

Abstract
Background and Aims
The clinical utility of two biomarkers, hepatitis B virus (HBV) RNA and hepatitis B core‐related antigen (HBcrAg), as compared to conventional markers of HBV replication and disease activity, is unclear.
Approach and Results
Untreated participants in the North American Hepatitis B Research Network Adult Cohort Study were categorized by chronic hepatitis B (CHB) phases based on HBsAg and HBeAg status and HBV DNA and alanine aminotransferase (ALT) levels. HBV RNA and HBcrAg were measured (Abbott HBV pgRNA Research Assay and Fujirebio Lumipulse Immunoassay, respectively), and cross‐sectional associations with conventional CHB markers were tested. Among 1,409 participants across all CHB phases, median HBV DNA was 3.8 log10 IU/mL and ALT was 34 U/L. HBV RNA was quantifiable in 99% of HBeAg+ and 58% of HBeAg− participants; HBcrAg was quantifiable in 20% of HBeAg+ (above linear range in the other 80%) and 51% of HBeAg− participants. Both markers differed across CHB phases (P < 0.001), with higher levels in the HBeAg+ and HBeAg− immune active phases. HBV RNA and HBcrAg correlated moderately strongly with HBV DNA in both HBeAg+ and HBeAg− phases (HBV RNA: e+ ρ = 0.84; e− ρ = 0.78; HBcrAg: e+ ρ = 0.66; e− ρ = 0.56; P for all, <0.001), but with HBsAg levels among HBeAg+ phases only (HBV RNA: e+ ρ = 0.71; P < 0.001; e− ρ = 0.18; P = 0.56; HBcrAg: e+ ρ = 0.51; P < 0.001; e− ρ = 0.27; P < 0.001). Associations of higher HBV RNA and HBcrAg levels with higher ALT, APRI, and Fibrosis‐4 levels were consistent in HBeAg−, but not HBeAg+, phases.
Conclusions
Despite clear relationships between HBV RNA and HBcrAg levels and CHB phases, these markers have limited additional value in differentiating CHB phases because of their strong association with HBV DNA and, to a lesser extent, with clinical disease indicators.
Abbreviations
- ALT
alanine aminotransferase
- APRI
AST to platelet ratio index
- BMI
body mass index
- cccDNA
covalently closed circular DNA
- CHB
chronic hepatitis B
- e−
HBeAg negative
- e+
HBeAg positive
- FIB‐4
Fibrosis‐4 marker
- HBcrAg
hepatitis B core‐related antigen
- HBRN
Hepatitis B Research Network
- IA(e−)
immune active HBeAg negative
- IA(e+)
immune active HBeAg positive
- IC(e−)
inactive carriers HBeAg negative
- IND(e−)DNA‐H
indeterminant HBV DNA high HBeAg negative
- IND(e−)DNA‐L
indeterminant HBV DNA low HBeAg negative
- IND(e+)
indeterminant HBeAg positive
- IQR
interquartile range
- IT(e+)
immune tolerant HBeAg positive
- pgRNA
pregenome RNA
- qHBeAg
quantitative HBeAg
- qHBsAg
quantitative HBsAg
- Rec(s−)
recovered HBsAg loss
- ULN
upper limit of normal
Chronic hepatitis B (CHB) is a dynamic infection with different phases reflecting the complex immune interaction between HBV and the host. Accurately identifying the phase of CHB is important for providing advice on prognosis, monitoring disease activity, and determining need for treatment.( 1 ) Typically, a combination of serological (HBsAg, HBeAg), virological (HBV virus DNA), and biochemical testing (alanine aminotransferase; ALT) are used to obtain these goals.( 1 ) However, these tests are not sufficient in discriminating the phases of CHB because many patients fall into gray zones or indeterminant phases.( 2 )
Two HBV serum biomarkers, HBV RNA and hepatitis B core‐related antigen (HBcrAg),( 3 ) provide an opportunity to better characterize CHB, beyond currently available HBV assays.( 4 , 5 , 6 , 7 ) Understanding how these biomarkers compare to other sero‐ and virological markers, as well as their utility in quantifying transcriptionally active covalently closed circular DNA (cccDNA), might yield valuable clinical insights.( 8 , 9 , 10 ) These markers might also be used to characterize the natural history of the chronic phases of infection, assess risk of disease reactivation after withdrawal of nucleos(t)ide analogs, and understand mechanisms of action of antiviral agents in development for achieving functional cure.
It is well known that HBV RNA can be detected in serum of patients with CHB. However, there is a controversy as to the source of this RNA. A recent in vitro study suggested that HBV RNA in serum represents partially reverse‐transcribed, encapsidated pregenome RNA (pgRNA) in virus‐like particles.( 11 ) Given that pgRNA is transcribed directly from cccDNA, levels of HBV RNA can potentially serve as a surrogate marker for transcriptionally active cccDNA.( 12 ) HBcrAg is a composite biomarker incorporating several viral antigens expressed from the pre‐Core/Core gene: the HBcAg, HBeAg, and p22 core‐related antigen.( 13 ) HBcrAg can be detected as a defective particle without a HBV genome, in virions containing pgRNA, circulating virus, and HBeAg. Serum HBcrAg has also been shown to correlate with cccDNA, particularly in HBeAg‐positive patients, and may reflect the amount of cccDNA in hepatocytes.( 14 )
Although several recent studies have reported on the clinical utility of these two virological biomarkers in differentiating phases of CHB, they have been limited by small sample size, omission of some phases of CHB, and inclusion of subjects with predominantly Asian or European genotypes.( 10 , 15 , 16 , 17 , 18 , 19 ) We took advantage of the Hepatitis B Research Network (HBRN) Cohort Study( 20 ) to perform a cross‐sectional analysis of levels of HBV RNA and HBcrAg across the entire spectrum of CHB phases, including genotypes A‐D, in a large North American sample of adults with active—as well as recovered—CHB, not receiving treatment. We evaluated the associations of these biomarkers with conventional biomarkers of HBV replication and disease activity among those with active CHB.
Participants and Methods
HBRN
The HBRN is a research network of 28 clinical sites throughout the USA and Canada, funded by the National Institutes of Health, initiated to study the natural history of CHB and conduct clinical trials in both children and adults. The Adult Cohort study (NCT01263587) enrolled HBsAg‐positive subjects aged ≥18 years, between 2012 and 2017, who were not currently on antiviral therapy.( 20 ) Participants underwent evaluation at entry, at weeks 12, 24, and every 24 weeks thereafter. Follow‐up ended in January 2020. The HBRN study protocols were approved by the institutional review boards (research ethics board in the case of the Toronto site) of each participating institution, and each participant provided written informed consent. Details of the study protocol were described.( 20 )
Participant Selection
Adult Cohort participants were selected for this report if they had serum available for HBV RNA and HBcrAg testing at a time point in which they tested HBsAg negative, or a time point within the first 48 weeks of study entry at which required test results necessary to categorize their phase of CHB were available. Participants’ first qualifying time point was selected. Participants without the required laboratory data or stored serum were excluded, as were participants with acute HBV, history of HCC, or coinfection with HIV, HCV, or HDV.
CHB Phase Definitions
Phase of disease was determined based on results of HBeAg status and HBV DNA level obtained from the same visit and an ALT level within 12 weeks of that study visit using predefined criteria developed by the HBRN.( 2 ) HBeAg‐positive (e+) participants were categorized as immune tolerant (IT(e+)) if HBV DNA ≥105 IU/mL and ALT were normal, immune active (IA(e+)) if HBV DNA ≥105 IU/mL and ALT were elevated, or indeterminant (IND(e+)) if HBV DNA <105 IU/mL, regardless of ALT level. HBeAg‐negative (e−) participants were categorized as immune active (IA(e−)) if HBV DNA >104 IU/mL and ALT level were elevated, inactive carriers (IC(e−)) if HBV DNA ≤104 IU/mL and ALT were normal, indeterminant HBV DNA low (IND(e−)DNA‐L) if HBV DNA ≤104 IU/mL and ALT were elevated, and indeterminant HBV DNA high (IND(e−)DNA‐H) if HBV DNA >104 IU/mL and ALT were normal. Participants with HBsAg loss during follow‐up were categorized as recovered (Rec(s−)).
