

Abstract
‘Genome-first’ approaches to analyzing rare variants can reveal new insights into human biology and disease. Because pathogenic variants are often rare, new discovery requires aggregating rare coding variants into ‘gene burdens’ for sufficient power. However, a major challenge is deciding which variants to include in gene burden tests. Pathogenic variants in MYBPC3 and MYH7 are well-known causes of hypertrophic cardiomyopathy (HCM), and focusing on these ‘positive control’ genes in a genome-first approach could help inform variant selection methods and gene burdening strategies for other genes and diseases. Integrating exome sequences with electronic health records among 41 759 participants in the Penn Medicine BioBank, we evaluated the performance of aggregating predicted loss-of-function (pLOF) and/or predicted deleterious missense (pDM) variants in MYBPC3 and MYH7 for gene burden phenome-wide association studies (PheWAS). The approach to grouping rare variants for these two genes produced very different results: pLOFs but not pDM variants in MYBPC3 were strongly associated with HCM, whereas the opposite was true for MYH7. Detailed review of clinical charts revealed that only 38.5% of patients with HCM diagnoses carrying an HCM-associated variant in MYBPC3 or MYH7 had a clinical genetic test result. Additionally, 26.7% of MYBPC3 pLOF carriers without HCM diagnoses had clear evidence of left atrial enlargement and/or septal/LV hypertrophy on echocardiography. Our study shows the importance of evaluating both pLOF and pDM variants for gene burden testing in future studies to uncover novel gene-disease relationships and identify new pathogenic loss-of-function variants across the human genome through genome-first analyses of healthcare-based populations.
Introduction
‘Genome-first’ approaches, in which genetic variants of interest are first identified and then analyzed for association with phenotypes, can be used to inform the genetic basis of human disease and reveal new insights into gene function and human biology (1). Particularly when applied to medical biobanks consisting of healthcare populations with DNA sequencing data linked to extensive electronic health record (EHR) phenotype data, genome-first approaches allow for agnostic phenome-wide association studies (PheWAS) to determine the clinical impact of specific genetic variants (2,3). With the increasing use of large-scale whole-exome sequencing (WES) and identification of many rare coding variants predicted to impact protein structure or function, studies are increasingly interrogating the cumulative effect of multiple rare variants in a gene (i.e. ‘gene burden’) to increase the statistical power of regression analyses and to enable gene-based association studies to describe the implications of mutated genes in human disease (4).
We have previously shown that gene burden PheWAS applied to large healthcare populations has the potential to uncover novel consequences of rare coding variants in the human disease phenome (5). One approach to gene burden PheWAS is to focus only on predicted loss-of-function (pLOF) variants, but this could lead to lack of power owing to their infrequency. Additional coding variation could be added to substantially increase the number of effect alleles, but a major challenge is deciding which variants to include in gene burden tests of association. Furthermore, there are many in silico algorithms that can predict the probability that a variant may have a deleterious effect on its gene product, as well as various filters that can be applied for variant selection based on variant type and frequency. However, very little large-scale functional data are available that can be used to annotate missense variants.
Application of the unbiased genome-first approach to ‘positive control’ genes with known phenotype associations represents a valuable system for comparison of variant selection methods and gene burdening strategies, as we previously showed for the gene LMNA and its association with dilated cardiomyopathy (6). Pathogenic variants in MYBPC3 and MYH7 are known to cause hypertrophic cardiomyopathy (HCM) and together account for up to 50% of all clinically recognized HCM cases and at least 75% of HCM cases for which a pathogenic variant is identified (7). We leveraged the Penn Medicine BioBank (PMBB, University of Pennsylvania), a large academic medical biobank with WES linked to EHR data, to evaluate in detail the performance of methods for aggregating pLOF and/or annotated predicted deleterious missense (pDM) variants in MYBPC3 and MYH7 for gene burden association studies. Additionally, we followed up on our genome-first approach with review of EHR charts to describe the clinical characteristics of variant carriers that were identified through this gene burden study.
Results
pLOF variants in MYBPC3 were strongly associated with HCM
Among 41 759 unrelated individuals with WES in PMBB (Table 1), we identified 45 individuals carrying one of 33 pLOF variants in MYBPC3, including 13 frameshift insertions/deletions, 9 gain of stop codon and 11 splicing variants disrupting canonical splice site dinucleotides (Fig. 1A, Supplementary Material, Table S1). PheWAS of the gene burden of pLOF variants in MYBPC3 showed phenome-wide significant associations with HCM and related cardiac phenotypes, such as cardiac conduction disorders, heart failure, heart transplant/surgery and use of cardiac defibrillator (Supplementary Material, Fig. S1, Table 2). Fifteen of the 45 individuals with a rare pLOF had a clinical diagnosis of HCM (Phecodes ‘Hypertrophic obstructive cardiomyopathy’ or ‘Other hypertrophic cardiomyopathy’) (Supplementary Material, Table S1). Twelve of 33 pLOFs were annotated in ClinVar as pathogenic or likely pathogenic (P/LP), and of the 18 carriers of these P/LP variants, 6 had a clinical diagnosis of HCM. Of the 21 pLOF variants without a P/LP classification in ClinVar, 9 were carried by a total of 9 individuals with diagnoses of HCM (Supplementary Material, Table S1).
Table 1.
PMBB whole exome-sequenced cohort characteristics
| Basic demographics | |
|---|---|
| Total population, N | 41 759 |
| Female, N (%) | 20 731 (49.6) |
| Median age, years | 63 |
| Birth year range | 1911–2003 |
| Genetically informed ancestry | |
| AFR, N (%) | 10 217 (24.5) |
| AMR, N (%) | 572 (1.4) |
| EAS, N (%) | 672 (1.6) |
| EUR, N (%) | 29 362 (70.3) |
| AS, N (%) | 564 (1.4) |
| Phecodes | |
| Primary/intrinsic cardiomyopathies, N (%) | 2912 (7.6) |
| Hypertrophic obstructive cardiomyopathy, N (%) | 199 (0.6) |
| Other HCM, N (%) | 184 (0.5) |
| Cardiac dysrhythmias, N (%) | 12 130 (35.5) |
| Atrial fibrillation, N (%) | 5885 (21.0) |
| Atrial flutter, N (%) | 2324 (9.5) |
| Paroxysmal ventricular tachycardia, N (%) | 1960 (8.2) |
| Ventricular fibrillation and flutter, N (%) | 467 (2.1) |
| Cardiac conduction disorders, N (%) | 5688 (20.5) |
| Cardiac pacemaker/device in situ, N (%) | 3177 (12.6) |
| Cardiac defibrillator in situ, N (%) | 1945 (8.1) |
| Congestive heart failure; non-hypertensive, N (%) | 6224 (16.9) |
| Heart transplant/surgery, N (%) | 792 (2.5) |
| Cardiac shunt/heart septal defect, N (%) | 547 (1.4) |
| Cardiogenic shock, N (%) | 188 (0.5) |
| Muscular wasting and disuse atrophy, N (%) | 52 (0.1) |
Note: Basic demographic characteristics and representative Phecodes identified by gene burden PheWAS for MYBPC3 and MYH7. Each characteristic is labeled with count data and percent prevalence where appropriate. Individuals were determined to be a case for a Phecode if they had the corresponding ICD diagnosis on two or more dates, while controls consisted of individuals who never had the ICD code. Individuals with an ICD diagnosis on only one date as well as those under control exclusion criteria based on Phecode mapping protocols were not considered. AFR, African; AMR, Mixed American; EAS, East Asian; EUR, European; SAS, South Asian.
