

Abstract
Epilepsy is a dynamic and heterogeneous neurological disease, and in long-term studies on prognosis, classically 5 basic patterns (early remission, late remission, relapsing-remitting, worsening, and non-remitting) have been identified. The most frequent pattern was relapsing-remitting course, and factors such as the presence of genetic etiology, rare seizures at the beginning of epilepsy and the absence of psychiatric comorbid diseases were found to be related with this pattern as well as reaching 5 years of remission in the follow-ups. Anti-seizure drug resistance (ASD-R) and factors affecting the presence of this resistance (such as symptomatic etiology, abnormal electroencephalographic findings, having multiple seizure types together, status epilepticus and febrile seizure history) decrease the chance of remission, while idiopathic/genetic etiology, generalized epilepsy, and absence of comorbid diseases seem to be associated with achieving long-term remission. Apart from these basic course patterns, there are some patients with an “atypical prognosis” such as drug-resistant juvenile myoclonic epilepsy (JME), benign hippocampal sclerosis-related mesial temporal lobe epilepsy (HS-MTLE), and severe childhood epilepsy with centro-temporal spikes (CESTS), in which the pathophysiological mechanisms underlying these patterns have not been clarified despite the suggestions of various hypotheses. The presence of comorbid diseases such as hormonal factors (as in catamenial epilepsy), autoimmune processes, thyroid disorders and metabolic and psychiatric diseases may also cause an atypical prognostic pattern by affecting the course of the disease. In this review, our aim is to provide the clinician with an up-to-date and questioning perspective on the prognostic markers of epilepsy, by examining in detail some specific epilepsy syndromes that may show atypical prognosis as well as the general prognostic features of epilepsy.
Keywords: Epilepsy, prognosis, atypical prognosis, remission, drug resistance, relapse
INTRODUCTION
Epilepsy is a chronic neurological disease affecting approximately 70 million individuals around the world; its reported average annual incidence is 50 (range: 40 to 70) in 100000 and its reported annual prevalence is 7.60 (range: 4 to 12) in 1000 individuals, with certain variations between countries and a strikingly higher prevalence in lower-income countries (1, 2). Epilepsy represents a serious challenge in healthcare owing to the burden associated with the disease at both individual and social levels and in economic terms (3). Today, the mainstay of treatment for epilepsy aims to reach seizure freedom (remission) with as little adverse effect as possible and to this end, relies on the use of anti-seizure drugs (ASD) that control seizures for improving quality of life. In developed societies, the long-term remission rate among recently diagnosed epileptic individuals is reported to be quite high, i.e. approximately 2/3, and this rate is attributed to the easier access to treatment for epilepsy provided in such countries when compared to that in lower-income societies (4). However, epileptic individuals are also known to go into remission after a while when left untreated and this fact reinforces the opinion that prognosis is somewhat independent of medication (5,6). Currently, there are certain questions left unanswered and in need of clarification with respect to which patient will go into remission and when and whether such remission will be followed by relapse, as well as the rate of re-remission following relapse and the rate of seizure freedom in long-term follow-up. Long-term studies seeking answers to these questions are required in order for every case of epilepsy to be addressed with the most compatible course of treatment for the individual concerned; for the patient to be provided with adequate and accurate prognostic information on the course of their disease; and for every epileptic individual to be able to plan their lives and, most importantly, to improve their quality of life in the long term.
Highlights
Epilepsy has 5 basic prognostic patterns, relapsing-remitting is the most common.
Spontaneous remission rates are high even among untreated patients with epilepsy.
Atypical courses may also exist within some well-known epilepsy syndromes.
Several comorbid diseases might lead to an atypical prognosis in epilepsy.
In the present review, we aim to offer clinicians an updated and inquisitive outlook to the prognostic characteristics of epilepsy, through a detailed examination of general characteristics of disease courses in epilepsy, as well as of certain specific epilepsy syndromes that can present with atypical prognostic patterns.
DISEASE COURSE IN EPILEPSY
Overview
The conventional approach to epilepsy considers the natural course of epilepsies treated with ASDs as “excellent” in 20-40%; as “good” in 30-40%; as “uncertain” in 10-20%; and as “poor” in 20% of cases. This classification relies upon the type and underlying etiology of the epilepsy syndrome affecting an individual and a case may be reassigned to a new group only after the patient has been started on a new ASD or has been offered a new treatment option such as surgical intervention (7). A more detailed classification of disease courses was established by Sillanpää and Schmidt, where 144 patients with childhood-onset epilepsy were followed up for 37 years, and the authors specified five basic disease courses (6) (Figure 1):
Figure 1.

Five basic disease courses defined for epilepsy
1) Early remission: Patients achieving remission through complete seizure control either immediately after or within the first year of diagnosis/medication
2) Late remission: Patients achieving and maintaining remission through seizure control after the first year of diagnosis/medication
3) “Remitting-relapsing” course: Patients initially achieving early or late remission and experiencing one or more episodes of relapse and remission during follow-up
4) Worsening course: Patients initially achieving early or late remission, but developing one or more relapses and failing to achieve terminal remission during follow-up
5) No remission: Patients that continue to experience seizures without any remission during follow-up (6)
The study reported that 45 patients (31%) achieved early remission and 23 (16%) of them progressed to terminal remission without experiencing any relapse, and late remission (with a delay of 9 years) was achieved by a further 72 patients (50%), 46 (32%) of which achieved terminal remission without any relapse. Moreover, in 48 patients (33%), remission was interrupted by one or more relapses, indicating a remitting-relapsing course of epilepsy and 28 (19%) of these patients achieved terminal remission. Twenty patients (14%) (8 patients from the early remission group and 12 patients from the late remission group) in total never entered terminal remission. In the total cohort 144 patients entered at least one remission period during the follow-up and the remaining 27 patients (19%) could never enter remission and were considered as the drug-resistant group. Consequently, the authors suggested that early remission was not a conclusive predictor of terminal remission, nor would an initial failure to enter early remission predict poor prognosis in the long term. Other noteworthy conclusions of the study include an emphasis on the importance of etiology in the prognosis of epilepsy syndromes and on the possibly more difficult challenge of achieving terminal remission in symptomatic and localisation-related epilepsies.
Another recent retrospective and multicentre study examining the results of 1006 children and adults with an average follow-up period of 16 years, offered the striking observation that 923 patients (91.7%) achieved 1-year remission and 2-year, 5-year and 10-year remissions were found in 89.5%, 77.1% and 44.4% of cases, respectively, whereas the most common disease course was remitting-relapsing (52.2%), followed by early remission (24.5%). Approximately 8.3% of the cases, however, never achieved remission. Having one or two seizures at diagnosis, presence of a generalised epilepsy diagnosis, no psychiatric comorbidity, and treatment with one or two ASDs were associated with a 5-years of remission in this study. In addition, the predictors of a remitting-relapsing course were reported as <6 seizures at diagnosis, a possible genetic etiology, and having no psychiatric comorbidity. Another natural implication here was that the shorter the time after remission is, the higher the likelihood of relapse (8).
Another retrospective study comparing the rates of early and late remission in 352 adult patients with an average follow-up period of 75 months reported that 272 patients (62.5%) entered one or more periods of 2-year remission, whereas 115 patients (32.7%) achieved early remission (immediately after the start of ASD treatment and with full seizure control for 2 years) and 38 patients (10.8%) achieved late remission (seizure control reached after 24 months of the beginning of ASD treatment or later). A total of 101 (87.8%) and 34 (89.5%) out of the patients achieving early and late remission, respectively, were still in remission at the time of the final visit. This study underlined that patients presenting with focal seizures might exhibit late remission, indicating that this finding could be a part of a “remitting-relapsing” pattern and the frequency of pre-treatment focal seizures could be a predictor of either early or late remission (9).
Strikingly, although ASD treatment does have a certain positive effect on short-term prognosis, there is no significant difference reported between the treated and untreated groups in terms of long-term prognosis (10). However, various studies established that post-remission termination of ASD treatment might lead to relapse in one-third of the cases and furthermore, even with the restart of previously effective medication, it might be impossible at times to achieve re-remission (10,11). Especially the latter case represents a challenge even for experienced clinicians and affects the lives of patients in remission in extremely negative and unexpected ways. This adverse clinical problem, which is not easily controllable, has not been addressed with a significant hypothesis, let alone a definite explanation.
