

Abstract
The protein α-Klotho acts as transmembrane co-receptor for fibroblast growth factor 23 (FGF23) and is a key regulator of phosphate homeostasis. However, α-Klotho also exists in a circulating form, with pleiotropic, but incompletely understood functions and regulation. Therefore, we undertook a genome-wide association study (GWAS) meta-analysis followed by Mendelian randomization (MR) of circulating α-Klotho levels.
Plasma α-Klotho levels were measured by enzyme-linked immunosorbent assay (ELISA) in the Ludwigshafen Risk and Cardiovascular Health and Avon Longitudinal Study of Parents and Children (mothers) cohorts, followed by a GWAS meta-analysis in 4376 individuals across the two cohorts.
Six signals at five loci were associated with circulating α-Klotho levels at genome-wide significance (P < 5 × 10−8), namely ABO, KL, FGFR1, and two post-translational modification genes, B4GALNT3 and CHST9. Together, these loci explained >9% of the variation in circulating α-Klotho levels. MR analyses revealed no causal relationships between α-Klotho and renal function, FGF23-dependent factors such as vitamin D and phosphate levels, or bone mineral density. The screening for genetic correlations with other phenotypes followed by targeted MR suggested causal effects of liability of Crohn’s disease risk [Inverse variance weighted (IVW) beta = 0.059 (95% confidence interval 0.026, 0.093)] and low-density lipoprotein cholesterol levels [−0.198 (−0.332, −0.063)] on α-Klotho.
Our GWAS findings suggest that two enzymes involved in post-translational modification, B4GALNT3 and CHST9, contribute to genetic influences on α-Klotho levels, presumably by affecting protein turnover and stability. Subsequent evidence from MR analyses on α-Klotho levels suggest regulation by mechanisms besides phosphate-homeostasis and raise the possibility of cross-talk with FGF19- and FGF21-dependent pathways, respectively. Significance statement: α-Klotho as a transmembrane protein is well investigated along the endocrine FGF23-α-Klotho pathway. However, the role of the circulating form of α-Klotho, which is generated by cleavage of transmembrane α-Klotho, remains incompletely understood. Genetic analyses might help to elucidate novel regulatory and functional mechanisms. The identification of genetic factors related to circulating α-Klotho further enables MR to examine causal relationships with other factors. The findings from the first GWAS meta-analysis of circulating α-Klotho levels identified six genome-wide significant signals across five genes. Given the function of two of the genes identified, B4GALNT3 and CHST9, it is tempting to speculate that post-translational modification significantly contributes to genetic influences on α-Klotho levels, presumably by affecting protein turnover and stability.
Introduction
α-Klotho is a transmembrane protein that serves with fibroblast growth factor receptors as a co-receptor for fibroblast growth factor 23 (FGF23) (1). The endocrine FGF23-α-Klotho pathway plays a critical role in regulating vitamin D metabolism and phosphate balance (2). Deletion of α-Klotho in mice results in disturbed phosphate homeostasis with an accelerated ageing phenotype, including a shortened lifespan, vascular calcification, infertility and osteoporosis (3). A soluble form of α-Klotho is generated by cleavage of transmembrane α-Klotho, which is readily detected in the circulation, and henceforth referred to as circulating α-Klotho (4). Though the role of circulating α-Klotho remains incompletely understood, it is thought to share functional similarities with the membrane-bound form and to contribute to the actions of its ligand, FGF23 (5). However, circulating α-Klotho also mediates effects independent of FGF23 (6). Previous studies have investigated the clinical utility of serum α-Klotho as a prognostic marker. For example, a recent meta-analysis found that serum levels of circulating α-Klotho are positively related to the estimated glomerular filtration rate (eGFR) in patients with chronic kidney disease (CKD) (7), suggesting a role of α-Klotho as a biomarker for CKD progression. In addition, α-Klotho supplementation is under investigation as a possible drug target for treatment in CKD (5,8).
Genetic studies may prove useful in identifying novel mechanisms which regulate α-Klotho. Several KL (α-klotho) gene polymorphisms have previously been reported in association with urolithiasis, cardiovascular disease, cancers and longevity (9,10). However, little is known of the genetic pathways which regulate levels of circulating α-Klotho. To our knowledge, a genome-wide association study (GWAS) of α-Klotho has not previously been undertaken. As well as helping to elucidate novel regulatory mechanisms, identification of genetic factors related to circulating α-Klotho enables Mendelian randomization (MR) to examine causal relationships with other factors. This approach can also be used to validate potential drug targets such as α-Klotho, on the basis that the target in question will only modify the outcome (e.g. CKD) in the presence of a causal relationship (11,12).
Therefore, to elucidate novel regulatory mechanisms and functional relationships of circulating α-Klotho, we performed, to our knowledge, the first GWAS meta-analysis of plasma α-Klotho. Subsequently, we used our GWAS output to examine causal relationships between plasma α-Klotho and CKD, other FGF23-dependent pathways [i.e. vitamin D, phosphate and bone mineral density (BMD)], as well as other characteristics with evidence of genetic correlation.
Results
GWAS meta-analysis results
A total of 4376 individuals provided results for the α-Klotho GWAS meta-analysis after combining Ludwigshafen Risk and Cardiovascular Health (LURIC) and Avon Longitudinal Study of Parents and Children (ALSPAC) mothers (Supplementary Material, Table S1). Six independent signals at five loci showed genome-wide significance (Fig. 1, Table 1 and Supplementary Material, Table S3) (median genomic inflation λ = 1.013). The top-associated single-nucleotide polymorphism (SNP), rs12607664 [standard deviations difference relative to mean in plasma α-Klotho per minor allele (MA) T: β = 0.24, standard error (SE) = 0.02, P = 2.3 × 10−27], mapped to the second intron of the gene CHST9 on chromosome 18. The second top-associated variant rs8176672 (MA T: β = 0.41, SE = 0.04, P = 2.1 × 10−23) mapped to the first intron of the ABO gene on chromosome 9. A strong association was also observed for another conditionally independent variant at this locus, rs532436, not in linkage disequilibrium (LD) with rs8176672 (r2 = 0.02 in the 1000 Genome Europeans). A variant at a further locus, rs1056008 (MA C: β = 0.18, SE = 0.02, P = 1.8 × 10−14), is located in the longest exon of B4GALNT3 on chromosome 12. We also identified SNPs rs7333961 (MA A: β = −0.33, SE = 0.05, P = 1.73 × 10−10), upstream of the gene coding for α-Klotho on chromosome 13, and rs881301 (MA C: β = −0.12, SE = 0.02, P = 2.2 × 10−08), upstream of FGFR1 on chromosome 8. Genetic associations were broadly similar in both cohorts, with rs8176672 and rs12607664 reaching genome-wide significance in each (Supplementary Material, Fig. S1). That said, Z-score test suggested differences between the two cohort in the case of rs12607664, effect size being stronger in ALSPAC. This did not appear to reflect sex differences between the two cohorts, as associations at this locus were identical in LURIC males and females, suggesting other differences are likely responsible such as age.
Figure 1.