Figure 1.
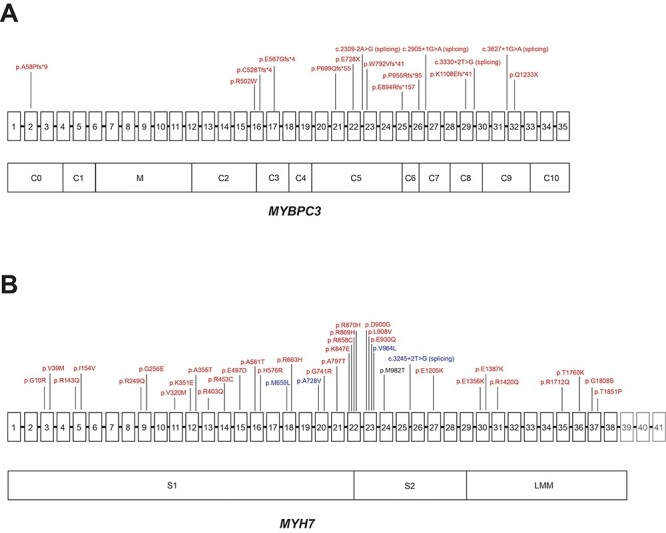
Distribution of disease-associated variants in MYH7 and MYBPC3. (A) Schematic of MYBPC3 gene with exons 1–35 (exons not to scale) above and domains below, with variants labeled in red denoting amino acid change (or location of splice variant) for pLOF or adjudicated missense variants that were associated with HCM in PMBB. (B) Schematic of MYH7 gene with exons 1–41 (exons not to scale) above and domains below. Variants are labeled in red denoting amino acid change for pDM variants with REVEL ≥ 0.5, which were associated with HCM in PMBB, and variants are labeled in blue denoting amino acid change (or location of splice variant) for pLOF or pDM variants with REVEL ≥ 0.5, which were associated with ‘Muscular wasting and disuse atrophy’ in PMBB. Note: exons 39–41, which do not encode for protein, are grayed out. Note 2: p.M982T in MYH7 was associated with both HCM and muscular wasting and disuse atrophy, but via different individuals, and is thus labeled in black.
Table 2.
Association of MYBPC3 and MYH7 gene burdens with HCM in PMBB
| Gene burden | P | Beta | SE | OR | MYBPC3 Carrier N | HCM N |
|---|---|---|---|---|---|---|
| ClinVar P/LP (pLOF only) | 4.70E-10 | 6.792 | 1.09 | 890.907 | 18 | 6 |
| ClinVar P/LP (missense only) | 3.43E-04 | 2.766 | 0.773 | 15.891 | 27 | 2 |
| ClinVar P/LP (pLOF + missense) | 3.15E-20 | 4.177 | 0.453 | 65.161 | 45 | 8 |
| pLOF only | 1.52E-35 | 5.112 | 0.411 | 166.008 | 45 | 15 |
| pDM (REVEL ≥ 0.6) only | 4.05E-03 | 0.908 | 0.316 | 2.48 | 628 | 10 |
| pDM (4/4 algorithms) only | 1.43E-01 | 0.514 | 0.351 | 1.671 | 859 | 8 |
| pLOF + pDM (REVEL ≥ 0.6) | 7.99E-20 | 1.949 | 0.214 | 7.023 | 673 | 25 |
| pLOF + adjudicated missense (SHaRe) | 1.03E-42 | 4.402 | 0.321 | 81.638 | 67 | 17 |
| Gene burden | P | Beta | SE | OR | MYH7 Carrier N | HCM N |
| ClinVar P/LP (pLOF only) | 9.78E-01 | −7.803 | 277.662 | 4.08E-04 | 2 | 0 |
| ClinVar P/LP (missense only) | 1.25E-55 | 4.767 | 0.303 | 117.543 | 75 | 22 |
| ClinVar P/LP (pLOF + missense) | 1.29E-55 | 4.737 | 0.301 | 114.039 | 77 | 22 |
| pLOF only | 9.73E-01 | −10.658 | 315.401 | 2.35E-05 | 27 | 0 |
| pDM (REVEL ≥ 0.5) only | 2.01E-29 | 2.059 | 0.183 | 7.839 | 858 | 37 |
| pDM (4/4 algorithms) only | 2.99E-19 | 2.146 | 0.239 | 8.553 | 442 | 22 |
| pLOF + pDM (REVEL ≥ 0.5) | 9.23E-29 | 2.033 | 0.183 | 7.635 | 885 | 37 |
Note: Summary statistics for gene burden associations with HCM (combining Phecodes ‘Hypertrophic obstructive cardiomyopathy’ and ‘Other hypertrophic cardiomyopathy’) in PMBB by using ClinVar P/LP, pLOF only, missense only (pDM and/or adjudicated) and pLOF + missense variants in MYBPC3 (top) and MYH7 (bottom). Each gene burden association is reported as beta, standard error (SE), odds ratio (OR), P-value, the number of carriers for variants included in the gene burden and the number of carriers having the HCM phenotype.
Review of clinical charts of the 15 pLOF carriers with a clinical diagnosis of HCM revealed that only 5 had a clinical genetic test report, all of which were concordant with the WES pLOF results. While all 5 individuals with clinical genetic testing had a family history of HCM, only 3 of 10 untested individuals had a family history of HCM noted in their charts. Chart review of the 30 MYBPC3 pLOF carriers without a clinical HCM diagnosis revealed that 17 had received at least one transthoracic echocardiogram, and 8 of these individuals had left atrial (LA) enlargement and/or hypertrophy of the septum or another segment within the left ventricle (LV). Additionally, 10 of 30 pLOF carriers without a diagnosis of HCM had a history of atrial fibrillation.
