

Abstract
Epidemic obesity is the most important risk factor for prediabetes and type 2 diabetes (T2D) in youth as it is in adults. Obesity shares pathophysiological mechanisms with T2D and is likely to share part of the genetic background. We aimed to test if weighted genetic risk scores (GRSs) for T2D, fasting glucose (FG) and fasting insulin (FI) predict glycaemic traits and if there is a causal relationship between obesity and impaired glucose metabolism in children and adolescents. Genotyping of 42 SNPs established by genome-wide association studies for T2D, FG and FI was performed in 1660 Italian youths aged between 2 and 19 years. We defined GRS for T2D, FG and FI and tested their effects on glycaemic traits, including FG, FI, indices of insulin resistance/beta cell function and body mass index (BMI). We evaluated causal relationships between obesity and FG/FI using one-sample Mendelian randomization analyses in both directions. GRS-FG was associated with FG (beta = 0.075 mmol/l, SE = 0.011, P = 1.58 × 10−11) and beta cell function (beta = −0.041, SE = 0.0090 P = 5.13 × 10−6). GRS-T2D also demonstrated an association with beta cell function (beta = −0.020, SE = 0.021 P = 0.030). We detected a causal effect of increased BMI on levels of FI in Italian youths (beta = 0.31 ln (pmol/l), 95%CI [0.078, 0.54], P = 0.0085), while there was no effect of FG/FI levels on BMI. Our results demonstrate that the glycaemic and T2D risk genetic variants contribute to higher FG and FI levels and decreased beta cell function in children and adolescents. The causal effects of adiposity on increased insulin resistance are detectable from childhood age.
Introduction
Childhood obesity is growing around the globe. In some developed countries, the disease incidence plateaued; nonetheless, the rate of severe obesity had increased worldwide (1). In Italy, the prevalence of overweight and 3rd grade obese children has decreased from 35.2% in 2008 to 30.6% in 2016, while the rate of severely obese has reached 2.1% (2). This caused the onset of health conditions previously considered exclusively adult diseases, such as prediabetes (impaired fasting glucose, IFG, and glucose tolerance, IGT) and type 2 diabetes (T2D), at an earlier age.
Obesity, especially in children, is a major risk factor of T2D and urges its study for diabetes prevention (3). Strong evidence suggests that pathogenic mechanisms are shared between obesity, prediabetes and T2D as well as causal effect of body adiposity on hyperinsulinaemia and T2D in adults (4). The rates of their occurrence differ among ethnicities, consistent with differences in genetic susceptibility to T2D, currently less characterized in youths than in adults. In Italy, 3% of adolescents with moderate and severe obesity present IFG and 5% IGT, while T2D is diagnosed in less than 0.5% of adolescents (5). An increased amount of adipose tissue leads to more severe insulin resistance (IR) via different mechanisms (i.e. lipotoxicity, release of some adipokines etc.), thus promoting the development of T2D (6). The early-life adiposity levels correlate with adulthood measures and might therefore represent a longitudinal causal risk factor for adult metabolic health deterioration, which requires further insight from studies dissecting health of young individuals (7).
To date, over 400 genetic variants have been implicated in the development of T2D and more than 500 genomic loci were uniquely associated with body mass index (BMI) (8–11). In general, these loci have small effect sizes, explaining approximately 6% of trait variance and disease susceptibility (12) when combined. Studies in children (13) and young adults (14–20) have demonstrated that genetic risk scores (GRSs) show stronger predictive ability in younger individuals than in older ones (21–25). Several obesity genes harbour loci that are associated with T2D but affect T2D susceptibility largely through their effect on BMI via increased IR (e.g. FTO and MC4R gene variants) (26). However, their effects on glucose homeostasis might be overestimated in the context of obesity (27).
Dissection of the genetic effects on the quantitative endophenotypes of T2D, including fasting glucose (FG), and insulin (FI), indices of beta cell function (HOMA-B) and IR (HOMA-IR) in individuals without diabetes, helps uncovering the disease pathophysiology (28). The Meta-Analyses of Glucose and Insulin-related traits Consortium (MAGIC) identified sixteen loci associated with FG/HOMA-B, and two loci associated with FI/HOMA-IR (29). Additional evidence was provided for 24 FG loci (12).
In this paper, we calculated GRS to combine the effects of multiple genetic variants to increase the power of the study. The choice of loci was based on reported associations of genetic variants with FG, FI and T2D (30).
The selection of SNPs for this study was based on prioritization of the variants with the largest effect size of the phenotype of interest. These variants, including TCF7L2, FTO, MTNR1B and G6PC2, were most frequently identified in the earliest GWAS studies (Supplementary Material, Table S1) and confirmed by multiple replications (28,31). This approach allowed us to detect the effect in individuals of young age, when genetic effects are usually smaller compared with adulthood. We hypothesized that the GRSs for FG, FI and T2D are associated with glycaemic traits, such as IR and beta cell function measured by HOMA-IR/HOMA-B, and adiposity (BMI) as a quantitative measure of obesity in children and adolescents. The aim of our study was to analyse 42 loci with the largest effects on T2D and glycaemic traits in a cohort of children enrolled at the Bambino Gesù Children’s Hospital in Rome, Italy, and to define the effects of these variants on glycaemic traits and indices of IR, beta cell function, and BMI as well as to evaluate the causal relationships between these traits using genetic variants as instruments in a one-sample Mendelian randomization (MR) framework.
Results
Our study included 1660 young European-descent individuals from Italy with a mean BMI of 20.72 kg/m2 (range 10.42–44.95 kg/m2) and a mean age of 9.09 years (range 2.02–18.93 years) (Table 1). Most (1638; 98.67%) of the participants had FG level below 5.6 mmol/l. Twenty (1.20%) participants had FG values between 5.6 and 6.1 mmol/l and two (0.12%) had values between 6.1 and 7.0 mmol/l. Most of them fell within the BMI range between −2 and 2 of gender- and age-specific standard deviation scores (SDS) of BMI units (Material and Methods) with the exception of 205 individuals (12.35%) who were obese (BMI values ≥2SD). These individuals with obesity were older and had higher FG, FI, HOMA-B and HOMA-IR values when compared with non-obese individuals.
Table 1.
Characteristics of the study sample
| Phenotype | Males (N = 889) | Females (N = 771) | P-value for difference* |
|---|---|---|---|
| Mean ± SD | Mean ± SD | ||
| Age (years) | 9.07 ± 3.74 | 9.12 ± 3.91 | 0.82 |
| BMI (kg/m2) | 20.83 ± 5.59 | 20.59 ± 5.47 | 0.37 |
| BMI SDS | 0.47 ± 1.52 | 0.47 ± 1.41 | 0.97 |
| FG (mmol/l) | 4.58 ± 0.48 | 4.47 ± 0.50 | 2.27 × 10−5 |
| FI (pmol/l) | 54.91 ± 45.23 | 62.51 ± 49.62 | 0.0012 |
| HOMA-B | 108.28 ± 55.93 | 123.87 ± 60.88 | 7.74 × 10−8 |
| HOMA-IR | 0.99 ± 0.80 | 1.13 ± 0.88 | 0.0013 |
BMI, body mass index; FG, fasting glucose; FI, fasting insulin; SDS, standard deviation score; T2D, type 2 diabetes.
*Difference between the mean values in male and female participants calculated by t-test.
