclusion
Exclusion
|
|
| Interventions |
|
|
| Outcomes | Primary (specified)
Primary (collected)
Secondary (specified)
Secondary (collected)
Time points reported: at 30 days after the start of the intervention |
|
| Notes |
|
|
| Item | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Yes | Quote "Concealed randomisation will be performed using a central, automated, electronic web‐based system." |
| Allocation concealment (selection bias) | Yes | Quote "Concealed randomisation will be performed using a central, automated, electronic web‐based system." |
| Blinding of participants and personnel (performance bias) | No | Quote "The study is open‐label with blinded outcomes adjudication" |
| Blinding of outcome assessment (detection bias) | Yes | Quote "The study is open‐label with blinded outcomes adjudication" |
| Incomplete outcome data (attrition bias) | Yes | There was one loss in the experimental group (1/311, 0.3%): one participant withdrew consent and declined to contribute data. |
| Selective reporting (reporting bias) | Yes | All prespecified outcomes were reported. |
| Other bias | Yes | We do not suspect any other bias related to this study. |
Rentsch 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
Inclusion criteria
Exclusion criteria
|
|
| Interventions |
|
|
| Outcomes | Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
Sadeghipour 2021.
| Study characteristics | ||
| Methods |
|
|
| Participants |
Inclusion criteria for anticoagulation hypothesis
Exclusion criteria for anticoagulation hypothesis
|
|
| Interventions |
|
|
| Outcomes | Primary (specified)
Primary (collected)
Secondary (specified)
Secondary (collected)
Time points reported: at 30 and 90 days after the start of the intervention |
|
| Notes |
|
|
| Item | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Yes | Quote "Randomization was done using an electronic web‐based system with permuted blocks of 4 and allocation sequence concealment." |
| Allocation concealment (selection bias) | Yes | Quote "For the first hypothesis, allocation sequence concealment and blinded endpoint adjudication." |
| Blinding of participants and personnel (performance bias) | No | Quote "an open‐label randomised clinical trial with blinded outcome adjudication." |
| Blinding of outcome assessment (detection bias) | Yes | Quote "an open‐label randomised clinical trial with blinded outcome adjudication." |
| Incomplete outcome data (attrition bias) | Yes | The losses were balanced between the groups (experimental = 24 (8%); comparator = 14 (4%)) |
| Selective reporting (reporting bias) | Yes | All prespecified outcomes were reported. |
| Other bias | Yes | We do not suspect any other bias related to this study. |
Santoro 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
Inclusion criteria
Exclusion criteria
|
|
| Interventions |
|
|
| Outcomes | Primary (specified)
Primary (collected)
Secondary (specified)
Secondary (collected)
Time point reported: during hospitalisation or up to 30 days |
|
| Notes |
|
|
Zarychanski 2021.
| Study characteristics | ||
| Methods |
|
|
| Participants |
Inclusion criteria
Exclusion criteria
|
|
| Interventions | Experimental: therapeutic‐dose LMWH or UFH was administered according to local protocols used for the treatment of acute VTE for up to 14 days or until recovery (defined as hospital discharge, or liberation from supplemental oxygen for ≥ 24 h)
Comparator: pharmacological thromboprophylaxis was administered according to local practice or with guidance from the trial protocol on maximum dosing, which included either standard low‐dose thromboprophylaxis or enhanced intermediate‐dose thromboprophylaxis
Concomitant therapy: NR |
|
| Outcomes | Primary (specified)
Primary (collected)
Secondary (specified)
Secondary (collected)
Time points reported: at 21, 28, or 90 (days after the start of the intervention |
|
| Notes |
|
|
| Item | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Yes | Quote: "Randomization was performed using central web‐based systems." Quote: "ACTIV‐4a randomised all participants in a 1:1 ratio. The other two platforms specified response‐adaptive randomisation; randomisation probabilities were updated in the severe patient group within REMAP‐CAP and ATTACC during the interim period between the mpRCT interim data cut and the halt of enrolment" |
| Allocation concealment (selection bias) | Yes | Quote: "Randomization was performed using central web‐based systems." Quote "The ATTACC and REMAP‐CAP designs specified the possibility for response‐adaptive randomisation, whereby blinded randomisation allocation ratios could be modified during the trial based on adaptive analyses to favor allocation of participants to the treatment arm demonstrating greater benefit." |
| Blinding of participants and personnel (performance bias) | No | Quote "One limitation of our trial is the open‐label design" |
| Blinding of outcome assessment (detection bias) | No | Not all outcomes were assessed in a blinding approach Quote "The open label strategy may also introduce systematic bias in the ascertainment of thrombotic events." Quote "All reported bleeding and thrombotic events were adjudicated in a blinded fashion by clinical endpoints committees using consensus definitions" |
| Incomplete outcome data (attrition bias) | No | There was an acceptable loss rate (critically ill: 1205 randomised, 1074 analysed, 10.8% lost; moderate‐severity: 2245 randomised, 2219 analysed, 1.1% lost). However, there was a high and imbalanced cross‐over rate (experimental/comparator): critically ill 92 (22.3%)/23 (5.3%), moderate‐severity 213 (20.4%)/8 (0.9%). Supplemental data |
| Selective reporting (reporting bias) | No | The trial protocol also planned to assess other outcomes of interest for this review. However, the available manuscript did not report 'quality of life' and reported 'hospitalisation time', 'necessity of additional respiratory support', 'thrombocytopenia', and 'DVT' only for moderate‐severity participants. The study authors planned to report data at 90 days of follow‐up but there are no available data at this time point. |
| Other bias | No | Although study authors declare that they harmonised their protocols into a "prospectively multiplatform uniformisation", they combined results from 3 different trials registries, with different centres of randomisation and documentation. It was reflected in an imbalance of losses to follow‐up (moderate‐severity: experimental = 19 losses (1.5%), comparator = 7 losses (0.6%)) Quote "may be imbalanced due to response adaptive randomisation" There was a factorial randomisation for antiplatelet agent intervention in one of the considered trials (REMAP‐CAP). Quote "A subset of participants enrolled in REMAP‐CAP were also randomised in the antiplatelet agent domain and in other domains of that trial." There is a possibility of additional heterogeneity in overall results when combining these 3 trials as a unique trial Although the trial authors merged their platforms, there was a change in the primary outcome specified in the registered protocols compared to the unique reported primary outcome. |
ACS: acute coronary syndrome; ACTIV‐4a: accelerating COVID‐19 therapeutic interventions and vaccines‐4 antithrombotics inpatient platform trial; ARDS: acute respiratory distress syndrome; aPTT: activated partial thromboplastin time; ASA: acetylsalicylic acid (aspirin); ATTACC: antithrombotic therapy to ameliorate complications of COVID‐19; BMI: body mass index; BP: blood pressure; CEC: clinical events committee; CKD: chronic kidney disease; COPD: chronic obstructive pulmonary disease; COI: conflict of interest; CPR: cardiopulmonary resuscitation; CrCl: creatinine clearance; CT: computed tomography; DIC: disseminated intravascular coagulation; DOACs: direct oral anticoagulants; DVT: deep vein thrombosis; ECMO: extracorporeal membrane oxygenation; FiO2: fractional inspired oxygen; GFR: glomerular filtration rate; HIT: heparin‐induced thrombocytopenia; ICU: intensive care unit; INR: international normalised ratio; IQR: interquartile range; ISTH: International Society on Thrombosis and Haemostasis; INR: international normalised ratio; IU: international unit; IV: intravenously; LMWH: low‐molecular‐weight heparin; μL: microlitre; MRI: magnetic resonance imaging; NR: not reported; NRS: non‐randomised study; PaO2: arterial blood oxygen partial pressure; PCR: polymerase chain reaction; PE: pulmonary embolism; RCT: randomised controlled trial; REMAP‐CAP: Randomised, embedded, multifactorial adaptive platform trial for community‐acquired pneumonia; RT‐PCR: reverse transcription polymerase chain reaction; SC: subcutaneous(ly); SD: standard deviation; SOFA: sequential organ failure assessment score; UFH: unfractionated heparin; ULN: upper limit of normal; VKA: vitamin K antagonists; VTE: venous thromboembolism; WHO: World Health Organization
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Al‐Samkari 2020 | Ineligible study design. Retrospective cohort study without a parallel comparator group of intervention |
| Artifoni 2020 | Ineligible study design. Retrospective cohort study without a comparator group (single‐arm study) |
| Ayerbe 2020 | Ineligible study design. Retrospective cohort study |
| ChiCTR2000034796 | Ineligible study design. Retrospective cohort study comparing people hospitalised with COVID‐19 receiving LMWH versus those not receiving LMWH |
| CTRI/2021/01/030373 | Ineligible participants. RCT of non‐hospitalised people with COVID‐19 |
| D'Ardes 2021 | Ineligible study design. Retrospective cohort study without a prospective parallel comparator group of intervention |
| DCTC 2021 | Ineligible study design. Prospective cohort study without a comparator group (single‐arm study) |
| Di Castelnuovo 2021 | Ineligible study design. Retrospective cohort study comparing people hospitalised with COVID‐19 receiving anticoagulation versus those not receiving anticoagulation |
| EUCTR2020‐001748‐24‐SE | Ineligible participants. The RCT did not assess the effects of any anticoagulant |
| EudraCT2020‐001823‐15 | Ineligible study design. Prospective cohort study without a comparator group (single‐arm study) |
| Falcone 2020 | Ineligible study design. Prospective cohort study without a comparator group (single‐arm study) |
| Frohlich 2021 | Ineligible study design. Retrospective cohort study without a comparator group (single‐arm study) |
| Helms 2020 | Ineligible study design. Prospective cohort study without an intervention purpose |
| Helms 2021 | Ineligible study design. Prospective cohort study comparing prophylactic and therapeutic anticoagulant |
| Ho 2021 | Ineligible study design. Retrospective cohort study comparing people hospitalised with COVID‐19 receiving anticoagulants versus people not receiving anticoagulants in USA |
| Huang 2020 | Ineligible study design. Case report |
| Ionescu 2020 | Ineligible study design. Prospective cohort study comparing people hospitalised with COVID‐19 receiving anticoagulants (higher dose) versus anticoagulants (lower dose) in USA |
| Jiménez‐Soto 2021 | Ineligible study design. Prospective cohort study comparing people hospitalised with COVID‐19 receiving anticoagulants (higher dose) versus anticoagulants (lower dose) in Mexico |
| Jonmarker 2020 | Ineligible study design. Prospective cohort study comparing people hospitalised with COVID‐19 receiving three different doses of anticoagulants in Sweden |
| Khider 2020 | Ineligible study design. Prospective cohort study without a parallel comparator group of intervention |
| Kodama 2020 | Ineligible study design. Retrospective cohort study comparing people hospitalised with COVID‐19 receiving 2 different doses of anticoagulants in USA |
| Kow 2020 | Ineligible study design. Letter to editor |
| Kukin 2020 | Ineligible participant. RCT comparing non‐pharmacological intervention for people without COVID‐19 |
| Liu 2020 | Ineligible study design. Retrospective cohort study |
| Mareev 2020 | Ineligible intervention. RCT comparing anti‐inflammatory pharmacological interventions. There is no comparison with anticoagulants |
| Martinelli 2020 | Ineligible study design. Prospective cohort study comparing people hospitalised with COVID‐19 receiving two different doses of anticoagulants |
| Maurer 2020 | Ineligible intervention. RCT comparing arterial line heparinisation versus no‐heparinisation in people hospitalised with COVID‐19 |
| NCT04354155 | Ineligible study design. Prospective cohort study without a parallel comparator group of intervention |
| NCT04359212 | Ineligible study design. Prospective cohort study without a parallel comparator group of intervention |
| NCT04365309 | Ineligible intervention. RCT of aspirin for COVID‐19. There is no difference between the intervention groups regarding anticoagulants. |
| NCT04368377 | Ineligible study design. Prospective cohort study without a comparator group (single‐arm study) |
| NCT04393805 | Ineligible study design. Retrospective cohort study |
| NCT04427098 | Ineligible study design. Prospective cohort study without a comparator group (single‐arm study) |
| NCT04483830 | Ineligible participants. RCT of non‐hospitalised people with COVID‐19 |
| NCT04492254 | Ineligible participants. RCT of non‐hospitalised people with COVID‐19 |
| NCT04504032 | Ineligible participants. RCT of non‐hospitalised people with COVID‐19 |
| NCT04516941 | Ineligible participants. RCT of non‐hospitalised people with COVID‐19 |
| NCT04662684 | Ineligible participants. RCT where the participants received the intervention after hospital discharge (non‐hospitalised people with COVID‐19) |
| NCT04673214 | Ineligible participants. RCT of non‐hospitalised people with COVID‐19 |
| NCT04715295 | Ineligible participants. RCT of non‐hospitalised people with COVID‐19 |
| NCT04736901 | Ineligible study design. Prospective cohort study comparing five different anticoagulant regimens |
| NCT04757857 | Ineligible participants. RCT of non‐hospitalised people with COVID‐19 |
| NCT04828772 | Ineligible study design. Retrospective cohort study comparing people hospitalised with COVID‐19 receiving anticoagulants versus people not receiving anticoagulants in Japan |
| Paranjpe 2020 | Ineligible study design. Retrospective cohort study |
| Piagnerelli 2020 | Ineligible study design. Letter to editor |
| Poulakou 2021 | Ineligible study design. Retrospective cohort study comparing different doses of anticoagulants in people hospitalised with COVID‐19 |
| Qin 2021 | Ineligible study design. Retrospective cohort study comparing different doses of anticoagulants in people hospitalised with COVID‐19 |
| Rosovsky 2020 | Ineligible study design. Prospective survey about physician practice. There was no participant with COVID‐19. |
| Russo 2020 | Ineligible study design. Retrospective cohort study |
| Secco 2020 | Ineligible study design. Retrospective cohort study comparing different doses of anticoagulants in people hospitalised with COVID‐19 |
| Shi 2020 | Ineligible study design. Retrospective cohort study |
| Sivaloganathan 2020 | Ineligible study design. Case‐control study comparing mortality in people hospitalised with COVID‐19 who previously used anticoagulants and antiplatelet agents versus those who did not |
| Smith 2020 | Ineligible study design. Prospective survey about physician practice. There was no participant with COVID‐19. |
| Stessel 2020 | Ineligible study design. Prospective before‐after cohort study without a parallel comparator group |
| Tacquard 2021 | Ineligible study design. Prospective cohort study comparing different doses of anticoagulants in people hospitalised with COVID‐19 |
| Tamargo 2021 | Ineligible study design. Editorial |
| Tang 2020 | Ineligible study design. Retrospective cohort study |
| Trinh 2020 | Ineligible study design. Retrospective cohort study |
| Zhang 2020 | Ineligible study design. Retrospective cases series. Description of 7 participants without a consistent comparator group |
LMWH: low molecular weight heparin, RCT: randomised controlled trial
Characteristics of ongoing studies [ordered by study ID]
ACTRN12620000517976.
| Study name | A randomised controlled trial of nebulised heparin in critically ill mechanically ventilated patients with COVID‐19 to assess the effect on the duration of mechanical ventilation |
| Starting date | 21 May 2020 |
| Contact information | Barry Dixon St Vincent’s Hospital, Melbourne, Australia +613439618815 | barry.dixon@svha.org.au |
| Methods | Prospective, multicentre, 2‐armed, parallel‐assignment RCT |
| Participants | 172 participants, ≥ 18 years, female and male Inclusion criteria
Exclusion criteria
|
| Interventions | Experimental: nebulised (vibrating mesh nebuliser) heparin sodium 25,000 IU in 5 mL 6‐hourly/day 10 while invasively ventilated in addition to standard care. The medication will be prescribed and administration documented in the medical record. Comparator: standard care represents the treatments routinely provided by the medical team managing the patient. Standard care will be at the discretion of the medical team. |
| Outcomes | Primary
Secondary
|
| Notes | ACTRN12620000517976p | No data provided |
Busani 2020.
| Study name | Steroids and unfractionated heparin in critically ill patients with pneumonia from COVID‐19 infection |
| Starting date | 25 November 2020 |
| Contact information | Massimo Girardis, PD ICU‐ University Hospital Modena, Modena, Italy, 41124 0594225878 ext 0039 | massimo.girardis@unimore.it |
| Methods | Multicentre RCT with 3 parallel arms, 1:1:1 |
| Participants | 210 participants; ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Drug: enoxaparin
Drug: methylprednisolone
Drug: unfractionated heparin
|
| Outcomes | Primary
Secondary
Other outcomes
|
| Notes | NCT04528888 | No data provided |
Chambers 2020.
| Study name | COVID‐19‐associated coagulopathy: safety and efficacy of prophylactic anticoagulation therapy in hospitalized adults with COVID‐19 |
| Starting date | 6 May 2020 |
| Contact information | Usha Perepu, MBBS Gundersen Health System La Crosse, Wisconsin, United States, 54601 319‐356‐2195 | usha-perepu@uiowa.edu |
| Methods | Multicentre, open‐label, 2‐armed, parallel‐assignment RCT |
| Participants | 170 participants, ≥ 18 years, female and male Inclusion criteria
Exclusion criteria
|
| Interventions | Interventional: intermediate‐dose enoxaparin (1 mg/kg SC daily if BMI < 30 kg/m² or 0.5 mg/kg SC twice daily if BMI ≥ 30 kg/m²) Comparator: standard care. Standard prophylactic dose enoxaparin (40 mg SC daily if BMI < 30 kg/m² and 30 mg SC twice daily or 40 mg SC twice daily if BMI ≥ 30 kg/m²) |
| Outcomes | Primary
Secondary
Other outcomes
|
| Notes | NCT04360824 | No data provided |
ChiCTR2000030700.
| Study name | An evaluative clinical study: efficacy and safety of Prolongin (enoxaparin sodium injection) in treatment of hospitalized adult patients with common novel coronavirus pneumonia (COVID‐19) |
| Starting date | 09 March 2020 |
| Contact information | Zhang Yu Union Hospital affiliated to Tongji Medical College of Huazhong University of Science and Technology, Wuhan, Hubei, China +86 13901849660 | whxhzy@163.com |
| Methods | Prospective, open‐label, 2‐armed, 1:1; parallel‐assignment RCT |
| Participants | 60 participants, ≥ 18 years, female and male Inclusion criteria
Exclusion criteria
|
| Interventions | Experimental: based on the standard treatment recommended in the guidelines, a combination of Prolongin (enoxaparin sodium injection) was used Comparator: follow the guidelines for standard treatment |
| Outcomes | Primary
Secondary
|
| Notes | ChiCTR2000030700 | No data provided |
ChiCTR2000030701.