Conventional Assays
Assessments during the study included detailed medical history, physical examination, health surveys, and routine blood tests, including HBV DNA level, and HBV serologies. Antibodies against HIV, HCV, and HDV were tested at enrollment. Quantitative HBeAg (qHBeAg) was tested every 24 weeks for those who were HBeAg positive at enrollment and quantitative HBsAg (qHBsAg) every 48 weeks. Local HBV serology testing was performed using commercially available ELISA assays. Standardized sex‐specific cut‐off values were chosen to define the upper limit of normal (ULN) for ALT: 30 U/L for men and 20 U/L for women, and categories were defined as ≤1.0, >1.0‐2.0, or >2.0 × ULN. Aspartate aminotransferase to platelet ratio index (APRI) and Fibrosis‐4 marker (FIB‐4) were calculated as described, and standard thresholds were applied (APRI: ≤0.5, 0.5‐2.0, or >2.0( 21 ) and FIB‐4: <1.45, 1.45‐3.25, or >3.25).( 22 )
HBV DNA, qHBeAg, and qHBsAg testing were performed at an HBRN‐funded virology laboratory (University of Washington, Seattle, WA), using research blood samples stored at −70°C.( 20 ) HBV DNA levels were determined using a real‐time PCR assay (COBAS Ampliprep/COBAS TaqMan HBV Test, v2.0; Roche Molecular Diagnostics, Branchburg, NJ), and qHBeAg and qHBsAg were determined using Roche Diagnostics’ Elecsys platform for research purposes (i.e., Elecsys HBeAg II Quant and Elecsys HBsAg II Quant assay; Roche Molecular Systems). Lower limit of quantification and detection for HBV DNA were 20 and 10 IU/mL, respectively; values below these thresholds were randomly imputed using uniform distributions (10‐<20 and 0‐<10 IU/mL, respectively). The lowest quantifiable/detectable value for qHBsAg was 0.05 IU/mL and for qHBeAg was 0.30 IU/mL; evaluation of qHBeAg and qHBsAg was limited to participants with detectable values. HBV DNA, qHBsAg, and qHBeAg are reported on the log10 scale. HBV genotype was determined based on mass spectrometry, at the Molecular Epidemiology and Bioinformatics Laboratory in the Division of Viral Hepatitis at the Centers for Disease Control and Prevention (Atlanta, GA).( 23 )
Assays
HBV RNA was isolated from plasma and amplified as described by Butler et al.,( 24 ) using the m2000 system (Abbott Molecular; Department of Infectious Diseases, Abbott Diagnostics, Abbott Park, IL), and results are presented as log10 U/mL. Levels below quantification (<1.65 log10 U/mL) were randomly imputed using a uniform distribution (0.01‐<1.65 log10 U/mL). Nondetected HBV RNA levels were set to 0 log10 U/mL. HBcrAg serum concentrations were measured using a chemiluminescence enzyme immunoassay (Lumipulse G HBcrAg assay by Fujirebio Europe, Gent, Belgium). The assay has a linear measurement range of 3.0‐6.8 log10 U/mL, with 3 log10 U/mL being the detection limit. Dilution was not performed for samples with concentrations >6.8 log10 U/mL. HBcrAg levels were categorized as <3, 3‐<4, 4‐<5, 5‐<6, 6‐<6.8, and ≥6.8 log10 U/mL.
Statistical Analysis
Demographic and clinical characteristics of participants are summarized overall and by HBV phase, as median and interquartile range (IQR; 25th‐75th percentiles) for continuous variables, and frequencies (percentage) for categorical variables. All analyses were stratified by HBeAg status and limited to HBsAg‐positive participants. Characteristics were compared across CHB phases using the Kruskal‐Wallis, chi‐square, or Fisher’s exact test, as appropriate. Box plots and stacked bar charts were used to visualize the distributions of HBV RNA (continuous) and HBcrAg (ordinal), respectively, by phases and by genotypes, and the Kruskal‐Wallis and Fisher’s exact tests were used for comparisons.
Scatter plots and box plots were used to visualize the distribution of HBV DNA, qHBeAg, and qHBsAg by HBV RNA and by HBcrAg, respectively. Associations were tested with Spearman’s rank correlation (ρ). Box plots and stacked bar charts were used to visualize the distributions of HBV RNA, HBcrAg, HBV DNA, and qHBsAg, respectively, by ALT, APRI, and FIB‐4 categories, and a series of multinomial logistic regression models were used to test the odds of higher ALT, APRI, or FIB‐4 categories, versus the lowest category, by HBV RNA, HBcrAg, HBV DNA, and qHBsAg, respectively. Modeling was repeated, adjusting for age and body mass index (BMI), with the exception that the FIB‐4 models, which were adjusted for BMI only because age is part of the FIB‐4 score. Finally, box plots were used to visualize the distribution of HBV RNA by HBcrAg categories, and the association was tested with Spearman’s rank correlation (ρ). For box plots, each box represents the first (lower end) to third (upper end) quartiles (IQR); the horizontal line in each box represents the median. The vertical line at either end of the box extends to the most extreme values or is cut off at 1.5 times the IQR. Analyses were conducted using SAS software (version 9.4; SAS Institute Inc., Cary, NC).
Results
Characteristics of the cohort
Among 2,018 adult participants, 1,409 (373 HBeAg positive, 978 HBeAg negative, and 58 who lost HBsAg in follow‐up) met inclusion criteria, 609 having been excluded (167 for acute HBV infection or coinfection with HIV, HCV, or HDV and 442 for lack of a research serum sample available at the qualifying time points; Supporting Fig. S1). Per exclusion criteria, no participants were currently taking antiviral therapy. However, 14% had previously received antiviral therapy; median (IQR) time between last use of antiviral therapy and assessment was 4.1 (1.3‐7.9) years. Median age of participants was 41 years; 49% were female, 76% Asian, 10% White, 11% Black, and 3% other/mixed race. Genotype distribution was: A, 16%; B, 40%; C, 34%; D, 7%; and other, 3%. Median HBV DNA was 3.8 log10 IU/mL, and median ALT was 34 U/L. CHB phase allocation is shown in Supporting Fig. S2, with the highest percentage (29%) in IND(e−)DNA‐L phase, roughly 20% in each of IA(e+), IA(e−), and IC(e−) phases, and 4% in Rec(s−) phase. Demographic and clinical characteristics of participants, overall and by phase, are reported in Table 1. Among HBeAg‐positive phases, there were significant differences in distributions of age, sex, ALT categories, platelets, APRI categories, and all viral makers (HBV DNA, qHBeAg, qHBsAg, HBV RNA, and HBcrAg), but not in race, treatment history, genotype, or FIB‐4 categories. Among HBeAg‐negative phases, there were significant differences in distributions of all examined factors except treatment history.
TABLE 1.
Demographics and Clinical Characteristics Among Adults With CHB, Overall and by Phase
| HBeAg+ Phases | HBeAg− Phases | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | IT(e+) | IA(e+) | IND(e+) | P Value | IA(e−) | IC(e−) | IND(e−)DNA‐L | IND(e−)DNA‐H | P Value | Rec(s−) | |