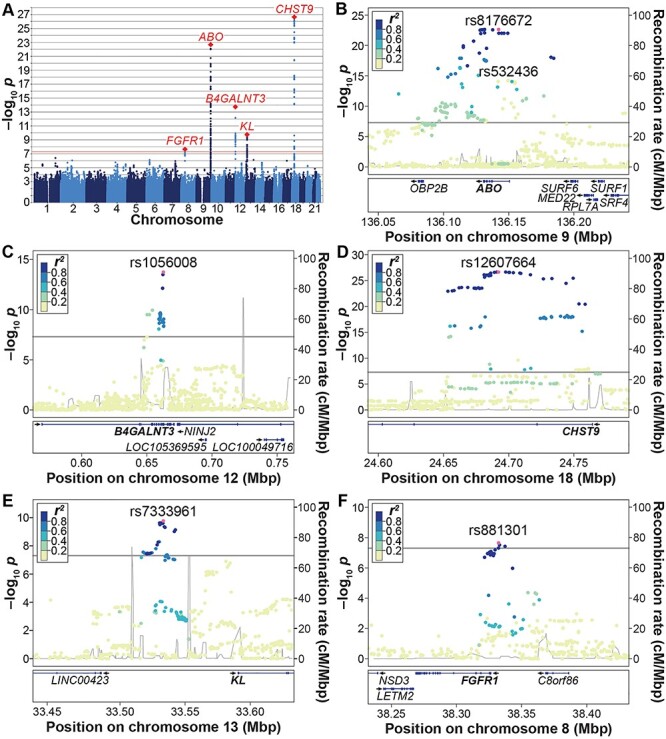
Results from the α-Klotho GWAS meta-analysis. (A) Manhattan plot. The x-axis indicates the chromosomal position of each SNP, whereas the y-axis denotes the evidence of association shown as −log10(P-value). The red line indicates genome-wide significance of association (P = 5 × 10−8). (B–F) Locus-specific Manhattan plots of the genome-wide significant loci at (B) the ABO locus, (C) the B4GALNT3 locus, (D) the CHST9 locus, (E) the KL locus and (F) the FGFR1 locus. The x-axis indicates the physical position of each SNP on the chromosome, the y-axis denotes the evidence of association as the −log10(P-value). The linkage disequilibrium (LD) r2 between SNPs, based on the 1000 Genomes EUR superpopulation, is shown in colour.
Table 1.
Top-associated SNPs of the meta-analysis of both GWAS on plasma α-Klotho levels
| SNP | Chr. | EA | OA | EAF | Gene | β | SE | P | Pair-wise Z | P_Z |
|---|---|---|---|---|---|---|---|---|---|---|
| rs12607664 | 18 | T | G | 31.61 | CHST9 | 0.243 | 0.022 | 2.28 × 10−27 | 3.337 | 0.001 |
| rs8176672 | 9 | T | C | 7.18 | ABO | 0.406 | 0.041 | 2.11 × 10−23 | 1.785 | 0.074 |
| rs532436 | 9 | G | A | 23.99 | ABO | 0.204 | 0.026 | 5.86 × 10−15 | 0.691 | 0.490 |
| rs1056008 | 12 | C | T | 26.82 | B4GALNT3 | 0.184 | 0.024 | 1.80 × 10−14 | 0.478 | 0.632 |
| rs7333961 | 13 | A | G | 4.62 | KL | −0.327 | 0.051 | 1.73 × 10−10 | 0.816 | 0.415 |
| rs881301 | 8 | C | T | 41.28 | FGFR1 | −0.119 | 0.021 | 2.23 × 10−08 | 0.087 | 0.931 |
The effect size (β) represents SD difference in α-Klotho relative to the mean (i.e. Z-score) per effect allele. Results show univariate/simple linear regression estimates for the six conditionally independent SNPs. The effect size (β) represents SD difference in α-Klotho relative to the mean (i.e. Z-scores) per effect allele. Chr., chromosome; EA, effect allele; OA, other allele; EAF, effect allele frequency in the pooled sample in percentage; SE, standard error. Pair-wise Z and P_Z are the pair-wise Z estimating the difference between two effect estimates (in LURIC and ALSPAC) and P-value of the Z-score.
LD score regression
We used our α-Klotho GWAS data in LD score regression to identify other traits with which α-Klotho might be functionally related, using P < 0.05 to indicate findings for potential follow-up, which applied to eight traits (Table 2). Of these, one metabolic trait was related to lipid levels, corresponding to the action of FGF21 (1), which, together with FGF19 and FGF23, comprise the endocrine FGFs (Table 2). In addition, one trait, Crohn’s disease, was related to the action of FGF19 (1).
Table 2.
LD score regression between α-Klotho and human traits (for associations with P < 0.05)
| Trait 1 | Trait 2 | r g | SE | Z | P |
|---|---|---|---|---|---|
| α-Klotho | Excessive frequent and irregular menstruation | 0.48 | 0.21 | 2.3 | 0.02 |
| α-Klotho | Fibroblastic disorders | −0.38 | 0.17 | −2.2 | 0.03 |
| α-Klotho | Triglycerides in small VLDL | −0.54 | 0.24 | −2.2 | 0.03 |
| α-Klotho | Chronotype | −0.17 | 0.08 | −2.1 | 0.03 |
| α-Klotho | Crohn’s disease | 0.25 | 0.12 | 2.1 | 0.03 |
| α-Klotho | Eczema | −0.48 | 0.23 | −2.1 | 0.04 |
| α-Klotho | Co-codamol | 0.44 | 0.21 | 2.1 | 0.04 |
| α-Klotho | Prostate cancer | 0.49 | 0.25 | 2.0 | 0.05 |
r g, genetic correlation; SE, standard error of the genetic correlation analysis; Z, corresponding Z-score; P, corresponding P-value; VLDL, very low-density lipoprotein.
MR analyses
Our α-Klotho instruments for MR were based on six SNPs that showed conditional independent effects on α-Klotho using GCTA conditional and joint analysis (COJO) (13), which together accounted for a relatively high proportion of variance (9.1%) (Supplementary Material, Table S4). An F-test revealed the instrument to have an acceptable instrumental strength for subsequent MR analyses (F-statistic = 74.8). Using these α-Klotho-associated SNPs as instruments of MR, IVW analyses showed no causal effect of α-Klotho on CKD risk (based on an eGFR <60 ml/min/1.73 m2) or eGFR derived from either creatinine or cystatin-C (Table 3). Bi-directional analyses using results from eGFR (creatinine) and eGFR (cystatin-C) GWASs as the exposures and our α-Klotho results as the outcome revealed no reverse causality. Null results were also observed in sensitivity analyses (Supplementary Material, Table S5). Similarly, MR analyses examining relationships between α-Klotho and outcomes related to the FGF23 pathway, namely BMD, vitamin D and phosphate levels, did not support causal relationships, including bi-directional and sensitivity analyses (Table 3 and Supplementary Material, Table S6).
Table 3.
Bi-directional MR results between α-Klotho- and FGF-related outcomes
| Outcome | MR of α-Klotho versus outcomes | Reverse MR of outcomes versus α-Klotho | ||||
|---|---|---|---|---|---|---|
| β | 95% CI | P | β | 95% CI | P | |
| Renal outcomes | ||||||
| CKD | −0.019 | −0.077 to 0.039 | 0.525 | −0.170 | −0.337 to −0.004 | 0.055 0.387 |
| eGFR (crea) | 0.000 | −0.003 to 0.004 | 0.871 | 0.405 | −0.512 to 1.322 | |
| eGFR (cys) | 0.005 | −0.291 to 0.300 | 0.976 | 0.381 | −0.436 to 1.198 | 0.361 |
| FGF23 pathway outcomes | ||||||
| eBMD | 0.020 | −0.017 to 0.056 | 0.288 | 0.022 | −0.068 to 0.113 | 0.626 |
| Phosphate | 0.009 | −0.007 to 0.025 | 0.288 | 0.085 | −0.073 to 0.243 | 0.291 |
| Vitamin D | −0.001 | −0.018 to 0.015 | 0.861 | 0.150 | −0.003 to 0.304 | 0.055 |
| FGF19 pathway outcomes | ||||||
| IBD | −0.030 | −0.109 to 0.049 | 0.454 | 0.059 | 0.026 to 0.093 | 5.4 × 10−4 |
| UC | −0.055 | −0.151 to 0.041 | 0.261 | 0.039 | −0.007 to 0.085 | 0.099 |
| CD | 0.011 | −0.102 to 0.124 | 0.848 | 0.044 | 0.003 to 0.086 | 0.034 |
| FGF21 pathway lipids/obesity | ||||||
| BMI | −0.010 | −0.029 to 0.009 | 0.304 | −0.080 | −0.228 to 0.067 | 0.286 |
| LDL-C | −0.097 | −0.237 to 0.044 | 0.178 | −0.198 | −0.332 to −0.063 | 3.9 × 10−3 |
| HDL-C | 0.018 | −0.033 to 0.070 | 0.490 | −0.063 | −0.133 to 0.007 | 0.078 |
| TG | 0.006 | −0.031 to 0.043 | 0.748 | 0.032 | −0.087 to 0.152 | 0.594 |
| Apo A1 | −0.006 | −0.068 to 0.056 | 0.843 | 0.004 | −0.030 to 0.037 | 0.833 |
| Apo B | −0.046 | −0.159 to 0.068 | 0.430 | −0.023 | −0.052 to 0.007 | 0.130 |
| FGF21 pathway insulin sensitivity | ||||||
| Fasting glucose | −0.010 | −0.040 to 0.020 | 0.520 | 0.186 | −0.108 to 0.481 | 0.215 |
| Fasting insulin | 0.009 | −0.008 to 0.026 | 0.296 | 0.829 | −0.702 to 2.360 | 0.288 |
| HbA1C | −0.019 | −0.043 to 0.005 | 0.120 | 0.158 | −0.266 to 0.583 | 0.465 |
| HOMA-B | 0.015 | −0.012 to 0.042 | 0.279 | −0.264 | −1.035 to 0.508 | 0.503 |
The effect size (β) represents SD change in outcome per SD change in exposure. eGFR (crea), estimated glomerular filtration rate by creatinine plasma concentration; eGFR (cys), estimated glomerular filtration rate by Cystatin C plasma concentration; BMD, bone mineral density; IBD, inflammatory bowel disease; UC, ulcerative colitis; CD, Crohn’s disease; BMI, body mass index; LDL, low-density lipoprotein; HDL, high-density lipoprotein; TG, triglycerides; ApoA1, Apolipoprotein A1; ApoB, Apolipoprotein B; HbA1C, glycated haemoglobin A; HOMA-I, homeostasis model assessment.
