

Summary
Modifications are present on many classes of RNA, including tRNA, rRNA, and mRNA. These modifications modulate diverse biological processes such as genetic recoding and mRNA export and folding. In addition, modifications can be introduced to RNA molecules using chemical probing strategies that reveal RNA structure and dynamics. Many methods exist to detect RNA modifications by short-read sequencing; however, limitations on read length inherent to short-read-based methods dissociate modifications from their native context, preventing single-molecule modification analysis. Here, we demonstrate direct RNA nanopore sequencing to detect endogenous and exogenous RNA modifications on long RNAs at the single-molecule level. We detect endogenous 2′-O-methyl and base modifications across E. coli and S. cerevisiae ribosomal RNAs as shifts in current signal and dwell times distally through interactions with the helicase motor protein. We further use the 2′-hydroxyl reactive SHAPE reagent acetylimidazole to probe RNA structure at the single-molecule level with readout by direct nanopore sequencing.
Keywords: RNA, nanopore, direct RNA nanopore sequencing, RNA modifications, RNA structure, long-read, SHAPE-MaP, 2′-O-methyl
Graphical abstract
Highlights
Nanopore sequencing detects endogenous RNA modifications at a single-molecule level
2′-O-methyl (Nm) and pseudouridine () alter nanopore translocation kinetics
AcIm is a small-adduct-generating SHAPE-MaP reagent
AcIm enables full-length single-molecule structural profiling on RNA
Stephenson et al. employ direct RNA nanopore sequencing to detect endogenous and exogenous modifications on single RNA molecules. The authors demonstrate detection of endogenous 2′-O-methylation (Nm) on native ribosomal RNAs, confirming known modification patterns. They describe the development of nanoSHAPE, a method that involves exogenously labeling RNA with a small-adduct-generating chemical probe that can reveal RNA structure using long-read sequencing.
Introduction
More than 100 distinct modifications of RNA have been identified, occurring on either the nucleobase or the ribose sugar. These modifications exhibit diverse effects on RNA structure and function, including modulation of stability, translation efficiency, structural dynamics, nuclear export, and translational recoding1, 2, 3 and, in some cases, are installed, read, or removed by modification “writers,” “readers,” and “erasers,” suggesting a dynamic model of post-transcriptional gene regulation.4,5 The 2′-O-methyl (Nm) modification occurs in the 5′ cap of eukaryotic mRNAs (m7GpppNmNm) and extensively in ribosomal RNAs (rRNA). Nm modifications have also been detected within coding regions of mRNA6 and appear to tune cognate tRNA selection during translation, thereby adjusting protein synthesis dynamics.7 Pseudouridine (), often referred to as the fifth base because of its widespread inclusion in diverse classes of RNA, is generated by isomerization of uracil to create a nucleoside with distinct hydrogen bonding and base-pairing properties and increased base-stacking propensity relative to uridine.8,9 The absence or reduction of RNA modifications has been implicated in multiple diseases, including cancer, heart disease, and genetic diseases.10, 11, 12 Comprehensive detection and localization of RNA modifications within their native context will improve our understanding of RNA modification function and regulation and their role in disease.
Current methods to detect post-transcriptional RNA modifications fall into three broad classes. Immunoprecipitation methods use antibodies specific to individual modifications to enrich short fragments of RNA with the modifications, which are then converted into cDNA and sequenced with short reads.13,14 These approaches can be applied genome-wide but do not always provide nucleotide resolution and are limited by the availability and specificity of the pull-down reagents. An alternative family of approaches takes advantage of the propensity of reverse transcriptase (RT) enzymes to either terminate cDNA synthesis or incorporate noncomplementary nucleotides when a modified RNA base is encountered.6,15, 16, 17 Finally, modified nucleosides can be directly detected by mass spectrometry (MALDI or LC-MSMS); unique mass to charge ratios (m/z) and peaks can provide information relating to precise chemical identities and abundances of modified nucleosides and their immediate sequence context.18,19 Each of these methods has proven useful in specific contexts, but all have limited resolution and typically only probe one modification at a time.
Exogenous modifications of RNA have been used as temporal tags to assay RNA dynamics, including stability, turnover, and splicing timing.20, 21, 22, 23 In addition, chemical probing is widely used to monitor RNA structure. Chemical probes modify either the nucleobase (for example, dimethyl sulfate [DMS]) or the ribose 2′-hydroxyl group, the site of modification in selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) strategies. The location of SHAPE reagent-induced modification is detected by exploiting RT termination at the modified residue or through detection of a mutation opposite the modified site during RT readthrough. This latter approach, termed selective 2′-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP),24 yields single-nucleotide-resolution reactivity patterns and can be employed transcriptome-wide.25,26 However, SHAPE-MaP technology is limited by current constraints of short-read sequencing. An ideal method for investigating both endogenous and exogenous RNA modifications would detect multiple classes of modifications and would allow observation of multiple modifications on the same RNA molecule.
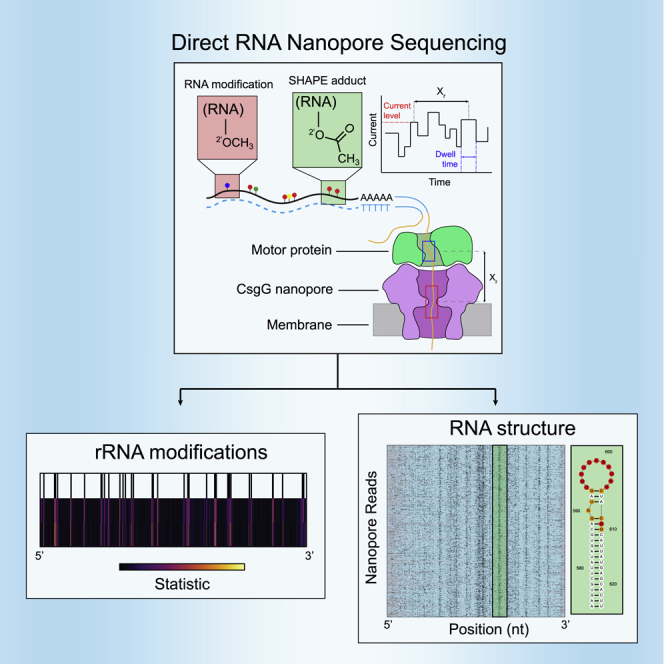
Direct RNA nanopore sequencing has emerged as a promising technology for full-length sequencing and analysis of both cell-derived and synthetic RNA molecules. In the commercial platform developed by Oxford Nanopore Technologies (ONT), RNA is translocated via a motor protein through a biological nanopore suspended in a membrane (Figure 1A). As the RNA transits through the pore under voltage bias, the observed changes in picoampere ionic current are characteristic of the chemical identity and sequence of the 5 nucleotides (“kmer”) positioned at the pore constriction.27,28
Figure 1.

Direct RNA nanopore sequencing and modification detection
(A) Scheme for direct RNA nanopore sequencing. rRNAs containing native modifications are poly(A) tailed before ligation of adapters and RT. Ionic current blockage events are characteristic of the kmer sequence of RNA transiting through the pore constriction.
(B) Read quality heatmap for IVT rRNA (left) and native rRNA (right) from E. coli. Dashed red lines indicated the expected lengths for 16S rRNA (1.5 kb) and 23S rRNA (2.9 kb)
(C) Normalized native (red) and IVT (black) current signal alignment for 16S rRNA from E. coli spanning positions 1395–1415 performed using Tombo. Sites of known modifications within this window are highlighted in blue.
(D) Positional Kolmogorov-Smirnov (KS) statistical testing of current signals across rRNA from E. coli and S. cerevisiae using both Tombo and Nanopolish. Modification positions described in the literature are indicated as black lines.
(E) Median current and dwell KS statistic profiles separated by modification type (base [excluding ], blue; 2′-O-methyl, red; and , green) and aligned by modification position from both Tombo and Nanopolish. Colored, shaded regions represent the standard deviation of the KS statistic.
Here, we adapt direct RNA sequencing on the ONT platform to detect both endogenous and exogenous RNA modifications. Importantly, we show that the raw current signal from nanopore sequencing detects RNA modifications, independent of the chemical nature of the modification. In addition, we describe modification-dependent signals in the time domain, relating to the translocation rate of single molecules through the nanopore. This time domain signal provides a complementary dimension of information that may be incorporated with current signal for de novo identification of nucleotide modification classes. Building on these insights, we developed nanoSHAPE, which combines long-read, direct RNA sequencing with a new SHAPE reagent that, by virtue of its high reactivity and small adduct size, enables full-length probing of structure in long RNAs.