| n = 1,409 | n = 62 | n = 284 | n = 27 | n = 260 | n = 274 | n = 401 | n = 43 | n = 58 | |||
| Age, years | 0.03 | 0.051 | |||||||||
| Median, percentile (25th‐75th) | 41 (33: 51) | 30 (25: 36) | 35 (26: 44) | 32 (27: 38) | 45 (38: 53) | 43 (34: 52) | 42 (34: 52) | 43 (35: 57) | 50 (44: 59) | ||
| Female, n (%) | 696 (49.4) | 42 (67.7) | 136 (47.9) | 19 (70.4) | 0.003 | 98 (37.7) | 141 (51.5) | 220 (54.9) | 22 (51.2) | <0.001 | 18 (31.0) |
| Race, n (%) | n = 1,408 | 0.20 | n = 273 | <0.001 | |||||||
| Asian | 1,065 (75.6) | 59 (95.2) | 235 (82.7) | 22 (81.5) | 214 (82.3) | 199 (72.9) | 263 (65.6) | 39 (90.7) | 34 (58.6) | ||
| Black | 161 (11.4) | 1 (1.6) | 18 (6.3) | 1 (3.7) | 25 (9.6) | 43 (15.8) | 59 (14.7) | 2 (4.7) | 12 (20.7) | ||
| White | 143 (10.2) | 0 (0.0) | 21 (7.4) | 3 (11.1) | 19 (7.3) | 23 (8.4) | 65 (16.2) | 2 (4.7) | 10 (17.2) | ||
| Other/mixed | 39 (2.8) | 2 (3.2) | 10 (3.5) | 1 (3.7) | 2 (0.8) | 8 (2.9) | 14 (3.5) | 0 (0.0) | 2 (3.4) | ||
| Treatment history, n (%) | 202 (14.3) | 7 (11.3) | 52 (18.3) | 4 (14.8) | 0.39 | 28 (10.8) | 35 (12.8) | 57 (14.2) | 7 (16.3) | 0.55 | 12 (20.7) |
| Genotype, n (%) | n = 1,319 | n = 61 | n = 283 | n = 26 | 0.14 | n = 257 | n = 244 | n = 361 | <0.001 | n = 44 | |
| A | 213 (16.1) | 1 (1.6) | 37 (13.1) | 4 (15.4) | 28 (10.9) | 50 (20.5) | 76 (21.1) | 3 (7.0) | 14 (31.8) | ||
| B | 526 (39.9) | 20 (32.8) | 96 (33.9) | 9 (34.6) | 135 (52.5) | 97 (39.8) | 124 (34.3) | 29 (67.4) | 16 (36.4) | ||
| C | 453 (34.3) | 39 (63.9) | 133 (47.0) | 12 (46.2) | 67 (26.1) | 72 (29.5) | 113 (31.3) | 7 (16.3) | 10 (22.7) | ||
| D | 87 (6.6) | 0 (0.0) | 13 (4.6) | 1 (3.8) | 17 (6.6) | 20 (8.2) | 29 (8.0) | 3 (7.0) | 4 (9.1) | ||
| E | 35 (2.7) | 1 (1.6) | 4 (1.4) | 0 (0.0) | 8 (3.1) | 5 (2.0) | 16 (4.4) | 1 (2.3) | 0 (0.0) | ||
| Other | 5 (0.4) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (0.8) | 0 (0.0) | 3 (0.8) | 0 (0.0) | 0 (0.0) | ||
| ALT (U/mL) | n = 1,409 | n = 62 | n = 284 | n = 27 | <0.001 | n = 260 | n = 274 | n = 401 | n = 43 | <0.001 | n = 58 |
| Median, percentile (25th‐75th) | 34 (22: 53) | 18 (15: 23) | 58 (37: 105.5) | 30 (21: 42) | 53 (38: 81) | 19 (15: 23) | 36 (28: 46) | 20 (17: 24) | 22 (14: 29) | ||
| ALT (× ULN), n (%) | n = 1,409 | n = 62 | n = 284 | n = 27 | <0.001 | n = 260 | n = 274 | n = 401 | n = 43 | <0.001 | n = 58 |
| ≤1 | 431 (30.6) | 62 (100.0) | 0 (0.0) | 8 (29.6) | 0 (0.0) | 274 (100.0) | 0 (0.0) | 43 (100.0) | 44 (75.9) | ||
| >1‐2 | 596 (42.3) | 0 (0.0) | 121 (42.6) | 13 (48.1) | 130 (50.0) | 0 (0.0) | 319 (79.6) | 0 (0.0) | 13 (22.4) | ||
| >2 | 382 (27.1) | 0 (0.0) | 163 (57.4) | 6 (22.2) | 130 (50.0) | 0 (0.0) | 82 (20.4) | 0 (0.0) | 1 (1.7) | ||
| Platelets (×103/mm3) | n = 1,212 | n = 57 | n = 255 | n = 24 | 0.04 | n = 221 | n = 231 | n = 344 | n = 34 | <0.001 | n = 46 |
| Median, percentile (25th‐75th) | 218 | ||||||||||
| (180: 257) | 231 | ||||||||||
| (199: 268) | 213 | ||||||||||
| (175: 248) | 218.5 | ||||||||||
| (173: 252.5) | 205 | ||||||||||
| (169: 246) | 223 | ||||||||||
| (185: 259) | 227 | ||||||||||
| (186: 267) | 229.5 | ||||||||||
| (187: 268) | 219.5 | ||||||||||
| (176: 254) | |||||||||||
| APRI, n (%) | n = 1,208 | n = 57 | n = 254 | n = 24 | <0.001 | n = 221 | n = 231 | n = 341 | n = 34 | <0.001 | n = 46 |
| ≤0.5 | 889 (73.6) | 56 (98.2) | 122 (48.0) | 17 (70.8) | 114 (51.6) | 224 (97.0) | 278 (81.5) | 34 (100.0) | 44 (95.7) | ||
| >0.5‐1.5 | 260 (21.5) | 1 (1.8) | 103 (40.6) | 7 (29.2) | 81 (36.7) | 5 (2.2) | 61 (17.9) | 0 (0.0) | 2 (4.3) | ||
| >1.5 | 59 (4.9) | 0 (0.0) | 29 (11.4) | 0 (0.0) | 26 (11.8) | 2 (0.9) | 2 (0.6) | 0 (0.0) | 0 (0.0) | ||
| FIB‐4, n (%) | n = 1,208 | n = 57 | n = 254 | n = 24 | 0.01 | n = 221 | n = 231 | n = 341 | n = 34 | <0.001 | n = 46 |
| <1.45 | 945 (78.2) | 52 (91.2) | 191 (75.2) | 23 (95.8) | 143 (64.7) | 189 (81.8) | 284 (83.3) | 28 (82.4) | 35 (76.1) | ||
| 1.45‐3.25 | 227 (18.8) | 5 (8.8) | 48 (18.9) | 1 (4.2) | 66 (29.9) | 39 (16.9) | 51 (15.0) | 6 (17.6) | 11 (23.9) | ||
| >3.25 | 36 (3.0) | 0 (0.0) | 15 (5.9) | 0 (0.0) | 12 (5.4) | 3 (1.3) | 6 (1.8) | 0 (0.0) | 0 (0.0) | ||
| qHBeAg (log10 IU/mL) among HBeAg+ | n = 372 | n = 62 | n = 283 | n = 27 | <0.001 | NA | NA | NA | NA | NA | |
| Median, percentile (25th‐75th) | 3.1 (1.6: 3.3) | 3.3 (2.8: 3.4) | 3.1 (1.9: 3.3) | 0.1 (−0.2: 0.4) | |||||||
| qHBsAg (log10 IU/mL) among HBsAg+ | n = 1,255 | n = 61 | n = 263 | n = 26 | <0.001 | n = 248 | n = 252 | n = 367 | n = 38 | 0.002 | NA |
| Median, percentile (25th‐75th) | 3.5 (2.8: 4.2) | 4.6 (4.3: 4.9) | 4.5 (3.7: 4.9) | 3.6 (3.0: 3.9) | 3.3 (2.9: 3.7) | 3.0 (2.1: 3.6) | 3.2 (2.5: 3.9) | 3.1 (2.6: 3.7) | |||
| HBV DNA (log10 IU/mL)† | <0.001 | <0.001 | |||||||||
| Median, percentile (25th‐75th) | 3.8 (2.6: 6.2) | 8.3 (7.9: 8.5) | 8.1 (7.1: 8.4) | 4.2 (3.1: 4.7) | 5.1 (4.6: 5.9) | 2.7 (2.1: 3.3) | 2.8 (2.1: 3.3) | 4.5 (4.3: 4.9) | 0.9 (0.5: 1.2) | ||
| HBV RNA quantifiable, n (%) | 938 (66.6) | 62 (100.0) | 284 (100.0) | 23 (85.2) | <0.001 | 254 (97.7) | 103 (37.6) | 170 (42.4) | 40 (93.0) | <0.001 | 2 (3.5) |
| HBV RNA (log10 U/mL) ‡ | <0.001 | <0.001 | |||||||||
| Median, percentile (25th‐75th) | 2.4 (1.1: 5.1) | 7.3 (6.2: 7.6) | 7.1 (6.3: 7.7) | 3.1 (2.0: 3.7) | 3.6 (3.0: 4.7) | 1.3 (0.2: 1.9) | 1.4 (0.3: 2.1) | 2.9 (2.3: 3.4) | 0.0 (0.0: 0.0) | ||
| HBV DNA/RNA ratio | n = 1,245 | n = 25 ‡ | n = 221 ‡ | n = 338 ‡ | n = 12 ‡ | ||||||
| Median, percentile (25th‐75th) | 1.3 (1.1: 1.9) | 1.1 (1.1: 1.2) | 1.1 (1.1: 1.2) | 1.2 (1.1: 1.5) | <0.001 | 1.4 (1.2: 1.6) | 1.9 (1.4: 3.0) | 1.7 (1.3: 3.0) | 1.6 (1.3: 2.0) | <0.001 | 0.8 (0.3: 1.2) |
| HBcrAg quantifiable, n (%) | 575 (40.8) | 11 (17.7) | 36 (12.7) | 26 (96.3) | <0.001 | 223 (85.8) | 82 (29.9) | 159 (39.7) | 31 (72.1) | <0.001 | 7 (12.1) |
| HBcrAg (log10 U/mL), n (%) | <0.001 | <0.001 | |||||||||
| Below LLQ (<3) | 516 (36.6) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 20 (7.7) | 192 (70.1) | 242 (60.3) | 11 (25.6) | 51 (87.9) | ||
| 3‐<4 | 293 (20.8) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 91 (35.0) | 64 (23.4) | 117 (29.2) | 16 (37.2) | 5 (8.6) | ||
| 4‐<5 | 142 (10.1) | 0 (0.0) | 1 (0.4) | 4 (14.8) | 70 (26.9) | 16 (5.8) | 40 (10.0) | 9 (20.9) | 2 (3.4) | ||
| 5‐<6 | 86 (6.1) | 4 (6.5) | 6 (2.1) | 20 (74.1) | 47 (18.1) | 2 (0.7) | 2 (0.5) | 5 (11.6) | 0 (0.0) | ||
| 6‐<6.8 | 54 (3.8) | 7 (11.3) | 29 (10.2) | 2 (7.4) | 15 (5.8) | 0 (0.0) | 0 (0.0) | 1 (2.3) | 0 (0.0) | ||
| Above ULQ (≥6.8) | 318 (22.6) | 51 (82.3) | 248 (87.3) | 1 (3.7) | 17 (6.5) | 0 (0.0) | 0 (0.0) | 1 (2.3) | 0 (0.0) | ||
Imputed values used when below level of quantification (<20 IU/mL) and detection (<10 IU/mL): Uniform imputation ranged from 10.0 to 19.9 and 0.0 to 9.9, respectively.