Given findings from LD score regression suggesting that α-Klotho might have functional relationships with other endocrine FGFs, we extended our MR analyses to traits related to FGF19 and FGF21. We found no causal effect of α-Klotho on inflammatory bowel disease. However, in bi-directional MR, IVW analyses revealed evidence of a positive relationship for genetic liability to inflammatory bowel disease and, particularly, Crohn’s disease on α-Klotho levels (Table 3), with similar findings in sensitivity analyses (Supplementary Material, Table S7).
IVW and sensitivity analyses revealed no causal relationship between α-Klotho and body mass index (BMI), lipids and traits related to insulin sensitivity (Tables 3, Supplementary Material, Tables S8 and S9). Bi-directional analyses likewise revealed no cause effect of BMI and traits related to insulin sensitivity on α-Klotho. However, IVW suggested a negative causal effect of low-density lipoprotein cholesterol (LDL-C) on α-Klotho, whereas no consistent causal effects were seen for high-density lipoprotein cholesterol (HDL-C) or triglycerides (TGs) (Table 3). The putative causal effect of LDL-C was strengthened when adjusting for HDL-C and TGs in multivariable MR analyses including all three variables (Model 1, Table 4). Additional models examined the contribution of apolipoproteins to these effects, in light of their role in mediating effects of lipoprotein lipids on coronary heart disease (14). In a further multivariable model to study the role of apolipoprotein B (ApoB) (Model 2), only ApoB showed a potentially causal relationship (Table 4). A final multivariable model to study the role of apolipoprotein A-I (ApoA1) (Model 3) found no additional causal effect of ApoA1.
Table 4.
Multivariable MR analyses for lipid traits on α-Klotho
| Model | Exposure | Outcome | N SNPs | β | SE | P |
|---|---|---|---|---|---|---|
| Model 1 | LDL-C | α-Klotho | 309 | −0.173 | 0.060 | 3.6 × 10−3 |
| HDL-C | α-Klotho | 309 | −0.069 | 0.043 | 0.106 | |
| TG | α-Klotho | 309 | 0.042 | 0.065 | 0.513 | |
| Model 2 | Low-density lipoprotein | α-Klotho | 365 | 0.070 | 0.236 | 0.766 |
| Triglyceride | α-Klotho | 365 | −0.227 | 0.216 | 0.295 | |
| Apolipoprotein B | α-Klotho | 365 | 0.114 | 0.056 | 0.041 | |
| Model 3 | High-density lipoprotein | α-Klotho | 459 | −0.070 | 0.135 | 0.601 |
| Apolipoprotein A-I | α-Klotho | 459 | 0.025 | 0.152 | 0.870 |
Model refers to the three multivariable MR models used to examine relationship between lipids and α-Klotho. The effect size (β) represents SD change in outcome per SD change in exposure.
N SNPs, the number of SNPs used in the genetic instrument for the exposure; LDL, low-density lipoprotein; HDL, high-density lipoprotein; TG, triglycerides; β, causal effect size of each lipid subtype on α-Klotho; SE respective standard error; P, respective p-value.
Functional follow-up of GWAS results
According to the expression quantitative trait loci (eQTL) databases GTEx v8 and eQTLGen, rs8176672 and rs532436 were strong cis-eQTLs for ABO in multiple tissues (Supplementary Material, Table S10). Likewise, rs1056008 and rs881301 were strong eQTLs for B4GALNT3 and FGFR1, respectively, in multiple tissues and both databases. SNP rs12607664 was, according to GTEx, an eQTL for CHST9 in the cerebellum only. Variant rs7333961 was an eQTL for KL in whole blood in the eQTLGen project. Co-localization analyses confirmed a common genetic signal in the case of plasma α-Klotho and B4GALNT3 eQTL data in whole blood, but the same was not observed for the other loci (Supplementary Material, Table S11). The B4GALNT3 SNP, rs1056008, had a RegulomeDB score of 1B, indicating a strong likelihood of affecting transcription factor binding and gene expression. No top association signal, or one in high LD, intersected with a DNase-hypersensitive site in the B4GALNT3 gene in kidney tubule cells.
To explore the contribution of B4GALNT3 to the regulation of circulating α-Klotho levels, α-Klotho levels were measured in B4galnt3-deficient mice. However, no clear differences were observed when comparing wild-type, heterozygous and homozygous animals (Supplementary Material, Fig. S2).
Discussion
Having performed a GWAS for circulating levels of α-Klotho, we identified six GWAS significant signals, mapping to five genes, which together explained over 9% of the variance. This provided genetic predictors of α-Klotho with acceptable strength to interrogate causal relationships. We applied a two-sample MR approach in large GWAS data sets. Despite the potential role of circulating α-Klotho as an early biomarker of CKD (7), MR analyses revealed no causal relationship between circulating α-Klotho and renal function. We also interrogated potential causal relationships between α-Klotho and other outcomes linked to the FGF23 pathway, such as BMD, phosphate and vitamin D, again with little MR evidence. Based on findings from genetic correlation analyses, we also examined causal relationships between α-Klotho and traits related to other endocrine FGFs. Of the six genome-wide significant signals identified in our GWAS meta-analysis, the cis-eQTL signal for B4GALNT3 co-localized with plasma α-Klotho, suggesting that higher levels of B4GALNT3 expression lead to greater α-Klotho levels. RegulomeDB predicted that our top variant at this locus, rs1056008, alters the binding affinity of activating transcription factors.
As to the mechanisms underlying this genetic association, B4GALNT3 expresses the enzyme beta-1,4-N-acetylgalactosaminyltransferase 3 (EC:2.4.1.244), localizing to the Golgi apparatus. This enzyme transfers N-acetylgalactosamine (GalNAc) onto N-acetylglucosamine-beta-benzyl to form GalNAcβ1,4-GlcNAc structures on protein epitopes, also known as N,N′-diacetyllactosediamine (LacdiNAc) (15). We previously found that B4GALNT3 is expressed at the highest levels in renal tissue (16), an important site of α-Klotho production (8). The presence of the LacdiNAc moiety on circulating proteins such as α-Klotho might influence protein levels in the circulation by altering their turnover and degradation. Such a mechanism may explain why mutations in another glycosylation enzyme, GALNT3, lead to heritable tumoural calcinosis as a consequence of FGF23 deficiency (17). On the other hand, we found that B4GALNT3 null mice showed no clear alteration in α-Klotho levels in an established immunoprecipitation–immunoblot (IP-IB) assay. One potential explanation for this apparent discrepant finding is that, rather than altering actual levels of α-Klotho, genetic alterations in LacdiNAc content affect the epitope binding with the human ELISA assay used in our study. Furthermore, different functional effects of B4GALNT3-dependent α-Klotho modification in mice and humans cannot be ruled out.