The dynamic and fluctuating, rather than stable, nature of treatment responsiveness in epilepsy naturally leads to various challenges in the definition of drug resistance as a concept. According to Kwan and Brodie’s conventional, but widely acknowledged definition, drug resistance is defined as the failure of two syndrome specific, appropriately chosen and well tolerated ASDs used at maximum tolerable dosage, either as monotherapy or in combination, to achieve sustained seizure freedom (12). Even though this definition is useful for clinicians as a description and for guidance, it is well-known that drug responsiveness in epilepsy varies in the long-term follow-up course and a patient identified as “drug-resistant” at one point may translate to the “drug-responsive” group later in the process. In the literature, approximately 1/3 of epileptic individuals were found to be drug-resistant (5, 13); however, approximately 1/4 of drug-resistant patients may enter periods of remission of 1 year or longer during their follow-up (14,15). Prospective studies indicated that approximately 4% of adult patients per year could achieve remission even if they were accepted as “drug-resistant” before and this finding was attributed to the dynamic nature of epilepsy; nevertheless, the underlying mechanisms are yet to be clarified (16, 17).
There are various factors identified as predictors of drug resistance. Among these factors, abnormal EEG findings (both slow wave activity and epileptiform abnormalities); etiology of epilepsy (increased risk for structural causes); history of status epilepticus; multiple seizure types; and febrile seizures are underlined as strong and proven risk factors for drug resistance, whereas disputed factors currently lacking consensus include initial treatment failure, neurodevelopmental delay, high initial seizure frequency, and a concomitant neurological deficit (18,19).
The rate of drug resistance was shown to be higher in symptomatic focal and generalised epilepsies than in the idiopathic group and in parallel, the rate of early or late remission was reported to be higher in idiopathic epilepsies (6). Certain studies undertaken with epileptic children reported that presence of mental retardation and symptomatic etiology might be associated with progressive development of drug resistance (20) and therefore, 4% of children initially responsive to drug treatment might develop drug resistance even after 10 years. This finding is striking and it demonstrates a dynamic and complex process of the concept of drug resistance.
Even if some of the patients initially considered to be drug-resistant might achieve remission of 1 year or longer, relevant studies report very high rates of relapse among such patients. Berg et al. conducted an observational study with 128 drug-resistant children and reported that 68% of the patients relapsed after 1-year of remission (17). In another prospective study, 1-year of remission was reported in 59 (24%) of 246 drug-resistant adult patients and 58% of these patients experienced a relapse later. In this study, such factors as developmental delay, generalised epilepsy with symptomatic etiology, duration of resistance, and the number of tried and failed ASDs were reported to decrease the likelihood of 1-year of remission significantly and more specifically, it was concluded that the number of tried and failed ASDs might be a negative predictor of remission in drug-resistant patients (15). Another prospective study added a third ASD to the treatment schedules of 403 adult patients, who were previously treated with two ASDs and failed, in order to analyse their seizure prognosis. This study showed that 212 (53%) of these patients never achieved remission, 16% followed a complex and fluctuating course, and 31% achieved early or late seizure freedom at the end of the study (21). See Figure 2 for factors reported to affect prognosis in epilepsy positively or negatively.
Figure 2.

Factors reported to affect prognosis in epilepsy positively or negatively
Abrreviations: ASD, anti-seizure drug; benign childhood epilepsy with centrotemporal spikes (BCECTS); CAE, childhood absence epilepsy; EE, epileptic encephalopathy; EEG, electroencephalography; GTCS, generalized tonic-clonic seizures; MTLE, mesial temporal lobe epilepsy
How Does Epilepsy Progress Naturally in Untreated Patients?
Studies examining the natural course of epilepsy in untreated patients mostly rely on limited data obtained from studies addressing either patient groups from low-income countries providing limited access to treatment or groups of patients exhibiting drug rejection on various grounds (22, 23).
It is known that around 80% of epileptic patients around the world live in middle- and low-income countries and accordingly, approximately 50-75% of patients are without access to treatment for epilepsy (24). A face-to-face survey study implemented in Ecuador interviewed 72,121 individuals and identified epilepsy in 1026 respondents. 56% of these patients had active epilepsy; %37 had never used any ASDs in their lives; and only 12% were under regular treatment at the time of the study. 44% of these patients were in remission and interestingly, 2/3 of the patients with inactive epilepsy had never used any ASD treatment during their lives (25). On the other hand, a more recent study conducted in Bolivia reported spontaneous remission independent of ASD treatment in 30% of patients diagnosed with chronic epilepsy (26). Consequently, the emerging data indicate that a considerable rate of spontaneous remission is observed even in untreated cases of epilepsy and ASD treatment does not represent a determining factor for remission by itself.
Predictors of Good Prognosis in Epilepsy and General Characteristics of Epilepsy Syndromes with Good Prognosis
There are a large number of studies undertaken with the aim of identifying the predictors of good prognosis in epilepsy; however, methodological variations among such studies have precluded the achievement of a full consensus on this matter.
Certain epilepsy syndromes, especially idiopathic generalised epilepsies (IGEs), have been long known to have a better prognosis comparing to the other syndromes (27). In general, the rates of remission in IGEs are reported between 64-82% and despite variations among studies, factors such as; history of febrile seizures, seizure onset before the age of 5, atypical absence seizures, myoclonic epilepsy, onset of GTC seizures before the age of 3 or after the age of 20, and asymmetrical discharges in EEG were found to be associated with negative prognosis (27, 28).
The rates of 1-year terminal remission in childhood absence epilepsy (CAE) are reported between 51-93%, with the range being this wide owing to the methodological differences and variations in diagnostic criteria among existing studies (29, 30). In the prospective study of Callenbach et al., with a follow-up duration of 15 years in children recently diagnosed with CAE, the rate of terminal remission of at least 1 year was 93%. Subdividing these children into three groups depending on their time of remission, namely those becoming seizure-free within (I) 1 month after enrolment; (II) 1-6 months after enrolment; and (III) more than 6 months after enrolment or having continuing seizures during follow-up, revealed that the rate of relapse was significantly higher in group III than in group I. The study did not establish any association between other epilepsy-specific basic clinical and electrophysiological characteristics and remission (29). Although some studies point out to a possible association between the development of myoclonic and GTC seizures and poor prognosis in children with CAE (30, 31), it is unclear whether this finding is of relevance to a possible evolution to juvenile myoclonic epilepsy (JME) (27). Late onset of seizures (>8 years), development of absence status, atypical EEG responses, photosensitivity, and family history of generalised seizures are other factors reported in various studies to be associated with a negative prognosis in CAE (30, 32).
When it comes to juvenile absence epilepsy (JAE), another epilepsy syndrome progressing with absence seizures, remission rates are reported between 21-89% (27, 33) and in their study on patients diagnosed with CAE and JAE, Trinka et al., found that the development of myoclonic and GTC seizures are associated with lower rates of remission (31). On the other hand, another recent retrospective study observed that 2-year remission rates in 36 patients diagnosed with JAE and 145 with JME were quite low, i.e. 39% and 22%, respectively. For these patients, a comparison was drawn between one group of patients who had stopped and one group of patients who had continued with their ASD treatment after remission of at least 2 years, and it was observed that, in the JAE, group 6 (43%) out of 14 patients who had stopped their ASD treatment without any subsequent seizure experienced a relapse at a later time, while such relapse was observed only in 2 (25%) out of 8 patients who had been seizure free, but were still on medication. The authors tried to draw attention to the association between the post-remission discontinuation of ASD treatment and negative prognosis in patients with JAE (33).
JME is an epileptic syndrome which older reviews recommended a lifelong ASD treatment for and where medication discontinuation is associated with high rates of relapse (34). However, under appropriate ASD treatment 75-90% of patients can achieve long-term remission. The highly heterogeneous nature of JME in semiologic terms leads to variations in the natural course of the disease and therefore, rates of remission may vary between the subtypes of JME (35). Martinez-Juarez et al. offered a classification in 2006 where they described 4 subtypes for JME (classic JME, CAE evolving to JME, JME with adolescent pyknoleptic absence, and JME with astatic seizures) and reported the best prognosis in the classic subtype and the poorest prognosis in CAE evolving to JME (34). Another retrospective long-term follow-up study found the prominent factors associated with a negative course in JME as; bilateral myoclonic seizures accompanied by GTC seizures; long-term but ineffective treatment; and polytherapy and established an additional association between photoparoxysmal response and increased likelihood of relapse after ASD discontinuation. The same study also reported that the disappearance of GTC seizures under ASD treatment was associated with long-term seizure freedom (36). In the Senf et al.’s study reporting the results of a follow-up of 45 years of 66 patients with JME including the patients of Dieter Janz who defined the JME syndrome, 5-year terminal remission was reported in 59.1% of the patients (39 patients); 28(71.8%) of who were still under ASD treatment and 11(28.2%) were followed-up without any medication for the last 5 years. The authors underlined the high rate of absence seizures in their patients without remission and concluded that concomitant absence seizures at the onset of JME were associated with negative prognosis in the long term (37). In recent years, various studies have also reported the concurrent development of three seizure types (myoclonic, absence, and GTC seizures altogether) as another factor for negative prognosis and a predictor of resistance (38-40), whereas absence seizures alone or the concurrent development of absence and myoclonic seizures had no relevance in terms of poor prognosis. In addition to these factors, one meta-analysis reported an association between early seizure onset, psychiatric comorbidities, and praxis-induced seizures (seizures triggered with complex cognitive activity and epileptiform discharges on EEG) with resistance in JME (41). The rate of relapse following medication discontinuation in JME is reported to be around 78% with this rate varying significantly between individual studies and it may be concluded that approximately 1/5 of patients remain seizure-free following medication discontinuation which is significantly lower when compared to those in other subtypes of epilepsy (42). The prognostic determinants of JME was given in detail in our other paper (35).