We performed the association analyses with FG, FI, HOMA-B, HOMA-IR and BMI SDS for the 42 variants, assuming an additive genetic model implemented in linear regression and detected nominally significant associations (P < 0.05) at eight FG, eight T2D and two FI loci with the phenotypes tested (Material and Methods, Supplementary Material, Table S1). For FG, we observed the most significant associations with rs560887 near G6PC2 (glucose-6-phosphatase 2) (P = 7.35 × 10−6), rs4607517 near GCK (glucokinase) (P = 3.24 × 10−5) and rs10830963 at MTNR1B (melatonin receptor 1B) (P = 4.37 × 10−5), of which the signals at G6PC2 and MTNRB1 were also associated with HOMA-B (P = 1.13 × 10−2 and P = 1.32 × 10−4, respectively). Additionally, signals within MAP Kinase Activating Death Domain (MADD) and Coiled-Coil-Helix-Coiled-Coil-Helix Domain Containing 9/transducin-like enhancer protein 4 (CHCHD9/TLE4) were nominally associated with FG. For HOMA-B, we further observed an association with rs10440833 at CDK5 regulatory subunit-associated protein 1-like 1 (CDKAL1, P = 9.72 × 10−3). For FI, the variant rs340874 at Prospero Homeobox 1 (PROX1) showed a nominal association (P = 2.37 × 10−2). Furthermore, variation at Peroxisome Proliferator Activated Receptor Gamma (PPARG) was associated with FI (P = 4.49 × 10−2), HOMA-B (P = 2.15 × 10−3) and HOMA-IR (P = 4.01 × 10−2). HOMA-IR was further associated with rs12970134 at MC4R (melanocortin 4 receptor) (P = 3.25 × 10−2). Variation at MC4R was also associated with age- and gender-standardized BMI (P = 8.45 × 10−3), as were rs9939609 at FTO (fat mass and obesity-associated) and rs11558471 at SLC30A8 (Solute Carrier Family 30 Member 8), (P = 1.81 × 10−2 and P = 2.91 × 10−2, respectively).
We calculated unweighted and weighted GRS for FG (20 SNPs), FI (5 SNPs) and T2D (36 SNPs) (Material and Methods, Supplementary Material, Table S1). Both unweighted and weighted GRSs were further multiplied by the proportion of successfully genotyped SNPs per individual. We compared the distributions of the number of trait-increasing risk alleles for FG, FI and T2D over the distributions of related phenotypes. As the number of FG risk alleles increased, concentrations of FG increased, whereas values of HOMA-B decreased (Fig. 1). However, contrary to our study hypothesis, the BMI SDS measurement remained relatively invariable compared with the number of FG risk alleles. For the number of FI risk alleles, no significant association was observed with FI, HOMA-IR or BMI SDS (Fig. 2). Similar results were obtained for the relationship between T2D risk alleles and FG, FI, HOMA-B, HOMA-IR and BMI SDS (Fig. 3).
Figure 1.
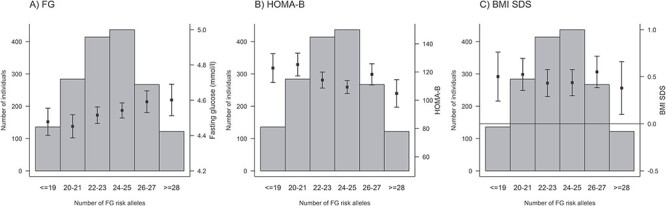
Relationships between the number of fasting glucose (FG) risk alleles and FG (A), HOMA-B (B) and age- and BMI SDS (C).
Figure 2.
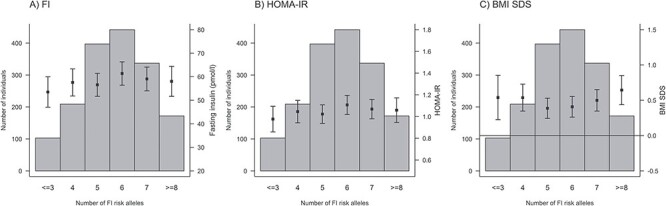
Relationships between the number of fasting insulin (FI) risk alleles and FI (A) HOMA-IR (B) and age- and BMI SDS (C).
Figure 3.
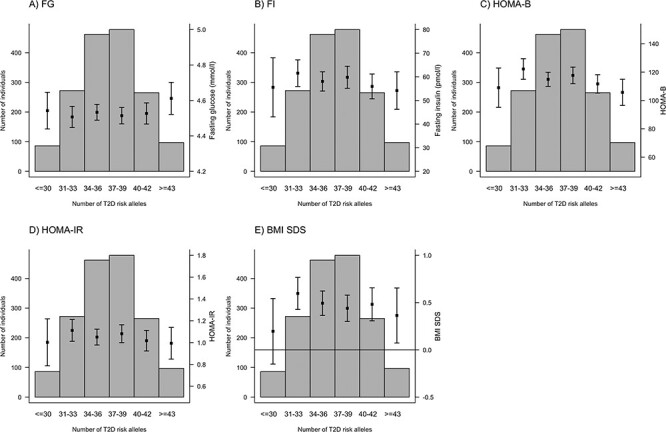
Relationships between the number of type 2 diabetes (T2D) risk alleles and FG (A), FI (B), HOMA-B (C), HOMA-IR (D), and BMI SDS (E).
When grouping the phenotype distributions according to percentiles, we observed that the individuals in the highest (>95%) percentile group of FG had a slightly increased number of FG risk alleles (meangroup1 = 22.86, meangroup2 = 23.45, meangroup3 = 23.84; P = 0.026 in the linear regression of the number of FG risk alleles on the percentile group), whereas the opposite was true for HOMA-B (meangroup1 = 23.84, meangroup2 = 23.46, meangroup3 = 22.61; P = 0.0050). No such effect was detected for the BMI SDS percentile groups (meangroup1 = 23.67, meangroup2 = 23.43, meangroup3 = 23.16; P = 0.24) (Fig. 4).
Figure 4.
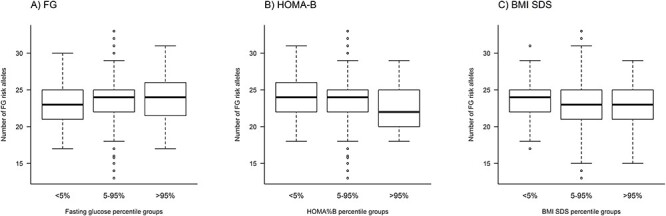
Relationship between the number of fasting glucose (FG) risk alleles and FG (A), HOMA-B (B), and BMI SDS (C). The results from the association analyses using linear regression of both GRS types, weighted and unweighted, confirmed the findings from the comparison of phenotypic distributions in relation to allele counts (Table 2).
A higher number of FG risk alleles was associated with elevated FG (beta = 0.075, 95%CI [0.053;0.097] mmol/l per unit increase in the weighted GRS) and lower HOMA-B values (beta = −0.041, 95%CI [−0.059;−0.024] ln(HOMA-B) units per unit increase in the weighted GRS) after adjustment for age, sex and BMI SDS (Material and Methods, Table 2). The weighted T2D GRS was associated with lower HOMA-B values (−0.020 [−0.038;−0.0019]) after adjusting for age, sex and BMI SDS (Table 2). The unweighted GRS provided mostly similar but weaker associations than the weighted GRS (Table 2), except for the unweighted FG GRS that showed a trend towards a negative association with FI levels (−0.024 [−0.050, 0.0029] ln(pmol/l), P = 0.082) and this was further strengthened (−0.028 [−0.055, −0.0016] ln(pmol/l), P = 0.038) when adding adjustment for family history of T1D and T2D. It is worth noticing that this additional adjustment had in general no noticeable effect on the other estimates (Material and Methods, Supplementary Material, Table S2). We did not observe any evidence for association between the FG and FI GRSs and BMI SDS or obesity status, including adjustments for sex, age, BMI SDS and family history of T1D and T2D (Table 3).
Table 2.