Imputed values used when below level of quantification (<1.65 log10 U/mL): Uniform imputation ranged from 0.01 and 1.64. Nondetected HBV RNA levels were set to 0.
The ratio is missing in participants with undetectable HBV RNA.
Abbreviations: IH, immunohistochemistry; LLQ, lower limit of quantification; NA, not applicable; ULQ, upper limit of quantification.
HBV RNA and HBcrAg levels and phases of CHB
HBV RNA was quantifiable in 99% of HBeAg‐positive, 58% of HBeAg‐negative, and 4% of HBsAg‐negative participants. HBcrAg was present within the quantifiable range of the assay in 20% of HBeAg‐positive (detectable but above limit of quantification in the other 80%), 51% of HBeAg‐negative, and 12% of HBsAg‐negative participants. HBV RNA and categories of HBcrAg levels were strongly correlated independent of HBeAg status (Supporting Fig. S3; e+: ρ = 0.65; P < 0.001; e−: ρ = 0.61; P < 0.001), and both differed across HBeAg‐positive as well as HBeAg‐negative phases (Fig. 1A,B). In general, HBV RNA and HBcrAg levels were higher among HBeAg‐positive than HBeAg‐negative participants and lowest in HBsAg‐negative participants. However, levels were similar in IND(e+) and IA(e−). Among HBeAg‐positive participants, median HBV RNA levels were similar among those in IT(e+) and IA(e+) phases (7.1 and 7.3 log10 U/mL, respectively) and markedly lower among those in IND(e+) phase (3.1 log10 U/mL). Among the HBeAg‐negative participants, HBV RNA levels were highest among those in IA(e−) phase (3.6 log10 U/mL), followed by those in IND(e−)DNA‐H phase (2.9 log10U/mL), and lowest among participants in IC(e−) and IND(e‐)DNA‐L phases (1.3 and 1.4 log10 U/mL, respectively). Median HBV DNA/HBV RNA ratios were ~1 among HBeAg‐positive participants, >1 among HBeAg‐negative participants with highest ratios among IC(e−) and IND(e−)DNA‐L phases (1.9 and 1.7, respectively), and <1 among HBsAg‐negative participants (Table 1; Supporting Fig. S4). Median HBV DNA/HBV RNA ratios were similar across genotypes A‐D irrespective of HBeAg status (Supporting Fig. S5).
FIG. 1.
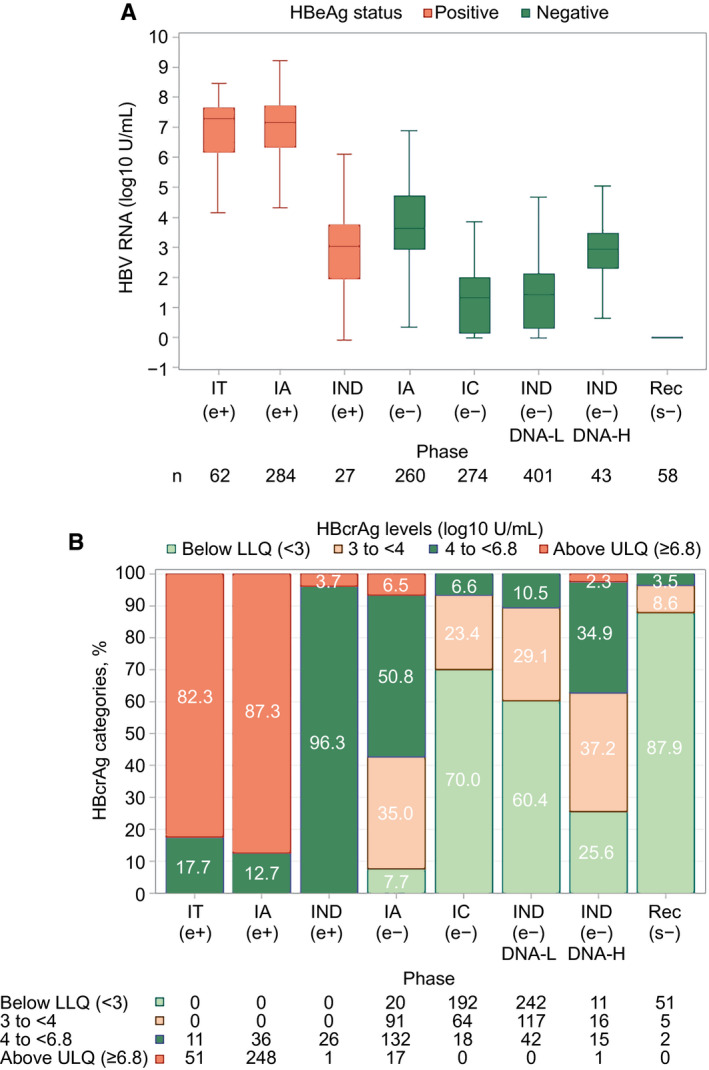
HBV RNA and HBcrAg categories by CHB phase. (A) HBV RNA by CHB phase. (B) HBcrAg categories by CHB phase. (A) HBeAg‐positive participants depicted by red boxes and HBeAg‐negative participants by blue boxes. In box‐whisker plots, upper and lower ends of the boxes = upper and lower quartiles, horizontal line = median, and upper and lower whiskers = highest and lowest observations. n = numbers of participants in each CHB phase. (B) Green bars = HBcrAg below lower limit of detection, brown bars = HBcrAg 3‐<4 U/L, blue bars = HBcrAg 4‐<6.8 U/L, and red bars = HBcrAg ≥6.8 U/L. Abbreviations: LLQ, lower limit of quantification; ULQ, upper limit of quantification.
Among the HBeAg‐positive participants, the majority of IT(e+) (82%) and IA(e+) (87%) participants had HBcrAg values above the upper limit of quantification (≥6.8 log10 U/mL) compared to only 4% of IND(e+) participants (Table 1). Among the HBeAg‐negative participants, the majority of IA(e−) (92%) participants had values above the lower limit of quantification, with most having values 3‐<6 log10 U/mL and only 7% with values above the upper limit of quantification. By contrast, the majority of those in IC(e−) (70%) and IND(e−)DNA‐L (60%) phases had values below the lower limit of quantification, and most of the remainder had values between 3 and <5 log10 U/mL (Table 1). IND(e−)DNA‐H participants had intermediate HBcrAg levels between that of IA (e−) and IC(e−)/IND(e‐)DNA‐L participants.
Associations Between HBV RNA and HBcrAg Levels and Quantitative HBV DNA, HBeAg, and HBsAg Levels
HBV RNA levels correlated strongly with HBV DNA levels independent of HBeAg status (Fig. 2A; HBeAg positive ρ = 0.84; P < 0.001; HBeAg negative ρ = 0.78; P < 0.001) and with qHBeAg (Supporting Fig. S6A; ρ = 0.55; P < 0.0001) and qHBsAg (Fig. 3A; HBeAg‐positive ρ = 0.71; P < 0.001) among HBeAg‐positive phases. Correlation between HBV RNA and qHBsAg was significant but weak among HBeAg‐negative phases (Fig. 3A; ρ = 0.18; P < 0.0001).
FIG. 2.
