

Abstract
Exonic (i.e. coding) variants in genes associated with disease can exert pathogenic effects both at the protein and mRNA level, either by altering the amino acid sequence or by affecting pre-mRNA splicing. The latter is often neglected due to the lack of RNA analyses in genetic diagnostic testing. In this study we considered both pathomechanisms and performed a comprehensive analysis of nine exonic nucleotide changes in OPA1, which is the major gene underlying autosomal dominant optic atrophy (DOA) and is characterized by pronounced allelic heterogeneity. We focused on the GTPase-encoding domain of OPA1, which harbors most of the missense variants associated with DOA. Given that the consensus splice sites extend into the exons, we chose a split codon, namely codon 438, for our analyses. Variants at this codon are the second most common cause of disease in our large cohort of DOA patients harboring disease-causing variants in OPA1. In silico splice predictions, heterologous splice assays, analysis of patient’s RNA when available, and protein modeling revealed different molecular outcomes for variants at codon 438. The wildtype aspartate residue at amino acid position 438 is directly involved in the dimerization of OPA1 monomers. We found that six amino acid substitutions at codon 438 (i.e. all substitutions of the first and second nucleotide of the codon) destabilized dimerization while only substitutions of the first nucleotide of the codon caused exon skipping. Our study highlights the value of combining RNA analysis and protein modeling approaches to accurately assign patients to future precision therapies.
Introduction
Splicing is the molecular processing of immature transcripts, i.e. the recognition and joining of coding elements (exons) and the concomitant removal of non-coding elements (introns) (1). The specificity of the splicing reaction depends heavily on the recognition of conserved sequences at the exon-intron boundaries. Approximately 99% of mammalian introns obey to the so-called GT-AG rule, which states that the first dinucleotide of an intron is almost invariably GT, while the last dinucleotide is almost always AG (2). However, consensus splice sites are not restricted to introns but extend into the exons—in particular the first and the last three exonic positions are an integral part of 3′ and 5′ splice site consensus sequences (3). The most preserved coding nucleotides are G at the first position of an exon and AG at the two last positions (3). It has also been shown that variants at the first and the last position of an exon are mostly incompatible with correct splicing (4). Moreover, computational analyses revealed that 22% of all variants within exons may affect splicing (5). Exonic variants away from the intron borders can affect splicing by creating cryptic splice sites or by either disrupting exonic splicing enhancers or creating exonic splicing silencer motifs. In contrast, aberrant splicing due to exonic variants near intron boundaries is rather caused by disrupted base-pairing with small nuclear ribonucleoproteins (3).
Exonic mutations that cause splicing alterations can be easily misclassified as synonymous, missense, or nonsense variants (6). This has immediate effects on variant interpretation and classification in genetic diagnostics. Synonymous and missense variants that have not been previously reported or functionally validated must be classified as variants of uncertain significance (VUS) according to the rules for variant classification suggested by the American College of Medical Genetics and Genomics (7). Inconclusive reports, in turn, contribute to patient uncertainty and may prolong their diagnostic odyssey. The situation is different for nonsense variants, which are mostly classified as pathogenic if loss of function is a known disease mechanism for the gene. Transcripts derived from alleles with nonsense variants will in most cases be targeted to nonsense-mediated decay (NMD) (8). NMD may also occur as the consequence of a splicing induced frameshift rather than the original nonsense variant recognized as premature termination codon (PTC) (9), but this does not influence the classification of nonsense variants. Regardless of its classification, the exact determination of a variant’s molecular mechanism is crucial for an accurate assignment of patients to precision therapies.
Variants acting on splicing considerably contribute to genetic disease etiology. As of its Professional Version 2021.1, the Human Gene Mutation Database (HGMD) (10) groups a total of 27 192 (8.6%) out of 314 707 disease-causing mutations into the category of splicing variants. This is certainly an underestimate since synonymous, missense, or nonsense variants acting on splicing as well as deletions and insertions interrupting splice sites for the most part are not included. A vast number of these variants remains to be characterized at the transcript level. However, genetic diagnostic testing is almost exclusively performed at the genomic DNA level, which means that any effect of variants on splicing must be inferred from general rules, such as deviation from the splice site consensus. Bioinformatic algorithms can aid in assessing the effect of a variant on splicing, but still have limitations because cis-regulatory elements other than the canonical splice sites show high nucleotide diversity and are therefore difficult to predict.
The OPA1 (optic atrophy type 1) gene is a suitable model to analyze the effect of exonic variants on splicing, since the gene is ubiquitously expressed. This means that direct transcript analysis is feasible by using easily accessible patient-derived cells such as blood leukocytes. OPA1 was the first gene to be described as an underlying cause of dominant optic atrophy (DOA, OMIM #165500) (11,12) characterized by progressive degeneration of retinal ganglion cells and optic nerve axons and subsequent vision loss. OPA1 is a dynamin-related GTPase which is imported into mitochondria and is responsible for a plethora of functions, including, but not limited to mitochondrial morphology (13,14), mitochondrial dynamics (15,16) and energy metabolism (16,17).
More than 500 unique disease-causing OPA1 variants have been reported to date (10,18), with the majority of them representing loss of function alleles (nonsense, frameshift and splice variants as well as structural variants), while missense variants account for approximately 28% of disease-causing variants (19). Most missense variants identified to date cluster to the GTPase domain of the protein encoded by exons 9–16 (based on GenBank accession numbers NM_015560.2 and NP_056375.2) (18), where they are thought to act via dominant-negative effects (20–22). This pathomechanism was suggested to result in reduced OPA1 function, prompted by the observation that oligomers of mutant and wildtype OPA1 decreased the GTPase activity of the dynamin, thereby reducing the mitochondrial fusion competency (15). Of note, patients with missense variants in the GTPase domain have a 2–3-fold increased risk of developing a severe syndromic phenotype (23).
In this study, we aimed to analyze the effects of nucleotide variants affecting a particular codon in the GTPase-encoding domain of OPA1, namely codon 438 (based on GenBank accession NP_056375.2). Variants at this codon represent the second most common cause of disease in our cohort of 279 DOA patients harboring pathogenic variants in OPA1. In this study, we thought to establish which variants cause missplicing and which exert a disease-causing effect by altering functional properties of the GTPase domain. More specifically, we assessed the effect of all theoretically possible nucleotide variants at codon 438 based on in silico splice prediction, heterologous splice assays, analysis of patient’s RNA when available, and protein modeling.
Results
Codon 438 is a split codon in all vertebrate orthologues of OPA1, meaning that there is an intron between its first and second nucleotides (Fig. 1). Depending on the species, the codon sequence is either GAT or GAC, in both cases encoding an aspartate residue. This aspartate is highly conserved not only in vertebrates but also in invertebrates (Supplementary Material, Fig. S1). In the human transcript NM_015560.2, codon 438 is composed of the last nucleotide of exon 13 and the first two nucleotides of exon 14 (Fig. 1). With respect to the consensus sequence of exon-intron boundaries, the last nucleotide of exon 13 is G and thereby consistent with the 5′ splice site consensus. In contrast, exon 14 does not correspond to the 3′ splice site consensus as its first nucleotide is A instead of G (3).
Figure 1.
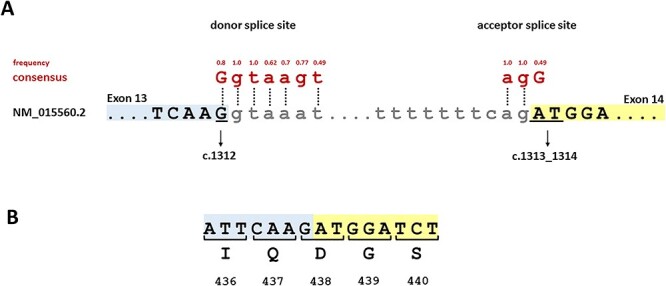
Schematic representation of split codon 438 and its immediate neighboring sequence. (A) Segment of genomic OPA1 sequence with the junction of exon 13 (blue) and intron 13 as well as the junction of intron 13 and exon 14 (yellow). Exonic and intronic nucleotides are given in upper and lower case letters, respectively. The nucleotides constituting codon 438 (c.1312_1314) are underlined. The consensus sequence for the 5′ and 3′ splice sites and the frequencies of the reference bases as determined by Zhang and colleagues (4) are shown above the OPA1 sequence in red. Vertical dotted lines indicate identity of the OPA1 sequence with the consensus sequence. (B) Segment of OPA1 cDNA with codon numbers and amino acid residues in 1-letter code. Numbering is based on NM_015560.2 and NP_056375.2, respectively.
We retrieved known human sequence variants at codon 438 from different public databases, namely dbSNP (24), ClinVar (25), HGMD (10), and the Leiden Open Variation Database (LOVD) for OPA1 (18) (Table 1). The only variant reported to affect the third position of codon 438 (i.e. the second nucleotide of exon 14) is a synonymous variant, namely c.1314 T > C, p.(D438D) listed in dbSNP (rs1219542370). All three possible variants affecting the second position of the codon (i.e. the first nucleotide of exon 14) have been identified in patients with DOA, namely c.1313A > C, p.(D438A) (26), c.1313A > G, p.(D438G) (27,28), and c.1313A > T, p.(D438V) (29). Variants affecting the first position of codon 438 (i.e. the last position of exon 13) have not been reported yet, with the exception of c.1312G > C, p.(D438H), which has been recently identified by us in a patient with DOA (see LOVD entry OPA1_000603). Eight of the nine theoretically possible variants of codon 438 are absent in the Genome Aggregation database (gnomAD) (30). Only variant c.1314 T > C, p.(D438D) was found at an extremely low frequency (reported once among a total of 251 184 alleles).