Association between the FG/FI/T2D genetic risk scores and unadjusted FG, FI, HOMA-B, HOMA-IR and age- and gender-standardized BMI
| Model | FG (mmol/l) | ln(FI (pmol/l)) | ln(HOMA-B) | ln(HOMA-IR) | BMI SDS | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| β (95% CI) | P-value | β (95% CI) | P-value | β (95% CI) | P-value | β (95% CI) | P-value | β (95% CI) | P-value | |
| Weighted GRS, adjusted | ||||||||||
| GRS_FG | 0.075 (0.053, 0.097) | 1.58 × 10 −11 | −0.011 (−0.038, 0.016) | 0.43 | −0.041 (−0.059, −0.024) | 5.13 × 10 −6 | −0.0049 (−0.017, 0.0072) | 0.43 | 0.034 (−0.033, 0.10) | 0.32 |
| GRS_FI | 0.0023 (−0.020, 0.024) | 0.84 | 0.020 (−0.0069, 0.046) | 0.15 | 0.012 (−0.0061, 0.030) | 0.20 | 0.0081 (−0.0040, 0.020) | 0.19 | 0.013 (−0.055, 0.080) | 0.71 |
| GRS_T2D | 0.0074 (−0.015, 0.029) | 0.51 | −0.025 (−0.052, 0.0016) | 0.066 | −0.020 (−0.038, −0.0019) | 0.030 | −0.0082 (−0.020, 0.0040) | 0.19 | −0.018 (−0.085, 0.050) | 0.61 |
| Weighted GRS, unadjusted | ||||||||||
| GRS_FG | 0.082 (0.059, 0.11) | 1.010 × 10 −11 | 0.013 (−0.025, 0.050) | 0.50 | −0.029 (−0.052, −0.0070) | 0.010 | 0.0052 (−0.011, 0.022) | 0.54 | 0.051 (−0.020, 0.12) | 0.16 |
| GRS_FI | 0.0016 (−0.022, 0.025) | 0.90 | 0.023 (−0.014, 0.060) | 0.23 | 0.014 (−0.0083, 0.037) | 0.22 | 0.0095 (−0.0072, 0.026) | 0.27 | 0.011 (−0.060, 0.082) | 0.76 |
| GRS_T2D | 0.0064 (−0.018, 0.030) | 0.60 | −0.026 (−0.063, 0.011) | 0.17 | −0.020 (−0.042, 0.0027) | 0.085 | −0.0086 (−0.025, 0.0081) | 0.31 | −0.017 (−0.087, 0.054) | 0.65 |
| Unweighted GRS, adjusted | ||||||||||
| GRS_FG | 0.045 (0.023, 0.067) | 5.31 × 10 −5 | −0.024 (−0.050, 0.0029) | 0.082 | −0.036 (−0.054, −0.018) | 7.15 × 10 −5 | −0.0089 (−0.021, 0.0032) | 0.15 | −0.020 (−0.088, 0.047) | 0.55 |
| GRS_FI | 0.0011 (−0.021, 0.023) | 0.92 | 0.016 (−0.011, 0.042) | 0.25 | 0.0097 (−0.0082, 0.028) | 0.29 | 0.0065 (−0.0056, 0.019) | 0.29 | 0.024 (−0.043, 0.092) | 0.48 |
| GRS_T2D | 0.018 (−0.0044, 0.040) | 0.12 | −0.013 (−0.040, 0.013) | 0.31 | −0.017 (−0.035, 0.0010) | 0.064 | −0.0029 (−0.015, 0.0093) | 0.65 | −0.0021 (−0.070, 0.066) | 0.95 |
| Unweighted GRS, unadjusted | ||||||||||
| GRS_FG | 0.048 (0.025, 0.072) | 6.83 × 10 −5 | −0.017 (−0.054, 0.020) | 0.37 | −0.033 (−0.056, −0.011) | 0.0036 | −0.0064 (−0.023, 0.010) | 0.45 | −0.0090 (−0.080, 0.062) | 0.80 |
| GRS_FI | 0.0022 (−0.022, 0.026) | 0.86 | 0.021 (−0.016, 0.059) | 0.26 | 0.013 (−0.0095, 0.035) | 0.26 | 0.0089 (−0.0078, 0.027) | 0.30 | 0.023 (−0.047, 0.094) | 0.52 |
| GRS_T2D | 0.017 (−0.0073, 0.040) | 0.17 | −0.011 (−0.049, 0.026) | 0.55 | −0.015 (−0.037, 0.0077) | 0.20 | −0.0019 (−0.019, 0.015) | 0.82 | 0.00036 (−0.070, 0.071) | 0.99 |
BMI, body mass index; FG, fasting glucose; FI, fasting insulin; GRS, genetic risk score; SDS, standard deviation score; T2D, type 2 diabetes. Model adjusted for age, sex and BMI SDS. BMI SDS adjustment not done when BMI SDS is the outcome of interest. BMI SDS adjustment not done when BMI SDS is the outcome of interest. Bold values denote statistical significance at the P < 1× 10−5.
Table 3.
Effects of FG/FI/T2D genetic risk scores on obesity
| Model | Obese (N = 205) versus non-obese (N = 1350) (control group 1) a | Obese (N = 205) versus non-overweight (N = 639) (control group 2) b | ||||||
|---|---|---|---|---|---|---|---|---|
| Unadjusted | Adjusted c | Unadjusted | Adjusted c | |||||
| OR (95% CI) c | P-value | OR (95% CI) c | P-value | OR (95% CI) c | P-value | OR (95% CI) c | P-value | |
| Weighted GRS | ||||||||
| GRS_FG | 1.00 (0.86–1.15) | 0.96 | 0.97 (0.83–1.13) | 0.74 | 1.00 (0.86–1.18) | 0.96 | 1.00 (0.84–1.19) | 0.99 |
| GRS_FI | 0.98 (0.85–1.13) | 0.78 | 0.98 (0.84–1.13) | 0.74 | 0.94 (0.81–1.10) | 0.48 | 0.93 (0.79–1.10) | 0.41 |
| GRS_T2D | 0.93 (0.80–1.07) | 0.31 | 0.91 (0.78–1.06) | 0.22 | 0.99 (0.85–1.15) | 0.89 | 0.99 (0.84–1.17) | 0.91 |
| Unweighted GRS | ||||||||
| GRS_FG | 0.95 (0.82–1.10) | 0.48 | 0.93 (0.80–1.09) | 0.36 | 0.94 (0.30–1.03) | 0.45 | 0.93 (0.79–1.11) | 0.44 |
| GRS_FI | 0.99 (0.86–1.15) | 0.93 | 0.99 (0.85–1.15) | 0.86 | 0.96 (0.82–1.11) | 0.60 | 0.95 (0.80–1.12) | 0.52 |
| GRS_T2D | 0.97 (0.84–1.12) | 0.68 | 0.96 (0.82–1.12) | 0.58 | 1.02 (0.88–1.20) | 0.77 | 1.02 (0.86–1.20) | 0.83 |
FG, fasting glucose; FI, fasting insulin; GRS, genetic risk score; SDS, standard deviation score; T2D, type 2 diabetes.
aObese individuals: BMI SDS > =2; non-obese individuals: −2 < BMI SDS < 2.
bObese individuals: BMI SDS > =2; non-overweight individuals: −1 < BMI SDS < 1.
cAdjusted for age, sex and family history of T1D and T2D.
We evaluated the associations between FG/FI and BMI for causality in a bi-directional one-sample MR framework (Material and Methods). We identified a positive causal effect of BMI on FI (P = 0.0085) (Table 4). The IV estimator indicated a causal effect of 0.31 ln(pmol/l) higher FI (95%CI [0.078;0.54]) per unit increase in BMI SDS. We did not observe a causal effect in the other direction. Contrary to the observed epidemiological associations, we did not find evidence for a causal effect of BMI on the levels of FG and vice versa. However, this could be due to low power as the epidemiological effect estimates between BMI and FG in either direction are lower than those between BMI and FI (Table 4). Additionally, the low F-statistic of the FI IV (Table 4) suggests weak instrument bias and low power of the MR analysis testing the effect of FI levels on BMI.