Results
Identification of specific modifications at defined locations within 16S rRNA
We first applied direct ONT RNA sequencing to rRNAs from Escherichia coli and Saccharomyces cerevisiae; these rRNA are highly abundant and harbor well-characterized modifications (Data S1). We generated in vitro transcripts, which are devoid of modifications, of the small- and large-subunit RNAs as controls (Figures S1A and S1B). These in vitro transcribed (IVT) controls were sequenced independently to provide a modification-free baseline against which native, cell-derived RNAs could be compared to identify putative sites of modification.29,30 IVT controls and native RNAs exhibited reads at the expected lengths for E. coli (16S: 1.5 kb, 23S: 2.9 kb) (Figure 1B) and S. cerevisiae (18S: 1.8 kb, 25S: 3.4 kb) (Figure S1C). Notably, median quality scores for full-length molecules from E. coli were lower than those for the native samples as compared to IVT controls (16S: −0.53, 23S: −0.31, t test p < 0.05); results were similar for S. cerevisiae (Figure S1D). This reduction likely reflects the effect that high modification levels have on read quality using the current iteration of modification un-aware base calling software. Coverage was lower toward the 5′ ends for all samples, including IVT controls (Figure S2) as expected from the configuration of direct RNA nanopore sequencing, which translocates RNA in the 3′-to-5′ direction.
Reads were processed using both Tombo31 and Nanopolish,32 which perform raw current-signal-level-to-sequence alignment. As an example, we highlight a region of current signal mapped with Tombo on 16S rRNA in E. coli containing two modifications in proximity: N4,2′-O-dimethylcytosine (m4Cm) at position 1402 and 5-methylcytosine (m5C) at position 1407 (Figure 1C). We observed a clear deviation in current signal distribution of the native sample at position 1402, whereas current signal deviation due to the chemically distinct m5C modification at position 1407 was less pronounced and spread over three positions, 1406–1408 (Figure 1C), highlighting the often-distributed nature of complex nanopore signals in the kmer context.
Comprehensive rRNA modification detection
We next examined whether nanopore sequencing could detect all known modifications in the small- and large-subunit rRNAs of both E. coli33 and S. cerevisiae (Data S1).34 Normalized current and dwell time differences between native and IVT samples were observed across the small- and large-subunit rRNAs consistent with the locations of known modifications (Figure S3). To more rigorously assess modification signals, which are highly dependent on kmer sequence context, we performed non-parametric Kolmogorov-Smirnov testing (KS) across all positions for current and dwell time from raw signal-aligned data using both Tombo and Nanopolish (STAR Methods; Figures 1D and S4). Peaks in the KS statistic profile indicated distributional differences between the IVT unmodified control sample and the native samples. The KS statistic for the current signal was strongly correlated between Tombo and Nanopolish (Pearson correlation, r = 0.75–0.79); however, the dwell time KS statistic profiles were only moderately correlated (Pearson correlation, r = 0.45–0.55). Generally, KS statistic peaks for current were observed within ±2 nt of the known modifications.
To assess modification class features, we collated all RNA modifications across the small and large ribosomal subunits from both E. coli and S. cerevisiae, aligning them by their known modification position, and considered the aggregate median KS statistic profile across modification class: Nm, , and base (e.g., base modifications excluding ). The median KS statistic for current of all modification classes had an appreciable signal at the site of modification (pore constriction) as expected (Figure 1E, top row). With respect to the dwell time, the median KS statistic profile exhibited two peaks for Nm and pseudouridine modifications but not for base modifications (Figure 1E, bottom row). The primary peak occurred at the nanopore constriction (relative modification position 0); however, secondary peaks were observed approximately 10 nt in the 3′ direction with Nm modifications exhibiting a larger median KS statistic than . This 10-nt distance is the “registration distance” (Xr) from the pore constriction where kmer currents are measured to the motor protein that sits atop the nanopore. The Xr in DNA nanopore sequencing experiments using a more terminally located pore constriction in MspA and a different motor protein suggested an Xr of ∼20 nt.35 In the R9.4.1 version nanopore system used here, a CsgG pore is used, which has a centrally located pore constriction, likely explaining the smaller Xr.27
Interestingly, these observations suggest that, at least in some sequence contexts, the motor protein kinetics are more sensitive to Nm than modifications. The 2′-O-methylation confers approximately −0.2 kcal/mol of stacking free energy to single-stranded RNA and favors the C3′-endo conformation of RNA.36 Thus, changes in RNA conformation or steric and chemical interactions with particular amino acid residues of the motor protein may perturb translocation times. Interestingly, all types of modifications exhibit a median KS statistic peak for dwell time above background at relative modification position 0, indicating that transit times through the nanopore constriction itself are also perturbed by many of the modifications studied here. However, we did not observe significant changes in deletion or insertion error frequencies as might be expected from these dwell time differences (Figure S5). The majority of errors observed because of or Nm modifications were mismatches centered at the modification site, with having the highest average error of 48% mismatch.
RNA translocation rate is sensitive to nucleotide modifications and sequence composition
We next investigated the dwell time as a function of position to ascertain whether modifications and/or sequence content perturb dwell times. Comparison of dwell times for both native and IVT samples at a distance of Xr in the 3′ direction from selected sites within E. coli 16S (1402 m4Cm), E. coli 23S (2552 Um), S. cerevisiae 18S (1428 Gm), and S. cerevisiae 25S (2220 Am) rRNAs revealed a significant increase (Mann-Whitney U test) in native samples relative to IVT controls (Figure 2A). To assess the extent of motor protein pausing due to sequence rather than the presence of modifications, we explored sequence similarities for the top 1% of ranked dwell times from all IVT samples in the region spanning the entire sequencing complex from upstream of the motor protein to the exit of the nanopore. We observed a strong guanosine enrichment approximately 9–11 nt upstream of the pore constriction (Figure 2B), consistent with the registration distance of 10 nt, that corresponds to the location of the active site of the motor protein. We quantified the representation of each nucleotide in the motor protein active site, defined as the three nucleotides at positions 9–11 3′ of the nanopore constriction, across dwell time percentiles for all IVT samples and observed overrepresentation of guanosine in the highest percentiles (Figure 2C). Collectively, these data indicate that even absent modifications, the motor protein used in direct RNA nanopore sequencing experiments has a tendency to pause on guanosine-rich sequences. These observations are consistent with pausing over guanosine-rich sequences by single-molecule picometer resolution nanopore tweezers (SPRNT) experiments with DNA (using a Hel308-based translocation mechanism).37 Pausing over guanosine-rich regions may be a general feature of enzyme-based translocation; these regions may sterically hinder the enzyme or higher-than-average single-stranded stacking energies might decelerate translocation.38
Figure 2.

Dependence of dwell time on modifications and sequence
(A) Dwell time comparison of native (red) and IVT (black) samples (Mann-Whitney U test, n = 1,000 reads) at selected Nm modification sites in 16S and 23S from E. coli and 18S and 25S from S. cerevisiae. Each box spans the interquartile range, the horizontal line within each box defines the median, and the whiskers extend out to the minimum and maximum values. The kmer is indicated below the x axis along with the position of the modification (red). Dwell times are from +Xr from the centered modification site kmer.
(B) Sequence motif from the top 1% of dwell times (IVT samples only, 16S, 23S, 18S, and 25S) spanning a 30-nt window encompassing the entire biomolecular sequencing complex.
(C) Nucleotide representation (fraction) within the trimer (positions −9, −10, and −11) from IVT samples as a function of dwell time percentile. The highest dwell time percentiles are enriched for guanosine within the trimer.
The 1-acetylimidazole reagent generates a compact SHAPE adduct
As nanopore sequencing was able to detect endogenous Nm modifications in rRNA, we hypothesized that we could detect 2′-O-adducts resulting from exposure of folded RNAs to electrophilic SHAPE reagents, which would then enable us to interrogate RNA structure using nanopore sequencing. A long-term advantage to exploiting nanopore sequencing for structural probing is the possibility of detection of multiple modifications per molecule, enabling analysis of phased and correlated structural information over long sequence distances.39,40 We initially attempted nanopore sequencing using RNA that had been modified with established SHAPE reagents 2-methylnicotinic acid imidazolide (NAI), 1-methyl-7-nitroisatoic anhydride (1M7), and N-methylisatoic anhydride (NMIA). In our hands, at the concentrations tested, readout of these experiments with nanopore sequencing resulted in few full-length reads and poor alignment accuracy as compared to an unmodified control sample (Figure S6). A similar poor alignment accuracy was reported recently when a short RNA modified with high concentrations of NAI (100 mM) was analyzed using nanopore sequencing.41 We observed that the alignment percentage and fraction of full-length reads were poor at both high (200 mM) and low (25 mM) concentrations of NAI, precluding the detection of multiple modifications on single, long RNA molecules. We hypothesized that the observed inefficient and incomplete translocation was due to the presence of multiple bulky 2′-O-aryl adducts that result from reaction of RNA with these SHAPE reagents.