CHST9 expresses the enzyme carbohydrate sulphotransferase 9, which catalyzes the transfer of a sulphate to terminal LacdiNAc sequences. The two enzymes, B4GALNT3 and CHST9, thus both contribute to generating the terminal SO4-4-GalNAcβ1,4GlcNAcβ structure (18). Several studies have shown that oligosaccharides terminating with this structure are recognized by asialoglycoprotein and mannose receptors and are rapidly degraded (19–22). Such specific receptors have been identified on hepatocytes and endothelial cells (20,21). Interestingly, the clearance rate mediated by the mannose receptor differs by the position of GalNAcβ1,4GlcNAcβ sulphation (23).
The identification of α-Klotho GWAS signals in the KL and FGFR1 genes, coding for the two components of the fibroblast growth factor receptor, was predictable and supports the overall validity of our findings. The ABO gene, also found to be associated with α-Klotho in our GWAS, codes for two proteins localizing to the Golgi apparatus, alpha 1–3-N-acetylgalactosaminyltransferase (EC:2.4.1.40) and alpha 1–3-galactosyltransferase (EC:2.4.1.37). These enzymes add UDP-N-acetyl-galactosamine and UDP-galactose, respectively, to glycoprotein fucosyl-galactosyl residuals (24). Conceivably, α-Klotho may serve as a target for these enzymes, thereby altering its turnover and degradation, as also postulated for B4GALNT3 and CHST9. However, associations with the ABO locus, observed in many previous GWASs including a recent GWAS of severe coronavirus disease 2019 with respiratory failure (25), may also have arisen as a result of population stratification despite our best attempts to adjust for this.
Whereas little causal effect was observed for α-Klotho, bi-directional analyses revealed causal effects of genetic liability of Crohn’s disease risk and LDL-C on circulating α-Klotho. Moreover, on multivariable MR of lipid indices, a causal relationship was only retained for ApoB, consistent with the suggestion from another recent multivariable MR that ApoB underlies the relationship between lipid traits and coronary heart disease risk (14). We are not aware of any previous reports linking genetic liability to Crohn’s disease risk or LDL-C to α-Klotho levels. That said, our results are consistent with previous findings that α-Klotho expression is downregulated in hyperlipidaemic mouse models and oxidized-LDL treated tubular cells (26,27). There was reasonably strong statistical evidence for the causal effects of Crohn’s disease which we observed, even when taking into account the multiple traits examined in our MR analyses and the bi-directional causal effects which were evaluated. On the other hand, statistical evidence with respect to causal effects of LDL-C was somewhat weaker having adjusted for multiple comparisons. In the absence of other sources of evidence, further confirmation is required in the case of both of these novel putative causal pathways for α-Klotho levels.
Crohn’s disease and metabolic traits were selected for MR analysis on the basis of their relationship with endocrine FGFs. However, FGF19 and FGF21 pathways (involved in Crohn’s disease and lipid metabolism, respectively) are mediated by β-Klotho, as opposed to α-Klotho (1). Though there is currently no other evidence linking α-Klotho to FGF19 and FGF21 pathways, the present findings certainly raise this as a possibility. Alternatively, relationships between Crohn’s disease susceptibility, LDL-C and circulating α-Klotho which we observed may be independent of endocrine FGFs. It is well recognized that circulating α-Klotho exerts a number of effects independently of FGF23, such as inhibition of insulin, Wingless and Int-1 or transforming growth factor-β signalling (6). In addition, as well as having anti-inflammatory effects by suppressing the transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells (28), α-Klotho renal expression has been found to be downregulated in mouse models of colitis, which was prevented by neutralizing antibodies against Tumor necrosis factor (TNF)-α (29). Accordingly, renal α-Klotho expression is reduced by tumor necrosis factor-like weak inducer of apoptosis and TNF-α (30), and circulating α-Klotho levels are negatively correlated with circulating markers of inflammation (31).
Many of the genetic factors associated with Crohn’s disease risk represent inflammatory mediators, such as IL-23 (32), which could conceivably also affect α-Klotho levels. This raises the possibility of horizontal pleiotropy, whereby genetic factor(s) related to Crohn’s disease affect α-Klotho levels directly, as opposed to via Crohn’s disease risk. That said, sensitivity analyses such as the MR–Egger intercept test did not suggest that pleiotropy contributed to our results, although we recognize that this test is often underpowered.
The main limitation of the present study is the relatively small GWAS sample size. That said, the genetic signals which we identified seemed plausible and provided a relatively strong genetic instrument for subsequent MR analyses. In addition, other characteristics which we examined in relation to α-Klotho were supported by well-powered GWASs that were derived from large data sets. In terms of other limitations, the commercial ELISA employed to measure α-Klotho in the GWAS samples has inferior performance compared with the IP-IB assay, though the latter is unsuitable for use in large cohorts due to its labour-intensive nature (33).
In conclusion, we present findings from the first GWAS of circulating α-Klotho levels, in which we identified six genome-wide significant signals across five genes. Given the function of two of the genes identified, B4GALNT3 and CHST9, it is tempting to speculate that post-translational modification contributes to genetic influences on α-Klotho levels, presumably by affecting protein turnover and stability. In subsequent MR analyses, we found no causal relationship between α-Klotho and CKD or FGF23-dependent pathways. However, there was evidence of a causal effect of Crohn’s disease risk and, to a lesser extent, LDL levels on α-Klotho levels, pointing to novel interactions which require further study.
Materials and Methods
LURIC
The LURIC study is a prospective cohort study of individuals with and without cardiovascular disease and was designed to investigate environmental and genetic risk factors for the development of cardiovascular diseases. Between July 1997 and January 2000, 3316 participants of German ancestry were enrolled in the cardiology unit of a tertiary care medical centre in Southwestern Germany. The inclusion criteria were defined as clinical stability except for acute coronary syndromes (ACS), German ancestry and availability of a coronary angiogram (indicated after standard clinical test diagnoses like chest pain and a positive, non-invasive stress test). Exclusion criteria were pre-specified as any acute illness other than ACS, any chronic disease where non-cardiac disease predominated and a history of malignancy within the past 5 years. The detailed study protocol has been published (34). Written informed consent was obtained from each participant prior to inclusion. The study was in accordance with the Declaration of Helsinki and approved by the ethics committee at the Medical Association of Rhineland-Palatinate (Ärztekammer Rheinland-Pfalz). Genotyping was conducted on the Affymetrix 6.0 platform and genotype calling using the algorithm Birdseed v2, both at the LURIC study non-profit LLC, Heidelberg. Quality control was performed in PLINK v1.90b3s (35), as described before (36). Genotype data were imputed to the 1000 Genomes Phase 1 reference panel using SHAPEIT2 and IMPUTE2 (37–39). The resulting data set contained 8 014 018 high-quality variants with an minor allele frequency (MAF) ≥1% and an information metric (INFO) metric ≥0.8 (Supplementary Material S1: Supplementary Methods: LURIC genotype data for more details).
ALSPAC
ALSPAC is a prospective birth cohort that recruited pregnant women with expected delivery dates between April 1991 and December 1992 from Bristol, UK. The initial number of pregnancies enrolled was 14 541 (for these, at least one questionnaire has been returned or a ‘Children in Focus’ clinic had been attended by July 19, 1999). Of these initial pregnancies, there was a total of 14 676 fetuses, resulting in 14 062 live births and 13 988 children who were alive at 1 year of age. Detailed information on the health and development of children and their parents were collected from regular clinic visits and completion of questionnaires (40,41). Ethical approval was obtained from the ALSPAC Law and Ethics Committee and the Local Ethics Committees. Please note that the study website contains details of all the data that is available through a fully searchable data dictionary (http://www.bristol.ac.uk/alspac/researchers/our-data/). Genotyping of ALSPAC samples was conducted on the Illumina Human660W-Quad platform and genotype calling using Illumina GenomeStudio. Quality control was carried out in PLINK v1.90 (35). Genotype data were imputed to the Haplotype Reference Consortium V1.0 reference panel using SHAPEIT v2 and IMPUTE v2.2.2 (37–39) The resulting data set contained 7 122 422 variants with an MAF ≥1% and INFO ≥0.8.
