

Abstract

Thermally activated delayed fluorescence (TADF) materials are commonly used in various apparatus, including organic light-emitting device-based displays, as they remarkably improve the internal quantum efficiencies. Although there is a wide range of donor–acceptor-based compounds possessing TADF properties, in this computational study, we investigated TADF and some non-TADF chromophores, containing benzophenone or its structural derivatives as the acceptor core, together with various donor moieties. Following the computational modeling of the emitters, several excited state properties, such as the absorption spectra, singlet–triplet energy gaps (ΔEST), natural transition orbitals, and the topological ΦS indices, have been computed. Along with the donor–acceptor torsion angles and spin-orbit coupling values, these descriptors have been utilized to investigate potential TADF efficiency. Our study has shown that on the one hand, our photophysical/structural descriptors and computational methodologies predict the experimental results quite well, and on the other hand, our extensive benchmark can be useful to pinpoint the most promising functionals and descriptors for the study of benzophenone-based TADF emitters.
1. Introduction
Organic light-emitting diodes (OLEDs) have attracted widespread attention since the invention of the first organic electroluminescent device in 1987.1 As compared to conventional LEDs and liquid crystal display systems, OLEDs do not require a backlight unit, as they are self-illuminating. Given this distinct feature, OLEDs offer several advantages, such as flexible device structures, decreased panel thickness, improved brightness, and reduced power consumption, rendering them most suitable for various devices.2 Nevertheless, OLEDs also display serious efficiency drawbacks, which are grounded in the fundamental spin statistics rule (Figure 1) because the population of the nonemissive triplet state comprises 75% of the generated excitons in the device, hence leading to the loss of two thirds of the applied energy.3 Phosphorescent organic light-emitting diodes (PhOLEDs), which contain heavy atoms, can improve the efficiencies of OLEDs with the help of enhanced intersystem crossing (ISC) and improved phosphorescence rates as a result of increased spin-orbit coupling (SOC).4,5 However, heavy metals instigate environmental issues while significantly increasing the cost of the device, thus limiting the commercial availability of PhOLEDs.6
Figure 1.

Schematic representation of ISC, RISC, TADF, and phosphorescence processes in a Jablonski diagram along with the possible design strategies for TADF emitters.
Thermally activated delayed fluorescence (TADF) materials, first proposed by Adachi et al. in 2011, represent a most suitable alternative to overcome spin statistics burdens and achieve high internal quantum efficiencies (IQEs) in OLED displays.7 In TADF processes, the population of triplet excitons undergoes a slow reverse intersystem crossing (RISC), re-populating the emissive first excited singlet state (S1) and hence avoiding the inclusion of heavy metals. To assure efficiency, RISC or the upconversion of the triplet states [up-intersystem crossing (UISC)] require a small S1-T1 energy gap (ΔEST). UISC can be achieved by promoting the population of intramolecular charge-transfer (ICT) states and is usually, albeit not necessarily, correlated with a small gap between the highest occupied molecular orbital and lowest unoccupied molecular orbital. To achieve efficient ICT, the molecular architecture requires the presence of a donor (D) moiety bridged to an acceptor (A) (Figure 1). The D and A groups can be separated by using bulky substituents to increase steric effects and maintain orthogonal molecular structures, hence minimizing conjugation and delocalization, or alternatively by π-bridges.8,9
Benzophenone is a widely used building block in the design of OLED devices, in part due to its strong and efficient electron accepting and efficient ultraviolet (UV) absorbing abilities, as well as its high ISC efficiency.10−13 Adachi and co-workers showed in 2014 that efficient deep blue TADF could be achieved by using benzophenone-based D-A-D frameworks.12 Thus far, many benzophenone-based TADF emitters possessing a small ΔEST have been reported to feature full-color delayed fluorescence emission, usually in a range from deep blue to green and with external quantum efficiencies up to 14.3%.14 Benzophenone-based luminogens can also be used to induce aggregation-induced delayed fluorescence.15,16
Nonetheless, the rather floppy phenyl moieties of benzophenone itself may induce intramolecular rotations, which enhance nonradiative decay and lead to relatively low reverse ISC rates (kRISC).17 Hence, more compact and rigid benzophenone derivatives, such as anthraquinone,18,19 xanthone,20−23 dibenzothiophene-benzoyl24,25 and phenylcarbazole-benzoyl,26 are deemed more promising in terms of their TADF efficiencies. Figure 2 depicts the molecular structures of benzophenone derivatives commonly used in TADF materials, together with some of the most widely used D groups.
Figure 2.

Electron-donating and -accepting moieties frequently used in the design of benzophenone-based TADF emitters.
We used molecular modeling to elucidate the relationship between the molecular structure and optimal photophysical properties that are essential for effective TADF emission. Various descriptors are used to define TADF properties, which include ΔEST, SOC, natural transition orbitals (NTOs), the topological ΦS index quantifying the charge-transfer amount, and the torsion angles between D and A moieties.27−33 The oscillator strength (f) is also a critical parameter to attain a reasonable radiative decay rate (kr) from the S1 state to the ground state. The oscillator strength is usually closer to 0 for orthogonal donor–acceptor compounds, where CT is prominent.29 Negligible oscillator strengths represent a challenge in TADF emitters because it is essential to balance a small ΔEST and a sufficiently large kr to reach high IQE in OLED devices.
In this study, a series of experimentally studied TADF emitters, employing benzophenone and its derivatives, have been investigated by quantum chemical calculations. Several non-TADF benzophenone chromophores have also been modeled for comparison. Absorption spectra of the selected compounds have been generated, including a sampling of the Franck–Condon region by Wigner distribution, to include dynamic effects. To determine the degree of charge separation in the excited state, NTOs and ΦS indices have been computed, as well as the ΔEST values. NTO and ΔEST calculations have been performed on both the ground state (S0) and the lowest triplet state (T1) equilibrium geometries because geometrical reorganization can also play a role in photophysical processes. Furthermore, the SOCs between singlet and triplet excited states have been computed to better estimate RISC probability.
The mentioned descriptors have proven to be insightful in determining potential TADF efficiency, as well as shedding light on the correlation between molecular structure and TADF performance. Indeed, the investigation of the photophysical characteristics of both the excited and ground state geometries, and also the assessment of the DFT functionals for the excited state calculations of TADF emitters, may help provide a better understanding for the photophysical processes related to TADF at a molecular level.
2. Methodology
All ground state geometry optimizations have been performed using the Gaussian 16 program package,34 and a comprehensive conformational search has been carried out. UV–vis absorption spectra and Boltzmann-weighted ΔEST calculations (Tables S1 and S2) showed that different conformations did not exhibit significant differences in their excitation energies and on the ordering of the excited states. Hence, T1 geometry optimizations and related excited state calculations were carried out solely on the most stable conformer. The M06-2X functional35 has been used together with the 6–31 + G(d,p) basis set for S0 and T1 geometry optimizations. This choice is justified because M06-2X is well known to correctly reproduce medium-range interactions, electronic energies, and equilibrium geometries of compounds with aromatic ring systems.35 However, a dispersion-corrected functional, B3LYP-D3, was also tested for the geometry optimization of rather larger compounds.36 In order to increase the accuracy, 6–311++G(3df,3pd)37 and 6–311++G(2d,2p)38 basis sets have been used for compounds including sulfur and phosphorus, respectively. Calculations have been performed taking into account the experimentally employed solvents by the polarizable continuum model in the integral equation formalism (IEF-PCM). CYLview software package has been used for visualization purposes.39
Similar to ground state calculations, excited state calculations have been carried out by using Gaussian 16 software package. Tamm–Dancoff approximation (TDA) has been used, as this approach avoids unphysically stable triplet states in CT molecules while maintaining a good description of the singlet excited states, hence yielding more accurate and balanced results for ΔEST calculations.40,41 The absorption spectra as well as the energies of the lowest lying singlet and triplet excited states have been computed with different functionals (B3LYP,42−45 BLYP,42,45,46 PBE0,43,45,47,48 M06-2X,42,45,49 CAM-B3LYP,42,45,50,51 and LC-ωPBE52−54) and 6–31 + G(d,p) basis set, and the performances of these functionals have been evaluated with respect to experimental data. Absorption spectra have been modeled, including the effects of vibrational and thermal motion via Wigner distribution sampling of the equilibrium region on the potential energy surface by generating 40 conformations via the Newton-X program55 on the equilibrium region of the PES.