We therefore searched for a reagent that would produce a smaller adduct, more chemically similar to native Nm modifications. We examined five carbonyl-imidazolide candidates for structure-selective 2′-O-acylation. We detected covalent adduct formation for NAI, as expected, and for 1-acetylimidazole (AcIm) (Figure S7). AcIm was previously identified as a 2′-hydroxyl-reactive reagent.42 The proposed reaction of AcIm with the 2′-hydroxyl of RNA (Figure 3A) results in the most-compact-possible acetyl adduct using an electrophilic carbonyl reagent.
Figure 3.

Acetylimidazole generates small adducts detectable by SHAPE-MaP
(A) Schematic of acylation of RNA with acetylimidazole (AcIm).
(B) Time dependence of hydrolysis for AcIm analyzed by changes in UV absorbance. Experiment performed at 37°C.
(C) SHAPE-MaP reactivity profiles using NMIA, NAI, and AcIm for E. coli 16S and 23S rRNAs.
(D) Two-dimensional kernel density estimates for NMIA, NAI, and AcIm adduct-induced mutation rates for E. coli 16S and 23S rRNAs with unmodified control and SHAPE-modified rates on the x and y axes, respectively. Separation between peak density distributions for paired (blue) versus unpaired (red) positions indicates reaction selectivity with respect to degree of nucleotide flexibility.
(E) Receiver operator characteristic curve and associated area under the curve (AUC) for MaP reactivities as a function of pooled nucleotide base-pairing status for SHAPE reagents. True positive rate: unpaired nucleotides with reactivity above a given threshold/total unpaired nucleotides. False positive rate: base-paired nucleotides with reactivity above a given threshold/total base-paired nucleotides.
The relative rate of reaction for a SHAPE reagent with the 2′-hydroxyl of RNA is mirrored by its rate of reaction with water.43 We investigated the timescale of AcIm reactivity by monitoring the change in absorbance of AcIm in reaction buffer; imidazole was monitored as a control. The AcIm signal, centered at 250 nm, decayed via a single exponential, with a half-life of 3 min at 37°C (Figure 3B), consistent with prior measurements of N-acetylimidazole hydrolysis.42,44 The AcIm spectrum decays to that observed for imidazole, supporting hydrolysis of AcIm into imidazole and non-absorbing acetate.
To assess AcIm reactivity with RNA, we extracted RNA from E. coli and treated total RNA with 100 mM NAI, 13 mM NMIA, 100 mM AcIm, or DMSO (as a vehicle control). We then obtained per-nucleotide reactivity profiles using mutational profiling (SHAPE-MaP)24 and aligned the resulting cDNAs to the 16S and 23S rRNAs (Figures 3C and S8). Reactivity profiles for the three reagents were highly correlated, indicating that AcIm is a robust SHAPE reagent. AcIm reacted with each of the four canonical RNA nucleotides and preferentially reacted with conformationally flexible (unpaired) nucleotides (Figures 3D and S9). Receiver operator characteristic curve analysis demonstrated that there was virtually no difference in discrimination between paired and unpaired nucleotides for NAI, NMIA, and AcIm (area under curve ∼0.8 for all probes) (Figures 3E and S10). In sum, AcIm generates small adducts, reacts broadly with all four ribonucleotides, and generates SHAPE-MaP reactivity profiles consistent with known reagents.
nanoSHAPE: Direct RNA nanopore sequencing of AcIm-modified RNA
We next assessed whether AcIm chemical probing could be used to guide RNA secondary structure modeling based on single-molecule direct RNA nanopore sequencing, a method we call nanoSHAPE. We focused on an in vitro transcribed pri-miRNA (primary transcript) of the miR-17∼92 cluster, which spans 951 nucleotides and folds to form a series of well-defined hairpin structures.45,46 The pri-miR-17∼92 is predicted to form a moderately structured complex (predicted ΔGcentroid = –298.80 kcal/mol) with a low number of isoenergetic suboptimal conformations (ensemble diversity [ED] = 152.9), making it a suitable substrate for folding studies. We first assessed the structure of pri-miR-17∼92 by SHAPE-MaP using both AcIm and NAI (Figure S11A). Reactivities for the two reagents were highly correlated (Spearman’s rho = 0.78). Furthermore, secondary structure modeling informed by the AcIm SHAPE-MaP reactivity profile produced a centroid structure consistent with the current pri-miR transcript structure models (Figure S11B).45 These experiments indicate that both chemical probes are suitable for investigating this structure and provide a control for comparison with nanoSHAPE. We next assessed the compatibility of AcIm with nanopore sequencing by performing a series of direct RNA nanopore sequencing experiments using either unmodified pri-miR-17∼92 or RNA modified with 5, 20, 50, 75, 100, 150, or 200 mM final concentrations of AcIm. Prior to AcIm modification, the terminal RNA nucleoside was modified through oxidation and beta-elimination to remove the 3′-nucleoside and leave a 3′-phosphate. After AcIm modification, the 3′-phosphate was removed by phosphatase treatment to leave a 3′-hydroxyl, allowing ligation with RNA-sequencing adapters (Figure 4A). At higher AcIm modification rates, we observed noticeable decreases in read quality and fraction of full-length reads obtained. This signal degradation reduced the percentage of reads successfully aligned by Tombo (Figures S12A and S12C–S1E; Table S1). The coverage was higher at the 3′ end at all concentrations, consistent with the 3′-to-5′ read direction of the RNA through the nanopore (Figure S12B). The coverage, fraction of full-length reads, and aligned read percentage became inadequate at the highest AcIm concentration (200 mM), so this condition was excluded from further analysis. The lower fraction of full-length reads observed after modification with AcIm suggested that reads were truncating at sites of AcIm modification. We mapped direct RNA nanopore sequencing read termini from unmodified RNA and RNA modified with 25 mM NAI or 150 mM AcIm to the pri-miR-17∼92 RNA to the parent sequence and compared to the SHAPE-MaP reactivity profile (Figure S13). The control and AcIm-modified data had similar read termini profiles (Spearman’s rho = 0.76), which in turn were similar to the SHAPE-MaP reactivity profile albeit shifted by approximately 10–15 nt in the 3′ direction, which implicates motor protein involvement in read truncation events. These observations suggest, first, that in the absence of SHAPE reagent modification, intrinsic RNA structures cause a small degree of truncation and, second, that modification with AcIm slightly accentuates read truncations in these same regions. Read termini in NAI-modified RNA had a different profile (Spearman’s rho = 0.12 and 0.19 for control and AcIm, respectively), with most termini mapping to the 3′ end of the pri-miR-17∼92 sequence, consistent with the very small fraction of full-length reads obtained from NAI-modified sequencing experiments. Thus, NAI is a poor nanoSHAPE probe, as these adducts pose serious problems for motor protein processing and processivity.
Figure 4.

Direct structural probing of a pri-miR-17∼92 transcript RNA using AcIm and nanopore sequencing
(A) Scheme for 3′ end modification, SHAPE probing, and 3′ end tailing.
(B) Normalized SHAPE-MaP reactivity (blue) and nanoSHAPE reactivity detected by changes in current (red) and dwell time (gray) for the pri-miR-17∼92 RNA.
(C) Heatmap of 1,000 nanopore reads of pri-miR-17∼92 modified with 150 mM AcIm. Modifications were determined by per-nucleotide Student’s t test using Fisher’s method context of ±1 of the current signal. Per-nucleotide p values were corrected using the Benjamini-Hochberg procedure and binarized. Nucleotides scored as modified and unmodified are shown in black and teal, respectively; unmapped regions are gray.
(D) Kernel density estimate of the number of called modifications per pri-miR-17∼92 molecule as a function of AcIm concentration. Called modifications correspond to an upper limit.
(E) Spearman’s rank-order correlation (rho) between nanoSHAPE and SHAPE-MaP as a function of the number of contributing pri-miR-17∼92 molecules across the AcIm concentrations tested.
(F) Spearman’s rho as a function of the distance from the 3′ end of the pri-miR-17∼92 RNA.
We next performed KS statistical testing for current and dwell time distributions across all AcIm concentrations as compared to the unmodified control (Figure S14A). KS peaks in current were observed in all profiles, primarily in single-stranded regions of pri-miR-17∼92. We determined Spearman’s rank-order correlations for both KS of current and KS of dwell time (shifted by Xr) against the AcIm SHAPE-MaP profile (Figure S14B). The correlation was greatest for current and maximized at 150 mM (current rho = 0.51, dwell time rho = 0.28). To assess the capability of single-molecule-based reconstruction of reactivity profiles, we performed per-read statistical testing and normalization for every nucleotide position within 1,000 individual full-length molecules of pri-miR-17∼92 across all AcIm concentrations (Figures 4B, 4C, and S15). The median mutation rates from SHAPE-MaP libraries derived from pri-miR-17∼92 modified with 25 mM and 200 nM AcIm were 0.03% and 0.1%, respectively, which corresponds to 0.285 and 0.951 detected adducts per full-length read, respectively. These low values suggest that the current MaP approach does not detect the AcIm adduct efficiently. Based on the single-molecule nanopore data, and after statistical testing and detection, we obtained approximately 105 called modification sites per full-length read at 150 mM AcIm (Figure 4D). This value is likely notably inflated because of the distributed nature of modification detection and because of the high level of noise intrinsic to current generation nanopores. Nonetheless, these comparisons suggest that nanopore detection is more efficient at detecting the compact 2′-ribose AcIm adduct than MaP-RT is.