| Study name | A randomized, parallel controlled open‐label trial to evaluate the efficacy and safety of Prolongin (enoxaparin sodium injection) in adult hospitalized patients with novel coronavirus pneumonia (COVID‐19) |
| Starting date | 10 March 2020 |
| Contact information | Cai Qingxian The Third People's Hospital of Shenzhen, Shenzhen, Guangdong, China +86 13901849660 | 41180423@qq.com |
| Methods | Single‐centre, open‐label, 2‐armed, parallel assignment RCT |
| Participants | 60 participants, ≥ 18 years, female and male Inclusion criteria
Exclusion criteria
|
| Interventions | Experimental: based on the standard treatment recommended in the guidelines, a combination of Prolongin (enoxaparin sodium injection) was used Comparison: follow the guidelines for standard treatment |
| Outcomes | Primary
Secondary
|
| Notes | ChiCTR2000030701 | No data provided |
ChiCTR2000030946.
| Study name | Effects of different VTE prevention methods on the prognosis of hospitalized patients with novel coronavirus pneumonia (COVID‐19) |
| Starting date | 10 February 2020 |
| Contact information | Chunli Liu The First Affiliated Hospital of Guangzhou Medical University, Guangzhou, Guangdong, China +86 13560158649 | chunli@gird.cn |
| Methods | Prospective cohort, non‐randomised, open‐label, 2 parallel and comparative arms |
| Participants | 120 participants, 18‐80 years, female and male Inclusion criteria
Exclusion criteria
|
| Interventions | Experimental: 7/5000 LMWH therapy Comparison: mechanical prevention |
| Outcomes | Primary: biochemical indicators Secondary: not described |
| Notes | ChiCTR2000030946 | No data provided |
CTRI/2020/06/026220.
| Study name | A study to evaluate the efficacy and safety of nafamostat mesilate in treatment of coronavirus infection |
| Starting date | 17 July 2020 |
| Contact information | Guruprasad Palekar Sun Pharma Laboratories Limited Sun House, 201 B/1, Western Express Highway, Goregaon (E), Mumbai 400063 390020 Mumbai, MAHARASHTRA India 02656612829 maulik.doshi@sunpharma.com |
| Methods | Multicentre, open‐label RCT with 3 parallel arms |
| Participants | 40 participants; ≥ 18 years and ≥ 65 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental: nafamostat mesilate injection 50 mg/100 mg vial: dissolve a daily dose of nafamostat mesilate in 1000 mL of 5% dextrose and to be infused at dose of 0.1 mg/kg/h for 24 h by continuous infusion for 10 days Comparator: standard care as per institutional practice Patients may be given prophylactic LMWH (e.g. enoxaparin 1mg/kg per day SC) as per investigator discretion. Before starting LMWH, investigator should check for bleeding tendency risk and presence of any contraindications. When nafamostat and LMWH are given concomitantly daily PT/INR and aPTT should be monitored |
| Outcomes | Primary
Secondary
|
| Notes | CTRI/2020/06/026220 | No data provided | Source(s) of monetary support: Sun Pharmaceutical Industries Limited Sun House, 201 B/1, Western Express Highway, Goregaon (E), Mumbai 400063 |
CTRI/2020/08/027033.
| Study name | SARS‐COV‐2 and COVID‐19 ‐ a randomized controlled trail |
| Starting date | 07 August 2020 |
| Contact information | Dr N Anbu Department of General Medicine, Government Siddha Medical College, Arumbakkam Chennai 106 600106 Chennai, TAMIL NADU India 9443279412 | nanbu.sumi@gmail.com |
| Methods | Interventional, RCT, multiple arm trial, 1:1; open label |
| Participants | 100 participants, ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental:
Comparator:
|
| Outcomes | Primary
Secondary
|
| Notes | CTRI/2020/08/027033 | No data provided | Source(s) of monetary support: Govt Kilpauk Medical College Hospital Chennai 600010 |
CTRI/2020/11/029175.
| Study name | Role of heparin inhalation in reducing the duration the patient is breathing with the help of a ventilator |
| Starting date | 17 November 2020 |
| Contact information | Shagufta Naaz Associate professor, department of Anaesthesiology, AIIMS Patna, Phulwarisharif Patna 801507 801507 Kancheepuram, BIHAR India 07765937919 | drshaguftanaaz@gmail.com |
| Methods | Interventional, RCT, 1:1, active controlled trial, participant blinded |
| Participants | 58 participants, ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental:
Comparator:
|
| Outcomes | Primary
Secondary
|
| Notes | CTRI/2020/11/029175 | No data provided | Source(s) of monetary support: AIIMS Patna |
CTRI/2020/11/029345.
| Study name | To determine efficacy, safety and optimal dosing of anticoagulant strategies to prevent adverse outcomes in hospitalized COVID‐19 patients |
| Starting date | 25 November 2020 |
| Contact information | Dr Viral Shah 401, 4th Floor Kshamalaya Building, 37 New Marine Lines. Mumbai MAHARASHTRA 400020 India 400020 Mumbai, MAHARASHTRA India 02240645101 | vshah@spectrumcr.com |
| Methods | Interventional, RCT, 1:1, open label |
| Participants | 3600 participants; ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental: nil
Comparator:
|
| Outcomes | Primary
Secondary
|
| Notes | CTRI/2020/11/029345 | No data provided | Source(s) of monetary support: Icahn School of Medicine at Mount Sinai One Gustave L. Levy Place, Box 1030 New York, NY 10029 |
EUCTR2020‐001302‐30‐AT.
| Study name | A multicenter, randomized, active controlled, open label, platform trial on the efficacy and safety of experimental therapeutics for patients with a lung disease caused by coronavirus infection ACOVACT (Austrian CoronaVirus Adaptive Clinical Trial) |
| Starting date | 09 April 2020 |
| Contact information | Medical University of Vienna Währinger Gürtel 18‐20 1090 Vienna Austria klin‐pharmakologie@meduniwien.ac.at |
| Methods | Interventional, multicentre, open‐label RCT with 4 parallel arms |
| Participants | 500 participants; ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Kaletra:
Xarelto
Asunercept
Veklury
Pentaglobin
|
| Outcomes | Primary
Secondary
|
| Notes | EUCTR2020‐001302‐30‐AT | No data provided | Source(s) of monetary support: Medical University of Vienna |
EUCTR2020‐001708‐41‐IT.
| Study name | Enoxaparin for thromboprophylaxis in hospitalized COVID‐19 patients: comparison of 40 mg o.d. versus 40 mg b.i.d. A randomized clinical trial |
| Starting date | 24 June 2020 |
| Contact information | Azienda Ospedaliera Ao Ospedale Niguarda Ca' Granda ASST Grande Ospedale Metropolitano Niguarda Segreteria Unità Intensive Cure Ca Piazza Ospedale Maggiore 3 Milano 0264442576 | ucict@ospedaleniguarda.it |
| Methods | Prospective, multicentre, open‐label, parallel‐assignment RCT |
| Participants | 2000 participants, ≥ 18 years and ≤ 64 years, 700 participants, ≥ 65 years, female and male Inclusion criteria
Exclusion criteria
|
| Interventions | Main objective of the trial
Secondary objectives of the trial
|
| Outcomes | Primary
Secondary
|
| Notes | EUCTR2020‐001708‐41‐IT | No data provided |
EUCTR2020‐001709‐21‐FR.
| Study name | Low‐molecular‐weight heparin to prevent venous thromboembolism in COVID‐19 patients: a randomized controlled trial of different doses |
| Starting date | 29 April 2020 |
| Contact information | Direction de la Recherche Clinique Bâtiment Recherche ‐ rue du Morvan 54511 Vandoeuvre lès Nancy France 33383 155285 | dripromoteur@chru‐nancy.fr |
| Methods | Open‐label, 2‐armed, parallel assignment RCT |
| Participants | 230 participants of 18‐64 years and 320 participants ≥ 65 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Enoxaparin (Lovenox or other specialties)
Tinzaparine (Innohep)
Dalteparin (Fragmine=E)
Nadroparin (Fraxiparine)
Enoxaparin (Lovenox or other specialities)
Tinzaparin (Innohep)
|
| Outcomes | Primary
Secondary
|
| Notes | EUCTR2020‐001709‐21‐FR | No data provided | Source(s) of monetary support: DGOS (being obtained), Région Grand Est (being obtained) |
EUCTR2020‐001891‐14‐ES.
| Study name | Impact of the use of low molecular weight heparins (LMWH), at prophylactic versus intermediate doses, on SARS‐CoV2 infection (COVID‐19) |
| Starting date | 04 May 2020 |
| Contact information | Alejandro Calle Editor José Manuel Lara, 28, 1B 41013 Sevilla Spain +34630157890 | secretaria@delosclinical.com |
| Methods | Open‐label, 2‐armed, parallel‐assignment RCT |
| Participants | 140 participants, ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Enoxaparin sodium
|
| Outcomes | Primary
Secondary
|
| Notes | EUCTR2020‐001891‐14‐ES | No data provided | Source(s) of monetary support: Fundación Neumosur |
EUCTR2020‐002234‐32‐IT.
| Study name | Efficacy and safety of edoxaban and or colchicine for patients with SARS‐CoV‐2 infection managed in the out of hospital setting (COVID 19) |
| Starting date | 28 December 2020 |
| Contact information | Cardiologia Freiburgstrasse, 8 3010 Bern Switzerland +41316325492 | marco.valgimigli@insel.ch |
| Methods | Prospective, multicentre, open‐label, 4‐armed RCT |
| Participants | 420 participants, ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Lixiana 30 mg
Colchicine
Lixiana 60 mg
|
| Outcomes | Primary
Secondary
|
| Notes | EUCTR2020‐002234‐32‐IT | No data provided | Source(s) of monetary support: Daiichi Sankyo Europe GmbH |
EUCTR2020‐002504‐39‐DE.
| Study name | Hamburg edoxaban for anticoagulation in COVID‐19 study |
| Starting date | 10 November 2020 |
| Contact information | Lilli Gerstenmaier Martinistrasse 64 20251 Hamburg Germany +49040524719216 | regulatory@ctc‐north.com |
| Methods | Prospective, single‐blind, 2‐armed, parallel‐assignment RCT |
| Participants | 172 participants, ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental:
Comparator:
|
| Outcomes | Primary
Secondary
|
| Notes | EUCTR2020‐002504‐39‐DE | No data provided | Source(s) of monetary support: Daiichi Sankyo Europe GmbH; University Medical Center Hamburg‐Eppendorf |
EUCTR2020‐003349‐12‐IE.
| Study name | This is a proof of principle/feasibility study aiming to evaluate the effect of nebulised unfractionated heparin on procoagulant markers related to acute respiratory distress syndrome in patients invasively ventilated for COVID‐19 lung disease |
| Starting date | 09 October 2020 |
| Contact information | Prof John Laffey University Road H91 TK33 Galway Ireland 35391524411 | john.laffey@nuigalway.ie |
| Methods | Prospective, open‐label, 1‐armed, parallel‐assignment RCT |
| Participants | 40 participants, ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental:
Comparator: nil |
| Outcomes | Primary
Secondary
|
| Notes | EUCTR2020‐003349‐12‐IE | No data provided | Source(s) of monetary support: Aerogen; CURAM/SFI |
Goldin 2020.
| Study name | Systemic anticoagulation with full dose low molecular weight heparin (LMWH) vs. prophylactic or intermediate dose LMWH in high risk COVID‐19 patients (HEP‐COVID Trial) |
| Starting date | 26 April 2020 |
| Contact information | Damian N Inlall Northwell Health, USA (516) 600‐1482 | dinlall@northwell.edu |
| Methods | Prospective, multicentre, triple‐blinded, 2‐armed, parallel‐assignment RCT |
| Participants | 308 participants, ≥ 18 years, female and male Inclusion criteria
Exclusion criteria
|
| Interventions | Experimental: full‐dose LMWH anticoagulation therapy Participants in this study arm will be treated with therapeutic doses of SC LMWH (enoxaparin). Enoxaparin 1 mg/kg SC twice a day for CrCl ≥ 30 mL/min (or enoxaparin 0.5 mg/kg SC twice a day for CrCl ≥ 15 mL/min and < 30 mL/min) during the course of their hospitalisation Comparator: prophylactic/intermediate‐dose LMWH or UFH therapy Participants in this study arm will be treated with local institutional standard care for prophylactic‐dose or intermediate‐dose UFH or LMWH. Regimens allowed are UFH up to 22,500 IU daily in twice daily or three times daily doses (i.e. UFH 5000 IU SC twice a day/three times a day or 7500 IU twice a day/three times a day), enoxaparin 30 mg and 40 mg SC daily or twice daily (the use of weight‐based enoxaparin i.e. 0.5 mg/kg SC twice a day for this arm is acceptable but strongly discouraged), dalteparin 2500 IU or 5000 IU a day |
| Outcomes | Primary
Secondary
|
| Notes | NCT04401293 | No data provided |
IRCT20200515047456N1.
| Study name | The role of anticoagulant and thrombolitic in treatment of COVID patients |
| Starting date | 17 June 2020 |
| Contact information | Farid Rashidi Daneshgah 5166614756 Iran (Islamic Republic of) +98 41 3336 4901 | fr2652@yahoo.com |
| Methods | Single‐blinded, 2‐armed, parallel‐assignment RCT |
| Participants | 15 participants, ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental:
Comparator: without placebo |
| Outcomes | Primary
|
| Notes | IRCT20200515047456N1 | No data provided | Source(s) of monetary support: Tabriz University of Medical Sciences |
ISRCTN14212905.
| Study name | Understanding how COVID‐19 leads to respiratory failure in COVID‐19 positive patients |
| Starting date | 03 July 2020 |
| Contact information | Annya Bruce Queen's Medical Research Institute Little France Crescent EH16 4TJ Edinburgh United Kingdom +44 (0)131 2429180 | Annya.Bruce@ed.ac.uk |
| Methods | Single‐centre RCT |
| Participants | 100 participants, ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Patients will be divided into cohorts
2 treatments will be compared to standard care. Nafamostat (anti‐viral and anti‐coagulant) and TD139 (galectin 3 inhibitor). For nafamostat, it is intended that the licensed dose (0.2 mg/kg/h) in Japan will be used. Patients randomised to nafamostat will receive a continuous IV infusion at 0.2 mg/kg/h for 7 days. If a participant is discharged from hospital or can no longer receive this treatment, the treatment will be stopped. For TD139, patients will inhale 5 mg x 2 (10 mg) twice daily for the first 48 h and then subsequently 5 mg x 2 (10 mg) once daily for the remaining 12 days. Unless a participant is discharged from hospital or can no longer use an inhaler – in which case treatment will be stopped at such time. Follow‐up will be at 30, 60 and 90 days post‐treatment |
| Outcomes | Primary
Secondary
|
| Notes | ISRCTN14212905 | No data provided | Source(s) of monetary support: Life Arc |
Kharma 2020.
| Study name | Anticoagulation in patients suffering from COVID‐19 disease‐The Anti‐Co Trial |
| Starting date | 28 June 2020 |
| Contact information | Marcus Lance, MD, PhD Hamad Medical Corporation Doha, Qatar 00974 ext 33530292 | mlance@hamad.qa |
| Methods | Triple‐blind, 2‐armed, parallel‐assignment RCT |
| Participants | 100 participants, ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental:
Comparator:
|
| Outcomes | Primary
Secondary
|
| Notes | NCT04445935 | No data provided |
Lasky 2021.
| Study name | A phase 2/3 study to evaluate the safety and eficacy of dociparstat sodium for the treatment of severe COVID‐19 in adults at high risk of respiratory failure |
| Starting date | 3 June 2020 |
| Contact information | Marion Morrison, MD University of Alabama at Birmingham Birmingham, Alabama, USA, 35294 919‐313‐2977 | mmorrison@chimerix.com |
| Methods | Double‐blind, 2‐armed, 2:1 parallel‐assignment RCT |
| Participants | 525 participants, ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental: dociparstat sodium (DSTAT)
Comparator: placebo
|
| Outcomes | Primary
Secondary
|
| Notes | NCT04389840 | No data provided |
Lins 2020.
| Study name | CoV‐Hep Study: randomized and paired clinical trial comparing regional anticoagulation modalities in continuous venous venous hemodialysis in patients with COVID‐19 |
| Starting date | 29 June 2020 |
| Contact information | Paulo Lins, MD University of São Paulo General Hospital São Paulo, SP, Brazil, 05403‐010 +55.11.98279‐2696 | paulo.lins@hc.fm.usp.br |
| Methods | Open‐label, 2‐armed, parallel RCT |
| Participants | 90 participants, ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental:
Comparator
|
| Outcomes | Primary
Secondary
|
| Notes | NCT04487990 | No data provided | |
Marietta 2020.
| Study name | Randomised controlled trial comparing high versus low LMWH dosages in hospitalised patients with severe COVID‐19 pneumonia and coagulopathy not requiring invasive mechanical ventilation |
| Starting date | 1 June 2020 |
| Contact information | Marco Marietta, MD Azienda Ospedaliero‐Universitaria di Modena, Italy 0594224640 ext +39 | marco.marietta@unimore.it |
| Methods | Multicentre, open‐label, investigator‐sponsored, 2‐arm, parallel‐assignment RCT |
| Participants | 300 participants, 18‐80 years, female and male Inclusion criteria (all required)
Exclusion criteria
|
| Interventions | Experimental: high‐dose LMWH: 70 IU/kg twice daily, other name: Inhixa Comparator: low‐dose LMWH: enoxaparin 4000 IU daily |
| Outcomes | Primary
Secondary
|
| Notes | NCT04408235 | EudraCT 2020‐001972‐13 | No data provided |
NCT04333407.
| Study name | Preventing cardiac complication of COVID‐19 disease with early acute coronary syndrome therapy: a randomised controlled trial |
| Starting date | 3 April 2020 |
| Contact information | Alena Marynina Charing Cross Hospital, London, UK 07776 224520 | alena.marynina@nhs.net |
| Methods | Multicentre, open‐label RCT with 2 parallel arms, 1:1 |
| Participants | 3170 participants, ≥ 18 years, female and male Inclusion criteria
Exclusion criteria
|
| Interventions | Experimental: active arm
Comparator: no intervention |
| Outcomes | Primary
Secondary
|
| Notes | NCT04333407 | No data provided |
NCT04344756.
| Study name | Cohort multiple randomized controlled trials open‐label of immune modulatory drugs and other treatments in COVID‐19 patients CORIMUNO‐COAG trial |
| Starting date | 20 April 2020 |
| Contact information | Tristan Mirault Assistance Publique ‐ Hôpitaux de Paris, France 1 56 09 50 41 ext 33 | tristan.mirault@aphp.fr |
| Methods | Randomised clinical trial with 2 parallel arms, 1:1, stratified on disease severity (ventilation or not) |
| Participants | 808 participants, ≥ 18 years, female and male Inclusion criteria
Exclusion criteria
|
| Interventions | Experimental: tinzaparin or UFH
Comparator: standard care
|
| Outcomes | Primary
Secondary
|
| Notes | NCT04344756 | APHP200389‐6 | No data provided |
NCT04345848.