NTOs and ΦS indices have been calculated for the S1 state by using the Gaussian 16 and Nancy_EX program packages,56 while hole and electron NTOs have been visualized with the Avogadro program package.57 ΦS index can be defined as the spatial overlap between attachment and detachment densities. Values closer to 1 indicate the presence of local excitation character, whereas values approaching 0 imply that the CT character is dominant.56 SOC values between S1 and T1, and in some cases between S1 and T2, have been calculated using the Amsterdam Density Functional software package by utilizing a DZP basis set.58
3. Results and Discussion
The benzophenone emitters investigated in this study have been grouped according to the type of A moieties used. As depicted in Figure 3, Group 1 emitters are in the form of D-A-D, and they contain benzophenone A cores (in red) bridged with various electron-donating groups. They can be further subdivided into two subgroups: symmetric and asymmetric emitters. Symmetric emitters Px2BP,12 DMAC-BP,59 Cz2BP,12 and CC2BP12 contain phenoxazine (PXZ), dimethylacridine (DMAC), and carbazole (Cz) donors (in blue), respectively. Asymmetric emitters, A-BP-TA60 and OPDPO,61 include D groups such as thianthrene, phenothiazene (PTZ), and diphenylphosphineoxide.
Figure 3.

Classification of TADF emitters modeled in this study.
Group 2 emitters have fused D-A ring structures where DBT-BZ-PXZ,25 DBT-BZ-PTZ,25 and DBT-BZ-DMAC24 bear the A unit dibenzothiophene-benzoyl (in red), while CP-BP-PXZ26 and CP-BP-DMAC26 have the A unit of phenylcarbazole-benzoyl (in red). D groups such as PXZ, PTZ, and DMAC have been employed in these emitters as well.
Group 3 emitters have para-substituted structures in which the A unit is anthraquinone.18 These include D-A-D type emitters a1-a4, in which the D units are diphenylamine (DPA), bis(4-biphenyl)amine, 3,6-di-tert-butylcarbazole, and DMAC, respectively. Similarly, in the D-π-A-π-D structures, b1 and b4, the D groups are DPA and DMAC.
Group 4 emitters have D-A type of structures in which the A unit is xanthone. DMAC, PXZ, and PTZ donors have been used in ACRXTN,23 3-PXZ-XO,20 and PTZ-XT,62 respectively. In MCz-XT,21 1,3,6,8-tetramethylcarbazole is present as the D unit.
Finally, seven non-TADF compounds have been selected from literature in an attempt to elucidate the main structural differences leading to TADF emission (Figure 4). While some of these non-TADF emitters have D-A type structures, including MC2,63 OPM,64 and p-Cz,65 others (ODFRCZ,66 ODBTCZ,67 C1,15 and C215) possess D-A-D type of structures.
Figure 4.

Non-TADF benzophenone emitters.
3.1. Benchmark Calculations and UV–vis Absorption Spectra
M06-2X/6–31 + G(d,p) and B3LYP-D3/6–31 + G(d,p) methods have been tested for the ground state geometry optimizations of a series of compounds selected from each group. Table S1 depicts the optimized geometries at both levels of theory, and Table S2 includes the D-A torsion angles for the optimized geometries. Tables S1 and S2 suggest that there is no significant change in the geometries optimized at both levels as both functionals yielded similar structures with very close D-A torsion angles. Because the compounds investigated in this study are relatively small in which long-range interactions are not so dominant, we decided to continue with M06-2X/6–31 + G(d,p) for the geometry optimizations because the latter is computationally more affordable.
A series of widely used DFT functionals have been chosen for excited state calculations, BLYP, B3LYP, PBE0, and M06-2X. More specifically, UV–vis absorption spectra have been generated for a group of emitters, and the results have been compared with the available experimental spectra.
The conformational space has been taken into account in the modeling of four compounds (Px2BP, Cz2BP, DBT-BZ-PXZ, and DBT-BZ-DMAC) (Table S1). Their absorption spectra have been obtained as the union of the spectra of all the conformers according to their Boltzmann weights. The merged spectra have then been compared to those obtained from the most stable conformation only. Table S3 clearly demonstrates that weighted conformations exhibit similar photophysical features and spectral shapes as the most stable conformation, the maximum deviation being around 3 nm for B3LYP (DBT-BZ-PXZ), 4 nm for PBE0 (DBT-BZ-PXZ), and 2 nm for M06-2X (Cz2BP). In addition to the UV–vis absorption spectra, different conformations of Cz2BP and CC2BP have very similar ΔEST values, and the Boltzmann-weighted ΔEST values of Cz2BP and CC2BP were found to be in good agreement with the experimental findings (Table S4). Thus, henceforth, the most stable conformer will solely be considered. In Tables S5–S7, the absorption spectra for Group 1 emitters and three emitters from Group 2 have been computed by using BLYP, B3LYP, PBE0, and M06-2X. The results suggest that the spectra obtained with M06-2X exhibit hypsochromic shift as compared to the experimental spectrum.
As opposed to M06-2X, BLYP produced bathochromic shifts and lower ΔEST values (energies are given in Table S8; histogram charts are depicted in Figures S1 and S2) when compared with the experimental data. This behavior is most probably due to well-known unphysical stabilization of CT states by LDA functionals.68 In fact, hybrid B3LYP and PBE0 functionals yield the best agreement with experimental spectra and ΔEST. In order to further confirm this finding, two long-range corrected functionals, CAM-B3LYP and LC-ωPBE, were tested for selected compounds. Table S9 depicts the ΔES1-T1 values (eV) calculated with BLYP, B3LYP, PBE0, CAM-B3LYP, and LC-ωPBE for the S0 and T1 optimized geometries of the compounds. Similar to M06-2X, the ΔEST values calculated with the long-range corrected CAM-B3LYP and LC-ωPBE functionals are significantly higher as compared with experiment and the other functionals. It is also noteworthy that BLYP gave results consistent with the experiment for a few molecules (Px2BP and DMAC-BP). This is probably due to the fact that, due to the relatively small size of the compounds, the CT states are not long-range and hence can be correctly reproduced by hybrid functionals as already observed for instance in some organometallic compounds, hence outperforming medium- or long-range corrected functionals.69
In an attempt to take into account the role of excitation on the molecular geometry, the ΔEST values have been computed from the S0, S1, and T1 geometries with B3LYP, PBE0, and BLYP functionals for a series of compounds, and the results have been reported in Table S10. The results clearly indicate that the ΔEST values computed from the S0 and S1 geometries are usually very similar. This can be explained by the similar molecular structures obtained for S0 and S1 geometries (Figure S3), as TADF efficiency relies on the molecular geometry to a great extent. Also, considering the computational cost of S1 geometry optimizations, for the rest of the calculations, the photophysical properties have been calculated solely with the S0 and T1 geometries.
The absorption spectra of all other molecules have hence been calculated with BLYP, B3LYP, and PBE0 functionals (Figures S4–S8). From the results, it can be deduced that compounds with more rigid electron-donating groups such as PXZ, PTZ, and DMAC possess a broadened band (∼350 to 500 nm) appearing after the high intensity local excitation band (∼300 nm), which can be ascribed to the presence of CT states.70−72 Nevertheless, the reasonably rigid emitters of Group 4 also have blue-shifted absorption spectra, which is most likely due to the less efficient nature of the specific D-A type architecture. Indeed, the presence of a unique D unit might decrease the CT character of the electronic transitions, and coherently reduced T1-S1 upconversion efficiency has been previously reported for some D-A type emitters.73
Oscillator strengths calculated with B3LYP, PBE0, and BLYP functionals for the transitions from S0 to S1, and the reorganization energies between S0 and S1 geometries have also been reported in Tables S11 and S12, respectively. While the latter remain low to moderate and are between around 15 and 6 kcal/mol, no systematic behavior between the different classes of molecules can be underlined, thus hampering the use of this parameter to preview TADF efficiency. Low to moderate geometrical distortion and reorganization energies may also be another reason for the very close ΔEST values calculated from the S0 and S1 geometries. The oscillator strengths were calculated to be near-zero for most of the TADF emitters with very low ΔEST values and ΦS indices, for which we assume that the CT characteristics are more pronounced. As is the case in this study, it was previously reported that the oscillator strengths computed at the TDA or full TD-DFT level might be erroneous, especially for the transitions involving CT states.74
3.2. D–A Torsion Angles
Several computational TADF descriptors were assessed in order to analyze and investigate the structural and photophysical phenomena related to TADF emissions. One of the most critical TADF descriptors is the torsion angle between the D and A units. The torsion angle must be close to 90° in an ideal TADF emitter because an effective CT configuration can be achieved by spatially separating the hole and electron densities and by breaking the conjugation pattern.75
The S0 and T1 optimized geometries of all emitters and the conformation energies (M06-2X/6–31 + G(d,p)) are given in Supporting Information (Tables S13–S17). Figure 5 demonstrates the torsion angles measured for all emitters investigated in this study.