We next examined the number of single-molecule reads that are required to obtain an optimal correlation with SHAPE-MaP. We subsampled full-length reads from n = 1 to n = 1,000 and calculated the normalized reactivity profiles from the per-read current statistical testing as a function of number of reads. The Spearman’s rank correlation of the normalized reactivity against the SHAPE-MaP reactivity profile reached 95% of the maximum correlation at around 200 reads for each AcIm concentration, with higher correlations with MaP data obtained at higher AcIm concentrations (Figure 4E). In the normalized reactivity profile derived from nanoSHAPE, we observe less distinctive reactivity features closer to the 5′ end of the pri-miR-17∼92 transcript. To explore this phenomenon, we calculated the Spearman’s rank correlation on a progressively shortened normalized reactivity profile, trimming from the 5′ end. This procedure revealed a maximum correlation at about 300 nucleotides from the 5′ end of the transcript (rho = 0.53, 150 mM AcIm) (Figure 4F), indicating that the 5′ end of RNAs may not be well resolved by this approach. Poor structural resolution at the 5′ end may be due to reduced coverage in this region resulting from incomplete reverse transcription due to the presence of AcIm adducts that can cause cDNA truncation. Reverse transcription is not strictly required for direct RNA sequencing using the nanopore platform; however, the presence of a cDNA is known to stabilize the sequenced RNA strand increasing overall yield and throughput, potentially favoring the recovery of RNA molecules with a full-length cDNA annealed.
nanoSHAPE facilitates RNA structure modeling
We performed secondary structure modeling using nanoSHAPE and SHAPE-MaP reactivities as pseudo-free energy constraints introduced into a nearest-neighbor RNA-folding algorithm.47,48 The two structural models differ in that the nanoSHAPE centroid structure includes fewer long-range base pairs and has larger loop sizes (specifically for hairpins 17 and 19a) than does the SHAPE-MaP-based structure (Figure 5A). Hairpin 17 and 19a are both predicted to have internal bulges (U151 and G435-U437) at the bases of their loops toward the 3′ side, based on both SHAPE-MaP and unconstrained modeling (Figure S11B).46 Reactivity at these positions, observed by nanoSHAPE, which features a 3′-to-5′ read direction, may over-detect reactivity at loop-closing base pairs, leading to prediction of larger loop sizes. Importantly, centroid structures for both SHAPE-MaP- and nanoSHAPE-constrained predictions contain the six miRNA hairpins expected to occur in the 17∼92 cluster.
Figure 5.

Comparison of RNA structure modeling based on SHAPE-MaP and nanoSHAPE reactivities
(A) Secondary structure models, visualized as arc diagrams, for the pri-miR-17∼92 RNA. Centroids for structures modeled using SHAPE-MaP (blue) or nanoSHAPE (green) constraints are shown. Secondary structure models for the constituent miRNA hairpins are shown with overlaid SHAPE-MaP and nanoSHAPE reactivities. SHAPE data correspond to the 25 mM AcIm concentration.
(B) Probabilities of all possible base pairs in pri-miR-17∼92 based on SHAPE-MaP (top) or nanoSHAPE (bottom) reactivity constraints, shown as a partition function dot plot. Probabilities are displayed as −log10(probability base pair (i,j)).
(C) Similarity in structure models, reported as relative positive predictive value (PPV) and sensitivity for minimum free energy (MFE) and centroid structures. Pairwise comparison was performed between SHAPE-MaP-constrained (MaP), nanoSHAPE-constrained, and no-probing-data (NPD) secondary structure models.
We next performed partition function calculations for RNA structures arising from SHAPE-MaP-constrained and nanoSHAPE-constrained pri-miR-17∼92 sequences. Partition function base pair probabilities between all possible nucleotides (i,j) exhibited generally concordant connectivity patterns, indicating broad agreement between the collective predicted structural ensembles (Figure 5B). We then benchmarked minimum free energy and centroid secondary structures predicted from nanoSHAPE versus predictions from SHAPE-MaP-constrained modeling relative to models obtained with no probing data (NPD). In general, MaP-constrained models were the most distinct, consistent with extensive prior work showing that SHAPE-MaP data generally substantially change RNA structure models relative to NPD models, in the direction of the correct structure.24,48 nanoSHAPE data also clearly perturbed the structural ensemble, relative to the NPD ensemble, to become more similar to the SHAPE-MaP-informed model (Figure 5C). We conclude that nanoSHAPE produces reactivity patterns and secondary structure predictions for the pri-miR-17∼92 sequence broadly consistent with high-throughput-sequencing-based RNA chemical probing and structural profiling approaches.
Discussion
The long-read direct RNA nanopore sequencing on the ONT platform is a promising tool for characterizing RNA at the single-molecule level. RNA molecules exhibit diverse chemical and structural states that serve as effectors and modulators of RNA function, interaction, and dynamics. We sought to use the direct measurement of RNA, rather than a cDNA copy, to examine RNA chemical modifications and secondary structure. Our direct RNA-sequencing approach was able to detect native modifications in ribosomal RNA from E. coli and S. cerevisiae at both the nucleobase () and backbone (Nm). The majority of these positions are modified stoichiometrically, making them good systems for benchmarking endogenous modification detection. In our dataset comparing endogenous rRNAs to in vitro transcribed controls, we performed raw-signal-to-sequence alignment with both Tombo31 and Nanopolish32 and identified rRNA modification positions to within ±2 nt of known modified sites.
Direct RNA nanopore sequencing is uniquely positioned to answer questions about the dynamics and ordering of modification installation on rRNA and, in principle, has the potential to address both quantification and long-range phasing of modifications. Signal discrimination remains an outstanding challenge for direct RNA nanopore sequencing for modification detection, which is a function of the kmer sequence context and ability to align raw current signal to sequence. Development of training sets consisting of known modifications in all possible kmer sequence contexts will be required for RNA modification identification without resorting to comparison with an IVT control. The ability to call modifications without an IVT control, coupled with increasing yield of direct RNA sequencing, should allow investigation of other, less-abundant cellular mRNAs and long non-coding RNAs.
In addition to current signal levels, we characterized changes in the current level dwell times in direct RNA sequencing. Ribose modifications, both the endogenous Nm and exogenous SHAPE reagent modifications, notably extend dwell times. Dwell time changes due to RNA modifications at the pore constriction have been recently reported.49 Here, we demonstrated that dwell time is dependent on motor protein translocation kinetics mediated at a registration distance (Xr ∼10 nt in the 5′ direction) from the pore constriction. Additionally, we observed that motor protein dwell times are influenced by primary sequence. Translocation rates have been shown to be sequence dependent for DNA using the Hel308 motor protein and a MspA nanopore.37 We found that average translocation rates varied across the nanopore array (Figure S16), complicating direct comparison of dwell times across array channels. However, we suggest that large dwell times observed in direct RNA nanopore sequencing experiments may be used to infer sites (at Xr distance) of Nm or modifications in the absence of IVT controls, provided that the sequence context around the putative modification is not G-rich. Full characterization and incorporation of this extra dimension of information will require channel- or even read-specific normalization to faithfully compare translocation rates across the nanopore array.
The detection of naturally occurring Nm modifications in rRNA suggested the possibility of applying this approach to detect experimentally introduced 2′-O adducts, as used in RNA structure probing methods. Key to our success was identifying a SHAPE reagent specifically tailored for nanopore sequencing. AcIm had favorable properties, including a short (but still experimentally manageable) half-life, small adduct size, detection by mutational profiling, and commercial availability. The small adduct (2′-O-acetyl) created by modifying RNA with AcIm is detectable in direct RNA nanopore sequencing experiments. High rates of AcIm modification do lower the number of full-length reads, overall yield, and alignment rates; however, the resulting yield and data quality are vastly superior to that obtained with reagents that yield bulkier adducts. Consistent with our analysis, an NAI analog (NAI-N3) was independently41 found to induce drastic decreases in yield. In that work, NAI-N3 yielded hit rates of 1%–2% over the readable fraction of RNA, whereas AcIm achieved median hit rates up to 11% on full-length single molecules. Application of nanoSHAPE with AcIm to the analysis of the structure of the pri-miR-17∼92 transcript revealed that nanoSHAPE-data-constrained modeling yielded RNA structures broadly similar to those obtained with SHAPE-MaP data.