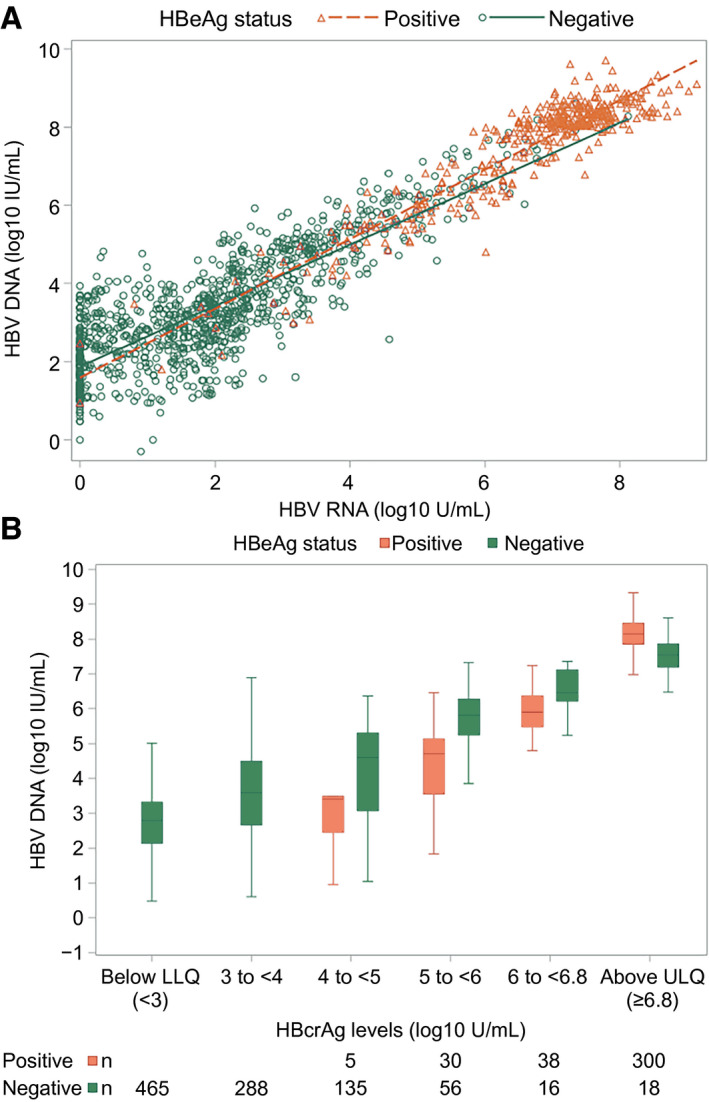
HBV DNA (log10 IU/mL) by HBV RNA (log10 U/mL) and HBcrAg (log10 U/mL) categories, respectively. (A) HBV DNA by RNA. HBV DNA by HBcrAg categories. (A) HBeAg‐positive participants represented by red triangles and HBeAg‐negative participants represented by blue circles. (B) HBeAg‐positive participants depicted by red boxes and HBeAg‐negative participants by blue boxes. In box‐whisker plots, upper and lower ends of the boxes = upper and lower quartiles, horizontal line = median, and upper and lower whiskers = highest and lowest observations. Abbreviations: LLQ, lower limit of quantification; ULQ, upper limit of quantification.
FIG. 3.
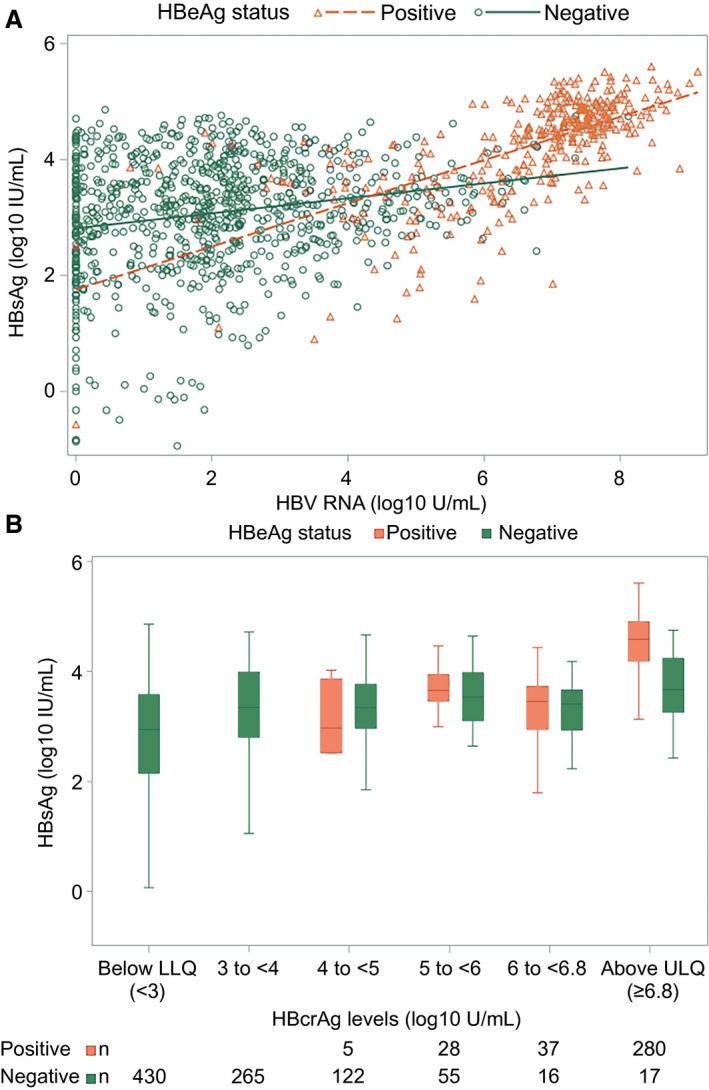
HBsAg (log10 IU/mL) by HBV RNA (log10 U/mL) and HBcrAg (log10 U/mL), respectively. (A) HBsAg by HBV RNA. HBsAg by HBcrAg categories. (A) HBeAg‐positive participants represented by red triangles and HBeAg‐negative participants represented by blue circles. (B) HBeAg‐positive participants depicted by red boxes and HBeAg‐negative participants by blue boxes. In box‐whisker plots, upper and lower ends of the boxes = upper and lower quartiles, horizontal line = median, and upper and lower whiskers = highest and lowest observations. *The lowest value of −3.0 HBsAg (log10 IU/mL) corresponds to 0.001 HBsAg (IU/mL). Abbreviations: LLQ, lower limit of quantification; ULQ, upper limit of quantification.
Similarly, HBcrAg levels correlated strongly with HBV DNA levels independent of HBeAg status (Fig. 2B; HBeAg positive ρ = 0.66; P < 0.001; HBeAg negative ρ = 0.56; P < 0.001) and with qHBeAg (Supporting Fig. S6B; ρ = 0.67; P < 0.0001) and moderately with qHBsAg among HBeAg‐positive phases (Fig. 3B; HBeAg positive ρ = 0.51; P < 0.001). Correlation between HBcrAg and qHBsAg was significant but weak among HBeAg‐negative phases (Fig. 2B; ρ = 0.27; P < 0.001).
Associations Between HBV RNA and HBcrAg Levels and Genotype
To assess whether the observed associations with virological markers (HBV DNA, qHBeAg, and qHBsAg) were influenced by genotype, we explored the HBV RNA and HBcrAg distributions by genotype. Among HBeAg‐positive participants, there were no significant differences in either HBV RNA (Supporting Fig. S7; P = 0.48) or HBcrAg (Supporting Fig. S8A; P = 0.43) levels by genotype. However, among HBeAg‐negative participants, there were significant differences in both HBV RNA and HBcrAg levels by genotype (Supporting Figs. S7 and S8B; P < 0.0001; P < 0.01, respectively), with higher median HBV RNA levels (log10 U/L) in genotype B (2.3) and lower levels in genotype D (1.5) compared to genotypes A, C, and E (all 1.9). HBcrAg levels also appeared highest in genotype B (e.g., 29.4% HBcrAg ≥4 log10 U/L), followed by C (24.7%), E (20.0%), A (15.9%), and D (14,5%). As a sensitivity analysis, among HBeAg‐negative participants, multivariable linear and ordinal logistic regression models evaluated whether genotype was related to HBV RNA and HBcrAg levels, respectively, independent of HBV DNA. Genotype was not independently related to HBV RNA (P = 0.28), but was independently related to HBcrAg (P < 0.01), with higher adjusted values in genotypes B and C versus A and D. HBV genotypes did not influence the association between HBV RNA and HBcrAg levels and other virological markers (data not shown).
Associations Between HBV RNA, HBcrAg, HBV DNA, and HBsAg Levels and Liver Disease Markers
Among HBeAg‐positive participants, there was a weak positive association between HBV RNA levels and ALT categories (P < 0.01), whereas associations with APRI (P = 0.25) and FIB‐4 (P = 0.56) categories were not significant (Fig. 4). HBcrAg levels were not associated with ALT (P = .08), APRI (P = 0.67), or FIB‐4 (P = 0.27; Supporting Fig. S9A,C,E), nor were HBV DNA levels (ALT, P = 0.06; APRI, P = 0.71; FIB‐4, P = 0.59). Finally, there was not a significant association between qHBsAg level (log10 U/mL) and ALT (P = 0.25), whereas associations with APRI (P < 0.001) and FIB‐4 (P < 0.001) were in the opposite direction, with a higher HBsAg level associated with lower odds of higher FIB‐4 categories (Supporting Fig. S11). In general, adjusting for age and BMI strengthened associations (Supporting Table S1); for example, HBV RNA was associated with higher APRI (P = 0.045), and HBcrAg and HBV DNA were associated with higher ALT (P = 0.01 and P < 0.01, respectively). However, qHBsAg was no longer inversely associated with APRI (P = 0.10).
FIG. 4.
