

Abstract
Immunotherapeutic strategies that combine oncolytic virus (OV) and immune checkpoint inhibitors have the potential to overcome treatment resistance in pancreatic ductal adenocarcinoma (PDAC), one of the least immunogenic solid tumors. Oncolytic viral chimera, CF33-hNIS-anti-PDL1 genetically modified to express anti-human PD-L1 antibody and CF33-hNIS-Δ without the anti-PD-L1 gene, were used to investigate the immunogenic effects of OVs and virus delivered anti-PD-L1 in PDAC in vitro. Western blot, flow cytometry, and immunofluorescence microscopy were used to evaluate the effects of CF33-hNIS-Δ and IFNγ on PD-L1 upregulation in AsPC-1 and BxPC-3 cells, and CF33-hNIS-antiPDL1 production of anti-PD-L1 and surface PD-L1 blockade of AsPC-1 and BxPC-3 with or without co-cultured activated T cells. The cytosolic and cell surface levels of PD-L1 in PDAC cell lines varied; only BxPC-3 showed high cell surface expression. Treatment of these cells with CF33-hNIS-Δ and IFNγ significantly upregulated PD-L1 expression and translocation of PD-L1 from the cytosol onto the cell surface. Following coculture of activated T cells and BxPC-3 with CF33-hNIS-antiPDL1, the cell surface PD-L1 blockade on BxPC-3 cells by virus delivered anti-PD-L1 antibody increased granzyme B release and prevented virus-induced decrease of perforin release from activated CD8+ T cells. Our results suggest that CF33-IOVs can prime immune checkpoint inhibition of PDAC and enhance antitumor immune killing.
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) poses one of the greatest challenges in cancer immunotherapy as a non-immunogenic solid tumor that remains obstinately therapy-resistant [1]. Standard of care multi-drug therapies such as FOLFIRINOX (Fluorouracil, leucovorin, irinotecan, and oxaliplatin) and gemcitabine plus nab-paclitaxel have achieved incremental improvements in patient outcomes, yet the median survival for unresectable PDAC remains less than a year in the United States [2–5]. PDAC is projected to become the second leading cause of cancer-related deaths worldwide by 2030 [ref.6]. A rapidly emerging new class of anti-cancer agents such as immune checkpoint inhibitors (ICI) targeting pathways of T-cell escape, namely anti-PD1 and anti-PD-L1 have shown great promise in several immunogenic tumors. However, in genomically stable and “cold” PDAC tumors, these agents have had limited efficacy as single agents and have failed to improve survival in patients [7–9]. Multimodal therapies that prime PDAC to express receptors for ICIs and activate its anti-tumoral adaptive immunity are currently under intense investigation to improve tumor response and patient outcomes.
A complex and dynamic tumor microenvironment (TME) protects PDAC from immune recognition and leads to T cell evasion. As mechanisms of T cell escape are elucidated, ICIs that target the programmed death receptor and ligand 1 (PD-1/PD-L1) axis have been effective in solid tumors such as melanoma, non-small-cell lung cancer, and gastric adenocarcinomas with high levels of PD-L1 and active lymphocytic infiltrates [10, 11]. Unfortunately, ICIs that depend on single targets have failed in PDAC patients [12, 13]. Even in patients with PD-L1 overexpression, single-agent ICIs have had a limited effect with no survival benefits for PDAC patients in clinical trials [12, 14]. Low antigen-recognition, limited cytotoxic T cell recruitment, and newly recognized aberrant signaling of co-stimulatory immune checkpoints have been identified as a key barrier to the success of immunotherapies in PDAC [15–17]. To optimize the anti-tumor efficacy of immunotherapies, strategies must overcome the inherent immune quiescence and protective immune TME of PDAC which persist as the main challenges to drug resistance [18, 19].
Oncolytic viruses (OVs) are a group of potent and adaptable antitumor agents capable of selective cancer-killing, immune activation, and gene delivery. OVs can be genetically engineered to express various functional proteins and have been shown to elicit robust immune responses in solid tumors. A combination of OVs and ICIs is a promising immunotherapeutic strategy for treating PDAC and is under intense investigation to overcome key immune barriers in PDAC therapy [20–22]. Currently, reovirus plus pembrolizumab and adenovirus plus atezolizumab in PDAC patients are in Phase I testing (NCT03723915, NCT0275096). However, OVs alone or in combination with other agents have yet to be optimized for their full anti-tumor potential. The shortcomings that continue to impede success include a complex array of dynamic virus-host-immune interactions contributing to viral clearance and residual tumor resistance.
To overcome these limitations, we have equipped a potent oncolytic viral chimera, CF33 with the capacity to express anti-PD-L1 antibody for the immune-enhanced killing of PDAC [ref.23]. In this study, we investigated the unique properties of CF33-hNIS-antiPDL1 for direct oncolysis, immunogenicity, and immune checkpoint inhibition to understand the dynamic immuno-OV (IOV)-cancer cell interaction in vitro. Results will inform future in vivo preclinical studies of immune-competent models and human clinical trials for therapy-resistant PDAC.
MATERIALS AND METHODS
Cell lines
Human pancreatic cancer cell lines AsPC-1, BxPC-3, MIA PaCa-2, and PANC-1 were purchased from ATCC (Manassas, VA, USA). Capan-2 cells were a kind gift from Dr. Teresa Ku’s lab (City of Hope, Duarte, CA, USA). All pancreatic cancer cell lines except Capan-2 were cultured in RPMI 1640 medium supplemented with 10% FBS and 1% antibiotic–antimycotic solution (Corning, NY, USA). Capan-2 cells were cultured in McCoy’s 5A medium with 10% FBS and 1% antibiotic-antimycotic solution. All cells were maintained in a humidified incubator at 37°C and 5% CO2 and the absence of mycoplasma contamination was confirmed. [23]
Antibodies, reagents and CF33-hNIS-Δ and CF33-hNIS-antiPDL1 viruses
Anti-human PD-L1/CD274, -PE and –APC (Clone# 29E.2A3, catalog#329745, 329706, 329708), anti-FLAG antibody (Clone# L5, catalog#637304), Alexa Fluor 555 goat anti-rat IgG antibody (Catalog#405420), Alexa Fluor 467 goat anti-rat IgG antibody(Catalog#405416), human IFNγ (Catalog#570208), human IFNα (Catalog#592704), human IL-2 (Catalog#589104), and Zombie UV dye (Catalog#77474) were purchased from Biolegend (San Diego, CA, USA). Human IFNβ (Catalog#8499-1F) was purchased from R&D Systems (Minneapolis, MN 55413). Heat Inactivated human AB serum was purchased from Innovative Research, Inc (Catalog# IPLA-SERAB-27146, MI 48377, USA). Dynabeads™ Human T-Activator CD3/CD28 for T Cell Expansion and Activation was purchased from ThermoFisher Scientific (Catalog# 11131D, Waltham, MA, USA). The CF33-hNIS-Δ (CF33-hNIS-ΔF14.5L) virus [24, 25], CF33-GFP [ref.26], and the CF33-hNIS-antiPDL1 virus [23] were generated in our lab as previously described. Anti-sodium/Iodide Symporter (hNIS) monoclonal antibody was purchased from EMD Millipore Corp (Catalog# MAB3564, Billerica, MA, USA). Goat anti-rabbit IgG (H+L) Alexa Fluor 555 antibody (Catalog# A21434) and Goat anti-mouse Alexa Fluor 488 antibody (Catalog# A11029) were purchased from Invitrogen Corporation (Carlsbad, CA, USA).
Immune cell isolation, differentiation, activation, and coculture with BxPC-3
CD3 T lymphocyte isolation was performed using the isolation protocol from Biolegend (MojoSort™ Human CD3 T Cell Isolation Kit, Catalog #480131, San Diego, CA, USA). T cell activation and proliferation were performed according to the manufacturer’s protocol of Dynabeads™ Human T-Activator CD3/CD28 for T Cell Expansion and Activation (Catalog#11131D, Thermo Fisher Scientific, Waltham, MA, USA). Coculture of activated T cells with BxPC-3 used a cell ratio of 1:1 in RPMI 1640 supplemented with 100 U/mL penicillin, 100 mg/mL streptomycin, 1 mM L-glutamine, and 10% FBS in the presence of CF33-hNIS-antiPDL1 (MOI = 3) for 5 days.
Flow cytometric analysis
Cell surface staining:
Cells were harvested, washed with PBS, blocked with 10% human serum in PBS, stained with isotype controls and specific antibodies described in the figures, washed thrice with 1% BSA PBS, and analyzed with a BD LSRFortessa Flow Cytometer (BD Biosciences, San Jose, CA, USA). For virus-treated cells, before performing flow cytometry, cells were fixed with 4% paraformaldehyde.