Measurement of α-Klotho levels
α-Klotho was measured in plasma samples from both cohorts using the human circulating α-Klotho assay kit (Immuno-Biological Laboratories Co., Ltd, Japan) (42). The lower detection limit was 6.15 pg/ml with a measurement range from 93.75 to 6000 pg/ml. The coefficient of variation was 11.4% at 165.47 pg/ml and 2.9% at 2903.01 pg/ml.
α-Klotho levels in B4GALNT3 null mice
B4galnt3 −/− mice were generated by breeding B4galnt3tm1c(EUCOMM)Wtsi male mice (Institut Clinique de la Souris, Illkirch, France), having LoxP sites introduced upstream of exon 8 and downstream of exon 9 of the B4galnt3 gene, with female mice expressing cre recombinase under the control of the phosphoglycerate kinase-1 promoter (PGKcre) (43). The offspring were heterozygous B4galnt3+/− mice. To generate B4galnt3−/− knockout mice, heterozygous B4galnt3+/−, and littermate wild type control mice, female and male B4galnt3+/− mice were mated. The mice were housed in a standard animal housing facility with a 12 h dark/light period. Food and water were available ad libitum. Before termination at 13 weeks of age, the mice were given an intraperitoneal injection with Ketalar (Pfizer, New York, NY, USA) and Dexdomitor (Orion Pharma, Esbo, Finland) before they were bled and euthanized with cervical dislocation. The animal experiments were approved by the Ethics Committee at University of Gothenburg, and the care of the animals was according to their guidelines. Circulating α-Klotho in mouse serum of B4galnt3 wild-type, heterozygous and knockout mice was measured by an established IP-IB assay at the UT Southwestern (TX, USA) (44). Differences between groups were analysed by the Steel–Dwass test.
GWAS meta-analysis
α-Klotho levels were transformed using rank-based inverse normal transformation before analysis. GWAS was conducted using linear regression on imputed probabilities in PLINK for LURIC and SNPTEST for ALSPAC data. Sex, age and the first eight ancestry principle components (PCs) were used as covariates in LURIC, where sex, age and the first 10 PCs were used as covariates in ALSPAC. The final sample sizes were n = 2234 for LURIC and n = 2142 for ALSPAC (Supplementary Material, Table S1). Results from both GWAS were combined using fixed-effects meta-analysis in METAL (45), the pooled data set containing 6 439 450 common variants and N = 4376 individuals. A threshold of P < 5 × 10−8 was used to denote genome-wide significance. A Z-score test, comparing mean estimates of associations between the two cohorts, was used to evaluate heterogeneity.
Conditional analysis
To detect multiple independent association signals at each of the genome-wide significant α-Klotho loci, we carried out an approximate conditional and joint genome-wide association analysis using the software package GCTA-COJO (13). SNPs in LD (LD r2 > 0.9) were ignored, and those situated >10 Mb away were assumed to be in complete linkage equilibrium. A reference sample of 8890 unrelated individuals of ALSPAC mothers was used to model patterns of LD between variants. The reference genotyping data set consisted of the same 6.44 million variants assessed in our GWAS meta-analysis. Conditionally independent variants that reached GWAS significance were annotated to the physically closest gene with the hg19 gene range list available in dbSNP (https://www.ncbi.nlm.nih.gov/SNP/).
LD score regression
Two analyses were conducted using LD score regression (46,47). First, we quantified the overall SNP-based heritability using LD score regression utilizing a subset of 1.2 million HapMap SNPs (with INFO >0.9 and MAF ≥1%). Second, we estimated the genetic correlation between plasma α-Klotho levels and 832 human traits implemented in the LD Hub database (48). This method uses the cross-products of summary test statistics from two GWASs and regresses them against a measure of how much variation each SNP tags (i.e. its LD score). Variants with high LD scores are more likely to contain more true signals and thus provide a greater chance of overlap with genuine signals between GWASs.
MR
We examined the evidence for a causal relationship between plasma α-Klotho levels and several renal phenotypes using two-sample MR (49). Primary analyses were performed using the inverse-variance weighted method, followed by sensitivity analyses [MR–Egger, weighted median, simple mode and weighted mode (50–52)]. Bi-directional analyses were also performed to examine reverse causality, where α-Klotho was considered as the outcome. In addition, MR analyses examined causal relationships between α-Klotho and other FGF23-dependent pathways and phenotypes, namely BMD, vitamin D and phosphate.
LD score regression suggested possible relationships with IBD and metabolic outcomes, with which the other endocrine FGFs, FGF19 and FGF21, have previously been implicated (1). Therefore, MR analyses also examined relationships between α-Klotho and inflammatory bowel disease, and between α-Klotho and metabolic outcomes. In total, 24 outcomes were selected for the MR analysis (Supplementary Material, Table S2 for GWAS sources). After adjusting the influence of correlations among the 24 outcomes using PhenoSpD (53), 14 independent variables/tests remained. Therefore, in this further set of MR analyses, the threshold corrected for multiple testing using Bonferroni’s method was defined as α = 0.05/14 = 0.004. This figure was then divided by two to account for the bi-directional analyses performed for each outcome, giving a final figure of 0.002.
In addition, given lipid traits are correlated with each other, multivariable MR (54) was conducted for five lipid traits on α-Klotho to control for such a correlation. Three multivariable MR models were used:
LDL-C, HDL-C and TG versus α-Klotho.
LDL-C, TG and APOB versus α-Klotho.
HDL-C and APOA1 versus α-Klotho.
Genetic functional analyses
Cis-acting effects of the genome-wide significant GWAS signals were examined by lookups in the GTEx database v8 (55) and the eQTLGen database (56). Regulatory elements in non-coding regions of the human genome were identified using RegulomeDB v1.1 (57). To further estimate whether the top association signals of α-Klotho and gene eQTLs of the cis-genes share the same causal variants within a 1 Mb window around the top signals of the α-Klotho GWAS, we used a Bayesian model (coloc) to estimate such posterior probabilities (PP) (58). The default priors for co-localization analyses were used (the prior probability a SNP is associated with α-Klotho was 1 × 10−4; the prior probability a SNP is associated with gene expression of the cis-gene was 1 × 10−4; and the prior probability a SNP is associated with both α-Klotho and gene expression was 1 × 10−5). A lack of evidence (i.e. a PP <80%) in the co-localization analysis goes against the hypothesis that the cis-gene mediates the effect of the genetic signal in question on α-Klotho levels. The gene expression data were extracted from the eQTLGen database. We treated co-localized findings (PP ≥80%) as ‘colocalised’ and other results that did not pass co-localization as ‘not colocalised’.
DNase-I hypersensitivity site data was obtained from the ENCODE database (59) or two replicates of human kidney tubule primary cell cultures (accessions: ENF428WYR and ENCFF711TUV). Called peaks were lifted-over to hg19 using the liftOver utility (60) with ‘minMatch = 0.1’, all other settings left to default. Peaks were considered replicable if present in both samples—the middles of overlapping peaks were padded to define 150 bp regions. Genome-wide significant SNPs (defined as reported P-value <5e-8) were intersected with regions using bedtools intersect (61). In addition, assay for transposase-accessible chromatin using sequencing open-chromatin regions from mouse embryonic (E15.5) distal femur growth plate (GEO accession GSM2687479) (62) were obtained and lifted-over to hg19 using the liftOver utility as above. Whether genome-wide significant SNPs overlapped with these regions was then examined using bedtools.
Authors’ Contribution
I.G., J.Z., T.F.M.A., N.M.S., D.R., L.F., S.M.S., C.O., W.M., J.V. and J.H.T. contributed to the concept, interpretation, critical writing and/or analysis of the data. All authors contributed to revision of the intellectual content and final approval of the version to be published.