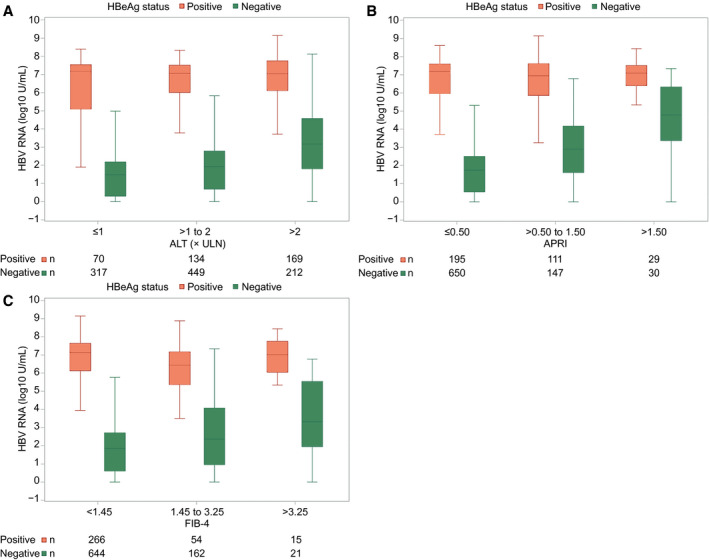
HBV RNA (log10 U/mL) by ALT (ULN), APRI, and FIB‐4 categories. (A) HBV RNA by ALT. (B) HBV RNA by APRI. HBV RNA by FIB‐4. (A,B,C) HBeAg‐positive particpants represented by red boxes and HBeAg‐negative participants by blue boxes. In box‐whisker plots, upper and lower ends of the boxes = upper and lower quartiles, horizontal line = median, and upper and lower whiskers = highest and lowest observations.
Among HBeAg‐negative participants, there were significant associations between higher HBV RNA levels (Fig. 4) and HBcrAg levels (Supporting Fig. S9B,D,F), respectively, with higher ALT, APRI, and FIB‐4 categories, respectively (P for all, <0.0001). Associations with HBV DNA level (log10 U/mL) mimicked those with HBV RNA (Supporting Fig. S10; P for all, <0.001). In contrast, the association between qHBsAg level (log10 U/mL) and ALT (P < 0.001) was weaker, not quite significant with APRI (P = 0.08), and in the opposite direction with FIB‐4 (P = .01), with a higher HBsAg level associated with a lower odds of a higher FIB‐4 category (Supporting Fig. S11). Adjusting these models for age and BMI had a negligible impact on associations (Supporting Table S2). However, the positive association between qHBsAg and APRI categories was strengthened with adjustment for age (P = 0.01).
Discussion
In this study of 1,409 North American participants with CHB, we examined whether the HBV biomarkers, HBV RNA and HBcrAg, which are claimed to be better surrogates of hepatic cccDNA transcriptional activity,( 12 , 14 ) can further discriminate CHB phase compared to conventional viral markers (HBeAg, HBV DNA, and quantitative HBsAg). We observed that although both HBV RNA and HBcrAg levels were significantly correlated with HBV DNA levels, they had little to no correlation with ALT levels in HBeAg‐positive participants. Although HBV RNA and HBcrAg levels were correlated with ALT levels in HBeAg‐negative participants, they did not provide substantial discriminating capability to separate IND(e−)DNA‐L, from the IC(e−) group given that they essentially mirrored HBV DNA levels in this cross‐sectional analysis. We postulate that many of these HBeAg‐negative indeterminant participants with low HBV DNA, yet elevated ALT, may have concomitant fatty liver disease, as reflected by higher BMI and higher prevalence of diabetes, but this will require longitudinal follow‐up to confirm. However, this large study, which includes all phases of chronic HBV infection and major HBV genotypes, provides valuable insights on our understanding of CHB.
First, similar to previous studies, we confirmed that there are strong associations between HBV RNA and HBV DNA levels among both HBeAg‐positive and ‐negative participants.( 15 , 16 , 17 , 25 ) Regardless of phase of CHB, HBV RNA levels mirrored those of HBV DNA, albeit 1‐2 logs lower, with the highest HBV RNA values observed in IT(e+) and IA(e+), intermediate in IA(e−), IND(e+), and IND(e−)DNA‐Hi and lowest in IC(e−) and IND(e−)DNA‐L. The ratio of HBV DNA/HBV RNA was ~1 among HBeAg‐positive, >1 among HBeAg‐negative, and <1 among HBsAg‐negative participants. The reason for an apparent increase in HBV DNA level over HBV RNA level among HBeAg‐negative participants is uncertain. It is possible that this represents integrated HBV DNA detected by the PCR assay. In HBeAg‐positive participants, the contribution of HBV DNA from integrated HBV DNA would likely be minimal, but may increase given that cccDNA levels decline following HBeAg loss. Alternatively, after HBeAg loss, viral transcription may be more repressed compared to replication.( 26 ) In contrast to the relationship with HBV DNA, the correlation between HBV RNA and qHBsAg levels was modest and limited to HBeAg‐positive CHB patients. The lack of correlation between HBsAg levels and HBV RNA in HBeAg‐negative patients supports the emerging concept that the source of circulating HBsAg in many HBeAg‐negative patients is integrated HBV DNA, not cccDNA.( 27 ) Inclusion of indeterminant phases is a noteworthy aspect of this study and shows that HBV RNA values add little beyond HBeAg and HBV DNA in determining assigned CHB phase.
Second, we found a small, but significant, difference in HBV RNA levels across genotypes A‐E among HBeAg‐negative, but not HBeAg‐positive, patients, with higher levels in HBV genotype B and lower levels for genotype D, compared to genotypes A, C, and E. This corroborates a previous study from Europe where patients with HBV genotype B had the highest HBV RNA levels compared to HBV genotype D, although these differences were no longer significant after adjustment for HBeAg status.( 28 ) Sequence differences between genotypes that affect the secondary structure of the pgRNA (epsilon), which binds the HBV polymerase, have been suggested as an explanation for the differential detection of HBV RNA by HBV genotype.( 25 ) However, differences in HBV RNA by genotype were no longer significant after adjustment for HBV DNA, indicating that the associations with genotype and HBV RNA may have been driven by differing HBV DNA levels (e.g., higher HBV DNA in genotype B). Conversely, genotype was associated with HBcrAg level in unadjusted and adjusted analysis (i.e., independent of HBV DNA). This finding is contrary to most other studies,( 9 , 14 , 29 ) though previous data are generally more limited. The reasons for the difference in results between HBV RNA and HBcrAg are not clear, but may be related to the strong correlation between HBV DNA and HBV RNA versus between HBV DNA and HBcrAg.
Third, we found that both HBV RNA and HBcrAg levels were consistently positively associated with liver disease markers (ALT, APRI, and FIB‐4) among HBeAg‐negative, but not HBeAg‐positive, participants, although some associations among HBeAg‐positive participants were stronger after adjustment for age and BMI. The exact reasons for these differences are unclear. As potential biomarkers of cccDNA transcription, they reflect and mirror the high viral replication rates among HBeAg‐positive participants, but do not provide information on the host immune response and therefore cannot discriminate between those with normal and elevated ALT levels. In contrast, among HBeAg‐negative patients where viral replication is more closely linked with disease activity, HBV RNA and HBcrAg levels might provide additional evidence of virally mediated liver disease. In this regard, it would be of interest to determine whether there are associations between the HBV transcriptional biomarkers and immunological correlates of disease activity, such as proinflammatory cytokines, in those with and without elevated ALT levels. Similarly, other studies have found that qHBsAg levels, which are a more indirect marker of cccDNA activity, provide additional prognostic information in HBeAg‐negative patients with low HBV DNA, but not in those with high HBV DNA levels.( 30 , 31 , 32 ) Additionally, the ability of HBV RNA and HBcrAg to differentiate HBV phases among HBeAg‐negative subjects is hampered by their limited sensitivity: 1.65 log U/mL for HBV RNA and 3.0 log U/mL for HBcrAg. Thus, in HBeAg‐positive patients with high HBV DNA levels, HBV RNA and HBcrAg levels provide more direct evidence of HBV replication. By contrast, in HBeAg‐negative patients, other markers of cccDNA activity—HBV RNA, HBcrAg, and qHBsAg—may provide additional prognostic information on HBV‐mediated liver disease.
Although the biomarkers HBV RNA and HBcrAg did not contribute additional information compared to conventional markers in classifying phases of CHB, they provide support for hypotheses that the IND(e+) group was probably on the way to spontaneous HBeAg clearance given lower HBV RNA and HBcrAg levels, whereas the IND(e−)DNA‐L group likely represent inactive carriers with concomitant fatty liver disease accounting for the elevated ALT levels, given that they not only had low HBV DNA, but also low HBV RNA, low HBcrAg levels, and similar HBV DNA/HBV RNA ratios to IC(e−). The two biomarkers also provided assurance that the HBsAg‐negative participants likely had low cccDNA activity because only 3% had quantifiable HBV RNA and only 12% had HBcrAg above the lower limit of quantification.
There were several limitations to this study. First, as a cross‐sectional analysis, we were unable to assess the role of the two markers to predict phase transitions. It is possible that changes in HBV RNA and HBcrAg levels may predate or predict phase transitions and provide better indications when antiviral treatment should be initiated or deferred. Future evaluation of longitudinal samples in our cohort will address these issues. Second, we were unable to examine the role of HBV RNA and HBcrAg in monitoring response during antiviral treatment or in predicting relapse after treatment withdrawal in this study given that all participants were required to be off treatment at study enrollment. Indeed, monitoring therapeutic responses appears to represent the major utility of HBV RNA and HBcrAg testing.( 3 , 12 ) Presently, these biomarkers should continue to be used as research tools until more studies are performed to confirm their clinical utility.
