

Abstract
G-quadruplexes are motifs found in DNA and RNA that can fold into tertiary structures. Until now, they have been studied experimentally mainly in humans and a few other species. Recently, predictions have been made with bacterial and archaeal genomes. Nevertheless, a global comparison of predicted G4s (pG4s) across and within the three living kingdoms has not been addressed. In this study, we aimed to predict G4s in genes and transcripts of all kingdoms of living organisms and investigated the differences in their distributions. The relation of the predictions with GC content was studied. It appears that GC content is not the only parameter impacting G4 predictions and abundance. The distribution of pG4 densities varies depending on the class of transcripts and the group of species. Indeed, we have observed that, in coding transcripts, there are more predicted G4s than expected for eukaryotes but not for archaea and bacteria, while in noncoding transcripts, there are as many or fewer predicted G4s in all species groups. We even noticed that some species with the same GC content presented different pG4 profiles. For instance, Leishmania major and Chlamydomonas reinhardtii both have 60% of GC content, but the former has a pG4 density of 0.07 and the latter 1.16.
INTRODUCTION
G-quadruplexes (G4s) are noncanonical tertiary structures that use Hoogsteen base pairing between four guanines to form G-tracks that stack on top of each other (1,2). G4s can fold into DNA and RNA. The G4 structure is highly stable, but the presence of a cation could improve its stability. The monocation with the highest stabilization potential is potassium (K+), while lithium (Li+) has a very poor stabilization potential (3). These two monocation conditions are often used to experimentally detect G4s, revealing the folding state of the sequence into a G4. Some G4s can still fold under the Li+ condition, even if they are less stable than under the K+ condition. Thus, other methods can be required to further confirm the folding of a G4 (for more information on G4 detection, see (4)).
G4s occur in G-rich sequences, often under the form of well-defined motifs. The first motif discovered is known as canonical G4 and follows the pattern GxN1-7GxN1-7GxN1-7Gx where x is equal or higher than 3 and N are any nucleotides. This motif has been used to build many prediction tools like Quadparser (5) and QGRSmapper (6). These tools yielded an initial broad view of the predicted G4 (pG4) sequences in the human genome (7). Over the years, G4s that did not fit into the definition of the canonical motif have been discovered. They are called noncanonical G4s. Four main types of noncanonical G4s have been discovered so far: loop >7 nucleotides, bulge in G-tracks, G-track of only 2 guanines, and a quartet with 3 guanines and another nucleotide to compensate for the last guanine (8–11). Later, it came to light that a low-cytosine environment is required for G4 folding (12). Indeed, there is a competition between the Watson and Crick pairing (G–C) and Hoogsteen pairing (G–G), which leads to unstable G4s. These discoveries helped improve prediction tools by modulating G4 motif parameters (13) or by using the GC content on short windows. Recently, new approaches have been developed using machine-learning approaches or by combining the motif and GC content (14–16).
Not only have new types of G4s been discovered over the years, but their diverse functions in the cellular cycle and their involvement in numerous diseases have also been reported. G4s can impact viral infections (17–20), cancer (by modulating oncogene expression) (21–23), and neurodegenerative diseases such as Alzheimer’s and Parkinson’s (for a recent review on the subject, see (24–27)). In coding transcripts, they can allow the formation of internal ribosome entry site (IRES) structures, which produce alternative proteins (28) or interfere in the polyadenylation process (29–31). In noncoding transcripts, many functions still need to be unraveled. Some are already known to impact miRNA processing (32–35).
As a consequence of the diverse and significant roles played by G4 structures, knowing their distribution in genomes and transcriptomes from all species’ kingdoms is important. The human species is the first to have been extensively studied using prediction tools. Some other species have been the focus of in silico genome-wide studies such as Saccharomyces cerevisiae and related species, Escherichia coli, Dictyostelium discoideum and many others (36–38). The development of experimental methods such as G4-seq, rG4-seq, and G4 ChIP-seq (39–41) allowed genome-wide and transcriptome-wide detection of G4s, first in humans and then in a few other species. In recent years, some studies predicted G4s in the genomes of diverse species to check pG4 conservation, but only in one specific kingdom of life at a time: for eukaryotes (42–44), archaea (45) and bacteria (46–48). More recently, one study looked at pG4 coevolution with viruses and their host species (49). According to these studies, G4 distribution differs in prokaryotes and eukaryotes, which raises the question of why G4 distribution would be different in different groups of species? This observation might be due to the fact that each living kingdom possesses different GC-content signatures, which could account for the different G4 distributions, as their presence is highly connected to GC content. In this study, we predicted G4s in all living kingdoms, from their genes to their transcripts. Looking at the distribution in each species group and each transcript class; we have shown that pG4 distribution depends on GC content as well as other parameters.
MATERIALS AND METHODS
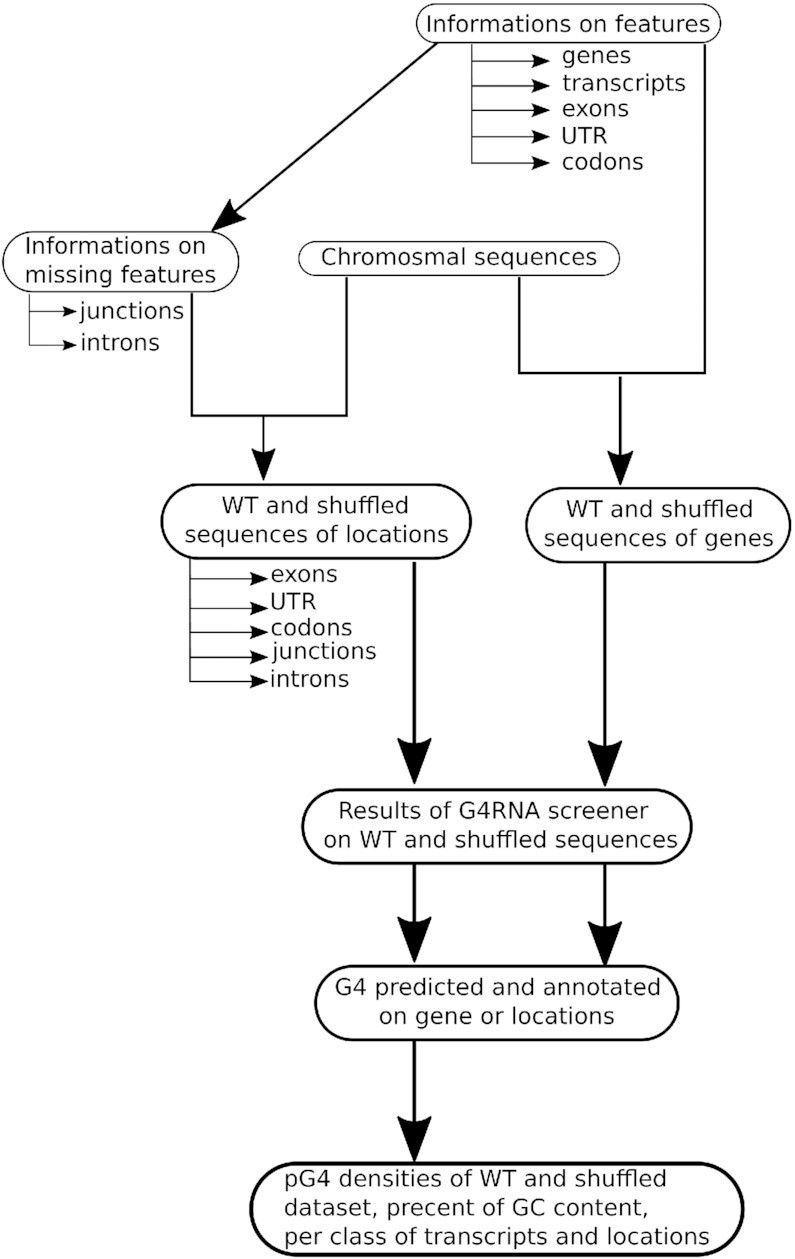
The methodological pipeline was performed using Snakemake, a workflow management system (50). Figure 1 presents the four steps in the pipeline. The first step consisted in retrieving data and generating sequences of all genes and genetic locations considered in this study for each species (see Figure 2). Location types are separated into two groups: segment locations (exons, introns, coding sequences (CDS), and untranslated regions (UTRs) and point locations (codons and junctions)), as shown in Figure 2. The second step was to predict G4s using the G4RNA screener predictor (14). The third step consisted in comparing the pG4 sets to experimental data to validate G4 prediction, when possible. In the fourth step, the pG4 densities for all types of locations and all species were computed. Based on these densities, descriptive statistics and statistical tests were carried out to analyze the results.