Intracellular staining:
Cells were washed with PBS, treated with a fixation/permeabilization solution kit (Catalog#554714, BD Biosciences, San Jose, CA, USA), and stained with the specific antibodies shown in the figures. Stained cells were assessed with a BD LSRFortessa Flow Cytometer and analyzed using Flowjo software. Results are shown as histograms and mean fluorescence intensity (MFI) [ref.27].
Immunofluorescence microscopy
Cells were harvested, fixed/permeabilized with BD fixation/permeabilization solution kit (BD Biosciences), and stained with primary antibodies for 30 min. After washing with washing buffer, the cells were stained with secondary Alexa 488 or 555 conjugated antibodies for 30 min. After washing, cells were mounted with Hard Set Mounting Medium with DAPI (Catalog# H-1500, Vector Laboratories, Inc., Burlingame, CA, USA). Images were acquired on a Zeiss LSM880 confocal microscope using Zen Black and using a 20x/0.8 NA PlanApochromatic objective at a spatial resolution of 0.42um/pixel and frame size 1024 × 1024. The excitation and emission were, Red 594nm excitation and 600nm-650nm emission, Green 488nm excitation and 500nm–550nm emission, and Blue 405nm excitation and 410nm-490nm emission via PMT detectors. Images were adjusted for brightness in a linear manner using Zen Blue v2.3 software and all images were adjusted identically. [28]
Beads based cytokine assay
The supernatants from each of the wells of the cell culture experiments were collected and transferred into a 96-well plate using Biolegend LEGENDplex Human CD8/NK Panel (13-plex, Catalog#740267) according to the manufacturer’s instructions. Samples were run in BD LSRFortessa Flow cytometer and results were acquired using Legendplex v8.0 software (Biolegend, San Diego, CA, USA) [24].
Immunoblot analysis
Cells were harvested and lysed in RIPA lysis buffer plus proteinase inhibitors (Catalog#89900 and 186180, ThermoFisher Scientific, Waltham, MA, USA). Protein concentration was determined using the Bio-Rad protein assay kit ( Catalog#23227, Bio-Rad, Hercules, CA, USA). Supernatant or total cell lysate protein (50 μg) was separated by SDS-gel polyacrylamide electrophoresis, transferred to PVDF membranes, and probed with rabbit anti-human CD274/PD-L1 (BioRad, catalog#AHP1703), anti-FLAG tag (Catalog# ab205606, rabbit monoclonal antibody, Abcam, Cambridge, MA, USA) or anti-GAPDH (Catalog#2118L, Cell Signaling Technology. Inc, Danvers, MA, USA) following donkey anti-rabbit antibody in iBind Flex FD solutions (Catalog#SLF2020, Invitrogen, Walsham, MA, USA) using iBind Flex Western Device (Thermo Fisher Scientific, Waltham, MA, USA) for 3 h. The membrane was then scanned using the Azure C600 scanner (Azure Biosystems, Dublin, CA, USA) [23].
Bioinformatics analysis
We further verified the gene expression profile of CD274 (PD-L1) and its association with survival in PDAC patients using publicly available GEO Affymetrix human U133A human GeneChip 2.0 ST microarray with 45 matched adjacent non-tumor tissues and pancreatic tumor samples (GSE62452) and identified that PD-L1 (CD274: Gene ID#29126) is significant highly expressed in pancreatic cancer tumor samples with fold change = 1.46 and p-value = 5.059e-06 [Appendix A] [ref.29]. We also correlated PD-L1 mRNA gene expression profile with survival outcome using pancreatic patient cohort data from The Cancer Genome Atlas (TCGA) [ref.30]. However, TCGA pancreatic patient (n = 170) public cohorts that we downloaded did not have sufficient normal tissue samples (n = 3) compared to tumor samples (n = 167).
Statistical analysis
Assay results were expressed as mean ±SEM and paired or unpaired Student’s t-tests were used for comparisons. All p-values are two-sided. Data were analyzed with GraphPad Prism software (version 7, GraphPad Software, San Diego, CA, USA). Group comparisons for continuous data were conducted using Student’s t-tests or χ2 tests, and quantitative variables were analyzed with the paired Wilcoxon signed-rank test or the Spearman rank correlation test. Kaplan-Meier survival analysis was used to determine the survival differences between “high” and “low” expression and these differences were visualized by Kaplan-Meier plots and compared using Cox regression analysis, with p-values calculated by log-rank test using the Survival package in R package version R 4.0.2 ( R Core Team, 2018). Statistical significance was set at p < 0.05. The results shown here are in whole or part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga.
RESULTS
CF33-hNIS-Δ leads to a time-dependent increase in both the upregulation of PD-L1 protein expression and/or cytosol to cell surface translocation of PD-L1 in human PDACs
Western blot analysis and intracellular staining of PD-L1 demonstrated varying levels of PD-L1 protein expression within the cytosol in all five PDAC cell lines tested - AsPC-1, PANC-1, Capan-2, BxPC-3, and MIA PaCa-2 (Fig.1A and 1B). On the contrary, minimal cell surface PD-L1 expression was observed for AsPC-1, Capan-2, MIA PaCa-2, and PANC-1, while BxPC-3 had a comparatively higher cell surface PD-L1 expression (Fig. 1C). These results indicate that PD-L1 expression varies in different types of PDACs.
Figure 1. PD-L1 is found mostly in the cytosol of human pancreatic ductal adenocarcinoma cells (PDACs).
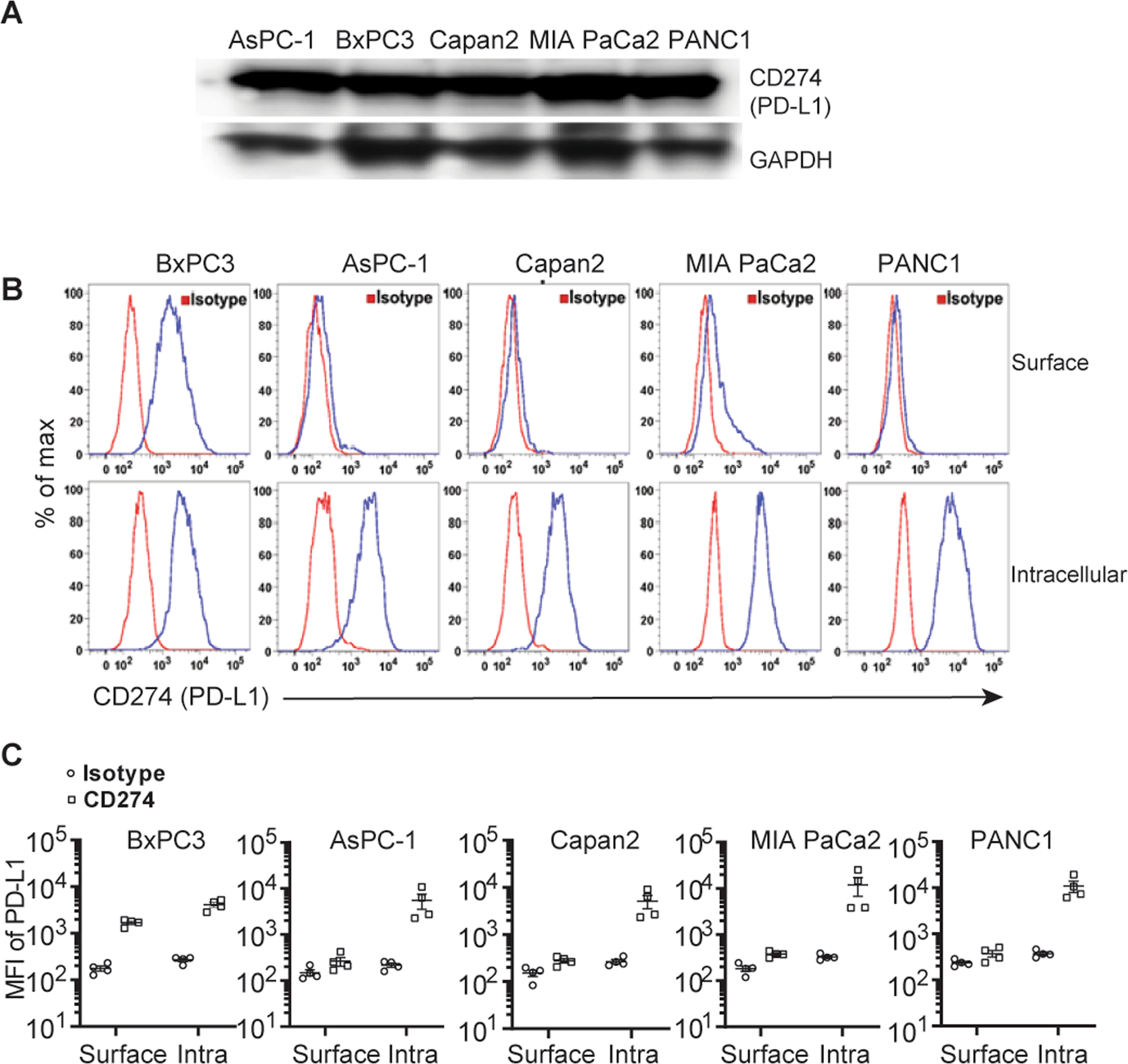
(A) Five human pancreatic carcinoma cell lines - AsPC-1, PANC-1, Capan-2, BxPC-3, and MIA PaCa-2 cells were lysed and blotted with anti-CD274/PD-L1 and GAPDH antibodies. (B) These cell lines were stained with PE-isotype control antibody or PE-anti-PD-L1 antibody (CD274, clone 29E.2A3, Biolegend) for surface staining and intracellular staining and analyzed by flow cytometry. PD-L1 expression levels were shown as a histogram (blue line) (representative of four independent experiments). (C) MFI (mean fluorescence intensity) of PD-L1 expression in comparison with isotype control is shown (n = 4) “Surface” - surface staining and “Intra” - intracellular staining.