Table 1.
Theoretically possible variants at OPA1 codon 438
| NM_015560.2 | NP_056375.2 | Database | Identifier | gnomAD MAF | Cases in our patient cohort* |
|---|---|---|---|---|---|
| c.1312G > A | p.(D438N) | - | - | - | - |
| c.1312G > C | p.(D438H) | LOVD | OPA1_000603 | - | 1 |
| c.1312G > T | p.(D438Y) | - | - | - | - |
| c.1313A > C | p.(D438A) | HGMD; LOVD |
CM126968; OPA1_000293 |
- | - |
| c.1313A > G | p.(D438G) | HGMD; LOVD |
CM066157; OPA1_000150 |
- | 2 |
| c.1313A > T | p.(D438V) | HGMD; LOVD |
CM012169; OPA1_000097 | - | 6 |
| c.1314 T > A | p.(D438E) | - | - | - | - |
| c.1314 T > C | p.(D438D) | dbSNP | rs1219542370 | 0.000003981 | - |
| c.1314 T > G | p.(D438E) | - | - | - | - |
MAF, minor allele frequency. *279 DOA cases harboring disease-causing variants in OPA1
Of note, we have recently published a mutation overview of our cohort of 755 unrelated DOA patients, 278 of which harbor putatively pathogenic variants in OPA1 (31). Adding the recently identified patient with the c.1312G > C, p.(D438H) variant, nine cases of our cohort harbor variants at codon 438 (Table 1). This makes variants at codon 438 the second most common cause of disease in our cohort, right after the common founder mutation c.2708_2711del (39 cases). All nine cases with codon 438 variants present with a DOA phenotype without syndromic involvement (data not shown).
We analyzed all nine theoretically possible variants at codon 438 (including those not reported yet) by means of in silico predictions using three different splice prediction algorithms embedded in the Alamut Visual Plus software (Interactive Biosoftware, Rouen, France). Although Alamut includes a fourth program in its splicing module, namely GeneSplicer (32), it was excluded from our analysis because of its high missing rate, which was also observed in other studies (33,34). Instead, we used the deep neural network SpliceAI for the prediction of splice site alterations (35). Table 2 shows the prediction scores for all variants at codon 438 obtained with SpliceSiteFinder(SSF)-like (36), MaxEntScan (37), NNSplice (38), and SpliceAI (35).
Table 2.
In silico splice site predictions for all theoretically possible variants at codon 438
| Splice donor site exon 13 | Splice acceptor site exon 14 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| c.1312 | c.1313 | c.1314 | ||||||||||
| Program | NNSplice | MaxEntScan | SSF-like | SpliceAI | NNSplice | MaxEntScan | SSF-like | SpliceAI | NNSplice | MaxEntScan | SSF-like | SpliceAI |
| Score range | [0–1] | [0–12] | [0–100] | [0–1] | [0–1] | [0–16] | [0–100] | [0–1] | [0–1] | [0–16] | [0–100] | [0–1] |
| Threshold | ≥0.4 | ≥0 | ≥70 | ≥0.5 | ≥0.4 | ≥0 | ≥70 | ≥0.5 | ≥0.4 | ≥0 | ≥70 | ≥0.5 |
| A | 0.75 | 4.06 | 75.41 | 1.0 | 10.70 | 94.80 | 0.99 | 8.82 | 94.80 | |||
| (−24.5%) | (−54.3%) | (−13.9%) | 0.78 | (−0.3%) | (−17.5%) | (=) | 0.01 | |||||
| C | 0.87 | 0.44 | 74.2 | 1.0 | 9.58 | 92.95 | 0.99 | 8.95 | 94.80 | |||
| (−13.2%) | (−95.1%) | (−15.2%) | 0.85 | (=) | (−10.4%) | (−1.9%) | 0.0 | (−0.2%) | (−16.3%) | (=) | 0.0 | |
| G | 1.0 | 8.88 | 87.54 | 1.0 | 11.42 | 98.71 | 0.99 | 9.09 | 94.80 | |||
| (=) | (+6.7%) | (+4.1%) | 0.0 | (−0.3%) | (−15.0%) | (=) | 0.01 | |||||
| T | 0.58 | 2.37 | 74.93 | 1.0 | 9.27 | 92.47 | 1.0 | 10.70 | 94.80 | |||
| (−41.5%) | (−73.4%) | (−14.4%) | 0.9 | (=) | (−13.3%) | (−2.5%) | 0.0 | |||||
Table cells for NNSplice, MaxEntScan, and SSF-like contain the scores as well as the relative percentage decrease or increase (in parentheses). SpliceAI predictions are given as Δscores (difference between reference and mutant scores).
Concordant results were obtained for alterations of the second and third nucleotide of codon 438, which were predicted by all algorithms to have either no effect or to decrease the score of the acceptor site of exon 14 less than 20%. In contrast, score changes for alterations of the first nucleotide of codon 438 were more variable. SSF-like and NNSplice calculated a decrease of the donor site of exon 13 ranging from 13–41%, while MaxEntScan and SpliceAI predicted a decrease ranging from 55–95%.
Following in silico analysis, we evaluated the effect on splicing of all nine variants by means of in vitro splice assays. Briefly, a genomic segment of 2673 bp encompassing exon 12 to exon 16 of the OPA1 gene was amplified from genomic DNA and cloned into the exon trap vector pSPL3. After verifying that this wildtype minigene construct generated normally spliced RNA, we used site-directed mutagenesis to introduce all nine theoretically possible variants at codon 438.
In line with the in silico predictions, none of the alterations of the second and third nucleotide of codon 438 exerted a splicing defect in the minigene assays. In contrast and again corresponding to the in silico predictions, any alteration of the first nucleotide of codon 438 induced the skipping of exon 13 from the transcript. Fig. 2 shows agarose gel-based analysis and sequencing of transcripts obtained after transfection of human embryonic kidney (HEK) 293 T cells with minigene constructs. Expression of both the wildtype minigene construct and all mutant constructs affecting the second or third nucleotides of codon 438 resulted in a single RT-PCR product of 623 bp (Fig. 2A). This product corresponds to correct splicing of exon 12, exon 13, exon 14, exon 15, and exon 16 between the resident exons of pSPL3, which was confirmed by sequencing (Fig. 2B and C). In contrast, expression of any mutant construct affecting the first nucleotide of codon 438 resulted in a single RT-PCR product of 523 bp that lacked exon 13 (Fig. 2D). The skipping of exon 13 is predicted to cause a frameshift and PTC (p.(T405Mfs*29)).
Figure 3.
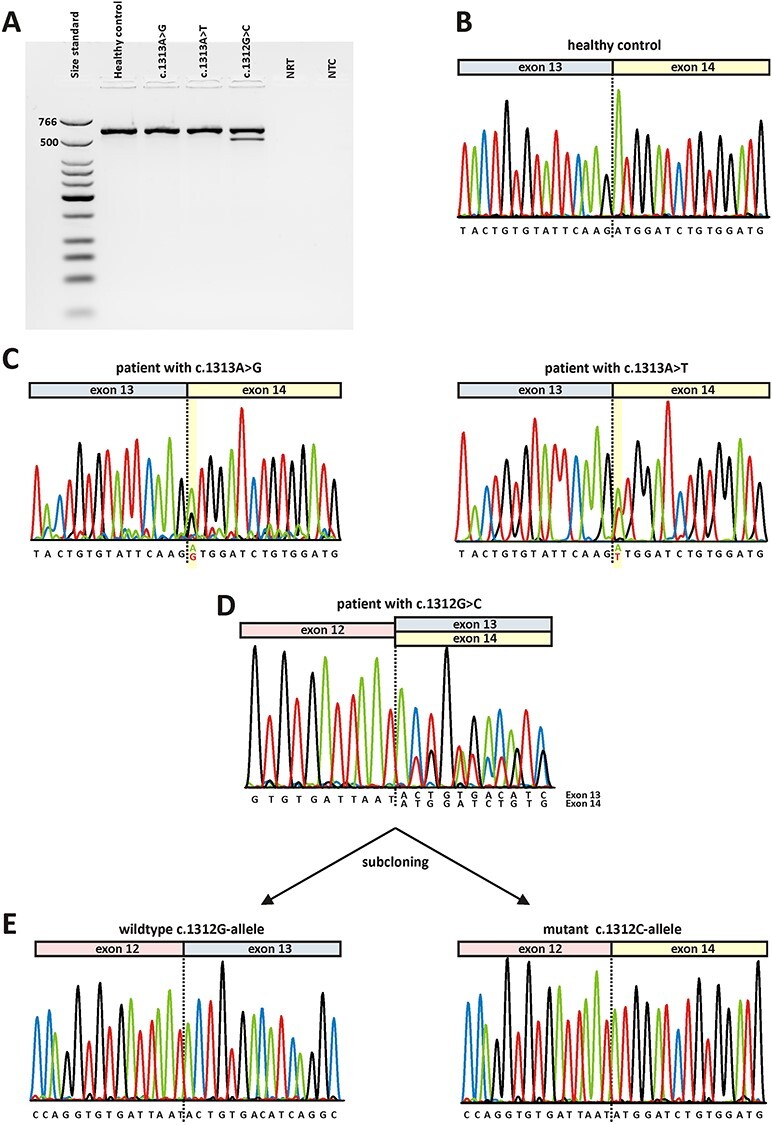
Direct transcript analysis from blood samples. Following cDNA synthesis with random hexamers, RT-PCR was performed using primers located in OPA1 exon 11 and exon 17. A) Agarose gel electrophoresis. Lane 1: size standard (low molecular weight DNA ladder, NEB); lane 2: RT-PCR from a healthy control person; lane 3: RT-PCR from a patient heterozygous for c.1313A > G; lane 4: RT-PCR from a patient heterozygous for c.1313A > T; lane 5: RT-PCR from a patient heterozygous for c.1312G > C; lane 6: no reverse transcriptase control (NRT); lane 7: no template control (NTC). Expected sizes of RT-PCR amplified products were 599 bp in the event of normal splicing and 499 bp in the event of exon 13 skipping. B) Sequence electropherogram of healthy control person shows correct splicing of exon 13 to exon 14. C) Sequence electropherograms relating to variants c.1313A > G (left) and c.131A > T (right) show correct splicing of exon 13 to exon 14. Both alleles have been amplified from cDNA as can be seen from an overlay of the wildtype nucleotide and the respective mutant nucleotide at position c.1313 (highlighted in yellow). D) Sequence analysis of RT-PCR products from a patient harboring the c.1312G > C variant. The overlay of sequence traces after exon 12 indicates amplification from both the correct transcript and a transcript lacking exon 13. E) Subcloned RT-PCR products confirm that the wildtype c.1312G-allele generated a correctly spliced transcript (left) while the transcript derived from the mutant c.1312C-allele lacks exon 13 (right).