Table 4.
Mendelian randomization analysis
| Causal relationship tested (exposure on outcome) | Epidemiological association | Causal effect, instrumental variable (IV) approach | |||
|---|---|---|---|---|---|
| β (95%CI) | P-value | F-stat (IV) | βIV(95%CI) | P-value | |
| FG on BMI | 0.68 (0.54, 0.82) | 6.70 × 10−21 | 46.98 | 0.61 (−0.22, 1.43) | 0.15 |
| FI on BMI | 0.96 (0.89, 1.043) | 1.75 × 10−109 | 0.79 | −2.55 (−11.15, 6.053) | 0.56 |
| BMI on FG | 0.077 (0.061, 0.093) | 6.70 × 10−21 | 15.72 | 0.049 (−0.12, 0.21) | 0.56 |
| BMI on FI | 0.27 (0.25, 0.29) | 1.75 × 10−109 | 15.72 | 0.31 (0.078, 0.54) | 0.0085 |
Mendelian randomization analyses using the Two-Stage-Least Squares method. FG, fasting glucose (mmol/l); BMI, body mass index (kg/m2); FI, fasting insulin (ln(pmol/l)). For the instrumental variable analyses, estimates are reported in units of the outcome as described in the previous sentence per amount of increase in the exposure attributable to one unit increase in the genetic instrument of the exposure. For the statistically significant causal relationship of BMI on FI, one unit increase in BMI GRS corresponds to 0.14 (95%CI [0.070;0.21]) standard deviation score increase in BMI.
Discussion
Our results confirm the ability of a GRS combining 20 independent genetic variants, associated with FG in previous GWAS (12,28,32,33), to predict values of fasting glucose and beta cell activity in children and adolescents already. Using a one-sample MR approach, we discovered that a causal effect of adiposity via BMI on FI levels is detectable as early as in childhood/teenage years.
Effects of adiposity on altered glucose metabolism
The present investigation expands on earlier findings in adults on FTO/MC4R-mediated adiposity’s effect on increased fasting insulin levels (34,35) to its earlier age manifestation in children and adolescents. Despite much larger sample sizes in earlier adult studies, the causal effect of adiposity on FI in our study is comparable to that in adults (35). While we were able to observe a causal effect in the combined cohort, we were underpowered to perform sex-stratified analyses and to validate the larger causal effect of BMI on FI reported in men compared with women (4). This study did not find evidence for a causal effect of adiposity on FG, possibly due to lack of statistical power. The lack of causal effect of BMI on FG is in contrast to the findings of Dale et al. and Xu et al. who report a causal effect of BMI on glucose levels with markedly lower causal effect size than that between BMI and insulin (36,37). Even though our causal analyses from BMI to FI and FG used only two variants, namely those within FTO and MC4R, to date, these are the most strongly associated with BMI and have been successfully used as instrumental variables to estimate causal effects previously (4,38). The F-statistic of the BMI instrument (Table 4) demonstrates that these two variants make a strong enough IV in this study. Our findings support a crucial role of adiposity in the development of IR in young individuals. During puberty, IR is changing drastically, when insulin sensitivity undergoes a decline of around 25–50% during puberty and improves when puberty ends (39,40). Visceral and subcutaneous adipose tissue secrete free fatty acids and pro-inflammatory cytokines into blood that contribute to IR (41). Changes related to increased adiposity affect the complex interplay between pathophysiological processes already in early age, which might increase the risk of early onset T2D and comorbidities.
Genetic variants associated with adiposity
Our findings are in line with previous studies that linked genetic variants near MC4R and FTO genes with adiposity traits and T2D (42). MC4R rs12970134 is associated with increased risk of T2D and higher BMI in both European and trans-ethnic studies (8,26). The Nord-Trøndelag Health (HUNT) study assessing gene–environment interactions of FTO and MC4R on obesity in people with extreme phenotypes observed age- and gender-dependent associations of rs9939609 (FTO) and rs17782313 (MC4R) loci with BMI (43). Notably, the effect sizes of FTO tended to be the highest in the youngest age group for both genders; for MC4R, the highest effect on BMI was observed in the youngest age group, dipping in the middle age and increasing again after the age of sixty, while in men, it peaked at 40–60 years and became negligible later in life (43). The FTO locus is not only strongly associated with T2D (42) and higher BMI (44), but also with higher FI and HOMA-IR (29). These observations are in agreement with BMI playing a role in the FTO association with T2D via IR.
Effects on altered glucose metabolism in adult and paediatric populations
In our analysis, the GRS for FG comprising 20 DNA variants explained 2.76% of the variability in FG (beta = 0.075 mmol/l, P = 1.58 × 10−11) and 0.34% of the variability in β-cell function (beta = 0.042, P = 5.13 × 10−6). A number of studies (13–20,45) evaluated the performance of glucose homeostasis GRSs as a useful tool to estimate the effects of multiple risk alleles predicting prevalent or incident cases of T2D.
Similarly, in cross-sectional studies of normal-weight and overweight/obese children, T2D and FI GRSs comprising 62 (23) and 53 (9) SNPs were associated with different glycaemic traits, particularly with FG and estimates of beta cell function.
Genetic variants associated with fasting glucose and β-cell function
We confirmed previously established associations of some of the T2D susceptibility variants with glucose metabolism traits (Supplementary Material, Table S1). Specifically, among 20 loci previously implicated in FG variability, G6PC2, GCK and MTNR1B variants showed effects on FG in Italian children consistent with those found in European children in MAGIC (28,29) and in adults (46). According to our results and previous studies, two of these genes, G6PC2 and MTNR1B, have been found to be associated with β-cell function in adults (47,48). Increased MTNR1B expression in individuals at risk of T2D suggests a direct inhibitory effect on beta cells (47). Our results demonstrate a significant association between MADD and PPARG loci and HOMA-B, consistent with previous reports (49,50). Our study provides evidence that CDKAL1 variants confer risk of T2D through reduced insulin secretion, which is also in line with the findings of the genome-wide association study in European and Hong Kong populations (51).
Study limitations
We acknowledge the limitations of our study of which the limited sample size and the wide age range are the most evident. Other limitations include the cross-sectional design; the weights taken from older populations and effect sizes for risk variants can vary between different age groups and might not provide as good a predictive ability; the limited number of variants investigated; the proxy estimation of fasting IR and β-cell function; the lack of information on pubertal status and the enrolment of individuals with exclusively European descent. In an attempt to mitigate the limitation of wide age range, we used gender- and age-specific standardized obesity indices (BMI SDS). Future investigations must consider the changing physiology and hormonal levels during pubertal transition; they should enrol individuals of non-European descent to better reflect the evolving multi-ethnic nature of populations in Italy and worldwide; and the FG GRS should be validated in longitudinal studies. Additionally, the GRSs in our study are constructed based on a relatively small number of variants from pioneer GWAS studies. Therefore, as more variants are characterized, improved GRSs should be constructed.
Conclusion
We report that T2D risk genetic variants contribute to higher FG levels and beta cell function in Italian children and adolescents. We provide novel evidence for a causal effect of childhood adiposity on higher FI levels validating previously published results in adults. Further larger studies in children are mandatory to expand present knowledge of the genetic overlap between childhood/adolescent age obesity and risk of T2D.
Materials and Methods
Study sample
The study sample comprised individuals referred to the Bambino Gesù Children’s Hospital in Rome, Italy, between July 2012 and July 2013 by general practitioners from the Metropolitan Area of Rome (Italy) to participate in ‘The Bambino Gesù Study: Profiling the genetic risk of complex diseases in the Italian population’. The primary aim of the study was to dissect the genetic architecture of glucose homeostasis in the Italian children and adolescents. The study was approved by the Ethics Committee of the Bambino Gesù Hospital, and written informed consent was obtained from the child’s parents or legal guardians in accordance with the Helsinki declaration (52).