| Study name | Preventing COVID‐19‐associated thrombosis, coagulopathy and mortality with low‐ and high‐dose anticoagulation: a randomized, open‐label clinical trial |
| Starting date | 28 April 2020 |
| Contact information | Marc Blondon University Hospital, Geneva, Switzerland +41.22.372.92.92 | marc.blondon@hcuge.ch |
| Methods | Prospective, multicentre, single‐blind (outcomes assessor), 2‐armed, parallel‐assignment RCT |
| Participants | 200 participants, ≥ 18 years, female and male Inclusion criteria Adult patient with COVID‐19 infections, admitted to:
Exclusion criteria
|
| Interventions | Experimental: therapeutic anticoagulation Participants will be treated with therapeutic doses of SC LMWH (enoxaparin) or IV UFH, from admission until the end of hospital stay or clinical recovery Comparator: prophylactic anticoagulation Participants will be treated with prophylactic doses of SC LMWH (enoxaparin) or UFH, from admission until the end of hospital stay or clinical recovery. If hospitalised in the ICU, they will receive an augmented thromboprophylaxis regimen as standard care. |
| Outcomes | Primary
Secondary
Other outcome
|
| Notes | NCT04345848 | No data provided |
NCT04352400.
| Study name | RAndomized clinical trial in COvid19 patients to assess the efficacy of the transmembrane protease serine 2 (TMPRSS2) inhibitor NAfamostat (RACONA Study) |
| Starting date | 1 April 2020 |
| Contact information | Gian Paolo Rossi University Hospital Padova, Italy 00390498217821 | gianpaolo.rossi@unipd.it |
| Methods | Multicentre, double‐blind, 2‐armed, parallel‐assignment RCT |
| Participants | 256 participants, 18‐85 years, female and male Inclusion criteria
Exclusion criteria
|
| Interventions | Experimental: nafamostat mesilate, administered IV as a continuous infusion Comparator: placebo, administered IV as a continuous infusion |
| Outcomes | Primary
Secondary
|
| Notes | NCT04352400 | No data provided |
NCT04366960.
| Study name | Enoxaparin for thromboprophylaxis in hospitalized COVID‐19 patients: comparison of 40 mg o.d. versus 40 mg b.i.d. a randomized clinical trial |
| Starting date | 14 May 2020 |
| Contact information | Nuccia Morici Azienda Socio Sanitaria Territoriale Grande Ospedale Metropolitano Niguarda, Milano, Italy +396444 ext 2565 | nuccia.morici@ospedaleniguarda.it |
| Methods | Prospective, multicentre, open‐label, 1:1, 2‐armed, parallel‐assignment RCT |
| Participants | 2712 participants, ≥ 18 years, female and male Inclusion criteria
Exclusion criteria
|
| Interventions | Experimental: 40 mg SC enoxaparin twice a day Comparator: 40 mg SC enoxaparin once a day |
| Outcomes | Primary
Secondary
|
| Notes | NCT04366960 | No data provided |
NCT04367831.
| Study name | Intermediate or prophylactic‐dose anticoagulation for venous or arterial thromboembolism in severe COVID‐19: a cluster based randomized selection trial (IMPROVE‐COVID) |
| Starting date | 2 May 2020 |
| Contact information | Sahil A. Parikh Columbia University, New York, New York, USA 212‐305‐7060 | sap2196@cumc.columbia.edu |
| Methods | Prospective, single‐centre, single‐blinded (outcomes assessor), 2‐armed, parallel‐assignment, cluster‐RCT |
| Participants | 100 participants, ≥ 18 years, female and male Inclusion criteria
Exclusion criteria
|
| Interventions | Experimental: intermediate‐dose anticoagulation UFH infusion at 10 units/kg/h with goal anti‐Xa 0.1 ‐0.3U/mL If estimated GFR ≥ 30 mL/min: enoxaparin 1 mg/kg SC daily Comparator: enoxaparin prophylactic dose following local guideline If estimated GFR ≥ 30 mL/min (stable kidney function):
UFH at 5000‐7500 units SC every 8 h |
| Outcomes | Primary
Secondary
|
| Notes | NCT04367831 | No data provided |
NCT04373707.
| Study name | Effectiveness of weight‐adjusted prophylactic low molecular weight heparin doses compared with lower fixed prophylactic doses to prevent venous thromboembolism in COVID‐2019. The multicenter randomized controlled open‐label trial COVI‐DOSE |
| Starting date | 13 May 2020 |
| Contact information | Yohann Bernard Central Hospital, Nancy, France +33.3.83.15.52.72 | y.bernard@chru‐nancy.Fr |
| Methods | Multicentre, open‐label, 2‐armed, parallel‐assignment RCT; stratified on disease severity (admission to ICU or not) |
| Participants | 602 participants, ≥ 18 years, female and male Inclusion criteria
Exclusion criteria
|
| Interventions | Experimental: weight‐adjusted prophylactic dose LMWH For example (enoxaparin):
Other names: tinzaparin, nadroparin, dalteparin Comparator: low prophylactic dose of LMWH For example (enoxaparin): from 4000 IU once a day in participants admitted in medical ward to 4000 IU twice a day in participants admitted in the ICU. In participants with severe renal insufficiency (GFR = 15‐30 mL/min/1.73 m²), LMWH doses will be reduced by 50%. Other names: tinzaparin, nadroparin, dalteparin |
| Outcomes | Primary
Secondary
|
| Notes | NCT04373707 | 2020‐001709‐21 | No data provided |
NCT04377997.
| Study name | A randomized, open‐label trial of therapeutic anticoagulation in COVID‐19 patients with an elevated D‐dimer |
| Starting date | 15 May 2020 |
| Contact information | Mazen Albaghdadi Massachusetts General Hospital, USA 617‐726‐7400 | MALBAGHDADI@mgh.harvard.edu |
| Methods | Open‐label, 2‐armed, parallel‐assignment RCT |
| Participants | 300 participants, ≥ 18 years, female and male Inclusion
Exclusion
|
| Interventions | Experimental: therapeutic anticoagulation group Higher dose (not described) of heparin (LMWH for most participants but UFH for those with morbid obesity or moderate to severe renal dysfunction) Comparator: standard care anticoagulation group There is no dose or drug description. |
| Outcomes | Primary
Secondary
|
| Notes | NCT04377997 | No data provided |
NCT04397510.
| Study name | Nebulized heparin vs. placebo for the treatment of COVID‐19 induced lung injury |
| Starting date | 1 June 2020 |
| Contact information | Thomas Smoot Frederick Health Hospital, Frederick, Maryland, USA |
| Methods | Multicentre, single‐masking (outcomes assessor), investigator‐sponsored, 2‐armed, parallel‐assignment RCT |
| Participants | 50 participants, ≥ 18 years, female and male Inclusion criteria
Exclusion criteria
|
| Interventions | Experimental: nebulised heparin 5000 units/mL IV formulation diluted with 3 mL of 0.9% sodium chloride. Dose: 10,000 units. Frequency: every 4 h. Duration: 10 days Comparator: placebo. 0.9% sodium chloride. Dose: 5 mL. Frequency: every 4 h. Duration: 10 days |
| Outcomes | Primary
Secondary
|
| Notes | NCT04397510 | FHHep518 | No data provided |
NCT04406389.
| Study name | InterMediate ProphylACtic versus Therapeutic dose anticoagulation in critically ill patients with COVID‐19: a prospective randomized study (The IMPACT Trial) |
| Starting date | 13 October 2020 |
| Contact information | Maria T DeSancho, MD, MSc Weill Cornell Medicine New York, New York, United States, 10065 646‐962‐2065 | mtd2002@med.cornell.edu |
| Methods | Open‐label, 2‐armed, 1:1, parallel‐assignment RCT |
| Participants | 186 participants, ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental: therapeutic‐dose anticoagulation Participants will receive 1 of the following interventions, at their physician's discretion
Comparator: intermediate‐dose prophylaxis Participants will receive 1 of the following interventions, at their physician's discretion
|
| Outcomes | Primary
Secondary
|
| Notes | NCT04406389 | No data provided |
NCT04409834.
| Study name | A multicenter, randomized‐controlled trial to evaluate the efficacy and safety of antithrombotic therapy for prevention of arterial and venous thrombotic complications in critically‐ill COVID‐19 patients |
| Starting date | 5 August 2020 |
| Contact information | Vivian Baird‐Zars Brigham and Women's Hospital Boston, Massachusetts, USA, 02459 800‐385‐4444 | vbaird-zars@bwh.harvard.edu |
| Methods | Multicentre, open‐label, parallel‐assignment RCT |
| Participants | 750 participants, ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
Patients who meet the following criterion are excluded from the second randomisation (antiplatelet therapy vs no antiplatelet therapy):
|
| Interventions | Experimental
Comparator
|
| Outcomes | Primary
Secondary
|
| Notes | NCT04409834 | No data provided |
NCT04416048.
| Study name | Effect of anticoagulation therapy on clinical outcomes in moderate to severe coronavirus disease 2019 (COVID‐19) |
| Starting date | 15 June 2020 |
| Contact information | Ulf Landmesser Charite University, Berlin, Germany +49 30 450 513 702 | ulf.landmesser@charite.de |
| Methods | Prospective, multicentre, event‐driven, 2‐armed, parallel‐assignment RCT |
| Participants | 400 participants, ≥ 18 years, female and male Inclusion criteria
Exclusion criteria:
|
| Interventions | Experimental: rivaroxaban Treatment with rivaroxaban 20 mg (15 mg for participants with an estimated GFR ≥ 30 mL/min/1.73 m² and < 50 mL/min/1.73 m²) once daily for at least 7 days. In case of hospitalisation for > 7 days, the therapeutic treatment with rivaroxaban will be continued for the duration of the hospital stay until discharge. After at least 7 days of therapeutic treatment with rivaroxaban or after hospital discharge, the study dose of rivaroxaban will be adjusted as follows:
Other Name: Xarelto Comparator: standard care Participants will receive standard care treatment including prophylactic LMWH or UFH, when considered appropriate according to the judgment of the treating physician. |
| Outcomes | Primary
Secondary
|
| Notes | NCT04416048 | 2020‐002282‐33 | No data provided |
NCT04420299.
| Study name | A randomized, single‐blind study with a parallel control group on the efficacy and safety of bemiparin at therapeutic dose vs. prophylactic dose in patients hospitalized for COVID‐19 |
| Starting date | 4 June 2020 |
| Contact information | Antonio Cubillo, MD Hospital Universitario HM Montepríncipe Recruiting Boadilla Del Monte, Madrid, Spain, 28660 +34 917567800 | secretaria@fundaciónhm.com |
| Methods | Single‐blind, parallel‐assignment RCT |
| Participants | 120 participants, ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental:
Comparator:
|
| Outcomes | Primary
Secondary
|
| Notes | NCT04420299 | No data provided |
NCT04444700.
| Study name | Utilização da enoxaparina em dose anticoagulante em pacientes hospitalizados com síndrome respiratória aguda grave por COVID‐19 |
| Starting date | 4 July 2020 |
| Contact information | Hassan Rahhal, MD Hospital das Clínicas da FMUSP São Paulo, SP, Brazil, 05402‐000 +551126619033 | hassan.r@hc.fm.usp.br |
| Methods | Open‐label, parallel‐assignment RCT |
| Participants | 462 participants, ≥ 18 years, female and male Inclusion criteria
Exclusion criteria:
|
| Interventions | Experimental: therapeutic anticoagulation
Comparator: standard care
|
| Outcomes | Primary
Secondary
|
| Notes | NCT04444700 | No data provided |
NCT04485429.
| Study name | Efficacy assessment of methylprednisolone and heparin in patients with COVID‐19 pneumonia: a randomized, controlled, 2x2 factorial study |
| Starting date | 20 July 2020 |
| Contact information | Eduardo M Rego, MD, PhD D'Or Institute for Research and Education Rio de Janeiro, Brazil 55 16 981110090 | edumrego@hotmail.com |
| Methods | Open‐label, 2:2, parallel‐assignment RCT |
| Participants | 268 participants, ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental:
Comparator: standard treatment
|
| Outcomes | Primary
Secondary
|
| Notes | NCT04485429 | No data provided |
NCT04508439.
| Study name | Effect of the use of anticoagulant therapy during hospitalization and discharge in patients with COVID‐19 infection |
| Starting date | 20 June 2020 |
| Contact information | Omar Ramos‐Peñafiel, MD, PhD Hospital Regional de Alta Especialidad de Ixtapaluca Mexico City, Ixtapaluca, Mexico, 56530 +525523351588 | christian.ramos.penafiel@gmail.com |
| Methods | Double‐blind, 1:1, parallel‐assignment RCT |
| Participants | 130 participants, ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental: prophylactic enoxaparin
Comparator: therapeutic enoxaparin
|
| Outcomes | Primary
|
| Notes | NCT04508439 | No data provided |
NCT04511923.
| Study name | Can nebulised heparin reduce acute lung injury in patients with SARS‐CoV‐2 requiring respiratory support in Ireland |
| Starting date | 23 December 2020 |
| Contact information | John Laffey University Hospital Galway Galway, Ireland +353 91 544074 | John.laffey@nuigalway.ie |
| Methods | Open‐label, parallel‐assignment RCT |
| Participants | 40 participants, ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria
|
| Interventions | Experimental: heparin
Comparator: standard care |
| Outcomes | Primary
Secondary
|
| Notes | NCT04511923 | No data provided |
NCT04512079.
| Study name | FREEDOM COVID anticoagulation strategy randomized trial |
| Starting date | 8 September 2020 |
| Contact information | Debra Fitzpatrick, MS Icahn School of Medicine at Mount Sinai Gustave L. Levy Pl, New York, NY 10029, USA 212‐659‐9151 | debra.fitzpatrick@mssm.edu |
| Methods | Prospective, multicentre, open‐label, 1:1:1, 3‐armed, parallel‐assignment RCT |
| Participants | 3600 participants, ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental:
Comparator:
|
| Outcomes | Primary
Secondary
|
| Notes | NCT04512079 | No data provided |
NCT04530578.
| Study name | Efficacy and safety study to evaluate the use of nebulized heparin in patients with severe acute respiratory syndrome COVID‐19 (SARS‐CoV‐2) |
| Starting date | 1 June 2020 |
| Contact information | ALICIA B VILASECA, DR Clinica San Camilo Ciudad Autonoma de Buenos Aire, Buenos Aires, Argentina, 1405 +5401148588144 ext 244 | avilaseca@clinicasancamilo.org.ar |
| Methods | Prospective, open‐label RCT |
| Participants | 200 participants, ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental: nebulised heparin
Comparator: enoxaparin
|
| Outcomes | Primary
Secondary
|
| Notes | NCT04530578 | No data provided |
NCT04542408.
| Study name | Hamburg edoxaban for anticoagulation in COVID‐19 study |
| Starting date | 12 November 2020 |
| Contact information | Stefan Kluge, MD Universitätsklinikum Düsseldorf Düsseldorf, Germany +49 40 7410 ext 57010 | s.kluge@uke.de |
| Methods | Prospective, multicentre, double‐blinded, 1:1, parallel assignment RCT |
| Participants | 172 participants, ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental: intensive anticoagulation strategy
Comparator: moderate anticoagulation strategy
|
| Outcomes | Primary
Secondary
|
| Notes | NCT04542408 | No data provided |
NCT04545541.
| Study name | Can nebulised heparin reduce mortality and time to extubation in patients with COVID‐19 requiring mechanical ventilation meta‐trial (CHARTER‐MT): protocol for an investigator‐initiated international meta‐trial of randomised studies |
| Starting date | 1 November 2020 |
| Contact information | Frank MP van Haren, MD, PhD Frederick Health Hospital Frederick, Maryland, USA, 21701 +61467051809 | fvanharen@me.com |
| Methods | Prospective, multicentre, double‐blinded, 1:1, parallel‐assignment RCT |
| Participants | 300 participants, ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental: nebulised heparin
Comparator:
|
| Outcomes | Primary
|
| Notes | NCT04545541 | No data provided |
NCT04584580.
| Study name | D‐dimer adjusted versus therapeutic dose low‐molecular‐weight heparin in patients with COVID‐19 pneumonia |
| Starting date | 1 August 2020 |
| Contact information | Ashraf Madkour Faculty of Medicine Ain Shams University Research Institute‐ Clinical Research Center Cairo, Non‐US, Egypt, 11566 +20 100 177 0703 | asfrah_madkour@yahoo.com |
| Methods | Single‐blinded, parallel‐assignment RCT |
| Participants | 50 participants, ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental:
Comparator: therapeutic‐dose LMWH
|
| Outcomes | Primary
Secondary
|
| Notes | NCT04584580 | No data provided |
NCT04600141.
| Study name | Clinical efficacy of heparin and tocilizumab in patients with severe COVID‐19 infection: a randomized clinical trial |
| Starting date | 10 November 2020 |
| Contact information | Ludhmila A Hajjar, MD, PhD Fundação São Francisco Xavier Ipatinga, Minas Gerais, Brazil 551126614177 | ludhmila@terra.com.br |
| Methods | Open‐label, parallel‐assignment RCT |
| Participants | 308 participants, ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental:
Comparator:
|
| Outcomes | Primary
Secondary
|
| Notes | NCT04600141 | No data provided |
NCT04604327.
| Study name | Ensayo clínico aleatorizado, abierto, para evaluar el efecto de dosis profilácticas o terapéuticas de bemiparina en pacientes con COVID‐19 |
| Starting date | 26 October 2020 |
| Contact information | Ramon Lecumberri, MD, PhD Hospital Rey Juan Carlos Móstoles, Madrid, Spain +34 948296397 | rlecumber@unav.es |
| Methods | Open‐label, parallel‐assignment RCT |
| Participants | 164 participants, ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria: nil |
| Interventions | Experimental: full therapeutic bemiparin (weight adjusted)
Comparator: prophylactic bemiparin (3500 IU/day)
|
| Outcomes | Primary
|
| Notes | NCT04604327 | No data provided |
NCT04623177.
| Study name | Thromboprophylaxis for patients in ICU With COVID‐19 |
| Starting date | 1 March 2020 |
| Contact information | Raquel Ferrandis, MD Hospital Universitario La Fe, Valencia, Spain phone and email not available |
| Methods | Prospective cohort, non‐randomised, open‐label, 3 parallel and comparative arms |
| Participants | 950 participants, ≥ 18 years, female and male Inclusion criteria
Exclusion criteria
|
| Interventions | Experimental: anticoagulant dose (≥ 150 IU/kg/24 h) of LMWH within the first 48 h after the ICU admission Experimental: prophylactic dose (lower than 150 IU/kg/24 h) of LMWH within the first 48 h after the ICU admission Comparator: no anticoagulant drug within the first 48 h after the ICU admission |
| Outcomes | Primary
Secondary
|
| Notes | NCT04623177 | No data provided |
NCT04640181.