Figure 2.
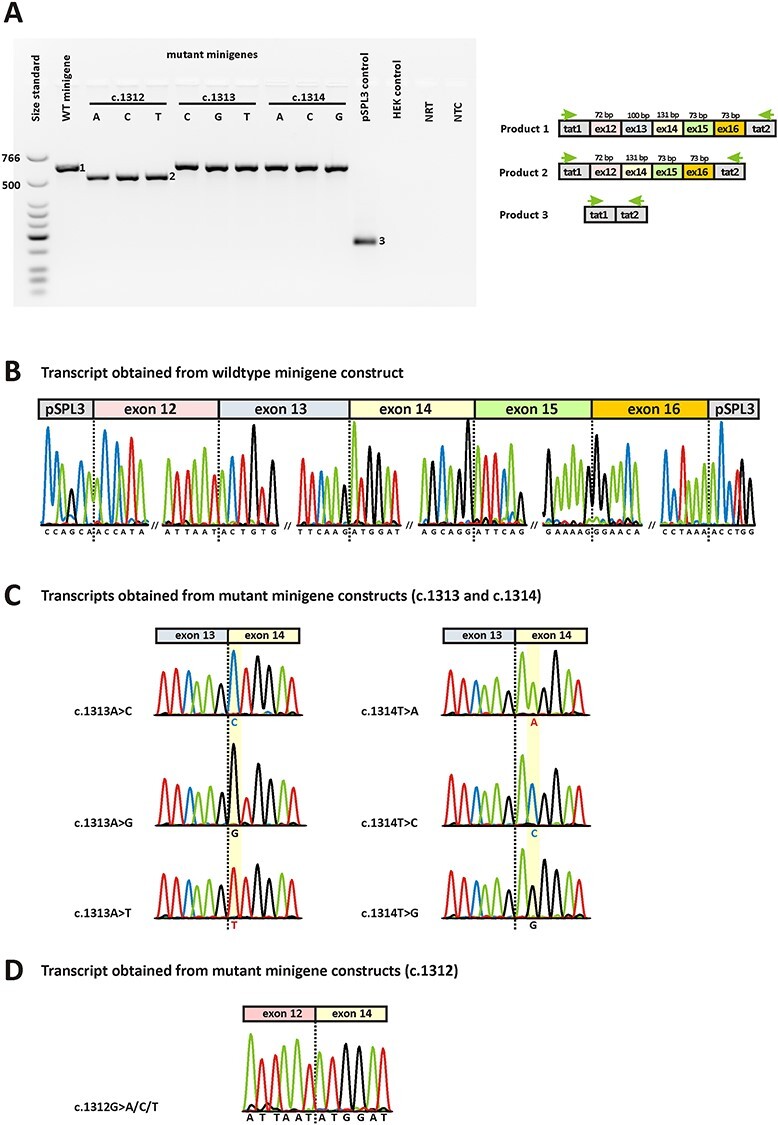
In vitro splice assays. A) Agarose gel electrophoresis of RT-PCR products. Gel loading is as follows: A size standard (low molecular weight DNA ladder, NEB) is loaded in the leftmost lane. The RT-PCR product derived from HEK293T cells transfected with the wildtype minigene construct is shown in lane 2, while the RT-PCR products obtained upon transfection with the nine mutant minigenes are shown in lane 3–11. RT-PCRs from transfection with empty pSPL3 vector (lane 12) and untransfected HEK293T cells (lane 13) served as controls. NRT (lane 14), no reverse transcriptase control; NTC (lane 15), no template control. Schemes of the amplified products are presented next to the agarose gel picture. Grey boxes represent pSPL3 resident exons tat1 and tat2, and colored boxes OPA1 exons, respectively. The green arrows indicate the location of the RT-PCR primers. B) Sequencing analysis of the RT-PCR product obtained upon transfection with the wildtype minigene revealed correct splicing. Only the exon junctions are shown. C) Sequencing analysis of RT-PCR products obtained upon transfection with mutant minigenes c.1313A > C, c.1313A > G, c.1313A > T, c.1314 T > A, c.1314 T > C, and c.1314 T > G revealed correct splicing. Only the relevant junction of exon 13 and exon 14 is shown. The respective mutant nucleotides in exon 14 are highlighted in yellow. D) Exemplary sequencing analysis of RT-PCR product obtained upon transfection with mutant minigenes c.1312G > A, c.1312G > C, and c.1312G > T revealed skipping of exon 13 from the transcript. Only the relevant junction of exon 12 and exon 14 is shown.
Since OPA1 is ubiquitously expressed, we aimed to characterize RNA isolated from patients´ blood cells to corroborate the in vitro data. However, only five out of nine possible alterations of codon 438 have been observed in human subjects so far. We had the opportunity to analyze RNA samples harboring three of those variants, namely c.1312G > C, p.(D438H), c.1313A > G, p.(D438G), and c.1313A > T, p.(D438V). All three RNA samples were from patients diagnosed with DOA and heterozygous for the respective variant. Briefly, following reverse transcription (RT) of patients´ RNA using random hexamers, RT-PCR was performed using a forward primer binding within OPA1 exon 11 and a reverse primer binding within exon 17. Agarose gel electrophoresis revealed a single RT-PCR product for the two patients harboring variants at position c.1313. Subsequent sequencing of RT-PCR products showed that both alleles were amplified from cDNA in both patients. The sequence electropherograms showed an overlay of the respective peaks at the position of the variant but revealed no trace of aberrant splicing (Fig. 3C). In contrast, the RT-PCR from the patient harboring the c.1312G > C variant revealed two distinct products (Fig. 3A). The sequence electropherogram showed an overlay of peaks starting directly after exon 12, indicating aberrant splicing for the mutant allele (Fig. 3D). This was confirmed by subcloning of RT-PCR products which revealed correct splicing for the wildtype allele and skipping of exon 13 for the mutant allele (Fig. 3E). In order to precisely determine the ratio between correct and mutant transcript we performed RT-PCR with a fluorescently labeled primer and separated the PCR products by capillary electrophoresis. From the area-under-the-curve (AUC) of fluorescence intensity for each fragment we calculated the relative abundance of correctly versus aberrantly spliced RT-PCR product, which was found to be 27% versus 73% (Supplementary Material, Fig. S2). Since the skipping of exon 13 is predicted to cause a frameshift and PTC, the decreased abundance of the mutant transcript is most probably due to NMD.
Figure 4.