In total, 1806 participants were enrolled in the study. We excluded participants below 2 or above 19 years of age (N = 10), non-Europeans (N = 135) and one individual with an FG value ≥7 mmol/l. The final study sample included 1660 (889 male) participants of European descent aged between 2 and 19 years (Supplementary Material, Fig. S1). None of the participants were following a weight loss diet or an intensive exercise program, and, at the time of enrolment, all study participants were healthy. Information on family history of diabetes in the first-degree relatives was obtained by a short questionnaire completed by both parents (53).
Anthropometric measurements and biochemical assays
Weight and height were measured using standard procedures (54). All participants were asked to refrain from intensive physical activity in the 3 days prior to the study. Fasting glucose was measured by glucose oxidase technique (Cobas Integra, Roche) and insulin by a chemiluminescent immunoassay method (ADVIA Centaur analyzer; Bayer Diagnostics).
Phenotypes
Body mass index (BMI) (kg/m2)
Gender- and age-specific standard deviation scores (SDS) of BMI were calculated (Supplementary Material, Fig. S2) with the Growth Analyser RCT tool (version 3.0, https://www.growthanalyser.org/; Dutch Growth Research Foundation, Rotterdam, the Netherlands). Within the Growth Analyser, BMI data of 2- to 20-year-olds from Italy were used as the reference (54). We also dichotomized the BMI-SDS scores into obese (SDS ≥ 2) and non-obese using two different definitions for the control groups: (1) non-obese (−2 < BMI SDS < 2) and (2) non-overweight (−1 < BMI SDS < 1). The distribution of the BMI SDS scores according to age of the participants is shown in Figure 5.
Figure 5.
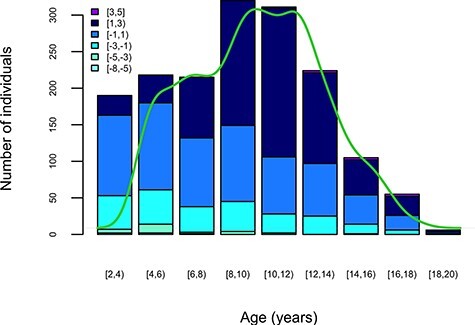
BMI SDS values of the Bambino Gesù study sample within their age distribution. Colours represent BMI SDS groups as displayed in the figure insert. The bars represent subject counts within each BMI SDS group by the age group (x-axis). The green line shows the age density of the study sample.
Fasting insulin (FI) and glucose (FG)
None of the included children had diabetes according to WHO criteria (55). We used different units for FG and FI (mmol/l and pmol/l, respectively) for the calculation of indices. Indices of insulin sensitivity and beta cell function, namely HOMA-IR and HOMA-B, were calculated using the HOMA calculator provided by the University of Oxford (https://www.dtu.ox.ac.uk/homacalculator/). To reduce skewness, FI, HOMA-IR and HOMA-B were natural logarithm transformed.
DNA extraction
DNA was extracted from 300 ul of whole blood using the QIAsymphony DSP DNA kit. The extraction was performed on the automated extractor QIAsymphony SP workstation Qiagen, Hilden, Germany) according to manufacturer procedure. DNA was eluted in 200 ul deionized water.
SNP genotyping
For the analysis, we selected 42 DNA variants reported in previous publications (Supplementary Material, Table S1) (12,28,33,56). The SNP genotyping was performed using Agena MassArray® System (Agena Bioscience, San Diego, USA). SNPs were assayed and typed using iPLEX® chemistry on a matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometer. All biochemical reactions were performed as recommended by the manufacturer. The iPLEX single base extension was spotted on 384-SpectroChips and analysed in the MassARRAY Analyzer. MassARRAY Typer 4.0 software was used for evaluating and managing the genotype results.
Quality control of genetic data
For the quality control (QC) purposes, missing rate per individuals and missing rate per SNP were calculated. In addition, for QC of SNP genotyping, positive and negative template control samples were included in each assay plate. Any assay found as positive in the negative template control was removed from the study. We kept SNPs with missingness ≤0.02, Hardy–Weinberg equilibrium test P-value < 1 × 10−6, SNP genotyping call rate ≥ 95% and minor allele frequency (MAF) > 1%.
Statistical analyses
All statistical analyses were performed using the software package R version 3.5.1 (57).
Genetic risk scores (GRSs)
We calculated unweighted and weighted GRS for FG (20 SNPs), FI (5 SNPs) and T2D (36 SNPs) (see Supplementary Material, Table S1 for the list of SNPs used). Effect sizes of genetic variants on each specific phenotype were obtained from large-scale consortia with mean age ranging from 31.0 to 73.4 (12,28,33,56). For the unweighted GRS, the numbers of effect alleles (0, 1, 2) for each SNP were added up, while for the calculation of the weighted GRS, each effect allele count for each SNP was multiplied by the reported effect size of the effect allele (beta for all, i.e. log(OR) for T2D). If the reported SNP was not available in our data, we used an SNP in linkage disequilibrium and further weighted the SNP by the r2 value (58). Both unweighted and weighted GRSs were further multiplied by the proportion of successfully genotyped SNPs per individual. The distributions of the GRSs for each respective set of established loci by phenotype were investigated against the distributions of the phenotypes of interest, e.g. FG, FI and T2D. For the association analyses described below, the GRSs were standardized to have a mean value of 0 and standard deviation of 1 to allow comparison of effect estimates across different GRSs and the outcome variables.
Association analysis
We performed the association analysis with FG, FI, HOMA-B, HOMA-IR and BMI SDS for the 42 genotyped SNPs assuming an additive genetic model using linear regression. Unweighted and weighted GRSs were also tested for association with FG, FI, HOMA-B, HOMA-IR and BMI SDS via linear regression. Logistic regression was used for the association analysis of GRS and obese versus non-obese individuals. For the linear regression, we report the effects as regression coefficients with their 95% confidence intervals (CIs), whereas for logistic regression, we provide estimates of odds ratios (OR) with their 95% CIs. We report unadjusted associations as well as analyses adjusted for (1) age, sex and BMI SDS (BMI adjustment not done when BMI or obesity is the outcome), and (2) age, sex, BMI SDS and family history of T1D and T2D. We applied a Bonferroni correction to adjust for multiple testing. The P-value thresholds for statistical significance after Bonferroni correction were P = 0.0025, 0.0014 and 0.008 for 5, 20 and 36 tests for FI, FG and T2D SNPs, respectively.
Mendelian randomization
We evaluated the casual relationship between FG/FI and BMI in a one-sample MR framework (Fig. 6) using Two-Stage-Least Squares (2SLS) as implemented in the ivreg(v.0.5–0) R package. In 2SLS, the first regression model regresses the exposure on the genetic instruments providing fitted exposure values independent of the confounders. The second stage of 2SLS regresses the outcome on the fitted values of the exposure. The genetic instrument for FG was the same as the GRS for FG described previously, whereas the instrument for FI comprised of four variants after excluding the FTO variant from the FI GRS. The genetic instrument for BMI was constructed from the FTO and MC4R variants, and we used the effect sizes as reported (8). Since Locke et al. reported different variants for these two loci, we further weighted the effect sizes by the r2 values between the reported lead variants and the variants used in the present study. We report F-statistics from the regression model of the exposure on the corresponding IV as a measure of instrument strength. Causal effects estimated in MR are only valid if the following core assumptions hold true: (1) the genetic instrument has a true effect on the exposure and that (2) it only affects the outcome through its effect on the exposure as well as (3) it is independent of any measured and unmeasured confounding factors of the exposure–outcome relationship.
Figure 6.