Figure 5.
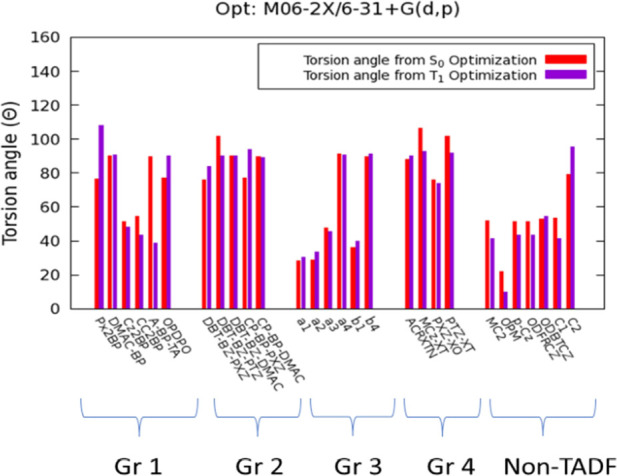
Donor–acceptor torsion angles for the S0 and T1 optimized geometries of the investigated emitters shown with stacked histograms (M06-2X/6–31 + G(d,p)).
Because symmetrical emitters have similar torsion angles for each D–A bond, only one of the torsion angles has been shown in Figure 5, while numerical values for torsion angles depicted in Figures S9–S13 are reported in Table S18. For asymmetrical emitters, the torsion angle of the electron-donating unit has been ascribed based on the NTO analysis discussed in the following sections. Similarly, if the electron–hole density is localized on a different electron-donating unit in the T1 equilibrium geometry, the corresponding torsion angle has been considered for the analysis of the T1 equilibrium geometries.
It is evident that the emitters containing rigid electron-donating moieties, including DMAC, PXZ, and PTZ, have significantly higher D–A torsion angles. While reasonably high torsion angles have been observed for Group 1, Group 2, and Group 4, Group 3 emitters bear relatively low torsion angles approximately in the range of 30–35°. This feature can be due to the presence of less rigid DPA donors whose low steric hindrance may lead to highly planar structure with extended π-conjugation patterns. The highest torsion angles belong to a4 and b4 among Group 3 emitters due to the presence of DMAC electron donors in these groups. Indeed, the additional dimethyl units in the acridine structure impose sterical constraints that restrict the free rotation of the D. Carbazole (Cz)-containing compounds also exhibit low torsion angles, around 55°, which are slightly higher than that for the DPA-bearing compounds. Interestingly, the majority of the non-TADF compounds, where the D unit is Cz, have notably lower torsion angles, along with two compounds from Group 1 emitters (Cz2BP and CC2BP). The torsion angles measured for Group 4 emitters are satisfactorily high and close to 90° regardless of the electron-donating unit. This situation can be attributed to the rigid xanthone skeleton that fixes the D–A torsion angle at a desirable position. This observation is in line with the study of Kreiza et al.17 Nonetheless, the obtained results suggest that such a descriptor can only provide a partial understanding of TADF efficiency because Group 3 compounds, which are known as TADF emitters, possess very low torsion angles, despite the presence of the rigid anthraquinone A. Hence, a finer analysis of the property and reorganization of the electronic density should be considered to improve TADF efficiency predictions.
While in most cases the torsion angles are similar for both T1 and S0 equilibrium geometries, there is a sharp decrease in the torsion angle for T1 equilibrium geometry in A-BP-TA. Indeed, the hole density in the triplet state is localized on the less rigid electron D thianthrene, causing a rather important planarization of the global structure. This will be better understood in the following section.
3.3. NTOs and ΦS Indices
NTOs and ΦS indices have been calculated for all emitters in an attempt to determine the electron–hole density reorganization. Accordingly, electron and hole NTOs describing electronic density reorganization in the S1 state are shown in Tables S19–S28. Only the NTOs generated from B3LYP/6–31 + G(d,p) densities are given because no remarkable difference has been observed with BLYP and PBE0. ΦS values describing the spatial overlap between attachment and detachment densities76 are also reported in Tables S29–S33.
In general, the overlap between electron and hole densities has been observed to be high for compounds bearing low D-A torsion angles. This is expected because low torsion angles cause high degrees of π-delocalization and a corresponding planarization of the molecular core. As a result, compounds bearing Cz and DPA donors, including Cz2BP, CC2BP, a1, a2, a3, b1, and most of the non-TADF compounds, show highly overlapping hole and electron NTOs. Interestingly, in non-TADF emitters ODBTCZ and ODFRCZ, the highly flexible dibenzothiophene and dibenzofuran units did not show electron-donating ability at S0 or T1 equilibrium geometries. Obviously, because high ΦS indices are indicative of spatially overlapping densities, the corresponding excited states have a more prominent local excitation character. On the other hand, emitters with more rigid D units (DMAC, PXZ, and PTZ) usually have low amounts of overlap between their NTOs, and consequently small ΦS values, even in the presence of small torsion angles. Hence, even if more computationally expensive, the explicit analysis of the excited state density is much more informative in predicting TADF potential.
It is also noteworthy that the structural reorganization may have an undeniable impact on the amount of CT from the accessible excited states, as in the case of excited state twisting.77 Indeed, some compounds, particularly those containing butterfly-shaped PXZ, PTZ, or thianthrene units, underwent drastic changes; more specifically, T1 equilibrium geometries exhibited planarization in the butterfly-shaped moieties (Table 1). This, in turn, is related to a change in the shape of the NTOs obtained from the T1 equilibrium geometries. The change in the NTO distribution is indicative of the fact that geometric relaxation leads, for some compounds, such as Px2BP, A-BP-TA, and b1, to the population of a different diabatic state. The most obvious change in the NTO localization pattern is seen in A-BP-TA (Figure 6), where at T1 equilibrium geometry, the hole NTO is localized mostly on the now planar thianthrene unit instead of the rigid DMAC unit. This effect also produces an increase in the ΦS indices at the T1 equilibrium geometry for Px2BP and its meta-substituted analogue C2 (Tables S19 and S28). The electron NTO localizes only on one of the PXZ units, which becomes planar at T1 equilibrium geometry; this feature is also indicative of a consistent pattern, which is observed in two other similar cases. In the π-bridged anthraquinone-based b1 and b4, similar changes have been observed in the transition from S0 to T1 equilibrium geometry (Table S24). These consist of slight rotations in the bridging phenyl links, which cause disruptions in orthogonality and give rise to an increased electron delocalization. For a better visualization, the hole and electron NTOs and ΦS indices, calculated at T1 and S0 equilibrium geometries for two TADF and one non-TADF compounds, are shown in Figure 6.
Table 1. S0 and T1 Geometries of Selected Emitters with Butterfly-Shaped Electron Donors.

Figure 6.

Hole–Electron levels and ΦS indices calculated with B3LYP/6–31 + G(d,p) for the molecules (a) ACRXTN, (b) MC2, and (c) A-BP-TA.
As expected, non-TADF compounds exhibit higher ΦS, which are due to the presence of weaker electron donors like Cz, dibenzofuran, and dibenzothiophene. However, despite the fact that the only noticeable structural difference is related to the position of the PXZ substituents, the meta-substituted non-TADF C2 also exhibits higher ΦS indices compared to its para-substituted analogue, Px2BP, in line with its classification as a non-TADF compound. ΦS indices calculated with B3LYP for all compounds are depicted in Figure 7, and the ΦS indices calculated with PBE0 and BLYP are given in Figures S14–S15. TADF behavior is clearly inferred from these values, hence making this descriptor quite promising for the discrimination and prediction of optical properties with TADF potential.
Figure 7.

ΦS indices for the TADF emitters calculated from S0 and T1 geometries with B3LYP/6–31 + G(d,p).