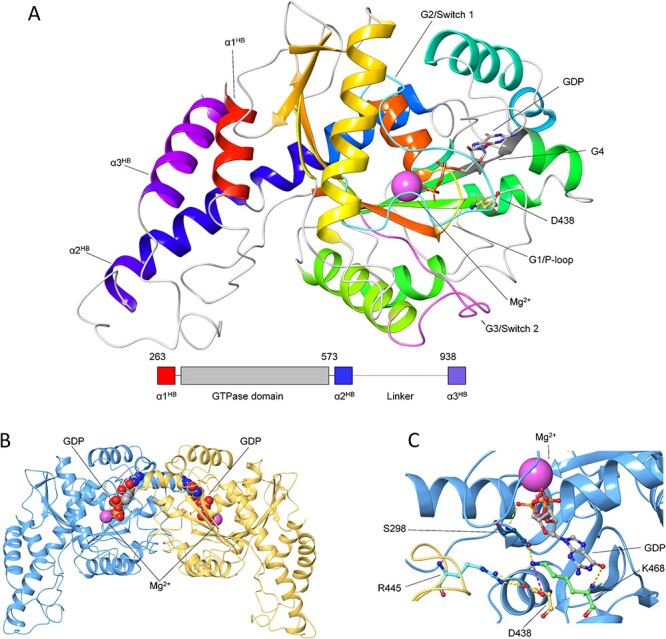
Structural organization of OPA1-MGD monomer and dimer. A) The three-dimensional structure of GDP-bound OPA1-MGD monomer is shown as cartoon, secondary structure elements are colored in a red-to-blue scheme according to the sequence, GDP is shown as sticks with C atoms in grey, O atoms in red, N atoms in blue and P atoms in orange, Mg2+-ion is represented as a pink sphere, residue D438 is shown as sticks with C atoms in yellow, N atoms in blue and O atoms in red. Functional element G1/P-loop is colored in yellow, G2/Switch 1 in cyan, G3/Switch 2 in magenta, G4 in dark grey. Inset shows the structural organization of OPA1-MGD with the three helices of the Helix Bundle colored as above. B) The three-dimensional structure of GDP-bound OPA1-MGD dimer is represented as cartoon with protomers in blue and yellow, Mg2+-ion is represented as a pink sphere, GDP is shown as spheres with C atoms in grey, N atoms in blue, O atoms in red, P atoms in orange. C) Intra-intermolecular interactions involving D438 residue. The three-dimensional structure of GDP-bound OPA1-MGD dimer is represented as cartoon with protomers in blue and yellow green, Mg2+-ion is represented as a pink sphere, GDP, S298, D438, R445 and K468 are shown as sticks with C atoms in grey, dark teal, yellow, cyan, and light green respectively, N atoms in blue, O atoms in red, P atoms in orange. H-bonds are represented by yellow dashed lines, salt bridges by magenta dashed lines.
The nine theoretically possible nucleotide variants at codon 438 are predicted to result in seven different amino acid substitutions (Table 1). More specifically, the wildtype aspartate residue can be converted to either asparagine, histidine, tyrosine, alanine, glycine, valine, or glutamate. We modeled the effects of the seven amino acid substitutions regarding the stability of the OPA1 monomer as well as the dimerization process.
Fig. 4A shows the structural organization of the OPA1 monomer. OPA1 is a transmembrane protein constituted by a N-terminal transmembrane domain, a GTPase domain (residues 263–573) and a long C-terminal helical region, where the last helix (α3HB, residues 938–960) forms, together with the first (α1HB) and last (α2HB) helix of the GTPase domain, a helix bundle (HB) necessary for GTPase activity (39). The overall fold of the GTPase domain, where most of OPA1 missense variants are located (18), is characterized by 9 β-sheets and 13 α-helices arranged in a Rossmann-like, 3-layer, alpha/beta/alpha sandwich fold. The GTPase domain and helix α3HB constitute the Minimal GTPase Domain (OPA1-MGD), which is known to undergo nucleotide-dependent dimerization required for GTP hydrolysis (39). Residue D438 is located on the β4-α3 loop (also called ‘trans stabilizing loop’) at the interface between monomers and is directly involved in dimerization through a double charge-reinforced H-bond with R445 (Fig. 4B) residing on the β4-α3 loop of the symmetric protomer, whose DOA-associated mutations are known to disrupt the dimeric assembly (20,39). Moreover, D438 forms an intramolecular salt-bridge with K468 (G4 loop, Fig. 4B), which interacts with the GDP both directly, via a backbone H-bond with the guanine carbonyl group, and indirectly via an H-bond with the backbone carbonyl of S298 (G1/P-loop) and another H-bond between the backbone NH and the O of the β-phosphate of GDP (Fig. 4B), thus implying an indirect involvement of D438 in GDP binding.
To evaluate the effects of D438 variants on the dimerization process, we reconstituted both monomeric OPA1-MGD using the same artificial linker (GSGSGSGGS) as described in (39) and the functional dimer through the symmetry operators defined in (39), introduced each variant by in silico mutagenesis and monitored the relative stability (∆∆Gfapp) and affinity (∆∆Gbapp) of the complexes both in homo- and heterozygosis.
The analysis of the stability of OPA1-MGD heterozygous dimer (with a single mutation in only one of the protomers) suggested that all substitutions but E438 destabilize complex structure, although to a different extent. N438 and V438 exhibited a milder destabilizing effect (∆∆Gfapp ~ 2 kcal/mol, Table 3) than A438, G438, and H438 (∆∆Gfapp > 11 kcal/mol), nevertheless all such variants displayed a loss of stabilizing electrostatic interactions (Table 3). On the other hand, Y438 exhibited the largest destabilization (∆∆Gfapp > 90 kcal/mol, Table 3), ascribable to major steric clashes of the bulky tyrosine residue in such a packed dimerization interface. At odds with all the other variants, the conservative substitution p.(D438E) exerted a slightly stabilizing effect on OPA1-MGD dimer (∆∆Gfapp ~ 1.4 kcal/mol), as the longer side chain increased the solvent accessibility.
Table 3.
Effects of variants on the apparent relative stability (∆∆Gfapp) and affinity (∆∆Gbapp) of OPA1-MGD dimer in hetero- and homozygosis. The largest positive contributions to ∆∆Gfapp and ∆∆Gbapp are reported together with the energetic term they represent (47). Mutations in italics have been described previously (39)
| Variant | ∆∆Gfapp (kcal/mol) | Largest contribution (kcal/mol) | ∆∆Gbapp (kcal/mol) | Largest contribution (kcal/mol) |
|---|---|---|---|---|
| Heterozygosis | ||||
| p.(D438A) | 11.34 | 40.61 (Electrostatic) | 6.50 | 17.62 (Electrostatic) |
| p.(D438N) | 2.36 | 5.89 (H-bond) | 9.13 | 19.86 (Electrostatic) |
| p.(D438E) | --1.43 | 20.20 (Electrostatic) | 0.31 | 5.69 (Electrostatic) |
| p.(D438G) | 16.71 | 40.87 (Electrostatic) | 6.81 | 17.47 (Electrostatic) |
| p.(D438H) | 50.07 | 35.67 (Electrostatic) | 8.59 | 16.27 (Electrostatic) |
| p.(D438Y) | 96.25 | 62.32 (Van der Waals) | 13.85 | 18.10 (Electrostatic) |
| p.(D438V) | 2.03 | 46.82 (Electrostatic) | 5.77 | 17.77 (Electrostatic) |
| p.(E320A) | 7.99 | 31.58 (Solvation) | 8.12 | 14.03 (Electrostatic) |
| p.(T407A) | 9.63 | 2.48 (Self-contact) | 10.65 | 4.21 (Lipophilic) |
| p.(D442A) | 7.25 | 42.52 (Electrostatic) | 1.67 | 5.09 (Van der Waals) |
| p.(E444A) | 16.21 | 31.32 (Electrostatic) | 8.08 | 10.32 (Solvation) |
| p.(R445A) | 41.48 | 22.09 (Solvation) | 29.59 | 19.41 (Solvation) |
| Homozygosis | ||||
| p.(D438A) | 23.73 | 96.16 (Electrostatic) | 14.08 | 50.18 (Electrostatic) |
| p.(D438N) | 5.64 | 16.19 (Electrostatic) | 19.24 | 54.18 (Electrostatic) |
| p.(D438E) | --3.99 | 41.55 (Electrostatic) | --0.41 | 12.53 (Electrostatic) |
| p.(D438G) | 34.56 | 96.60 (Electrostatic) | 14.79 | 49.79 (Electrostatic) |
| p.(D438H) | 109.36 | 94.98 (Electrostatic) | 16.86 | 58.69 (Electrostatic) |
| p.(D438Y) | 151.42 | 112.36 (Van der Waals) | 22.59 | 48.69 (Electrostatic) |
| p.(D438V) | 5.43 | 108.73 (Electrostatic) | 12.94 | 50.63 (Electrostatic) |
| p.(E320A) | 16.12 | 63.17 (Solvation) | 16.38 | 31.93 (Electrostatic) |
| p.(T407A) | 19.25 | 4.97 (Self-contact) | 21.27 | 8.40 (Lipophilic) |
| p.(D442A) | 14.90 | 85.06 (Electrostatic) | 3.73 | 10.16 (Van der Waals) |
| p.(E444A) | 32.63 | 62.63 (Electrostatic) | 16.37 | 15.52 (Solvation) |
| p.(R445A) | 83.09 | 44.08 (Solvation) | 59.28 | 19.55 (Van der Waals) |
Homozygous dimers harboring mutation on both protomers exhibited a similar behavior in terms of the overall effect of the mutations, although both stabilizing and destabilizing effects were more than 1.5-fold larger (Table 3). As far as dimer affinity is concerned, again all but p.(D438E) variants substantially decreased complex affinity with respect to native OPA1, both in heterozygosis (∆∆Gbapp > 5.77 kcal/mol, Table 3) and in homozygosis (∆∆Gbapp > 12.94 kcal/mol, Table 3). The substitution of the negatively charged aspartate residue in position 438 not only affected locally the H-bonds network by disrupting the charge-reinforced H-bonds with R445 of the symmetric protomer, but also exerted long-range effects. Indeed, the electrostatic potential mapped on the molecular surface of both monomer and dimer (Fig. 5) highlighted the loss of shape and, most importantly, charge complementarity at the protomer interface, as the positively charged R445 remained unbalanced and in proximity of its counterpart on the other protomer, thus resulting in electrostatic repulsion between protomers. Homozygous dimers displayed 1.5-fold larger decrease in affinity, similarly to the effect found on the stability (Table 3). The conservative substitution p.(D438E) displayed again a peculiar behavior, since while the stability of the complex was slightly enhanced with respect to the WT, the affinity showed a minor decrease in the presence of a single mutation and a minor increase in the case of the homozygous dimer. Nevertheless, the overall effect of the p.(D438E) variant on dimer association is almost negligible and consistent with the conservation of the physico-chemical properties of the residue.