Figure 1.
Overview of the methodology.
Figure 2.

Schema of locations generated and used to predict pG4s. For point locations, 40 nucleotides upstream and downstream of the junctions or codon were also retrieved.
Step 1: Data retrieval and preprocessing
Gene-structure information was retrieved from release 46 of the Ensembl Compara database (51). This version brings together many species from all living kingdoms (eukaryotes, archaea and bacteria). Chromosome sequences were retrieved for all selected species (see Supplementary Table S1). Then, GTF files containing gene-structure information were used to generate sequences of all genes and genetic locations. In GTF files, some location—such as introns and junctions—are not annotated and were generated using exon information. All point locations (codons and junctions) were extended by adding 40 nucleotides upstream and downstream. The set of sequences thus generated is referred to as the WT dataset.
A control dataset was also generated in parallel with the WT sequences. For each WT sequence, a shuffled counterpart was generated by shuffling the order of nucleotides while preserving the composition in nucleotides (shown in Figure 2). The shuffled sequences were used to compute the expected pG4 number and densities. Ten runs of shuffled datasets were generated in order to compute an average number of shuffled pG4s and avoid biased results (see Supplementary Figure S1). Note that this control process by shuffling sequences might be less adequate for short locations such as point locations which are between 20 and 80 nucleotides long.
In our analysis, species trees are used to order species. These trees were built using super tree methods to combine several species trees from studies in references (52–56). The resulting trees are used as relation indicators between the species.
Some genome features were also analyzed. For these features, data were not retrieved via Ensembl. Microsatellite sequences were retrieved with MSDB (57). The predictions were made only on repeats >20 nt. The TREP database (58) was used for transposable elements. Lastly, centromere sequences were easily obtainable via NCBI but only for Homo sapiens and Saccharomyces cerevisiae. Other species centromere locations were available with NCBI, but the corresponding sequences contained a high number of unresolved nucleotides (N), which made prediction impossible.
It is important to mention that most of our sequences were retrieved from databases containing large amounts of information that is often updated and subject to correction. Our work aimed at looking at all three species kingdoms at the same time. In few years, more data might be available, mainly for noncoding transcripts. In that case, this study should be repeated to update our knowledge.
Step 2: G4 prediction
We extended our previous pipeline for pG4 prediction used on the human genome (59) to 61 species (see Table 1 and Supplementary Table S1 for more details). G4RNA screener (14) was used to predict G4s. This tool computes three scores for each candidate sequence that indicate the propensity of the sequence to fold into a G4. The first score—G4NN—is a score returned by an artificial neural network, which is trained based on data from a G4RNA database (60). The literature was manually curated to create this database, which contains human RNA G4s tested experimentally. These G4 sequences have been proven to fold into a G4 or not. Thereby, G4NN is a score of similarity with sequences known to fold into a G4 and its threshold is 0.5. The second score is G4Hunter (61), which puts positive weights on consecutive guanines and negative weights on consecutive cytosines. The threshold used for this score is 0.9. The last score—cGcC (62)—is a measure of competition between guanine and cytosine content, and it has a threshold of 4.5. Several tests were performed on human data to detect the default parameters of G4 screener, as described in Garant et al. (14). These tests could not be reproduced with other species because of the lack of experimentally validated data. Thus, we kept the same parameters and applied them to all species. Despite the potential bias in using the same parameters for all species, changing them arbitrarily without testing would only modify the number of false positive or false negatives, without any means to evaluate the bias.
Table 1.
Number of species for each kingdom and total number of species used in this study
| Kingdom | Number of species |
|---|---|
| Eukaryota | 25 |
| Archaea | 12 |
| Bacteria | 24 |
| Total | 61 |
G4RNA screener uses a sliding overlapping window system, in which windows are 60 nucleotides long and separated by a step of 10 nucleotides. Some locations can be <60 nucleotides, so a minimum length of 20 nucleotides was required to keep them. All positives overlapping windows were merged to generate sets of predicted G4 regions (pG4rs).
Step 3: Validation of G4 predictions by comparison with experimental data
To validate the prediction of G4s, the pG4r sets were compared to experimentally detected G4s from Marsico et al. (44). In this study, G4s from 12 species were detected genome-wide. There were nine species shared between these two studies. Marsico et al. (44) detected G4s in two conditions: K+, which is more physiological, and K+ with PDS (hereafter PDS condition) in which the G4s are highly stabilized. Experimentally detected G4s and pG4s were filtered to only keep G4s and pG4s in common genes, as well as to remove G4s in intergenic regions. Even if G4s are experimentally detected, G4seq is prone to errors. Some false positive G4s can be detected, such as some sequences which do not contain enough G to form a G4 but still give a signal, or on the contrary some stable G4s might fold transiently in Li+ condition and thus be missed due to the comparison Li+ / K+. Comparing the G4s predicted by our method with the G4s detected with G4seq allows to approximate the percent of true/false positive that might be generated. Yet, G4seq cannot be considered as a ground truth. But, to our knowledge, it is currently the best experimental genome-wide method for G4 detection with available predictions across multiple genomes. We used blastn (v2.6.0+) to compare the G4 sets from the Marsico et al. study and our pG4 sets used to map pG4s and G4s against a reference genome for each species (63). Overlapping hits from both datasets were considered as common G4s. Note that we could not compare G4s and pG4s simply by using their locations on genomes because of different genome assemblies in the two studies.