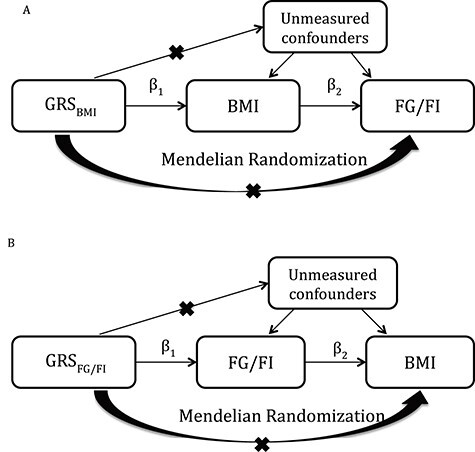
Mendelian randomization analysis to explore causality between BMI and FG/FI. (A) IV estimator is calculated as the beta coefficient from the association of GRSBMI with FG or FI divided by the beta coefficient from the association of GRSBMI with BMI (IV estimator = 0.31 pmol/l/BMI unit). The IV estimator is equivalent to what is seen when FI is regressed on BMI. These results are supportive of a causal, non-confounded relationship. (B) The relationship of FG or FI with BMI.
Supplementary Material
Acknowledgements
The authors are thankful to all the patients and their families, care providers and clinicians at the Bambino Gesù Children's Hospital for their contribution to the study.
Conflict of Interest statement. The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Contributor Information
Zhanna Balkhiyarova, Section of Statistical Multi-Omics, Department of Clinical and Experimental Medicine, University of Surrey, Guildford GU2 7XH, UK; Institute of Biochemistry and Genetics, Ufa Federal Research Centre Russian Academy of Sciences, Ufa 450008, Russian Federation; Department of Endocrinology, Bashkir State Medical University, Ufa 450054, Russian Federation.
Rosa Luciano, Research Area for Multifactorial Disease, Bambino Gesù Children’s Hospital, IRCCS, Rome 00146, Italy; Department of Laboratory Medicine, Bambino Gesù Children’s Hospital, IRCCS, Rome 00146, Italy.
Marika Kaakinen, Section of Statistical Multi-Omics, Department of Clinical and Experimental Medicine, University of Surrey, Guildford GU2 7XH, UK; Section of Genetics and Genomics, Department of Metabolism, Digestion and Reproduction, Imperial College London, London SW7 2AZ, UK.
Anna Ulrich, Section of Statistical Multi-Omics, Department of Clinical and Experimental Medicine, University of Surrey, Guildford GU2 7XH, UK; Section of Genetics and Genomics, Department of Metabolism, Digestion and Reproduction, Imperial College London, London SW7 2AZ, UK.
Aleksey Shmeliov, Section of Statistical Multi-Omics, Department of Clinical and Experimental Medicine, University of Surrey, Guildford GU2 7XH, UK.
Marzia Bianchi, Research Area for Multifactorial Disease, Bambino Gesù Children’s Hospital, IRCCS, Rome 00146, Italy.
Laura Chioma, Unit of Endocrinology, Bambino Gesù Children's Hospital, IRCCS, Rome 00146, Italy.
Bruno Dallapiccola, Genetics and Rare Diseases Research Division, Bambino Gesù Children's Hospital, IRCCS, Rome 00146, Italy.
Inga Prokopenko, Section of Statistical Multi-Omics, Department of Clinical and Experimental Medicine, University of Surrey, Guildford GU2 7XH, UK; Institute of Biochemistry and Genetics, Ufa Federal Research Centre Russian Academy of Sciences, Ufa 450008, Russian Federation; UMR 8199—EGID, Institut Pasteur de Lille, CNRS, University of Lille, Lille 59000, France.
Melania Manco, Research Area for Multifactorial Disease, Bambino Gesù Children’s Hospital, IRCCS, Rome 00146, Italy.
Data availability
The datasets generated during and/or analysed during the current study are not publicly available for reasons related to privacy and participant consent but are available from the corresponding author on reasonable request.
Funding
This work has been supported by the grant from `Regione Lombardia' to BD `Sviluppare profili genetici di rischio e trasferirli alla Sanità pubblica, in Italia' and by FPRC 5xmille 2019 Ministero della Salute to MM. This research was in part funded by the World Cancer Research Fund (WCRF UK) and World Cancer Research Fund International (2017/1641), the Wellcome Trust (WT205915/Z/17/Z), Royal Society (IEC\R2\181075), the European Union's Horizon 2020 research and innovation programme (LONGITOOLS, H2020-SC1-2019-874739), the Ministry of Science and Higher Education of Russian Federation (075-15-2021-595), Agence Nationale de la Recherche (PreciDIAB, ANR-18-IBHU-0001), the European Union through the `Fonds européen de développement regional' (FEDER), the `Conseil Régional des Hauts-de-France' (Hauts-de-France Regional Council) and the `Métropole Européenne de Lille' (MEL, European Metropolis of Lille). M.K. was in part sponsored by the European Foundation for the Study of Diabetes (EFSD) Albert Renold Travel Fellowship. Z.B. was in part funded through the Medical Research Council (MRC) UK (MR/R010676/1).The funding bodies named had no role in the design and conduct of the study, collection management, analysis and interpretation of the data, preparation review or approval of the manuscript.
Contribution statement
Z.B., M.K., A.U., M.M. and I.P. wrote the manuscript and researched the data. M.K., A.U. and A.S. performed the statistical analyses. M.M., I.P. and B.D. designed the study and reviewed and edited the manuscript. R.L., M.B. and L.C. contributed to data/sample collection and genotyping and reviewed and edited the manuscript. B.D. obtained funding for the study. All authors approved the final version of the manuscript.