pDM variants in MYBPC3 were not associated with HCM
Based on ‘phenotype-first’ presentations of HCM, most pathogenic variants in MYBPC3 are pLOF variants (8). ClinVar shows that 482 frameshift, nonsense or splicing variants in MYBPC3 are classified as P/LP, whereas only 54 missense variants are annotated as P/LP (9). In PMBB, the gene burden of all non-synonymous coding variants in MYBPC3 classified by ClinVar as P/LP (N = 45 heterozygous carriers) was strongly associated with HCM as expected (Supplementary Material, Fig. S2, Table 2). However, while a gene burden of just the 12 pLOF variants classified as P/LP (N = 18 heterozygous carriers) also showed phenome-wide significant associations with HCM Phecodes (Supplementary Material, Fig. S3A), a gene burden of the 17 missense variants classified as P/LP (N = 27 heterozygous carriers) showed a much weaker association with HCM that was not phenome-wide significant (P = 9.50E-05) (Supplementary Material, Fig. S3B). We previously have shown that prediction of deleteriousness for missense variants using the ensemble tool REVEL (10) is highly correlated with clinical annotations for missense variants in LMNA (6). We similarly applied REVEL to missense variants in MYBPC3 and found through an analysis of variance on ClinVar-annotated variants that REVEL scores showed essentially no correlation with annotations of clinical pathogenicity for MYBPC3 (Supplementary Material, Table S2). We experimented with REVEL score thresholds in bins of 0.05 to evaluate the optimal score cutoff for capturing the most robust association of missense variants in MYBPC3 associated with HCM. We found that all REVEL cutoff thresholds showed very weak association with HCM, although a REVEL threshold score of 0.6 was relatively optimal (Supplementary Material, Fig. S4, Table 2). Additionally, application of the aggregation of pDM variants chosen by a consensus of algorithms (SIFT, PolyPhen2 HumDiv, PolyPhen2 HumVar and MutationTaster)—one of the standard approaches for predicting the deleteriousness of missense variants—also showed only weak association with HCM (Supplementary Material, Fig. S4, Table 2).
Given the weak or insignificant association of missense variants in MYBPC3 with HCM based on algorithms predicting deleteriousness of missense variants alone, we overlapped a list of 19 expert-adjudicated pathogenic missense variants in MYBPC3 from the SHaRe Database to identify high-confidence pathogenic missense variants in PMBB (11). We identified 6 of these high-confidence pathogenic missense variants in MYBPC3 carried by a total of 22 individuals among the PMBB WES cohort (Supplementary Material, Table S3). A gene burden including just these six missense variants showed a similar degree of association with HCM compared with a gene burden of ClinVar P/LP missense variants and was more strongly associated with HCM compared with a burden of pDM variants based on all REVEL score thresholds as well as 4/4 algorithms (Supplementary Material, Fig. S4, Table 2). Two of the 22 carriers were diagnosed with HCM, and chart review of the 20 carriers without an HCM diagnosis revealed 5 individuals with mild concentric hypertrophy or dilated atria noted on transthoracic echocardiography.
Including pDMs together with pLOFs in a gene burden has the potential to increase power for finding phenotype associations. When we combined pLOFs with pDMs having REVEL ≥ 0.6 (at which pDMs in MYBPC3 had the relatively lowest P-value for association with HCM), we noted no improvement in the association of the gene burden with HCM (Table 2). Only when the adjudicated pathogenic missense variants were included with pLOFs did we see a modestly stronger association with HCM compared with pLOFs alone (Fig. 2, Table 2). We found that individuals with a clinical diagnosis of HCM who carry one of these HCM-associated pLOF or adjudicated pathogenic missense variants in MYBPC3 had a median [interquartile range (IQR)] age at HCM diagnosis of 50 (32, 56). By contrast, individuals without a clinical diagnosis of HCM who carry one of these pLOF or pathogenic missense variants in MYBPC3 had a median (IQR) age of 58.5 (36.75, 71.25). We further interrogated this MYBPC3 gene burden combining pLOFs and adjudicated pathogenic missense variants by analyzing available echocardiography data in PMBB regardless of HCM diagnoses. Carriers of pLOF and adjudicated pathogenic missense variants in MYBPC3 on average had increased left ventricular posterior wall (LVPW) diastolic thickness, interventricular septum (IVS) diastolic thickness, LA volume index and left ventricular outflow tract (LVOT) peak gradient (Table 3) compared with non-carriers, although not always at clinically relevant thresholds.
Figure 2.
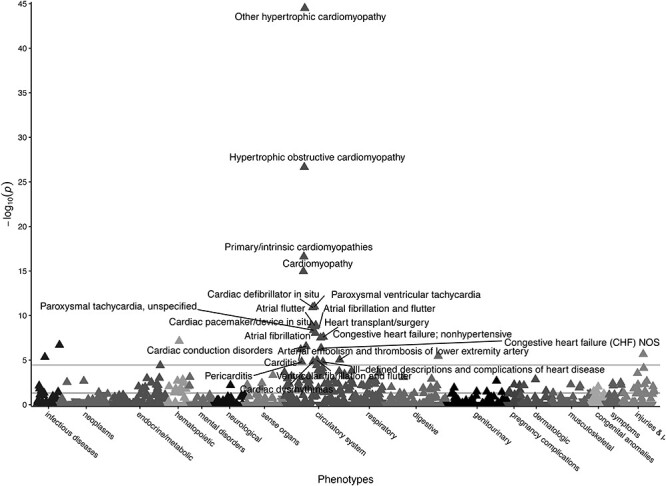
Gene burden PheWAS of pLOF and adjudicated pathogenic missense variants in MYBPC3. Gene burden PheWAS of pLOF variants (N = 45, Supplementary Material, Table S1) and adjudicated pathogenic missense variants from SHaRE (N = 22, Supplementary Material, Table S3) in MYBPC3. Phecodes are plotted along the x-axis to represent the phenome, and the association of the gene burden with each Phecode is plotted along the y-axis representing –log10(P-value). The directionality of each triangular point represents the direction of effect. The top horizontal line represents the Bonferroni-corrected significance threshold to adjust for multiple testing (P = 3.58E-05), and the bottom horizontal line represents a nominal significance threshold (P = 0.05).
Table 3.