3.4. Singlet–Triplet Energy Gaps (ΔEST)
The energy difference between the first singlet and triplet excited states is an important descriptor for predicting the possibility of TADF emission because the upconversion of T1 to S1 can be achieved only if the energy gap is low enough to be overcome by thermal energy. According to previous studies, ΔEST values above 0.3 eV usually decrease the likelihood of the RISC process, whereas the upconversion of the triplet becomes more likely if the ΔEST value is below 0.1 eV.5
Even though the RISC process usually takes place from the T1 state, upper lying triplet excited states may also influence the efficiency of RISC if they are in close proximity to T1. Spin-vibronic coupling may cause state mixing between triplet energy levels, giving rise to strongly coupled S1 and T1 states. Therefore, the strong impact of internal conversion on ISC and RISC processes cannot be underestimated.78 Thus, the energy differences between S1-T2 (ΔES1-T2) states have also been reported along with the ΔES1-T1 values when the T2 state lies below S1 for any given compound. The calculated values together with the experimental findings are reported in Tables S34–S38 and in Figure 8.
Figure 8.

ΔEST values for TADF emitters calculated from S0 and T1 geometries with B3LYP/6–31 + G(d,p). (Experimental ΔEST values also included).
Figure 8 depicts the ΔES1-T1 values calculated with B3LYP for almost all groups except for Group 3 because ΔES1-T2 values calculated for them are in a better agreement with the experimental findings. It is evident that the lowest ΔEST values have been obtained from the BLYP calculations, which is consistent with the lowest ΦS indices computed with this functional. On the other hand, ΔEST values calculated with B3LYP and PBE0 are closer to the experimental ones. Overall, the calculated ΔEST values are usually consistent with the experimental observation, especially for the S0 geometries. The ΔEST values calculated with PBE0 and BLYP are also given in Figures S16 and S17.
It is noteworthy that ΔEST values are closely correlated with torsion angles and ΦS indices because the compounds possessing low torsion angles and high ΦS indices, such as Cz2BP, CC2BP, most of the Group 3 compounds, and non-TADF compounds, also have relatively higher ΔES1-T2 and in some cases even high ΔES1-T1 values. Freely rotating donors DPA and sterically less hindered donors Cz lead to higher ΔEST values because of enhanced geometrical relaxation. Despite having DPA donors, b1 has been observed to possess relatively low ΔEST values, which may have been acquired through the phenyl bridges that lead to a higher spatial separation of electron and hole densities. Also, as expected, emitters with DMAC, PXZ, or PTZ donors have satisfying ΔEST values usually at both S0 and T1 equilibrium geometries due to the induced orthogonality. There is a sharp increase in the ΔEST values obtained from the T1 equilibrium geometry of A-BP-TA, where the electron-donating group is the less rigid thianthrene moiety. This is also consistent with the NTO calculations previously mentioned, pointing out the close relationship between ΔEST and electron–hole separation. This result for A-BP-TA also further proves the reliability of NTO calculations and ΦS index in elucidating the excited state properties by relating them to the molecular structure.
Almost all of the non-TADF compounds have high ΔEST values due to higher amounts of density overlap introduced by freely rotating D–A moieties, which planarize to enhance conjugation. However, C2 is an exception as it exhibits notably lower ΔEST values in spite of its high ΦS index and overlapping NTOs. The calculated ΔEST value is also lower as compared with its experimental ΔEST value, clearly identifying this compound as an outlier. Still, out of 28 benzophenone derivatives investigated in this study, C2 is the only emitter in which an inverse relationship between ΔEST values and ΦS indices was observed, and this can also be resulting from the insufficiency of our methodology in reproducing excited state energies for this compound.
3.5. Spin-Orbit Couplings
The last descriptor analyzed in this study is the SOC. The mixing of the wavefunctions of singlet and triplet energy levels are indeed strongly dictated by SOC.79 Therefore, by performing SOC calculations, crucial information regarding the feasibility of ISC or RISC processes is obtained. Indeed, SOC matrix elements significantly different from zero are necessary to attain RISC and, hence, TADF emission. However, SOC usually increases with atomic number (Z); thus small organic chromophores are usually less prone to ISC and RISC as compared to heavy-metal containing complexes.80 Additionally, the coupling of CT states produce negligible SOC values as a consequence of El-Sayed’s rule, which highlights the need for local triplet excited states for an effective RISC process.79 Efficient TADF emitters usually have lower SOC values due to their orthogonal D–A type molecular backbones, where CT character is more pronounced; this unfavorable factor is, however, compensated by small singlet–triplet energy gaps, which are inversely proportional to the (R)ISC probability. Hence, SOC between the S1 and T1 states of TADF emitters could also indicate the presence of CT states. Because we are dealing with RISC, calculations were solely performed on equilibrium T1 geometries. SOC between S1 and T2 states have also been computed from the T1 geometries if the T2 lies below the S1 state, in a similar approach adopted for ΔEST calculations. The calculated SOC values have been reported in Tables S39–S43. For a better comprehension, the SOC values calculated with B3LYP, BLYP, and PBE0 functionals are shown in Figure 9.
Figure 9.

SOC values for TADF emitters calculated with B3LYP, BLYP, and PBE0 using the T1 geometries.
The results indicate that the compounds possessing lower ΔEST values and ΦS indices, such as most of Group 1 emitters (Px2BP, DMAC-BP, and OPDPO), Group 2 emitters, a4 and b4, and Group 4 emitters, exhibit extremely low SOC values as a result of the higher CT character of their S1 and T1 geometries.
Higher SOC values have been obtained for compounds with less steric hindrance, including Cz2BP, CC2BP, most of Group 3 emitters, and all non-TADF emitters, because local excitations become more favorable in these emitters. A-BP-TA also exhibits relatively high SOC values because its hole density becomes localized on the flexible thianthrene unit in its T1 geometry, reducing the CT character in a similar manner observed in its ΔEST values and ΦS index. Additionally, higher SOC constants obtained for S1-T2 interactions may indicate that the triplet upconversion might be taking place from the T2 rather than the T1 state.
Table 2 summarizes the D–A torsion angle, ΦS index, ΔEST, and SOC results of the TADF and non-TADF emitters investigated, showing a consistent behavior for all descriptors studied, pointing toward the successful accuracy of NTOs and ΦS in inferring the TADF behavior. More importantly, in most cases, the TADF capability may be correctly predicted by calculating the excited state indicators at the Franck–Condon geometry, hence avoiding rather expensive excited state optimizations.
Table 2. D–A Torsion Angles (°), ΦS Indices (Lowdin Charge Population), ΔEST Values (eV), and SOC Constants for the TADF and Non-TADF Emitters Calculated from the T1 Geometries (TDA: B3LYP/6–31 + G(d,p)).
| group | compound | torsion angle | ΦS index | ΔEST | SOC |
|---|---|---|---|---|---|
| group 1 | Px2BP | 107.90 | 0.6097 | 0.104 | 0.073 |
| DMAC-BP | 90.91 | 0.1991 | 0.013 | 0.037 | |
| Cz2BP | 48.19 | 0.4603 | 0.278 | 0.465 | |
| CC2BP | 43.33 | 0.5206 | 0.284 | 0.942 | |
| A-BP-TA | 38.90 | 0.4509 | 0.225 | 0.999 | |
| OPDPO | 90.06 | 0.1009 | 0.004 | 0.004 | |
| group 2 | DBT-BZ-PXZ | 83.84 | 0.1279 | 0.018 | 0.042 |
| DBT-BZ-PTZ | 90.21 | 0.1480 | 0.005 | 0.002 | |
| DBT-BZ-DMAC | 90.11 | 0.1967 | 0.004 | 0.026 | |
| CP-BP-PXZ | 93.80 | 0.1494 | 0.011 | 0.026 | |
| CP-BP-DMAC | 88.90 | 0.1514 | 0.008 | 0.009 | |
| group 3 | a1 | 30.54 | 0.6839 | 0.481 | 0.144 |
| a2 | 33.32 | 0.6992 | 0.428 | 0.120 | |
| a3 | 45.42 | 0.5689 | 0.272 | 0.134 | |
| a4 | 90.67 | 0.0546 | 0.007 | 0.025 | |
| b1 | 39.68 | 0.3627 | 0.346 | 0.097 | |
| b4 | 91.00 | 0.0544 | 0.001 | 0.000 | |
| group 4 | ACRXTN | 90.21 | 0.0595 | 0.008 | 0.025 |
| MCz-XT | 92.89 | 0.1199 | 0.011 | 0.026 | |
| 3-PXZ-XO | 74.04 | 0.3243 | 0.066 | 0.060 | |
| PTZ-XT | 91.49 | 0.0721 | 0.018 | 0.022 | |
| non-TADF | MC2 | 41.45 | 0.5879 | 0.651 | 0.617 |
| OPM | 21.76 | 0.6566 | 0.944 | 2.070 | |
| p-Cz | 43.66 | 0.5675 | 0.613 | 0.246 | |
| ODFRCZ | 43.69 | 0.5936 | 0.553 | 0.345 | |
| ODBTCZ | 52.73 | 0.4911 | 0.591 | 1.078 | |
| C1 | 41.19 | 0.4369 | 0.283 | 0.202 | |
| C2 | 95.14 | 0.4208 | 0.016 | 0.015 |
4. Conclusions
In this study, the photophysical and structural properties of 21 benzophenone-based TADF emitters and 7 benzophenone-based non-TADF emitters have been investigated by using computational descriptors with the aim to elucidate the factors causing RISC. The main descriptors have been identified as the torsion angle between the D and A, NTOs, ΦS indices, ΔEST values, and SOC constants.