Supplementary Material
Acknowledgements
The authors extend their appreciation to the participants of the LURIC study. The authors thank the LURIC study team, which was either temporarily or permanently involved in patient recruitment, sample and data handling. The authors also would like to thank the laboratory staff at the Ludwigshafen General Hospital, Universities of Freiburg, Heidelberg, Ulm (Germany) and Graz (Austria).
The authors are extremely grateful to all the families who took part in the ALSPAC study, the midwives for their help in recruiting them and the whole ALSPAC team, which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists and nurses.
Conflict of Interest. I.G. reports employment with Boehringer Ingelheim International GmbH, outside the submitted work. B.K.K. reports lecture fees and/or advisory board memberships and/or study participation from Astellas, Bayer, Boehringer Ingelheim, Chiesi, Riepharm, Pfizer, Sanofi, Servier and Vifor Pharma, all outside the submitted work. W.M. reports employment with Synlab Holding Deutschland GmbH, received grants from Abbott Diagnostics, grants and personal fees from Aegerion Pharmaceuticals, grants and personal fees from AMGEN, grants and personal fees from AstraZeneca, grants and personal fees from BASF, grants and personal fees from Danone Research, personal fees from MSD, grants and personal fees from Sanofi, grants and personal fees from Siemens Diagnostics, personal fees from Synageva, all outside the submitted work. The IP-sb48/IB-KM2076 Klotho assay is supported by the O’Brien Kidney Research Center at the University of Texas Southwestern Medical Center (grant P30-DK079328).
All other authors declare that they have no competing interests.
Contributor Information
Ingrid Gergei, Vth Department of Medicine (Nephrology, Hypertensiology, Rheumatology, Endocrinology, Diabetology), University Medical Center, Medical Faculty Mannheim, University of Heidelberg, Mannheim 69117, Germany; Therapeutic Area Cardiovascular Medicine, Boehringer Ingelheim International GmbH, Ingelheim 06877, Germany.
Jie Zheng, MRC Integrative Epidemiology Unit (IEU), Bristol Medical School, University of Bristol, Oakfield House, Oakfield Grove, Bristol BS8 2BN, UK; Population Health Science, Bristol Medical School, University of Bristol, Bristol BS8 2BN, UK.
Till F M Andlauer, Max Planck Institute of Psychiatry, Munich 80804, Germany; Department of Neurology, Klinikum rechts der Isar, School of Medicine, Technical University of Munich, Munich 80333, Germany.
Vincent Brandenburg, Department of Cardiology and Nephrology, Rhein-Maas Klinikum Würselen, Würselen 52146, Germany.
Nazanin Mirza-Schreiber, Institute of Neurogenomics, Helmholtz Zentrum München, Neuherberg 85764, Germany.
Bertram Müller-Myhsok, Max Planck Institute of Psychiatry, Munich 80804, Germany; Munich Cluster for Systems Neurology (SyNergy), Munich 2145, Germany; Institute of Translational Medicine, University of Liverpool, Liverpool 11341, UK.
Bernhard K Krämer, Vth Department of Medicine (Nephrology, Hypertensiology, Rheumatology, Endocrinology, Diabetology), University Medical Center, Medical Faculty Mannheim, University of Heidelberg, Mannheim 69117, Germany; European Center for Angioscience ECAS, Medical Faculty Mannheim, University of Heidelberg, Mannheim 69117, Germany; Center for Preventive Medicine and Digital Health Baden-Württemberg (CPDBW), Medical Faculty Mannheim, Heidelberg University, Mannheim 69117, Germany.
Daniel Richard, Department of Human Evolutionary Biology, Harvard University, Cambridge 02138, MA, USA.
Louise Falk, MRC Integrative Epidemiology Unit (IEU), Bristol Medical School, University of Bristol, Oakfield House, Oakfield Grove, Bristol BS8 2BN, UK.
Sofia Movérare-Skrtic, Department of Internal Medicine and Clinical Nutrition, University of Gothenburg, Sahlgrenska Osteoporosis Centre, CBAR, Institute of Medicine, Gothenburg 41296, Sweden.
Claes Ohlsson, Department of Internal Medicine and Clinical Nutrition, University of Gothenburg, Sahlgrenska Osteoporosis Centre, CBAR, Institute of Medicine, Gothenburg 41296, Sweden; Department of Drug Treatment, Region Västra Götaland, Sahlgrenska University Hospital, Gothenburg 7163, Sweden.
George Davey Smith, MRC Integrative Epidemiology Unit (IEU), Bristol Medical School, University of Bristol, Oakfield House, Oakfield Grove, Bristol BS8 2BN, UK; Population Health Science, Bristol Medical School, University of Bristol, Bristol BS8 2BN, UK.
Winfried März, Vth Department of Medicine (Nephrology, Hypertensiology, Rheumatology, Endocrinology, Diabetology), University Medical Center, Medical Faculty Mannheim, University of Heidelberg, Mannheim 69117, Germany; SYNLAB Academy, SYNLAB Holding Deutschland GmbH, Mannheim 24496, Germany; Clinical Institute of Medical and Chemical Laboratory Diagnostics, Medical University of Graz, Graz 8010, Austria.
Jakob Voelkl, Institute for Physiology and Pathophysiology, Johannes Kepler University Linz, Linz 4040, Austria; Department of Nephrology and Medical Intensive Care, Charité-Universitätsmedizin Berlin, Berlin 10117, Germany; DZHK (German Centre for Cardiovascular Research), Partner Site Berlin, Berlin 10623, Germany.
Jonathan H Tobias, MRC Integrative Epidemiology Unit (IEU), Bristol Medical School, University of Bristol, Oakfield House, Oakfield Grove, Bristol BS8 2BN, UK; Musculoskeletal Research Unit, Translational Health, Learning and Research Building, Level 1 , Southmead Hospital, Bristol BS10 5NB, UK.
Funding
ALSPAC data collection was supported by the Wellcome Trust (grants WT092830M, WT088806, WT102215/2/13/2), UK Medical Research Council (G1001357) and University of Bristol. The UK Medical Research Council and the Wellcome Trust (ref: 102215/2/13/2) and the University of Bristol provide core support for ALSPAC.
J.Z. is supported by the UK Medical Research Council Integrative Epidemiology Unit (MC_UU_00011/1 and MC_UU_00011/4). J.Z. is funded by the Vice-Chancellor Fellowship from the University of Bristol. J.Z. is supported by Shanghai Thousand Talents Program. J.Z. is supported by the Academy of Medical Sciences (AMS) Springboard Award, the Wellcome Trust, the Government Department of Business, Energy and Industrial Strategy (BEIS), the British Heart Foundation and Diabetes UK (SBF006\1117).