Echocardiographic analyses for MYBPC3 and MYH7 in PMBB
| Echo parameter | MYBPC3 median (IQR) | Control median (IQR) | Beta | SE | P | MYBPC3 N | Control N |
|---|---|---|---|---|---|---|---|
| LVPW diastolic thickness, max (cm) | 1.320 (1.160, 1.525) | 1.100 (0.951, 1.284) | 0.257 | 0.043 | 2.86E-09 | 31 | 11 928 |
| IVS diastolic thickness, max (cm) | 1.510 (1.203, 1.871) | 1.150 (0.989, 1.340) | 0.414 | 0.050 | 7.68E-17 | 30 | 11 854 |
| LA volume index, max (ml/m2) | 48.593 (40.224, 52.374) | 33.725 (24.929, 44.948) | 12.567 | 3.744 | 7.89E-04 | 22 | 8128 |
| LVOT peak gradient, max (mmHg) | 6.554 (4.162, 9.000) | 4.410 (3.254, 6.052) | 2.605 | 0.500 | 1.91E-07 | 29 | 8807 |
| Echo parameter | MYH7 median (IQR) | Control median (IQR) | Beta | SE | P | MYH7 N | Control N |
| LVPW diastolic thickness, max (cm) | 1.130 (0.934, 1.285) | 1.100 (0.951, 1.284) | 0.045 | 0.014 | 1.63E-03 | 303 | 11 928 |
| IVS diastolic thickness, max (cm) | 1.190 (1.001, 1.390) | 1.150 (0.989, 1.340) | 0.071 | 0.016 | 1.09E-05 | 300 | 11 854 |
| LA volume index, max (ml/m2) | 36.796 (27.763, 50.182) | 33.725 (24.929, 44.948) | 5.235 | 1.242 | 2.51E-05 | 214 | 8128 |
| LVOT peak gradient, max (mmHg) | 4.801 (3.467, 6.554) | 4.410 (3.254, 6.052) | 0.458 | 0.187 | 1.41E-02 | 222 | 8807 |
Note: Top: comparison of echocardiographic parameters representative of HCM with gene burden of pLOF variants and adjudicated pathogenic missense variants from SHaRE in MYBPC3 versus non-carriers. Carriers of pLOF variants and pDM variants with REVEL ≥ 0.5 in MYH7 were removed from this analysis. Median and IQR ranges for each echo parameter are listed for each test group, and association results are listed as beta, SE and P-value. The number of individuals with each echo parameter available is also listed per test group. The median (IQR) age for the MYBPC3 carrier group was 64 (60, 72) (N = 31). Bottom: comparison of echocardiographic parameters representative of HCM with gene burden of pLOF variants and pDM variants with REVEL ≥ 0.5 in MYH7 versus non-carriers. Carriers of pLOF variants and adjudicated pathogenic missense variants from SHaRE in MYBPC3 were removed from this analysis. Median and IQR ranges for each echo parameter are listed for each test group, and association results are listed as beta, SE and P-value. The number of individuals with each echo parameter available is also listed per test group. The median (IQR) age for the MYH7 carrier group was 67 (54, 76) (N = 303).
pLOF variants in MYH7 were not significantly associated with HCM but had suggestive associations with other cardiac phenotypes
We identified 27 individuals carrying one of 23 pLOF variants in MYH7, including 5 frameshift insertion/deletions, 13 gain of stop codon and 5 splicing variants (Supplementary Material, Table S4). Of note, none of these pLOF variants were in the last exon of MYH7. In contrast to MYBPC3, PheWAS of the MYH7 pLOF only gene burden showed no association with HCM (P = 0.973) (Supplementary Material, Fig. S5, Table 2). We confirmed through chart review that none of the 27 pLOF carriers had an HCM diagnosis (Supplementary Material, Table S4). Interestingly, however, this pLOF gene burden had weak evidence for association with ‘Cardiac shunt/heart septal defect’ (P = 1.12E-04, N = 3), suggesting the possibility of a cardiac phenotype among heterozygous carriers for pLOF variants in MYH7. Detailed review of the clinical charts of the 27 MYH7 pLOF carriers confirmed a history of patent foramen ovale or atrial septal defect in the 3 pLOF carriers with ‘Cardiac shunt/heart septal defect’ diagnoses and also revealed that 13 of 27 had received echocardiograms of whom 5 individuals had mild concentric left ventricular hypertrophy.
pDM variants in MYH7 were strongly associated with HCM
Most known pathogenic variants in MYH7 are missense (12), as exemplified by the ClinVar database which has classified 272 missense variants in MYH7 as P/LP, whereas there are only 20 frameshift, nonsense or splicing variants annotated as P/LP (9). In PMBB, we found a total of 43 missense variants annotated by ClinVar as P/LP (N = 75 heterozygous carriers) as well as just two pLOFs annotated as P/LP (N = 2 carriers). As expected, a gene burden comprised only of these ClinVar P/LP non-synonymous variants had a very strong association with HCM (Supplementary Material, Fig. S6, Table 2). Of the 75 carriers with ClinVar P/LP missense variants, 22 had a diagnosis of HCM, while neither of the two carriers for pLOFs annotated in ClinVar as P/LP had an HCM diagnosis.
We then asked how a computational approach to selecting MYH7 missense variants would perform and explored in detail the association of pDM variants in MYH7 with HCM and other phenotypes. We applied REVEL to missense variants in MYH7 and found through an analysis of variance on ClinVar-annotated variants that REVEL scores were highly correlated with annotations of clinical pathogenicity for MYH7 (Supplementary Material, Table S5). We then experimented with REVEL score thresholds in bins of 0.05 to evaluate the optimal score cutoff for capturing the most robust association of missense variants in MYH7 with HCM, with the goal of potentially capturing deleterious missense variants in MYH7 that lack a pathogenic ClinVar classification (i.e. no classification available or having a non-P/LP classification). We found that a REVEL cutoff score of 0.5 had the optimal P-value for association of MYH7 pDM variants with HCM (Supplementary Material, Fig. S7, Table 2). Of note, this REVEL-based association was more strongly associated with HCM compared with the aggregation of pDM variants predicted deleterious by a consensus of algorithms (SIFT, PolyPhen2 HumDiv, PolyPhen2 HumVar and MutationTaster), even while identifying more variants (303 variants with REVEL ≥ 0.5 vs. 166 variants passing consensus of algorithms) (Supplementary Material, Fig. S7, Table 2). The HCM-prevalent missense variants were concentrated among the globular S1 head and coiled-coil S2 domains of MYH7 (Fig. 1B, Supplementary Material, Table S6). Among 858 total carriers for pDM variants in MYH7 with REVEL ≥ 0.5, 37 heterozygous carriers for 33 different pDM variants had a diagnosis of HCM (Supplementary Material, Table S6). Fifteen MYH7 pDM variants with REVEL ≥ 0.5 lacking a pathogenic ClinVar classification were carried by individuals with a diagnosis of HCM (Supplementary Material, Table S6). Chart review of these 37 carriers confirmed the diagnosis of HCM and indicated that only 15 of the 37 carriers had clinical genetic testing for HCM in their chart (all were concordant with the WES MYH7 variant). While 13 of 15 individuals with a clinical genetic test in the chart noted a family history of HCM, only half of untested individuals had a documented family history. Individuals with a clinical diagnosis of HCM who carry one of these HCM-associated pDM variants in MYH7 had a median (IQR) age at HCM diagnosis of 42 (27, 54.5). By contrast, individuals without a clinical diagnosis of HCM who carry pDM variants in MYH7 with REVEL ≥ 0.5 had a median (IQR) age of 62 (46, 72).
Gene burden of pLOF and pDM variants in MYH7 was also associated with a skeletal muscle phenotype
In order to increase power for finding associations, we aggregated the 23 pLOF variants in MYH7 (N = 27 carriers) with the 303 pDM variants with REVEL ≥ 0.5 in MYH7 (N = 858 carriers) into a gene burden. PheWAS of this combined MYH7 gene burden showed, as expected, phenome-wide significant associations with HCM (Fig. 3, Table 2), but the strength of this association was not greater than with a pDM gene burden alone. This expanded gene burden was also significantly associated with related cardiac phenotypes ‘Heart transplant/surgery’ and ‘Cardiac defibrillator in situ’ (Fig. 3). We also linked the expanded gene burden with available echocardiography data in PMBB (including both those with and without HCM diagnoses) and found that carriers of pLOF and pDM variants in MYH7 on average had increased LVPW diastolic thickness, IVS diastolic thickness, LA volume index and LVOT peak gradient (Table 3) compared with non-carriers, although not always at clinically relevant thresholds.