In summary, HBV RNA and HBcrAg, at a single time point, offer limited advantages over currently approved assays in characterizing the phase of chronic HBV infection, but may have a role in assessing the efficacy of antiviral agents in development. Characterization of CHB phases may be relevant in the rapidly evolving arena of HBV therapeutics, where potentially aligning subtypes of patients based on these markers with specific therapeutic approaches may be envisioned. The detailed virological and clinical characterization of CHB phases provided by this representative North American cohort study provides a solid foundation for such future studies.
Author Contributions
Study concept and design: M.G.G., W.C.K., A.S.F.L., N.T., H.L.A.J., M.K., M.L.M., R.K.S. Acquisition of data: all authors. Analysis/interpretation: M.G.G., W.C.K., A.S.F.L., N.T., H.L.A.J., M.K., M.L.M., R.K.S. Drafting: M.G.G., W.C.K., A.S.F.L., N.T., H.L.A.J., M.K., M.L.M. Critical revision: All authors. Statistical analysis: W.C.K. The Hepatitis B Research Network (HBRN): Harvard Consortium: Raymond T. Chung, M.D. (Massachusetts General Hospital, Boston, MA). Minnesota Alliance for Research in Chronic Hepatitis B Consortium: Lewis R. Roberts, M.B., Ch.B., Ph.D. (Mayo Clinic Rochester, Rochester, MN). Midwest Hepatitis B Consortium: Mauricio Lisker‐Melman, M.D. (Washington University School of Medicine, St. Louis, MO). University of Toronto Consortium: David K. Wong, M.D. (Toronto General Hospital, Toronto, Ontario, Canada); Joshua Juan, M.D. (Toronto General Hospital, Toronto, Ontario, Canada); Colina Yim, N.P., M.N. (Toronto General Hospital, Toronto, Ontario, Canada); Keyur Patel, M.D. (Toronto General Hospital, Toronto, Ontario, Canada). HBV CRN North Texas Consortium: William M. Lee, M.D. (Division of Digestive and Liver Diseases, University of Texas Southwestern Medical Center at Dallas); Robert Perrillo, M.D. (Baylor University Research Institute); Son Do, M.D. (University of Texas Southwestern Medical Center), Dallas, TX. Los Angeles Hepatitis B Consortium: Steven‐Huy B. Han, M.D. (David Geffen School of Medicine, UCLA, Los Angeles, CA); Tram T. Tran, M.D. (Cedars Sinai Medical Center, Los Angeles, CA). San Francisco Hepatitis B Research Group Consortium: Stewart L. Cooper, M.D. (Division of General and Transplant Hepatology, California Pacific Medical Center, San Francisco, CA). Michigan Hawaii Consortium: Robert J. Fontana, M.D. (University of Michigan, Ann Arbor, MI); Naoky Tsai, M.D. (The Queen’s Medical Center, University of Hawaii, Honolulu, HI); Barak Younoszai, D.O. (The Queen’s Medical Center, University of Hawaii, Honolulu, HI). Chapel Hill, NC Consortium: Andrew Muir, M.D. (Duke University Medical Center, Durham, NC); Donna Evon, Ph.D. (University of North Carolina at Chapel Hill, Chapel Hill, NC); Jama M. Darling, M.D. (University of North Carolina at Chapel Hill, NC). PNW/Alaska Clinical Center Consortium: Robert C. Carithers, M.D. (University of Washington Medical Center, Seattle, WA); Kris V. Kowdley, M.D. (Virginia Mason Medical Center, Seattle, WA); Chia C. Wang, M.D. (Virginia Mason Medical Center, Seattle, WA). Virginia Commonwealth University Medical Center: Velimir A. Luketic, M.D. (Virginia Commonwealth University Health System, Richmond, VA). Liver Diseases Branch, NIDDK: T. Jake Liang, M.D. (National Institutes of Health, Bethesda, MD). Liver Disease Research Branch, NIDDK: Jay H. Hoofnagle, M.D. (National Institutes of Health, Bethesda, MD); Edward Doo, M.D. (National Institutes of Health, Bethesda, MD). Immunology Center: Kyong‐Mi Chang, M.D. (University of Pennsylvania Perelman School of Medicine, Philadelphia, PA); Jang‐June Park, Ph.D. (University of Pennsylvania Perelman School of Medicine, Philadelphia, PA). Data Coordinating Center: Abdus Wahed, Ph.D. (Graduate School of Public Health, University of Pittsburgh, Pittsburgh, PA); Wendy C. King, Ph.D. (Graduate School of Public Health, University of Pittsburgh, Pittsburgh, PA). Central Pathology: David Kleiner, M.D., Ph.D. (Center for Cancer Research, National Cancer Institute, NIH, Bethesda, MD).
Supporting information
Supplementary Material
Acknowledgment
The authors acknowledge the use of HBRN samples and data as the sole contribution of the HBRN. The HBRN was funded as a Cooperative Agreement between the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and the following investigators: Lewis R. Roberts, M.B., Ch.B., Ph.D. (U01‐DK082843); Anna Suk‐Fong Lok, M.D. (U01‐DK082863); Steven H. Belle, Ph.D., M.Sc.Hyg. (U01‐DK082864); Kyong‐Mi Chang, M.D. (U01‐DK082866); Michael W. Fried, M.D. (U01‐DK082867); Adrian M. Di Bisceglie, M.D. (U01‐DK082871); William M. Lee, M.D. (U01‐DK082872); Harry L.A. Janssen, M.D., Ph.D. (U01‐DK082874); Daryl T.‐Y. Lau, M.D., M.P.H. (U01‐DK082919); Richard K. Sterling, M.D., M.Sc. (U01‐DK082923); Steven‐Huy B. Han, M.D. (U01‐DK082927); Robert C. Carithers, M.D. (U01‐DK082943); Mandana Khalili, M.D. (U01‐DK082944); an interagency agreement with NIDDK: Lilia M. Ganova‐Raeva, Ph.D. (A‐DK‐3002‐001); and support from the intramural program, NIDDK, NIH: Marc G. Ghany, M.D., Intramural Research Program, NIDDK, NIH. Additional funding to support this study was provided to Kyong‐Mi Chang, M.D., the Immunology Center, (NIH/NIDDK Center of Molecular Studies in Digestive and Liver Diseases P30DK50306, NIH Public Health Service Research Grant M01‐RR00040), Richard K. Sterling, M.D., M.Sc. (UL1TR000058, NCATS (National Center for Advancing Translational Sciences, NIH), Mandana Khalili, M.D., M.A.S. (CTSA Grant Number UL1TR000004), Michael W. Fried, M.D. (CTSA Grant Number UL1TR001111), and Anna Suk‐Fong Lok (CTSA Grant Numbers UL1RR024986, U54TR001959). Additional support was provided by Gilead Sciences, Inc. and Roche Molecular Systems by a CRADA through the NIDDK. The authors also acknowledge the contributions of Jeffrey Gersch and Mark Anderson, Abbott Diagnostics, who performed the HBV RNA and HBcrAg testing.
This study was funded by Abbott Diagnostics as an ancillary study of the Hepatitis B Research Network to Dr. Richard K. Sterling.
Potential conflict of interest: Dr. Chung received grants from Gilead, AbbVie, Merck, Bristol‐Myers Squibb, Boehringer Ingelheim, Roche, Janssen, and GlaxoSmithKline. Dr. King received grants from AbbVie. Dr. Lisker‐Melman is on the speakers’ bureau for AbbVie and Gilead. Dr. Lok received grants from Bristol‐Myers Squibb, Gilead, and TARGET. Dr. Terrault consults for EXIGO, Entourage, and PPD Pharma. She received grants from Gilead, GlaxoSmithKline, and Roche‐Genentech. Dr. Janssen consults for and received grants from Arbutus, Gilead, Janssen, Merck, and Roche. He consults for Aligos, Arena, Eiger, Enyo, GlaxoSmithKline, Regulus, VBI Vaccines, Vir and Viroclinics. He received grants from AbbVie and Bristol‐Myers Squibb. Dr. Khalili consults for and received grants from Gilead. She received a grant from Intercept. Dr. Lee consults for Genentech, Karuna, Forma, SeaGen, and Cortexyme. He received grants from Merck, Gilead, Intercept, Celgene, Cumberland, Eiger, and Alexion. Dr. Lau consults for and received grants from Abbott. She advises for and received grants from Gilead. Dr. Cloherty is employed by and owns stock in Abbott. Dr. Sterling received grants from Abbott, Roche, AbbVie, and Gilead. He is on the data security monitoring board for Pfizer and AskBio.