Childhood epilepsy with centro-temporal spikes, another epilepsy syndrome featuring better prognosis, is a focal epilepsy syndrome of childhood with excellent prognosis. Remission often occurs within 2-4 years following onset and seizures resolve before the age of 16 (43). Only 2% of patients were reported to experience absence seizures and GTC seizures later in the adulthood (44). Rarely (<1%) evolution to syndromes entailing poorer prognosis might be seen such as , evolution to Landau-Kleffner syndrome, atypical focal epilepsy syndrome of childhood or epilepsy with continuous spikes and waves during sleep (ESES) (45). There is no clear hypothesis that explain the exact factors of relevance for such course and progressive follow-up studies are needed in this respect.
These syndromes are considered to entail good prognosis and lead physicians and patients to higher expectations of remission with or without medication; therefore, it is especially important that any indication of poorer prognosis should be examined more carefully; possible genetic markers be identified; and/or possible mistakes in treatment planning should be recognised/resolved for patients diagnosed with these syndromes. In recent years, it has been demonstrated that the prognosis of IGE is poorer among women than men due to the decreased use of valproic acid (VPA) which has a risk for teratogenicity in women of childbearing potential and this finding has underlined the importance of ASD selection in this context and revealed the necessity of clarifying the underlying mechanisms in cases dependent on VPA (46). Furthermore, clarifications of the mechanisms behind additional factors including thyroid disease and psychiatric comorbidities, as observed in some resistant cases of JME, will allow for an optimised treatment approach for these patients (38).
EVALUATION OF HIDDEN UNDERLYING MECHANISMS OF PROGNOSTIC PATTERNS IN THE LIGHT OF OPPOSITE EXAMPLES OF ATYPICAL PROGNOSTIC PROCESSES
There are expected disease patterns for most of the epilepsy syndromes, in the light of long-term follow-up studies however, it is known that some prognostic variability (atypical or unusual course) may exist, at times, in a single epilepsy syndrome. Forms of atypical course may be seen in such common epilepsy syndromes as JME, mesial temporal lobe epilepsy with hippocampal sclerosis (HS-MTLE), childhood epilepsy with centro-temporal spikes (CECTS), and IGE, and this fact represents a remarkable challenge for treatment planning in daily clinical practice. Such cases should bring to mind the possibility of distinct endophenotypes in a given epilepsy syndrome as underlined above and a detailed genetic study planned in this context will offer guidance through the demonstration of several genetic variations that may be associated with different treatment responses and prognoses (34, 35).
CECTS, as an example of syndromes with an atypical course, is an epilepsy syndrome expected to follow a very good course as mentioned above; however, disease forms with an atypical course were also defined in a small group of patients. There are certain findings that point out to CECTS with an atypical course including early age of onset, atypical seizure characteristics, only daytime seizures, post-ictal Todd’s paralysis, prolonged seizures or status epilepticus, atypical EEG findings (irregular baseline activity, spikes in atypical locations and morphologies, absence-like spike-wave discharges, etc.), and neurocognitive disorders. Findings that may help clinicians predict an atypical course at early stages of the disease include early age of onset and evolution to more severe types of seizures (atypical absence, negative myoclonus, and diurnal seizures, etc.) (47). Despite the availability of neuroimaging studies that corroborate the presence of cognitive and structural brain abnormalities starting with disease onset and progressed thereafter again in CECTS, the functional implications of such abnormalities are still not fully known and are considered to be potentially linked to cognitive and behavioural outcomes in cases presenting with an atypical course (48). Cases of the atonic variant of CECTS, a rare subtype of CECTS with an atypical course, exhibit an earlier age of seizure onset and more frequent and prolonged focal seizures than classic cases; in addition, another striking finding in these cases relates to how atonic seizures in patients may worsen with carbamazepine (CBZ) and benzodiazepines (49). There are also studies indicating that CBZ may cause language problems and speech disorders in certain typical cases (50). Prats et al. studied 6 children with CECTS evolving to an atypical form with ESES and neuropsychological abnormalities and found that the condition of 4 out of 6 patients worsened under treatment with CBZ and later improved upon the discontinuation of this medication (51). All of these studies lead us to consider that ASDs used in the treatment of epilepsy may trigger the evolution of the disease to a resistant form or lead to distinct disease courses by giving rise to certain modulations on ion channels and neuronal networks in particular syndromes or in the presence of underlying genetic variants.
HS-MTLE is known in general as a subtype of epilepsy that is resistant to ASDs; however, the literature has also defined its “benign” subtypes. Various relevant studies report a wide range, i.e. 5-42%, for the benign subgroup in all HS-MTLE cases (52, 53). In Varoğlu et al’s. retrospective study on 287 patients with HS-MTLE, the rate of patients that achieved seizure freedom (good prognosis) with medical treatment at the end of 1 year was 9.1%. The authors indicated that early age of onset, comorbid mental retardation, female sex, and history of head trauma at <5 years might be associated with poor prognosis (54). There are also other studies that report later age of onset and good response to ASDs associated with the subgroup of patients with the benign form of the disease and although the etiology has not been clarified, the underlying genetic factors are blamed (55). Even though the presence of HS in magnetic resonance imaging (MRI) is generally associated with resistance and poor prognosis in MTLE, the presence of HS identified with MRI in certain cases of familial MTLE of a mild course supported how this might not necessarily be the case (56, 57). Studies addressing familial MTLE show that endophenotypes may vary in a family (such as seizure frequency and semiology, history of febrile seizures, drug resistance, HS on MRI, and comorbid pathologies including head trauma) and such variation supports the conception of rather a complex genotype-phenotype association (58,59). A recent study reporting 5 patients with benign MTLE of a mild course, underlined that whereas these patients experience fewer seizures and responded better to ASDs, certain seizures (such as nocturnal seizures) and auras might not be clinically recognised and might be confounded with psychiatric and/or gastrointestinal diseases and therefore, these individuals might be faced with such potential dangers as under-treatment and sudden unexpected death in epilepsy (SUDEP) (55). Consequently, it is of great prognostic importance for individuals with HS-MTLE, to be followed up by clinicians closely provided with appropriately regimented treatment, and be cautioned about the risk factors in daily life, even though they belong to a subgroup defined with a “benign” form.
IGEs or genetic generalised epilepsies (GGEs) are generally considered to show good prognosis; however, it is known that drug resistance may be observed in approximately 7-12% of the cases (60). Gesche et al. implemented an incidence and prevalence study with a cohort of GGE patients on the Island of Funen in Denmark and found quite a low rate of drug-resistant GGE, i.e. 4.5%, and reported that the presence of absence seizures was closely associated with the development of drug resistance (61). In another study undertaken at a tertiary centre with a follow-up of 27 years, 199 patients with GGE were evaluated and the most common disease course was reported as remitting-relapsing pattern (40.2%) and persistent drug resistance was found in 14.6% of the patients. In the same study, history of febrile seizures, presence of specific EEG patterns (such as generalised paroxysmal fast activity), VPA resistance, and JME as the syndromic diagnosis were proposed as predictors of drug resistance. Accordingly, heterogeneous clinical manifestations and varied drug responsiveness observed in GGEs may be assumed to represent different pieces of a wide spectrum and to appear as a result of polygenic effects (62). Additionally, there are studies that support the presence of generalised paroxysmal fast activity as a negative prognostic finding on EEG especially in cases of GGE presenting with absence seizures (63, 64). In our relevant study, it was hypothesised that GGEs represented a disease of “network diffusion” and generalised paroxysmal activity might be the result of functional changes affecting the reticulothalamocortical network during the epileptic process especially in certain genetically predisposed patients presenting with typical absence seizures (65). VPA responsiveness/dependency suggested as another prognostic predictor, is suggested to be potentially linked to the regulating, as well as the anti-seizure effects of this drug on the diffuse neuronal networks involved in GGEs (65). As a result, despite high rates of remission and positive prognosis in GGEs, both certain endophenotypic differences within the syndrome (such as absence seizures accompanying JME as a third seizure type or generalised paroxysmal fast activity on EEG in absence epilepsy) in this group and certain implications of currently unclear genetic factors on the phenotype point out to the existence of a “resistant” subgroup.