Figure 3.
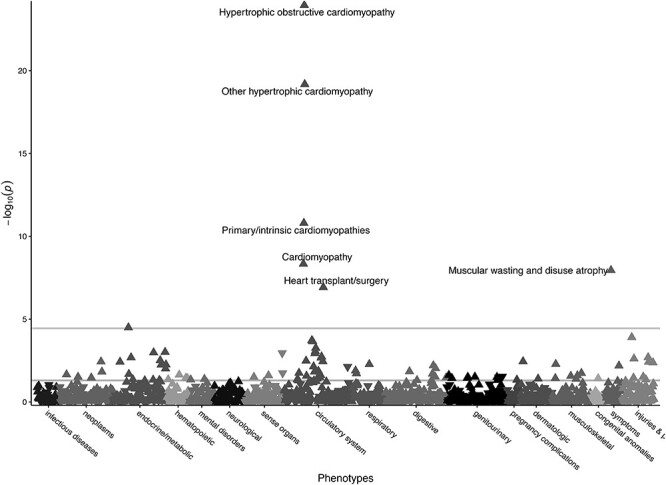
Gene burden PheWAS of pLOF + pDM variants in MYH7. Gene burden PheWAS of pLOF variants (N = 27, Supplementary Material, Table S4) and pDM variants with REVEL ≥ 0.5 (N = 858, Supplementary Material, Table S6) in MYH7. Phecodes are plotted along the x-axis to represent the phenome, and the association of the gene burden with each Phecode is plotted along the y-axis representing –log10(P-value). The directionality of each triangular point represents the direction of effect. The top horizontal line represents the Bonferroni-corrected significance threshold to adjust for multiple testing (P = 3.58E-05), and the bottom horizontal line represents a nominal significance threshold (P = 0.05).
Additionally, we found a phenome-wide significant association with ‘Muscular wasting and disuse atrophy’ with this expanded gene burden (Fig. 3) that was not seen with the gene burden limited to ClinVar P/LP variants alone (Supplementary Material, Fig. S6). We identified one pLOF variant and four missense variants with REVEL ≥ 0.5, which were carried by a total of 7 individuals who had the Phecode of ‘Muscular wasting and disuse atrophy’ (Fig. 1B, Supplementary Material, Tables S4 and S6); only 36 individuals in our entire PMBB PheWAS study dataset (N = 10 217 AFR + 29 362 EUR ancestry individuals) had this Phecode. Of note, all five MYH7 variants lacked a P/LP classification in ClinVar, and none of the seven individuals had a diagnosis for HCM. Chart review of these individuals was not suggestive of a primary myopathy diagnosis, and of those individuals with electromyography performed (N = 3), none revealed a primary neuromuscular disorder. We also compared available serum creatine kinase (CK) measurements between carriers for pLOF variants or pDM with REVEL ≥ 0.5 in MYH7 (total of 73 carriers with CK available) versus all non-carriers, and there was no significant association between the MYH7 gene burden and CK concentrations (P = 0.549).
Discussion
Gene burden PheWAS applied to large healthcare-based biobanks has the potential to elucidate the medical consequences of gene variants on the human disease phenome (5). A logical first step is to perform PheWAS focused only on pLOF variants, which has the advantage of interrogating the largest effect sizes that a gene burden may have on associated phenotypes but could lead to lack of power owing to their infrequency. In aggregating pLOF variants in MYBPC3 and MYH7 for gene burden PheWAS in PMBB, only the MYBPC3 pLOF gene burden was associated with HCM, while the MYH7 pLOF gene burden was associated with other cardiac phenotypes and not HCM. This is an expected finding given that truncating variants account for >90% of cases of MYBPC3 HCM (13), while only missense MYH7 variants have a demonstrated association with HCM (14). Furthermore, detailed review of clinical charts confirmed a diagnosis of HCM among a subset of MYBPC3 pLOF carriers while revealing mild concentric left ventricular hypertrophy and patent foramen ovale or atrial septal defect among a subset of MYH7 pLOF carriers. Functional studies have shown that pLOF variants in MYBPC3 promote nonsense-mediated decay pathways to contribute to the pathogenesis of HCM through haploinsufficiency (8,15). pLOF variants in MYH7, on the other hand, are not associated with HCM; however, recently, pLOF variants in MYH7 have been associated with left ventricular non-compaction cardiomyopathy, which has low diagnostic accuracy on echocardiography and thus could have been missed in our review of clinical charts (16).
In principle, missense variants could be combined with pLOFs to substantially increase the number of effect alleles and power for novel discovery, but a major challenge is deciding which variants to include in gene burden tests of association. Including pDM variants based on in silico prediction algorithms in addition to pLOFs in gene burdens increases power for disease associations in some genes, but the performance of such algorithms for missense variants may not be consistent across all genes. Our interrogation of missense variants based on prediction algorithms like REVEL serves as a testament to this notion; while pDM variants in MYH7 showed strong correlation with clinical annotations of pathogenicity as well as strong associations with the expected HCM phenotype in PMBB, predictions of deleteriousness for missense variants in MYBPC3 did not correlate with clinical annotations of pathogenicity and were not associated with HCM. The difference in performance may be owing to different mechisms of HCM pathogenesis. For example, MYH7 missense variants exert dominant negative effects on sarcomeric function, while haploinsufficiency and allelic imbalance in MYBPC3 is the major mechanism leading to HCM (17,18).
Importantly, these differences show that aggregating only pLOFs for gene burden association testing may be more appropriate for some genes like MYBPC3, while inclusion of additional pDM variants chosen based on in silico predictions may be more beneficial for increasing the number of effect alleles for others like MYH7, as we have similarly shown for LMNA (6). Additionally for MYBPC3, in which in silico prediction of missense variants performed poorly, we show that expert-adjudication of annotations of pathogenicity for missense variants based on in vitro and in silico analyses of missense variants in SHaRe, the largest comprehensive HCM cohort assembled to date, is superior in selecting missense variants for addition to gene burden testing (11,13). For other genes in which in silico predictions perform poorly for missense variants, we suggest that high-confidence pathogenic missense variants be added after expert adjudication to a degree that essentially parallels a saturated mutagenesis experiment.