It was observed that the orthogonality of the molecular backbone is an important factor for achieving a successful RISC process. The compounds adopting a rigid molecular structure where free rotations are more restricted tend to have lower ΔEST values and ΦS indices due to increased ICT through well separated electron and hole densities. Conversely, the emitters possessing less rigid and more freely rotating D-A arrangements were observed to have higher ΔEST values and ΦS indices due to increased local excitation character induced by high amounts of overlap between the hole and electron and enhanced π-delocalization. Although molecular rigidity relies on both the molecular structures of the D and A, it was shown that the role of D units is more dominant, as freely rotating electron donors such as Cz, DPA, or thianthrene disrupt the orthogonality of the compounds, which leads to, in most cases, lower torsion angles (∼30 to 55°). The more rigid electron donors, such as DMAC, PXZ, or PTZ, enhance TADF efficiency through increased orthogonality and electron/hole separation. However, the A rigidity must also be taken into account. The compounds presenting more rigid and planar benzophenone derivatives, such as xanthone, as the A moiety, exhibit satisfying TADF characteristics, regardless of the electron donors. On the contrary, the emitters bearing the benzophenone A, in which the loose phenyl rings have more free rotation, exhibit less satisfying TADF properties. The presence of a π-bridge may also favorably affect the TADF properties owing to the increased spatial separation of D and A units as observed in compounds b1 and b4. Moreover, SOC values were calculated to be lower for the TADF compounds when the presence of highly twisted D-A frameworks induces lower ΔEST values due to enhanced electron/hole separation.
Excited state calculations were carried out using both S0 and T1 geometries because excited state processes, such as RISC, should take place from the T1 equilibrium geometry. In some cases, a significant geometrical relaxation was observed, in particular for the butterfly-shaped PXZ, PTZ, or thianthrene units, which become planar in T1 geometry. In these cases, different indicators were obtained for calculations performed at S0 or T1 geometries. However, in general, the outcome of the photophysical properties can be correctly inferred by solely considering the Franck–Condon geometry. The benchmark studies revealed that B3LYP and PBE0 can reproduce the excited state energies and UV–vis absorption spectra correctly for almost all emitters, while BLYP yielded satisfying results for several compounds only.
In summary, this study gives a comprehensive outlook on the relationship between molecular structure and photophysical features of a series of benzophenone-based TADF and non-TADF compounds, employing computational tools and several descriptors. It can be concluded that these descriptors, especially the topological ΦS index, can be safely used to investigate TADF properties of different classes of compounds, as they predict the experimental findings quite accurately.
Acknowledgments
The computations were mainly performed using TUBITAK ULAKBIM High Performance and Grid Computing Center (TRUBA) and LPCT local computing resources. We also acknowledge the support from TUBITAK (Project Number:118Z914) and BAP-M (Project Number:16863). V.A. acknowledges the resources of the National Center for High Performance Computing of Turkey (UHeM) under Grant Number 5005832018. A.M. thanks ANR (Agence Nationale de la Recherche) and CGI (Commissariat à l’Investissement d’Avenir) for their financial support of this work through Labex SEAM (Science and Engineering for Advanced Materials and devices) ANR 11 LABX 086 and ANR 11 IDEX 05 02. The support of the IdEx ″Université Paris 2019″ ANR-18-IDEX-0001 and of the Platform P3MB is gratefully acknowledged.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpca.1c08320.
3D representations of the optimized geometries at M06-2X/6–31 + G(d,p) and B3LYP-D3/6–31 + G(d,p) levels, investigation of the conformational effects, benchmark calculations, UV–vis absorption spectra, investigated donor–acceptor torsion angles, hole and electron NTOs, ΦS indices, low lying singlet–triplet energy gaps, spin-orbit coupling values, and S0, T1, and S1 cartesian coordinates of the most stable conformations optimized at the M06-2X/6–31 + G(d,p) level of theory (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Tang C. W.; VanSlyke S. A. Organic Electroluminescent Diodes. Appl. Phys. Lett. 1987, 51, 913–915. 10.1063/1.98799. [DOI] [Google Scholar]
- Huang Y.; Hsiang E. L.; Deng M. Y.; Wu S. T. Mini-LED, Micro-LED and OLED Displays: Present Status and Future Perspectives. Light-Sci. Appl. 2020, 9, 105. 10.1038/s41377-020-0341-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H.; Shizu K.; Miyazaki H.; Adachi C. Efficient Green Thermally Activated Delayed Fluorescence (TADF) from a Phenoxazine–Triphenyltriazine (PXZ–TRZ) Derivative. Chem. Commun. 2012, 48, 11392–11394. 10.1039/c2cc36237f. [DOI] [PubMed] [Google Scholar]
- Xiao L.; Chen Z.; Qu B.; Luo J.; Kong S.; Gong Q.; Kido J. Recent Progresses on Materials for Electrophosphorescent Organic Light-Emitting Devices. Adv. Mater. 2011, 23, 926–952. 10.1002/adma.201003128. [DOI] [PubMed] [Google Scholar]
- Dias F. B.; Bourdakos K. N.; Jankus V.; Moss K. C.; Kamtekar K. T.; Bhalla V.; Santos J.; Bryce M. R.; Monkman A. P. Triplet Harvesting with 100% Efficiency by Way of Thermally Activated Delayed Fluorescence in Charge Transfer OLED Emitters. Adv. Mater. 2013, 25, 3707–3714. 10.1002/adma.201300753. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Li J.; Shizu K.; Huang S.; Hirata S.; Miyazaki H.; Adachi C. Design of Efficient Thermally Activated Delayed Fluorescence Materials for Pure Blue Organic Light Emitting Diodes. J. Am. Chem. Soc. 2012, 134, 14706–14709. 10.1021/ja306538w. [DOI] [PubMed] [Google Scholar]
- Endo A.; Sato K.; Yoshimura K.; Kai T.; Kawada A.; Miyazaki H.; Adachi C. Efficient Up-Conversion of Triplet Excitons into a Singlet State and Its Application for Organic Light Emitting Diodes. Appl. Phys. Lett. 2011, 98, 2009–2012. 10.1063/1.3558906. [DOI] [Google Scholar]
- Masui K.; Nakanotani H.; Adachi C. Analysis of Exciton Annihilation in High-Efficiency Sky-Blue Organic Light-Emitting Diodes with Thermally Activated Delayed Fluorescence. Org. Electron. 2013, 14, 2721–2726. 10.1016/j.orgel.2013.07.010. [DOI] [Google Scholar]
- Uoyama H.; Goushi K.; Shizu K.; Nomura H.; Adachi C. Highly Efficient Organic Light-Emitting Diodes from Delayed Fluorescence. Nature 2012, 492, 234–238. 10.1038/nature11687. [DOI] [PubMed] [Google Scholar]