G4RNA screener was developed to predict RNA G4s, thus some pG4s might not be predicted in RNA, while they could fold in DNA. The rate of these false negatives can be partly estimated with the percent of G4s detected experimentally that do not match any pG4.
Step 4: Computation and comparison of pG4 distributions
There is a direct relationship between sequence length and the chances of predicting a pG4 in the sequence. To avoid this bias, pG4 numbers were normalized by the total length of the sequences in the type of location in which they were detected:
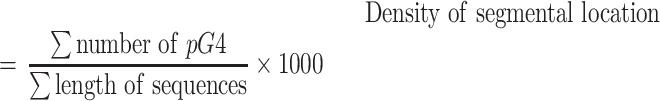 |
For point locations like junctions and codons—which have limited length—the density was computed with the number of locations in the type of location:
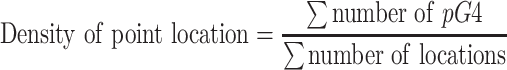 |
Densities were computed at different levels: for all genes, all transcripts (introns + exons locations), and each type of location. Densities were also computed for transcript classes and subclasses.
WT and shuffle densities were compared in two ways. Either by subtracting WT and shuffle densities, referred as densities differences. This allows for the estimation of the WT density without pG4s that would be forming by chance. This is done either by dividing them to get the relative differences between the two types of densities, which shows how many time pG4 are predicted more or less than by chance. In the latter case, a logarithm transformation was applied to the quotient. This gave a positive value when the WT density was higher than the shuffle density. Yet, for species with no pG4s in one of the density types, either a division by 0 cannot be done, so the species were removed, or the log returned a minus infinite value, which is not plottable, so species were removed.
Boxplots were used to compare distributions, and statistical tests were used to find significant differences between distributions. Densities did not often follow a normal distribution. Thus, the Mann–Whitney–Wilcoxon test was used in most cases. All graphics were generated using a Jupyter notebook script which is available on ‘github’ with all the other scripts developed in the methodological pipeline.
RESULTS AND DISCUSSIONS
Overall, our study provides in-depth insights on pG4 densities in genes, transcripts, and annotated coding locations. The prediction of G4s in WT datasets and shuffled datasets in diverse species points out that there are more or fewer pG4s depending on the GC content, but also on species’ kingdoms, transcript classes and locations. pG4s were first analyzed at the gene level, then at the level of whole transcriptomes and transcript classes: coding, long noncoding (longNC), short noncoding (shortNC) and pseudogene (see Figure 3A–F and Supplementary Figure S2). For each of these levels, the analysis started by looking at density differences (WT minus shuffled) to get an overview of density distribution. Next, the correlation between GC content and density differences was analyzed. Afterward, statistical differences were computed between all WT densities versus all shuffled densities of a species’ kingdom. Globally, densities were similar between genes, overall transcripts and coding transcripts, but different distribution profiles were found for longNC, shortNC and pseudogene transcripts (see Figure 3).
Figure 3.

pG4 densities differences in genes and all transcripts. (A–F) are pG4 densities in genes, all transcripts, coding, longNC, shortNC and pseudogene transcripts, respectively. All species kingdoms are shown in the figure with eukaryotes in triangles, bacteria in circles and archaea in squares. The densities shown in these dot plots are the WT densities minus the shuffle densities. If the density is over 0, the WT density is higher than the shuffled density.
Comparison of G4 prediction with G4seq data
For 9 species in our study, K+ G4s had already been experimentally detected in genes by Marsico et al. (44). In (44), the authors used G4seq, a high-throughput method which uses DNA polymerase stalling to find folded G4. G4seq improved a lot our knowledge on G4 existence and propensity in genomes. This method is of great value without being considered as a ground truth since errors in the G4 detection has been demonstrated (see (64)).
The number of pG4s detected in our study is similar to the numbers of K+ G4s detected experimentally (Figure 4), except for some species such as S. cerevisiae and Danio rerio, which yielded lower pG4 numbers. It would appear that all G4s of most species should be predictable.
Figure 4.

Results of the comparison between pG4s and G4s detected experimentally in the K+ or PDS condition. (A) Number of G4s for each dataset on a log scale. (B) and (C) Percent of G4s that mapped at the same location of the genome.
As a result of the detailed comparison between our datasets and the experimentally detected K+ G4s, our predictions were consistent with experimental data. All G4s were mapped against the human genome to retrieve overlapping hits, that is, G4s common to both studies. Surprisingly, there were few common G4s with the K+ condition. Since PDS is a ligand that stabilizes strongly G4s, we did not expect the overlap to be higher in K+ condition compared to K+ and PDS, but we expected to find a higher correspondence than ∼20% in most species. Yet, in almost all species, 67% of pG4s corresponded to experimental data with the K+ condition and the presence of PDS (which is a ligand that stabilizes G4 structures) (see Figure 4C). Thus only 37% of pG4s were false negatives. The results are satisfying given the fact that G4s are hard to predict and that G4NN is based on human data. On the other hand, only 20% of experimentally detected G4s were found in pG4s, which means that our method failed to predict many G4s detected experimentally. It highlights a limitation in our study’s predictive capacity, which potentially underestimates the number of pG4s. Many reasons can account for this. First, G4RNA screener is built to predict RNA G4s, and G4 structures are more constrained in RNA than DNA (65). Thus, some detected G4s might fold in genomes but not in transcripts. Second, the three scores are used in G4Screener to predict G4s in order to avoid false positives and thus increase the number of false negatives. Moreover, even if the high-throughput sequencing of DNA G4s is an important step forward and has been improved, it may still miss some G4s when compared to the Li+ condition and K+ condition in the absence or presence of PDS. Lastly, as discussed in (64), the G4-seq method produces false positive predictions in AT-rich sequences. For instance, in E. coli, which is AT rich (i.e. only 39% GC), G4seq detected a high rate of G4s, while G4RNA screener predicted low rates of pG4, which results in the lowest pG4 matching among all species.
In addition to past results, no bias was detected towards humans. A bias toward the human species was expected, as G4NN is built on human data. Indeed, E. coli, which is the most distant species from H. sapiens, got the lowest correspondence with detected G4s. Yet, this is the only bacterium present in the two studies. This result cannot be generalized to other species. In addition, more pG4s matched the experimental data in Drosophila melanogaster than in Homo sapiens (i.e. 73% for D. melanogaster and 67% for H. sapiens). Moreover, many other species had similar results in this section as H. sapiens. Thus, a bias might exist but cannot be confirmed.
Overall, the predictions were validated by comparison to experimental data, despite the highlighted drawbacks, such as the fact that the prediction might underestimate the real distribution of G4s in genes.
Gene densities: the correlation between densities differences and GC content is not the same for all species’ kingdoms
G4s were predicted in 61 species (see Supplementary Table S1), in WT, and shuffled sequences. To compare both density types, WT and shuffled densities of all species were subtracted and plotted (Figure 3A). Densities of species from the same kingdom were clustered together. Indeed, eukaryote densities were above 0 (WT density superior to the shuffled one), while the bacterial and archaeal densities were 0 or under 0 (shuffled densities slightly superior of the WT one). This would mean that WT pG4s were more present than expected in eukaryotes, but less than expected in bacteria and archaea. In Supplementary Figure S2 A, pG4s appear to be between one and fourfold higher than expected in eukaryotes, compared to 0 or fewer pG4s than expected in archaea and bacteria (i.e. between 0 and -2).