A key goal in precision medicine initiatives is to promote a platform by which healthcare providers can make accurate diagnoses based on a wide variety of personalized health data, including the individuals’ genetic information. Genetic testing of patients with HCM can identify the precise genetic cause of disease, improve diagnostic accuracy in a patient with an ambivalent diagnosis and allow for cascade screening in family members (19). However, pathogenic HCM-causing variants found on more comprehensive sequencing can also be missed in clinical practice, which makes the genome-first approach applied to WES for review of clinical charts for carriers of pathogenic and potentially deleterious variants crucial (20). Our review of clinical charts revealed that a substantial number of patients clinically diagnosed with HCM did not appear to have had genetic testing, including those who were found in WES to have a pathogenic HCM-associated variant in MYBPC3 or MYH7. In addition to supporting previous suggestions that pathogenic variants in MYBPC3 may be associated with later disease onset compared with those in MYH7 (21), we found that about a third of these HCM individuals without a genetic diagnosis were diagnosed with HCM before age 30. We also noted that patients with HCM without a genetic test in their chart had a significantly lower rate of a family history of HCM versus those who received genetic testing. While genetic testing is now routinely offered in specialized cardiomyopathy clinics, it is still broadly underutilized and patients may benefit from referral to specialized HCM clinics for genetic diagnosis and family screening (22). Additionally, we found that on average carriers for P/LP or predicted deleterious variants without a diagnosis of HCM had increased LA size and LV wall thickness compared with non-carriers, which are subclinical features that may warrant clinical follow-up when noted on echocardiography. Importantly, many carriers for P/LP or predicted deleterious variants without a diagnosis of HCM had no clinical or echocardiographic evidence of cardiac disease despite not being younger than the ages at which carriers were diagnosed with HCM. Thus, while HCM is a progressive disease that often manifests later in life, large-scale studies of this nature show the value of a genome-first approach for estimating penetrance of single-gene Mendelian disorders like HCM.
A major advantage of the genome-first approach to conducting gene burden PheWAS is the potential to capture pleiotropy. We found that the pLOF + pDM (REVEL ≥ 0.5) gene burden for MYH7 had phenome-wide significant associations with both HCM and ‘Muscular wasting and disuse atrophy’. Importantly, while MYBPC3 is specifically expressed in the heart, MYH7 is also expressed in skeletal muscle (23), and MYH7-related myopathies are an emerging and underdiagnosed group of muscle diseases of childhood and adulthood (24). It has been reported that variants in MYH7 which cause skeletal muscle disorders may cluster in the distal regions of the rod domain (light meromyosin domain, LMM with or without cardiac involvement) (24). We found that myopathy-associated variants in MYH7 were located in the neck and hinge (distal S1 and S2 domains) of MYH7 from exons 18–24, suggesting that variants which affect skeletal muscle function may not be limited to the LMM domain. Of note, patients diagnosed with ‘Muscular wasting and disuse atrophy’ who carried a predicted deleterious MYH7 variant did not have a primary myopathy diagnosis but rather had secondary muscular wasting, indicating that MYH7-related myopathy may be a ‘second hit’ phenomenon that can be unmasked by comorbidities.
In conclusion, we used a genome-first approach to include pLOFs and selected missense variants in MYBPC3 and MYH7 in gene burdens to show their significant associations with HCM as well as the MYH7-specific association with myopathy. We demonstrate gene-specific differences in appropriate coding variant selection for gene burden testing to uncover important clinical and subclinical features relevant to associated diseases implicated by predicted deleterious variants. Our approach also suggests an expanded opportunity for clinical genetic evaluation and referral to multidisciplinary HCM centers. Importantly, our study demonstrates the value of assessing both pLOF and pDM variants for gene burden testing in future studies to uncover novel gene-disease relationships and to identify novel pathogenic loss-of-function variants across the human genome through genome-first analyses of healthcare-based populations.
Materials and Methods
Setting and study participants
All individuals recruited for the PMBB are patients of clinical practice sites of the University of Pennsylvania Health System. Appropriate consent was obtained from each participant regarding storage of biological specimens, genetic sequencing and access to all available EHR data. This study was approved by the Institutional Review Board of the University of Pennsylvania and complied with the principles set out in the Declaration of Helsinki.
Whole-exome sequencing
This study included a subset of 43 731 individuals in the PMBB who had undergone WES. We extracted DNA from stored buffy coats and then mapped exome sequences as generated by the Regeneron Genetics Center (Tarrytown, NY) to GRCh38 as previously described (6). Samples with low exome sequencing coverage, high missingness (i.e. >5% of targeted bases), dissimilar reported and genetically determined sex and genetic evidence of sample duplication were not included in this subset (5,6). For subsequent phenotypic association analyses, we removed samples with evidence of first- and second-degree relatedness, leading to a total of sample size of 41 759 for analysis.
Variant annotation and selection for gene burden association testing
For PMBB, variants were annotated using ANNOVAR (25) as pLOF or missense variants based on RefSeq reference sequences NM_000256.3 (MYBPC3) and NM_000257.4 (MYH7). pLOFs were defined as frameshift insertions or deletions, gain of stop codon and disruption of canonical splice site dinucleotides. For splicing variants, we removed those with SpliceAI scores <0.2 for loss or gain of acceptor or donor site (26). Several approaches to inclusion of rare (minor allele frequency < 0.1% in gnomAD v2) variants in the gene burden were applied, including pLOFs only, additional ClinVar pathogenic variants (annotated using ClinVar version 20 200 316 via ANNOVAR) and inclusion of pDM variants that were scored deleterious by 4/4 algorithms—SIFT (27), PolyPhen2 HumDiv and Polyphen2 HumVar (28) and MutationTaster (29). To capture additional individuals with potentially pathogenic missense variants, we utilized an ensemble method for predicting the pathogenicity of missense variants called REVEL (10) to score rare missense variants in MYBPC3 and MYH7 on a scale from 0 to 1, where higher scores indicate a higher probability of a missense variant having a damaging effect on the gene product. Of note, selection of pDM variants via these in silico algorithmic filters was independent of ClinVar annotation. Finally, we overlapped a list of all 19 expert-adjudicated pathogenic missense variants in MYBPC3 from the SHaRe Database to identify high-confidence pathogenic missense variants in PMBB (11).
Clinical data collection
All International Classification of Diseases Ninth Revision (ICD-9) and Tenth Revision (ICD-10) diagnosis codes, clinical imaging and laboratory measurements were extracted from the patients’ EHR. All ICD diagnosis codes and outpatient laboratory measurements available up to July 2020 were extracted for PMBB participants. Inpatient and outpatient echocardiography measurements were extracted if available for participants from September 2005 until November 2018. Outliers for each echocardiographic parameter (values >10 median absolute deviations from the median) were removed. Minimum, median and maximum measurements of each quantitative trait were recorded per individual.