Limitations of the study
Direct RNA nanopore sequencing, and by extension nanoSHAPE, has limitations. The method requires high concentrations of target RNA and a free 3′-hydroxyl for ligation to the 5′ phosphate of the first adaptor required for nanopore sequencing. Electrophilic SHAPE reagents, AcIm included, covalently modify the 3′-hydroxyl, preventing ligation. We ameliorated this challenge by chemical treatment to create a terminal phosphate; after modification, the RNA was then treated with a phosphatase to enable poly(A) tailing and ligation to the sequencing adaptor. If nanoSHAPE is to be properly extended transcriptome-wide and to in-cell structural probing experiments with high single-molecule modification rates, novel methods for selection and enrichment of target RNAs and protection of the 3′-hydroxyl (or enrichment of molecules with ligate-able 3′-hydroxyl ends) in cells may be required to ensure sufficient yield with the current direct RNA nanopore sequencing method.
nanoSHAPE is also limited by the poor resolution of reactivity profiles at the 5′ ends of longer RNA molecules. This loss of resolution is likely due to multiple factors, including coverage bias inherent to the 3′-to-5′ direction of RNA nanopore sequencing and the difficulty of translocation through a highly structured RNA, like pri-miR-17∼92. It is also possible that cDNA synthesis on highly AcIm-modified RNA, which is an optional step that facilitates translocation, was incomplete in our experiments.
Finally, nanoSHAPE is limited by the method of signal analysis used to identify intrinsic posttranscriptional modifications and SHAPE adduct sites. In this work, we used a comparative approach, comparing current signals of modified RNAs to those of unmodified RNAs of the same sequence to identify sites of difference. Modification detection may be improved by using methods that employ trained models for signal classification. However, de novo methods are reliant on a training or ground truth set containing the modification in all kmer sequence contexts. A second challenge in adduct detection is distinguishing authentic chemical modifications from current signal and dwell time changes induced by the underlying RNA structure. Extensive benchmarking with native RNAs of known structure would inform deconvolution of adduct versus structure effects.
Despite these challenges, nanoSHAPE demonstrates significant promise. Long-read single-molecule sequencing will permit investigation of RNA structural ensembles for long RNAs directly. Adduct detection and sequencing throughput are poised to improve as direct RNA nanopore sequencing technology and analyses mature. Direct sequencing of AcIm-modified RNA will be crucial to deciphering RNA energy landscapes, alternative folding pathways, and phasing of distal RNA structural elements.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| E. coli K-12 MG1655 | ATCC | 47076 |
| S. cerevisiae (S288C) | ATCC | 204508 |
| Chemicals, peptides, and recombinant proteins | ||
| 1-acetylimidazole | Millipore Sigma | 157864 |
| N-Methylisatoic Anhydride | Invitrogen | M25 |
| 2-methylnicotinic acid imidazole | Millipore Sigma | 913839 |
| 1,1¢-carbonyldiimidazole | Millipore Sigma | 115533 |
| YPD | Millipore Sigma | Y1500 |
| M9 minimal salts, 5X | Millipore Sigma | M6030 |
| LB Broth (Luria low salt) | Millipore Sigma | L3397 |
| Critical commercial assays | ||
| YeaStar RNA kit | Zymo Research | R1002 |
| Direct RNA nanopore sequencing kit | Oxford Nanopore Technologies | SQK-RNA002 |
| MinION flow cell | Oxford Nanopore Technologies | FLO-MIN106D |
| Flongle flow cell | Oxford Nanopore Technologies | FLO-FLG001 |
| HiScribe T7 Quick High Yield RNA Synthesis Kit | New England Biolabs | E2050S |
| Deposited data | ||
| Raw nanopore sequence data | NCBI SRA | PRJNA634693, https://www.ncbi.nlm.nih.gov/bioproject/634693 |
| Modification detection output, raw absorbance data, SHAPE-MaP profile data | GitHub | https://github.com/physnano/rRNA_nanoSHAPE |
| Oligonucleotides | ||
| ec16f_t7p: 5′-TAATACGACTCACTATAGGGAAATTGAA GAGTTTGATCATGGCTC-3′ |
This Paper | N/A |
| ec16r: 5′-TAAGGAGGTGATCCAACCG CAGG-3′ |
This Paper | N/A |
| ec23f_t7p: 5′-TAATACGACTCACTATAGGGGGTTAA GCGACTAAGCGTACACGGT-3′ |
This Paper | N/A |
| ec23r: 5′-AAGGTTAAGCCTCACGGTTC ATTAG-3′ |
This Paper | N/A |
| sc18f_t7p: 5′- TAATACGACTCACTATAGGGTATCT GGTTGATCCTGCCAGTAGTC-3′ |
This Paper | N/A |
| sc18r: 5′- TAATGATCCTTCCGCAGGTTCACCTAC-3′ | This Paper | N/A |
| sc25f_t7p: 5′- TAATACGACTCACTATAGGGGTTTGA CCTCAAATCAGGTAGGAGTA-3′ |
This Paper | N/A |
| sc25r: 5′- ACAAATCAGACAACAAAGGCTTAATCTC-3′ | This Paper | N/A |
| Software and algorithms | ||
| nanopolish v0.11.1 | Simpson Lab: https://simpsonlab.github.io/ | https://github.com/jts/nanopolish |
| tombo v1.5.1 | Stoiber et al.31 | https://github.com/nanoporetech/tombo |
| Jupyter notebooks | Project Jupyter | https://jupyter.org/ |
| Analysis and plotting scripts | This Paper | https://github.com/physnano/rRNA_nanoSHAPE |
| guppy v3.1.5 | Oxford Nanopore Technologies | https://nanoporetech.com/ |
| ont_fast5_api | Oxford Nanopore Technologies | https://github.com/nanoporetech/ont_fast5_api |
| R-chie | Lai et al.50 | https://www.e-rna.org/r-chie/ |
| RNAstructure v6.2 | Mathews Lab | https://rna.urmc.rochester.edu/RNAstructure.html |
| RNAfold v2.4.13 | RNAfold WebServer | http://rna.tbi.univie.ac.at//cgi-bin/RNAWebSuite/RNAfold.cgi |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Dr. William Stephenson, stephenson.william@gene.com.
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
E. coli and S. Cerevisiae
E. coli (K-12 MG1655) cells were grown in an overnight culture at 37°C in 5% CO2 in 10 mL of freshly prepared Luria Bertani broth (LB) or 10 mL M9 minimal salts. 75 mL of pre-warmed media was inoculated with 1 mL of overnight culture. E. coli cells were grown to OD600 = 0.5, typically over 3 - 4 h at 37°C.
S. cerevisiae (S288C), colonies were picked from an agar plate and incubated at 30°C in ∼7 mL of YPD broth for two days.
Method details
rRNA extraction (E. coli and S. cerevisiae)
25 mL of E. coli cells were pelleted at 3280 × g at 4°C for 12 min. Cells were lysed in 16.5 mL lysis buffer (15mM Tris pH 8, 450 mM sucrose, 8 mM EDTA, 0.4 mg/mL lysozyme) for 5 min at room temperature then 10 min at 4°C. The pellet was collected at 3280 × g for 5 min and then resuspended in 2 mL proteinase K buffer (50 mM HEPES, 200 mM NaCl, 5 mM MgCl2, 1.5% SDS, 0.2 mg/mL Proteinase K). The solution was vortexed for 10 s and incubated at room temperature for 5 min, and then 4°C for 10 min. Nucleic acids were extracted twice with 1 volume of phenol:chloroform:isoamyl alcohol (25:24:1) followed by two subsequent chloroform extractions prior to ethanol precipitation and resuspension in 88 μl RNase-free water. Purified nucleic acids were treated with Turbo DNase (10 μl Turbo DNase buffer [10X] and 2 μl Turbo DNase) at 37°C for 1 h. Finally, RNA was purified with 0.8X vol AmpureXP beads.
For S. cerevisiae, RNA extraction was carried out using the YeaStar RNA kit (Zymo Research) according to instructions.
Generation of rRNA IVT controls
gDNA was extracted from E. coli using the method described for RNA above up to and including the ethanol precipitation step. After resuspension in 88 μl RNase-free water, purified nucleic acids were treated with 1-5 μl 1 mg/mL RNaseA (QIAGEN) for 45 min at 37°C to degrade RNA. gDNA was then purified with 0.5X SPRI or subsequent ethanol precipitation. gDNA was purified from S. cerevisiae using the YeaStar Genomic DNA kit (Zymo Research) according to instructions. Primers for amplifying rDNA amplicons, which include a T7 transcription promoter for subsequent in vitro transcription (IVT) are detailed in Table S2. Amplicons were generated by PCR using Kapa HiFi DNA polymerase and purified by SPRI. T7 transcription templates were transcribed using HiScribe T7 Quick High Yield RNA Synthesis Kit (New England Biolabs). Reactions were cleaned up using the MEGAClear Transcription Clean-up Kit (Invitrogen) before nanopore sequencing library preparation.