Figure 5.
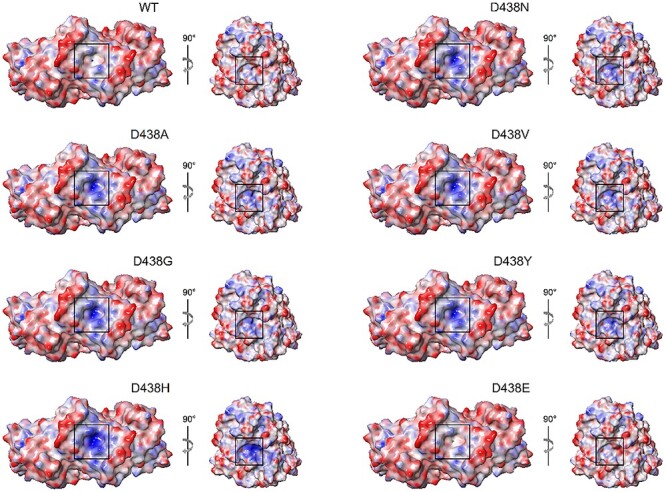
Electrostatic potential mapped on the molecular surface of WT OPA1-MGD dimer (left) and monomer (right) and D438 variants. The molecular surface of the protomers is colored in a blue-to-red scale from −12.5 to 12.5 kT/e. The right side of each panel shows a single protomer rotated clockwise along the z-axis, the surface of residue 438 is framed in black.
To validate the effects of D438 substitutions on the dimerization, we performed the same structural analysis on the Ala variants described in Yu et al. (39), namely p.(E320A), p.(T407A), p.(D442A), p.(E444A) and p.(E445A), which were experimentally proven to abolish dimer formation under conditions that mimic the physiological ones. All Ala substitutions resulted in a significant destabilization of the dimeric assembly and displayed a marked decrease of dimer affinity both in hetero and homozygosis (Table 3), though to a different extent. The ∆∆Gbapp values of Ala variants ranged between 1.67 and 29.59 kcal/mol under heterozygous conditions and between 3.73 and 59.28 kcal/mol in homozygosis (Table 3). Since all ∆∆Gbapp values of D438 variants (with the exception of the p.(D438E) substitution) were found to fall within such ranges, we conclude that non-conservative missense mutations of D438 are likely to prevent the formation of the dimeric complex.
Discussion
In this study, we have analyzed all theoretically possible variants at codon 438 of the OPA1 mRNA transcript variant 1 using in silico splice predictions, heterologous splice assays, analysis of patient’s RNA when available, and protein modeling.
Since we have analyzed only few variants in a single gene, we cannot determine which algorithm is the best performing splice prediction tool. The main purpose of splice prediction programs, of course, is to accurately predict the effect of a putative splice variant, thereby eliminating the need for functional analysis. To do so, cut-off values in % score change need to be defined for each algorithm. However, it is difficult to establish consensus cut-off values. Studies that evaluated sensitivity and specificity of different algorithms have proposed specific cut-off values. For instance, the use of 15% and 5% as cut-off values have been suggested for MaxEntScan and SSF-like, respectively (40). These seem to be valid cut-off values as SSF-like predicted all variants at positions c.1313 and c.1314 with score changes below 5%. However, MaxEntScan predicted all three variants at position c.1314 with score changes ranging from 15.0–17.5%, which rather supports the use of 20% as a cut-off value in our study. Our results once more demonstrate how important it is to use different algorithms for in silico splice prediction, as they have individual strengths and weaknesses. In any case, experimental validation of putative splice variants is still needed, as it is not recommended to use bioinformatic predictions as the sole source of evidence for variant classification (7).
The key result of our study is that variants at the split codon 438 in OPA1 act either by missplicing (variants at the first codon position) or by an alteration of the amino acid sequence leading to impaired GTPase function (variants at the second and third codon position). Because of the small number of patients, it is not possible to draw any conclusions with respect to genotype–phenotype correlations. We can only state that, in our study, codon 438 variants at the second position (c.1313A > C, c.1313A > G, c.1313A > T), which act via amino acid exchange do not lead to a more severe (i.e. syndromic) phenotype than the codon variant at the first position (c.1312G > C), which leads to exon skipping. The question remains why only variants of the first nucleotide of codon 438 act on splicing.
It is well known that not only the invariant dinucleotides at both ends of an intron contribute to splicing. A pioneering study on the influence of nonessential sites near splice junctions was performed by Zhang and colleagues who could demonstrate that intronic positions +3 to +6 as well as the first and the last position of an exon are significantly intolerant of mutations, with the last position of an exon being even more intolerant of substitutions than variants at the first position of an exon (4). Moreover, they could show that this intolerance is nucleotide specific, meaning that the consensus nucleotide at these positions is less tolerant to mutational changes than the other three nucleotides at the same positions. Their findings are in line with our observation that all variants at the first nucleotide of codon 438, which is consistent with the consensus, lead to missplicing. Overall, the entire donor splice site of exon 13 is highly consistent with the consensus which is reflected by the high scores obtained by splice site prediction algorithms (Supplementary Material, Table S2). In contrast, the acceptor splice site of exon 13 obtained lower scores. There are no consensus thresholds for scores that would allow a simple categorization of splice sites into ‘strong’ or ‘weak’. In this study, we defined a splice site as strong if its scores reached more than 75% of the respective maximum score in at least two splice site prediction algorithms. Following this approach, we considered the acceptor site of exon 13 as weak but its donor as strong. Accordingly, both splice sites of exon 14 were considered strong (Supplementary Material, Table S2).
In a recent study, Khan and colleagues have addressed splice site interdependency and could demonstrate that the recognition of an exon (i.e. its inclusion in the transcript) strongly depends on the combined strengths of its acceptor and donor splice sites (41). More specifically, they were able to rescue exon skipping effects of several splicing variants by increasing the strength of the other splice site of the affected exon. One conclusion that can be drawn from the study of Khan and colleagues is that exons with a weak splice site can still be recognized by the splicing machinery if their other splice site is strong. As a consequence, a decrease of the strong splice site by mutational alteration will impair exon recognition and result in skipping of the exon from the transcript. In an attempt to replicate the findings of Khan and colleagues, we aimed to analyse the effects of a strengthened acceptor splice site of exon 13 on exon skipping. Two nucleotide substitutions, namely c.1213-3A > T and c.1213A > G were predicted in silico to strengthen the acceptor splice site, in particular the combination of both substitutions (Supplementary Material, Table S3). We used the wildtype as well as the c.1312G > C minigene construct to introduce these substitutions by site-directed mutagenesis, either alone or in combination. Transfection of HEK293T cells and subsequent RT-PCR revealed a small amount (10%) of correctly spliced product, but only in the presence of both acceptor modifications (Supplementary Material, Fig. S3). The effect (i.e. restoration of correct splicing) in our study was much smaller than the effects obtained by Khan and colleagues, who analysed five non-canonical splice site variants that could be completely rescued (41). Apparently, the interdependence of splice sites of OPA1 exon 13 is too complex for a straightforward restoration of splicing that has been demonstrated by Khan and colleagues.
In contrast to the skipping of exon 13 caused by variants at the first position of codon 438, variants at the second and third position did not result in the skipping of exon 14 in our minigene assays. This could be confirmed in vivo when patient RNA was available. Both splice sites of exon 14 can be considered strong, even though the first nucleotide of exon 14 is not consistent with the consensus (Fig. 1). The nucleotide specificity demonstrated by Zhang and colleagues (4) could explain why in our study mutational alterations of the non-consensus reference nucleotide at the first exonic position did not affect splicing. The pathogenicity of these variants is solely due to the amino acid substitutions they cause. Previous attempts to model amino acid substitutions at codon 438 have shown that an exchange of aspartate with glycine (p.(D438G)) or valine (p.(D438V)) abolishes the possibility to form hydrogen bonds through their side chains, thereby possibly causing a misalignment of GTP in the GTP-binding pocket of OPA1’s GTPase domain (27). In our study, based on the experimental structure of the OPA1-MGD dimer in its GDP-bound form, such predictions on the effects of G438 and V438 were confirmed to destabilize the structure of the protomer and decrease the affinity of the dimeric complex (Table 3). Moreover, also the other possible mutation at the second position of the codon (c.1313A > C), resulting in the p.(D438A) variant, was found to exert comparable effects on dimer affinity and stability (Table 3), with a similar molecular mechanism consisting of unfavorable charge coupling leading to electrostatic repulsion between the protomers (Fig. 5). Such decrease in affinity is comparable to that exhibited by Ala variants (Table 3) preventing dimer formation (39), thus pointing towards a similar effect on protomer association exerted by D438 mutants. Interestingly, the only missense mutation involving the third nucleotide of codon 438 (c.1314 T > A/G, p.(D438E)) improved complex stability with minor alterations of the affinity depending on the genetic nature of the assembly, that is in hetero- or homozygosis (Table 3). Nevertheless, it is worth noticing that, so far, the p.(D438E) variant has not been found in patients diagnosed with DOA.