GC content also appears to impact pG4 density, although this impact varies depending on the species’ kingdom. For bacteria, unless the GC is over 45%, there is almost no pG4. Only the shuffled density seems to increase with GC content. For eukaryotes, most of the species seem to have increasing WT densities as the GC content increases. While for archaea, it seems to depend on species (Supplementary Figure S3A–C). In order to confirm the impact of the GC content on densities, a linear regression was performed to check the correlation between densities differences after removing the outliers using the interquartile range method (IQR method, Supplementary Figure S3D–F). The linear regressions were all significant, except for archaea. For archaea and bacteria, the correlations were negative, meaning that, as the GC content rose, fewer WT pG4s than the expected pG4s were found. As archaea correlation is not significant, this can mean that either there is no correlation with the GC content or that there is a tendency for an insignificant negative correlation.
Aside from these results, densities seem to be homogeneous within each kingdom, but there are some exceptions. In the case of eukaryotes, Chlamydomonas reinhardtii and Leishmania major have comparable GC contents, but different densities. In the case of bacteria, the same observation can be made with Mycobacterium tuberculosis, Thermus thermophilus and Myxococcus xanthus. For these three bacteria, shuffling the sequences led to similar densities, but the shuffled densities of C. reinhardtii and L.major remained different. The nucleotide content of those species was inspected (see Figure 5). C. reinhardtii had a higher percent of guanine than cytosine, while the guanine and cytosine contents were similar in L. major. It highlights that, given equivalent GC content, pG4 densities are expected to be similar in shuffled sequences. To confirm this, we also applied a shuffling preserving tri-nucleotides usage with ushuffle (66). The tri-nucleotide shuffling randomizes a sequence while minimizing the distance between the original and the shuffled sequences (67). In particular, the content in G-rich subsequences is more preserved in tri-nucleotide shuffling than in mono-nucleotide shuffling. By applying a tri-nucleotide shuffling, we hypothesized that the pG4 density would come back close to the WT density. The results in Supplementary Figure S1D confirm this hypothesis. For all tested species, the WT density was close to the tri-nucleotide shuffling density, probably because the use of guanosine trimers (GGG) is more conserved than in the mono-nucleotide content. The only exception was once again C. reinhardtii. For the latter, the G and C contents of positive and negative windows for G4RNAscreener were investigated (Supplementary Table S2). In WT positive windows, the C content was relatively low at ∼15%, while the G content was relatively high at ∼55%. None of the shuffled sequences succeeded in retaining these properties, which could explain the low mono- and tri-nucleotide shuffling densities. In other species, the G and C contents of positive windows were close to the WT sequences and the tri-nucleotide sequences.
Figure 5.

Special case nucleotide content, correlation with either the temperature or the pathogenicity of the species and pG4 density in genomic features. (A) and (B) are data for archaea; (C–E) are for bacteria. (A) and (C) represent the correlation between the G4 densities and the environment temperature of the species. (B) and (C) show the correlation between the GC content and the temperature. (E) is a dot plot with the pathogenicity of each bacterium, featuring the density differences (WT and shuffle) and the GC content. (G–I) correspond, respectively, to dot plots of the difference between the WT density and the shuffle density for microsatellites, transposable elements and centromeres.
Moreover, Marsico et al. (48) recently reported that bacteria living in hot environments return more pG4s than bacteria living under normal conditions with the same GC content. We observed this with Thermus thermophilus living at around 65°C with a density of 0.8, compared to Myxococcus xanthus and Mycobacterium tuberculosis, which live, respectively, at around 20 and 37°C and have a density of about 0.17. The hypothesis behind this difference is that bacteria lack the machinery to unfold G4s, so a negative pressure of selection would be globally applied in bacteria. In contrast, in species living at high temperature, G4s would naturally fold and unfold, and so lead to a lower pressure of selection. The correlation between pG4 density and temperature was further investigated (Figure 5B–E). It is known that normally, the higher the environmental temperature, the higher the GC content. Thus, this correlation was also explored. In Archaea, densities tended to be higher with temperature, yet a small negative correlation was found between GC content and temperature, which means that our species selection was not the best suited to look at temperature correlation. In the case of Bacteria, there was a tendency for a positive correlation between densities, GC content and temperature. Most of the selected bacteria are human pathogens and therefore live in environments of around 37°C, as shown in Figure 5F. There is increasing evidence of the involvement of G4s in pathogenicity and virulence (64,68,69), but the distributions of pathogenic and nonpathogenic bacteria are similar. Thus, as for temperature correlation, the selected bacteria were mainly pathogenic, which does not allow us to draw conclusions.
Genomic structures such as microsatellites, transposable elements and centromeres were also investigated (see Figure 5, panels G–I). Microsatellites are unstable structures with high mutation rates. While we anticipated finding as many pG4s as expected but pG4 densities differences were all under zero in all tested species (Supplementary Table S3), which means, that regardless of the GC content of microsatellites, there is an unknown mechanism that prevents G4 sequences from folding. The GC content in transposable elements (TEs) varied only between ∼40% and ∼50%, while a wide range of pG4 densities were observed. Some species had more pG4s than expected; others had fewer pG4s. This result is not surprising since our data on transposable elements is not extensive, and pG4 distribution varies depending on the type of transposable elements (see (70–72)). Globally, in TEs, the pG4 densities do not seem to be correlated to GC content and show a wide range of densities. Lastly, we checked the pG4 densities in centromeres for which easily accessible data were only available for H. sapiens and S. cerevisiae. We observe that either there were fewer pG4s than expected or no pG4s in the other species. Currently, a large number of high-quality genomes are being sequenced and assembled thanks to the progress in long-read sequencing technologies. Therefore, more resolved data on centromeres and telomeres might be available soon, which will allow for in-depth studies of the G4 structures they contain. Note that we did not perform study of telomeres because it is already known that several G4s are folding in these regions and even have known biological functions such as telomerase binding (73–76).
Overall, GC content impacted pG4 density, as was expected. Yet the densities within species’ kingdoms were homogeneous but did not have the same distributions. This would mean that GC content is not the only factor affecting the presence or absence of pG4s. Thus, in genes with high GC content, the G and C nucleotides were non-randomly distributed in sequences so as to avoid G4 formation.
Global transcript densities: densities at the transcriptome level were similar to gene densities
The distribution of different pG4 densities differences and quotients among species and domains was relatively similar in transcriptomes compared to genes (Figure 3A and B, and Supplementary Figure S2A and B). This result was expected as from genes to transcripts, redundancy was added for each transcript, which was corrected by normalization with sequence length. The correlation between density differences and GC content were also similar (Supplementary Figure S4D–F). Yet, the level of density difference changed for C. reinhardtii. The WT density of C. reinhardtii was similar to the gene WT density (1.16 in genes and 1.10 in transcripts), but the shuffled one ranged from 0.57 in genes to 0.83 in transcripts. Supplementary Figure S1 shows that the standard deviation between shuffled runs is low. This difference can be due to the fact that, to predict gene densities, the entire sequence was passed through G4Screener, but, for transcripts, only specific locations (presented in Figure 2) were used and the transcript densities were computed from exon and intron densities. Therefore, more shuffling possibilities are available in genes that are longer than exon/intron locations.