Next, we analyzed the effect of viral infection on the translocation and upregulation of PD-L1. At 24 h after treatment of AsPC-1 and BxPC-3 with CF33-hNIS-Δ (MOI = 3), the cell surface PD-L1 expression slightly increased in the AsPC-1 cell line while no change was observed in the BxPC-3 cell line (data not shown). Five days after treatment, the virus killed 71% of AsPC-1 cells and 86.4% of BxPC-3 cells (Fig. 2A). The surviving cells expressed significantly higher levels of PD-L1 on the surface (Fig. 2B). Confocal immunofluorescence microscopy indicated that CF33-hNIS-Δ induced total PD-L1 protein upregulation and cytosol to cell surface translocation of treatment-resistant AsPC-1 cells by day 5 after treatment of the virus. For BxPC-3 cells, translocation of PD-L1 onto the cell surface was observed without total PD-L1 protein upregulation (Fig. 2C). These findings suggest that virus infection can induce translocation and/or upregulation of cell surface PD-L1 expression onto the cell surface in both PDAC cell lines.
Figure 2. CF33-hNIS-Δ increases the translocation/expression of PD-L1 in human PDAC cell lines.
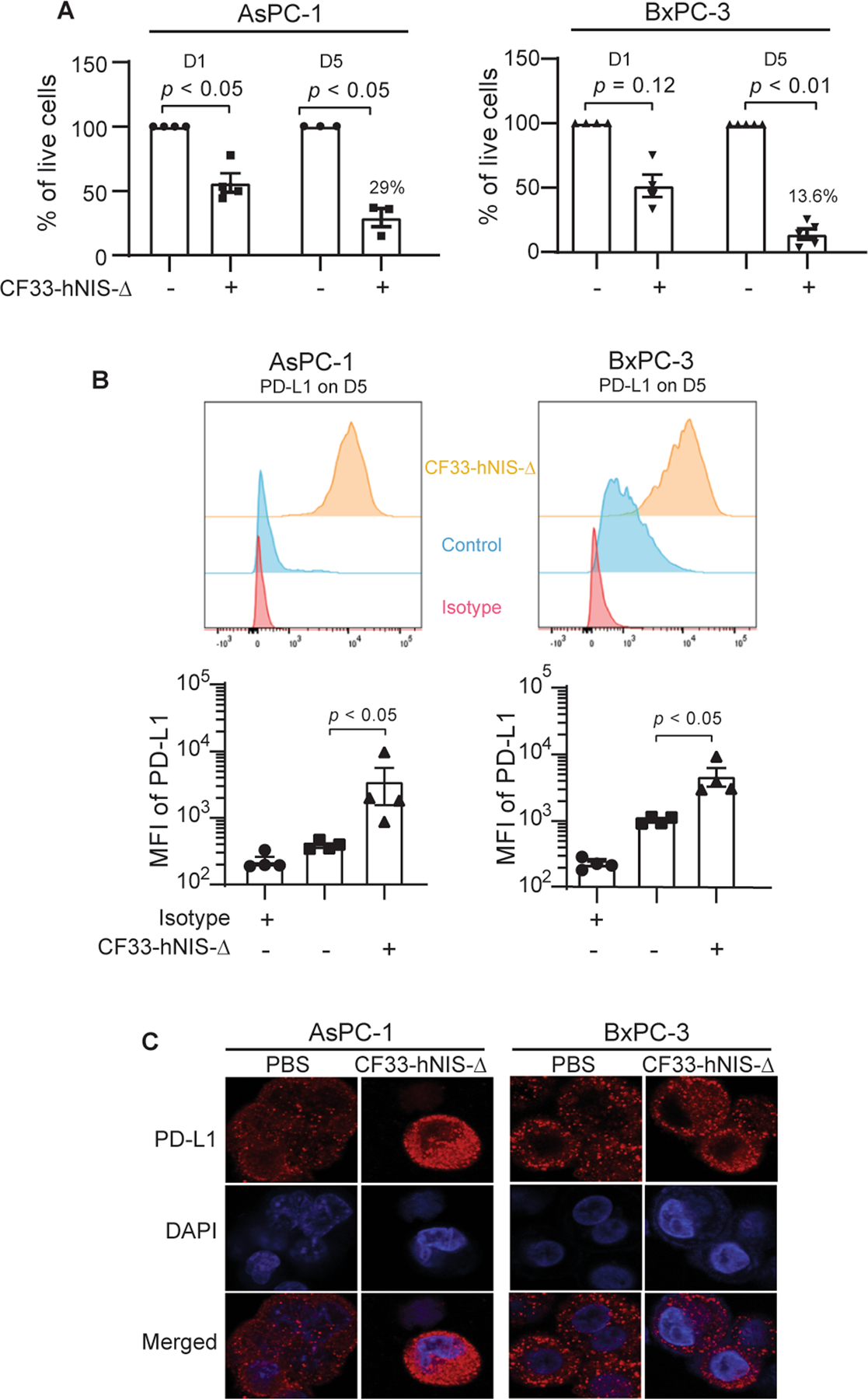
Human pancreatic carcinoma cell lines AsPC-1 and BxPC-3 were treated with CF33-hNIS-Δ (MOI = 3) and analyzed for (A) percent cell survival at days 1 and 5. (B) surface expression of PD-L1 at day 5 (representative of four independent experiments) analyzed by flow cytometry (anti-human CD274, clone 29E.2A3, Biolegend). (C) Cells were fixed/permeabilized, stained with anti-PD-L1 antibody and DAPI, and imaged using Zeiss LSM 880. D1= day 1; D5= day 5; MOI = multiplicity of infection.
IFNγ induces the upregulation of PD-L1 expression and/or increases cytosol to cell surface translocation of PD-L1 in human PDACs
Since OV infection can induce the release of proinflammatory cytokines such as interferons (IFNs) that play an important role in anti-viral response, [31] the effects of their subsets, IFNα, IFNβ, and IFNγ were evaluated on PD-L1 expression. IFNα and IFNβ in the dose range of 1 to 10 ng/mL only affected PD-L1 translocation onto the cellular surface in BxPC-3 but did not affect this translocation on AsPC-1 cells (Supplementary Fig. 1). However, IFNγ significantly increased PD-L1 translocation onto the cell surface from the cytosol in AsPC-1 cells without the change of total PD-L1 protein expression (Fig. 3A and 3B). For BxPC-3 cells, both upregulation of PD-L1 and increased translocation onto the cell surface were observed by distinguishing surface staining and intracellular staining with flow cytometry (Fig. 3A and 3B). The translocation of PD-L1 onto the cell surface of AsPC-1 and translocation/upregulation of PD-L1 in BxPC-3 cells following IFNγ treatment was also observed using confocal microscopy (Fig. 3C). These results demonstrate that IFNγ induces the upregulation of PD-L1 expression and/or the increased translocation of PD-L1 on PDAC cell lines.
Figure 3. IFNγ induces PD-L1 translocation/upregulation from the cytosol onto the surface of human PDAC cell lines.