Currently, there is no treatment or therapy for OPA1-associated DOA. Haploinsufficiency is considered the major disease mechanism, which is supported by genetic evidence such as excess of loss-of-function mutations as well as reduced protein levels in patient-derived fibroblasts (17,19). Hence, gene supplementation approaches seem to be the method of choice. However, they are hampered by the presence of a variety of functionally important OPA1 isoforms and the requirement of a well-balanced protein expression (15,16). In addition, gene supplementation is not suited for those patients that harbor missense variants acting in a dominant negative fashion. The mutation spectrum of OPA1 also comprises a considerable number of variants acting on splicing. Hence, techniques that focus on a restoration of splicing (e.g. by modified splice factors or trans-splicing), are considered a promising therapeutic approach as well. However, restoration of splicing may not be sufficient if the splicing defect is caused by a coding exonic variant. In this study, we could demonstrate that variants at the first position of OPA1 codon 438 cause missplicing. In addition, the predicted amino acid substitutions p.(D438N), p.(D438H), and p.(D438Y) were shown to destabilize the dimeric complex with detrimental effects on the affinity, mainly ascribable to electrostatic repulsion among protomers (Fig. 5 and Table 3). In detail, while the decrease in affinity was comparable between the three variants (8.59 to 13.85 kcal/mol in heterozygosis and 16.86 to 22.59 kcal/mol in homozygosis), the destabilizing effect was > 25-fold higher for variants p.(D438Y) and p.(D438H) (96.25 and 50.07 kcal/mol in heterozygosis; 151.42 and 109.36 kcal/mol in homozygosis, respectively) with respect to the N substitution (2.36 and 5.64 kcal/mol in hetero- and homozygosis, respectively). These predictions imply that, even if splicing could be restored, the mutant alleles would still exert a negative effect due to the altered amino acid sequence. For such cases, gene editing approaches that specifically change single nucleotides (i.e. base editors) may be the only possible gene therapeutic approach.
To conclude, we analyzed nine individual nucleotide changes at a frequently mutated split codon in the GTPase-encoding domain of OPA1 from their effect on splicing to their impact on enzyme function and protein dimerization. While gene therapy is not yet available as a treatment option for OPA1-associated DOA patients, a pre-clinical study in a mouse model for DOA has raised new hopes (42). Quite likely, one therapeutic approach will not be sufficient to address all patients because the mutation spectrum and the underlying pathomechanisms in OPA1-associated DOA are so diverse. RNA analyses are essential for understanding the functional outcome of variants, especially for missense variants that may exert a pathogenic effect through a splicing defect rather than through an amino acid change. Until bioinformatic tools are able to make unambiguous predictions, functional analyses to determine the exact molecular mechanism of a variant are essential for a correct assignment of patients to future precision therapies.
Materials and Methods
Clinical diagnosis
The diagnosis of DOA was based on a history of gradual, bilateral loss of vision associated with the presence of central or caeco-central scotoma on visual field evaluation and symmetric temporal or diffuse optic disc pallor (31). Considering the known reduced penetrance of OPA1-associated DOA, a pedigree demonstrating autosomal dominant inheritance was not a prerequisite for diagnosis.
Patient Enrollment and retrieval of blood samples
Patient blood samples were recruited in accordance with the principles of the Declaration of Helsinki. All patients provided informed written consent to use their medical records and samples for research purposes. Specifically, this study was approved by the institutional review board of the Ethics Committee of the University Hospital of Tübingen under the study numbers 112/2001, 598/2011BO1 and 637/2017BO1.
Genomic DNA was extracted from venous blood samples applying standard salting-out procedure or by using the chemagic™ MSM1 system and the chemagic™ DNA Blood 7 k Kit (Chemagen, Baesweiler, Germany). For RNA isolation venous blood was collected in PAXgene blood RNA tubes and RNA was isolated using the PAXgene blood RNA Kit (Qiagen, Hilden, Germany). Alternatively, leukocytes were isolated from venous blood by Ficoll-Paque density centrifugation (Pharmacia Biotech, Freiburg, Germany) and total RNA was extracted with Trizol reagent (Life Technologies, Eggenstein, Germany).
Variant nomenclature
Variant nomenclature in this study is in accordance with Human Genome Variation Society recommendations (43) and based on GenBank accession numbers NM_015560.2 and NP_056375.2, respectively. Transcript NM_015560.2 (also often designated as transcript 1) differs from transcript NM_130837.2 (also often designated as variant 8), which represents the longest transcript and comprises two alternatively spliced exons. We refer to transcript NM_015560.2 throughout the manuscript. Consecutive exon numbering for this transcript is 1–28. It encodes an isoform of 960 amino acids. For the sake of brevity, in the following, we refer to the codon defined by nucleotides c.1312_1314 as codon 438. The first nucleotide of the codon corresponds to nucleotide position c.1312 and thus to the last position of exon 13 (Fig. 1). The second and the third position of the codon correspond to nucleotide positions c.1313 and c.1314, and thus to the first and second position of exon 14, respectively (Fig. 1).
In silico analysis
Splice site predictions were preformed using the Alamut visual software (v.2.12, Interactive Biosoftware, Rouen, France) using default settings, and the SpliceAI (35) lookup tool from the Broad Institute (https://spliceailookup.broadinstitute.org/). Orthologous gene sequences were downloaded from NCBI (https://www.ncbi.nlm.nih.gov/), and amino acid sequences aligned using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) (44).
Mutant minigene generation and in vitro splice assays
In vitro splice assays were performed as described previously (45). Briefly, a genomic segment of the OPA1 gene (GrCh37/hg19 3: 193,361,041-193,363,713; corresponding to exons 12–16 and flanking intronic sequences) was amplified from genomic DNA from a healthy control subject using a proofreading polymerase and cloned into the pSPL3 minigene plasmid vector. After verifying that this wildtype minigene construct generated normally spliced RNA, all nine theoretically possible variants at codon 438 were introduced by site-directed mutagenesis. For rescue experiments, two nucleotide substitutions of the acceptor site of exon 13 were introduced. Mutagenesis primers were designed following the guidelines of the QuikChange® Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, United States). The mutagenesis PCR reaction mixture (50 μL) contained 0.5 μM of each forward and reverse primer, 0.2 mM dNTPs, 1 unit Pfu Ultra High Fidelity Polymerase (Agilent, Waldbronn, Germany), 1× 10× Pfu Ultra HF reaction buffer, and 20 ng wild-type entry clone. All PCRs were performed using the following thermocycling conditions: initial denaturation at 95°C for 2 min; 16 cycles of denaturation at 95°C for 30 sec each, annealing at 55°C for 30 sec each, and extension at 68°C for 20 min each, with a final extension at 68°C for 20 min. PCR-amplified products were incubated with DpnI restriction enzyme (New England Biolabs, Frankfurt, Germany) for 2 hr to remove the wild-type plasmid DNA. Digested products were run on a 1% (w/v) agarose gel to check for the presence of a single linear fragment, corresponding to the mutant construct. Sanger sequencing was used to verify the presence of the introduced substitution and the integrity of the minigene. The resulting minigene constructs in their wildtype and mutant versions were used to transfect HEK293T/17 cells (ATCC® CRL-11268™), which were then analyzed with respect to splicing of minigene-derived transcripts using reverse transcription polymerase chain reaction (RT-PCR). Ratios of misspliced and correctly spliced RT-PCR products were determined by densitometric analysis using Image J software after gel electrophoresis as described elsewhere (46). Primers for site-directed mutagenesis, PCR amplification, cDNA synthesis, and RT-PCR are given in Supplementary Material, Table S1.
Direct mRNA analysis from blood
Four hundred ng of total RNA isolated from blood samples was used for cDNA synthesis using random hexamers and the Maxima H Minus Reverse Transcriptase Kit according to the manufacturer’s protocol (Thermo Fisher Scientific, Carlsbad, USA). RT-PCR was performed using 2 μL cDNA, a forward primer located in exon 11 and a reverse primer located in exon 17 (see Supplementary Material, Table S1). For quantitative analysis, RT-PCR was performed using the same primers, but the reverse primer was 5′ FAM (6-carboxyfluorescein) labeled.
FAM-labeled RT-PCR products were mixed with 0.5 μL of GeneScan ROX500 size standard (Life Technologies, Darmstadt, Germany) and 8.5 μL of Hi-Di Formamide (Life Technologies) in a total volume of 10 μL. Mixes were separated by capillary electrophoresis on an ABI 3130XL Genetic Analyzer instrument (Life Technologies). The area-under-the-curve (AUC) was calculated with GeneMapper 5 software (Life Technologies). Ratios of RT-PCR products were determined as the AUC for individual peaks divided by the sum of AUC of all peaks.