Phenome-wide association studies
A PheWAS approach was used to determine the phenotypes associated with predicted deleterious variants in MYBPC3 or MYH7 carried by individuals in PMBB (30). ICD-10 encounter diagnoses were mapped to ICD-9 via the Center for Medicare and Medicaid Services 2017 General Equivalency Mappings (https://www.cms.gov/Medicare/Coding/ICD10/2017-ICD-10-CM-and-GEMs.html) and manual curation. Phenotypes for each individual were then determined by mapping ICD-9 codes to distinct disease entities (i.e. Phecodes) using the R package ‘PheWAS’ (31). Patients were determined to be a case for a certain Phecode if they had the corresponding ICD diagnosis on two or more dates, while controls consisted of individuals who never had the ICD code. Individuals with an ICD diagnosis on only one date as well as individuals meeting control exclusion criteria based on default PheWAS phenotype mapping protocols were not considered in statistical analyses.
Each disease phenotype was tested for association with a gene burden of pLOF and/or pDM variants using a logistic regression model adjusted for age, sex and the first 10 principal components of genetic ancestry. We used an additive genetic model to aggregate variants into gene burdens as previously described (5). PheWAS analyses were performed separately by African and European genetic ancestry and were then combined with inverse variance weighted meta-analysis. Our association analyses considered only disease phenotypes with at least 20 cases based on a prior simulation study for power analysis of rare variant gene burden PheWAS (5). This led to the interrogation of 1396 total Phecodes, and we used a Bonferroni correction to adjust for multiple testing (P = 0.05/1396 = 3.58E-05).
Statistical analyses
We also created a single HCM phenotype by combining Phecodes ‘Hypertrophic obstructive cardiomyopathy’ and ‘Other hypertrophic cardiomyopathy’ for association with various gene burdens in conjunction with PheWAS by using a logistic regression model adjusted for age, sex and the first 10 principal components of genetic ancestry. These HCM-specific analyses were performed separately by African and European genetic ancestry and were combined with inverse variance weighted meta-analysis. Additionally, to compare available echocardiographic and serum laboratory measurements between carriers of predicted deleterious variants and genotypic controls, we used linear regression adjusted for age, sex and the first 10 principal components of genetic ancestry. These analyses were performed separately by African and European genetic ancestry and were combined with inverse variance weighted meta-analysis. For echocardiographic comparison of MYBPC3 pLOF and pathogenic missense variant carriers versus controls, MYH7 pLOF and REVEL-informed pDM variant carriers were removed from controls. Likewise, for echocardiographic comparison of carriers of pLOF and pDM with REVEL ≥ 0.5 variants in MYH7 versus controls, carriers of MYBPC3 pLOF and pathogenic missense variants were removed from controls. All statistical analyses were completed using R version 3.5 (Vienna, Austria).
Review of clinical charts
We reviewed the clinical charts of patients with HCM who also carry a pLOF or pDM variant in MYBPC3 or MYH7 to assess the prevalence of clinical genetic testing for a molecular diagnosis of carrying a pathogenic MYBPC3 or MYH7 variant. We also reviewed the clinical charts of MYH7 pLOF carriers to characterize a cardiac phenotype among these individuals without an HCM diagnosis. Finally, we interrogated the clinical charts of cases for ‘Muscular wasting and disuse atrophy’, who also carried a pLOF or pDM variant in MYH7, to assess the prevalence of clinical genetic testing for a molecular diagnosis of MYH7-related myopathy.
Data Availability
Up-to-date summary data for genetic variants captured via WES in PMBB can be accessed via the PMBB Genome Browser (https://pmbb.med.upenn.edu/biobank/allele-frequency/). Individual-level data are not made publicly available owing to research participant privacy concerns; however, requests from accredited researchers for access to individual-level data relevant to this manuscript can be made by contacting the corresponding author.
Supplementary Material
Acknowledgements
We thank JoEllen Weaver, Stephanie DerOhannessian, Marjorie Risman, Anurag Verma, Anastasia Lucas and the Regeneron Genetics Center. Conflict of Interest statement. None declared.
Contributor Information
Joseph Park, Department of Genetics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA; Department of Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA; Institute for Biomedical Informatics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA.
Elizabeth A Packard, Department of Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA.
Michael G Levin, Department of Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA.
Renae L Judy, Department of Surgery, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA.
Regeneron Genetics Center, Regeneron Genetics Center, Regeneron Pharmaceuticals, Tarrytown, NY 10591, USA.
Scott M Damrauer, Department of Surgery, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA.
Sharlene M Day, Department of Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA.
Marylyn D Ritchie, Department of Genetics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA; Institute for Biomedical Informatics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA.
Daniel J Rader, Department of Genetics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA; Department of Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA; Institute for Translational Medicine and Therapeutics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA.
Funding
The PMBB is funded by the Perelman School of Medicine at the University of Pennsylvania, a gift from the Smilow family and the National Center for Advancing Translational Sciences of the National Institutes of Health under CTSA award number UL1TR001878. Research reported in this paper was supported by grants from the Blavatnik Family Foundation and National Human Genome Research Institute of the National Institutes of Health under award number F30HG010442 (to J.P.).
References
- 1. Stessman, H.A., Bernier, R. and Eichler, E.E. (2014) A genotype-first approach to defining the subtypes of a complex disease. Cell, 156, 872–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dewey, F.E., Murray, M.F., Overton, J.D., Habegger, L., Leader, J.B., Fetterolf, S.N., O'Dushlaine, C., Van Hout, C.V., Staples, J., Gonzaga-Jauregui, C.et al. (2016) Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science, 354, aaf6814. [DOI] [PubMed] [Google Scholar]
- 3. Bush, W.S., Oetjens, M.T. and Crawford, D.C. (2016) Unravelling the human genome-phenome relationship using phenome-wide association studies. Nat. Rev. Genet., 17, 129–145. [DOI] [PubMed] [Google Scholar]