Poly(A) tailing of RNA
Oxford Nanopore Technologies direct RNA sequencing requires a poly(A) tail for first adaptor ligation. Both rRNA and pri-miR-17∼92 (951 nt) samples were poly(A) tailed. Briefly, 0.5 – 1.0 ug of RNA was poly(A) tailed with 2 ul ATP [10mM], 1 ul [5U] of E. coli Poly(A) polymerase (EPAP) (New England Biolabs) and 2 ul EPAP reaction buffer [2X] in a final volume of 20 ul. Reactions were carried out for 15 min and quenched with 0.5 ul of 0.5 M EDTA before purification with 1X SPRI.
Nanopore library preparation
Direct RNA sequencing was performed using the Oxford Nanopore Technologies kit (SQK-RNA002) as directed with the RCS control RNA. Sequencing was performed on the MinION device using either standard flowcells (FLO-MIN106D) for rRNA experiments or a mixture of standard and flongle flowcells (FLO-FLG001) for pri-miR-17∼92 experiments. Sequencing was carried out until the number of active nanopores dropped below 5% of the initial total number of pores, typically 12-36 h.
Reagents
All standard laboratory reagents, including AcIm, were purchased from Millipore-Sigma, with the exception of NMIA purchased from Invitrogen/Thermo Fisher Scientific. NAI was synthesized from 2-methylnicotinic acid and 1,1′-carbonyldiimidazole, as described.51 Briefly, 137 mg (1mmol) 2-methylnicotinic acid was dissolved in 0.5 mL anhydrous DMSO. A solution of 162 mg (1 mmol) 1,1′-carbonyldiimidazole in 0.5 mL anhydrous DMSO was added dropwise over 5 min. The resulting solution was stirred at room temperature using a PTFE coated micromagnet until gas evolution was complete and then stirred at room temperature for 1 h further. The resulting solution was used as a 1.0 M stock solution (assuming complete conversion) containing a 1:1 mixture of the desired compound and imidazole. The NAI stock solution was aliquoted and frozen at −80°C when not in use. The reagent is stable for several months if stored in anhydrous DMSO at −80°C. The stock solution should be warmed to room temperature prior to opening.
AcIm hydrolysis
AcIm hydrolysis was tracked at 37°C by time resolved UV absorbance using a Nanodrop 2000 spectrophotometer in [1x] modification buffer (100 mM HEPES pH 8.0, 100 mM NaCl, 10mM MgCl2) every 2 min for 40 min. Imidazole spectra were collected every 2 min in [1x] modification buffer for 40 min.
SHAPE-MaP on rRNA
Extracted rRNA was treated with NAI [100 mM final], AcIm [100 mM final], NMIA [13 mM final] or DMSO (unmodified control). All SHAPE-MaP experiments were performed with 10% volume fraction of DMSO. Modification was carried out at 37°C for 3 half-lives of the chemical probe used. For mutational profiling RT, 1 μl of nonamer primer [200 ng/μl or 2μM] was added to 1-3 μg of rRNA in 10 μl nuclease free water. The samples were incubated at 65°C for 5 min then cooled on ice. 8 μl of [2.5x] MaP buffer (125 mM Tris pH 8.0, 187.5 mM KCl, 15 mM MnCl2, 25 mM DTT, and 1.25 mM dNTPs) was added and incubated at 42°C for 2 min. 1 μl of SuperScript II reverse transcriptase was added and mixed well before incubating the reaction at 42°C for 2-3 h, and then at 70°C to inactivate the polymerase. cDNA was exchanged into water using G-50 columns (GE Life Sciences) the volume increased to 68 μl using nuclease free water. Second strand synthesis was carried out (Second Strand Synthesis Enzyme mix; New England Biolabs) and the dsDNA was used to generate a Nextera library for sequencing on an Illumina MiSeq, as described.52
RNA modification (pri-miR-17∼92)
In order to protect the 3′-OH of pri-miR-17∼92 RNA from modification with acylating reagents, the terminal 3′ nucleotide was oxidized followed by a beta-elimination reaction to remove the terminal nucleotide leaving a terminal phosphate. Then RNA modification was carried out prior to dephosphorylation and nanopore library preparation. Briefly, pri-miR-17∼92 was incubated at 37°C for 30 min with shaking in oxidation buffer (NaIO4 [20mM], Lysine-HCl [200mM] pH 8.5, final volume: 40 μl). The reaction was quenched with 2 μl of ethylene glycol then purified using 1x SPRI, eluting into beta-elimination buffer (Sodium borate [33.75mM], boric acid [50mM], pH 9.5) incubating at 45°C for 45 min. RNA was again purified by 1x SPRI. 1-2.5 μg of IVT pri-miR-17∼92 RNA was diluted into 7 μl water and heated to 95°C for 2 min and immediately placed on ice (2 min). 6μl of folding buffer [3.3x] (333 mM Tris-HCl pH 8.0, 333 mM NaCl and 33 mM MgCl2) and 5 μl HEPES pH 8.0 [200 mM] were added and the RNA was allowed to fold for 20 min at 37°C. 2 μl of DMSO (control) or SHAPE reagent (NAI or AcIm) were added to a new tube, then folded RNA was added and mixed by pipetting. Modification was carried out for at least 3 half-lives at 37°C. RNA was then dephosphorylated by adding 22 μl RNase-free water, 5 μl Antarctic Phosphatase reaction buffer [10x], 2 μl Antarctic Phosphatase [5k U/mL] (NEB) and 1 μl RNase inhibitor and incubating at 37°C for 30 min with shaking. The phosphatase was inactivated by incubating the reaction at 65°C for 5 min. Finally, the RNA was purified using 1x SPRI prior to poly(A) tailing.
SHAPE-MaP on pri-miR-17∼92
After pri-miR-17∼92 RNA was dephosphorylated, mutational profiling reverse transcription (RT) was performed. 1 μl of nonamer primer [200 ng/μl or 2μM] was added to 1-3 μg of RNA in 10 μl nuclease free water. The samples were incubated at 65°C for 5 min then cooled on ice. 8 μl of [2.5x] MaP buffer (125 mM Tris pH 8.0, 187.5 mM KCl, 15 mM MnCl2, 25 mM DTT, and 1.25 mM dNTPs) was added and incubated at 42°C for 2 min. 1 μl of SuperScript II reverse transcriptase was added and mixed well before incubating the reaction at 42°C for 2-3 h, and then at 70°C to inactivate the polymerase. cDNA was exchanged into water using G-50 columns (GE Life Sciences) the volume increased to 68 μl using nuclease free water. Second strand synthesis was carried out (Second Strand Synthesis Enzyme mix; New England Biolabs) and the dsDNA was used to generate a Nextera library for sequencing on an Illumina MiSeq, as described.52
Quantification and statistical analysis
Nanopore data processing
Multi-fast5 reads were basecalled using guppy (v3.1.5). Base called multi-fast5 reads were then converted to single read fast5s using the Oxford Nanopore Technologies API, ont_fast5 (v1.0.1). Fastqs were mapped to their respective transcriptomes for E. coli (GenBank: NC_000913.3) and S. cerevisiae (Saccharomyces Genome Database: R1-1-1_19960731.fsa) using minimap2 (v2.11).
Nanopolish and Tombo analysis of data
Tombo (v1.5.1) and Nanopolish (v0.11.1) were both used to detect native modifications in rRNA datasets as well as detect modifications deposited from SHAPE reagents. Comparisons were performed between native and IVT samples for the rRNA datasets and between modified (at indicated concentrations) and unmodified samples for pri-miR-17∼92. Nanopolish eventalign module was used to align current intensities and dwell times to reference sequences. Kolmogorov–Smirnov (KS) statistical testing was performed in order to detect modified nucleotides. Using Tombo, raw signal squiggles were assigned to reference sequences using resquiggle. Next, modified base detection was carried out using the detect_modifications model_sample_compare method. Per-read statistical testing (AcIm modified RNA) was performed with a ± 1 nucleotide Fisher’s method context adjustment. The requisite text output was obtained using text_output browser_files method. Reactivity profiles from Tombo per-read statistical testing were further adjusted using Benjamini-Hochberg procedure for multiple testing. Adjusted per-read reactivity profiles were used to calculate percentage modification per genomic position. This percentage profile was then normalized using the normalization procedure described in SHAPE-MaP method.52 Single molecule positional current, standard deviation of current, and dwell time data were extracted as numpy arrays directly from single read fast5 data using custom written python scripts.