Subcloning of RT-PCR products was performed using the NEB® PCR Cloning Kit (New England Biolabs, Frankfurt, Germany) according to manufacturer’s instructions.
Molecular modeling of OPA1 GTPase domain and structural analysis of D438 variants
The molecular modeling and structural analysis of the Minimal OPA1 GTPase domain (OPA1-MGD) was carried out on Maestro/Bioluminate (Schroedinger, New York, USA) suite. The three-dimensional structure of GDP-bound human OPA1-MGD was modeled on the 6JTG template (39) with a resolution of 2.4 Å, containing both the atomic coordinates and the symmetric operators which allowed the reconstitution of the dimeric biological unit. OPA1-MGD structure was prepared following the Protein Preparation tool protocol. In detail, bond orders were assigned according to the Chemical Components Dictionary database (www.pdb.org, wwPDB Foundation, Piscataway NJ, USA), H-atoms were added, zero-order bonds to metals were created and the artificial linker GSGSGSGGS (39) connecting the C-terminus of helix α2HB (residue N580) and the N-terminus of helix α3HB of the helix bundle (HB) domain (residue D938) was modeled by Prime, together with the missing sidechains. The structure was refined by sampling water orientation, prediction of the protonation state of heteroatoms by Epik and of ionizable residues by PROPKA at pH 7.5, H-bonds assignment and optimization using crystal symmetry for protomer-protomer interactions. Protein energy minimization was finally carried out using OPLS4 forcefield (Schroedinger, New York, USA) by setting the threshold of Root-Mean Square Deviation of heavy atoms to 0.3 Å.
All seven possible aminoacidic substitutions corresponding to D438 nucleotide variants, namely A, V, G, N, Y, E, and H, were introduced using Bioluminate’s Residue scanning tool in either one of the two protomers, mimicking the heterozygous condition, or in both protomers, mimicking the homozygous assembly. For each variant the most probable rotamer was automatically selected prior to minimization, which was carried out with the same parameter as for the wildtype.
For each OPA1 dimer carrying one of the seven theoretical missense mutations, we calculated the effects of amino acid replacement on both folding (∆∆Gfapp) and binding (∆∆Gbapp) in terms of variation of the Gibbs free energy with respect to the wildtype. Computations were done with Molecular Mechanics/Generalized Born and Surface Area Continuum solvation (MM/GBSA) method and comparisons with the wildtype made use of the variant-specific thermodynamic cycle. As the free energy is calculated using MM forcefield, which does not consider the explicit contribution rising from conformational changes, the free energy variations (∆∆Gapp in Table 3, expressed in kcal/mol) should be considered ‘apparent’ values and not precise thermodynamic quantities. Nevertheless, such values allow a reliable comparison of the variation in free energy associated with each mutant. Dimer affinity variations (∆∆Gbapp) were calculated for each of the two protomers against the other, but since the dimer is symmetric, numerical differences were below the precision of the calculation in almost all cases, therefore the standard deviation is not reported in Table 3. The largest positive contribution to either ∆∆Gfapp or ∆∆Gbapp reported in Table 3 refers to the forcefield term yielding the largest effect on ∆∆Gfapp and ∆∆Gbapp. The electrostatic potential mapped on the molecular surface of OPA1 variants shown in Figure 5 was calculated by Bioluminate’s Poisson-Boltzmann Electrostatic Potential Surface tool, using the default parameters, namely solute dielectric constant = 1, solvent dielectric constant = 80, solvent radius = 1.4 Å, temperature = 298 K, grid extension = 5 Å.
Supplementary Material
Acknowledgements
This work was supported by the University of Verona [Joint project 2018, grant no. JPVR184ZZ5 to D.DO] and the Italian Ministry of Education, University and Research [PRIN 2017 201744NR8S ‘Integrative tools for defining the molecular basis of the diseases’ to D.DO].
Conflict of Interest statement. The authors declare no conflicts of interest.
Contributor Information
Nicole Weisschuh, Institute for Ophthalmic Research, Centre for Ophthalmology, University of Tübingen, Tübingen 72076, Germany.
Valerio Marino, Department of Neurosciences, Biomedicine and Movement Sciences, Section of Biological Chemistry, University of Verona, Verona 37134, Italy.
Karin Schäferhoff, Institute of Medical Genetics and Applied Genomics, University of Tübingen, Tübingen 72076, Germany.
Paul Richter, University Eye Hospital, Centre for Ophthalmology, University of Tübingen, Tübingen 72076, Germany.
Joohyun Park, Institute of Medical Genetics and Applied Genomics, University of Tübingen, Tübingen 72076, Germany.
Tobias B Haack, Institute of Medical Genetics and Applied Genomics, University of Tübingen, Tübingen 72076, Germany; Centre for Rare Diseases, University of Tübingen, Tübingen 72076, Germany.
Daniele Dell’Orco, Department of Neurosciences, Biomedicine and Movement Sciences, Section of Biological Chemistry, University of Verona, Verona 37134, Italy.
References
- 1. Gilbert, W. (1978) Why genes in pieces? Nature, 271, 501. [DOI] [PubMed] [Google Scholar]
- 2. Burset, M., Seledtsov, I.A. and Solovyev, V.V. (2000) Analysis of canonical and non-canonical splice sites in mammalian genomes. Nucleic Acids Res., 28, 4364–4375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cartegni, L., Chew, S.L. and Krainer, A.R. (2002) Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat. Rev. Genet., 3, 285–298. [DOI] [PubMed] [Google Scholar]
- 4. Zhang, S., Samocha, K.E., Rivas, M.A., Karczewski, K.J., Daly, E., Schmandt, B., Neale, B.M., Mac Arthur, D.G. and Daly, M.J. (2018) Base-specific mutational intolerance near splice sites clarifies the role of nonessential splice nucleotides. Genome Res., 28, 968–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lim, K.H., Ferraris, L., Filloux, M.E., Raphael, B.J. and Fairbrother, W.G. (2011) Using positional distribution to identify splicing elements and predict pre-mRNA processing defects in human genes. Proc. Natl. Acad. Sci. U. S. A., 108, 11093–11098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Abramovicz, A. and Gos, M. (2018) Splicing mutations in human genetic disorders: examples, detection, and confirmation. J. Appl. Genet., 59, 253–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., Grody, W.W., Hegde, M., Lyon, E., Spector, E.et al. (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med., 17, 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Frischmeyer, P.A. and Dietz, H.C. (1999) Nonsense-mediated mRNA decay in health and disease. Hum. Mol. Genet., 8, 1893–1900. [DOI] [PubMed] [Google Scholar]
- 9. Mort, M., Sterne-Weiler, T., Li, B., Ball, E.V., Cooper, D.N., Radivojac, P., Sanford, J.R. and Mooney, S.D. (2014) MutPred splice: machine learning-based prediction of exonic variants that disrupt splicing. Genome Biol., 15, R19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stenson, P.D., Mort, M., Ball, E.V., Chapman, M., Evans, K., Azevedo, L., Hayden, M., Heywood, S., Millar, D.S., Phillips, A.D.et al. (2020) The human gene mutation database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum. Genet., 139, 1197–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alexander, C., Votruba, M., Pesch, U.E., Thiselton, D.L., Mayer, S., Moore, A., Rodriguez, M., Kellner, U., Leo-Kottler, B., Auburger, G.et al. (2000) OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet., 26, 211–215. [DOI] [PubMed] [Google Scholar]
- 12. Delettre, C., Lenaers, G., Griffoin, J.M., Gigarel, N., Lorenzo, C., Belenguer, P., Pelloquin, L., Grosgeorge, J., Turc-Carel, C., Perret, E.et al. (2000) Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet., 26, 207–210. [DOI] [PubMed] [Google Scholar]
- 13. Olichon, A., Baricault, L., Gas, N., Guillou, E., Valette, A., Belenguer, P. and Lenaers, G. (2003) Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem., 278, 7743–7746. [DOI] [PubMed] [Google Scholar]
- 14. Frezza, C., Cipolat, S., Martins de Brito, O., Micaroni, M., Beznoussenko, G.V., Rudka, T., Bartoli, D., Polishuck, R.S., Danial, N.N., De Strooper, B.et al. (2006) OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell, 126, 177–189. [DOI] [PubMed] [Google Scholar]
- 15. Olichon, A., Guillou, E., Delettre, C., Landes, T., Arnauné-Pelloquin, L., Emorine, L.J., Mils, V., Daloyau, M., Hamel, C., Amati-Bonneau, P.et al. (2006) Mitochondrial dynamics and disease, OPA1. Biochim. Biophys. Acta, 1763, 500–509. [DOI] [PubMed] [Google Scholar]