I.P. and M.M. are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
References
- 1. Shashaj, B., Bedogni, G., Graziani, M.P., Tozzi, A.E., DiCorpo, M.L., Morano, D., Tacconi, L., Veronelli, P., Contoli, B. and Manco, M. (2014) Origin of cardiovascular risk in overweight preschool children: a cohort study of cardiometabolic risk factors at the onset of obesity. JAMA Pediatr., 168, 917–924. [DOI] [PubMed] [Google Scholar]
- 2. Lauria, L., Spinelli, A., Buoncristiano, M. and Nardone, P. (2019) Decline of childhood overweight and obesity in Italy from 2008 to 2016: results from 5 rounds of the population-based surveillance system. BMC Public Health, 19, 618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen, L., Magliano, D.J. and Zimmet, P.Z. (2011) The worldwide epidemiology of type 2 diabetes mellitus--present and future perspectives. Nat. Rev. Endocrinol., 8, 228–236. [DOI] [PubMed] [Google Scholar]
- 4. Fall, T., Hägg, S., Ploner, A., Mägi, R., Fischer, K., Draisma, H.H., Sarin, A.P., Benyamin, B., Ladenvall, C., Åkerlund, M.et al. (2015) Age- and sex-specific causal effects of adiposity on cardiovascular risk factors. Diabetes, 64, 1841–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Di Bonito, P., Pacifico, L., Chiesa, C., Valerio, G., Del Giudice, E.M., Maffeis, C., Morandi, A., Invitti, C., Licenziati, M. and Loche, S. (2017) Impaired fasting glucose and impaired glucose tolerance in children and adolescents with overweight/obesity. J. Endocrinol. Investig., 40, 409–416. [DOI] [PubMed] [Google Scholar]
- 6. Day, C. and Bailey, C.J. (2011) Obesity in the pathogenesis of type 2 diabetes. Br. J. Diabetes Vasc. Dis., 11, 55–61. [Google Scholar]
- 7. Cousminer, D.L. and Freathy, R.M. (2020) Genetics of early growth traits. Hum. Mol. Genet., 29, R66–R72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Locke, A.E., Kahali, B., Berndt, S.I., Justice, A.E., Pers, T.H., Day, F.R., Powell, C., Vedantam, S., Buchkovich, M.L., Yang, J.et al. (2015) Genetic studies of body mass index yield new insights for obesity biology. Nature, 518, 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mahajan, A., Taliun, D., Thurner, M., Robertson, N.R., Torres, J.M., Rayner, N.W., Payne, A.J., Steinthorsdottir, V., Scott, R.A., Grarup, N.et al. (2018) Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet., 50, 1505–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ingelsson, E. and McCarthy, M.I. (2018) Human genetics of obesity and type 2 diabetes mellitus. Circ. Genom. Precis. Med., 11, e002090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Barroso, I. and McCarthy, M.I. (2019) The genetic basis of metabolic disease. Cell, 177, 146–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Morris, A.P., Voight, B.F., Teslovich, T.M., Ferreira, T., Segre, A.V., Steinthorsdottir, V., Strawbridge, R.J., Khan, H., Grallert, H. and Mahajan, A. (2012) Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat. Genet., 44, 981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Graae, A.-S., Hollensted, M., Kloppenborg, J.T., Mahendran, Y., Schnurr, T.M., Appel, E.V.R., Rask, J., Nielsen, T.R., Johansen, M.Ø. and Linneberg, A. (2018) An adult-based insulin resistance genetic risk score associates with insulin resistance, metabolic traits and altered fat distribution in Danish children and adolescents who are overweight or obese. Diabetologia, 61, 1769–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stančáková, A., Kuulasmaa, T., Kuusisto, J., Mohlke, K.L., Collins, F.S., Boehnke, M. and Laakso, M. (2017) Genetic risk scores in the prediction of plasma glucose, impaired insulin secretion, insulin resistance and incident type 2 diabetes in the METSIM study. Diabetologia, 60, 1722–1730. [DOI] [PubMed] [Google Scholar]
- 15. Lotta, L.A., Gulati, P., Day, F.R., Payne, F., Ongen, H., Van De Bunt, M., Gaulton, K.J., Eicher, J.D., Sharp, S.J. and Rolfe, E.D.L. (2017) Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat. Genet., 49, 17. [DOI] [PubMed] [Google Scholar]
- 16. Vassy, J.L., Hivert, M.-F., Porneala, B., Dauriz, M., Florez, J.C., Dupuis, J., Siscovick, D.S., Fornage, M., Rasmussen-Torvik, L.J. and Bouchard, C. (2014) Polygenic type 2 diabetes prediction at the limit of common variant detection. Diabetes, 63, 2172–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vaxillaire, M., Yengo, L., Lobbens, S., Rocheleau, G., Eury, E., Lantieri, O., Marre, M., Balkau, B., Bonnefond, A. and Froguel, P. (2014) Type 2 diabetes-related genetic risk scores associated with variations in fasting plasma glucose and development of impaired glucose homeostasis in the prospective DESIR study. Diabetologia, 57, 1601–1610. [DOI] [PubMed] [Google Scholar]
- 18. Jensen, A.C., Barker, A., Kumari, M., Brunner, E.J., Kivimäki, M., Hingorani, A.D., Wareham, N.J., Tabák, A.G., Witte, D.R. and Langenberg, C. (2011) Associations of common genetic variants with age-related changes in fasting and postload glucose: evidence from 18 years of follow-up of the Whitehall II cohort. Diabetes, 60, 1617–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Andersson, E.A., Allin, K.H., Sandholt, C.H., Borglykke, A., Lau, C.J., Ribel-Madsen, R., Sparsø, T., Justesen, J.M., Harder, M.N. and Jørgensen, M.E. (2013) Genetic risk score of 46 type 2 diabetes risk variants associates with changes in plasma glucose and estimates of pancreatic β-cell function over 5 years of follow-up. Diabetes, 62, 3610–3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Go, M.J., Lee, Y., Park, S., Kwak, S.H., Kim, B.-J. and Lee, J. (2016) Genetic-risk assessment of GWAS-derived susceptibility loci for type 2 diabetes in a 10 year follow-up of a population-based cohort study. J. Hum. Genet., 61, 1009–1012. [DOI] [PubMed] [Google Scholar]
- 21. Lyssenko, V., Jonsson, A., Almgren, P., Pulizzi, N., Isomaa, B., Tuomi, T., Berglund, G., Altshuler, D., Nilsson, P. and Groop, L. (2008) Clinical risk factors, DNA variants, and the development of type 2 diabetes. N. Engl. J. Med., 359, 2220–2232. [DOI] [PubMed] [Google Scholar]
- 22. de Miguel-Yanes, J.M., Shrader, P., Pencina, M.J., Fox, C.S., Manning, A.K., Grant, R.W., Dupuis, J., Florez, J.C., D'Agostino, R.B. and Cupples, L.A. (2011) Genetic risk reclassification for type 2 diabetes by age below or above 50 years using 40 type 2 diabetes risk single nucleotide polymorphisms. Diabetes Care, 34, 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vassy, J., Durant, N., Kabagambe, E., Carnethon, M.R., Rasmussen-Torvik, L.J., Fornage, M., Lewis, C., Siscovick, D. and Meigs, J. (2012) A genotype risk score predicts type 2 diabetes from young adulthood: the CARDIA study. Diabetologia, 55, 2604–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vassy, J.L., DasMahapatra, P., Meigs, J.B., Schork, N.J., Magnussen, C.G., Chen, W., Raitakari, O.T., Pencina, M.J., Jamal, S.M. and Berenson, G.S. (2012) Genotype prediction of adult type 2 diabetes from adolescence in a multiracial population. Pediatrics, 130, e1235–e1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Walford, G.A., Porneala, B.C., Dauriz, M., Vassy, J.L., Cheng, S., Rhee, E.P., Wang, T.J., Meigs, J.B., Gerszten, R.E. and Florez, J.C. (2014) Metabolite traits and genetic risk provide complementary information for the prediction of future type 2 diabetes. Diabetes Care, 37, 2508–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mahajan, A., Go, M.J., Zhang, W., Below, J.E., Gaulton, K.J., Ferreira, T., Horikoshi, M., Johnson, A.D., Ng, M.C., Prokopenko, I.et al. (2014) Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat. Genet., 46, 234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Florez, J. (2008) Newly identified loci highlight beta cell dysfunction as a key cause of type 2 diabetes: where are the insulin resistance genes? Diabetologia, 51, 1100. [DOI] [PubMed] [Google Scholar]
- 28. Dupuis, J., Langenberg, C., Prokopenko, I., Saxena, R., Soranzo, N., Jackson, A.U., Wheeler, E., Glazer, N.L., Bouatia-Naji, N., Gloyn, A.L.et al. (2010) New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat. Genet., 42, 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Scott, R.A., Lagou, V., Welch, R.P., Wheeler, E., Montasser, M.E., Luan, J., Magi, R., Strawbridge, R.J., Rehnberg, E., Gustafsson, S.et al. (2012) Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat. Genet., 44, 991–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Grarup, N., Sandholt, C.H., Hansen, T. and Pedersen, O. (2014) Genetic susceptibility to type 2 diabetes and obesity: from genome-wide association studies to rare variants and beyond. Diabetologia, 57, 1528–1541. [DOI] [PubMed] [Google Scholar]