Along with the previous observation, eukaryotes densities differences are higher than those of archaea and bacteria. According to figure 3B, it seems there is more pG4 than expected in Eukaryotes, and less pG4 in Archaea and Bacteria. To confirm this observation, WT densities and shuffled densities were compared for each species kingdom. All statistical tests for Supplementary Figure S4G–I were obtained with the nonparametric Mann–Whitney–Wilcoxon test, because the densities did not follow a normal distribution. Supplementary Figure S4G–I confirm that there were more pG4s than expected in eukaryotes (WT densities were significantly higher than shuffled ones), while there was no significant difference between the WT dataset and the shuffled one for archaea and bacteria. Lastly, the density differences between species’ kingdoms were investigated. Supplementary Figure S5 shows that the eukaryote densities were significantly higher than those of archaea and bacteria, while archaea and bacteria presented no significant differences. Hence, there were significantly more pG4s than expected in eukaryotes than in prokaryotes. It seems that there was a shift between eukaryotes and prokaryotes with respect to pG4 densities.
Densities of coding transcripts and noncoding transcripts: distribution of density changes depending on the transcript class
Coding transcripts: As for transcriptomes, coding densities differences and quotients were similar to gene densities (see Figure 3A–C and Supplementary Figure S2A and C). Annotated transcripts were mainly protein-coding transcripts, which could explain why the results for genes and transcriptomes were so similar. Indeed, >90% (see Supplementary Figure S6) of archaeal and bacterial transcripts were annotated as coding in our dataset, which might account for the high similarity between densities at the transcriptome and coding levels. In the Eukaryote kingdom, coding transcripts varied between 75% and 90% depending on the species. Densities in this kingdom changed the most even if the densities were still highly similar between the whole transcriptome and coding transcripts. To further confirm the similarity of results, correlations and differences between datasets of coding transcripts were investigated and are also identical to those of all transcripts (see Supplementary Figure S7). The comparisons between species kingdoms are still similar: eukaryotes densities differences were significantly higher than those of archaea and bacteria (see Supplementary Figure S8).
Long noncoding transcripts: Among long noncoding transcripts (longNC), the distribution of pG4 densities differed from those of coding transcripts (see Figure 3C and D). In this class of transcripts, it seems that densities of all species groups are not clearly separated as for coding transcripts. For each species kingdom, densities differences and quotients were distributed above, around and <0. It means that the predicted G4s were more or less than expected, independently of the species kingdom. This is confirmed by Supplementary Figure S9, which shows no significative differences between the WT and shuffled datasets for eukaryotes and archaea. Nevertheless, bacteria had significantly fewer pG4s than expected. There is a null or a small negative correlation between pG4 density differences and GC content for eukaryotes and archaea, but this correlation is not significant. In other words, there was a trend of fewer pG4s at high GC contents. As for the relationship between species kingdoms, Supplementary Figure S9A–C indicates that eukaryote densities were significantly higher than those of archaea and bacteria, although there were no significant differences between them.
Short noncoding transcripts: ShortNC transcripts exhibited almost no densities over 0 (see Figure 3E). The densities were distributed under 0, which suggests fewer pG4s than expected. Supplementary Figure S10 shows that the shuffled densities were significantly higher than the WT ones in all species’ kingdoms. Thus, the first observation is confirmed. This result is in accordance with the global observation that there are fewer G4s in shortNC due to their highly structured sequences, and thus in all species’ kingdoms. Supplementary Figure S11 also shows that the GC content did not influence densities. This means that for shortNC transcripts, pG4s were negatively selected in almost all species and all GC content. As for the relation between species’ kingdoms, often eukaryote densities were higher than those of archaea (see Supplementary Figure S10D). Bacteria pG4 densities were almost all removed because of the IQR method to remove outliers, preventing us from checking the correlation and comparing bacteria to archaea and eukaryotes.
Pseudogene transcripts: Some pseudogene transcripts were annotated too but in fewer species than other transcript classes. All densities were close to 0 or a bit above, which could mean that there were fewer pG4s than expected. This observation is confirmed by the lack of significant differences between the shuffled densities and the WT density (see Supplementary Figure S12). In addition, there was a strong negative correlation (the higher the GC, the fewer predicted G4s than expected) for bacteria, while eukaryote exhibited no significant negative correlations. No correlations could be computed for archaea because of lacking annotation.
To summarize, the distribution of pG4 densities in coding transcripts were similar to those of all transcriptomes, while, in longNC, there were as many pG4s as expected and, in shortNC, fewer pG4s than expected. Those results still show that pG4 densities and expectations do not always depend on GC content; other parameters might influence the presence or absence of G4s. For shortNC, the parameter might be the competition between Hoogsteen and Watson and Crick base pairing, due the tight relation between the function and the secondary structure. This observation not only was known in humans but also seems to exist throughout all living domains. For coding and longNC, more possibilities can be considered. Through RNA-binding proteins (RBPs), G4s can fold (with chaperon) to play some roles or remain unfolded as a result of helicase activity. G4s can also recruit RBPs for translation in coding or for sponging in longNC. All together, these possibilities seem highly dependent on RBP systems.
Eukaryote species had more pG4s than expected in 3’UTR and introns of coding transcript
In coding transcripts, the annotation allowed for investigating transcript locations but not for UTR in archaea and bacteria. In addition, the splicing mechanism was more developed and annotated in eukaryotes than in prokaryotes because splicing is rare in the latter. To see the densities of these regions, we provide heatmaps to make the results more readable (see Figure 6 and Supplementary Figure S13).
Figure 6.

pG4 densities in all location types. Each dot plot represents density differences between the WT and Shuffle dataset. (A–E) correspond to segmental locations with respectively: (A) and (B) show exon and intron location; (C–E) correspond to 5’UTR, CDS and 3’UTR. Point locations from (F) to (J) are, respectively, donor, acceptor and junction in (F–H), while (I) and (J) represent the start and stop codons. (K) is a heat map with P-value indicating if there is a significant difference between WT densities and shuffled densities. The numbers are rounded P-values. Blank boxes correspond to non-annotated locations.
Eukaryotes had more pG4s than expected in introns than in exons (see Figure 6A and B). Moreover, G4s were predicted more than expected in UTRs compared to CDSs, although only introns and 3’UTR in eukaryotes exhibited a significant difference between WT and shuffled densities (see Figure 6C–E). In 5’UTR, the tetrapod subgroup had higher densities than in other species. This can be explained by a lack of annotation in the UTRs in some species or a real change in the pressure of selection depending on species’ subgroup, which can be due a molecular mechanism unique to tetrapods. In CDSs, fewer G4s were predicted than expected in almost all selected species, which agrees with past studies (77). In all point locations, there were as many pG4s as expected (data not shown) or fewer pG4s than expected (see Figure 6G and H). For most segment locations, however, there was no significant difference between WT and shuffled datasets, except for junctions where there were significantly fewer pG4s than expected.