What are the Possible Underlying Mechanisms in Processes of an Atypical Course?
As mentioned and prominent examples were given above, “unexpected disease courses” may be observed in epileptic patients. Developing drug resistance plays an important role in processes that may be culminate in a poor disease course. Currently, relevant authorities are not yet explaining drug resistance mechanisms with a full consensus and there are varying opinions on the matter (66).
A “polygenic complex inheritance pattern”, which may be defined as different genes defining the phenotype through interaction, is among the factors that indicate how certain genetic variants may be responsible for drug-resistant subgroups exhibiting an atypical course in GGEs (67). Mutations in subunits of the ion channels were again pointed out as genetic disorders that potentially lead to drug resistance in these patients and additionally, gene pathologies outside the channel were reported in various studies to have a potential role in processes of an atypical course (66).
Another mechanism with an alleged role here is defined as the “epigenetic hypothesis”. Epigenome refers to the molecules that regulate gene expression along a genome and its extremely dynamic and variable structure makes it difficult to study the contribution of the epigenome to drug resistance in human epilepsy. Histone modification and long and short non-coding RNAs and microRNAs are among the mechanisms with contributions to the epigenome and their roles in drug resistance are still unclear (67).
Besides the genetic mechanisms suggested in relevance to drug resistance in epilepsies summarised briefly above; there are also other proposed mechanisms suggested to be of significance such as factors related to the drug treatment itself (changes in the area of the brain targeted by a drug, changes arising from variations in the carriers operating at the entry of the drug to the brain, and changes in the peripheral pharmacokinetics of the drug, etc.), the neural network hypothesis (structural changes in the brain associated with epilepsy), and disruption of the blood-brain barrier under the neuroinflammation hypothesis (68).
The neural network hypothesis argues that, changes observed in synaptic networks as a result of synaptic reorganisation arising from neuronal degeneration, necrosis, gliosis, and axonal branching disrupt seizure supressing mechanisms in the brain and complicate the transmission of ASDs to their target areas. It is possible, then, that the seizures that cannot be controlled well create permanent changes in brain plasticity, thereby paving the way for the formation of abnormal neuronal networks and for the development of drug-resistant epilepsy (69).
According to the neuroinflammation hypothesis, inflammation may contribute to the development of drug resistance by disrupting the permeability of the blood-brain barrier (BBB) and increasing the expression of P-glycoprotein (Pgp). Patients with chronic epilepsy are known to exhibit increased BBB permeability at the epileptic focus. The BBB, once thus disrupted, may allow for the access of exogenous inflammatory mediators such as leucocytes and serum proteins (including interleukins (IL), interferons (IFN), tumour necrosis factors (TNF), and growth factors (GF)) to brain tissue, thereby lowering the seizure threshold and contributing to epileptogenesis (70). Brain tissue samples collected from patients diagnosed with reportedly drug-resistant epilepsy such as temporal lobe epilepsy (TLE) or epilepsy associated with cortical dysplasia have been demonstrated to contain inflammatory mediators and the development of resistant focal seizures may be a product of chronic inflammation (71). The identification of neuronal autoantibodies in serum among certain cases of status epilepticus of unknown cause, is among the data that point out to an association between resistance and autoimmunity (72).
What are the Effects of Comorbid Diseases on the Course of Epilepsy?
Hormonal changes and seizure frequency are prominent among the factors that potentially affect prognosis and lead to a fluctuating disease course. A complex and bidirectional association is mentioned to exist between sex steroid hormones and epilepsy. Sex hormones are known to have an effect on cerebral excitability and changes in hormone levels might lead to changes in seizure frequency. The subtype of epilepsy that is characterised by changes in seizure frequency in correlation with menstruation, known as catamenial epilepsy, represents a typical, albeit short-cycle, example for this phenomenon. In individuals with catamenial epilepsy, changes in seizure frequency during menstruation are potentially attributed to the sudden withdrawal of neurosteroids (steroid hormones affecting neuronal tissue) (73). Among these hormones, oestrogen is a proconvulsant by nature and the anticonvulsant effect of progesterone has been confirmed in a large number of studies (74). Specifically, the withdrawal of progesterone is considered to act as a seizure trigger in individuals with catamenial epilepsy (75). Another factor implicated here relates to the changes in the genes coding the enzymes that are instrumental in the metabolization of sex hormones (76). There are three distinct patterns defined for increases in seizure frequency in catamenial epilepsy: Perimenstrual (C1: between day -3 and day +3), periovulatory (C2: increase during the ovulatory phase in a normal cycle; between day +10 and day -13), and during inadequate luteal phase (C3: between day +10 of the initial cycle and day +3 of the next cycle in women with inadequate luteal phase). The most common one is the perimenstrual form (77). A recent multicentre observational study evaluated 121 drug-resistant and 468 drug-responsive patients and reported a close association between drug resistance and catamenial epilepsy, as well as seizure type and history of psychiatric disorders, indicating that catamenial epilepsy gave rise to a 4-fold increase in drug resistance (78). As it is known that most individuals with catamenial epilepsy have already used their first or second choice of ASD and still and suffered from an increase in their seizure frequency despite their ASD treatment, catamenial epilepsy is regarded as a drug-resistant subtype of epilepsy (79). Another reason reinforcing the association between catamenial epilepsy in individuals with GGE and drug resistance may be related with the decreased use of VPA in these patients due to the recent restrictions on its use in this population (78).
Thyroid hormones, another group of steroid hormones, play an important role in the maintenance of brain development and functions in both the intrauterine period and during childhood and adulthood (80). Thyroid hormone disorders present with mitochondrial dysfunction, increased reactive oxygen radicals, disruptions in the formation and regulation of GABAergic interneurons, and changes in the oxidative capacity of the brain due to the modulation in antioxidant enzymes. Considering the epilepsy onset mechanisms of mitochondrial dysfunction, oxidative stress, and excitatory glutamatergic/inhibitor GABAergic neurotransmission disorders along with the possible effects of thyroid hormones on these mechanisms, it is possible to establish an association between thyroid disorders and the pathogenesis of epilepsy (81). A bidirectional association is thought to exist between thyroid hormones and seizure onset mechanisms, similar to the one with reproductive hormones, and in addition to the effects of thyroid hormones on seizure onset, epileptic seizures themselves are considered to have an impact on the hypothalamus-hypophysis-thyroid axis modulated by the GABAergic system (82). Certain studies on children established that increased levels of thyroid-stimulating hormone (TSH) lead to prolonged seizure durations (83). Similarly, one study reported on an adult case where thyroid encephalopathy presented with focal status epilepticus (84). It is also known that the ASDs used in the treatment of epilepsy lead to changes in thyroid hormone levels in 1/3 of patients (increased TSH levels, development of subclinical hypothyroidism, and decreased T3, T4 levels, etc.), thereby affecting seizures and such effects resolve after the discontinuation of the ASD (85). Therefore, subclinical fluctuations in the levels of thyroid hormones or undiagnosed pathologies of relevance may be considered to cause fluctuations in the course of seizures.
It has been long known that autoimmune processes also affect prognosis in epilepsy. It is currently believed that in the course of common systemic autoimmune comorbidities such as Hashimoto’s thyroiditis, systemic lupus erythematosus and diabetes mellitus, resistant seizures may be added to the course and create a negative effect on prognosis. Hereditary immune dysfunction and related numerous hereditary immune-mediated inflammatory mediators, have been demonstrated to lower the threshold for neuronal excitability and cause seizures (86). In certain specific epilepsy syndromes (Rasmussen’s encephalitis and paraneoplastic encephalitis with antibodies targeting intracellular antigens, etc.), the role of adaptive immunity besides this immune dysfunction was proven through the demonstration of immune cells affected by encephalitis in pathological brain samples (87). Antibody-associated encephalitis causes acute symptomatic seizures, whereas antineuronal antibodies at up to 22.5% were detected in chronic epilepsies (88, 89). The presence of these antineuronal antibodies in serum was demonstrated in diseases such as systemic lupus erythematosus (SLE) and type-1 diabetes (DM-1); however, the exact mechanisms allowing these antibodies to induce epilepsy are currently unknown (90, 91). Especially in type-1 DM, 80% of patients have anti-glutamic acid decarboxylase antibodies (anti-GAD ab) and it is emphasised that these antibodies may be observed up to 6% in epileptic patients which might be regarded as a common risk factor (92).