| Study name | A phase 2‐3, multi‐center, randomized trial to study the potential benefit of factor Xa inhibitor (rivaroxaban) versus standard of care low molecular weight heparin (Lovenox) in hospitalized patients with COVID‐19 (XACT) |
| Starting date | 1 December 2020 |
| Contact information | Matt Cowperthwaite, PhD St. David's Medical Center Austin, Texas, USA, 78705 512‐544‐2626 | info@stdavidsresearch.com |
| Methods | Prospective, multicentre, open‐label, 1:1, parallel‐assignment RCT |
| Participants | 150 participants, ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
Vital signs
Laboratory
Medications
Other COVID‐19 drug studies or trials
|
| Interventions | Experimental: adaptive dosing: rivaroxaban
Comparator: adaptive dosing: enoxaparin
|
| Outcomes | Primary
Secondary
|
| Notes | NCT04640181 | No data provided |
NCT04646655.
| Study name | Enoxaparin at prophylactic or therapeutic doses with monitoring of outcomes in subjects infected with COVID‐19: a pilot study on 300 cases enrolled at ASST‐FBF‐Sacco |
| Starting date | 27 July 2020 |
| Contact information | Maddalena A Wu, M.D. ASST Fatebenefratelli Sacco Milan, Italy, 20157 +390239041 | maddalena.ale.wu@gmail.com |
| Methods | Single‐centre, open‐label, 2‐armed, parallel assignment RCT |
| Participants | 300 participants, ≥ 18 years and ≤ 80 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental: enoxaparin at therapeutic dose
Comparator: enoxaparin at prophylactic dose
|
| Outcomes | Primary
Secondary
|
| Notes | NCT04646655 | No data provided |
NCT04655586.
| Study name | Assessing safety, hospitalization and efficacy of rNAPc2 in COVID‐19 (ASPEN‐COVID‐19) |
| Starting date | 10 December 2020 |
| Contact information | Jennifer Meriwether ARCA Investigational Site Fairhope, Alabama, USA, 36532 720‐940‐2132 | jennifer.meriwether@arcabio.com |
| Methods | Multicentre, double‐blind, parallel‐assignment RCT |
| Participants | 100 participants, ≥ 18 years and ≤ 90 years, female and male |
| Interventions | Experimental:
Comparator: heparin
|
| Outcomes | Primary
Secondary
|
| Notes | NCT04655586 | No data provided |
NCT04723563.
| Study name | Inhaled unfractionated heparin for the treatment of hospitalized patients with COVID‐19 pneumonia |
| Starting date | 21 February 2021 |
| Contact information | Thomas Smoot, PharmD Frederick Health Hospital Frederick, Maryland, USA, 21701 |
| Methods | Quadruple‐blind, parallel‐assignment RCT |
| Participants | 50 participants, ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental: nebulised heparin
Comparator: placebo
|
| Outcomes | Primary
Secondary
|
| Notes | NCT04723563 | No data provided |
NCT04730856.
| Study name | Standard vs high prophylactic doses or anticoagulation in patients with high risk of thrombosis admitted with COVID‐19 pneumonia (PROTHROMCOVID) |
| Starting date | 1 February 2021 |
| Contact information | ANGEL PUEYO Hospital Universitario Infanta Leonor Gran Vía del Este, 80, 28031 Madrid, Spain +34 618 448 807 | angel.pueyo@salud.madrid.org |
| Methods | Open‐label, parallel‐assignment RCT |
| Participants | 600 participants, ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental: tinzaparin 4500 UI/day
Comparator:
|
| Outcomes | Primary
Secondary
|
| Notes | NCT04730856 | No data provided |
NCT04743011.
| Study name | Nebulized enriched heparin to treat no critical patients with SARS‐CoV‐2 ‐ triple blind clinical trial |
| Starting date | 1 June 2021 |
| Contact information | Matheus Bertanha, PhD School of Medicine at Botucatu‐ Paulista State University‐ UNESP, São Paulo, Brazil Botucatu, SP, Brazil, 18607030 +55(14)3880‐1444 | matheusbertanha@gmail.com |
| Methods | Triple‐blind, parallel‐assignment RCT |
| Participants | 50 participants, ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental: heparin sodium
Comparator: placebo
|
| Outcomes | Primary
Secondary
|
| Notes | NCT04743011 | No data provided |
NCT04745442.
| Study name | Pilot study of antithrombin as prophylaxis of acute respiratory distress syndrome in patients with COVID‐19 |
| Starting date | 27 April 2020 |
| Contact information | Maimónides Biomedical Research Institute of Córdoba Hospital Universitario Reina Sofía Córdoba, Spain, 14004 |
| Methods | Single‐centre, open‐label, parallel‐assignment RCT |
| Participants | 48 participants, ≥ 18 to ≤ 85 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental: best available treatment + antithrombin
Active comparator: best available treatment
|
| Outcomes | Primary
Secondary
|
| Notes | NCT04745442 | No data provided |
PACTR202007606032743.
| Study name | Nebulized heparin in patients with mainly moderate coronavirus disease 2019: randomized controlled trial. COVID‐19 |
| Starting date | 15 June 2020 |
| Contact information | Tarek Ismail Al Gamaa, Al Masaken Al Iqtisadeyah, Qism Helwan Cairo Egypt 00201112277417 | tareksalem00@gmail.com |
| Methods | RCT |
| Participants | 100 participants, ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | Standard care plus nebulised heparin, every 6 h started 24 h after randomisation and will continue for 1 week |
| Outcomes | Primary
Secondary
|
| Notes | PACTR202007606032743 | No data provided |
RBR‐7y8j2bs.
| Study name | Enriched heparin anti COVID‐19 trial |
| Starting date | 03 January 2021 |
| Contact information | Matheus Bertanha Departamento de Cirurgia Vascular do Hospital das Clínicas de Botucatu: Avenida Professor Emérito Mário Rubens Guimarães Montenegro, s/nº 18618687 Botucatu Brazil +551438801444 | matheusbertanha@gmail.com |
| Methods | Triple‐blind RCT |
| Participants | ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | High molecular weight inhalational heparin (HMWH) (250 µg/mL 0.9% normal saline), who will receive care with the experimental treatment with inhaled HMWH applied in 4‐4 h, for 7 days |
| Outcomes | Safety: related to the use of inhalational high molecular weight heparin in patients with SARS‐COV‐2 through the assessment of haemorrhagic events of any nature, alteration of the coagulogram that indicates an increase in aPTT > 1, 5 and HIT Efficacy: relative to the proposed treatment, through the analysis of the viral load of the SARS‐COV‐2 virus in the participants treated by the sequential evaluation of the viral load in RT‐PCR of nasal swab |
| Notes | RBR‐7y8j2bs | No data provided |
Sholzberg 2021a.
| Study name | Coagulopathy of COVID‐19: a pragmatic randomized controlled trial of therapeutic anticoagulation versus standard care as a rapid response to the COVID‐19 pandemic (RAPID COVID COAG) |
| Starting date | 11 May 2020 |
| Contact information | Michelle Sholzberg St. Michael's Hospital, Toronto, Ontario, Canada 416‐864‐5389 | Michelle.Sholzberg@unityhealth.to |
| Methods | Multicentre, quadruple masking (participant, care provider, investigator, outcomes assessor), investigator‐sponsored, 2‐armed, parallel‐assignment RCT |
| Participants | 462 participants, ≥ 18 years, female and male Inclusion criteria
Exclusion criteria
|
| Interventions | Experimental: therapeutic anticoagulation
Comparison: standard care
|
| Outcomes | Primary
Secondary
|
| Notes | NCT04362085 | No data provided |
Vanassche 2020.
| Study name | A randomized, open‐label, adaptive, proof‐of‐concept clinical trial of modulation of host thromboinflammatory response in patients with COVID‐19: the DAWn‐Antico study |
| Starting date | 25/jun/2020 |
| Contact information | Caroline Devooght Herestraat 49 3000 Leuven Belgium caroline.devooght@uzleuven.be |
| Methods | Multicentre, open‐label, randomised clinical trial |
| Participants | 210 participants, ≥ 18 years, female and male Male or non‐pregnant female adult ≥ 18 years of age at the time of enrolment, participants eligible for inclusion are hospitalised, adult patients with confirmed and severe COVID‐19 |
| Interventions | We compare LMWHs at 50 IU anti‐Xa/kg twice daily—or 75 IU anti‐Xa twice daily for ICU patients—in combination with aprotinin to standard thromboprophylaxis in hospitalised COVID‐19 patients |
| Outcomes | Primary
|
| Notes | EUCTR2020‐001739‐28‐BE | No data provided |
Van Haren 2020.
| Study name | Nebulised heparin in patients with severe COVID‐19 (CHARTER‐MT) |
| Starting date | 01 November 2020 |
| Contact information | Frank MP van Haren, MD, PhD Australian National University Helwan University Clinica San Camilo, Argentina +61467051809 | frank.vanharen@anu.edu.au |
| Methods | Randomised clinical trial |
| Participants | 712 participants, ≥ 18 years, female and male
|
| Interventions | Nebulised UFH (25,000 Units in 5 mL) will be administered 6‐hourly via an Aerogen Solo vibrating mesh nebuliser while patients receive invasive mechanical ventilation in ICU and for a maximum of 10 days |
| Outcomes | Primary
|
| Notes | NCT04635241 | No data provided |
Wilkinson 2020.
| Study name | A platform to investigate the safety and effectiveness of several new medicines for the treatment of COVID‐19 in hospitalised patients |
| Starting date | 08 May 2020 |
| Contact information | Tom Wilkinson Southampton University Faculty of Medicine Mailpoint 810, Level F, South Block Southampton General Hospital SO16 6YD Southampton UK +44 (0)2381 205341 | accord@uhs.nhs.uk |
| Methods | Multicentre, open‐label RCT |
| Participants | 1800 participants, ≥ 18 years, female and male Inclusion criteria:
Exclusion criteria:
|
| Interventions | The study consists of 2 stages:
|
| Outcomes | Primary
Secondary Measured from patient records unless otherwise noted:
|
| Notes | ISRCTN57085639 | No data provided |
aPTT: activated partial thromboplastin time; ACS: acute coronary syndrome; ACCT: Adaptive COVID‐19 Treatment Trial; AE: adverse event; AKI: acute kidney injury; AKIN: Acute Kidney Injury Network; ALT: alanine aminotransferase; ANC: absolute neutrophil count; ARDS: acute respiratory distress syndrome; ARs: adverse reactions; AST: aspartate aminotransferase; BARC: Bleeding Academic Research Consortium; BiPAP: bilevel positive airway pressure; BMI: body mass index; BP: blood pressure; CKI‐EPI: Chronic Kidney Disease Epidemiology Collaboration; CNS: central nervous system; COPD: chronic obstructive pulmonary disease; CPAP: continuous positive airway pressure; CPR: cardiopulmonary resuscitation; CrCl: creatinine clearance; CUS: serial compression ultrasonography; CT: computed tomography; CVVHD: continuous veno‐venous haemodialysis; DBP: diastolic blood pressure; DIC: disseminated intravascular coagulation; DMARDs: disease‐modifying antirheumatic drugs; DOAC: direct oral anticoagulant; DVT: deep vein thrombosis; ECG: electrocardiogram; ECMO: extracorporeal membrane oxygenation; ELISA: enzyme‐linked immunosorbent assay; ESKD: end‐stage kidney disease; FiO2: fraction of inspired oxygen concentration; FSH: follicle‐stimulating hormone; GFR: glomerular filtration rate; GI: gastrointestinal; GGT: glutamyltransferases; HCG: human chorionic gonadotropin; HFOV: high‐frequency oscillatory ventilation; HIT: heparin‐induced thrombocytopenia ICU: intensive care unit; INR: international normalised ratio; ISTH: International Society on Thrombosis and Haemostasis; IV: intravenous(ly); JAKi: Janus kinase inhibitors; LMWH: low molecular weight heparin; MOHFW: Ministry of Health and Family Welfare; MVTE: Major vascular thrombotic events; NEWS: National Early Warning Score; NIV: non‐invasive ventilation; NOACS: novel oral anticoagulants; NSAIDs: non‐steroidal antiflammatory drugs; NYHA: New York Heart Association; PaO2: partial pressure of oxygen; PCR: polymerase chain reaction; PD: Pharmacodynamic; PE: pulmonary embolism; PK: Pharmacokinetic; PRCB: packed red blood cell; PT(T): partial thromboplastin (time); RAS: Renin‐Angiotensin System; RCT: randomised controlled trial; RT‐PCR: reverse transcription polymerase chain reaction; SAE: serious adverse event; SARS: severe acute respiratory syndrome; SBP: systolic blood pressure; SC: subcutaneous(ly);SIC: sepsis‐induced coagulopathy;SOFA: sequential organ failure assessment; SpO2: ratio of oxygen saturation in the blood; TB: tuberculosis; TIA: transient ischaemic attack; UFH: unfractionated heparin; ULN: upper limit of normal; VKA: vitamin K antagonists; VTE: venous thromboembolism; WBC: white blood cells; WHO: World Health Organization
Differences between protocol and review
Title and Objectives
We amended the title from 'Prophylactic anticoagulants for people hospitalised with COVID‐19' at the protocol stage (Flumignan 2020a) and the previous version of this review (Flumignan 2020b) to 'Anticoagulants for people hospitalised with COVID‐19' in this version to better reflect the purposes of the review. The objectives were amended from 'To assess the effects of prophylactic anticoagulants versus active comparator, placebo or no intervention, on mortality and the need for respiratory support in people hospitalised with COVID‐19' in the protocol and previous version to 'To assess the benefits and harms of anticoagulants versus active comparator, placebo or no intervention in people hospitalised with COVID‐19.' in this version.
Types of studies
At the protocol stage, we had planned to narratively describe skewed data reported as medians and interquartile ranges. However, in our review, we estimated the mean difference (MD) using the method reported by Wan 2014 to convert median and interquartile range (IQR) into MD and confidence intervals (CI). When this was not possible, we narratively described the skewed data as originally planned. We planned to limit our primary analyses to specific studies, that is, randomised controlled trials (RCTs) and quasi‐RCTs, but we performed meta‐analyses for all included studies (RCTs or non‐randomised studies (NRS)) with available data to follow Chapter 24 of the Cochrane Handbook for Systematic Reviews of Interventions (Reeves 2021).
Subgroup analysis and investigation of heterogeneity
At the protocol stage, we had planned subgroup analysis but did not include illness severity. However, in this review version, we performed a subgroup analysis by illness severity when possible. Since venous thromboembolism is more prevalent in more severely ill people with COVID‐19, we considered it clinically relevant to present the results by this subgroup analysis.
Data extraction and management
Assessment of risk of bias in included studies
We planned to include only studies that used statistical adjustment for baseline factors using multivariate analyses for the following confounding factors in our protocol (Flumignan 2020a): participants already using anticoagulants (e.g. atrial fibrillation); participants who underwent surgery during the hospitalisation; active cancer treatment; concomitant antiplatelet use; and history of venous thromboembolism. However, we included all prospective NRS that met our inclusion criteria, irrespective of the 'statistical adjustment for baseline factors' and assessed the confounders at the 'bias due to confounding' domain of the ROBINS‐I tool in this review (Sterne 2016).
Summary of findings and assessment of the certainty of the evidence
Although we had already included major bleeding as a particular outcome, we amended the summary of findings tables to include adverse events (minor bleeding) in this review version.
Authors' contributions
Some review authors (Marcelly S Cossi, Maria ICD Fernandes, Isabelle KF Costa, and Larissa Souza) were not available to contribute to this review version; therefore, we moved them to the 'Acknowledgment' section. In addition, we incorporated Vinicius T Civile into the review authors' team due to his amount of contribution.
Contributions of authors
RLGF: clinical and methodological expertise, development of the search strategy and conception and writing of the review VTC: methodological expertise and advice JDST: clinical and methodological expertise and advice PIFP: clinical expertise and advice LLA: development of the search strategy CM: clinical expertise and advice BT: methodological expertise and advice VFMT: methodological expertise and advice ANA: clinical and methodological expertise and advice LCUN: clinical and methodological expertise and writing of the review
Sources of support
Internal sources
-
Division of Vascular and Endovascular Surgery, Universidade Federal de São Paulo, Brazil
Non‐financial internal sources.
-
Cochrane Brazil, Brazil
Non‐financial internal sources.
External sources
No sources of support provided
Declarations of interest
RLGF: none known VTC: none known JDST: none known PIFP: none known LLA: none known CM: none known BT: none known VFMT: none known ANA: none known LCUN: none known
New search for studies and content updated (conclusions changed)
References
References to studies included in this review
Albani 2020 {published data only}
- Albani F, Sepe L, Fusina F, Prezioso C, Baronio M, Caminiti F, et al.Thromboprophylaxis with enoxaparin is associated with a lower death rate in patients hospitalized with SARS-CoV-2 infection. A cohort study. EClinicalMedicine 2020;27:100562. [DOI: 10.1016/j.eclinm.2020.100562] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lemos 2020 {published data only (unpublished sought but not used)}
- Bikdeli B.Anticoagulation in COVID-19: randomized trials should set the balance between excitement and evidence. Thrombosis research 2020;196:638-40. [DOI: 10.1016/j.thromres.2020.09.033] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos AC, do Espírito Santo DA, Salvetti MC, Gilio RN, Agra LB, Pazin-Filho A, et al.Therapeutic versus prophylactic anticoagulation for severe COVID-19: a randomized phase II clinical trial (HESACOVID). Thrombosis Research 2020;196:359‐66. [DOI: 10.1016/j.thromres.2020.09.026] [DOI] [PMC free article] [PubMed] [Google Scholar]
- RBR-949z6v.Full versus prophylactic heparinization for the treatment of severe forms of SARS-COVID-19: clinical, randomized, open and controlled study - HeSAcovid trial. ictrptest.azurewebsites.net/Trial2.aspx?TrialID=RBR-949z6v (first received 23 February 2021).
- RBR-949z6v.RBR-949z6v Full versus prophylactic anticoagulation for the treatment of severe forms of COVID-19. ensaiosclinicos.gov.br/rg/RBR-949z6v/ (first received 6 April 2020).
Lopes 2021 {published data only}
- Bavry AA, Bhatt DL.AnticoagulatIon coronavirus - ACTION. acc.org/Latest-in-Cardiology/Clinical-Trials/2021/05/14/03/11/ACTION#.YKGzh1LrUW8.linkedin (assessed 16 May 2021).