References
- 1. Li, X. (2019) The FGF metabolic axis. Front. Med., 13, 511–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kuro, O.M. (2019) The Klotho proteins in health and disease. Nat. Rev. Nephrol., 15, 27–44. [DOI] [PubMed] [Google Scholar]
- 3. Lang, F., Leibrock, C., Pelzl, L., Gawaz, M., Pieske, B., Alesutan, I. and Voelkl, J. (2018) Therapeutic interference with vascular calcification-lessons from Klotho-hypomorphic mice and beyond. Front. Endocrinol. (Lausanne), 9, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen, C.D., Podvin, S., Gillespie, E., Leeman, S.E. and Abraham, C.R. (2007) Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM17. Proc. Natl. Acad. Sci. U. S. A., 104, 19796–19801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhong, X., Jagarlapudi, S., Weng, Y., Ly, M., Rouse, J.C., McClure, K., Ishino, T., Zhang, Y., Sousa, E., Cohen, J.et al. (2020) Structure-function relationships of the soluble form of the antiaging protein Klotho have therapeutic implications for managing kidney disease. J. Biol. Chem., 295, 3115–3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dalton, G.D., Xie, J., An, S.W. and Huang, C.L. (2017) New insights into the mechanism of action of soluble Klotho. Front. Endocrinol. (Lausanne), 8, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang, Q., Su, W., Shen, Z. and Wang, R. (2018) Correlation between soluble α-Klotho and renal function in patients with chronic kidney disease: a review and meta-analysis. Biomed. Res. Int., 2018, 9481475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zou, D., Wu, W., He, Y., Ma, S. and Gao, J. (2018) The role of Klotho in chronic kidney disease. BMC Nephrol., 19, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhu, Z., Xia, W., Cui, Y., Zeng, F., Li, Y., Yang, Z. and Hequn, C. (2019) Klotho gene polymorphisms are associated with healthy aging and longevity: evidence from a meta-analysis. Mech. Ageing Dev., 178, 33–40. [DOI] [PubMed] [Google Scholar]
- 10. Davey Smith, G. and Ebrahim, S. (2003) ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int. J. Epidemiol., 32, 1–22. [DOI] [PubMed] [Google Scholar]
- 11. Evans, D.M. and Davey Smith, G. (2015) Mendelian randomization: new applications in the coming age of hypothesis-free causality. Annu. Rev. Genomics Hum. Genet., 16, 327–350. [DOI] [PubMed] [Google Scholar]
- 12. Zheng, J., Haberland, V., Baird, D., Walker, V., Haycock, P.C., Hurle, M.R., Gutteridge, A., Erola, P., Liu, Y., Luo, S.et al. (2020) Phenome-wide Mendelian randomization mapping the influence of the plasma proteome on complex diseases. Nat. Genet., 52, 1122–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang, J., Ferreira, T., Morris, A.P., Medland, S.E., Genetic Investigation of ANthropometric Traits (GIANT) Consortium, DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium, Madden, P.A., Heath, A.C., Martin, N.G., Montgomery, G.W.et al. (2012) Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet., 44, S361–S363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Richardson, T.G., Sanderson, E., Palmer, T.M., Ala-Korpela, M., Ference, B.A., Davey Smith, G. and Holmes, M.V. (2020) Evaluating the relationship between circulating lipoprotein lipids and apolipoproteins with risk of coronary heart disease: a multivariable Mendelian randomisation analysis. PLoS Med., 17, e1003062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sato, T., Gotoh, M., Kiyohara, K., Kameyama, A., Kubota, T., Kikuchi, N., Ishizuka, Y., Iwasaki, H., Togayachi, A., Kudo, T.et al. (2003) Molecular cloning and characterization of a novel human beta 1,4-N-acetylgalactosaminyltransferase, beta 4GalNAc-T3, responsible for the synthesis of N,N'-diacetyllactosediamine, galNAc beta 1-4GlcNAc. J. Biol. Chem., 278, 47534–47544. [DOI] [PubMed] [Google Scholar]
- 16. Zheng, J., Maerz, W., Gergei, I., Kleber, M., Drechsler, C., Wanner, C., Brandenburg, V., Reppe, S., Gautvik, K.M., Medina-Gomez, C.et al. (2019) Mendelian randomization analysis reveals a causal influence of circulating sclerostin levels on bone mineral density and fractures. J. Bone Miner. Res., 34, 1824–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Garringer, H.J., Fisher, C., Larsson, T.E., Davis, S.I., Koller, D.L., Cullen, M.J., Draman, M.S., Conlon, N., Jain, A., Fedarko, N.S., Dasgupta, B. and White, K.E. (2006) The role of mutant UDP-N-acetyl-alpha-D-galactosamine-polypeptide N-acetylgalactosaminyltransferase 3 in regulating serum intact fibroblast growth factor 23 and matrix extracellular phosphoglycoprotein in heritable tumoral calcinosis. J. Clin. Endocrinol. Metab., 91, 4037–4042. [DOI] [PubMed] [Google Scholar]
- 18. Fiete, D., Beranek, M. and Baenziger, J.U. (2012) Peptide-specific transfer of N-acetylgalactosamine to O-linked glycans by the glycosyltransferases β1,4-N-acetylgalactosaminyl transferase 3 (β4GalNAc-T3) and β4GalNAc-T4. J. Biol. Chem., 287, 29204–29212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Baenziger, J.U., Kumar, S., Brodbeck, R.M., Smith, P.L. and Beranek, M.C. (1992) Circulatory half-life but not interaction with the lutropin/chorionic gonadotropin receptor is modulated by sulfation of bovine lutropin oligosaccharides. Proc. Natl. Acad. Sci. U. S. A., 89, 334–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fiete, D., Srivastava, V., Hindsgaul, O. and Baenziger, J.U. (1991) A hepatic reticuloendothelial cell receptor specific for SO4-4GalNAc beta 1,4GlcNAc beta 1,2Man alpha that mediates rapid clearance of lutropin. Cell, 67, 1103–1110. [DOI] [PubMed] [Google Scholar]
- 21. Fiete, D., Beranek, M.C. and Baenziger, J.U. (1997) The macrophage/endothelial cell mannose receptor cDNA encodes a protein that binds oligosaccharides terminating with SO4-4-GalNAcbeta1,4GlcNAcbeta or Man at independent sites. Proc. Natl. Acad. Sci. U. S. A., 94, 11256–11261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fiete, D.J., Beranek, M.C. and Baenziger, J.U. (1998) A cysteine-rich domain of the ‘mannose’ receptor mediates GalNAc-4-SO4 binding. Proc. Natl. Acad. Sci. U. S. A., 95, 2089–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Roseman, D.S. and Baenziger, J.U. (2001) The mannose/N-acetylgalactosamine-4-SO4 receptor displays greater specificity for multivalent than monovalent ligands. J. Biol. Chem., 276, 17052–17057. [DOI] [PubMed] [Google Scholar]
- 24. Patenaude, S.I., Seto, N.O., Borisova, S.N., Szpacenko, A., Marcus, S.L., Palcic, M.M. and Evans, S.V. (2002) The structural basis for specificity in human ABO(H) blood group biosynthesis. Nat. Struct. Biol., 9, 685–690. [DOI] [PubMed] [Google Scholar]
- 25. The Severe Covid-19 GWAS Group, Ellinghaus, D., Degenhardt, F., Bujanda, L., Buti, M., Albillos, A., Invernizzi, P., Fernandez, J., Prati, D., Baselli, G.et al. (2020) Genomewide association study of severe Covid-19 with respiratory failure. N. Engl. J. Med., 383, 1522–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nagai, R., Saito, Y., Ohyama, Y., Aizawa, H., Suga, T., Nakamura, T., Kurabayashi, M. and Kuroo, M. (2000) Endothelial dysfunction in the Klotho mouse and downregulation of Klotho gene expression in various animal models of vascular and metabolic diseases. Cell. Mol. Life Sci., 57, 738–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sastre, C., Rubio-Navarro, A., Buendia, I., Gomez-Guerrero, C., Blanco, J., Mas, S., Egido, J., Blanco-Colio, L.M., Ortiz, A. and Moreno, J.A. (2013) Hyperlipidemia-associated renal damage decreases Klotho expression in kidneys from ApoE knockout mice. PLoS One, 8, e83713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhao, Y., Banerjee, S., Dey, N., LeJeune, W.S., Sarkar, P.S., Brobey, R., Rosenblatt, K.P., Tilton, R.G. and Choudhary, S. (2011) Klotho depletion contributes to increased inflammation in kidney of the db/db mouse model of diabetes via RelA (serine)536 phosphorylation. Diabetes, 60, 1907–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thurston, R.D., Larmonier, C.B., Majewski, P.M., Ramalingam, R., Midura-Kiela, M., Laubitz, D., Vandewalle, A., Besselsen, D.G., Mühlbauer, M., Jobin, C., Kiela, P.R. and Ghishan, F.K. (2010) Tumor necrosis factor and interferon-gamma down-regulate Klotho in mice with colitis. Gastroenterology, 138, 1384–13941394.e1381–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Moreno, J.A., Izquierdo, M.C., Sanchez-Nino, M.D., Suarez-Alvarez, B., Lopez-Larrea, C., Jakubowski, A., Blanco, J., Ramirez, R., Selgas, R., Ruiz-Ortega, M.et al. (2011) The inflammatory cytokines TWEAK and