- 4. Lee, S., Abecasis, G.R., Boehnke, M. and Lin, X. (2014) Rare-variant association analysis: study designs and statistical tests. Am. J. Hum. Genet., 95, 5–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Park, J., Lucas, A.M., Zhang, X., Chaudhary, K., Cho, J.H., Nadkarni, G., Dobbyn, A., Chittoor, G., Josyula, N.S., Katz, N.et al. (2021) Exome-wide evaluation of rare coding variants using electronic health records identifies new gene-phenotype associations. Nat. Med., 27, 66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Park, J., Levin, M.G., Haggerty, C.M., Hartzel, D.N., Judy, R., Kember, R.L., Reza, N., Regeneron Genetics, C, Ritchie, M.D., Owens, A.T.et al. (2019) A genome-first approach to aggregating rare genetic variants in LMNA for association with electronic health record phenotypes. Genet. Med., 22, 102–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Burke, M.A., Cook, S.A., Seidman, J.G. and Seidman, C.E. (2016) Clinical and mechanistic insights into the genetics of cardiomyopathy. J. Am. Coll. Cardiol., 68, 2871–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Helms, A.S., Tang, V.T., O’Leary, T.S., Friedline, S., Wauchope, M., Arora, A., Wasserman, A.H., Smith, E.D., Lee, L.M., Wen, X.W.et al. (2020) Effects of MYBPC3 loss-of-function mutations preceding hypertrophic cardiomyopathy. JCI Insight, 5, e133782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Landrum, M.J., Lee, J.M., Benson, M., Brown, G.R., Chao, C., Chitipiralla, S., Gu, B., Hart, J., Hoffman, D., Jang, W.et al. (2018) ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res., 46, D1062–D1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ioannidis, N.M., Rothstein, J.H., Pejaver, V., Middha, S., McDonnell, S.K., Baheti, S., Musolf, A., Li, Q., Holzinger, E., Karyadi, D.et al. (2016) REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet., 99, 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thompson, A.D., Helms, A.S., Kannan, A., Yob, J., Lakdawala, N.K., Wittekind, S.G., Pereira, A.C., Jacoby, D.L., Colan, S.D., Ashley, E.A.et al. (2021) Computational prediction of protein subdomain stability in MYBPC3 enables clinical risk stratification in hypertrophic cardiomyopathy and enhances variant interpretation. Genet. Med., 23, 1281–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sabater-Molina, M., Perez-Sanchez, I., Hernandez Del Rincon, J.P. and Gimeno, J.R. (2018) Genetics of hypertrophic cardiomyopathy: a review of current state. Clin. Genet., 93, 3–14. [DOI] [PubMed] [Google Scholar]
- 13. Helms, A.S., Thompson, A.D., Glazier, A.A., Hafeez, N., Kabani, S., Rodriguez, J., Yob, J.M., Woolcock, H., Mazzarotto, F., Lakdawala, N.K.et al. (2020) Spatial and functional distribution of MYBPC3 pathogenic variants and clinical outcomes in patients with hypertrophic cardiomyopathy. Circ. Genom. Precis. Med., 13, 396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Walsh, R., Thomson, K.L., Ware, J.S., Funke, B.H., Woodley, J., McGuire, K.J., Mazzarotto, F., Blair, E., Seller, A., Taylor, J.C.et al. (2017) Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet. Med., 19, 192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Seeger, T., Shrestha, R., Lam, C.K., Chen, C., McKeithan, W.L., Lau, E., Wnorowski, A., McMullen, G., Greenhaw, M., Lee, J.et al. (2019) A premature termination codon mutation in MYBPC3 causes hypertrophic cardiomyopathy via chronic activation of nonsense-mediated decay. Circulation, 139, 799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Klaassen, S., Probst, S., Oechslin, E., Gerull, B., Krings, G., Schuler, P., Greutmann, M., Hurlimann, D., Yegitbasi, M., Pons, L.et al. (2008) Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation, 117, 2893–2901. [DOI] [PubMed] [Google Scholar]
- 17. Witjas-Paalberends, E.R., Piroddi, N., Stam, K., vanDijk, S.J., Oliviera, V.S., Ferrara, C., Scellini, B., Hazebroek, M., tenCate, F.J., vanSlegtenhorst, M.et al. (2013) Mutations in MYH7 reduce the force generating capacity of sarcomeres in human familial hypertrophic cardiomyopathy. Cardiovasc. Res., 99, 432–441. [DOI] [PubMed] [Google Scholar]
- 18. Glazier, A.A., Thompson, A. and Day, S.M. (2019) Allelic imbalance and haploinsufficiency in MYBPC3-linked hypertrophic cardiomyopathy. Pflugers Arch., 471, 781–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cirino, A.L., Seidman, C.E. and Ho, C.Y. (2019) Genetic testing and counseling for hypertrophic cardiomyopathy. Cardiol. Clin., 37, 35–43. [DOI] [PubMed] [Google Scholar]
- 20. Bagnall, R.D., Ingles, J., Dinger, M.E., Cowley, M.J., Ross, S.B., Minoche, A.E., Lal, S., Turner, C., Colley, A., Rajagopalan, S.et al. (2018) Whole genome sequencing improves outcomes of genetic testing in patients with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol., 72, 419–429. [DOI] [PubMed] [Google Scholar]
- 21. Biagini, E., Olivotto, I., Iascone, M., Parodi, M.I., Girolami, F., Frisso, G., Autore, C., Limongelli, G., Cecconi, M., Maron, B.J.et al. (2014) Significance of sarcomere gene mutations analysis in the end-stage phase of hypertrophic cardiomyopathy. Am. J. Cardiol., 114, 769–776. [DOI] [PubMed] [Google Scholar]
- 22. Mital, S., Musunuru, K., Garg, V., Russell, M.W., Lanfear, D.E., Gupta, R.M., Hickey, K.T., Ackerman, M.J., Perez, M.V., Roden, D.M.et al. (2016) Enhancing literacy in cardiovascular genetics: a scientific statement from the American Heart Association. Circ. Cardiovasc. Genet., 9, 448–467. [DOI] [PubMed] [Google Scholar]
- 23. Consortium, G.T (2013) The genotype-tissue expression (GTEx) project. Nat. Genet., 45, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fiorillo, C., Astrea, G., Savarese, M., Cassandrini, D., Brisca, G., Trucco, F., Pedemonte, M., Trovato, R., Ruggiero, L., Vercelli, L.et al. (2016) MYH7-related myopathies: clinical, histopathological and imaging findings in a cohort of Italian patients. Orphanet J. Rare Dis., 11, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang, K., Li, M. and Hakonarson, H. (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res., 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jaganathan, K., Kyriazopoulou Panagiotopoulou, S., McRae, J.F., Darbandi, S.F., Knowles, D., Li, Y.I., Kosmicki, J.A., Arbelaez, J., Cui, W., Schwartz, G.B.et al. (2019) Predicting splicing from primary sequence with deep learning. Cell, 176, 535–548.e24. [DOI] [PubMed] [Google Scholar]
- 27. Kumar, P., Henikoff, S. and Ng, P.C. (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc., 4, 1073–1081. [DOI] [PubMed] [Google Scholar]
- 28. Adzhubei, I.A., Schmidt, S., Peshkin, L., Ramensky, V.E., Gerasimova, A., Bork, P., Kondrashov, A.S. and Sunyaev, S.R. (2010) A method and server for predicting damaging missense mutations. Nat. Methods, 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schwarz, J.M., Cooper, D.N., Schuelke, M. and Seelow, D. (2014) MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods, 11, 361–362. [DOI] [PubMed] [Google Scholar]
- 30. Denny, J.C., Bastarache, L., Ritchie, M.D., Carroll, R.J., Zink, R., Mosley, J.D., Field, J.R., Pulley, J.M., Ramirez, A.H., Bowton, E.et al. (2013) Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat. Biotechnol., 31, 1102–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Carroll, R.J., Bastarache, L. and Denny, J.C. (2014) R PheWAS: data analysis and plotting tools for phenome-wide association studies in the R environment. Bioinformatics, 30, 2375–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Up-to-date summary data for genetic variants captured via WES in PMBB can be accessed via the PMBB Genome Browser (https://pmbb.med.upenn.edu/biobank/allele-frequency/). Individual-level data are not made publicly available owing to research participant privacy concerns; however, requests from accredited researchers for access to individual-level data relevant to this manuscript can be made by contacting the corresponding author.