- Reineke S.; Baldo M. A. Room Temperature Triplet State Spectroscopy of Organic Semiconductors. Sci. Rep. 2014, 4, 3797. 10.1038/srep03797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marazzi M.; Mai S.; Roca-Sanjuán D.; Delcey M. G.; Lindh R.; González L.; Monari A. Benzophenone Ultrafast Triplet Population: Revisiting the Kinetic Model by Surface-Hopping Dynamics. J. Phys. Chem. Lett. 2016, 7, 622–626. 10.1021/acs.jpclett.5b02792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. Y.; Yasuda T.; Yang Y. S.; Zhang Q.; Adachi C. Luminous Butterflies: Efficient Exciton Harvesting by Benzophenone Derivatives for Full-Color Delayed Fluorescence OLEDs. Angew. Chem., Int. Ed. 2014, 53, 6402–6406. 10.1002/anie.201402992. [DOI] [PubMed] [Google Scholar]
- Aloïse S.; Ruckebusch C.; Blanchet L.; Réhault J.; Buntinx G.; Huvenne J. P. The Benzophenone S1 (n, π*)→ T1 (n, π*) States Intersystem Crossing Reinvestigated by Ultrafast Absorption Spectroscopy and Multivariate Curve Resolution. J. Phys. Chem A 2008, 112, 224–231. 10.1021/jp075829f. [DOI] [PubMed] [Google Scholar]
- Huang B.; Ban X.; Sun K.; Ma Z.; Mei Y.; Jiang W.; Lin B.; Sun Y. Thermally Activated Delayed Fluorescence Materials Based on Benzophenone Derivative as Emitter for Efficient Solution-Processed Non-Doped Green OLED. Dyes Pigm. 2016, 133, 380–386. 10.1016/j.dyepig.2016.06.025. [DOI] [Google Scholar]
- Keruckiene R.; Keruckas J.; Cekaviciute M.; Volyniuk D.; Lee P. H.; Chiu T. L.; Lee J. H.; Grazulevicius J. V. Meta-Substituted Benzophenones as Multifunctional Electroactive Materials for OLEDs. Dyes Pigm. 2020, 174, 108058 10.1016/j.dyepig.2019.108058. [DOI] [Google Scholar]
- Gan S.; Zhou J.; Smith T. A.; Su H.; Luo W.; Hong Y.; Zhao Z.; Tang B. Z. New AIEgens with Delayed Fluorescence for Fluorescence Imaging and Fluorescence Lifetime Imaging of Living Cells. Mater. Chem. Front. 2017, 1, 2554–2558. 10.1039/C7QM00286F. [DOI] [Google Scholar]
- Kreiza G.; Banevičius D.; Jovaišaitė J.; Maleckaitė K.; Gudeika D.; Volyniuk D.; Gražulevičius J. V.; Juršėnas S.; Kazlauskas K. Suppression of Benzophenone-Induced Triplet Quenching for Enhanced TADF Performance. J. Mater. Chem. C 2019, 7, 11522–11531. 10.1039/C9TC02408E. [DOI] [Google Scholar]
- Zhang Q.; Kuwabara H.; Potscavage W. J.; Huang S.; Hatae Y.; Shibata T.; Adachi C. Anthraquinone-Based Intramolecular Charge-Transfer Compounds: Computational Molecular Design, Thermally Activated Delayed Fluorescence, and Highly Efficient Red Electroluminescence. J. Am. Chem. Soc. 2014, 136, 18070–18081. 10.1021/ja510144h. [DOI] [PubMed] [Google Scholar]
- Bin H.; Ji Y.; Li Z.; Zhou N.; Jiang W.; Feng Y.; Lin B.; Sun Y. Simple Aggregation–Induced Delayed Fluorescence Materials Based on Anthraquinone Derivatives for Highly Efficient Solution–Processed Red OLEDs. J. Lumin. 2017, 187, 414–420. 10.1016/j.jlumin.2017.03.038. [DOI] [Google Scholar]
- Zhang Y.; Ma H.; Wang S.; Li Z.; Ye K.; Zhang J.; Liu Y.; Peng Q.; Wang Y. Supramolecular Structure-Dependent Thermally-Activated Delayed Fluorescence (TADF) Properties of Organic Polymorphs. J. Phys. Chem. C 2016, 120, 19759–19767. 10.1021/acs.jpcc.6b05537. [DOI] [Google Scholar]
- Lee J.; Aizawa N.; Numata M.; Adachi C.; Yasuda T. Versatile Molecular Functionalization for Inhibiting Concentration Quenching of Thermally Activated Delayed Fluorescence. Adv. Mater. 2017, 29, 1604856 10.1002/adma.201604856. [DOI] [PubMed] [Google Scholar]
- Aizawa N.; Tsou C. J.; Park I. S.; Yasuda T. Aggregation-Induced Delayed Fluorescence from Phenothiazine-Containing Donor-Acceptor Molecules for High-Efficiency Non-Doped Organic Light-Emitting Diodes. Polym. J. 2017, 49, 197–202. 10.1038/pj.2016.82. [DOI] [Google Scholar]
- Nakanotani H.; Higuchi T.; Furukawa T.; Masui K.; Morimoto K.; Numata M.; Tanaka H.; Sagara Y.; Yasuda T.; Adachi C. High-Efficiency Organic Light-Emitting Diodes with Fluorescent Emitters. Nat. Commun. 2014, 5, 4016. 10.1038/ncomms5016. [DOI] [PubMed] [Google Scholar]
- Guo J.; Li X. L.; Nie H.; Luo W.; Gan S.; Hu S.; Hu R.; Qin A.; Zhao Z.; Su S. J.; et al. Achieving High-Performance Nondoped OLEDs with Extremely Small Efficiency Roll-Off by Combining Aggregation-Induced Emission and Thermally Activated Delayed Fluorescence. Adv. Funct. Mater. 2017, 27, 1606458 10.1002/adfm.201606458. [DOI] [Google Scholar]
- Guo J.; Li X. L.; Nie H.; Luo W.; Hu R.; Qin A.; Zhao Z.; Su S. J.; Tang B. Z. Robust Luminescent Materials with Prominent Aggregation-Induced Emission and Thermally Activated Delayed Fluorescence for High-Performance Organic Light-Emitting Diodes. Chem. Mater. 2017, 29, 3623–3631. 10.1021/acs.chemmater.7b00450. [DOI] [Google Scholar]
- Huang J.; Nie H.; Zeng J.; Zhuang Z.; Gan S.; Cai Y.; Guo J.; Su S. J.; Zhao Z.; Tang B. Z. Highly Efficient Nondoped OLEDs with Negligible Efficiency Roll-Off Fabricated from Aggregation-Induced Delayed Fluorescence Luminogens. Angew. Chem., Int. Ed. 2017, 56, 12971–12976. 10.1002/anie.201706752. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Wu Z.; Ni F.; Zhong C.; Zeng W.; Wei D.; An K.; Ma D.; Yang C. Emitters with a Pyridine-3,5-Dicarbonitrile Core and Short Delayed Fluorescence Lifetimes of about 1.5 Μs: Orange-Red TADF-Based OLEDs with Very Slow Efficiency Roll-Offs at High Luminance. J. Mater. Chem. C 2018, 6, 6543–6548. 10.1039/C8TC01698D. [DOI] [Google Scholar]
- Olivier Y.; Sancho-Garcia J. C.; Muccioli L.; D’Avino G.; Beljonne D. Computational Design of Thermally Activated Delayed Fluorescence Materials: The Challenges Ahead. J. Phys. Chem. Lett. 2018, 9, 6149–6163. 10.1021/acs.jpclett.8b02327. [DOI] [PubMed] [Google Scholar]
- Mewes J. M. Modeling TADF in Organic Emitters Requires a Careful Consideration of the Environment and Goingbeyond the Franck-Condon Approximation. Phys. Chem. Chem. Phys. 2018, 20, 12454–12469. 10.1039/C8CP01792A. [DOI] [PubMed] [Google Scholar]
- Kang S.; Jeon S. H.; Cho Y. M.; Kim Y. J.; Kim T.; Lee J. Y. The Key Role of Acceptor Moieties on the Structural and the Electronic Properties of Thermally Activated Delayed Fluorescence Emitters in Excited States: A Computational Study. Org. Electron. 2020, 78, 105595 10.1016/j.orgel.2019.105595. [DOI] [Google Scholar]
- Sanz-Rodrigo J.; Olivier Y.; Sancho-García J. C. Computational Studies of Molecular Materials for Unconventional Energy Conversion: The Challenge of Light Emission by Thermally Activated Delayed Fluorescence. Molecules 2020, 25, 1006. 10.3390/molecules25041006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q.; Guo X.; Zhang J. Theoretical Study on Photophysical Properties of a Series of Functional Pyrimidine-Based Organic Light-Emitting Diodes Emitters Presenting Thermally Activated Delayed Fluorescence. J. Comput. Chem. 2019, 40, 1578–1585. 10.1002/jcc.25808. [DOI] [PubMed] [Google Scholar]