- Lopes RD, Silva PG, Furtado RH, Macedo AVS, Ramacciotti E, Damini LP, et al.Randomized clinical trial to evaluate a routine full anticoagulation strategy in patients with coronavirus Infection (SARS-CoV2) admitted to hospital: rationale and design of the ACTION (AntiCoagulaTlon cOroNavirus)-Coalition IV trial. American Heart Journal 2021;238:1-11. [DOI: 10.1016/j.ahj.2021.04.005] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes RD, Silva PG, Furtado RH, Macedo AV, Bronhara B, Damiani LP, et al.Therapeutic versus prophylactic anticoagulation for patients admitted to hospital with COVID-19 and elevated D-dimer concentration (ACTION): an open-label, multicentre, randomised, controlled trial. Lancet 2021;397(10291):2253-63. [DOI: 10.1016/S0140-6736(21)01203-4] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes RD.ACC.21 Presentation Slides | ACTION. acc.org/education-and-meetings/image-and-slide-gallery/media-detail?id=DC46FDC6BBA84D9B85FC064F0D73DAF2 (assessed 16 May 2021).
- Lopes RD.Details about your trial data [ACTION trial] [personal communication]. Email to: RL Flumignan 22 May 2021.
- NCT04394377.Randomized clinical trial to evaluate a routine full anticoagulation strategy in patients with coronavirus (COVID-19) - COALIZAO ACTION Trial. clinicaltrials.gov/ct2/show/NCT04394377 (first received 8 May 2020).
Rentsch 2020 {published data only}
- Rentsch CT, Beckman JA, Tomlinson L, Gellad WF, Alcorn C, Kidwai-Khan F, et al.Early initiation of prophylactic anticoagulation for prevention of COVID-19 mortality: a nationwide cohort study of hospitalized patients in the United States. medRxiv [Preprint}. [DOI: 10.1101/2020.12.09.20246579] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sadeghipour 2021 {published data only (unpublished sought but not used)}
- Al-Samkari H.Finding the optimal thromboprophylaxis dose in patients with COVID-19. JAMA 2021;325(16):1613-15. [DOI: 10.1001/jama.2021.4295] [DOI] [PubMed] [Google Scholar]
- Bikdeli B, Talasaz AH, Rashidi F, Bakhshandeh H, Rafiee F, Matin S, et al.Intermediate vs standard-dose prophylactic anticoagulation in patients with COVID-19 admitted to ICU: ninety-day results from the INSPIRATION Trial. Thrombosis and Haemostasis 2022;122(1):131-41. [DOI: 10.1055/a-1485-2372] [DOI] [PubMed] [Google Scholar]
- Bikdeli B, Talasaz AH, Rashidi F, Sharif-Kashani B, Farrokhpour M, Bakhshandeh H, et al.Intermediate versus standard-dose prophylactic anticoagulation and statin therapy versus placebo in critically-ill patients with COVID-19: rationale and design of the INSPIRATION/INSPIRATION-S studies. Thrombosis Research 2020;196:382‐94. [DOI: 10.1016/j.thromres.2020.09.027] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bikdeli B.Details about your trial data [INSPIRATION trial] [personal communication]. Email to: RLG Flumignan 4 June 2021.
- NCT04486508.Intermediate-dose vs standard prophylactic anticoagulation and statin vs placebo in ICU patients with COVID-19. clinicaltrials.gov/ct2/show/NCT04486508 (first received 22 July 2020).
- NCT04486508.Intermediate-dose vs standard prophylactic anticoagulation and statin vs placebo in ICU patients with COVID-19 INSPIRATION. ictrptest.azurewebsites.net/Trial2.aspx?TrialID=NCT04486508 (first received 22 July 2020).
- Sadeghipour P, Talasaz AH, Rashidi F, Sharif-Kashani B, Beigmohammadi MT, Farrokhpour M, et al.Effect of intermediate-dose vs standard-dose prophylactic anticoagulation on thrombotic events, extracorporeal membrane oxygenation treatment, or mortality among patients with COVID-19 admitted to the intensive care unit: the INSPIRATION randomized clinical trial. JAMA 2021;325(16):1620-30. [DOI: 10.1001/jama.2021.4152] [DOI] [PMC free article] [PubMed] [Google Scholar]
Santoro 2020 {published data only (unpublished sought but not used)}
- Núñez-Gil IJ, Estrada V, Fernández-Pérez C, Feltes G, Vedia O, Vergara-Uzcategui CE.Health outcome predictive evaluation for COVID 19 international registry (HOPE COVID-19), rationale and design. Contemporary Clinical Trials Communication 2020;20:100654. [DOI: 10.1016/j.conctc.2020.100654] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT04334291.International COVID19 clinical evaluation registry, (HOPE COVID 19). clinicaltrials.gov/ct2/show/NCT04334291 (first received 1 April 2020).
- NCT04334291.International COVID-19 clinical evaluation registry 2: HOPE 2. hopeprojectmd.com/en/ (first received 1 April 2020).
- Rivera-Caravaca JM, Núñez-Gil IJ, Vivas D, Viana-Llamas MC, Uribarri A, Becerra-Muñoz VM, et al.Clinical profile and prognosis in patients on oral anticoagulation before admission for COVID-19. European Journal of Clinical Investigation 2021;51(1):e13436. [DOI: 10.1111/eci.13436] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro F, Nunez I, Pepe M, Estrada V, Brunetti ND.A289: Anticoagulation therapy in patients with COVID-19. Results from a multi-center international prospective registry (HOPE-COVID19). Giornale Italiano di Cardiologia 2020;21(12 Suppl 2):e90. [Google Scholar]
- Santoro F, Nunez I, Pepe M, Estrada V, Brunetti ND.Anticoagulation therapy in patients with COVID-19. Results from a multi-center international prospective registry (HOPE-COVID19). Critical Care Medicine 2021;49(6):e624-e633. [DOI: 10.1097/CCM.0000000000005010] [DOI] [PubMed] [Google Scholar]
Zarychanski 2021 {published data only (unpublished sought but not used)}
- EUCTR2015-002340-14-NL.Randomized, embedded, multifactorial, adaptive platform trial for community-acquired pneumonia (REMAP-CAP). (COVID-19) - REMAP-CAP. ictrptest.azurewebsites.net/Trial2.aspx?TrialID=EUCTR2015-002340-14-NL (first received 16 September 2015).
- EUCTR2020-004285-19.A multicenter, adaptive, randomized controlled platform trial of the safety and efficacy of antithrombotic strategies in hospitalized adults with COVID-19. clinicaltrialsregister.eu/ctr-search/trial/2020-004285-19/IT (first received 23 November 2020).
- Houston BL, Lawler PR, Goligher EC, Farkouh ME, Bradbury C, Carrier M, et al.Anti-thrombotic therapy to ameliorate complications of COVID-19 (ATTACC): study design and methodology for an international, adaptive Bayesian randomized controlled trial. Clinical Trials 2020;17(5):491‐500. [DOI: 10.1177/1740774520943846] [DOI] [PubMed] [Google Scholar]
- Hunt BJ, De Paula EV, McLintock C, Dumantepe M.Prophylactic anticoagulation for patients in hospital with COVID-19. BMJ 2021;372:n487. [DOI: 10.1136/bmj.n487] [DOI] [PubMed] [Google Scholar]
- Lawler P, Goligher E, Berger J, Neal M, McVerry B, Nicolau J, et al.Therapeutic anticoagulation in non-critically Ill patients with COVID-19. medRxiv [Preprint]. [DOI: 10.1101/2021.05.13.21256846] [DOI] [Google Scholar]
- NCT02735707.Randomized, embedded, multifactorial adaptive platform trial for community-acquired pneumonia (REMAP-CAP). clinicaltrials.gov/ct2/show/NCT02735707 (first received 13 April 2016).
- NCT04359277.A randomized trial of anticoagulation strategies in COVID-19. clinicaltrials.gov/ct2/show/NCT04359277 (first received 20 April 2020).
- NCT04372589.Antithrombotic therapy to ameliorate complications of COVID-19 (ATTACC), in collaboration with accelerating COVID-19 therapeutic interventions and vaccines (ACTIV-4). clinicaltrials.gov/ct2/show/NCT04372589 (first received 24 April 2020).
- NCT04505774.Anti-thrombotics for adults hospitalized with COVID-19 (ACTIV-4). clinicaltrials.gov/ct2/show/NCT04505774 (first received 6 August 2020).
- No authors listed.Erratum for [Prophylactic anticoagulation for patients in hospital with COVID-19]. BMJ 2021;372:n538. [PMID: 10.1136/bmj.n538] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker PM, Ortel TL, Hochman JS.Equipoise, trust, and the need for cardiologists to randomly assign patients Into anticoagulation trials in the time of COVID. Circulation 2020;142(24):2296-8. [DOI: 10.1161/CIRCULATIONAHA.120.052069] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarychanski R, Goligher EC, Bradbury CA, McVerry BJ, Lawler PR, Berger JS, et al.Therapeutic anticoagulation in critically ill patients with COVID-19 – preliminary report. medRxiv [Preprint]. [DOI: 10.1101/2021.03.10.21252749] [DOI] [Google Scholar]
- Zarychanski R.Details about your trial data [Zarychanski R, Goligher EC, Bradbury CA, McVerry BJ, Lawler PR, Berger JS, et al. Therapeutic anticoagulation in critically ill patients with COVID-19 – Preliminary Report. medRxiv [Preprint]. DOI: 10.1101/2021.03.10.21252749]. Email to: RLG Flumignan 23 April 2021.
References to studies excluded from this review
Al‐Samkari 2020 {published data only}
- Al-Samkari H, Karp Leaf RS, Dzik WH, Carlson JC, Fogerty AE, Waheed A, et al.COVID-19 and coagulation: bleeding and thrombotic manifestations of SARS-CoV-2 infection. Blood 2020;136(4):489-500. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Artifoni 2020 {published data only}
- Artifoni M, Danic G, Gautier G, Gicquel P, Boutoille D, Raffi F, et al.Systematic assessment of venous thromboembolism in COVID-19 patients receiving thromboprophylaxis: incidence and role of D-dimer as predictive factors. Journal of Thrombosis and Thrombolysis 2020;50(1):211-6. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ayerbe 2020 {published data only}
- Ayerbe L, Risco C, Ayis S.The association between treatment with heparin and survival in patients with COVID-19. Journal of Thrombosis and Thrombolysis 2020;50(2):298-301. [DOI: 10.1007/s11239-020-02162-z] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayerbe L, Risco C, Ayis S.The association between treatment with heparin and survival in patients with COVID-19. medRxiv [Preprint]. [DOI: 10.1101/2020.05.27.20114694] [DOI] [PMC free article] [PubMed]
ChiCTR2000034796 {published data only}
- ChiCTR2000034796.The efficacy and safety of heparin in the treatment of novel coronavirus pneumonia (COVID-19): a retrospective single center clinical trial. ictrptest.azurewebsites.net/Trial2.aspx?TrialID=ChiCTR2000034796 (first received 18 January 2020).
CTRI/2021/01/030373 {published data only}
- CTRI/2021/01/030373.ETHIC trial: early LMWH in symptomatic COVID-19 positive patients. ictrptest.azurewebsites.net/Trial2.aspx?TrialID=CTRI/2021/01/030373 (first received 11 January 2021).
D'Ardes 2021 {published data only}
- D'Ardes D, Carrarini C, Russo M, Dono F, Speranza R, Digiovanni A, et al.Low molecular weight heparin in COVID-19 patients prevents delirium and shortens hospitalization. Neurological Sciences 2021;42(4):1527-30. [DOI: 10.1007/s10072-020-04887-4] [DOI] [PMC free article] [PubMed] [Google Scholar]
DCTC 2021 {published data only}
- Dutch COVID & Thrombosis Coalition.Early effects of unfractionated heparin on clinical and radiological signs and D-dimer levels in patients with COVID-19 associated pulmonary embolism: an observational cohort study. Thrombosis Research 2021;200:130-2. [DOI: 10.1016/j.thromres.2021.01.023] [DOI] [PMC free article] [PubMed] [Google Scholar]
Di Castelnuovo 2021 {published data only}
- Di Castelnuovo A, Costanzo S, Antinori A, Berselli N, Blandi L, Bonaccio M, et al.Heparin in COVID-19 patients is associated with reduced in-hospital mortality: the multicenter Italian CORIST study. Thrombosis and Haemostasis 2021;121(8):1054-65. [DOI: 10.1055/a-1347-6070] [DOI] [PubMed] [Google Scholar]
EUCTR2020‐001748‐24‐SE {published data only}
- EUCTR2020-001748-24-SE.A multi-center, randomized, open-label study in patients with COVID-19 and respiratory distress not requiring mechanical ventilation, to compare standard-of-care with anakinra and tocilizumab treatment. The immunomodulation-CoV assessment (ImmCoVA) study. ictrptest.azurewebsites.net/Trial2.aspx?TrialID=EUCTR2020-001748-24-SE (first received 20 April 2020).
EudraCT2020‐001823‐15 {published data only}
- EudraCT2020-001823-15.Evaluation of the concentration-effect relationship of enoxaparin for thromboembolic prevention in COVID-19 resuscitation patients. COV-ENOX study. clinicaltrialsregister.eu/ctr-search/trial/2020-001823-15/FR (first received 15 April 2020).
Falcone 2020 {published data only}
- Falcone M, Tiseo G, Barbieri G, Galfo V, Russo A, Virdis A, et al.Role of low-molecular-weight heparin in hospitalized patients with severe acute respiratory syndrome coronavirus 2 pneumonia: a prospective observational study. Open Forum Infectious Diseases 2020;7(12):ofaa563. [DOI: 10.1093/ofid/ofaa563] [DOI] [PMC free article] [PubMed] [Google Scholar]
Frohlich 2021 {published data only}
- Frohlich GM, Jeschke E, Eichler U, Thiele H, Alhariri L, Reinthaler M, et al.Impact of oral anticoagulation on clinical outcomes of COVID-19: a nationwide cohort study of hospitalized patients in Germany. Clinical Research in Cardiology 2021;110(7):1041-50. [PMID: 10.1007/s00392-020-01783-x] [DOI] [PMC free article] [PubMed] [Google Scholar]
Helms 2020 {published data only}
- Helms J, Tacquard C, Severac F, Leonard-Lorant I, Ohana M, Delabranche X, et al.High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Medicine 2020;46(6):1089-98. [DOI: 10.1007/s00134-020-06062-x] [DOI] [PMC free article] [PubMed] [Google Scholar]
Helms 2021 {published data only}
- Helms J, Severac F, Merdji H, Schenck M, Clere-Jehl R, Baldacini M, et al.Higher anticoagulation targets and risk of thrombotic events in severe COVID-19 patients: bi-center cohort study. Annals of Intensive Care 2021;11(1):14. [DOI: 10.1186/s13613-021-00809-5] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ho 2021 {published data only}
- Ho G, Dusendang JR, Schmittdiel J, Kavecansky J, Tavakoli J, Pai A.Association of chronic anticoagulant and antiplatelet use on disease severity in SARS-CoV-2 infected patients. Journal of Thrombosis and Thrombolysis 2021;52(2):476-81. [DOI: 10.1007/s11239-021-02383-w] [DOI] [PMC free article] [PubMed] [Google Scholar]
Huang 2020 {published data only}
- Huang CT, Hsu SY, Chang KW, Huang CG, Yang CT, Cheng MH.Heparin-induced thrombocytopenia and thrombosis in a patient with COVID-19. Thrombosis Research 2020;196:11-4. [DOI: 10.1016/j.thromres.2020.07.056] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ionescu 2020 {published data only}
- Ionescu F, Grasso-Knight G, Castillo E, Naeem E, Petrescu I, Imam Z, et al.Therapeutic anticoagulation delays death in COVID-19 patients: cross-sectional analysis of a prospective cohort. TH open 2020;4(3):e263-70. [PMID: 10.1055/s-0040-1716721] [DOI] [PMC free article] [PubMed] [Google Scholar]
Jiménez‐Soto 2021 {published data only}
- Jiménez-Soto R, Aguilar-Soto M, Rodríguez-Toledo CA, Camiro-Zúñiga A, Demichelis R.The impact of different prophylactic anticoagulation doses on the outcomes of patients with COVID-19. Thrombosis Research 2021;202:14-6. [DOI: 10.1016/j.thromres.2021.02.031] [DOI] [PMC free article] [PubMed] [Google Scholar]
Jonmarker 2020 {published data only}
- Jonmarker S, Hollenberg J, Dahlberg M, Stackelberg O, Litorell J, Everhov ÅH, et al.Dosing of thromboprophylaxis and mortality in critically ill COVID-19 patients. Intensive Care Medicine Experimental 2020;8 Suppl 2:73. [DOI: 10.1186/s40635-020-00354-8] [DOI] [PMC free article] [PubMed] [Google Scholar]
Khider 2020 {published data only}
- Khider L, Gendron N, Goudot G, Chocron R, Hauw-Berlemont C, Cheng C, et al.Curative anticoagulation prevents endothelial lesion in COVID-19 patients. Journal of Thrombosis and Haemostasis 2020;18(9):2391-9. [DOI: 10.1111/jth.14968] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kodama 2020 {published data only}
- Kodama R, Kalsi A, Singh A, Vishnuvardhan N, Nimkar N, Paul S, et al.Outcomes in COVID-19 patients on treatment dose anti-coagulation compared to prophylactic dose anti-coagulation. Blood 2020;136 Suppl 1:40-1. [DOI: 10.1182/blood-2020-142552] [DOI] [Google Scholar]
Kow 2020 {published data only}
- Kow CS, Hasan SS.Use of low-molecular-weight heparin in COVID-19 patients. Journal of Vascular Surgery 2020;8(5):900-1. [DOI: 10.1016/j.jvsv.2020.06.006] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kukin 2020 {published data only}
- Kukin A, Gale SE, Reed BN, Devabhakthuni S, Watson K, Noel Z.Effect of video-assisted counseling versus traditional counseling on patient comprehension of prescribed direct oral anticoagulants. Journal of the American College of Clinical Pharmacy 2020;3(8):1538‐9. [DOI: 10.1002/jac5.1351] [DOI] [Google Scholar]
Liu 2020 {published data only}
- Liu X, Zhang X, Xiao Y, Gao T, Wang G, Wang Z, et al.Heparin-induced thrombocytopenia is associated with a high risk of mortality in critical COVID-19 patients receiving heparin-involved treatment. medRxiv [Preprint]. [DOI: 10.1101/2020.04.23.20076851] [DOI] [Google Scholar]
Mareev 2020 {published data only}
- Mareev VY, Orlova YA, Pavlikova EP, Akopyan ZA, Matskeplishvili ST, Plisyk AG, et al.Proactive anti-inflammatory and anticoagulant therapy in the treatment of advanced stages of novel coronavirus infection (COVID-19). Case series and study design: coLchicine versus ruxolitinib and secukinumab in open prospective randomIzed trial (COLORIT). Kardiologiia 2020;60(9):4‐21. [DOI: 10.18087/cardio.2020.9.n1338] [DOI] [PubMed] [Google Scholar]
- NCT04403243.Colchicine versus ruxolitinib and secukinumab in open prospective randomized trial (COLORIT). clinicaltrials.gov/ct2/show/NCT04403243 (first received 20 May 2020).