- 16. Del Dotto, V., Mishra, P., Vidoni, S., Fogazza, M., Maresca, A., Caporali, L., McCaffery, J.M., Cappelletti, M., Baruffini, E., Lenaers, G.et al. (2017) OPA1 isoforms in the hierarchical Organization of Mitochondrial Functions. Cell Rep., 19, 2557–2571. [DOI] [PubMed] [Google Scholar]
- 17. Zanna, C., Ghelli, A., Porcelli, A.M., Karbowski, M., Youle, R.J., Schimpf, S., Wissinger, B., Pinti, M., Cossarizza, A., Vidoni, S.et al. (2008) OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain, 131, 352–367. [DOI] [PubMed] [Google Scholar]
- 18. Le Roux, B., Lenaers, G., Zanlonghi, X., Amati-Bonneau, P., Chabrun, F., Foulonneau, T., Caignard, A., Leruez, S., Gohier, P., Procaccio, V.et al. (2019) OPA1: 516 unique variants and 831 patients registered in an updated centralized Variome database. Orphanet J. Rare Dis., 14, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lenaers, G., Neutzner, A., Le Dantec, Y., Jüschke, C., Xiao, T., Decembrini, S., Swirski, S., Kieninger, S., Agca, C., Kim, U.S.et al. (2020) Dominant optic atrophy: culprit mitochondria in the optic nerve. Prog. Retin. Eye Res., 100935. [DOI] [PubMed] [Google Scholar]
- 20. Amati-Bonneau, P., Valentino, M.L., Reynier, P., Gallardo, M.E., Bornstein, B., Boissière, A., Campos, Y., Rivera, H., de laAleja, J.G., Carroccia, R.et al. (2008) OPA1 mutations induce mitochondrial DNA instability and optic atrophy 'plus' phenotypes. Brain, 131, 338–351. [DOI] [PubMed] [Google Scholar]
- 21. Del Dotto, V., Fogazza, M., Musiani, F., Maresca, A., Aleo, S.J., Caporali, L., La Morgia, C., Nolli, C., Lodi, T., Goffrini, P.et al. (2018) Deciphering OPA1 mutations pathogenicity by combined analysis of human, mouse and yeast cell models. Biochim. Biophys. Acta Mol. basis Dis., 1864, 3496–3514. [DOI] [PubMed] [Google Scholar]
- 22. Chao de la Barca, J.M., Fogazza, M., Rugolo, M., Chupin, S., Del Dotto, V., Ghelli, A.M., Carelli, V., Simard, G., Procaccio, V., Bonneau, D.et al. (2020) Metabolomics hallmarks OPA1 variants correlating with their in vitro phenotype and predicting clinical severity. Hum. Mol. Genet., 29, 1319–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yu-Wai-Man, P., Griffiths, P.G., Gorman, G.S., Lourenco, C.M., Wright, A.F., Auer-Grumbach, M., Toscano, A., Musumeci, O., Valentino, M.L., Caporali, L.et al. (2010) Multi-system neurological disease is common in patients with OPA1 mutations. Brain, 133, 771–786s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sherry, S.T., Ward, M.H., Kholodov, M., Baker, J., Phan, L., Smigielski, E.M. and Sirotkin, K. (2001) dbSNP: the NCBI database of genetic variation. Nucleic Acids Res., 29, 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Landrum, M.J., Lee, J.M., Benson, M., Brown, G.R., Chao, C., Chitipiralla, S., Gu, B., Hart, J., Hoffman, D., Jang, W.et al. (2018) ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res., 46, D1062–D1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Almind, G.J., Ek, J., Rosenberg, T., Eiberg, H., Larsen, M., Lucamp, L., Brøndum-Nielsen, K. and Grønskov, K. (2012) Dominant optic atrophy in Denmark - report of 15 novel mutations in OPA1, using a strategy with a detection rate of 90%. BMC Med. Genet., 13, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dadgar, S., Hagens, O., Dadgar, S.R., Haghighi, E.N., Schimpf, S., Wissinger, B. and Garshasbi, M. (2006) Structural model of the OPA1 GTPase domain may explain the molecular consequences of a novel mutation in a family with autosomal dominant optic atrophy. Exp. Eye Res., 83, 702–706. [DOI] [PubMed] [Google Scholar]
- 28. Mei, S., Huang, X., Cheng, L., Peng, S., Zhu, T., Chen, L., Wang, Y. and Zhao, J. (2019) A missense mutation in OPA1 causes dominant optic atrophy in a Chinese family. J. Ophthalmol., 2019, 1424928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pesch, U.E., Leo-Kottler, B., Mayer, S., Jurklies, B., Kellner, U., Apfelstedt-Sylla, E., Zrenner, E., Alexander, C. and Wissinger, B. (2001) OPA1 mutations in patients with autosomal dominant optic atrophy and evidence for semi-dominant inheritance. Hum. Mol. Genet., 10, 1359–1368. [DOI] [PubMed] [Google Scholar]
- 30. Karczewski, K.J., Francioli, L.C., Tiao, G., Cummings, B.B., Alföldi, J., Wang, Q., Collins, R.L., Laricchia, K.M., Ganna, A., Birnbaum, D.P.et al. (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Weisschuh, N., Schimpf-Linzenbold, S., Mazzola, P., Kieninger, S., Xiao, T., Kellner, U., Neuhann, T., Kelbsch, C., Tonagel, F., Wilhelm, H.et al. (2021) Mutation spectrum of the OPA1 gene in a large cohort of patients with suspected dominant optic atrophy: identification and classification of 48 novel variants. PLoS One, 16, e0253987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pertea, M., Lin, X. and Salzberg, S.L. (2001) GeneSplicer: a new computational method for splice site prediction. Nucleic Acids Res., 29, 1185–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jian, X., Boerwinkle, E. and Liu, X. (2014) In silico tools for splicing defect prediction: a survey from the viewpoint of end users. Genet. Med., 16, 497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Moles-Fernández, A., Duran-Lozano, L., Montalban, G., Bonache, S., López-Perolio, I., Menéndez, M., Santamariña, M., Behar, R., Blanco, A., Carrasco, E.et al. (2018) Computational tools for splicing defect prediction in breast/ovarian cancer genes: how efficient are they at predicting RNA alterations? Front. Genet., 9, 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jaganathan, K., Kyriazopoulou Panagiotopoulou, S., McRae, J.F., Darbandi, S.F., Knowles, D., Li, Y.I., Kosmicki, J.A., Arbelaez, J., Cui, W., Schwartz, G.B.et al. (2019) Predicting splicing from primary sequence with deep learning. Cell, 176, 535–548.e24. [DOI] [PubMed] [Google Scholar]
- 36. Shapiro, M.B. and Senapathy, P. (1987) RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res., 15, 7155–7174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yeo, G. and Burge, C.B. (2004) Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol., 11, 377–394. [DOI] [PubMed] [Google Scholar]
- 38. Reese, M.G., Eeckman, F.H., Kulp, D. and Haussler, D. (1997) Improved splice site detection in genie. J. Comput. Biol., 4, 311–323. [DOI] [PubMed] [Google Scholar]
- 39. Yu, C., Zhao, J., Yan, L., Qi, Y., Guo, X., Lou, Z., Hu, J. and Rao, Z. (2020) Structural insights into G domain dimerization and pathogenic mutation of OPA1. J. Cell Biol., 219, e201907098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Houdayer, C., Caux-Moncoutier, V., Krieger, S., Barrois, M., Bonnet, F., Bourdon, V., Bronner, M., Buisson, M., Coulet, F., Gaildrat, P.et al. (2012) Guidelines for splicing analysis in molecular diagnosis derived from a set of 327 combined in silico/in vitro studies on BRCA1 and BRCA2 variants. Hum. Mutat., 33, 1228–1238. [DOI] [PubMed] [Google Scholar]
- 41. Khan, M., Cornelis, S.S., Sangermano, R., Post, I.J.M., Groesbeek, A.J., Amsu, J., Gilissen, C., Garanto, A., Collin, R.W.J. and Cremers, F.P.M. (2020) In or out? New insights on exon recognition through splice-site interdependency. Int. J. Mol. Sci., 21, 2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sarzi, E., Seveno, M., Piro-Mégy, C., Elzière, L., Quilès, M., Péquignot, M., Müller, A., Hamel, C.P., Lenaers, G. and Delettre, C. (2018) OPA1 gene therapy prevents retinal ganglion cell loss in a dominant optic atrophy mouse model. Sci. Rep., 8, 2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. den Dunnen, J.T., Dalgleish, R., Maglott, D.R., Hart, R.K., Greenblatt, M.S., McGowan-Jordan, J., Roux, A.F., Smith, T., Antonarakis, S.E. and Taschner, P.E. (2016) HGVS recommendations for the description of sequence variants: 2016 update. Hum. Mutat., 37, 564–569. [DOI] [PubMed] [Google Scholar]
- 44. Sievers, F., Wilm, A., Dineen, D.G., Gibson, T.J., Karplus, K., Li, W., Lopez, R., McWilliam, H., Remmert, M., Söding, J.et al. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal omega. Mol. Syst. Biol., 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Weisschuh, N., Wissinger, B. and Gramer, E. (2012) A splice site mutation in the PAX6 gene which induces exon skipping causes autosomal dominant inherited aniridia. Mol. Vis., 18, 751–757. [PMC free article] [PubMed] [Google Scholar]
- 46. Schneider, C.A., Rasband, W.S. and Eliceiri, K.W. (2012) NIH image to ImageJ: 25 years of image analysis. Nat. Methods, 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li, J., Abel, R., Zhu, K., Cao, Y., Zhao, S. and Friesner, R.A. (2011) The VSGB 2.0 model: a next generation energy model for high resolution protein structure modeling. Proteins, 79, 2794–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.