The most typical example indicating how metabolic disorders may cause epilepsy through neuronal damage is DM. The processes occurring during diabetes including hyperglycaemia, recurrent hypoglycaemic attacks, and diabetic ketoacidosis may, in time, give rise to the development of epilepsy through their morphological and functional damage to the brain (93). The risk of epilepsy is 2 to 6 times higher in patients with type-1 DM than the general population and this finding is thought to be the result of metabolic changes such as hypo- or hyperglycaemia and secondary cerebrovascular changes, as well as genetic and autoimmune mechanisms (94). It was argued that fluctuations in the metabolic state might lead to frequent seizures by lowering the threshold for focal seizures in patients with type-1 DM, whereas autoimmune mechanisms are associated more closely with generalised seizures (95). Furthermore, as is the case in GLUT1 deficiency syndrome, most congenital diseases of metabolic-genetic origin progress with resistant epilepsy typically and with possible fluctuations may follow a relatively good prognosis, therefore it is possible to speculate that latent forms of these presentations might have currently unknown effects on disease course and prognosis in epilepsy (96, 97).
Psychiatric comorbidities are observed in approximately 20-30% of epileptic patients with rather higher rates in focal epilepsies (98). In patients with psychiatric disorders diagnosed before the onset of epilepsy, it was demonstrated that drug-resistant epilepsy might often develop in the following period and it was further proven that concomitant psychiatric conditions might disrupt the natural course of epilepsy and the responsiveness to ASDs (99). In a study on 780 patients with newly diagnosed epilepsy, development of drug resistant epilepsy was 2 folds higher in the patients with psychiatric disease history and comorbid psychiatric diseases than the other group (99). Comorbid psychiatric conditions were also identified to be associated with ASD resistance in patients diagnosed with JME (38). Long-term follow-up studies have demonstrated how anxiety disorder, stress, and depression are associated with increased seizure frequency and depression comes out as the culprit in these studies as the comorbidity that leads to the highest increase in seizure frequency. The effects of depression on seizure frequency are considered to arise from the resulting increase in cortical hyperexcitability through multiple mechanisms (by increasing the activity of the hypothalamic-hypophysis-adrenal axis and disrupting the integrity of cortical and subcortical structures, and through neurotransmitter abnormalities and immunological abnormalities) (100). However, there is currently no definitive consensus on this question and prospective and controlled follow-up studies are needed. Consequently, it is possible to state that an additional psychiatric condition appearing before the onset of epilepsy or during the disease course may result in a course of poor prognosis by causing fluctuations in seizure frequency and resistance in follow-up (Figure 3).
Figure 3.
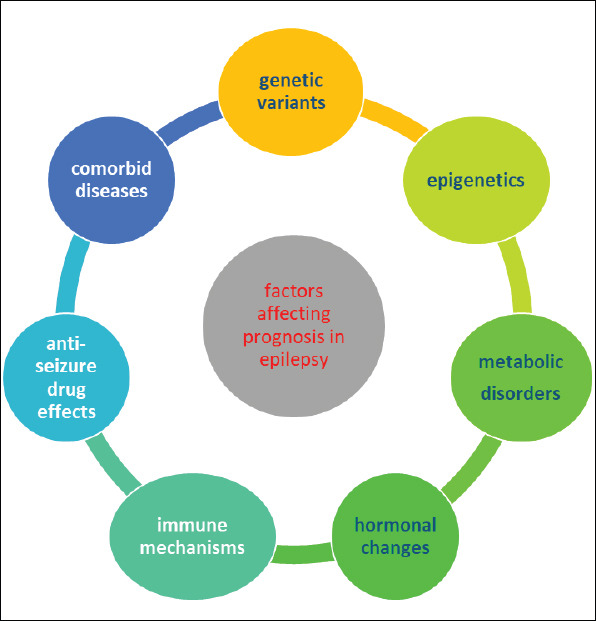
Factors affecting prognosis in epilepsy.
CONCLUSION
Epilepsy is a disease that directly affects the daily lives of individuals and brings along remarkable psychological, sociological, and economic burdens and it is of great importance for clinicians facing newly diagnosed epileptic individuals to enlighten their patients on disease prognosis and course. Physicians dealing with epilepsy may gather detailed information on general disease courses and possible atypical disease courses associated with common epilepsy syndromes in an effort to provide guidance in the planning of patient treatment and possible alternatives and in the education of their patients. Investigations into the underlying causes of unusual disease courses and into external and internal biomarkers triggering fluctuations in disease course will allow us to take further steps towards the unknown in epilepsy.
Footnotes
Peer-review: Externally peer-reviewed.
Author Contributions: Concept- BB; Design- BB, AÇA; Supervision- BB; Resource- AÇA; Materials- BB, AÇA; Data Collection and/or Processing- BB, AÇA; Analysis and/or Interpretation- BB, AÇA; Literature Search- AÇA; Writing- BB, AÇA; Critical Reviews- BB, AÇA.
Conflict of Interest: The authors have no conflict of interest.
Financial Disclosure: The authors did not receive any financial support.
REFERENCES
- 1.Fiest KM, Sauro KM, Wiebe S, Patten SB, Kwon CS, Dykeman J, et al. Prevalence and incidence of epilepsy: A systematic review and meta-analysis of international studies. Neurology. 2017;88:296–303. doi: 10.1212/WNL.0000000000003509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Onal AE, Tumerdem Y, Ozturk MK, Gurses C, Baykan B, Gokyigit A, et al. Epilepsy prevalence in a rural area in Istanbul. Seizure. 2002;11:397–401. doi: 10.1053/seiz.2001.0665. [DOI] [PubMed] [Google Scholar]
- 3.Beghi E. Addressing the burden of epilepsy: Many unmet needs. Pharmacol Res. 2016;107:79–84. doi: 10.1016/j.phrs.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 4.Thijs RD, Surges R, O'Brien TJ, Sander JW. Epilepsy in adults. Lancet. 2019;393:689–701. doi: 10.1016/S0140-6736(18)32596-0. [DOI] [PubMed] [Google Scholar]
- 5.Kwan P, Sander JW. The natural history of epilepsy: an epidemiological view. J Neurol Neurosurg Psychiatry. 2004;75:1376–81. doi: 10.1136/jnnp.2004.045690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sillanpää M, Schmidt D. Natural history of treated childhood-onset epilepsy: prospective, long-term population-based study. Brain. 2006;129:617–624. doi: 10.1093/brain/awh726. [DOI] [PubMed] [Google Scholar]
- 7.Sander JWAS, Sillanpää M. Natural history prognosis. In: Engels J, Pedley TA, editors. Epilepsy. A comprehensive textbook. I/III. New York: Raven Press; 1997. pp. 69–86. [Google Scholar]
- 8.Beghi E, Beretta S, Carone D, Zanchi C, Bianchi E, Pirovano M, et al. PRO-LONG Study Group. Prognostic patterns and predictors in epilepsy: a multicentre study (PRO-LONG) J Neurol Neurosurg Psychiatry. 2019;90:1276–1285. doi: 10.1136/jnnp-2019-320883. [DOI] [PubMed] [Google Scholar]
- 9.Del Felice A, Beghi E, Boero G, La Neve A, Bogliun G, De Palo A, et al. Early versus late remission in a cohort of patients with newly diagnosed epilepsy. Epilepsia. 2010;51:37–42. doi: 10.1111/j.1528-1167.2009.02141.x. [DOI] [PubMed] [Google Scholar]
- 10.Keränen T, Riekkinen P. Remission of seizures in untreated epilepsy. BMJ. 1993;307:483. doi: 10.1136/bmj.307.6902.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sillanpää M, Schmidt D. Prognosis of seizure recurrence after stopping antiepileptic drugs in seizure-free patients. A long-term population-based study of childhood-onset epilepsy. Epilepsy Behav. 2006;8:713–719. doi: 10.1016/j.yebeh.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 12.Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Allen Hauser W, Mathern G, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia. 2010;51:1069–1077. doi: 10.1111/j.1528-1167.2009.02397.x. [DOI] [PubMed] [Google Scholar]