Martinelli 2020 {published data only}
- Martinelli I, Ciavarella A, Abbattista M, Aliberti S, De Zan V, Folli C, et al.Increased doses of low-molecular-weight heparin in hospitalized patients with COVID-19. Research and Practice in Thrombosis and Haemostasis 2020;4 Suppl 2:15-6. [DOI: 10.1002/rth2.12413] [DOI] [Google Scholar]
Maurer 2020 {published data only}
- Maurer L, Luckhurst C, Hamidi A, Newman K, Barra M, El Hechi M, et al.A low dose heparin protocol is associated with improved duration of arterial line patency in critically ill COVID-19 patients. Research and Practice in Thrombosis and Haemostasis 2020;4 Suppl 2:8-9. [DOI: 10.1002/rth2.12413] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer LR, Luckhurst CM, Hamidi A, Newman KA, Barra ME, El Hechi M, et al.A low dose heparinized saline protocol is associated with improved duration of arterial line patency in critically ill COVID-19 patients. Journal of Critical Care 2020;60:253-9. [DOI: 10.1016/j.jcrc.2020.08.025] [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT04354155 {published data only}
- NCT04354155.COVID-19 anticoagulation in children - thromboprophlaxis (COVAC-TP) trial. clinicaltrials.gov/ct2/show/NCT04354155 (first received 21 April 2020).
NCT04359212 {published data only}
- NCT04359212.Increased risk of venous thromboembolism and higher hypercoagulable state in patients recovered in intensive care unit and in medical ward for coronavirus disease 2019 (COVID-19). clinicaltrials.gov/ct2/show/NCT04359212 (first received 21 April 2020).
NCT04365309 {published data only}
- NCT04365309.Protective effect of aspirin on COVID-19 patients (PEAC). clinicaltrials.gov/ct2/show/NCT04365309 (first received 28 April 2020).
NCT04368377 {published data only}
- NCT04368377.Platelet inhibition with GP IIb/IIIa inhibitor in critically ill patients with coronavirus disease 2019 (COVID-19). A compassionate use protocol. clinicaltrials.gov/ct2/show/NCT04368377 (first received 29 April 2020).
NCT04393805 {published data only}
- NCT04393805.Heparins for thromboprophylaxis in COVID-19 patients: HETHICO study in Veneto. clinicaltrials.gov/ct2/show/NCT04393805 (first received 16 May 2020).
NCT04427098 {published data only}
- 2020-001308-40.Intermediate dose enoxaparin in hospitalized patients with moderate-severe COVID-19: a pilot phase II single-arm study, INHIXACOVID19. clinicaltrialsregister.eu/ctr-search/trial/2020-001308-40/IT (first received 24 June 2020). [DOI] [PMC free article] [PubMed]
- NCT04427098.Intermediate dose enoxaparin in hospitalized patients with moderate-severe COVID19: a pilot phase II single-arm study, INHIXACOVID19. clinicaltrials.gov/ct2/show/study/NCT04427098 (first received 11 June 2020).
NCT04483830 {published data only}
- NCT04483830.Suloexide in the treatment of early stages of COVID-19 (SulES-COVID). clinicaltrials.gov/ct2/show/NCT04483830 (first received 22 July 2020) (first received 22 July 2020).
NCT04492254 {published data only}
- NCT04492254.Early prophylactic low-molecular-weight heparin (LMWH) in symptomatic COVID-19 positive patients (ETHIC). clinicaltrials.gov/ct2/show/NCT04492254 (first received 22 July 2020).
NCT04504032 {published data only}
- EUCTR2020-005395-35.A randomized, controlled, Phase 2b study to evaluate safety and efficacy of rivaroxaban (Xarelto®) for high risk people with mild COVID-19. clinicaltrialsregister.eu/ctr-search/trial/2020-005395-35/GB (first received 26 November 2020).
- NCT04504032.A trial to evaluate safety and efficacy of rivaroxaban (COVID-19). clinicaltrials.gov/ct2/show/NCT04504032 (first received 5 August 2020).
NCT04516941 {published data only}
- NCT04516941.Corona virus edoxaban colchicine (CONVINCE) COVID-19 (CONVINCE). clinicaltrials.gov/ct2/show/NCT04516941 (first received 5 August 2020).
NCT04662684 {published data only}
- NCT04662684.Medically Ill hospitalized patients for COVID-19 thrombosis extended prophylaxis with rivaroxaban therapy: the MICHELLE Trial. clinicaltrials.gov/ct2/show/NCT04662684 (first received 8 December 2020).
NCT04673214 {published data only}
- NCT04673214.Evaluation of prognostic modification in COVID-19 patients in early intervention treatment. clinicaltrials.gov/ct2/show/NCT04673214 (first received 16 December 2020).
NCT04715295 {published data only}
- NCT04715295.Safety and efficacy of doxycycline and rivaroxaban in COVID-19 (DOXYCOV). clinicaltrials.gov/ct2/show/NCT04715295 (first received 2 October 2020).
NCT04736901 {published data only}
- NCT04736901.Effect of prophylactic and therapeutic anticoagulants in Egyptian patients with COVID-19. clinicaltrials.gov/ct2/show/results/NCT04736901?view=results (first received 3 February 2021).
NCT04757857 {published data only}
- NCT04757857.COVID-19 antithrombotic rivaroxaban evaluation (CARE). clinicaltrials.gov/ct2/show/NCT04757857 (first received 8 February 2021).
NCT04828772 {published data only}
- NCT04828772.A study of anticoagulation treatment patterns and outcomes of participants hospitalized with coronavirus disease 2019 (COVID-19) in Japan. clinicaltrials.gov/ct2/show/NCT04828772 (first received 25 March 2021).
Paranjpe 2020 {published data only}
- Paranjpe I, Fuster V, Lala A, Russak AJ, Glicksberg BS, Levin MA, et al.Association of treatment dose anticoagulation with in-hospital survival among hospitalized patients with COVID-19. Journal of the American College of Cardiology 2020;76(1):122-4. [PMID: ] [DOI] [PMC free article] [PubMed]
Piagnerelli 2020 {published data only}
- Piagnerelli M, Cauchie P, Wautrecht JC.Optimizing the risk-benefit balance of thromboprophylaxis in critically ill patients with coronavirus disease 2019. Critical Care Medicine 2020;48(10):e988-9. [DOI: 10.1097/CCM.0000000000004509] [DOI] [PMC free article] [PubMed] [Google Scholar]
Poulakou 2021 {published data only}
- Poulakou G, Dimakakos E, Kollias A, Kyriakoulis KG, Rapti V, Trontzas I, et al.Beneficial effects of intermediate dosage of anticoagulation treatment on the prognosis of hospitalized COVID-19 patients: the ETHRA Study. In Vivo (Athens, Greece) 2021;35(1):653-61. [DOI: 10.21873/invivo.12305] [DOI] [PMC free article] [PubMed] [Google Scholar]
Qin 2021 {published data only}
- Qin W, Dong F, Zhang Z, Hu B, Chen S, Zhu Z, et al.Low molecular weight heparin and 28-day mortality among patients with coronavirus disease 2019: a cohort study in the early epidemic era. Thrombosis Research 2021;198:19-22. [DOI: 10.1016/j.thromres.2020.11.020] [DOI] [PMC free article] [PubMed] [Google Scholar]
Rosovsky 2020 {published data only}
- Rosovsky RP, Sanfilippo KM, Wang TF, Rajan SK, Shah S, Martin KA, et al.Anticoagulation practice patterns in COVID-19: a global survey. Research and Practice in Thrombosis and Haemostasis 2020;4(6):969‐83. [DOI: 10.1002/rth2.12414] [DOI] [PMC free article] [PubMed] [Google Scholar]
Russo 2020 {published data only}
- Russo V, Di Maio M, Attena E, Silverio A, Scudiero F, Celentani D, et al.Clinical impact of pre-admission antithrombotic therapy in hospitalized patients with COVID-19: a multicenter observational study. Pharmacological Research 2020;159:104965. [DOI: 10.1016/j.phrs.2020.104965] [DOI] [PMC free article] [PubMed] [Google Scholar]
Secco 2020 {published data only}
- Secco E, Pasqualetto MC, Bombardini T, Picano E, Rigo F.A possible benefit from therapeutic anticoagulation in patients with coronavirus disease 2019: the Dolo hospital experience in Veneto, Italy. Kardiologia Polka 2020;78(9):919-21. [DOI: 10.33963/KP.15489] [DOI] [PubMed] [Google Scholar]
Shi 2020 {published data only}
- Shi C, Wang C, Wang H, Yang C, Cai F, Zeng F, et al.The potential of low molecular weight heparin to mitigate cytokine storm in severe COVID-19 patients: a retrospective clinical study. medRxiv [Preprint]. [DOI: 10.1101/2020.03.28.20046144] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sivaloganathan 2020 {published data only}
- Sivaloganathan H, Ladikou EE, Chevassut T.COVID-19 mortality in patients on anticoagulants and antiplatelet agents. British Journal of Haematology 2020;190(4):e192‐e195. [DOI: 10.1111/bjh.16968] [DOI] [PMC free article] [PubMed] [Google Scholar]
Smith 2020 {published data only}
- Smith SR, Waite A, Johnston BW, Welters ID.P520 Survey of current practice in management of anticoagulation in adult critically ill patients with COVID-19 in the northwest of England. Critical Care 2020;24 Suppl 2:P520. [PMID: 10.1186/s13054-020-03187-9] [DOI] [Google Scholar]
Stessel 2020 {published data only}
- NCT04394000.Impact of implementation of an intensified thromboprofylaxis protocol in critically ill ICU patients with COVID-19: a longitudinal controlled before-after study. clinicaltrials.gov/ct2/show/record/NCT04394000 (first received 19 May 2020).
- Stessel B, Vanvuchelen C, Bruckers L, Geebelen L, Callebaut I, Vandenbrande J, et al.Impact of implementation of an individualised thromboprophylaxis protocol in critically ill ICU patients with COVID-19: a longitudinal controlled before-after study. Thrombosis Research 2020;194:209-15. [DOI: 10.1016/j.thromres.2020.07.038] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tacquard 2021 {published data only}
- NCT04405869.Thromboembolic events in severe COVID-19 patients (COVICLOT). clinicaltrials.gov/ct2/show/NCT04405869 (first received 25 May 2020).
- Tacquard C, Mansour A, Godon A, Godet J, Poissy J, Garrigue D, et al.Impact of high-dose prophylactic anticoagulation in critically ill patients with coronavirus disease 2019 pneumonia. Chest 2021;S0012-3692(21):00047-7. [DOI: 10.1016/j.chest.2021.01.017] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tamargo 2021 {published data only}
- Tamargo J.Does anticoagulation reduce mortality in patients with atrial fibrillation who later developed a COVID-19 infection? International Journal of Cardiology 2021;331:340-1. [DOI: 10.1016/j.ijcard.2021.01.019] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tang 2020 {published data only}
- Tang N, Bai H, Chen X, Gong J, Li D, Sun Z.Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. Journal of Thrombosis and Haemostasis : JTH 2020;18(5):1094-9. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Trinh 2020 {published data only}
- Trinh M, Chang DR, Govindarajulu US, Kane E, Fuster V, Kohli-Seth R, et al.Therapeutic anticoagulation is associated with decreased mortality in mechanically ventilated COVID-19 patients. medRxiv [Preprint]. [DOI: 10.1101/2020.05.30.20117929] [DOI] [Google Scholar]
Zhang 2020 {published data only}
- Zhang Y, Cao W, Xiao M, Li YJ, Yang Y, Zhao J, et al.Clinical and coagulation characteristics of 7 patients with critical COVID-2019 pneumonia and acro-ischemia. Zhonghua Xue Ye Xue za Zhi 2020;41(0):E006. [PMID: ] [DOI] [PubMed] [Google Scholar]
References to ongoing studies
ACTRN12620000517976 {published data only}
- ACTRN12620000517976.A randomised controlled trial of nebulised heparin in critically ill mechanically ventilated patients with COVID-19 to assess the effect on the duration of mechanical ventilation. anzctr.org.au/Trial/Registration/TrialReview.aspx?ACTRN=12620000517976 (first received 27 April 2020).
- ACTRN12620000517976.Can nebulised heparin reduce time to extubation in SARS CoV 2. The CHARTER study protocol. apps.who.int/trialsearch/Trial2.aspx?TrialID=ACTRN12620000517976 (first received 27 April 2020).
- Dixon B, Smith RJ, Artigas A, Laffey J, McNicholas B, Schmidt E, et al.Can nebulised heparin reduce time to extubation in SARS CoV 2. The CHARTER study protocol. medRxiv [Preprint]. [DOI: 10.1101/2020.04.28.20082552] [DOI] [Google Scholar]
Busani 2020 {published data only}
- Busani S, Tosi M, Mighali P, Vandelli P, D'Amico R, Marietta M, et al.Multi-centre, three arm, randomized controlled trial on the use of methylprednisolone and unfractionated heparin in critically ill ventilated patients with pneumonia from SARS-CoV-2 infection: a structured summary of a study protocol for a randomised controlled trial. Trials 2020;21:724. [DOI: 10.1186/s13063-020-04645-z] [DOI] [PMC free article] [PubMed] [Google Scholar]
- EUCTR2020-001921-30.Steroids and unfractionated heparin in critically-ill patients with pneumonia from COVID-19 infection. A multicenter, interventional, randomized, three arms study design. clinicaltrialsregister.eu/ctr-search/trial/2020-001921-30/IT (first received 26 June 2020).
- NCT04528888.Steroids and unfractionated heparin in critically-ill patients with pneumonia from COVID-19 infection. A multicenter, interventional, randomized, three arms study design. clinicaltrials.gov/ct2/show/NCT04528888 (first received 21 August 2020).
Chambers 2020 {published data only}
- Chambers I, Dayal S, Eyck PT, Eustes A, Sutamtewagul G, Rosenstein L, et al.COVID-19-associated coagulopathy: safety and efficacy of prophylactic anticoagulation therapy in hospitalized adults with COVID-19. Blood 2020;136 Suppl 1:11. [10.1182/blood-2020-141951] [Google Scholar]
- NCT04360824.COVID-19-associated coagulopathy: safety and efficacy of prophylactic anticoagulation therapy in hospitalized adults with COVID-19. clinicaltrials.gov/ct2/show/NCT04360824 (first received 24 April 2020).
ChiCTR2000030700 {published data only}
- ChiCTR2000030700.Study for the efficacy and safety of prolongin (enoxaparin sodium injection) in treatment of novel coronavirus pneumonia (COVID-19) adult common patients. apps.who.int/trialsearch/Trial2.aspx?TrialID=ChiCTR2000030700 (first received 10 March 2020).
- ChiCTR2000030700.Study for the efficacy and safety of prolongin (enoxaparin sodium injection) in treatment of novel coronavirus pneumonia (COVID-19) adult common patients. www.chictr.org.cn/showprojen.aspx?proj=50786 (first received 10 March 2020).
ChiCTR2000030701 {published data only}
- ChiCTR2000030701.A randomized, parallel controlled open-label trial for the efficacy and safety of prolongin (enoxaparin sodium injection) in the treatment of adult patients with novel coronavirus pneumonia (COVID-19). chictr.org.cn/showprojen.aspx?proj=50795 (first received 10 March 2020).
- ChiCTR2000030701.A randomized, parallel controlled open-label trial to evaluate the efficacy and safety of Prolongin (enoxaparin sodium injection) in adult hospitalized patients with novel coronavirus pneumonia (COVID-19). ictrptest.azurewebsites.net/Trial2.aspx?TrialID=ChiCTR2000030701 (first received 10 March 2020).
ChiCTR2000030946 {published data only}
- ChiCTR2000030946.Effects of different VTE prevention methods on the prognosis of hospitalized patients with novel coronavirus pneumonia (COVID-19). chictr.org.cn/showprojen.aspx?proj=51265 (first received 19 March 2020).
CTRI/2020/06/026220 {published data only}
- CTRI/2020/06/026220.An open label, randomized, multicenter, controlled clinical study to evaluate the efficacy and safety of nafamostat mesilate in the treatment of moderate COVID-19 disease. ictrptest.azurewebsites.net/Trial2.aspx?TrialID=CTRI/2020/06/026220 (first received 17 July 2020).
CTRI/2020/08/027033 {published data only}
- CTRI/2020/08/027033.SARS-COV-2 and COVID-19 - a randomized controlled trail (unblinded). ictrptest.azurewebsites.net/Trial2.aspx?TrialID=CTRI/2020/08/027033 (first received 07 August 2020).
CTRI/2020/11/029175 {published data only}
- CTRI/2020/11/029175.Role of heparin inhalation in reducing the duration the patient is breathing with the help of a ventilator. ictrptest.azurewebsites.net/Trial2.aspx?TrialID=CTRI/2020/11/029175 (first received 17 November 2020).
CTRI/2020/11/029345 {published data only}
- CTRI/2020/11/029345.To determine efficacy, safety and optimal dosing of anticoagulant strategies to prevent adverse outcomes in hospitalized COVID-19 patients. ictrptest.azurewebsites.net/Trial2.aspx?TrialID=CTRI/2020/11/029345 (first received 25 November 2020).
EUCTR2020‐001302‐30‐AT {published data only}
- EUCTR2020-001302-30-AT.A multicenter, randomized, active controlled, open label, platform trial on the efficacy and safety of experimental therapeutics for patients with a lung disease caused by coronavirus infection ACOVACT (Austrian CoronaVirus Adaptive Clinical Trial). ictrptest.azurewebsites.net/Trial2.aspx?TrialID=EUCTR2020-001302-30-AT (first received 09 April 2020).
EUCTR2020‐001708‐41‐IT {published data only}
- EUCTR2020‐001708‐41‐IT.Enoxaparin for thromboprophylaxis in hospitalized COVID-19 patients: comparison of 40 mg o.d. versus 40 mg b.i.d. A randomized clinical trial. clinicaltrialsregister.eu/ctr-search/trial/2020-001708-41/IT (first received 24 June 2020).
EUCTR2020‐001709‐21‐FR {published data only}
- EUCTR2020‐001709‐21‐FR.Effectiveness of low molecular weight heparin at increased doses prophylaxis weight-adjusted, compared with lower doses prophylaxis (intermediate or standard), on the onset of venous thromboembolism in coronavirus disease 2019 (COVID-19) hospitalized patients: the randomized multicentric controlled open-label trial COVI-DOSE. clinicaltrialsregister.eu/ctr-search/trial/2020-001709-21/FR (first received 24 March 2020).