RNA structure modeling
Centroid structures and free energies were obtained using the RNAfold (v2.4.13) (Vienna) web server. (http://rna.tbi.univie.ac.at//cgi-bin/RNAWebSuite/RNAfold.cgi) Options were to avoid isolated base pairs and temperature = 37°C. R-chie50 was used for displaying base pairing (arc) of centroid structures. The RNAstructure (v6.2) software suite53 was used for partition function calculation and associated dot plot visualization. The following options were used for partition function calculation: maximum percent energy difference = 10%, maximum number of structures = 50, window size = 3, temperature = 37°C.
Positive predictive value (PPV) and sensitivity
PPV and sensitivity were determined by performing pairwise comparison between the respective minimum free energy (MFE) and centroid structures of miR-17∼92 for sequence alone prediction, SHAPE-MaP and nanoSHAPE constrained experiments. CT files from constrained and unconstrained RNAfold predictions were used as input to the RNAstructure scorer function (https://rna.urmc.rochester.edu/RNAstructureWeb/Servers/scorer/scorer.html) to determine PPV and sensitivity. PPV corresponds to the percentage of predicted base pairs that are in the “accepted” structure and sensitivity corresponds to the percentage of known base pairs correctly predicted in the “accepted” structure.
Acknowledgments
This work was supported by the National Institutes of Health (HG010538 to W.T. and GM122532 to K.M.W.). The authors would like to acknowledge sequencing support from Oxford Nanopore Technologies (NYC). Additionally, we would like to thank Marcus Stoiber and Jared T. Simpson for assistance with Tombo and Nanopolish, respectively.
Author contributions
Conceptualization, W.S.; methodology W.S., P.S., W.T., and K.M.W.; investigation, W.S.; formal analysis, W.S., R.R., S.B., K.M.W., and W.T.; resources, W.T. and P.S.; writing – original draft, W.S., P.S., and W.T.; writing – review & editing, W.S., R.R., W.T., K.M.W., and P.S.; supervision, W.T., K.M.W., and P.S.; funding acquisition W.T., K.M.W., and P.S.
Declaration of interests
W.T. has two patents (8,748,091 and 8,394,584) licensed to ONT. W.S. and W.T. received reimbursement for travel, accommodation, and/or conference fees to speak at events organized by ONT. K.M.W. is an advisor to and holds equity in Ribometrix.
Published: February 9, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2022.100097.
Supplemental information
Data and code availability
-
•
Sequencing data have been deposited at the NCBI SRA and are publicly available as of the date of publication. The accession number is listed in the key resources table.
-
•
All computer code to reproduce the analyses and figures in this work has been deposited to GitHub are publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Gilbert W.V., Bell T.A., Schaening C. Messenger RNA modifications: Form, distribution, and function. Science. 2016;352:1408–1412. doi: 10.1126/science.aad8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jiang J., Seo H., Chow C.S. Post-transcriptional Modifications Modulate rRNA Structure and Ligand Interactions. Acc. Chem. Res. 2016;49:893–901. doi: 10.1021/acs.accounts.6b00014. [DOI] [PubMed] [Google Scholar]
- 3.Eyler D.E., Franco M.K., Batool Z., Wu M.Z., Dubuke M.L., Dobosz-Bartoszek M., Jones J.D., Polikanov Y.S., Roy B., Koutmou K.S. Pseudouridinylation of mRNA coding sequences alters translation. Proc. Natl. Acad. Sci. USA. 2019;116:23068–23074. doi: 10.1073/pnas.1821754116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patil D.P., Pickering B.F., Jaffrey S.R. Reading m6A in the Transcriptome: m6A-Binding Proteins. Trends Cell Biol. 2018;28:113–127. doi: 10.1016/j.tcb.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zaccara S., Ries R.J., Jaffrey S.R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019;20:608–624. doi: 10.1038/s41580-019-0168-5. [DOI] [PubMed] [Google Scholar]
- 6.Dai Q., Moshitch-Moshkovitz S., Han D., Kol N., Amariglio N., Rechavi G., Dominissini D., He C. Nm-seq maps 2′-O-methylation sites in human mRNA with base precision. Nat. Methods. 2017;14:695–698. doi: 10.1038/nmeth.4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi J., Indrisiunaite G., DeMirci H., Ieong K.W., Wang J., Petrov A., Prabhakar A., Rechavi G., Dominissini D., He C., et al. 2′-O-methylation in mRNA disrupts tRNA decoding during translation elongation. Nat. Struct. Mol. Biol. 2018;25:208–216. doi: 10.1038/s41594-018-0030-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harcourt E.M., Kietrys A.M., Kool E.T. Chemical and structural effects of base modifications in messenger RNA. Nature. 2017;541:339–346. doi: 10.1038/nature21351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwartz S., Bernstein D.A., Mumbach M.R., Jovanovic M., Herbst R.H., León-Ricardo B.X., Engreitz J.M., Guttman M., Satija R., Lander E.S., et al. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell. 2014;159:148–162. doi: 10.1016/j.cell.2014.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jonkhout N., Tran J., Smith M.A., Schonrock N., Mattick J.S., Novoa E.M. The RNA modification landscape in human disease. RNA. 2017;23:1754–1769. doi: 10.1261/rna.063503.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gonzales B., Henning D., So R.B., Dixon J., Dixon M.J., Valdez B.C. The Treacher Collins syndrome (TCOF1) gene product is involved in pre-rRNA methylation. Hum. Mol. Genet. 2005;14:2035–2043. doi: 10.1093/hmg/ddi208. [DOI] [PubMed] [Google Scholar]
- 12.Yu Y.T., Meier U.T. RNA-guided isomerization of uridine to pseudouridine--pseudouridylation. RNA Biol. 2014;11:1483–1494. doi: 10.4161/15476286.2014.972855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Helm M., Motorin Y. Detecting RNA modifications in the epitranscriptome: predict and validate. Nat. Rev. Genet. 2017;18:275–291. doi: 10.1038/nrg.2016.169. [DOI] [PubMed] [Google Scholar]
- 14.Li X., Xiong X., Yi C. Epitranscriptome sequencing technologies: decoding RNA modifications. Nat. Methods. 2016;14:23–31. doi: 10.1038/nmeth.4110. [DOI] [PubMed] [Google Scholar]
- 15.Carlile T.M., Rojas-Duran M.F., Zinshteyn B., Shin H., Bartoli K.M., Gilbert W.V. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature. 2014;515:143–146. doi: 10.1038/nature13802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meyer K.D. DART-seq: an antibody-free method for global m6A detection. Nat. Methods. 2019;16:1275–1280. doi: 10.1038/s41592-019-0570-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schaefer M., Pollex T., Hanna K., Lyko F. RNA cytosine methylation analysis by bisulfite sequencing. Nucleic Acids Res. 2009;37:e12. doi: 10.1093/nar/gkn954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rose R.E., Quinn R., Sayre J.L., Fabris D. Erratum: Profiling ribonucleotide modifications at full-transcriptome level: a step toward MS-based epitranscriptomics. RNA. 2015;21:2143. doi: 10.1261/rna.054908.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang N., Shi S., Jia T.Z., Ziegler A., Yoo B., Yuan X., Li W., Zhang S. A general LC-MS-based RNA sequencing method for direct analysis of multiple-base modifications in RNA mixtures. Nucleic Acids Res. 2019;47:e125. doi: 10.1093/nar/gkz731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herzog V.A., Reichholf B., Neumann T., Rescheneder P., Bhat P., Burkard T.R., Wlotzka W., von Haeseler A., Zuber J., Ameres S.L. Thiol-linked alkylation of RNA to assess expression dynamics. Nat. Methods. 2017;14:1198–1204. doi: 10.1038/nmeth.4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drexler H.L., Choquet K., Churchman L.S. Splicing Kinetics and Coordination Revealed by Direct Nascent RNA Sequencing through Nanopores. Mol. Cell. 2020;77:985–998.e8. doi: 10.1016/j.molcel.2019.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kawata K., Wakida H., Yamada T., Taniue K., Han H., Seki M., Suzuki Y., Akimitsu N. Metabolic labeling of RNA using multiple ribonucleoside analogs enables the simultaneous evaluation of RNA synthesis and degradation rates. Genome Res. 2020;30:1481–1491. doi: 10.1101/gr.264408.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meng L., Guo Y., Tang Q., Huang R., Xie Y., Chen X. Metabolic RNA labeling for probing RNA dynamics in bacteria. Nucleic Acids Res. 2020;48:12566–12576. doi: 10.1093/nar/gkaa1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siegfried N.A., Busan S., Rice G.M., Nelson J.A.E., Weeks K.M. RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP) Nat. Methods. 2014;11:959–965. doi: 10.1038/nmeth.3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wan Y., Qu K., Zhang Q.C., Flynn R.A., Manor O., Ouyang Z., Zhang J., Spitale R.C., Snyder M.P., Segal E., Chang H.Y. Landscape and variation of RNA secondary structure across the human transcriptome. Nature. 2014;505:706–709. doi: 10.1038/nature12946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mustoe A.M., Busan S., Rice G.M., Hajdin C.E., Peterson B.K., Ruda V.M., Kubica N., Nutiu R., Baryza J.L., Weeks K.M. Pervasive Regulatory Functions of mRNA Structure Revealed by High-Resolution SHAPE Probing. Cell. 2018;173:181–195.e18. doi: 10.1016/j.cell.2018.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garalde D.R., Snell E.A., Jachimowicz D., Sipos B., Lloyd J.H., Bruce M., Pantic N., Admassu T., James P., Warland A., et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods. 2018;15:201–206. doi: 10.1038/nmeth.4577. [DOI] [PubMed] [Google Scholar]