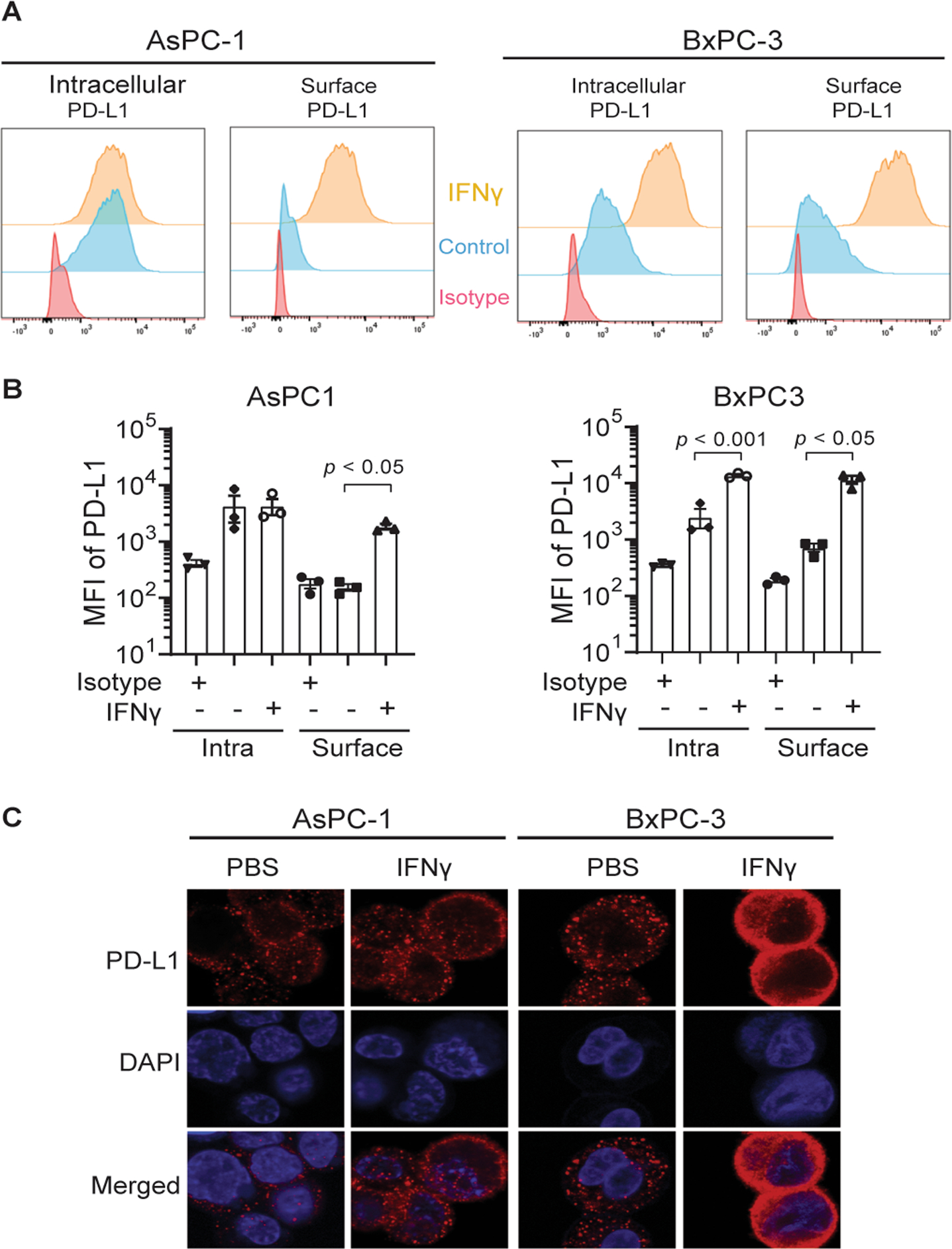
(A, B) Human pancreatic carcinoma cell lines AsPC-1 and BxPC-3 were treated with IFNγ (10 ng/mL) for 3 days and analyzed by flow cytometry for intracellular and surface PD-L1 expression (anti-human PD-L1/CD274, clone 29E.2A3, Biolegend). (C) Cells were also fixed/permeabilized, stained with anti-PD-L1/CD274 antibody and DAPI, and imaged using Zeiss LSM 880. Surface = Surface staining; Intra = Intracellular staining; n = 3.
CF33-hNIS-antiPDL1 infection of AsPC-1 and BxPC-3 cells leads to anti-PD-L1 antibody production within 60 minutes
Western blot analysis using a FLAG tag (DDDDK), anti-PD-L1 antibody, showed the presence of anti-PD-L1 protein as early as 60 min following infection with CF33-hNIS-antiPDL1 in both AsPC-1 and BxPC-3 cells and this expression continued to increase up to 360 min (Fig. 4A). Furthermore, confocal immunofluorescence microscopy showed hNIS (human sodium/iodide symporter) and PD-L1 antibody (FLAG tag) expression at 360 min post CF33-hNIS-antiPDL1 infection (MOI = 3). hNIS protein was observed following infection of both CF33-hNIS-Δ and CF33-hNIS-antiPDL1 at 360 min, while anti-PD-L1 expression was observed only following infection with CF33-hNIS-antiPDL1 (Fig. 4B). Cell culture supernatants and lysates were collected and blotted with anti-FLAG tag antibody at 360 min post virus infection and anti-PD-L1 antibodies were detected in both following CF33-hNIS-antiPDL1 infection (Fig. 4C). These results reveal the time-dependent production of virus-delivered anti-PD-L1 antibody and hNIS in situ.
Figure 4. Expression of anti-PD-L1 antibody encoded by CF33-hNIS-antiPDL1 in PDACs over time.
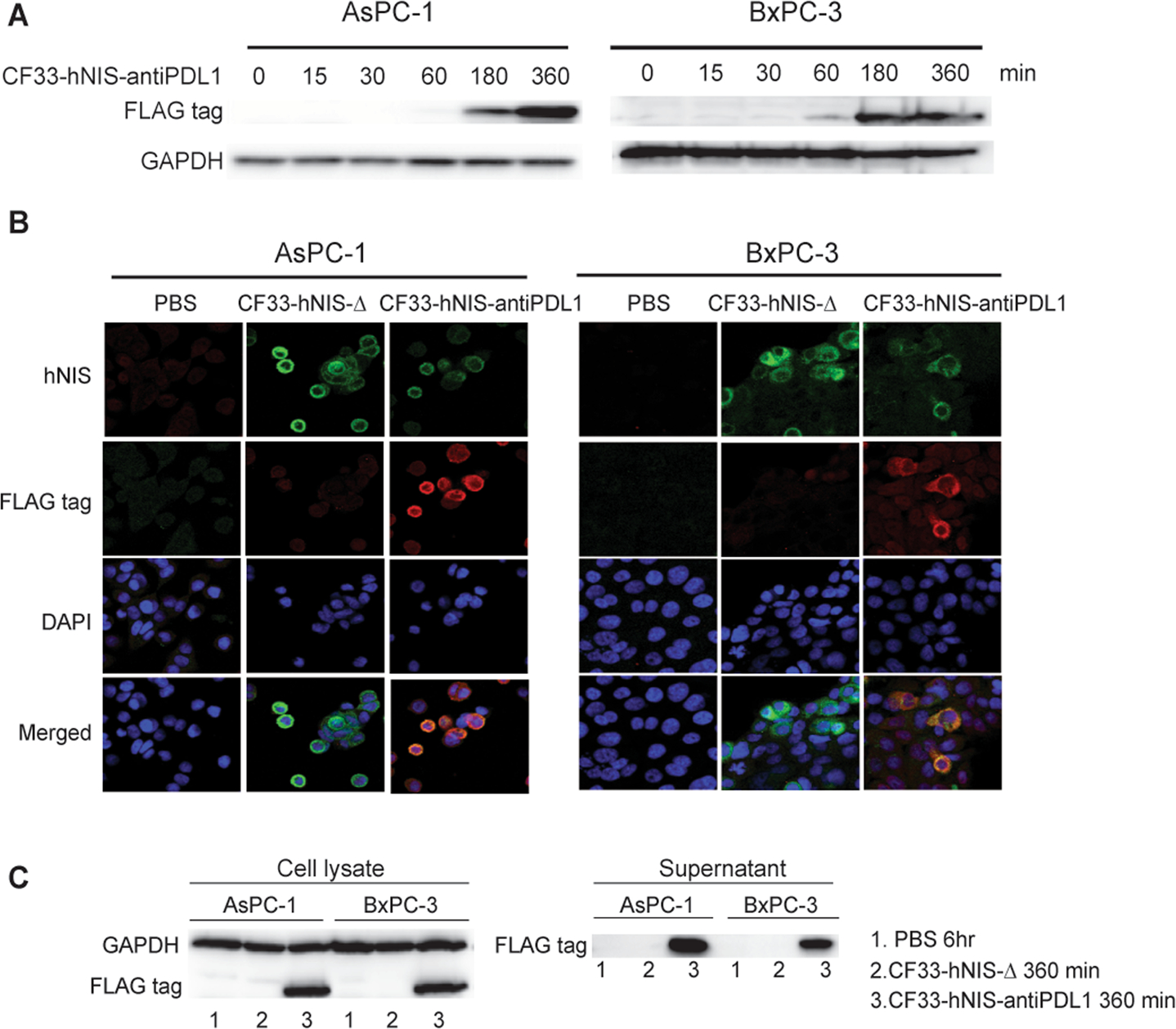
AsPC-1 and BxPC-3 cell lines were treated with CF33-hNIS-antiPDL1 (MOI = 3) and evaluated at 0-, 15-, 30-, 60-, 180- and 360-min time points. (A) Virus encoded-anti-PD-L1 antibody (FLAG tag) in cell lysate was detected using Western blot. (B) Virus encoded-hNIS and anti-PD-L1 antibody (FLAG tag) were observed using Zeiss LSM 880. (C) Virus encoded-anti-PD-L1 (FLAG tag) was detected using Western blot from both cell lysates and supernatants at 360 min following infection with the viruses.