Based on these results, pG4s present diverse density profiles depending on their locations, and thus independently of GC content. As for transcript classes, the different profiles might be due to pG4 functions and RBP interactions. In the 5’UTR, G4s are known to stabilize other secondary structures or to interact with RBPs or to produce some steric hindrance. In the CDS, it seems that G4s were found less than expected because of codon usage. In the 3’UTR, they can impact the polyadenylation of mRNA (29–31). Lastly, for splicing junctions, G4s are known to play a role in alternative splicing. All those functions, which often depend on G4 locations, might partly account for their different distributions. For each type of transcript location, there was a different pressure of selection to maintain its function.
For noncoding transcripts and pseudogenes, just few introns were annotated in some species, which limits their analysis. Exon densities were similar to the entire transcript class, while, for introns and their related locations, there were no G4s predicted (data not shown).
pG4 densities are not only driven by the GC content. There is also variable pressure of selection depending not only on species kingdom and transcripts class. Currently, parameters that influence the pressure of selection on pG4s are unknown. Further leads need be explored such as coevolution with RBPs, with transposable elements, or with viruses. Nevertheless, the evolution of G4s themselves still eludes us, since they cannot be easily aligned using conventional sequence alignment tools, given that the sequences in loops are often not conserved.
CONCLUDING REMARKS
Overall, our study provides more in-depth insights on pG4 densities in genes, transcripts and annotated coding locations. The prediction of G4s in a real dataset and a shuffled dataset in diverse species point out that GC content influenced pG4s, but that species’ kingdom, transcript class and locations also played a role. Most of the pG4 enrichment known in humans seems conserved in most eukaryotes. It could underlie a common mechanism in eukaryotes that might not exist in prokaryotes.
DATA AVAILABILITY
All information to retrieve the data and the scripts used for the analysis are available on the CoBIUS lab GitHub (https://github.com/UdeS-CoBIUS/G4Conservation).
Supplementary Material
Contributor Information
Anaïs Vannutelli, Department of Computer Science, Faculté des sciences, Université de Sherbrooke, QC, J1K 2R1, Canada; Department of Biochemistry and Functional Genomics, Faculté de médecine et des sciences de la santé, Université de Sherbrooke, QC J1E 4K8, Canada.
Jean-Pierre Perreault, Department of Computer Science, Faculté des sciences, Université de Sherbrooke, QC, J1K 2R1, Canada.
Aïda Ouangraoua, Department of Computer Science, Faculté des sciences, Université de Sherbrooke, QC, J1K 2R1, Canada.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
Canada Research Chairs [950–230577 to A.O.]; Chaire de recherche de l’Universite de Sherbrooke en Structure et Genomique de l’ARN (to J.P.P.); Fonds de Recherche du Quebec - Nature et Technologies (FRQ-NT); Natural Sciences and Engineering Research Council of Canada (NSERC) [RGPIN-155219–17 to J.P.P.; RGPIN-05552–17 to A.O.]; Centre de Recherche du CHUS (to J.P.P.); Universite de Sherbrooke.
Conflict of interest statement. None Declared.
REFERENCES
- 1. Kim J., Cheong C., Moore P.B.. Tetramerization of an RNA oligonucleotide containing a GGGG sequence. Nature. 1991; 351:331–332. [DOI] [PubMed] [Google Scholar]
- 2. Cheong C., Moore P.B.. Solution structure of an unusually stable RNA tetraplex containing G- and U-quartet structures. Biochemistry. 1992; 31:8406–8414. [DOI] [PubMed] [Google Scholar]
- 3. Juskowiak B., Galezowska E., Zawadzka A., Gluszynska A., Takenaka S.. Fluorescence anisotropy and FRET studies of G-quadruplex formation in presence of different cations. Spectrochim. Acta Part A: Mol. Biomol. Spectr. 2006; 64:835–843. [DOI] [PubMed] [Google Scholar]
- 4. Kwok C.K., Merrick C.J.. G-quadruplexes: prediction, characterization, and biological application. Trends Biotechnol. 2017; 35:997–1013. [DOI] [PubMed] [Google Scholar]
- 5. Huppert J.L., Balasubramanian S.. Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 2005; 33:2908–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kikin O., D’Antonio L., Bagga P.S. QGRS mapper: a web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res. 2006; 34(Suppl. 2):W776–W682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Todd A.K., Johnston M., Neidle S.. Highly prevalent putative quadruplex sequence motifs in human DNA. Nucleic Acids Res. 2005; 33:2901–2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bolduc F., Garant J.-M., Allard F., Perreault J.-P.. Irregular G-quadruplexes found in the untranslated regions of human mRNAs influence translation. J. Biol. Chem. 2016; 291:21751–21760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mukundan V.T., Phan A.T.. Bulges in G-quadruplexes: broadening the definition of G-quadruplex-forming sequences. J. Am. Chem. Soc. 2013; 135:5017–5028. [DOI] [PubMed] [Google Scholar]
- 10. Lim K.W., Amrane S., Bouaziz S., Xu W., Mu Y., Patel D.J., Luu K.N., Phan A.T.. Structure of the human telomere in K+ solution: a stable basket-type G-quadruplex with only two G-tetrad layers. J. Am. Chem. Soc. 2009; 131:4301–4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lim K.W., Alberti P., Guédin A., Lacroix L., Riou J.-F., Royle N.J., Mergny J.-L., Phan A.T.. Sequence variant (CTAGGG)n in the human telomere favors a G-quadruplex structure containing a G·C·G·C tetrad. Nucleic Acids Res. 2009; 37:6239–6248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Beaudoin J.-D., Perreault J.-P.. 5′-UTR G-quadruplex structures acting as translational repressors. Nucleic Acids Res. 2010; 38:7022–7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hon J., Martínek T., Zendulka J., Lexa M.. pqsfinder: an exhaustive and imperfection-tolerant search tool for potential quadruplex-forming sequences in R. Bioinformatics. 2017; 33:3373–3379. [DOI] [PubMed] [Google Scholar]