- 28.Workman R.E., Tang A.D., Tang P.S., Jain M., Tyson J.R., Razaghi R., Zuzarte P.C., Gilpatrick T., Payne A., Quick J., et al. Nanopore native RNA sequencing of a human poly(A) transcriptome. Nat. Methods. 2019;16:1297–1305. doi: 10.1038/s41592-019-0617-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith A.M., Jain M., Mulroney L., Garalde D.R., Akeson M. Reading canonical and modified nucleobases in 16S ribosomal RNA using nanopore native RNA sequencing. PLoS ONE. 2019;14:e0216709. doi: 10.1371/journal.pone.0216709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grünberger F., Knüppel R., Jüttner M., Fenk M., Borst A., Reichelt R., Hausner W., Soppa J., Ferreira-Cerca S., Grohmann D. Exploring prokaryotic transcription, operon structures, rRNA maturation and modifications using Nanopore-based native RNA sequencing. bioRxiv. 2020 doi: 10.1101/2019.12.18.880849. [DOI] [Google Scholar]
- 31.Stoiber M., Quick J., Egan R., Lee J.E., Celniker S.E., Neely R.K., Loman N., Pennacchio L.A., Brown J. De novo Identification of DNA Modifications Enabled by Genome-Guided Nanopore Signal Processing. bioRxiv. 2017 doi: 10.1101/094672. [DOI] [Google Scholar]
- 32.Simpson J.T., Workman R.E., Zuzarte P.C., David M., Dursi L.J., Timp W. Detecting DNA cytosine methylation using nanopore sequencing. Nat. Methods. 2017;14:407–410. doi: 10.1038/nmeth.4184. [DOI] [PubMed] [Google Scholar]
- 33.Siibak T., Remme J. Subribosomal particle analysis reveals the stages of bacterial ribosome assembly at which rRNA nucleotides are modified. RNA. 2010;16:2023–2032. doi: 10.1261/rna.2160010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taoka M., Nobe Y., Yamaki Y., Yamauchi Y., Ishikawa H., Takahashi N., Nakayama H., Isobe T. The complete chemical structure of Saccharomyces cerevisiae rRNA: partial pseudouridylation of U2345 in 25S rRNA by snoRNA snR9. Nucleic Acids Res. 2016;44:8951–8961. doi: 10.1093/nar/gkw564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manrao E.A., Derrington I.M., Laszlo A.H., Langford K.W., Hopper M.K., Gillgren N., Pavlenok M., Niederweis M., Gundlach J.H. Reading DNA at single-nucleotide resolution with a mutant MspA nanopore and phi29 DNA polymerase. Nat. Biotechnol. 2012;30:349–353. doi: 10.1038/nbt.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prusiner P., Yathindra N., Sundaralingam M. Effect of ribose O(2′)-methylation on the conformation of nucleosides and nucleotides. Biochim. Biophys. Acta. 1974;366:115–123. doi: 10.1016/0005-2787(74)90325-6. [DOI] [PubMed] [Google Scholar]
- 37.Craig J.M., Laszlo A.H., Nova I.C., Brinkerhoff H., Noakes M.T., Baker K.S., Bowman J.L., Higinbotham H.R., Mount J.W., Gundlach J.H. Determining the effects of DNA sequence on Hel308 helicase translocation along single-stranded DNA using nanopore tweezers. Nucleic Acids Res. 2019;47:2506–2513. doi: 10.1093/nar/gkz004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown R.F., Andrews C.T., Elcock A.H. Stacking free energies of all DNA and RNA nucleoside pairs and dinucleoside-monophosphates computed using recently revised AMBER parameters and compared with experiment. J. Chem. Theory Comput. 2015;11:2315–2328. doi: 10.1021/ct501170h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Homan P.J., Favorov O.V., Lavender C.A., Kursun O., Ge X., Busan S., Dokholyan N.V., Weeks K.M. Single-molecule correlated chemical probing of RNA. Proc. Natl. Acad. Sci. USA. 2014;111:13858–13863. doi: 10.1073/pnas.1407306111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sengupta A., Rice G.M., Weeks K.M. Single-molecule correlated chemical probing reveals large-scale structural communication in the ribosome and the mechanism of the antibiotic spectinomycin in living cells. PLoS Biol. 2019;17:e3000393. doi: 10.1371/journal.pbio.3000393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aw J.G.A., Lim S.W., Wang J.X., Lambert F.R.P., Tan W.T., Shen Y., Zhang Y., Kaewsapsak P., Li C., Ng S.B., et al. Determination of isoform-specific RNA structure with nanopore long reads. Nat. Biotechnol. 2021;39:336–346. doi: 10.1038/s41587-020-0712-z. [DOI] [PubMed] [Google Scholar]
- 42.Habibian M., Velema W.A., Kietrys A.M., Onishi Y., Kool E.T. Polyacetate and Polycarbonate RNA: Acylating Reagents and Properties. Org. Lett. 2019;21:5413–5416. doi: 10.1021/acs.orglett.9b01526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Merino E.J., Wilkinson K.A., Coughlan J.L., Weeks K.M. RNA structure analysis at single nucleotide resolution by selective 2¢-hydroxyl acylation and primer extension (SHAPE) J. Am. Chem. Soc. 2005;127:4223–4231. doi: 10.1021/ja043822v. [DOI] [PubMed] [Google Scholar]
- 44.Gour-Salin B.J. Hydrolysis rates of some acetylimidazole derivatives. Can. J. Chem. 1983;61:2059. [Google Scholar]
- 45.Chaulk S.G., Thede G.L., Kent O.A., Xu Z., Gesner E.M., Veldhoen R.A., Khanna S.K., Goping I.S., MacMillan A.M., Mendell J.T., et al. Role of pri-miRNA tertiary structure in miR-17∼92 miRNA biogenesis. RNA Biol. 2011;8:1105–1114. doi: 10.4161/rna.8.6.17410. [DOI] [PubMed] [Google Scholar]
- 46.Chakraborty S., Mehtab S., Patwardhan A., Krishnan Y. Pri-miR-17-92a transcript folds into a tertiary structure and autoregulates its processing. RNA. 2012;18:1014–1028. doi: 10.1261/rna.031039.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deigan K.E., Li T.W., Mathews D.H., Weeks K.M. Accurate SHAPE-directed RNA structure determination. Proc. Natl. Acad. Sci. USA. 2009;106:97–102. doi: 10.1073/pnas.0806929106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weeks K.M. SHAPE Directed Discovery of New Functions in Large RNAs. Acc. Chem. Res. 2021;54:2502–2517. doi: 10.1021/acs.accounts.1c00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leger A., Amaral P.P., Pandolfini L., Capitanchik C., Capraro F., Miano V., Migliori V., Toolan-Kerr P., Sideri T., Enright A.J., et al. RNA modifications detection by comparative Nanopore direct RNA sequencing. Nat. Commun. 2021;12:7198. doi: 10.1038/s41467-021-27393-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lai D., Proctor J.R., Zhu J.Y.A., Meyer I.M. R-CHIE: a web server and R package for visualizing RNA secondary structures. Nucleic Acids Res. 2012;40:e95. doi: 10.1093/nar/gks241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Spitale R.C., Crisalli P., Flynn R.A., Torre E.A., Kool E.T., Chang H.Y. RNA SHAPE analysis in living cells. Nat. Chem. Biol. 2013;9:18–20. doi: 10.1038/nchembio.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smola M.J., Rice G.M., Busan S., Siegfried N.A., Weeks K.M. Selective 2′-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile and accurate RNA structure analysis. Nat. Protoc. 2015;10:1643–1669. doi: 10.1038/nprot.2015.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bellaousov S., Reuter J.S., Seetin M.G., Mathews D.H. RNAstructure: web servers for RNA secondary structure prediction and analysis. Nucleic Acids Res. 2010;41:W471–W474. doi: 10.1093/nar/gkt290. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Sequencing data have been deposited at the NCBI SRA and are publicly available as of the date of publication. The accession number is listed in the key resources table.
-
•
All computer code to reproduce the analyses and figures in this work has been deposited to GitHub are publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.