Anti-PD-L1 antibodies encoded by the virus block virus- and IFNγ-induced surface PD-L1
By 18 h after infection, the anti-PD-L1 antibody encoded by CF33-hNIS-antiPDL1 blocked virus-induced cell surface expression of PD-L1 in the AsPC-1 and BxPC-3 cell lines (Fig. 5A). Compared to CF33-hNIS-Δ treated controls, the cells treated with CF33-hNIS-antiPDL1 demonstrated no cell surface PD-L1 expression by flow cytometric analysis in BxPC-3 cells which express a high level of surface PD-L1 (both p < 0.01). Immunofluorescence microscopy showed that virally induced PD-L1 was blocked by CF33-hNIS-antiPDL1 secreted anti-PD-L1 antibody in the BxPC-3 cells (Fig.5B).
Figure 5. Anti-PD-L1 antibody encoded by the virus blocks CF33-hNIS-Δ-induced surface PD-L1/CD274 binding of PDACs.
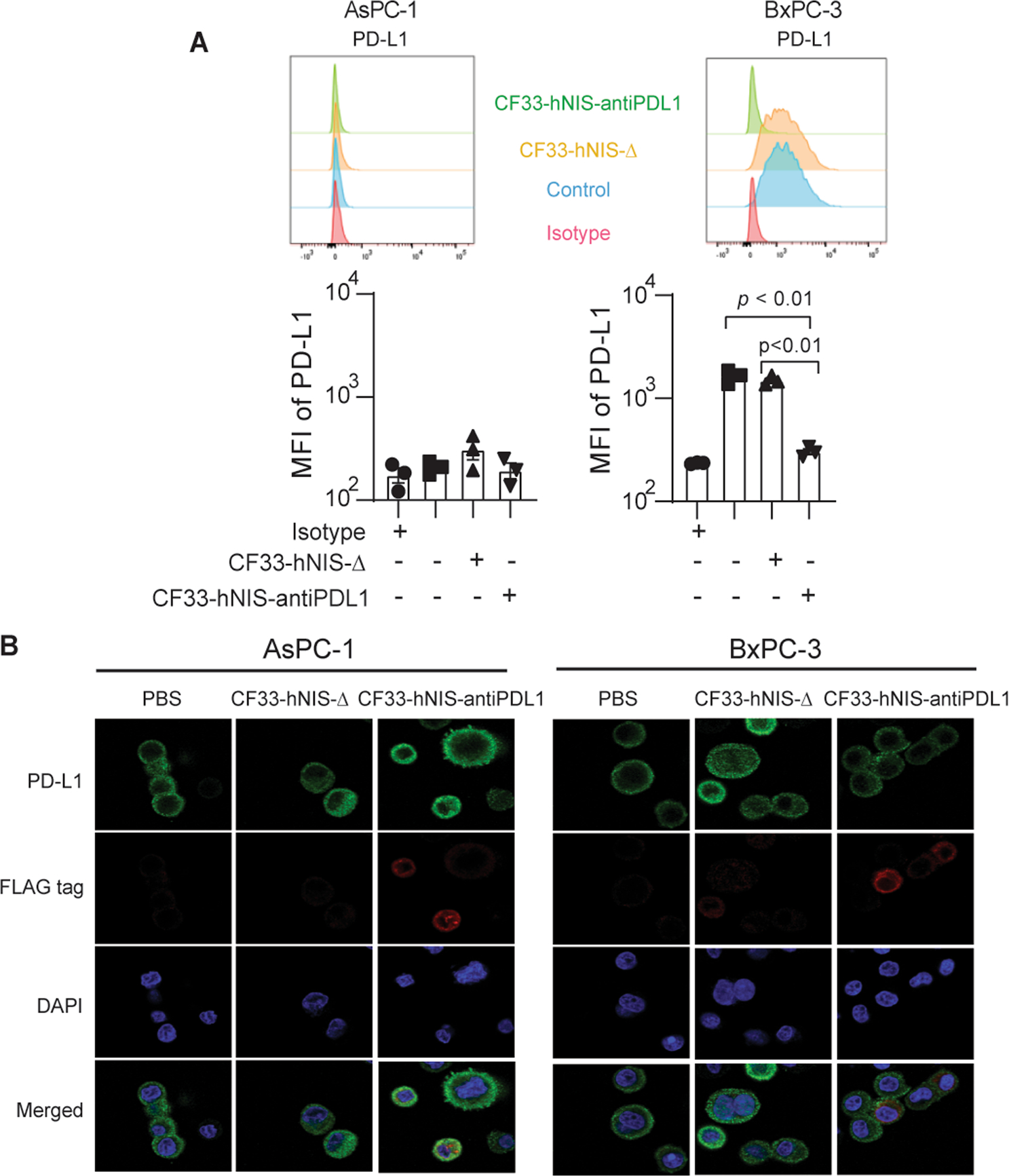
AsPC-1 and BxPC-3 cell lines were treated with CF33-hNIS-Δ or CF33-hNIS-antiPDL1 (MOI = 3) for 18 h. (A) PD-L1 mean fluorescence intensities (MFI) were observed by using flow cytometry or (B) with a Zeiss LSM 880. All experiments were performed in triplicate.
For IFNγ-induced PD-L1 translocation/upregulation, surface PD-L1 was blocked at 18 h post CF33-hNIS-antiPDL1 infection in both AsPC-1 and BxPC-3 cell lines (Fig. 6A and Fig 6B, all p < 0.01). Blockade bioassay of surface PD-L1 binding was analyzed using flow cytometry in the presence of an exogenous anti-PD-L1 antibody. The result showed that the concentration of anti-PD-L1 antibody in the supernatant was almost equal to 10 μg/mL compared to exogenous anti-PD-L1 antibody concentration (anti-PD-L1 mAb, Clone# 29E.2A3, Biolegend) (Fig. 6B). At the same time point of 18 h, immunofluorescence images correlated with flow cytometry results and did not show any free PD-L1 expression on the PDAC cell surfaces (Fig. 6C). These results thus show that CF33-hNIS-antiPDL1 can produce in situ functional blocking anti-PD-L1 antibody to bind the cell surface PD-L1 on PDAC cell surfaces.
Figure 6. Virus-encoded anti-PD-L1 antibody blocks IFNγ-induced surface PD-L1 binding of PDACs.