- 13.Picot MC, Baldy-Moulinier M, Daurès JP, Dujols P, Crespel A. The prevalence of epilepsy and pharmacoresistant epilepsy in adults: a population-based study in a Western European country. Epilepsia. 2008;49:1230–1238. doi: 10.1111/j.1528-1167.2008.01579.x. [DOI] [PubMed] [Google Scholar]
- 14.Neligan A, Bell GS, Elsayed M, Sander JW, Shorvon SD. Treatment changes in a cohort of people with apparently drug-resistant epilepsy: an extended follow-up. J Neurol Neurosurg Psychiatry. 2012;83:810–813. doi: 10.1136/jnnp-2011-302085. [DOI] [PubMed] [Google Scholar]
- 15.Callaghan B, Schlesinger M, Rodemer W, Pollard J, Hesdorffer D, Allen Hauser W, et al. Remission and relapse in a drug-resistant epilepsy population followed prospectively. Epilepsia. 2011;52:619–626. doi: 10.1111/j.1528-1167.2010.02929.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schiller Y. Seizure relapse and development of drug resistance following longterm seizure remission. Arch Neurol. 2009;66:1233–1239. doi: 10.1001/archneurol.2009.211. [DOI] [PubMed] [Google Scholar]
- 17.Berg AT, Levy SR, Testa FM, D'Souza R. Remission of epilepsy after two drug failures in children: a prospective study. Ann Neurol. 2009;65:510–519. doi: 10.1002/ana.21642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xue-Ping W, Hai-Jiao W, Li-Na Z, Xu D, Ling L. Risk factors for drug-resistant epilepsy: A systematic review and meta-analysis. Medicine (Baltimore) 2019;98:e16402. doi: 10.1097/MD.0000000000016402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med. 2000;342:314–319. doi: 10.1056/NEJM200002033420503. [DOI] [PubMed] [Google Scholar]
- 20.Camfield PR, Camfield CS, Gordon K, Dooley JM. If a first antiepileptic drug fails to control a child's epilepsy, what are the chances of success with the next drug? J Pediatr. 1997;131:821–824. doi: 10.1016/s0022-3476(97)70027-1. [DOI] [PubMed] [Google Scholar]
- 21.Choi H, Hayat MJ, Zhang R, Hirsch LJ, Bazil CW, Mendiratta A, et al. Drug-resistant epilepsy in adults: Outcome trajectories after failure of two medications. Epilepsia. 2016;57:1152–6110. doi: 10.1111/epi.13406. [DOI] [PubMed] [Google Scholar]
- 22.Mbuba CK, Ngugi AK, Newton CR, Carter JA. The epilepsy treatment gap in developing countries: a systematic review of the magnitude, causes, and intervention strategies. Epilepsia. 2008;49:1491–1503. doi: 10.1111/j.1528-1167.2008.01693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Diop AG, Hesdorffer DC, Logroscino G, Hauser WA. Epilepsy and mortality in Africa: a review of the literature. Epilepsia. 2005;46:33–35. doi: 10.1111/j.1528-1167.2005.00405.x. [DOI] [PubMed] [Google Scholar]
- 24.Singh G, Sander JW. The global burden of epilepsy report: Implications for low- and middle-income countries. Epilepsy Behav. 2020;105:106949. doi: 10.1016/j.yebeh.2020.106949. [DOI] [PubMed] [Google Scholar]
- 25.Placencia M, Sander JW, Roman M, Madera A, Crespo F, Cascante S, et al. The characteristics of epilepsy in a largely untreated population in rural Ecuador. J Neurol Neurosurg Psychiatry. 1994;57:320–325. doi: 10.1136/jnnp.57.3.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nicoletti A, Sofia V, Vitale G, Bonelli SI, Bejarano V, Bartalesi F, Tran DS, Preux PM, Zappia M, Bartoloni A. Natural history and mortality of chronic epilepsy in an untreated population of rural Bolivia: a follow-up after 10 years. Epilepsia. 2009;50:2199–2206. doi: 10.1111/j.1528-1167.2009.02174.x. [DOI] [PubMed] [Google Scholar]
- 27.Seneviratne U, Cook M, D'Souza W. The prognosis of idiopathic generalized epilepsy. Epilepsia. 2012;53:2079–2090. doi: 10.1111/j.1528-1167.2012.03723.x. [DOI] [PubMed] [Google Scholar]
- 28.Mohanraj R, Brodie MJ. Outcomes of newly diagnosed idiopathic generalized epilepsy syndromes in a non-pediatric setting. Acta Neurol Scand. 2007;115:204–208. doi: 10.1111/j.1600-0404.2006.00791.x. [DOI] [PubMed] [Google Scholar]
- 29.Callenbach PM, Bouma PA, Geerts AT, Arts WF, Stroink H, Peeters EA, et al. Long-term outcome of childhood absence epilepsy: Dutch Study of Epilepsy in Childhood. Epilepsy Res. 2009;83:249–256. doi: 10.1016/j.eplepsyres.2008.11.011. [DOI] [PubMed] [Google Scholar]
- 30.Wirrell EC, Camfield CS, Camfield PR, Gordon KE, Dooley JM. Long-term prognosis of typical childhood absence epilepsy: remission or progression to juvenile myoclonic epilepsy. Neurology. 1996;47:912–918. doi: 10.1212/wnl.47.4.912. [DOI] [PubMed] [Google Scholar]
- 31.Trinka E, Baumgartner S, Unterberger I, Unterrainer J, Luef G, Haberlandt E, et al. Long-term prognosis for childhood and juvenile absence epilepsy. J Neurol. 2004;251:1235–1241. doi: 10.1007/s00415-004-0521-1. [DOI] [PubMed] [Google Scholar]
- 32.Baykan B, Matur Z, Gürses C, Aykutlu E, Gökyiğit A. Typical absence seizures triggered by photosensitivity. Epilepsia. 2005;46:159–163. doi: 10.1111/j.0013-9580.2005.67303.x. [DOI] [PubMed] [Google Scholar]
- 33.Healy L, Moran M, Singhal S, O'Donoghue MF, Alzoubidi R, Whitehouse WP. Relapse after treatment withdrawal of antiepileptic drugs for Juvenile Absence Epilepsy and Juvenile Myoclonic Epilepsy. Seizure. 2018;59:116–122. doi: 10.1016/j.seizure.2018.05.015. [DOI] [PubMed] [Google Scholar]
- 34.Martinez-Juarez IE, Alonso ME, Medina MT, Duron RM, Bailey JN, Lopez-Ruiz M, et al. Juvenile myoclonic epilepsy subsyndromes: Family studies and longterm follow-up. Brain. 2006;129:1269–1280. doi: 10.1093/brain/awl048. [DOI] [PubMed] [Google Scholar]
- 35.Baykan B, Wolf P. Juvenile myoclonic epilepsy as a spectrum disorder: A focused review. Seizure. 2017;49:36–41. doi: 10.1016/j.seizure.2017.05.011. [DOI] [PubMed] [Google Scholar]
- 36.Geithner J, Schneider F, Wang Z, Berneiser J, Herzer R, Kessler C, et al. Predictors for long-term seizure outcome in juvenile myoclonic epilepsy: 25–63 years of follow-up. Epilepsia. 2012;53:1379–1386. doi: 10.1111/j.1528-1167.2012.03526.x. [DOI] [PubMed] [Google Scholar]
- 37.Senf P, Schmitz B, Holtkamp M, Janz D. Prognosis of juvenile myoclonic epilepsy 45 years after onset: seizure outcome and predictors. Neurology. 2013;81:2128–2233. doi: 10.1212/01.wnl.0000437303.36064.f8. [DOI] [PubMed] [Google Scholar]
- 38.Baykan B, Altindag EA, Bebek N, Ozturk AY, Aslantas B, Gurses C, et al. Myoclonic seizures subside in the fourth decade in juvenile myoclonic epilepsy. Neurology. 2008;70:2123–2129. doi: 10.1212/01.wnl.0000313148.34629.1d. [DOI] [PubMed] [Google Scholar]
- 39.Guaranha MSB, de Araujo Filho GM, Lin K, Guilhoto LMFF, Caboclo LOSF, Yacubian EMT. Prognosis of juvenile myoclonic epilepsy is related to endophenotypes. Seizure. 2011;20:42–48. doi: 10.1016/j.seizure.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 40.Baykan B, Martínez-Juárez IE, Altindag EA, Camfield CS, Camfield PR. Lifetime prognosis of juvenile myoclonic epilepsy. Epilepsy Behav. 2013;28:S18–524. doi: 10.1016/j.yebeh.2012.06.036. [DOI] [PubMed] [Google Scholar]
- 41.Stevelink R, Koeleman BPC, Sander JW, Jansen FE, Braun KPJ. refractory juvenile myoclonic epilepsy: a meta-analysis of prevalence and risk factors. Eur J Neurol. 2019;26:856–864. doi: 10.1111/ene.13811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lamberink HJ, Otte WM, Geerts AT, Pavlovic M, Ramos-Lizana J, Marson AG, et al. Individualised prediction model of seizure recurrence and longterm outcomes after withdrawal of antiepileptic drugs in seizure-free patients: a systematic review and individual participant data meta-analysis. Lancet Neurol. 2017;16:523–531. doi: 10.1016/S1474-4422(17)30114-X. [DOI] [PubMed] [Google Scholar]