- 14. Garant J.-M., Perreault J.-P., Scott M.S.. Motif independent identification of potential RNA G-quadruplexes by G4RNA screener. Bioinformatics (Oxford, England). 2017; 33:3532–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yano M., Kato Y.. Using hidden Markov models to investigate G-quadruplex motifs in genomic sequences. BMC Genomics. 2014; 15:S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Belmonte-Reche E., Morales J.C.. G4-iM grinder: when size and frequency matter. G-Quadruplex, i-Motif and higher order structure search and analysis tool. NAR Genom. Bioinform. 2020; 2:lqz005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ruggiero E., Richter S.N.. G-quadruplexes and G-quadruplex ligands: targets and tools in antiviral therapy. Nucleic Acids Res. 2018; 46:3270–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Perrone R., Lavezzo E., Palù G., Richter S.N.. Conserved presence of G-quadruplex forming sequences in the long terminal repeat promoter of lentiviruses. Sci. Rep. 2017; 7:2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Artusi S., Perrone R., Lago S., Raffa P., di Iorio E., Palù G., Richter S.N.. Visualization of DNA G-quadruplexes in herpes simplex virus 1-infected cells. Nucleic Acids Res. 2016; 44:10343–10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Perrone R., Nadai M., Poe J.A., Frasson I., Palumbo M., Palù G., Smithgall T.E., Richter S.N.. Formation of a unique cluster of G-quadruplex structures in the HIV-1 nef coding region: implications for antiviral activity. PLoS ONE. 2013; 8:e73121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kumari S., Bugaut A., Huppert J.L., Balasubramanian S.. An RNA G-quadruplex in the 5’ UTR of the NRAS proto-oncogene modulates translation. Nat. Chem. Biol. 2007; 3:218–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu Y., Brosh R.M.. G-quadruplex nucleic acids and human disease. FEBS J. 2010; 277:3470–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu J., Jiang R., He H., Ma C., Tang Z.. Recent advances on G-quadruplex for biosensing, bioimaging and cancer therapy. TrAC Trends Anal. Chem. 2021; 139:116257. [Google Scholar]
- 24. Shioda N., Yabuki Y., Asamitsu S.. The potential of G-quadruplexes as a therapeutic target for neurological diseases. Folia Pharmacol. Jpn. 2019; 154:294–300. [DOI] [PubMed] [Google Scholar]
- 25. Wu Y.-Y., Kuo H.-C.. Functional roles and networks of non-coding RNAs in the pathogenesis of neurodegenerative diseases. J. Biomed. Sci. 2020; 27:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tassinari M., Richter S.N., Gandellini P.. Biological relevance and therapeutic potential of G-quadruplex structures in the human noncoding transcriptome. Nucleic Acids Res. 2021; 49:3617–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang E., Thombre R., Shah Y., Latanich R., Wang J.. G-quadruplexes as pathogenic drivers in neurodegenerative disorders. Nucleic Acids Res. 2021; 49:4816–4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jodoin R., Carrier J.C., Rivard N., Bisaillon M., Perreault J.P.. G-quadruplex located in the 5’UTR of the BAG-1 mRNA affects both its cap-dependent and cap-independent translation through global secondary structure maintenance. Nucleic Acids Res. 2019; 47:10247–10266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bagga P.S., Ford L.P., Chen F., Wilusz J.. The G-rich auxiliary downstream element has distinct sequence and position requirements and mediates efficient 3′ end pre-mRNA processing through a trans-acting factor. Nucleic Acids Res. 1995; 23:1625–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dalziel M., Nunes N.M., Furger A.. Two G-rich regulatory elements located adjacent to and 440 nucleotides downstream of the core poly(a) site of the intronless melanocortin receptor 1 gene are critical for efficient 3′ end processing. Mol. Cell. Biol. 2007; 27:1568–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Decorsière A., Cayrel A., Vagner S., Millevoi S.. Essential role for the interaction between hnRNP H/F and a G-quadruplex in maintaining p53 pre-mRNA 3′-end processing and function during DNA damage. Genes Develop. 2011; 25:220–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rouleau S.G., Garant J.M., Bolduc F., Bisaillon M., Perreault J.P.. G-quadruplexes influence pri-microRNA processing. RNA Biol. 2018; 15:198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Imperatore J.A., Then M.L., McDougal K.B., Mihailescu M.R.. Characterization of a G-quadruplex structure in Pre-mirna-1229 and in its Alzheimer’s disease-associated variant rs2291418: implications for miRNA-1229 maturation. Int. J. Mol. Sci. 2020; 21:767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kwok C.K., Sahakyan A.B., Balasubramanian S.. Structural analysis using SHALiPE to reveal RNA G-quadruplex formation in human precursor microRNA. Angew. Chem. 2016; 128:9104–9107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chan K.L., Peng B., Umar M.I., Chan C.-Y., Sahakyan A.B., Le M.T.N., Kwok C.K.. Structural analysis reveals the formation and role of RNA G-quadruplex structures in human mature microRNAs. Chem. Commun. 2018; 54:10878–10881. [DOI] [PubMed] [Google Scholar]
- 36. Hershman S.G., Chen Q., Lee J.Y., Kozak M.L., Yue P., Wang L.-S., Johnson F.B.. Genomic distribution and functional analyses of potential G-quadruplex-forming sequences in Saccharomyces cerevisiae. Nucleic Acids Res. 2008; 36:144–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rawal P., Kummarasetti V.B.R., Ravindran J., Kumar N., Halder K., Sharma R., Mukerji M., Das S.K., Chowdhury S.. Genome-wide prediction of G4 DNA as regulatory motifs: role in Escherichia coli global regulation. Genome Res. 2006; 16:644–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Saad M., Guédin A., Amor S., Bedrat A., Tourasse N.J., Fayyad-Kazan H., Pratviel G., Lacroix L., Mergny J.-L.. Mapping and characterization of G-quadruplexes in the genome of the social amoeba dictyostelium discoideum. Nucleic Acids Res. 2019; 47:4363–4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chambers V.S., Marsico G., Boutell J.M., di Antonio M., Smith G.P., Balasubramanian S.. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat. Biotechnol. 2015; 33:877–881. [DOI] [PubMed] [Google Scholar]
- 40. Kwok C.K., Marsico G., Sahakyan A.B., Chambers V.S., Balasubramanian S.. rG4-seq reveals widespread formation of G-quadruplex structures in the human transcriptome. Nat. Methods. 2016; 13:841–844. [DOI] [PubMed] [Google Scholar]
- 41. Hänsel-Hertsch R., Spiegel J., Marsico G., Tannahill D., Balasubramanian S.. Genome-wide mapping of endogenous G-quadruplex DNA structures by chromatin immunoprecipitation and high-throughput sequencing. Nat. Protoc. 2018; 13:551–564. [DOI] [PubMed] [Google Scholar]
- 42. Wu F., Niu K., Cui Y., Li C., Lyu M., Ren Y., Chen Y., Deng H., Huang L., Zheng S.et al.. Genome-wide analysis of DNA G-quadruplex motifs across 37 species provides insights into G4 evolution. Commun. Biol. 2021; 4:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Puig Lombardi E., Holmes A., Verga D., Teulade-Fichou M.P., Nicolas A., Londoño-Vallejo A.. Thermodynamically stable and genetically unstable G-quadruplexes are depleted in genomes across species. Nucleic Acids Res. 2019; 47:6098–6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Marsico G., Chambers V.S., Sahakyan A.B., McCauley P., Boutell J.M., di Antonio M., Balasubramanian S.. Whole genome experimental maps of DNA G-quadruplexes in multiple species. Nucleic Acids Res. 47:3862–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brázda V., Luo Y., Bartas M., Kaura P., Porubiaková O., Šťastný J., Pečinka P., Verga D., da Cunha V., Takahashi T.S.et al.. G-quadruplexes in the archaea domain. Biomolecules. 2020; 10:1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dey U., Sarkar S., Teronpi V., Yella V.R., Kumar A.. G-quadruplex motifs are functionally conserved in cis-regulatory regions of pathogenic bacteria: an in-silico evaluation. Biochimie. 2021; 184:40–51. [DOI] [PubMed] [Google Scholar]