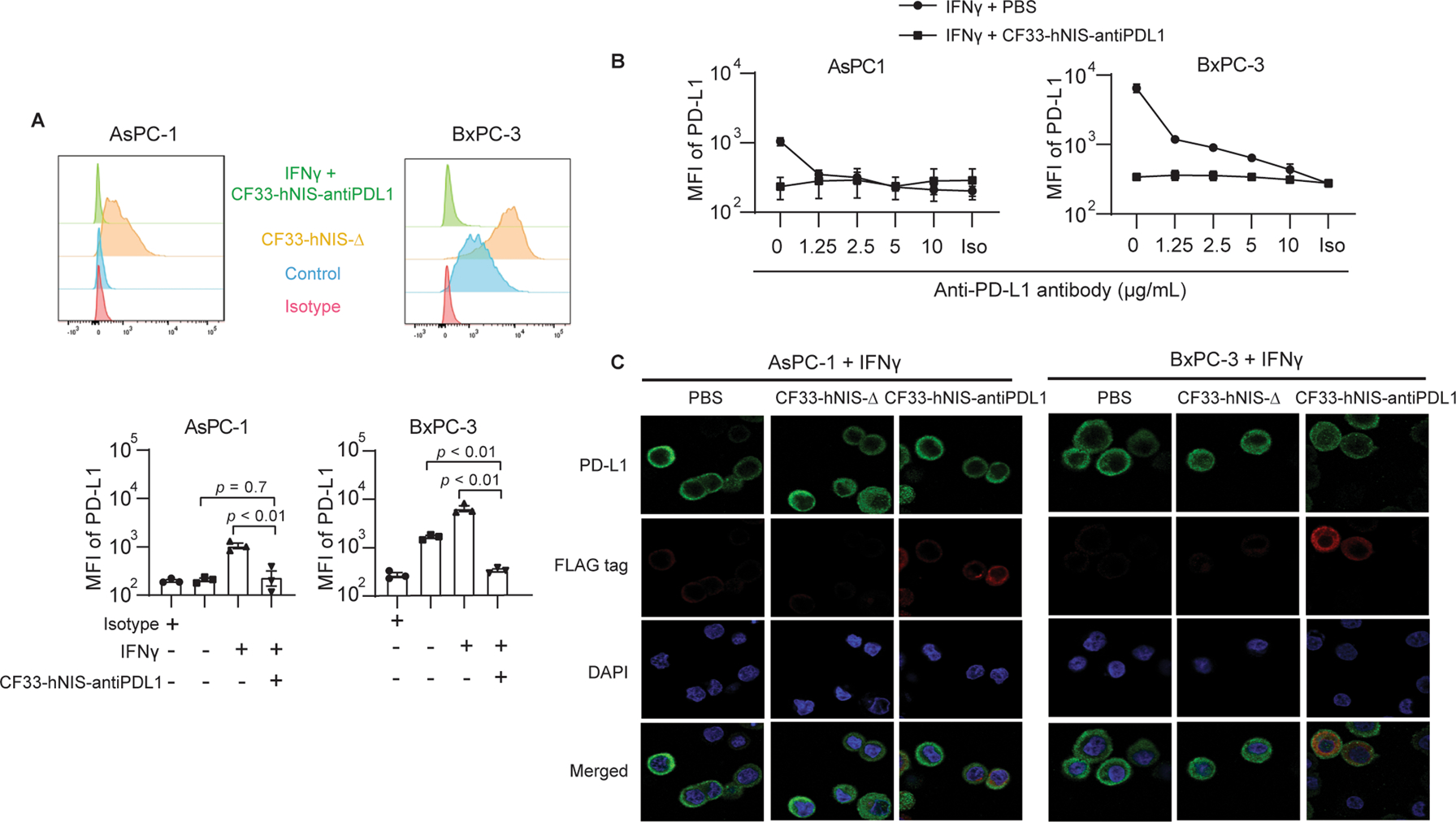
AsPC-1 and BxPC-3 cell lines were treated with IFNγ (10 ng/mL) or IFNγ (10 ng/mL) + CF33-hNIS-antiPDL1 (MOI = 3) for 18 h. (A) PD-L1 mean fluorescence intensities (MFI) were observed by using flow cytometry. (B) Blockade bioassay of surface PD-L1 binding was performed and analyzed by flow cytometry in the presence of exogenous anti-PD-L1 antibody and (C) observed by immunofluorescence microscopy with Zeiss LSM 880.
CF33-hNIS-antiPDL1 induces granzyme B and perforin release in activated T cells
In BxPC-3 cells cocultured with activated T cells, we observed significant changes in the cellular subsets following 5 days of treatment with CF33-hNIS-antiPDL1 (MOI = 3). Greater cytotoxicity was seen in the cocultured group treated with IOV compared to controls without IOV treatment (Fig. 7A). Among the immune cell subsets of cocultured cells, CD4+ T cell percentage significantly increased (p = 0.03) with a trend toward a significant increase in CD8+ T cell percentage (p = 0.15) (Fig. 7B). Flow cytometric analysis showed that PD-L1 on BxPC-3 cells treated with CF33-hNIS-antiPDL1 was completely blocked by virus-encoded anti-PD-L1 antibody compared to the control group which showed high cell surface expression of PD-L1 (Fig. 7C and Fig. 7D, p < 0.05). Furthermore, under coculture conditions, CF33-hNIS-antiPDL1 treatment led to a significant increase in the production of granzyme B and preventing virus-induced decrease of perforin release - the two most important factors of activated CD8+ T cells in tumor cell cytotoxicity (Fig. 7E). It is noted that CF33-GFP itself significantly decreases perforin release (p < 0.05), but CF33-hNIS-antiPDL1 can prevent it. These results suggest that CF33-hNIS-antiPDL1 can enhance anti-tumor T cell response by priming PDAC cells that escape the initial direct oncolysis of the virus.
Figure 7. CF33-hNIS-antiPDL1 induces BxPC-3 cell killing and increases T cell granzyme B and perforin release in coculture with activated T cells in vitro.
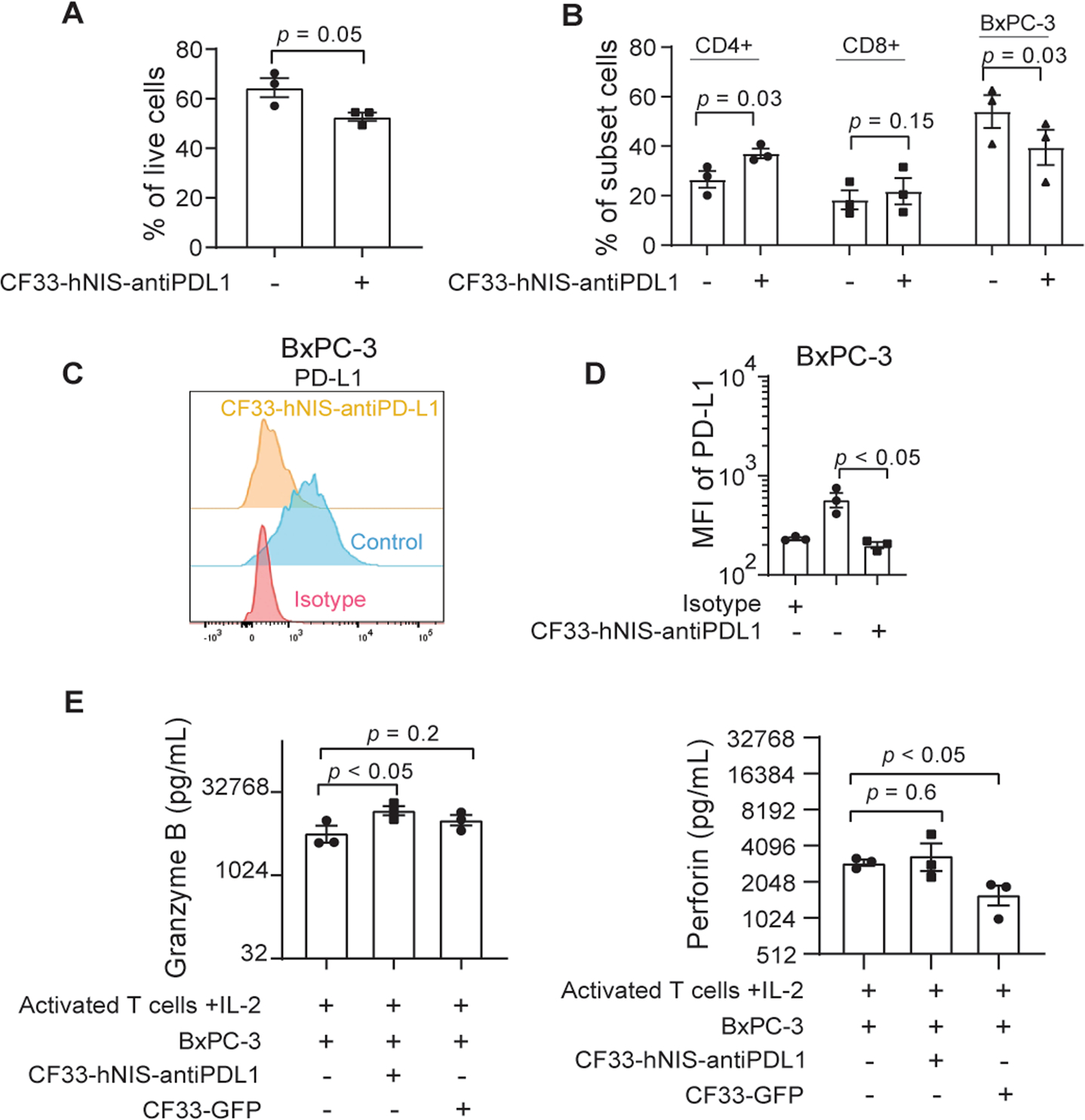
BxPC-3 cells cocultured with activated T cells + IL-2 were treated with CF33-hNIS-antiPDL1 (MOI = 3) or PBS (control) for 5 days. Cells were harvested and analyzed by flow cytometry. Percentage of (A) total live cells and (B) cell subsets are shown. (C) CD274 expression levels were detected using an anti-CD274 antibody and shown as a histogram (representative of three independent experiments), and (D) mean fluorescence intensities (MFI). (E) Granzyme B and perforin were measured in the supernatant of activated T cells cocultured with BxPC-3 in the presence or absence of CF33-hNIS-antiPDL1 for 5 days. (n = 3, MOI = 3, CF33-GFP was used as a virus control).