- 31. Voight, B.F., Scott, L.J., Steinthorsdottir, V., Morris, A.P., Dina, C., Welch, R.P., Zeggini, E., Huth, C., Aulchenko, Y.S., Thorleifsson, G.et al. (2010) Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat. Genet., 42, 579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Scott, R.A., Chu, A.Y., Grarup, N., Manning, A.K., Hivert, M.-F., Shungin, D., Tönjes, A., Yesupriya, A., Barnes, D. and Bouatia-Naji, N. (2012) No interactions between previously associated 2-hour glucose gene variants and physical activity or BMI on 2-hour glucose levels. Diabetes, 61, 1291–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Voight, B.F., Scott, L.J., Steinthorsdottir, V., Morris, A.P., Dina, C., Welch, R.P., Zeggini, E., Huth, C., Aulchenko, Y.S. and Thorleifsson, G. (2010) Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat. Genet., 42, 579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Freathy, R.M., Timpson, N.J., Lawlor, D.A., Pouta, A., Ben-Shlomo, Y., Ruokonen, A., Ebrahim, S., Shields, B., Zeggini, E., Weedon, M.N.et al. (2008) Common variation in the FTO gene alters diabetes-related metabolic traits to the extent expected given its effect on BMI. Diabetes, 57, 1419–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fall, T., Hagg, S., Magi, R., Ploner, A., Fischer, K., Horikoshi, M., Sarin, A.P., Thorleifsson, G., Ladenvall, C., Kals, M.et al. (2013) The role of adiposity in cardiometabolic traits: a Mendelian randomization analysis. PLoS Med., 10, e1001474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xu, L., Borges, M.C., Hemani, G. and Lawlor, D.A. (2017) The role of glycaemic and lipid risk factors in mediating the effect of BMI on coronary heart disease: a two-step, two-sample Mendelian randomisation study. Diabetologia, 60, 2210–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dale, C.E., Fatemifar, G., Palmer, T.M., White, J., Prieto-Merino, D., Zabaneh, D., Engmann, J.E.L., Shah, T., Wong, A., Warren, H.R.et al. (2017) Causal associations of adiposity and body fat distribution with coronary heart disease, stroke subtypes, and type 2 diabetes mellitus: a Mendelian randomization analysis. Circulation, 135, 2373–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hägg, S., Fall, T., Ploner, A., Mägi, R., Fischer, K., Draisma, H.H., Kals, M., deVries, P.S., Dehghan, A., Willems, S.M.et al. (2015) Adiposity as a cause of cardiovascular disease: a Mendelian randomization study. Int. J. Epidemiol., 44, 578–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Whincup, P.H., Gilg, J.A., Papacosta, O., Seymour, C., Miller, G.J., Alberti, K.G. and Cook, D.G. (2002) Early evidence of ethnic differences in cardiovascular risk: cross sectional comparison of British south Asian and white children. BMJ (Clinical Research ed.), 324, 635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tagi, V.M., Giannini, C. and Chiarelli, F. (2019) Insulin resistance in children. Front. Endocrinol., 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Preis, S.R., Massaro, J.M., Robins, S.J., Hoffmann, U., Vasan, R.S., Irlbeck, T., Meigs, J.B., Sutherland, P., D'Agostino, R.B., Sr., O'Donnell, C.J.et al. (2010) Abdominal subcutaneous and visceral adipose tissue and insulin resistance in the Framingham heart study. Obesity (Silver Spring), 18, 2191–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Scott, R.A., Scott, L.J., Magi, R., Marullo, L., Gaulton, K.J., Kaakinen, M., Pervjakova, N., Pers, T.H., Johnson, A.D., Eicher, J.D.et al. (2017) An expanded genome-wide association study of type 2 diabetes in Europeans. Diabetes, 66, 2888–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bjørnland, T., Langaas, M., Grill, V. and Mostad, I.L. (2017) Assessing gene-environment interaction effects of FTO, MC4R and lifestyle factors on obesity using an extreme phenotype sampling design: results from the HUNT study. PLoS One, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Speliotes, E.K., Willer, C.J., Berndt, S.I., Monda, K.L., Thorleifsson, G., Jackson, A.U., Lango Allen, H., Lindgren, C.M., Luan, J.a., Mägi, R.et al. (2010) Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat. Genet., 42, 937–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Morandi, A., Bonnefond, A., Lobbens, S., Yengo, L., Miraglia del Giudice, E., Grandone, A., Lévy-Marchal, C., Weill, J., Maffeis, C. and Froguel, P. (2016) Associations between type 2 diabetes-related genetic scores and metabolic traits, in obese and normal-weight youths. J. Clin. Endocrinol. Metabol., 101, 4244–4250. [DOI] [PubMed] [Google Scholar]
- 46. Prokopenko, I., Langenberg, C., Florez, J.C., Saxena, R., Soranzo, N., Thorleifsson, G., Loos, R.J., Manning, A.K., Jackson, A.U., Aulchenko, Y.et al. (2009) Variants in MTNR1B influence fasting glucose levels. Nat. Genet., 41, 77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lyssenko, V., Nagorny, C.L., Erdos, M.R., Wierup, N., Jonsson, A., Spégel, P., Bugliani, M., Saxena, R., Fex, M., Pulizzi, N.et al. (2009) Common variant in MTNR1B associated with increased risk of type 2 diabetes and impaired early insulin secretion. Nat. Genet., 41, 82–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. THart, L.M., Simonis-Bik, A.M., Nijpels, G., vanHaeften, T.W., Schäfer, S.A., Houwing-Duistermaat, J.J., Boomsma, D.I., Groenewoud, M.J., Reiling, E., vanHove, E.C.et al. (2010) Combined risk allele score of eight type 2 diabetes genes is associated with reduced first-phase glucose-stimulated insulin secretion during hyperglycemic clamps. Diabetes, 59, 287–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gupta, D., Kono, T. and Evans-Molina, C. (2010) The role of peroxisome proliferator-activated receptor γ in pancreatic β cell function and survival: therapeutic implications for the treatment of type 2 diabetes mellitus. Diabetes Obes. Metab., 12, 1036–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li, L.-c., Wang, Y., Carr, R., Haddad, C.S., Li, Z., Qian, L., Oberholzer, J., Maker, A.V., Wang, Q. and Prabhakar, B.S. (2014) IG20/MADD plays a critical role in glucose-induced insulin secretion. Diabetes, 63, 1612–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Steinthorsdottir, V., Thorleifsson, G., Reynisdottir, I., Benediktsson, R., Jonsdottir, T., Walters, G.B., Styrkarsdottir, U., Gretarsdottir, S., Emilsson, V., Ghosh, S.et al. (2007) A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat. Genet., 39, 770–775. [DOI] [PubMed] [Google Scholar]
- 52.(2013) World medical association declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA, 310, 2191–2194. [DOI] [PubMed] [Google Scholar]
- 53. Shashaj, B., Luciano, R., Contoli, B., Morino, G.S., Spreghini, M.R., Rustico, C., Sforza, R.W., Dallapiccola, B. and Manco, M. (2016) Reference ranges of HOMA-IR in normal-weight and obese young Caucasians. Acta Diabetol., 53, 251–260. [DOI] [PubMed] [Google Scholar]
- 54. Cacciari, E., Milani, S., Balsamo, A., Spada, E., Bona, G., Cavallo, L., Cerutti, F., Gargantini, L., Greggio, N., Tonini, G.et al. (2006) Italian cross-sectional growth charts for height, weight and BMI (2 to 20 yr). J. Endocrinol. Investig., 29, 581–593. [DOI] [PubMed] [Google Scholar]
- 55. WHO/IDF (2006) Definition and Diagnosis of Diabetes Mellitus and Intermediate Hyperglycaemia : Report of a WHO/IDF Consultation. World Health Organization, Geneva. [Google Scholar]
- 56. Scott, R.A., Lagou, V., Welch, R.P., Wheeler, E., Montasser, M.E., Mägi, R., Strawbridge, R.J., Rehnberg, E., Gustafsson, S. and Kanoni, S. (2012) Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat. Genet., 44, 991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. R Core Team . (2021) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria.
- 58. Belsky, D.W., Moffitt, T.E., Sugden, K., Williams, B., Houts, R., McCarthy, J. and Caspi, A. (2013) Development and evaluation of a genetic risk score for obesity. Biodemography Soc. Biol., 59, 85–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analysed during the current study are not publicly available for reasons related to privacy and participant consent but are available from the corresponding author on reasonable request.