

Abstract
We report an investigation of the complexation between a water soluble pillararene host (WP6) and a panel of hydrophobic cationic guests (G1 – G20) by a combination of 1H NMR spectroscopy and isothermal titration calorimetry in phosphate buffered saline. We find that WP6 forms 1:1 complexes with Ka values in the 104 – 109 M−1 range driven by favorable enthalpic contributions. This thermodynamic dataset serves as blinded data for the SAMPL9 challenge.
Introduction
A crucial step in the drug development process is the discovery and optimization of small molecule ligands that bind to their target proteins in aqueous solution. Experimentally, this process is very expensive and time consuming because it requires an iterative process of chemical synthesis and the measurement of binding affinity.1 Accordingly, the development of computational methods that successfully rank ligands by relative affinity and deliver binding free energies with errors below 1 kcal mol−1 are highly sought by the computational chemistry community and pharmaceutical industry. Validation is an important step in the development of such computational methods. However, testing of new methods on protein•ligand systems can be computationally expensive and time consuming because proteins are large and complex entities which require that extensive conformational sampling to ensure convergence. To address this issue, a group of computational chemists has organized a series of Statistical Assessment of the Modeling of Proteins and Ligands (SAMPL) challenges2 to assess and improve the state-of-the-art. Over the years, SAMPL challenges relied upon unpublished blinded datasets including small molecule solvation free energies, HIV integrase inhibitors binding free energy, and pKa and octanol-water partition coefficient predictions.3 Supramolecular chemists are also deeply involved in the fundamentals and applications of host•guest binding and measurement of the binding free energies.4 Given that supramolecular hosts are typically smaller and conformationally more homogenous than proteins and that some supramolecular systems achieve binding affinities and selectivities that rival Nature suggested that host•guest systems (Figure 1) should be included in the SAMPL challenges.5
Figure 1.
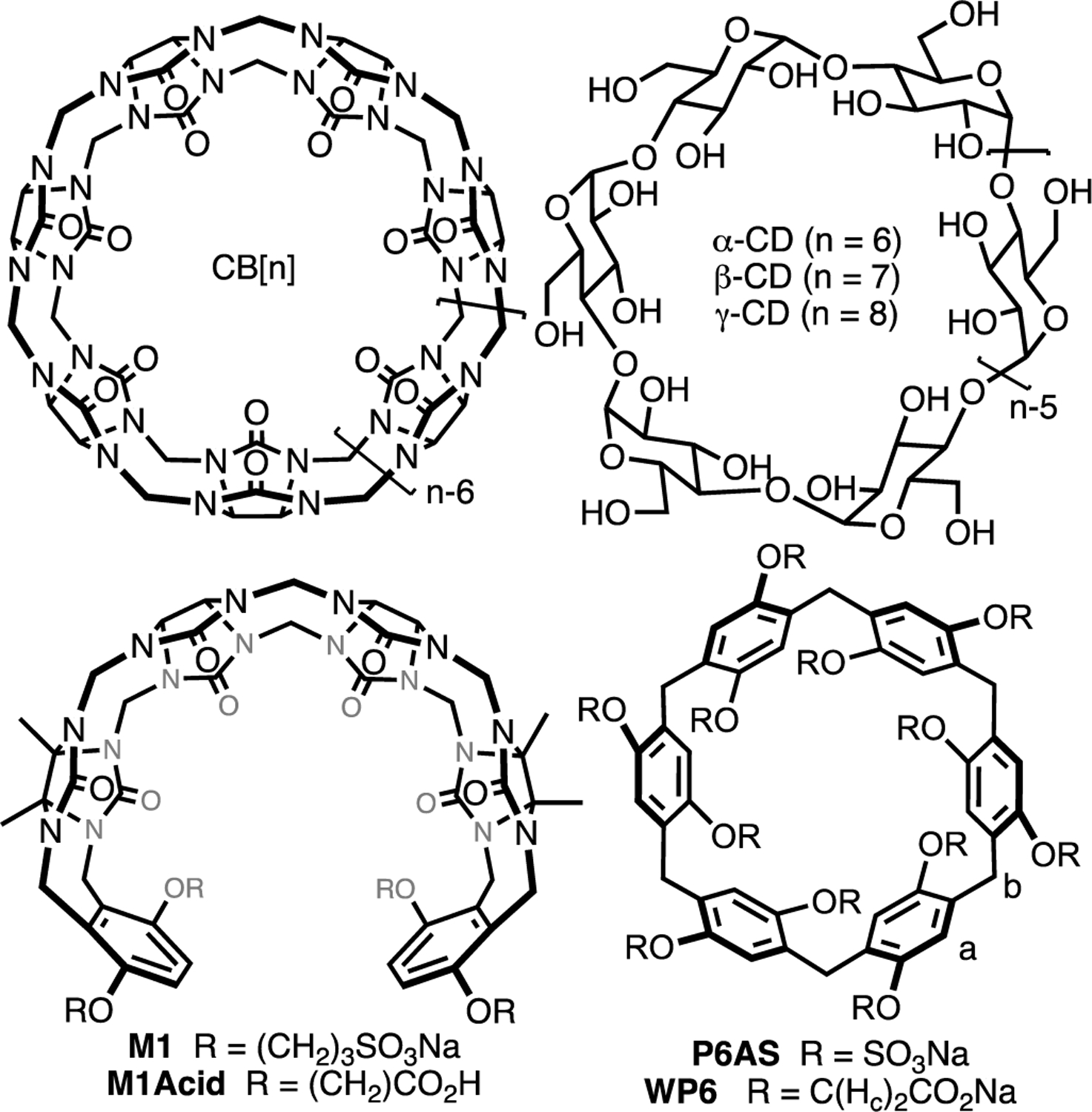
Structures of (acyclic) CB[n], cyclodextrins, and pillararenes.