TNFalpha reduce renal klotho expression through NFkappaB. J. Am. Soc. Nephrol., 22, 1315–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Martin-Nunez, E., Donate-Correa, J., Ferri, C., Lopez-Castillo, A., Delgado-Molinos, A., Hernandez-Carballo, C., Perez-Delgado, N., Rodriguez-Ramos, S., Cerro-Lopez, P., Tagua, V.G., Mora-Fernandez, C. and Navarro-Gonzalez, J.F. (2020) Association between serum levels of Klotho and inflammatory cytokines in cardiovascular disease: a case-control study. Aging, 12, 1952–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Duerr, R.H., Taylor, K.D., Brant, S.R., Rioux, J.D., Silverberg, M.S., Daly, M.J., Steinhart, A.H., Abraham, C., Regueiro, M., Griffiths, A.et al. (2006) A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science, 314, 1461–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Neyra, J.A., Moe, O.W., Pastor, J., Gianella, F., Sidhu, S.S., Sarnak, M.J., Ix, J.H. and Drew, D.A. (2019) Performance of soluble Klotho assays in clinical samples of kidney disease. Clin. Kidney J., 13, 235–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Winkelmann, B.R., Marz, W., Boehm, B.O., Zotz, R., Hager, J., Hellstern, P., Senges, J. and LURIC Study Group (2001) Rationale and design of the LURIC study--a resource for functional genomics, pharmacogenomics and long-term prognosis of cardiovascular disease. Pharmacogenomics, 2, S1–S73. [DOI] [PubMed] [Google Scholar]
- 35. Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M.A., Bender, D., Maller, J., Sklar, P., deBakker, P.I., Daly, M.J. and Sham, P.C. (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet., 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Andlauer, T.F.M., Buck, D., Antony, G., Bayas, A., Bechmann, L., Berthele, A., Chan, A., Gasperi, C., Gold, R., Graetz, C.et al. (2016) Novel multiple sclerosis susceptibility loci implicated in epigenetic regulation. Sci. Adv., 2, e1501678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Howie, B., Fuchsberger, C., Stephens, M., Marchini, J. and Abecasis, G.R. (2012) Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat. Genet., 44, 955–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Howie, B.N., Donnelly, P. and Marchini, J. (2009) A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet., 5, e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Delaneau, O., Zagury, J.F. and Marchini, J. (2013) Improved whole-chromosome phasing for disease and population genetic studies. Nat. Methods, 10, 5–6. [DOI] [PubMed] [Google Scholar]
- 40. Boyd, A., Golding, J., Macleod, J., Lawlor, D.A., Fraser, A., Henderson, J., Molloy, L., Ness, A., Ring, S. and Davey Smith, G. (2013) Cohort Profile: the ‘children of the 90s’--the index offspring of the Avon Longitudinal Study of Parents and Children. Int. J. Epidemiol., 42, 111–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fraser, A., Macdonald-Wallis, C., Tilling, K., Boyd, A., Golding, J., Davey Smith, G., Henderson, J., Macleod, J., Molloy, L., Ness, A.et al. (2013) Cohort Profile: the Avon Longitudinal Study of Parents and Children: ALSPAC mothers cohort. Int. J. Epidemiol., 42, 97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yamazaki, Y., Imura, A., Urakawa, I., Shimada, T., Murakami, J., Aono, Y., Hasegawa, H., Yamashita, T., Nakatani, K., Saito, Y.et al. (2010) Establishment of sandwich ELISA for soluble alpha-Klotho measurement: age-dependent change of soluble alpha-Klotho levels in healthy subjects. Biochem. Biophys. Res. Commun., 398, 513–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lallemand, Y., Luria, V., Haffner-Krausz, R. and Lonai, P. (1998) Maternally expressed PGK-Cre transgene as a tool for early and uniform activation of the Cre site-specific recombinase. Transgenic Res., 7, 105–112. [DOI] [PubMed] [Google Scholar]
- 44. Barker, S.L., Pastor, J., Carranza, D., Quiñones, H., Griffith, C., Goetz, R., Mohammadi, M., Ye, J., Zhang, J., Hu, M.C.et al. (2014) The demonstration of αKlotho deficiency in human chronic kidney disease with a novel synthetic antibody. Nephrol. Dial. Transplant., 30, 223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Willer, C.J., Li, Y. and Abecasis, G.R. (2010) METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics, 26, 2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bulik-Sullivan, B.K., Loh, P.R., Finucane, H.K., Ripke, S., Yang, J., Patterson, N., Daly, M.J., Price, A.L. and Neale, B.M. (2015) LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet., 47, 291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bulik-Sullivan, B., Finucane, H.K., Anttila, V., Gusev, A., Day, F.R., Loh, P.R., Duncan, L., Perry, J.R., Patterson, N., Robinson, E.B.et al. (2015) An atlas of genetic correlations across human diseases and traits. Nat. Genet., 47, 1236–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zheng, J., Erzurumluoglu, A.M., Elsworth, B.L., Kemp, J.P., Howe, L., Haycock, P.C., Hemani, G., Tansey, K., Laurin, C., Genetics, E.et al. (2016) LD Hub: a centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics, 33, 272–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Davey Smith, G. and Hemani, G. (2014) Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum. Mol. Genet., 23, R89–R98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bowden, J., Davey Smith, G. and Burgess, S. (2015) Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int. J. Epidemiol., 44, 512–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bowden, J., Del Greco, M.F., Minelli, C., Davey Smith, G., Sheehan, N.A. and Thompson, J.R. (2016) Assessing the suitability of summary data for two-sample Mendelian randomization analyses using MR-Egger regression: the role of the I2 statistic. Int. J. Epidemiol., 45, 1961–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hartwig, F.P., Davey Smith, G. and Bowden, J. (2017) Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int. J. Epidemiol., 46, 1985–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zheng, J., Richardson, T.G., Millard, L.A.C., Hemani, G., Elsworth, B.L., Raistrick, C.A., Vilhjalmsson, B., Neale, B.M., Haycock, P.C., Smith, G.D. and Gaunt, T.R. (2018) PhenoSpD: an integrated toolkit for phenotypic correlation estimation and multiple testing correction using GWAS summary statistics. Gigascience, 7, giy090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sanderson, E., Davey Smith, G., Windmeijer, F. and Bowden, J. (2019) An examination of multivariable Mendelian randomization in the single-sample and two-sample summary data settings. Int. J. Epidemiol., 48, 713–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Battle, A., Brown, C.D., Engelhardt, B.E. and Montgomery, S.B. (2017) Genetic effects on gene expression across human tissues. Nature, 550, 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Võsa, U., Claringbould, A., Westra, H.-J., Bonder, M.J., Deelen, P., Zeng, B., Kirsten, H., Saha, A., Kreuzhuber, R., Kasela, S.et al. (2018) Unraveling the polygenic architecture of complex traits using blood eQTL metaanalysis. bioRxiv, 447367. [Google Scholar]
- 57. Boyle, A.P., Hong, E.L., Hariharan, M., Cheng, Y., Schaub, M.A., Kasowski, M., Karczewski, K.J., Park, J., Hitz, B.C., Weng, S., Cherry, J.M. and Snyder, M. (2012) Annotation of functional variation in personal genomes using RegulomeDB. Genome Res., 22, 1790–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Giambartolomei, C., Vukcevic, D., Schadt, E.E., Franke, L., Hingorani, A.D., Wallace, C. and Plagnol, V. (2014) Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet., 10, e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Davis, C.A., Hitz, B.C., Sloan, C.A., Chan, E.T., Davidson, J.M., Gabdank, I., Hilton, J.A., Jain, K., Baymuradov, U.K., Narayanan, A.K.et al. (2018) The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res., 46, D794–D801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kuhn, R.M., Haussler, D. and Kent, W.J. (2013) The UCSC genome browser and associated tools. Brief. Bioinform., 14, 144–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Quinlan, A.R. and Hall, I.M. (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics, 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Guo, M., Liu, Z., Willen, J., Shaw, C.P., Richard, D., Jagoda, E., Doxey, A.C., Hirschhorn, J. and Capellini, T.D. (2017) Epigenetic profiling of growth plate chondrocytes sheds insight into regulatory genetic variation influencing height. Elife, 5, e29329. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.