CD274 mRNA expression patterns vary in human PDACs
Lastly, we determined the clinical significance of PD-L1 expression patterns in PDAC. We evaluated the differences in expression levels of PD-L1 (CD274) between normal and tumor samples by analyzing mRNA levels of CD274 comparing 45 matched adjacent non-tumor vs PDAC samples. Results indicate significantly higher expression of CD274 in tumor samples with fold change = 1.46 (log2); p = 5.059e-06 compared to normal samples (Supplementary Fig.2A). RNA-seq human genomics deep sequencing analysis of 167 TCGA pancreatic cancer patients showed that high levels of CD274 mRNA-seq expression correlated with poor survival outcomes, with Logrank test p = 0.092 consistent with the current understanding of PD-L1 expression levels in most solid tumors (Supplementary Fig. 2B) [ref.32].
DISCUSSION
In this study, we provide key insights into the multimodal cancer-killing effectiveness of CF33-hNIS-antiPDL1 in vitro. We show for the first time, the mechanisms by which dynamic immune modulation and check-point inhibition of PD-L1 by CF33-hNIS-antiPDL1 lead to the elimination of PDAC cells that escape initial viral oncolysis. We demonstrate that during the initial period of viral oncolytic activity, CF33-IOVs increase the translocation of PD-L1 to PDAC cell surface and that viral replication and transgene expression of CF33-hNIS-antiPDL1 delivers a sufficient amount of functional anti-PD-L1 antibody to inhibit the PD-L1 induced immunosuppressive state. Coculture experiments of BxPC-3 and activated T cells with CF33-hNIS-antiPDL1 virus, showed that virus-encoded anti-PD-L1 effectively inhibited PD-L1 binding on BxPC-3 cells and increased granzyme B level, and prevented virus-induced decrease of perforin release. Granzyme B and perforin are released using the granule exocytosis pathway and play an important role in activating CD8+ T cells to kill cancer cells. Perforin is a granule protein that promotes granzyme B delivery to the tumor cell cytosol and mediates target cell apoptosis, primarily via caspase activation [33, 34]. CF33-hNIS-antiPDL1 could prime PDAC for time-dependent enhanced anti-tumor T cell activation as a single agent OV for effective multimodal cancer destruction.
CF33-hNIS-antiPDL1 is a novel chimeric IOV that combines the anti-tumor effects of targeted viral oncolysis and ICI by the local delivery of anti-PD-L1 antibody. CF33-based IOVs have demonstrated robust and exclusive oncolytic activity against PDAC in both immunosuppressed and immunocompetent preclinical small animal models [23]. In vitro, CF33-hNIS (CF33 modified with single-gene deletion and insertion of the human sodium iodide symporter transgene), CF33-hNIS-ΔF14.5, and CF33-hNIS-antiPDL1 cause oncolysis in nearly 50% of PDAC cells by 24 h and maximum cancer cell killing within 5 to 8 days [23]. Like other OVs, repeat dosing of CF33-hNIS in immunocompetent models decreased the effectiveness of subsequent doses due to the host’s antiviral immunity, whereas the combined CF33-hNIS plus exogenous anti-PD-L1, increased tumor destruction compared with either agent alone in syngeneic mouse models of triple-negative breast cancer [24]. Safety and effectiveness of CF33-hNIS-antiPDL1 in immunocompromised mouse models bearing subcutaneous and peritoneal PDAC tumors were also previously established [23] to demonstrate that intraperitoneal delivery of CF33-hNIS-antiPDL1 could target both subcutaneous and peritoneal tumors and improved survival over intravenous IOV and PBS treatments. The understanding of the immune mechanisms that modulate the effectiveness of CF33-IOVs in this study adds valuable insight that will guide the timing and route of therapy in designing both preclinical studies and clinical trials.
Immune quiescence, excessive desmoplasia, and the lack of consensus expression of PD-L1 in PDAC tumors continue to be barriers to effective immune therapy in PDAC patients [15, 35]. PD-L1 is the protein encoded by the CD274 gene present on many cancer and immune cells. It is known to suppress the host’s immune response to cancer by binding to its receptors and suppressing T cell migration, proliferation, and secretion of cytotoxic mediators [13, 14]. Our analysis of the TCGA and GEO dataset of PDAC patients showed that tumor cells overexpressing the CD274 mRNA correlated with poor survival in these patients [Supplementary Figure 2B] [ref.29]. ICIs such as pembrolizumab (anti-PD-1), nivolumab (anti-PD-1), and atezolizumab (anti-PD-L1) which block PD-L1 mediated T cell escape of solid tumors have recently achieved remarkable responses in a variety of cancers [13, 36]. Unlike immunogenic tumors such as malignant melanoma and lung cancer, PDAC tumors have not responded to single-agent anti-PD-L1 therapy, and inhibiting the PD-1/PD-L1 interaction has not shown a survival benefit in PDAC patients. For example, in Phase I clinical trials, the monoclonal anti-PD-L1 antibody, BMS-93655 failed to demonstrate any clinically relevant tumor response in patients with advanced pancreatic cancer [13]. Our results suggest that primary cytosolic expression of PD-L1 in PDAC cells contributes to the failure of single-agent or duel agent immune checkpoint inhibition and increasing cell surface PD-L1 expression is needed to effectively target these otherwise immune quiescent cells. Thus, therapeutic strategies that combine OV-ICI to increase PD-L1 cell surface expression by OV infection to prime PDAC cells to take over the immunosuppressive state for immune checkpoint inhibition can be applied to effectively target and kill PDAC.
Such combination strategies show encouraging results in both preclinical models, as well as clinical trials, and the majority of clinical trials for OVs are now conducted as combination therapies with one or two ICIs [37]. For example, the efficacy of combining the Measles virus with anti-CTLA-4 and PD-L1 antibody was assessed in a xenograft model of human melanoma. Measles virus treatment groups showed a significant tumor reduction and survival benefit [38]. The combination of T-VEC with ipilimumab and pembrolizumab has significant preclinical support and has been evaluated in clinical trials for patients with advanced melanoma [39]. In a Phase Ib clinical trial, T-VEC combined with ipilimumab for patients with stage IIIB–IVM1c melanoma shows a superior efficacy and safety profile than a single agent treatment [39]. However, very few trials include patients with PDAC.
The main shortcoming of this study is the lack of in vivo studies in immunocompetent models of PDAC. We limited our study to in vitro experiments to better define the immune effects of the IOV at the cellular level including the time points that would best serve to optimize future in vivo experiments. Moreover, due to the dynamic immune changes within the patient and in the TME, in vitro experiments and even in vivo animal models cannot comprehensively represent clinically relevant time points. Additional in vivo experiments and clinical investigations are required to determine how the dynamic IOV-ICI-cancer cell interplay results in a clinically relevant improvement in PDAC patients.
In summary, our study shows that the CF33-hNIS-antiPDL1 virus can prime immune quiescent PDAC cells for anti-PD-L1 therapy. Its synergistic action of viral oncolysis, upregulation of cell surface PD-L1, effective immune checkpoint blockade, and increase in anti-tumor T cell activity provides support for CF33-hNIS-antiPDL1 as a single-agent IOV-ICI combination therapy against otherwise therapy-resistant tumors such as PDAC.
Supplementary Material
Acknowledgements:
The authors are grateful to Imugene Limited for providing approval to use their licensed oncolytic virus CF33 in the studies reported in this paper. Research reported in this publication included work performed in the Pathology Core, Flow Cytometry Core, Light Microscopy Core, and Integrative Genomics and Bioinformatics core facilities supported by the National Cancer Institute of the National Institutes of Health under grant number P30CA033572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors would also like to thank Dr. Jinhui Wang in Integrative Genomics Core, Lucy Brown in Flow Cytometry core, and Dr. Brian Armstrong in the Light Microscopy Core of City of Hope for supporting the work.