- 43.Panayiotopoulos CP, Michael M, Sanders S, Valeta T, Koutroumanidis M. Benign childhood focal epilepsies: assessment of established and newly recognized syndromes. Brain. 2008;131:2264–2286. doi: 10.1093/brain/awn162. [DOI] [PubMed] [Google Scholar]
- 44.Koutroumanidis M, Aggelakis K, Panayiotopoulos CP. Idiopathic epilepsy with generalized tonic-clonic seizures only vs. idiopathic epilepsy with phantom absences and generalized tonic-clonic seizures: one or two syndromes? Epilepsia. 2008;49:2050–2062. doi: 10.1111/j.1528-1167.2008.01702.x. [DOI] [PubMed] [Google Scholar]
- 45.Fejerman N, Caraballo R, Tenembaum SN. Atypical evolutions of benign localization-related epilepsies in children: are they predictable? Epilepsia. 2000;41:380–390. doi: 10.1111/j.1528-1157.2000.tb00177.x. [DOI] [PubMed] [Google Scholar]
- 46.Cerulli Irelli E, Cocchi E, Morano A, Casciato S, Fanella M, Albini M, Fisco G, Barone FA, Orlando B, Mascia A, Manfredi M, Fattouch J, Giallonardo AT, Di Gennaro G, Di Bonaventura C. Valproate impact and sex-dependent seizure remission in patients with idiopathic generalized epilepsy. J Neurol Sci. 2020;415:116940. doi: 10.1016/j.jns.2020.116940. [DOI] [PubMed] [Google Scholar]
- 47.Parisi P, Paolino MC, Raucci U, Ferretti A, Villa MP, Trenite DK-N. “Atypical forms”of benign epilepsy with centrotemporal spikes (BECTS): How to diagnose and guide these children. A practical/scientific approach. Epilepsy Behav. 2017;75:165–169. doi: 10.1016/j.yebeh.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 48.Garcia-Ramos C, Jackson DC, Lin JJ, Dabbs K, Jones JE, Hsu DA, et al. Cognition and brain development in children with benign epilepsy with centrotemporal spikes. Epilepsia. 2015;56:1615–1622. doi: 10.1111/epi.13125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cherian A, Baheti NN, Menon RN, Iyer RS, Rathore C, Radhakrishnan A. Atonic variant of benign childhood epilepsy with centrotemporal spikes (atonic-BECTS): a distinct electro-clinical syndrome. Brain Dev. 2012;34:511–519. doi: 10.1016/j.braindev.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 50.Park JI, Kim SJ, Kim HG. Acoustic effects of carbamazepine in benign rolandic epilepsy. Epilepsy Behav. 2005;7:468–471. doi: 10.1016/j.yebeh.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 51.Prats JM, Garaizar C, García-Nieto ML, Madoz P. Antiepileptic drugs and atypical evolution of idiopathic partial epilepsy. Pediatr Neurol. 1998;18:402–406. doi: 10.1016/s0887-8994(97)00223-3. [DOI] [PubMed] [Google Scholar]
- 52.Semah F, Lamy C, Demeret S. Hippocampal sclerosis and other hippocampal abnormalities in the early identification of candidates for epilepsy surgery. Arch. Neurol. 2002;59:1042–1043. doi: 10.1001/archneur.59.6.1042-a. [DOI] [PubMed] [Google Scholar]
- 53.Stephen LJ, Kwan P, Brodie MJ. Does the cause of localization related epilepsy influence the response to antiepileptic drug treatment? Epilepsia. 2011;43:357–362. doi: 10.1046/j.1528-1157.2001.29000.x. [DOI] [PubMed] [Google Scholar]
- 54.Varoglu AO, Saygi S, Acemoglu H, Ciger A. Prognosis of patients with mesial temporal lobe epilepsy due to hippocampal sclerosis. Epilepsy Res. 2009;85:206–211. doi: 10.1016/j.eplepsyres.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 55.Leong ECS, Seneviratne U. “Benign”temporal lobe epilepsy with hippocampal sclerosis: A forgotten entity? Epilepsy Behav Rep. 2020;14:100407. doi: 10.1016/j.ebr.2020.100407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kobayashi E, D'Agostino MD, Lopes-Cendes I, Berkovic SF, Li ML, Andermann E, et al. Hippocampal atrophy and T2-weighted signal changes in familial mesial temporal lobe epilepsy. Neurology. 2003;60:405–409. doi: 10.1212/wnl.60.3.405. [DOI] [PubMed] [Google Scholar]
- 57.Gambardella A, Labate A, Giallonardo AT, Aguglia U. Familial mesial temporal lobe epilepsies: clinical and genetic features. Epilepsia. 2009;50:55–57. doi: 10.1111/j.1528-1167.2009.02123.x. [DOI] [PubMed] [Google Scholar]
- 58.Cvetkovska E, Kuzmanovski I, Babunovska M, Boshkovski B, Cangovska TC, Trencevska GK. Phenotypic spectrum in families with mesial temporal lobe epilepsy probands. Seizure. 2018;58:13–16. doi: 10.1016/j.seizure.2018.03.019. [DOI] [PubMed] [Google Scholar]
- 59.Striano P, Gambardella A, Coppola A, Di Bonaventura C, Bovo G, Diani E, et al. Familial mesial temporal lobe epilepsy (FMTLE): a clinical and genetic study of 15 Italian families. J Neurol. 2008;255:16–23. doi: 10.1007/s00415-007-0653-1. [DOI] [PubMed] [Google Scholar]
- 60.Nilo A, Gelisse P, Crespel A. Genetic/idiopathic generalized epilepsies: Not so good as that! Rev Neurol (Paris) 2020;176:427–438. doi: 10.1016/j.neurol.2020.03.018. [DOI] [PubMed] [Google Scholar]
- 61.Gesche J, Christensen J, Hjalgrim H, Rubboli G, Beier CP. Epidemiology and outcome of idiopathic generalized epilepsy in adults. Eur J Neurol. 2020;27:676–684. doi: 10.1111/ene.14142. [DOI] [PubMed] [Google Scholar]
- 62.Cerulli Irelli E, Morano A, Barone FA, Fisco G, Fanella M, Orlando B, et al. Persistent treatment resistance in genetic generalized epilepsy: A long-term outcome study in a tertiary epilepsy center. Epilepsia. 2020;61:2452–2460. doi: 10.1111/epi.16708. [DOI] [PubMed] [Google Scholar]
- 63.Aydin-Özemir Z, Matur Z, Bebek N, Gürses C, Gökyiğit A, Baykan B. Long-term follow-up of adult patients with genetic generalized epilepsy with typical absence seizures and generalized paroxysmal fast activity in their EEG. Seizure. 2014;23:607–615. doi: 10.1016/j.seizure.2014.04.017. [DOI] [PubMed] [Google Scholar]
- 64.Bansal L, Vargas Collado L, Pawar K, Nagesh D, Ilyas M, Hall A, et al. Electroclinical Features of Generalized Paroxysmal Fast Activity in Typical Absence Seizures. J Clin Neurophysiol. 2019;36:36–44. doi: 10.1097/WNP.0000000000000535. [DOI] [PubMed] [Google Scholar]
- 65.Gesche J, Khanevski M, Solberg C, Beier CP. Resistance to valproic acid as predictor of treatment resistance in genetic generalized epilepsies. Epilepsia. 2017;58:64–e69. doi: 10.1111/epi.13702. [DOI] [PubMed] [Google Scholar]
- 66.Weston MC. A tRNA Variant Translates Into Seizure Resistance. Epilepsy Curr. 2021;21:126–128. doi: 10.1177/1535759721990043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Löscher W. Animal models of drug-resistant epilepsy. Novartis Found Symp. 2002;243:149–59. discussion 159–66, 180–5. https: //pubmed.ncbi.nlm.nih.gov/11990774/ [PubMed] [Google Scholar]
- 68.Fang M, Xi ZQ, Wu Y, Wang XF. A new hypothesis of drug refractory epilepsy:neural network hypothesis. Med Hypotheses. 2011;76:871–876. doi: 10.1016/j.mehy.2011.02.039. [DOI] [PubMed] [Google Scholar]