- EUCTR2020‐001709‐21‐FR.Low-molecular-weight heparin to prevent venous thromboembolism in COVID-19 patients: a randomized controlled trial of different doses. ictrptest.azurewebsites.net/Trial2.aspx?TrialID=EUCTR2020-001709-21-FR (first received 29 April 2020).
EUCTR2020‐001891‐14‐ES {published data only}
- EUCTR2020-001891-14-ES.Impact of the use of low molecular weight heparins (LMWH), at prophylactic versus intermediate doses, on SARS-CoV2 infection (COVID-19). ictrptest.azurewebsites.net/Trial2.aspx?TrialID=EUCTR2020-001891-14-ES (first received 05 May 2020).
EUCTR2020‐002234‐32‐IT {published data only}
- EUCTR2020-002234-32-IT.Efficacy and safety of edoxaban and or colchicine for patients with SARS-CoV-2 infection managed in the out of hospital setting (COVID 19) - CorONa Virus edoxabaN ColchicinE (CONVINCE). ictrptest.azurewebsites.net/Trial2.aspx?TrialID=EUCTR2020-002234-32-IT (first received 4 January 2021).
EUCTR2020‐002504‐39‐DE {published data only}
- EUCTR2020-002504-39-DE.Hamburg edoxaban for anticoagulation in COVID-19 study - HERO-19. ictrptest.azurewebsites.net/Trial2.aspx?TrialID=EUCTR2020-002504-39-DE (first received 14 August 2020).
EUCTR2020‐003349‐12‐IE {published data only}
- EUCTR2020-003349-12-IE.This is a proof of principle / feasibility study aiming to evaluate the effect of nebulised unfractionated heparin on procoagulant markers related to acute respiratory distress syndrome in patients invasively ventilated for COVID 19 lung disease - Charter Trial. ictrptest.azurewebsites.net/Trial2.aspx?TrialID=EUCTR2020-003349-12-IE (first received 14 August 2020).
Goldin 2020 {published data only}
- Goldin M, Giannis D, Diab W, Wang J, Khanijo S, Sharifova G, et al.Treatment-dose LMWH versus prophylactic/intermediate dose heparins in high risk COVID-19 inpatients: rationale and design of the HEP-COVID Trial. Thrombosis and Haemostasis 2021;121(12):1684-95. [DOI: 10.1055/a-1475-2351] [DOI] [PubMed] [Google Scholar]
- NCT04401293.Systemic anticoagulation with full dose low molecular weight heparin (LMWH) vs. prophylactic or intermediate dose LMWH in high risk COVID-19 patients (HEP-COVID Trial). clinicaltrials.gov/ct2/show/NCT04401293 (first received 26 May 2020).
IRCT20200515047456N1 {published data only}
- IRCT20200515047456N1.The role of anticoagulant and thrombolitic in treatment of COVID patients. ictrptest.azurewebsites.net/Trial2.aspx?TrialID=IRCT20200515047456N1 (first received 28 June 2020).
ISRCTN14212905 {published data only}
- ISRCTN14212905.Understanding how COVID-19 leads to respiratory failure in COVID-19 positive patients. ictrptest.azurewebsites.net/Trial2.aspx?TrialID=ISRCTN14212905 (first received 03 July 2020).
Kharma 2020 {published data only}
- Kharma N, Roehrig S, Shible AA, Elshafei MS, Osman D, Elsaid IM, et al.Anticoagulation in critically ill patients on mechanical ventilation suffering from COVID-19 disease, The ANTI-CO trial: a structured summary of a study protocol for a randomised controlled trial. Trials 2020;21(1):769. [PMID: ] [DOI] [PMC free article] [PubMed]
- NCT04445935.Anticoagulation in patients suffering from COVID-19 disease the ANTI-CO Trial. clinicaltrials.gov/ct2/show/NCT04445935 (first received 23 June 2020).
Lasky 2021 {published data only}
- Lasky JA, Fuloria J, Morrison ME, Lanier R, Naderer O, Brundage T, et al.Design and rationale of a randomized, double-blind, placebo-controlled, phase 2/3 study evaluating dociparstat in acute lung injury associated with severe COVID-19. Advances in Therapy 2021;38(1):782‐91. [DOI: 10.1007/s12325-020-01539-z] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT04389840.Dociparstat for the treatment of severe COVID-19 in adults at high risk of respiratory failure. clinicaltrials.gov/ct2/show/NCT04389840 (first received 13 May 2020).
Lins 2020 {published data only}
- Lins PR, Albuquerque CC, Assis CF, Rodrigues BC, e Siqueira Campos BP, Oliveira Valle E, et al.CoV-Hep study: heparin in standard anticoagulation based on citrate for continuous veno-venous hemodialysis in patients with COVID-19: a structured summary of a study protocol for a randomized controlled trial. Trials 2020;21(1):920. [DOI: 10.1186/s13063-020-04814-0] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT04487990.CoV-Hep study: comparative study between different anti-coagulation strategies in continuous hemodialysis in COVID-19 patients. clinicaltrials.gov/ct2/show/NCT04487990 (first received 23 July 2020).
Marietta 2020 {published data only}
- EUCTR2020-001972-13-IT.High versus low LMWH dosages in hospitalized patients with severe COVID-19 pneumonia and coagulopathy COVID-19 HD. ictrptest.azurewebsites.net/Trial2.aspx?TrialID=NCT04408235 (first received 26 May 2020).
- Marietta M, Vandelli P, Mighali P, Vicini R, Coluccio V, D'Amico R.Randomised controlled trial comparing efficacy and safety of high versus low low-molecular weight heparin dosages in hospitalized patients with severe COVID-19 pneumonia and coagulopathy not requiring invasive mechanical ventilation (COVID-19 HD). Blood Transfusion 2020;18 Suppl 4:S490. [DOI: 10.2450/2020.S4] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marietta M, Vandelli P, Mighali P, Vicini R, Coluccio V, D'Amico R.Randomised controlled trial comparing efficacy and safety of high versus low molecular weight heparin dosages in hospitalized patients with severe COVID-19 pneumonia and coagulopathy not requiring invasive mechanical ventilation (COVID-19 HD): a structured summary of a study protocol. Trials 2020;21(1):574. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT04408235.High versus low LMWH dosages in hospitalized patients with severe COVID-19 pneumonia and coagulopathy (COVID-19 HD). clinicaltrials.gov/ct2/show/NCT04408235 (first received 29 May 2020).
NCT04333407 {published data only}
- NCT04333407.Preventing cardiac complication of COVID-19 disease with early acute coronary syndrome therapy: a randomised controlled trial. clinicaltrials.gov/ct2/show/NCT04333407 (first received 3 April 2020).
NCT04344756 {published data only}
- NCT04344756.Trial evaluating efficacy and safety of anticoagulation in patients with COVID-19 infection, nested in the Corimmuno-19 cohort (CORIMMUNO-COAG). clinicaltrials.gov/ct2/show/study/NCT04344756 (first received 14 April 2020).
NCT04345848 {published data only}
- NCT04345848.Preventing COVID-19-associated thrombosis, coagulopathy and mortality with low- and high-dose anticoagulation: a randomized, open-label clinical trial. clinicaltrials.gov/ct2/show/NCT04345848 (first received 15 April 2020).
NCT04352400 {published data only}
- NCT04352400.Efficacy of nafamostat in COVID-19 patients (RACONA Study) (RACONA). clinicaltrials.gov/ct2/show/NCT04352400 (first posted 20 April 2020).
NCT04366960 {published data only}
- NCT04366960.Comparison of two doses of enoxaparin for thromboprophylaxis in hospitalized COVID-19 patients. clinicaltrials.gov/ct2/show/NCT04366960 (first received 29 April 2020).
NCT04367831 {published data only}
- NCT04367831.Intermediate or prophylactic-dose anticoagulation for venous or arterial thromboembolism in severe COVID-19: a cluster based randomized selection trial (IMPROVE-COVID). clinicaltrials.gov/ct2/show/NCT04367831 (first received 27 April 2020).
NCT04373707 {published data only}
- NCT04373707.Effectiveness of weight-adjusted prophylactic low molecular weight heparin doses compared with lower fixed prophylactic doses to prevent venous thromboembolism in COVID-2019. The multicenter randomized controlled open-label trial COVI-DOSE. clinicaltrials.gov/ct2/show/record/NCT04373707 (first received 4 May 2020).
NCT04377997 {published data only}
- NCT04377997.A randomized, open-label trial of therapeutic anticoagulation in COVID-19 patients with an elevated D-dimer. clinicaltrials.gov/ct2/show/NCT04377997 (first received 7 May 2020).
NCT04397510 {published data only}
- NCT04397510.Nebulized heparin vs. placebo for the treatment of COVID-19 induced lung injury. clinicaltrials.gov/ct2/show/NCT04397510 (first received 21 May 2020).
NCT04406389 {published data only}
- NCT04406389.Anticoagulation in critically ill patients with COVID-19 (The IMPACT Trial). www.clinicaltrials.gov/ct2/show/NCT04406389 (first received 26 May 2020).
NCT04409834 {published data only}
- NCT04409834.Prevention of arteriovenous thrombotic events in critically-ill COVID-19 patients trial (COVID-PACT). clinicaltrials.gov/ct2/show/NCT04409834 (first received 28 May 2020).
NCT04416048 {published data only}
- EUCTR2020-002282-33-DE.Effect of anticoagulation therapy on clinical outcomes in COVID-19 COVID-PREVENT. ictrptest.azurewebsites.net/Trial2.aspx?TrialID=NCT04416048 (first received 30 November 2020).
- NCT04416048.Effect of anticoagulation therapy on clinical outcomes in moderate to severe coronavirus disease 2019 (COVID-19). clinicaltrials.gov/ct2/show/NCT04416048 (first received 4 June 2020).
NCT04420299 {published data only}
- NCT04420299.Clinical trial on the efficacy and safety of bemiparin in patients hospitalized because of COVID-19. clinicaltrials.gov/ct2/show/NCT04420299 (first received 5 June 2020).
NCT04444700 {published data only}
- NCT04444700.A pragmatic randomized controlled trial of therapeutic anticoagulation versus standard care as a rapid response to (SARS-CoV-2) COVID-19 pandemic (RAPID-BRAZIL). clinicaltrials.gov/ct2/show/NCT04444700 (first received 22 June 2020).
NCT04485429 {published data only}
- NCT04485429.Efficacy assessment of methylprednisolone and heparin in patients with COVID-19 Pneumonia. clinicaltrials.gov/ct2/show/NCT04485429 (first received 20 July 2020).
NCT04508439 {published data only}
- NCT04508439.Effect of the use of anticoagulant therapy during hospitalization and discharge in patients with COVID-19 infection. clinicaltrials.gov/ct2/show/NCT04508439 (first received 5 August 2020).
NCT04511923 {published data only}
- NCT04511923.Nebulised heparin to reduce COVID-19 induced acute lung injury (CHARTER-Irl). clinicaltrials.gov/ct2/show/NCT04511923 (first received 10 August 2020).
NCT04512079 {published data only}
- NCT04512079.Freedom COVID-19 anticoagulation strategy (FREEDOM COVID). clinicaltrials.gov/ct2/show/NCT04512079 (first received 11 August 2020).
NCT04530578 {published data only}
- NCT04530578.Nebulized heparin in severe scute respiratory syndrome COVID-19 (NEBUHEPA). clinicaltrials.gov/ct2/show/NCT04530578 (first received 17 August 2020).
NCT04542408 {published data only}
- NCT04542408.Hamburg edoxaban for anticoagulation in COVID-19 study. clinicaltrials.gov/ct2/show/NCT04542408 (first received 6 September 2020).
NCT04545541 {published data only}
- NCT04545541.Nebulised heparin in patients with severe COVID-19 (CHARTER-MT). clinicaltrials.gov/ct2/show/NCT04545541 (first received 5 September 2020).
NCT04584580 {published data only}
- NCT04584580.D-dimer adjusted versus therapeutic dose low-molecular-weight heparin in patients with COVID-19 pneumonia. clinicaltrials.gov/ct2/show/NCT04584580 (first received 6 October 2020).
NCT04600141 {published data only}
- NCT04600141.Clinical efficacy of heparin and tocilizumab in patients with severe COVID-19 infection: a randomized clinical trial. clinicaltrials.gov/ct2/show/NCT04600141 (first received 22 October 2020).
NCT04604327 {published data only}
- NCT04604327.Comparison of two different doses of bemiparin in COVID-19 (BEMICOP). clinicaltrials.gov/ct2/show/NCT04604327 (first received 23 October 2020).
NCT04623177 {published data only}
- NCT04623177.Thromboprophylaxis for patients in ICU with COVID-19. clinicaltrials.gov/ct2/show/NCT04623177 (first received 02 November 2020).
NCT04640181 {published data only}
- NCT04640181.Factor Xa inhibitor versus standard of care heparin in hospitalized patients with COVID-19 (XACT). clinicaltrials.gov/ct2/show/NCT04640181 (first received 19 November 2020).
NCT04646655 {published data only}
- NCT04646655.Enoxaparin at prophylactic or therapeutic doses in COVID-19 (EMOS-COVID). clinicaltrials.gov/ct2/show/NCT04646655?term=NCT04646655&draw=2&rank=1 (first received 30 November 2020).
NCT04655586 {published data only}
- NCT04655586.Assessing safety, hospitalization and efficacy of rNAPc2 in COVID-19 (ASPEN). clinicaltrials.gov/ct2/show/NCT04655586?term=Assessing+Safety%2C+Hospitalization+and+Efficacy+of+rNAPc2+in+COVID-19&draw=2&rank=1 (first received 7 December 2020).
NCT04723563 {published data only}
- NCT04723563.Nebulized heparin for the treatment of COVID-19 (INHALE-HEP). clinicaltrials.gov/ct2/show/NCT04723563 (first received 22 January 2021).
NCT04730856 {published data only}
- NCT04730856.Standard vs high prophylactic doses or anticoagulation in patients with high risk of thrombosis admitted with COVID-19 pneumonia (PROTHROMCOVID). clinicaltrials.gov/ct2/show/NCT04730856 (first received 14 January 2021).
NCT04743011 {published data only}
- NCT04743011.Enriched heparin anti COVID-19 trial (EnHanCed). clinicaltrials.gov/ct2/show/NCT04743011 (first received 3 February 2021).
NCT04745442 {published data only}
- NCT04745442.Pilot study of antithrombin as prophylaxis of acute respiratory distress syndrome in patients with COVID-19. clinicaltrials.gov/ct2/show/NCT04745442 (first received 8 February 2021).
PACTR202007606032743 {published data only}
- PACTR202007606032743.Nebulized heparin in patients with mainly moderate coronavirus disease 2019: randomized controlled trial. COVID-19. ictrptest.azurewebsites.net/Trial2.aspx?TrialID=PACTR202007606032743 (first received 15 June 2020).
RBR‐7y8j2bs {published data only}
- RBR-7y8j2bs.Inhalational high molecular weight heparin for the treatment of SARS-Cov-2. ictrptest.azurewebsites.net/Trial2.aspx?TrialID=RBR-7y8j2bs (first received 23 February 2021).
Sholzberg 2021a {published data only}
- NCT04362085.Coagulopathy of COVID-19: a pragmatic randomized controlled trial of therapeutic anticoagulation versus standard care as a rapid response to the COVID-19 pandemic (RAPID COVID COAG). clinicaltrials.gov/ct2/show/NCT04362085 (first received 24 April 2020).
- Sholzberg M, Tang GH, Negri E, Rahhal H, Kreuziger LB, Pompilio CE, et al.Coagulopathy of hospitalised COVID-19: a pragmatic randomised controlled trial of therapeutic anticoagulation versus standard care as a rapid response to the COVID-19 pandemic (RAPID COVID COAG - RAPID Trial): a structured summary of a study protocol for a randomised controlled trial. Trials 2021;22(1):202. [DOI: 10.1186/s13063-021-05076-0] [DOI] [PMC free article] [PubMed] [Google Scholar]
Vanassche 2020 {published data only}
- EUCTR2020-001739-28-BE.A randomized, open-label, adaptive, proof-of-concept clinical trial of modulation of host thromboinflammatory response in patients with COVID-19. ictrptest.azurewebsites.net/Trial2.aspx?TrialID=EUCTR2020-001739-28-BE (first received 10 April 2020).
- Vanassche T, Engelen MM, Van Thillo Q, Wauters J, Gunst J, Wouters C, et al.A randomized, open-label, adaptive, proof-of-concept clinical trial of modulation of host thromboinflammatory response in patients with COVID-19: the DAWn-Antico study. Trials 2020;21:1005. [DOI: 10.1186/s13063-020-04878-y] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanassche T, Engelen MM, Van Thillo Q, Wauters J, Gunst J, Wouters C, et al.Correction to: A randomized, open-label, adaptive, proof-of-concept clinical trial of modulation of host thromboinflammatory response in patients with COVID-19: the DAWn-Antico study. Trials 2020;21:1033. [DOI: 10.1186/s13063-020-04991-y] [DOI] [PMC free article] [PubMed] [Google Scholar]
Van Haren 2020 {published data only}
- NCT04635241.Inhaled heparin for hospitalised COVID-19 patients (INHALE-HEP). clinicaltrials.gov/ct2/show/NCT04635241 (first received 19 November 2020).
- Van Haren FMP, Richardson A, Yoon H-J, Artigas A, Laffey JG, Dixon B, et al.Inhaled nebulised unfractionated heparin for the treatment of hospitalised patients with COVID-19 (INHALE-HEP): protocol and statistical analysis plan for an investigator-initiated international metatrial of randomised studies. British Journal of Clinical Pharmacology 2020;88(8):3075-91. [DOI: 10.1111/bcp.14714] [DOI] [PubMed] [Google Scholar]
Wilkinson 2020 {published data only}
- EudraCT2020-001736-95.ACCORD 2: a multicentre, seamless, phase 2 adaptive randomisation platform study to assess the efficacy and safety of multiple candidate agents for the treatment of COVID 19 in hospitalised patients. clinicaltrialsregister.eu/ctr-search/trial/2020-001736-95/GB (first received 23 April 2020). [DOI] [PMC free article] [PubMed]
- ISRCTN57085639.A platform to investigate the safety and effectiveness of several new medicines for the treatment of COVID-19 in hospitalised patients. ictrptest.azurewebsites.net/Trial2.aspx?TrialID=ISRCTN57085639 (first received 24 September 2020).