- Tavakoli M.; Ahmadvand H.; Alaei M.; Ranjbari M. A. Ab-Initio Search for Efficient Red Thermally Activated Delayed Fluorescence Molecules for Organic Light Emitting Diodes. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2021, 246, 118952 10.1016/j.saa.2020.118952. [DOI] [PubMed] [Google Scholar]
- Frisch G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; et al. Gaussian 16, Rev. A.03.; Gaussian, Inc.: Wallingford, CT, 2016. [Google Scholar]
- Josa D.; Rodríguez-Otero J.; Cabaleiro-Lago E. M.; Rellán-Piñeiro M. Analysis of the Performance of DFT-D, M05-2X and M06-2X Functionals for Studying π···π Interactions. Chem. Phys. Lett. 2013, 557, 170–175. 10.1016/j.cplett.2012.12.017. [DOI] [Google Scholar]
- Darvish Ganji M.; Hosseini-khah S. M.; Amini-tabar Z. Theoretical Insight into Hydrogen Adsorption onto Graphene: A First-principles B3LYP-D3 Study. Phys. Chem. Chem. Phys. 2015, 17, 2504–2511. 10.1039/C4CP04399E. [DOI] [PubMed] [Google Scholar]
- Kolleth A.; Müller S.; Lumbroso A.; Tanriver G.; Catak S.; Sulzer-Mossé S.; De Mesmaeker A. Access to 3-Aminobenzothiophenes and 3-Aminothiophenes Fused to 5-Membered Heteroaromatic Rings through 6π-Electrocyclization Reaction of Keteniminium Salts. Tetrahedron Lett. 2018, 59, 3242–3248. 10.1016/j.tetlet.2018.06.049. [DOI] [Google Scholar]
- Zabaradsti A.; Kakanejadifard A.; Ghasemian M. Theoretical Study of Molecular Interactions of Phosphorus Ylide with Hypohalous Acids HOF HOCl and HOBr. Comput. Theor. Chem. 2012, 989, 1–6. 10.1016/j.comptc.2012.02.017. [DOI] [Google Scholar]
- Legault C. Y.CYLview 1.0b; Université de Sherbrooke, 2009. [Google Scholar]
- Hirata S.; Head-Gordon M. Time-Dependent Density Functional Theory within the Tamm-Dancoff Approximation. Chem. Phys. Lett. 1999, 314, 291–299. 10.1016/S0009-2614(99)01149-5. [DOI] [Google Scholar]
- Chantzis A.; Laurent A. D.; Adamo C.; Jacquemin D. Is the Tamm–Dancoff Approximation Reliable for the Calculation of Absorption and Fluorescence Band Shapes?. J. Chem. Theory Comput. 2013, 9, 4517–4525. 10.1021/ct400597f. [DOI] [PubMed] [Google Scholar]
- Liang K.; Zheng C.; Wang K.; Liu W.; Guo Z.; Li Y.; Zhang X. Theoretical Investigation of the Singlet-Triplet Splittings for Carbazole-Based Thermally Activated Delayed Fluorescence Emitters. Phys. Chem. Chem. Phys. 2016, 18, 26623–26629. 10.1039/C6CP04545F. [DOI] [PubMed] [Google Scholar]
- Lu J.; Zheng Y.; Zhang J. Rational Design of Phenoxazine-Based Donor–Acceptor–Donor Thermally Activated Delayed Fluorescent Molecules with High Performance. Phys. Chem. Phys. Chem. 2015, 17, 20014–20020. 10.1039/C5CP02810H. [DOI] [PubMed] [Google Scholar]
- Wang F.; Hu J.; Cao X.; Yang T.; Tao Y.; Mei L.; Zhang X.; Huang W. A Low-Cost Phenylbenzoimidazole Containing Electron Transport Material for Efficient Green Phosphorescent and Thermally Activated Delayed Fluorescent OLEDs. J. Mater. Chem. C 2015, 3, 5533–5540. 10.1039/C5TC00350D. [DOI] [Google Scholar]
- Cardeynaels T.; Paredis S.; Deckers J.; Brebels S.; Vanderzande D.; Maes W.; Champagne B. Finding the Optimal Exchange–Correlation Functional to Describe the Excited State Properties of Push–Pull Organic Dyes Designed for Thermally Activated Delayed Fluorescence. Phys. Chem. Phys. Chem. 2020, 22, 16387–16399. 10.1039/D0CP02409K. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee I.; Acharya N.; Ray D. Thermally Activated Delayed Fluorescence and Room-Temperature Phosphorescence in Naphthyl Appended Carbazole–Quinoline Conjugates, and Their Mechanical Regulation. Chem. Commun. 2019, 55, 1899–1902. 10.1039/C8CC09220F. [DOI] [PubMed] [Google Scholar]
- Sagara Y.; Shizu K.; Tanaka H.; Miyazaki H.; Goushi K.; Kaji H.; Adachi C. Highly Efficient Thermally Activated Delayed Fluorescence Emitters with a Small Singlet–Triplet Energy Gap and Large Oscillator Strength. Chem. Lett. 2015, 44, 360–362. 10.1246/cl.141054. [DOI] [Google Scholar]
- Alipour M.; Karimi N. Dissecting the Accountability of Parameterized and Parameter-Free Single-Hybrid and Double-Hybrid Functionals for Photophysical Properties of TADF-Based OLEDs. J. Chem. Phys. 2017, 146, 234304. 10.1063/1.4986777. [DOI] [PubMed] [Google Scholar]
- Shizu K.; Uejima M.; Nomura H.; Sato T.; Tanaka K.; Kaji H.; Adachi C. Enhanced Electroluminescence from a Thermally Activated Delayed-Fluorescence Emitter by Suppressing Nonradiative Decay. Phys. Rev. Applied 2015, 3, 014001 10.1103/PhysRevApplied.3.014001. [DOI] [Google Scholar]
- Tanaka H.; Shizu K.; Lee J.; Adachi C. Effect of Atom Substitution in Chalcogenodiazole-Containing Thermally Activated Delayed Fluorescence Emitters on Radiationless Transition. J. Phys. Chem. C 2015, 119, 2948–2955. 10.1021/jp510751n. [DOI] [Google Scholar]
- Woo S. J.; Kim J. J. TD-DFT and Experimental Methods for Unraveling the Energy Distribution of Charge-Transfer Triplet/Singlet States of a TADF Molecule in a Frozen Matrix. J. Phys. Chem. A 2021, 125, 1234–1242. 10.1021/acs.jpca.0c11322. [DOI] [PubMed] [Google Scholar]
- Kim H. S.; Lee J. Y.; Shin S.; Jeong W.; Lee S. H.; Kim S.; Lee J.; Suh M. J.; Yoo S. Enhancement of Reverse Intersystem Crossing in Charge-Transfer Molecule through Internal Heavy Atom Effect. Adv. Funct. Mater. 2021, 31, 2104646 10.1002/adfm.202104646. [DOI] [Google Scholar]
- Fan J.; Cai L.; Lin L.; Wang C. -K. Excited State Dynamics for Hybridized Local and Charge Transfer State Fluorescent Emitters with Aggregation-Induced Emission in the Solid Phase: A QM/MM Study. Phys. Chem. Chem. Phys. 2017, 19, 29872–29879. 10.1039/C7CP05009G. [DOI] [PubMed] [Google Scholar]
- Kim D. H.; Inada K.; Zhao L.; Komino T.; Matsumoto N.; Ribierre J. C.; Adachi C. Organic Light Emitting Diodes with Horizontally Oriented Thermally Activated Delayed Fluorescence Emitters. J. Mater. Chem. C 2017, 5, 1216–1223. 10.1039/C6TC04786F. [DOI] [Google Scholar]
- Barbatti M.; Ruckenbauer M.; Plasser F.; Pittner J.; Granucci G.; Persico M.; Lischka H. Newton-X: A Surface-Hopping Program for Nonadiabatic Molecular Dynamics. WIREs. Comput. Mol. Sci. 2014, 4, 26–33. 10.1002/wcms.1158. [DOI] [Google Scholar]
- Etienne T.; Assfeld X.; Monari A. New Insight into the Topology of Excited States through Detachment/Attachment Density Matrices-Based Centroids of Charge. J. Chem. Theory Comput. 2014, 10, 3906–3914. 10.1021/ct500400s. [DOI] [PubMed] [Google Scholar]