- 69.Bazhanova ED, Kozlov AA, Litovchenko AV. Mechanisms of Drug Resistance in the Pathogenesis of Epilepsy:Role of Neuroinflammation. A Literature Review. Brain Sci. 2021;11:663. doi: 10.3390/brainsci11050663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shimada T, Takemiya T, Sugiura H, Yamagata K. Role of inflammatory mediators in the pathogenesis of epilepsy. Mediators Inflamm. 2014;2014:901902. doi: 10.1155/2014/901902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liimatainen S, Lehtimäki K, Palmio J, Alapirtti T, Peltola J. Immunological perspectives of temporal lobe seizures. J Neuroimmunol. 2013;263:1–7. doi: 10.1016/j.jneuroim.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 72.Atmaca MM, Tuzun E, Erdag E, Bebek N, Baykan B, Gurses C. Investigation of anti-neuronal antibodies in status epilepticus of unknown etiology:a prospective study. Acta Neurol Belg. 2017;117:841–848. doi: 10.1007/s13760-017-0796-5. [DOI] [PubMed] [Google Scholar]
- 73.Wang M, Backstrom T, Sundstrom I, Wahlström G, Olsson T, Zhu D, et al. Neuroactive steroids and central nervous system disorders. Int Rev Neurobiol. 2001;46:421–459. doi: 10.1016/s0074-7742(01)46071-5. [DOI] [PubMed] [Google Scholar]
- 74.Reddy DS. The role of neurosteroids in the pathophysiology and treatment of catamenial epilepsy. Epilepsy Res. 2009;85:1–30. doi: 10.1016/j.eplepsyres.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Herzog AG, Frye CA. Seizure exacerbation associated with inhibition of progesterone metabolism. Ann Neurol. 2003;53:390–391. doi: 10.1002/ana.10508. [DOI] [PubMed] [Google Scholar]
- 76.Grover S, Talwar P, Gourie-Devi M, Gupta M, Bala K, Sharma S, et al. Genetic polymorphisms in sex hormone metabolizing genes and drug response in women with epilepsy. Pharmacogenomics. 2010;11:1525–1534. doi: 10.2217/pgs.10.120. [DOI] [PubMed] [Google Scholar]
- 77.Verrotti A, Laus M, Coppola G, Parisi P, Mohn A, Chiarelli F. Catamenial epilepsy:hormonal aspects. Gynecol Endocrinol. 2010;26:783–790. doi: 10.3109/09513590.2010.490606. [DOI] [PubMed] [Google Scholar]
- 78.Choi H, Detyniecki K, Bazil C, Thornton S, Crosta P, Tolba H, et al. EPIGEN Consortium. Development and validation of a predictive model of drug-resistant genetic generalized epilepsy. Neurology. 2020;95:e2150–e2160. doi: 10.1212/WNL.0000000000010597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Reddy DS, Rogawski MA. Neurosteroid replacement therapy for catamenial epilepsy. Neurotherapeutics. 2009;6:392–401. doi: 10.1016/j.nurt.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schroeder AC, Privalsky ML. Thyroid hormones, t3 and t4, in the brain. Front Endocrinol (Lausanne) 2014;5:40. doi: 10.3389/fendo.2014.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tamijani SM, Karimi B, Amini E, Golpich M, Dargahi L, Ali RA, et al. Thyroid hormones:Possible roles in epilepsy pathology. Seizure. 2015;31:155–164. doi: 10.1016/j.seizure.2015.07.021. [DOI] [PubMed] [Google Scholar]
- 82.Wallis K, Sjögren M, van Hogerlinden M, Silberberg G, Fisahn A, Nordström K, et al. Locomotor deficiencies and aberrant development of subtype-specific GABAergic interneurons caused by an unliganded thyroid hormone receptor alpha1. J Neurosci. 2008;28:1904–1915. doi: 10.1523/JNEUROSCI.5163-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Han JY, Lee IG, Shin S, Park J. Seizure duration may increase thyroid-stimulating hormone levels in children experiencing a seizure. J Int Med Res. 2020;48:300060519888401. doi: 10.1177/0300060519888401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Aydin-Ozemir Z, Tüzün E, Baykan B, Akman-Demir G, Ozbey N, Gürses C, et al. Autoimmune thyroid encephalopathy presenting with epilepsia partialis continua. Clin EEG Neurosci. 2006;37:204–209. doi: 10.1177/155005940603700308. [DOI] [PubMed] [Google Scholar]
- 85.Hamed SA. The effect of antiepileptic drugs on thyroid hormonal function:causes and implications. Expert Rev Clin Pharmacol. 2015;8(6):741–750. doi: 10.1586/17512433.2015.1091302. [DOI] [PubMed] [Google Scholar]
- 86.Vezzani A, Balosso S, Ravizza T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat Rev Neurol. 2019;15:459–472. doi: 10.1038/s41582-019-0217-x. [DOI] [PubMed] [Google Scholar]
- 87.Bauer J, Bien CG. Neuropathology of autoimmune encephalitides. Handb Clin Neurol. 2016;133:107–20. doi: 10.1016/B978-0-444-63432-0.00007-4. [DOI] [PubMed] [Google Scholar]
- 88.Ekizoglu E, Tuzun E, Woodhall M, Lang B, Jacobson L, Icoz S, et al. Investigation of neuronal autoantibodies in two different focal epilepsy syndromes. Epilepsia. 2014;55:414–422. doi: 10.1111/epi.12528. [DOI] [PubMed] [Google Scholar]
- 89.Vanli-Yavuz EN, Erdag E, Tuzun E, Ekizoglu E, Baysal-Kirac L, Ulusoy C, et al. Neuronal autoantibodies in mesial temporal lobe epilepsy with hippocampal sclerosis. J Neurol Neurosurg Psychiatry. 2016;87:684–692. doi: 10.1136/jnnp-2016-313146. [DOI] [PubMed] [Google Scholar]
- 90.Karaaslan Z, Ekizoğlu E, Tektürk P, Erdağ E, Tüzün E, Bebek N, et al. Investigation of neuronal auto-antibodies in systemic lupus erythematosus patients with epilepsy. Epilepsy Res. 2017;129:132–137. doi: 10.1016/j.eplepsyres.2016.12.006. [DOI] [PubMed] [Google Scholar]
- 91.Steriade C, Titulaer MJ, Vezzani A, Sander JW, Thijs RD. The association between systemic autoimmune disorders and epilepsy and its clinical implications. Brain. 2021;144:372–390. doi: 10.1093/brain/awaa362. [DOI] [PubMed] [Google Scholar]
- 92.Keezer MR, Sisodiya SM, Sander JW. Comorbidities of epilepsy:current concepts and future perspectives. Lancet Neurol. 2016;15:106–115. doi: 10.1016/S1474-4422(15)00225-2. [DOI] [PubMed] [Google Scholar]
- 93.Cameron FJ, Scratch SE, Nadebaum C, Northam EA, Koves I, Jennings J, et al. DKA Brain Injury Study Group. Neurological consequences of diabetic ketoacidosis at initial presentation of type 1 diabetes in a prospective cohort study of children. Diabetes Care. 2014;37:1554–1562. doi: 10.2337/dc13-1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mastrangelo M, Tromba V, Silvestri F, Costantino F. Epilepsy in children with type 1 diabetes mellitus:Pathophysiological basis and clinical hallmarks. Eur J Paediatr Neurol. 2019;23:240–247. doi: 10.1016/j.ejpn.2018.12.006. [DOI] [PubMed] [Google Scholar]
- 95.Verrotti A, Scaparrotta A, Olivieri C, Chiarelli F. Seizures and type 1 diabetes mellitus:current state of knowledge. Eur J Endocrinol. 2012;167:749e58. doi: 10.1530/EJE-12-0699. [DOI] [PubMed] [Google Scholar]
- 96.Leen WG, Taher M, Verbeek MM, Kamsteeg EJ, van de Warrenburg BP, Willemsen MA. GLUT1 deficiency syndrome into adulthood:a follow-up study. J Neurol. 2014;261:589–599. doi: 10.1007/s00415-014-7240-z. [DOI] [PubMed] [Google Scholar]
- 97.Sharma S, Prasad AN. Inborn Errors of Metabolism and Epilepsy:Current Understanding, Diagnosis, and Treatment Approaches. Int J Mol Sci. 2017;18:1384. doi: 10.3390/ijms18071384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lu E, Pyatka N, Burant CJ, Sajatovic M. Systematic Literature Review of Psychiatric Comorbidities in Adults with Epilepsy. J Clin Neurol. 2021;17:176–186. doi: 10.3988/jcn.2021.17.2.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hitiris N, Mohanraj R, Norrie J, Sills GJ, Brodie MJ. Predictors of pharmacoresistant epilepsy. Epilepsy Res. 2007;75:192–196. doi: 10.1016/j.eplepsyres.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 100.Kanner AM. Can neurobiological pathogenic mechanisms of depression facilitate the development of seizure disorders? Lancet Neurol. 2012;11:1093–1102. doi: 10.1016/S1474-4422(12)70201-6. [DOI] [PubMed] [Google Scholar]