- Wilkinson T, Dixon R, Page C, Carroll M, Griffiths G, Ho L-P, et al.ACCORD: a multicentre, seamless, phase 2 adaptive randomisation platform study to assess the efficacy and safety of multiple candidate agents for the treatment of COVID-19 in hospitalised patients: a structured summary of a study protocol for a randomised controlled trial. Trials 2020;21(1):691. [DOI: 10.1186/s13063-020-04584-9] [DOI] [PMC free article] [PubMed] [Google Scholar]
Additional references
Abdel‐Maboud 2021
- Abdel-Maboud M, Menshawy A, Elgebaly A, Bahbah EI, El Ashal G, Negida A.Should we consider heparin prophylaxis in COVID-19 patients? a systematic review and meta-analysis. Journal of Thrombosis and Thrombolysis 2021;51(3):830-2. [PMID: ] [DOI] [PMC free article] [PubMed]
Ackermann 2020
- Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al.Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in COVID-19. New England Journal of Medicine 2020;383(2):120-8. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Alquwaizani 2013
- Alquwaizani M, Buckley L, Adams C, Fanikos J.Anticoagulants: a review of the pharmacology, dosing, and complications. Current Emergency and Hospital Medicine Reports 2013;1(2):83-97. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Amaral 2020
- Amaral FC, Baptista-Silva JC, Nakano LC, Flumignan RL.Pharmacological interventions for preventing venous thromboembolism in patients undergoing bariatric surgery. Cochrane Database of Systematic Reviews 2020, Issue 7. Art. No: CD013683. [DOI: 10.1002/14651858.CD013683] [DOI] [PMC free article] [PubMed] [Google Scholar]
AVF 2020
- American Venous Forum.Considerations in prophylaxis and treatment of VTE in COVID-19 patients. www.veinforum.org/wp-content/uploads/2020/04/COVID-19-White-Paper-04-17-2020-FINAL-1.pdf (accessed 3 July 2020).
Becker 2020
- Becker RC.COVID-19 update: COVID-19-associated coagulopathy. Journal of Thrombosis and Thrombolysis 2020;50(1):54-67. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Biagioni 2020
- Biagioni RB, Lopes RD, Agati LB, Sacilotto R, Wolosker N, Sobreira ML, et al.Rationale and design for the study apixaban versus clopidogrel on a background of aspirin in patients undergoing infrapopliteal angioplasty for critical limb ischemia: AGRIPPA trial. American Heart Journal 2020;227:100-6. [DOI: 10.1016/j.ahj.2020.06.010] [DOI] [PubMed] [Google Scholar]
Bikdeli 2020
- Bikdeli B, Madhavan M, Jimenez D, Chuich T, Dreyfus I, Driggin E, et al.COVID-19 and thrombotic or thromboembolic disease: implications for prevention, antithrombotic therapy, and follow-up: JACC state of the art review. Journal of American College of Cardiology 2020;75(23):2950-73. [DOI: 10.1016/j.jacc.2020.04.031] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bilaloglu 2020
- Bilaloglu S, Aphinyanaphongs Y, Jones S, Iturrate E, Hochman J, Berger JS.Thrombosis in hospitalized patients with COVID-19 in a New York City health system. JAMA 2020;324(8):799-801. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Clezar 2020
- Clezar CN, Cassola N, Flumignan CD, Nakano LC, Trevisani VF, Flumignan RL.Pharmacological interventions for asymptomatic carotid stenosis. Cochrane Database of Systematic Reviews 2020, Issue 4. Art. No: CD013573. [DOI: 10.1002/14651858.CD013573] [DOI] [PMC free article] [PubMed] [Google Scholar]
COMET 2020
- Core outcome set developers’ response to COVID-19. comet-initiative.org/Studies/Details/1538 (accessed 04 August 2020).
Covidence [Computer program]
- Covidence.Version accessed 1 August 2020. Melbourne, Australia: Veritas Health Innovation. Available at covidence.org.
COVIDSurg 2021
- COVIDSurg Collaborative, GlobalSurg Collaborative.SARS-CoV-2 infection and venous thromboembolism after surgery: an international prospective cohort study. Anaesthesia 2021 Aug 24 [Epub ahead of print]. [DOI: 10.1111/anae.15563] [DOI] [PMC free article] [PubMed]
Cuker 2021
- Cuker A, Tseng EK, Nieuwlaat R, Angchaisuksiri P, Blair C, Dane K, et al.American Society of Hematology 2021 guidelines on the use of anticoagulation for thromboprophylaxis in patients with COVID-19. Blood Advances 2021;5(3):872-88. [PMID: 10.1182/bloodadvances.2020003763] [DOI] [PMC free article] [PubMed] [Google Scholar]
Deeks 2021
- Deeks JJ, Higgins JP, Altman DG, editor(s).Chapter 10: Analysing data and undertaking meta-analyses. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page M, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Cochrane, 2021. Available from www.training.cochrane.org/handbook.
Dias 2021
- Dias SV, Flumignan RL, Iared W.Evidence from Cochrane systematic reviews for effects of antithrombotic drugs for lower-limb revascularization. A narrative review. Sao Paulo Medical Journal 2021 Aug 13 [Epub ahead of print]. [DOI: 10.1590/1516-3180.2020.0640.110321] [DOI] [PMC free article] [PubMed]
Dolhnikoff 2020
- Dolhnikoff M, Duarte-Neto AN, Almeida Monteiro RA, da Silva LF, Oliveira EP, Saldiva PH, et al.Pathological evidence of pulmonary thrombotic phenomena in severe COVID-19. Journal of Thrombosis and Haemostasis : JTH 2020;18(6):1517-9. [PMID: ] [DOI] [PMC free article] [PubMed]
Fox 2020
- Fox SE, Akmatbekov A, Harbert JL, Li G, Quincy-Brown J, Vander-Heide RS.Pulmonary and cardiac pathology in African American patients with COVID-19: an autopsy series from New Orleans. Lancet Respiratory Medicine 2020;8(7):681-6. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Giannis 2020
- Giannis D, Ziogas IA, Gianni P.Coagulation disorders in coronavirus infected patients: COVID-19, SARS-CoV-1, MERS-CoV and lessons from the past. Journal of Clinical Virology 2020;127:104362. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADEpro GDT [Computer program]
- GRADEpro GDT.Version accessed 1 August 2020. Hamilton (ON): McMaster University (developed by Evidence Prime). Available at gradepro.org.
Hasan 2020
- Hasan SS, Radford S, Kow CS, Zaidi ST.Venous thromboembolism in critically ill COVID-19 patients receiving prophylactic or therapeutic anticoagulation: a systematic review and meta-analysis. Journal of Thrombosis and Thrombolysis 2020;50(4):814-21. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG.Measuring inconsistency in meta-analyses. BMJ 2003;326:557-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2017
- Higgins JP, Altman DG, Sterne JA, editor(s).Chapter 8: Assessing risk of bias in included studies. In: Higgins JP, Churchill R, Chandler J, Cumpston MS, editor(s). Cochrane Handbook for Systematic Reviews of Interventions. Version 5.2.0 (updated June 2017). Cochrane, 2017. Available from training.cochrane.org/handbook/archive/v5.2.
Higgins 2020a
- Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al (editors).Cochrane Handbook for Systematic Reviews of Interventions version 6.1 (updated September 2020). Cochrane, 2020. Available from www.training.cochrane.org/handbook.
Higgins 2020b
- Higgins JP, Eldridge S, Li T, editor(s).Chapter 23: Including variants on randomized trials. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.1 (updated September 2020). Cochrane, 2020. Available from www.training.cochrane.org/handbook.
Kamel 2021
- Kamel AM, Sobhy M, Magdy N, Sabry N, Farid S.Anticoagulation outcomes in hospitalized COVID-19 patients: a systematic review and meta-analysis of case-control and cohort studies. Reviews in Medical Virology 2021;31(3):e2180. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Klok 2020a
- Klok FA, Kruip MJ, Van der Meer NJ, Arbous MS, Gommers DA, Kant KM, et al.Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thrombosis Research 2020;191:145-7. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Klok 2020b
- Klok FA, Kruip MJ, Van der Meer NJ, Arbous MS, Gommers D, Kant KM, et al.Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: an updated analysis. Thrombosis Research 2020;191:148-50. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lai 2020
- Lai CC, Wang CY, Wang YH, Hsueh SC, Ko WC, Hsueh PR.Global epidemiology of coronavirus disease 2019 (COVID-19): disease incidence, daily cumulative index, mortality, and their association with country healthcare resources and economic status. International Journal of Antimicrobial Agents 2020;55(4):105946. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lazaridis 2021
Lefebvre 2021
- Lefebvre C, Glanville J, Briscoe S, Littlewood A, Marshall C, Metzendorf M-I, et al.Chapter 4: Searching for and selecting studies. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Cochrane, 2021. Available from www.training.cochrane.org/handbook.
Li 2020
- Li Y, Li M, Wang M, Zhou Y, Chang J, Xian Y, et al.Acute cerebrovascular disease following COVID-19: a single center, retrospective, observational study. Stroke and Vascular Neurology 2020;5(3):279-84. [DOI: 10.1136/svn-2020-000431] [DOI] [PMC free article] [PubMed] [Google Scholar]
Liu 2019
- Liu Y, Mu S, Li X, Liang Y, Wang L, Ma X.Unfractionated heparin alleviates sepsis-induced acute lung injury by protecting tight junctions. Journal of Surgical Research 2019;238:175-85. [PMID: ] [DOI] [PubMed] [Google Scholar]
Long 2020
- Long B, Brady WJ, Koyfman A, Gottlieb M.Cardiovascular complications in COVID-19. American Journal of Emergency Medicine 2020;38(7):1504-7. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Marini 2020
- Marini JJ, Gattinoni L.Management of COVID-19 respiratory distress. JAMA 2020;323(22):2329-30. [DOI: 10.1001/jama.2020.6825] [PMID: ] [DOI] [PubMed] [Google Scholar]
Matli 2021
- Matli K, Farah R, Maalouf M, Chamoun N, Costanian C, Ghanem G.Role of combining anticoagulant and antiplatelet agents in COVID-19 treatment: a rapid review. Open Heart 2021;8(1):e001628. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
McBane 2020
- McBane RD 2nd, Torres Roldan VD, Niven AS, Pruthi RK, Franco PM, Linderbaum JA, et al.Anticoagulation in COVID-19: a systematic review, meta-analysis, and rapid guidance from Mayo Clinic. Mayo Clinic Proceedings 2020;95(11):2467-86. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
McGuinness 2020
- McGuinness LA, Higgins JP.Risk-of-bias visualization (robvis): an R package and Shiny web app for visualizing risk-of-bias assessments. Research Synthesis Methods 2020;12(1):55-61. [DOI: 10.1002/jrsm.1411] [DOI] [PubMed] [Google Scholar]
Moonla 2021
- Moonla C, Sosothikul D, Chiasakul T, Rojnuckarin P, Uaprasert N.Anticoagulation and in-hospital mortality from coronavirus disease 2019: a systematic review and meta-analysis. Clinical and Applied Thrombosis/hemostasis 2021;27:10760296211008999. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
NHS 2020
- NHS England and NHS Improvement.Clinical guide for the management of anticoagulant services during the coronavirus pandemic. www.england.nhs.uk/coronavirus/wp-content/uploads/sites/52/2020/03/C0077-Specialty-guide_Anticoagulant-services-and-coronavirus-v1-31-March.pdf (accessed 3 July 2020).
Obe 2020
- Obe BH, Retter A, McClintock C.Practical guidance for the prevention of thrombosis and management of coagulopathy and disseminated intravascular coagulation of patients infected with COVID-19. thrombosisuk.org/downloads/T&H%20and%20COVID.pdf (accessed 3 July 2020).
Oxley 2020
- Oxley TJ, Mocco J, Majidi S, Kellner CP, Shoirah H, Singh IP, et al.Large-vessel stroke as a presenting feature of COVID-19 in the young. New England Journal of Medicine 2020;382(20):e60. [DOI: 10.1056/NEJMc2009787] [DOI] [PMC free article] [PubMed] [Google Scholar]
Page 2021
- Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al.The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ 2021;372:n71. [DOI: 10.1136/bmj.n71] [DOI] [PMC free article] [PubMed] [Google Scholar]
Panigada 2020
- Panigada M, Bottino N, Tagliabue P, Grasselli G, Novembrino C, Chantarangkul V, et al.Hypercoagulability of COVID-19 patients in intensive care unit: a report of thromboelastography findings and other parameters of hemostasis. Journal of Thrombosis and Haemostasis 2020;18(7):1738-42. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Parisi 2021
- Parisi R, Costanzo S, Di Castelnuovo A, Gaetano G, Donati MB, Iacoviello L.Different anticoagulant regimens, mortality, and bleeding in hospitalized patients with COVID-19: a systematic review and an updated meta-analysis. Seminars in Thrombosis and Hemostasis 2021;47(4):372-91. [PMID: ] [DOI] [PubMed] [Google Scholar]
Patell 2021
- Patell R, Chiasakul T, Bauer E, Zwicker JI.Pharmacologic thromboprophylaxis and thrombosis in hospitalized patients with COVID-19: a pooled analysis. Thrombosis and Haemostasis 2021;121(1):76-85. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Perepu 2021
- Perepu US, Chambers I, Wahab A, Eyck PT, Wu C, Dayal S, et al.Standard prophylactic versus intermediate dose enoxaparin in adults with severe COVID-19: a multi-center, open-label, randomized controlled trial. Journal of Thrombosis and Haemostasis 2021;19(9):2225-34. [DOI: 10.1111/jth.15450] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ramacciotti 2020
- Ramacciotti E, Macedo AS, Biagioni RB, Caffaro RA, Lopes RD, Guerra JC, et al.Evidence-based practical guidance for the antithrombotic management in patients with coronavirus disease (COVID-19) in 2020. Clinical and Applied Thrombosis/hemostasis 2020;26:1076029620936350. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Reeves 2021
- Reeves BC, Deeks JJ, Higgins JP, Shea B, Tugwell P, Wells GA.Chapter 24: Including non-randomized studies on intervention effects. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Cochrane, 2021. Available from www.training.cochrane.org/handbook.
Review Manager 2020 [Computer program]
- Review Manager 5 (RevMan 5).Version 5.4. Copenhagen: The Cochrane Collaboration, 2020.
Robvis [Computer program]
- Robvis (visualization tool).Version accessed 20 August 2020. Bristol, UK: Luke McGuinness. Available at mcguinlu.shinyapps.io/robvis/.
Schulman 2010
- Schulman S, Angerås U, Bergqvist D, Eriksson B, Lassen MR, Fisher W.Definition of major bleeding in clinical investigations of antihemostatic medicinal products in surgical patients. Journal of Thrombosis and Haemostasis : JTH 2010;8(1):202-4. [PMID: ] [DOI] [PubMed] [Google Scholar]
Schünemann 2013
- Schünemann H, Brożek J, Guyatt G, Oxman A, editor(s).Handbook for grading the quality of evidence and the strength of recommendations using the GRADE approach (updated October 2013). GRADE Working Group, 2013. Available from gdt.gradepro.org/app/handbook/handbook.html.
Schünemann 2021
- Schünemann HJ, Higgins JP, Vist GE, Glasziou P, Akl EA, Skoetz N, et al.Chapter 14: Completing ‘Summary of findings’ tables and grading the certainty of the evidence. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Cochrane, 2021. Available from www.training.cochrane.org/handbook.
Sholzberg 2021b
- Sholzberg M, Tang GH, Rahhal H, AlHamzah M, Kreuziger LB, Ní Áinle F, et al.Effectiveness of therapeutic heparin versus prophylactic heparin on death, mechanical ventilation, or intensive care unit admission in moderately ill patients with COVID-19 admitted to hospital: RAPID randomised clinical trial. BMJ 2021;375:n2400. [DOI: 10.1136/bmj.n2400] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sobreira 2020
- Sobreira ML, Marques MA.Anticoagulants as panacea in COVID-19 infection. Jornal Vascular Brasileiro 2020;19:e20200063. [DOI: 10.1590/1677-5449.200063] [DOI] [PMC free article] [PubMed] [Google Scholar]
Spyropoulos 2021
- Spyropoulos AC, Goldin M, Giannis D, Diab W, Wang J, Khanijo S, et al.Efficacy and safety of therapeutic-dose heparin vs standard prophylactic or intermediate-dose heparins for thromboprophylaxis in high-risk hospitalized patients with COVID-19: the HEP-COVID randomized clinical trial. JAMA Internal Medicine 2021 October 7 [Epub ahead of print]. [DOI: 10.1001/jamainternmed.2021.6203] [DOI] [PMC free article] [PubMed]
Sterne 2016
- Sterne JA, Hernán MA, Reeves BC, Savović J, Berkman ND, Viswanathan M, et al.ROBINS-I: a tool for assessing risk of bias in non-randomised studies of interventions. BMJ (Clinical Research Ed.) 2016;355:i4919. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Talasaz 2021
- Talasaz AH, Sadeghipour P, Kakavand H, Aghakouchakzadeh M, Kordzadeh-Kermani E, Van Tassell BW, et al.Recent randomized trials of antithrombotic therapy for patients with COVID-19: JACC state-of-the-art review. Journal of the American College of Cardiology 2021;77(15):1903-21. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wan 2014
- Wan X, Wang W, Liu J, Tong T.Estimating the sample mean and standard deviation from the sample size, median, range and/or interquartile range. BMC Medical Research Methodology 2014;14:135. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ware 1992
- Ware JE Jr, Sherbourne CD.The MOS 36-item short-form health survey (SF-36). I. Conceptual framework and item selection. Medical Care 1992;30(6):473-83. [PMID: ] [PubMed] [Google Scholar]
Young 2008
- Young E.The anti-inflammatory effects of heparin and related compounds. Thrombosis Research 2008;122(6):743-52. [PMID: ] [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Flumignan 2020a
- Flumignan RL, Tinôco JD, Pascoal PI, Areias LL, Cossi MS, Fernandes MI, et al.Prophylactic anticoagulants for patients hospitalised with COVID-19 (Protocol). available from doi.org/10.17605/OSF.IO/8PRXW (registered 7 August 2020).
Flumignan 2020b
- Flumignan RL, Tinôco JD, Pascoal PI, Areias LL, Cossi MS, Fernandes MI, et al.Prophylactic anticoagulants for people hospitalised with COVID‐19. Cochrane Database of Systematic Reviews 2020, Issue 10. Art. No: CD013739. [DOI: 10.1002/14651858.CD013739] [DOI] [PMC free article] [PubMed] [Google Scholar]
Flumignan 2021
- Flumignan RL, Tinôco JD, Pascoal PI, Areias LL, Cossi MS, Fernandes MI, et al.Prophylactic anticoagulants for people hospitalized with COVID-19: systematic review. British Journal of Surgery 2021 Jun 10 [Epub ahead of print]. [DOI: 10.1093/bjs/znab197] [DOI] [PMC free article] [PubMed]