- Hanwell M. D.; Curtis D. E.; Lonie D. C.; Vandermeersch T.; Zurek E.; Hutchison G. R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. Aust. J. Chem. 2012, 4, 1–17. 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- te Velde G.; Bickelhaupt F. M.; Baerends E. J.; Fonseca Guerra C.; van Gisbergen S. J. A.; Snijders J. G.; Ziegler T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. 10.1002/jcc.1056. [DOI] [Google Scholar]
- Zhang Q.; Tsang D.; Kuwabara H.; Hatae Y.; Li B.; Takahashi T.; Lee S. Y.; Yasuda T.; Adachi C. Nearly 100% Internal Quantum Efficiency in Undoped Electroluminescent Devices Employing Pure Organic Emitters. Adv. Mater. 2015, 27, 2096–2100. 10.1002/adma.201405474. [DOI] [PubMed] [Google Scholar]
- Tomkeviciene A.; Matulaitis T.; Guzauskas M.; Andruleviciene V.; Volyniuk D.; Grazulevicius J. V. Thianthrene and Acridan-Substituted Benzophenone or Diphenylsulfone: Effect of Triplet Harvesting via TADF and Phosphorescence on Efficiency of All-Organic OLEDS. Org. Electron. 2019, 70, 227–239. 10.1016/j.orgel.2019.04.025. [DOI] [Google Scholar]
- Chen X.; Yang Z.; Xie Z.; Zhao J.; Yang Z.; Zhang Y.; Aldred M. P.; Chi Z. An Efficient Yellow Thermally Activated Delayed Fluorescence Emitter with Universal Applications in Both Doped and Non-Doped Organic Light-Emitting Diodes. Mater. Chem. Front. 2018, 2, 1017–1023. 10.1039/C8QM00064F. [DOI] [Google Scholar]
- Lee I. H.; Song W.; Lee J. Y. Aggregation-Induced Emission Type Thermally Activated Delayed Fluorescent Materials for High Efficiency in Non-Doped Organic Light-Emitting Diodes. Org. Electron. 2016, 29, 22–26. 10.1016/j.orgel.2015.11.019. [DOI] [Google Scholar]
- Wei Q.; Kleine P.; Karpov Y.; Qiu X.; Komber H.; Sahre K.; Kiriy A.; Lygaitis R.; Lenk S.; Reineke S.; Voit B. Conjugation-Induced Thermally Activated Delayed Fluorescence (TADF): From Conventional Non-TADF Units to TADF-Active Polymers. Adv. Funct. Mater. 2017, 27, 1605051 10.1002/adfm.201605051. [DOI] [Google Scholar]
- Chen C.; Huang R.; Batsanov A. S.; Pander P.; Hsu Y.-T.; Chi Z.; Dias F. B.; Bryce M. R. Intramolecular Charge Transfer Controls Switching Between Room Temperature Phosphorescence and Thermally Activated Delayed Fluorescence. Angew. Chem. Int. Ed. 2018, 130, 16645–16649. 10.1002/ange.201809945. [DOI] [PubMed] [Google Scholar]
- Mao Z.; Yang Z.; Xu C.; Xie Z.; Jiang L.; Gu F. L.; Zhao J.; Zhang Y.; Aldred M. P.; Chi Z. Two-Photon-Excited Ultralong Organic Room Temperature Phosphorescence by Dual-Channel Triplet Harvesting. Chem. Sci. 2019, 10, 7352–7357. 10.1039/C9SC02282A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L.; Liu L.; Li X.; Hu H.; Chen M.; Yang Q.; Ma Z.; Jia X. Crystal-State Photochromism and Dual-Mode Mechanochromism of an Organic Molecule with Fluorescence, Room-Temperature Phosphorescence, and Delayed Fluorescence. Angew. Chem. Int. Ed. 2019, 131, 16597–16602. 10.1002/ange.201908567. [DOI] [PubMed] [Google Scholar]
- Huang L.; Liu J.; Liu L.; Yang Q.; Ma Z.; Jia X. A D-A-D’ Type Organic Molecule with Persistent Phosphorescence Exhibiting Dual-Mode Mechanochromism. Dyes Pigm. 2020, 173, 107963 10.1016/j.dyepig.2019.107963. [DOI] [Google Scholar]
- Wanko M.; García-Risueño P.; Rubio A. Excited States of the Green Fluorescent Protein Chromophore: Performance of Ab Initio and Semi-Empirical Methods. Phys. Status Solidi B Basic Res. 2012, 249, 392–400. 10.1002/pssb.201100536. [DOI] [Google Scholar]
- Very T.; Despax S.; Hébraud P.; Monari A.; Assfeld X. Spectral Properties of Polypyridyl Ruthenium Complexes Intercalated in DNA: Theoretical Insights into the Surrounding Effects of [Ru(Dppz)(Bpy) 2]2+. Phys. Chem. Chem. Phys. 2012, 14, 12496–12504. 10.1039/c2cp40935f. [DOI] [PubMed] [Google Scholar]
- Laguitton-Pasquier H.; Pansu R.; Chauvet J.-P.; Collet A.; Faure J.; Lapouyade R. The Charge Transfer State of Excited Bianthryl and a Derivative: Solvatochromism, Emission CT Spectra Broadening in Homogeneous Solvents. Chem. Phys. 1996, 212, 437–455. 10.1016/S0301-0104(96)00139-5. [DOI] [Google Scholar]
- Banerjee S.; Both A. K.; Sarkar M. Probing the Aggregation and Signaling Behavior of Some Twisted 9,9′-Bianthryl Derivatives: Observation of Aggregation-Induced Blue-Shifted Emission. ACS Omega 2018, 3, 15709–15724. 10.1021/acsomega.8b02232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numata M.; Yasuda T.; Adachi C. High Efficiency Pure Blue Thermally Activated Delayed Fluorescence Molecules Having 10H-Phenoxaborin and Acridan Units. Chem. Commun. 2015, 51, 9443–9446. 10.1039/C5CC00307E. [DOI] [PubMed] [Google Scholar]
- Chen X. K.; Zhang S. F.; Fan J. X.; Ren A. M. Nature of Highly Efficient Thermally Activated Delayed Fluorescence in Organic Light-Emitting Diode Emitters: Nonadiabatic Effect between Excited States. J. Phys. Chem. C 2015, 119, 9728–9733. 10.1021/acs.jpcc.5b00276. [DOI] [Google Scholar]
- Olivier Y.; Moral M.; Muccioli L.; Sancho-García J. C. Dynamic Nature of Excited States of Donor–Acceptor TADF Materials for Oleds: How Theory Can Reveal Structure–Property Relationships. J. Mater. Chem. C 2017, 5, 5718–5729. 10.1039/C6TC05075A. [DOI] [Google Scholar]
- Meng G.; Chen X.; Wang X.; Wang N.; Peng T.; Wang S. Isomeric Bright Sky-Blue TADF Emitters Based on Bisacridine Decorated DBNA: Impact of Donor Locations on Luminescent and Electroluminescent Properties. Adv. Opt. Mater. 2019, 7, 1900130 10.1002/adom.201900130. [DOI] [Google Scholar]
- Etienne T.; Gattuso H.; Michaux C.; Monari A.; Assfeld X.; Perpète E. A. Fluorene-Imidazole Dyes Excited States from First-Principles Calculations—Topological Insights. Theor. Chem. Acc. 2016, 135, 1900130 10.1002/adom.201900130. [DOI] [Google Scholar]
- Gulbinas V.; Kodis G.; Jursenas S.; Valkunas L.; Gruodis A.; Mialocq J. C.; Pommeret S.; Gustavsson T. Charge Transfer Induced Excited State Twisting of N,N-Dimethylaminobenzylidene-1,3-Indandione in Solution. J. Phys. Chem. A 1999, 103, 3969–3980. 10.1021/jp9845468. [DOI] [Google Scholar]
- Huang R.; Avó J.; Northey T.; Chaning-Pearce E.; Dos Santos P. L.; Ward J. S.; Data P.; Etherington M. K.; Fox M. A.; Penfold T. J.; Berberan-Santos M. N.; Lima J. C.; et al. The Contributions of Molecular Vibrations and Higher Triplet Levels to the Intersystem Crossing Mechanism in Metal-Free Organic Emitters. J. Mater. Chem. C 2017, 5, 6269–6280. 10.1039/C7TC01958K. [DOI] [Google Scholar]
- Huang T.; Jiang W.; Duan L. Recent Progress in Solution Processable TADF Materials for Organic Light-Emitting Diodes. J. Mater. Chem. C 2018, 6, 5577–5596. 10.1039/C8TC01139G. [DOI] [Google Scholar]
- Marian C. M. Spin-Orbit Coupling and Intersystem Crossing in Molecules. WIREs. Comput. Mol. Sci 2012, 2, 187–203. 10.1002/wcms.83. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.