The authors would like to thank Byungwook Kim, Martha Magallanes, Seonah Kang, and Dr. Maria Hahn in our laboratory and Dr. Chunyan Zhang in Department of Immuno-Oncology for technical support, and Supriya Deshpande, PhD for assistance with manuscript editing.
Drs. Warner and Chaurasiya are supported through the generosity of Natalie and David Roberts. These authors wish to thank them for their philanthropy. The authors would also like to thank Samuel Kuo and Grace Liu of Samson Holding, Ltd. for their generosity.
Funding:
Department of Defense E01 Award W81XWH-19-1-0225 (CA180425).
Footnotes
Conflict of interest: The authors declare no conflict of interest.
Availability of data and material: All data relevant to the study are provided in the article or uploaded as supplementary information.
REFERENCES
- 1.Mizrahi JD, Surana R, Valle JW, Shroff RT. Pancreatic cancer. Lancet 2020; 395(10242): 2008–2020. [DOI] [PubMed] [Google Scholar]
- 2.Tempero MA. NCCN Guidelines Updates: Pancreatic Cancer. J Natl Compr Canc Netw 2019; 17(5.5): 603–605. [DOI] [PubMed] [Google Scholar]
- 3.Moore A, Donahue T. Pancreatic Cancer. JAMA 2019; 322(14): 1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang L, Jansen L, Balavarca Y, Babaei M, van der Geest L, Lemmens V et al. Stratified survival of resected and overall pancreatic cancer patients in Europe and the USA in the early twenty-first century: a large, international population-based study. BMC Med 2018; 16(1): 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Balsano R, Tommasi C, Garajova I. State of the Art for Metastatic Pancreatic Cancer Treatment: Where Are We Now? Anticancer Res 2019; 39(7): 3405–3412. [DOI] [PubMed] [Google Scholar]
- 6.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 2014; 74(11): 2913–21. [DOI] [PubMed] [Google Scholar]
- 7.Sunami Y, Kleeff J. Immunotherapy of pancreatic cancer. Progress in molecular biology and translational science 2019; 164: 189–216. [DOI] [PubMed] [Google Scholar]
- 8.Singh RR, O’Reilly EM. New Treatment Strategies for Metastatic Pancreatic Ductal Adenocarcinoma. Drugs 2020; 80(7): 647–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanmamed MF, Chen L. A Paradigm Shift in Cancer Immunotherapy: From Enhancement to Normalization. Cell 2018; 175(2): 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 2015; 27(4): 450–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kyi C, Postow MA. Immune checkpoint inhibitor combinations in solid tumors: opportunities and challenges. Immunotherapy 2016; 8(7): 821–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feng M, Xiong G, Cao Z, Yang G, Zheng S, Song X et al. PD-1/PD-L1 and immunotherapy for pancreatic cancer. Cancer Lett 2017; 407: 57–65. [DOI] [PubMed] [Google Scholar]
- 13.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012; 366(26): 2455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014; 515(7528): 563–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barrueto L, Caminero F, Cash L, Makris C, Lamichhane P, Deshmukh RR. Resistance to Checkpoint Inhibition in Cancer Immunotherapy. Transl Oncol 2020; 13(3): 100738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dougall WC, Kurtulus S, Smyth MJ, Anderson AC. TIGIT and CD96: new checkpoint receptor targets for cancer immunotherapy. Immunol Rev 2017; 276(1): 112–120. [DOI] [PubMed] [Google Scholar]
- 17.Dempke WCM, Fenchel K, Uciechowski P, Dale SP. Second- and third-generation drugs for immuno-oncology treatment-The more the better? Eur J Cancer 2017; 74: 55–72. [DOI] [PubMed] [Google Scholar]
- 18.Zeng S, Pottler M, Lan B, Grutzmann R, Pilarsky C, Yang H. Chemoresistance in Pancreatic Cancer. Int J Mol Sci 2019; 20(18):4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Balachandran VP, Beatty GL, Dougan SK. Broadening the Impact of Immunotherapy to Pancreatic Cancer: Challenges and Opportunities. Gastroenterology 2019; 156(7): 2056–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sivanandam V, LaRocca CJ, Chen NG, Fong Y, Warner SG. Oncolytic Viruses and Immune Checkpoint Inhibition: The Best of Both Worlds. Mol Ther Oncolytics 2019; 13: 93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vijayakumar G, McCroskery S, Palese P. Engineering Newcastle Disease Virus as an Oncolytic Vector for Intratumoral Delivery of Immune Checkpoint Inhibitors and Immunocytokines. J Virol 2020; 94(3)e01677–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Z, Ravindranathan R, Kalinski P, Guo ZS, Bartlett DL. Rational combination of oncolytic vaccinia virus and PD-L1 blockade works synergistically to enhance therapeutic efficacy. Nat Commun 2017; 8: 14754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woo Y, Zhang Z, Yang A, Chaurasiya S, Park AK, Lu J et al. Novel Chimeric Immuno-Oncolytic Virus CF33-hNIS-antiPDL1 for the Treatment of Pancreatic Cancer. J Am Coll Surg 2020; 230(4): 709–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chaurasiya S, Yang A, Kang S, Lu J, Kim SI, Park AK et al. Oncolytic poxvirus CF33-hNIS-DeltaF14.5 favorably modulates tumor immune microenvironment and works synergistically with anti-PD-L1 antibody in a triple-negative breast cancer model. Oncoimmunology 2020; 9(1): 1729300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Warner SG, Kim SI, Chaurasiya S, O’Leary MP, Lu J, Sivanandam V et al. A Novel Chimeric Poxvirus Encoding hNIS Is Tumor-Tropic, Imageable, and Synergistic with Radioiodine to Sustain Colon Cancer Regression. Mol Ther Oncolytics 2019; 13: 82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chaurasiya S, Chen NG, Lu J, Martin N, Shen Y, Kim SI et al. A chimeric poxvirus with J2R (thymidine kinase) deletion shows safety and anti-tumor activity in lung cancer models. Cancer Gene Ther 2020; 27(3–4): 125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Z, Shively JE. Generation of novel bone forming cells (monoosteophils) from the cathelicidin-derived peptide LL-37 treated monocytes. PLoS One 2010; 5(11): e13985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Z, Le K, La Placa D, Armstrong B, Miller MM, Shively JE. CXCR2 specific endocytosis of immunomodulatory peptide LL-37 in human monocytes and formation of LL-37 positive large vesicles in differentiated monoosteophils. Bone Rep 2020; 12: 100237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang S, He P, Wang J, Schetter A, Tang W, Funamizu N et al. A Novel MIF Signaling Pathway Drives the Malignant Character of Pancreatic Cancer by Targeting NR3C2. Cancer Res 2016; 76(13): 3838–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018; 173(2): 400–416 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levy DE, Marie IJ, Durbin JE. Induction and function of type I and III interferon in response to viral infection. Curr Opin Virol 2011; 1(6): 476–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016; 531(7592): 47–52. [DOI] [PubMed] [Google Scholar]
- 33.Cullen SP, Brunet M, Martin SJ. Granzymes in cancer and immunity. Cell Death Differ 2010; 17(4): 616–23. [DOI] [PubMed] [Google Scholar]
- 34.Brennan AJ, Chia J, Trapani JA, Voskoboinik I. Perforin deficiency and susceptibility to cancer. Cell Death Differ 2010; 17(4): 607–15. [DOI] [PubMed] [Google Scholar]
- 35.Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov 2019; 18(3): 197–218. [DOI] [PubMed] [Google Scholar]
- 36.Atkins MB, Larkin J. Immunotherapy Combined or Sequenced With Targeted Therapy in the Treatment of Solid Tumors: Current Perspectives. J Natl Cancer Inst 2016; 108(6): djv414. [DOI] [PubMed] [Google Scholar]
- 37.Russell L, Peng KW, Russell SJ, Diaz RM. Oncolytic Viruses: Priming Time for Cancer Immunotherapy. BioDrugs 2019; 33(5): 485–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Engeland CE, Grossardt C, Veinalde R, Bossow S, Lutz D, Kaufmann JK et al. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol Ther 2014; 22(11): 1949–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Puzanov I, Milhem MM, Minor D, Hamid O, Li A, Chen L et al. Talimogene Laherparepvec in Combination With Ipilimumab in Previously Untreated, Unresectable Stage IIIB-IV Melanoma. J Clin Oncol 2016; 34(22): 2619–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.