References
- 1. Terrault NA, Lok ASF, McMahon BJ, Chang KM, Hwang JP, Jonas MM, et al. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology 2018;67:1560‐1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Di Bisceglie AM, Lombardero M, Teckman J, Roberts L, Janssen HL, Belle SH, et al. Determination of hepatitis B phenotype using biochemical and serological markers. J Viral Hepat 2017;24:320‐329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Coffin CS, Zhou K, Terrault NA. New and old biomarkers for diagnosis and management of chronic hepatitis b virus infection. Gastroenterology 2019;156:355‐368.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wong DK, Tanaka Y, Lai CL, Mizokami M, Fung J, Yuen MF. Hepatitis B virus core‐related antigens as markers for monitoring chronic hepatitis B infection. J Clin Microbiol 2007;45:3942‐3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wong DH, Seto WK, Cheung KS, Chong CK, Huang FY, Fung J, et al. Hepatitis B virus core‐related antigen as a surrogate marker for covalently closed circular DNA. Liver Int 2017;37:995‐1001. [DOI] [PubMed] [Google Scholar]
- 6. Giersch K, Allweiss L, Volz T, Dandri M, Lutgehetmann M. Serum HBV pgRNA as a clinical marker for cccDNA activity. J Hepatol 2017;66:460‐462. [DOI] [PubMed] [Google Scholar]
- 7. Chen EQ, Feng S, Wang ML, Liang LB, Zhou LY, Du LY, et al. Serum hepatitis B core‐related antigen is a satisfactory surrogate marker of intrahepatic covalently closed circular DNA in chronic hepatitis B. Sci Rep 2017;7:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van Bommel F, Bartens A, Mysickova A, Hofmann J, Kruger DH, Berg T, et al. Serum hepatitis B virus RNA levels as an early predictor of hepatitis B envelope antigen seroconversion during treatment with polymerase inhibitors. Hepatology 2015;61:66‐76. [DOI] [PubMed] [Google Scholar]
- 9. Riveiro‐Barciela M, Bes M, Rodríguez‐Frías F, Tabernero D, Ruiz A, Casillas R, et al. Serum hepatitis B core‐related antigen is more accurate than hepatitis B surface antigen to identify inactive carriers, regardless of hepatitis B virus genotype. Clin Microbiol Infect 2017;23:860‐867. [DOI] [PubMed] [Google Scholar]
- 10. Maasoumy B, Wiegand SB, Jaroszewicz J, Bremer B, Lehmann P, Deterding K, et al. Hepatitis B core‐related antigen (HBcrAg) levels in the natural history of hepatitis B virus infection in a large European cohort predominantly infected with genotypes A and D. Clin Microbiol Infect 2015;21:e601‐e610. [DOI] [PubMed] [Google Scholar]
- 11. Wang J, Shen T, Huang X, Kumar GR, Chen X, Zeng Z, et al. Serum hepatitis B virus RNA is encapsidated pregenome RNA that may be associated with persistence of viral infection and rebound. J Hepatol 2016;65:700‐710. [DOI] [PubMed] [Google Scholar]
- 12. Liu S, Zhou B, Valdes JD, Sun J, Guo H. Serum hepatitis B virus RNA: a new potential biomarker for chronic hepatitis B virus infection. Hepatology 2019;69:1816‐1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mak LY, Wong DK, Cheung KS, Seto WK, Lai CL, Yuen MF. Review article: hepatitis B core‐related antigen (HBcrAg): an emerging marker for chronic hepatitis B virus infection. Aliment Pharmacol Ther 2018;47:43‐54. [DOI] [PubMed] [Google Scholar]
- 14. Testoni B, Lebossé F, Scholtes C, Berby F, Miaglia C, Subic M, et al. Serum hepatitis B core‐related antigen (HBcrAg) correlates with covalently closed circular DNA transcriptional activity in chronic hepatitis B patients. J Hepatol 2019;70:615‐625. [DOI] [PubMed] [Google Scholar]
- 15. Liu Y, Jiang M, Xue J, Yan H, Liang X. Serum HBV‐RNA quantification: useful for monitoring natural history of chronic hepatitis B infection. BMC Gastroenterol 2019;19:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang J, Yu Y, Li G, Shen C, Li J, Chen S, et al. Natural history of serum HBV‐RNA in chronic HBV infection. J Viral Hepat 2018;25:1038‐1047. [DOI] [PubMed] [Google Scholar]
- 17. Mak LY, Cloherty G, Wong DK, Gersch J, Seto WK, Fung J, et al. HBV‐RNA profiles in chronic hepatitis B patients under different disease phases and anti‐viral therapy. Hepatology 2021;73:2167‐2179. [DOI] [PubMed] [Google Scholar]
- 18. Gou Y, Zhao Y, Rao C, Feng S, Wang T, Li D, et al. Predictive value of hepatitis B core‐related antigen (HBcrAg) during the natural history of hepatitis B virus infection. Clin Lab 2017;63:1063‐1070. [DOI] [PubMed] [Google Scholar]
- 19. Seto WK, Wong DK, Fung J, Huang FY, Liu KS, Lai CL, et al. Linearized hepatitis B surface antigen and hepatitis B core‐related antigen in the natural history of chronic hepatitis B. Clin Microbiol Infect 2014;20:1173‐1180. [DOI] [PubMed] [Google Scholar]
- 20. Ghany MG, Perrillo R, Li R, Belle SH, Janssen HLA, Terrault NA, et al. Characteristics of adults in the hepatitis B research network in North America reflect their country of origin and hepatitis B virus genotype. Clin Gastroenterol Hepatol 2015;13:183‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wai CT, Greenson JK, Fontana RJ, Kalbfleisch JD, Marrero JA, Conjeevaram HS, et al. A simple noninvasive index can predict both significant fibrosis and cirrhosis in patients with chronic hepatitis C. Hepatology 2003;38:518‐526. [DOI] [PubMed] [Google Scholar]
- 22. Sterling RK, Lissen E, Clumeck N, Sola R, Correa MC, Montaner J, et al. Development of a simple noninvasive index to predict significant fibrosis in patients with HIV/HCV coinfection. Hepatology 2006;43:1317‐1325. [DOI] [PubMed] [Google Scholar]
- 23. Ganova‐Raeva L, Ramachandran S, Honisch C, Forbi JC, Zhai X, Khudyakov Y. Robust hepatitis B virus genotyping by mass spectrometry. J Clin Microbiol 2010;48:4161‐4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Butler EK, Gersch J, McNamara A, Luk KC, Holzmayer V, de Medina M, et al. Hepatitis B virus serum DNA and RNA levels in nucleos(t)ide analog‐treated or untreated patients during chronic and acute infection. Hepatology 2018;68:2106‐2117. [DOI] [PubMed] [Google Scholar]
- 25. Prakash K, Rydell GE, Larsson SB, Andersson M, Norkrans G, Norder H, et al. High serum levels of pregenomic RNA reflect frequently failing reverse transcription in hepatitis B virus particles. Virol J 2018;15:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Suslov A, Meier MA, Ketterer S, Wang X, Wieland S, Heim MH. Transition to HBeAg‐negative chronic hepatitis B virus infection is associated with reduced cccDNA transcriptional activity. J Hepatol 2021;74:794‐800. [DOI] [PubMed] [Google Scholar]
- 27. Wooddell CI, Yuen MF, Chan HY, Gish RG, Locarnini SA, Chavez D, et al. RNAi‐based treatment of chronically infected patients and chimpanzees reveals that integrated hepatitis B virus DNA is a source of HBsAg. Sci Transl Med 2017;9:eaan0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. van Campenhout MJH, van Bömmel F, Pfefferkorn M, Fischer J, Deichsel D, Boonstra A, et al. Host and viral factors associated with serum hepatitis B virus RNA levels among patients in need for treatment. Hepatology 2018;68:839‐847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chang XJ, Sun C, Chen Y, Li XD, Yu ZJ, Dong Z, et al. On‐treatment monitoring of liver fibrosis with serum hepatitis B core‐related antigen in chronic hepatitis B. World J Gastroenterol 2019;25:4764‐4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cornberg M, Wong VW, Locarnini S, Brunetto M, Janssen HLA, Chan HL. The role of quantitative hepatitis B surface antigen revisited. J Hepatol 2017;66:398‐411. [DOI] [PubMed] [Google Scholar]
- 31. Brouwer WP, Chan HY, Brunetto MR, Martinot‐Peignoux M, Arends P, Cornberg M, et al.; Good Practice in using HBsAg in Chronic Hepatitis B Study Group (GPs‐CHB Study Group) . Repeated measurements of hepatitis B surface antigen identify carriers of inactive HBV during long‐term follow‐up. Clin Gastroenterol Hepatol 2016;14:1481‐1489.e5. [DOI] [PubMed] [Google Scholar]
- 32. Brunetto MR, Oliveri F, Colombatto P, Moriconi F, Ciccorossi P, Coco B, et al. Hepatitis B surface antigen serum levels help to distinguish active from inactive hepatitis B virus genotype D carriers. Gastroenterology 2010;139:483‐490. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material