- 47. Bartas M., Čutová M., Brázda V., Kaura P., Šťastný J., Kolomazník J., Coufal J., Goswami P., Červeň J., Pečinka P.. The presence and localization of G-Quadruplex forming sequences in the domain of bacteria. Molecules. 2019; 24:1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ding Y., Fleming A.M., Burrows C.J.. Case studies on potential G-quadruplex-forming sequences from the bacterial orders deinococcales and thermales derived from a survey of published genomes. Sci. Rep. 2018; 8:15679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bohálová N., Cantara A., Bartas M., Kaura P., Šťastný J., Pečinka P., Fojta M., Brázda V.. Tracing dsDNA virus–host coevolution through correlation of their G-Quadruplex-Forming sequences. Int. J. Mol. Sci. 2021; 22:3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Koster J., Rahmann S.. Snakemake–a scalable bioinformatics workflow engine. Bioinformatics. 2012; 28:2520–2522. [DOI] [PubMed] [Google Scholar]
- 51. Vilella A.J., Severin J., Ureta-Vidal A., Heng L., Durbin R., Birney E.. EnsemblCompara genetrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 2009; 19:327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cavalier-Smith T. The phagotrophic origin of eukaryotes and phylogenetic classification of Protozoa. Int. J. Syst. Evol. Micr. 2002; 52:297–354. [DOI] [PubMed] [Google Scholar]
- 53. Battistuzzi F.U., Hedges S.B.. A major clade of prokaryotes with ancient adaptations to life on land. Mol. Biol. Evol. 2009; 26:335–343. [DOI] [PubMed] [Google Scholar]
- 54. Letunic I., Bork P.. Interactive tree of life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 2019; 47:W256–W259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pignatelli M., Vilella A.J., Muffato M., Gordon L., White S., Flicek P., Herrero J.. ncRNA orthologies in the vertebrate lineage. Database. 2016; 2016:bav127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Munoz R., Yarza P., Ludwig W., Euzeby J., Amann R., Schleifer K.H., Glöckner F.O., Rossello-Mora R.. Release LTPs104 of the all-species living tree. Syst. Appl. Microbiol. 2011; 34:169–170. [DOI] [PubMed] [Google Scholar]
- 57. Avvaru A.K., Saxena S., Sowpati D.T., Mishra R.K.. MSDB: a comprehensive database of simple sequence repeats. Genome Biol. Evol. 2017; 9:1797–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wicker T., Matthews D.E., Keller B.. TREP: a database for triticeae repetitive elements. Trends Plant Sci. 2002; 7:561–562. [Google Scholar]
- 59. Vannutelli A., Belhamiti S., Garant J.-M., Ouangraoua A., Perreault J.-P.. Where are G-quadruplexes located in the human transcriptome?. NAR Genomics Bioinform. 2020; 2:lqaa035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Garant J.-M., Perreault J.-P., Scott M.S.. G4RNA screener web server: User focused interface for RNA G-quadruplex prediction. Biochimie. 2018; 151:115–118. [DOI] [PubMed] [Google Scholar]
- 61. Bedrat A., Lacroix L., Mergny J.-L.. Re-evaluation of G-quadruplex propensity with G4Hunter. Nucleic Acids Res. 2016; 44:1746–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Beaudoin J.-D., Jodoin R., Perreault J.-P.. New scoring system to identify RNA G-quadruplex folding. Nucleic Acids Res. 2014; 42:1209–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Altschul S.F., Madden T.L., Schäffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J.. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997; 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gazanion E., Lacroix L., Alberti P., Gurung P., Wein S., Cheng M., Mergny J.-L., Gomes A.R., Lopez-Rubio J.-J.. Genome wide distribution of G-quadruplexes and their impact on gene expression in malaria parasites. PLoS Genetics. 2020; 16:e1008917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Halder Kangkan, Hartig JorgS.. RNA quadruplexes. Metal Ions Life Sci. 2011; 9:125–139. [DOI] [PubMed] [Google Scholar]
- 66. Jiang M., Anderson J., Gillespie J., Mayne M.. uShuffle: a useful tool for shuffling biological sequences while preserving the k-let counts. BMC Bioinformatics. 2008; 9:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Altschul S.F., Erickson B.W.. Significance of nucleotide sequence alignments: a method for random sequence permutation that preserves dinucleotide and codon usage. Mol. Biol. Evol. 1985; 2:526–538. [DOI] [PubMed] [Google Scholar]
- 68. Harris L.M., Merrick C.J.. G-Quadruplexes in pathogens: a common route to virulence control?. PLoS Pathog. 2015; 11:e1004562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Saranathan N., Vivekanandan P.. G-Quadruplexes: more than just a kink in microbial genomes. Trends Microbiol. 2019; 27:148–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kejnovsky E., Tokan V., Lexa M.. Transposable elements and G-quadruplexes. Chromosome Res. 2015; 23:615–623. [DOI] [PubMed] [Google Scholar]
- 71. Lexa M., Steflova P., Martinek T., Vorlickova M., Vyskot B., Kejnovsky E.. Guanine quadruplexes are formed by specific regions of human transposable elements. BMC Genomics. 2014; 15:1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hanna R., Flamier A., Barabino A., Bernier G.. G-quadruplexes originating from evolutionary conserved L1 elements interfere with neuronal gene expression in alzheimer's disease. Nat. Commun. 2021; 12:1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Henderson E., Hardin C.C., Walk S.K., Tinoco I., Blackburn E.H.. Telomeric DNA oligonucleotides form novel intramolecular structures containing guanine·guanine base pairs. Cell. 1987; 51:899–908. [DOI] [PubMed] [Google Scholar]
- 74. Moye A.L., Porter K.C., Cohen S.B., Phan T., Zyner K.G., Sasaki N., Lovrecz G.O., Beck J.L., Bryan T.M.. Telomeric G-quadruplexes are a substrate and site of localization for human telomerase. Nat. Commun. 2015; 6:7643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Jansson L.I., Hentschel J., Parks J.W., Chang T.R., Lu C., Baral R., Bagshaw C.R., Stone M.D.. Telomere DNA G-quadruplex folding within actively extending human telomerase. Proc. Natl. Acad. Sci. 2019; 116:9350–9359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lin C., Yang D. Human telomeric G-quadruplex structures and G-quadruplex-interactive compounds. Methods Mol. Biol. 2017; 1587:171–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Mirihana Arachchilage G., Hetti Arachchilage M., Venkataraman A., Piontkivska H., Basu S.. Stable G-quadruplex enabling sequences are selected against by the context-dependent codon bias. Gene. 2019; 696:149–161. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All information to retrieve the data and the scripts used for the analysis are available on the CoBIUS lab GitHub (https://github.com/UdeS-CoBIUS/G4Conservation).