The Isaacs group has a longstanding interest in the cucurbit[n]uril (CB[n]) family of molecular containers6 and has been involved in the elucidation of the mechanism of CB[n] formation as a means to create new CB[n]-type receptors and in the delineation of their host•guest recognition properties.7 We discovered that CB[n] bind tightly and with high selectivity toward hydrophobic cations in water (Ka typically 106 – 1012 M−1).8 The origin of the tight binding was traced to the presence of intracavity waters that lack a full complement of H-bonds that are released upon complexation.9 We, and others, have used CB[n]-type receptors as in vivo sequestration agents and for (targeted) drug delivery applications.5f, 10 Since SAMPL3, the Isaacs group has provided unpublished Ka values for guests toward various CB[n]-type receptors (e.g. CB[n] (n = 7, 8), acyclic CB[n] (e.g. M1Acid), and glycoluril derived molecular clips).11 The groups of Bruce Gibb and Michael Gilson have supplied blinded datasets for deep cavity cavitands and cyclodextrin derivatives, respectively.12 One issue the computational chemists encountered in previous SAMPL challenges with acyclic CB[n]-type receptors was conformational sampling. Recently, we have become interested in the pillararene family13 of molecular containers (e.g. WP6, P6AS) as sequestration agents.5f, 14 Pillararenes are macrocyclic and display high affinity toward cationic guests like viologens in water which makes them ideally suited as an alternative scaffold for the SAMPL challenges.5c, 15 Herein, we describe the binding of WP615 – which is a water soluble derivative of pillar[6]arene – toward a series of hydrophobic cations which serves as a blinded dataset for the SAMPL9 challenge.
Results and Discussion
This results and discussion section is organized as follows. First, we present the selection of the host (WP6) and guests (G1 – G20) used in the study. Subsequently, we present a qualitative investigation of the host•guest complexation by analysis of complexation induced changes in 1H NMR chemical shift and multiplicity. Thereafter, we present the determination of host•guest binding affinity and enthalpy by isothermal titration calorimetry (ITC). Finally, we discuss the thermodynamic parameters as a function of guest structure and offer come conclusions.
Selection of Host and Guests.
Previous SAMPL challenges have featured macrocyclic CB[7] and CB[8],2b, 11b, 11f glycoluril derived molecular clips and acyclic CB[n] that feature carboxylate or sulfonate groups,11a, c, e deep cavity cavitands,12a, b and cyclodextrins.12c In previous challenges, issues relating to the conformational flexibility of acyclic CB[n] hosts and the degree of deprotonation of ionizable functional groups have arisen. Accordingly, for SAMPL9 challenge we decided to select WP6 as host because it is more defined conformationally and is known to undergo strong host•guest complexation in water.14b, 15 Most studies of host•guest complexation of WP6 use less competitive media (e.g. unbuffered water or buffered water). To make the SAMPL9 challenge more biologically relevant, we elected to perform our studies in phosphate buffered saline (PBS) at physiological pH (pH 7.4). Given that WP6 is anionic at neutral pH, we knew that binding of cationic guests would be favored. Accordingly, we selected guests G1 – G20 (Figure 2) which are mono- and diammonium ions which were available from our previous studies of CB[n]•guest complexation events.8, 11b, 11d, 16 Guests G1 – G20 feature different numbers of cationic residues, different alkylation states (e.g. 1˚, 2˚, 3˚, 4˚), and different sized hydrophobic residues. Given that WP6 is highly negatively charged at neutral pH, we expected that G1 – G20 would form WP6•G complexes whose Ka values would span several orders of magnitude thereby making it easier for the computationalists to predict changes in binding free energy as a function of guest structure.
Figure 2.
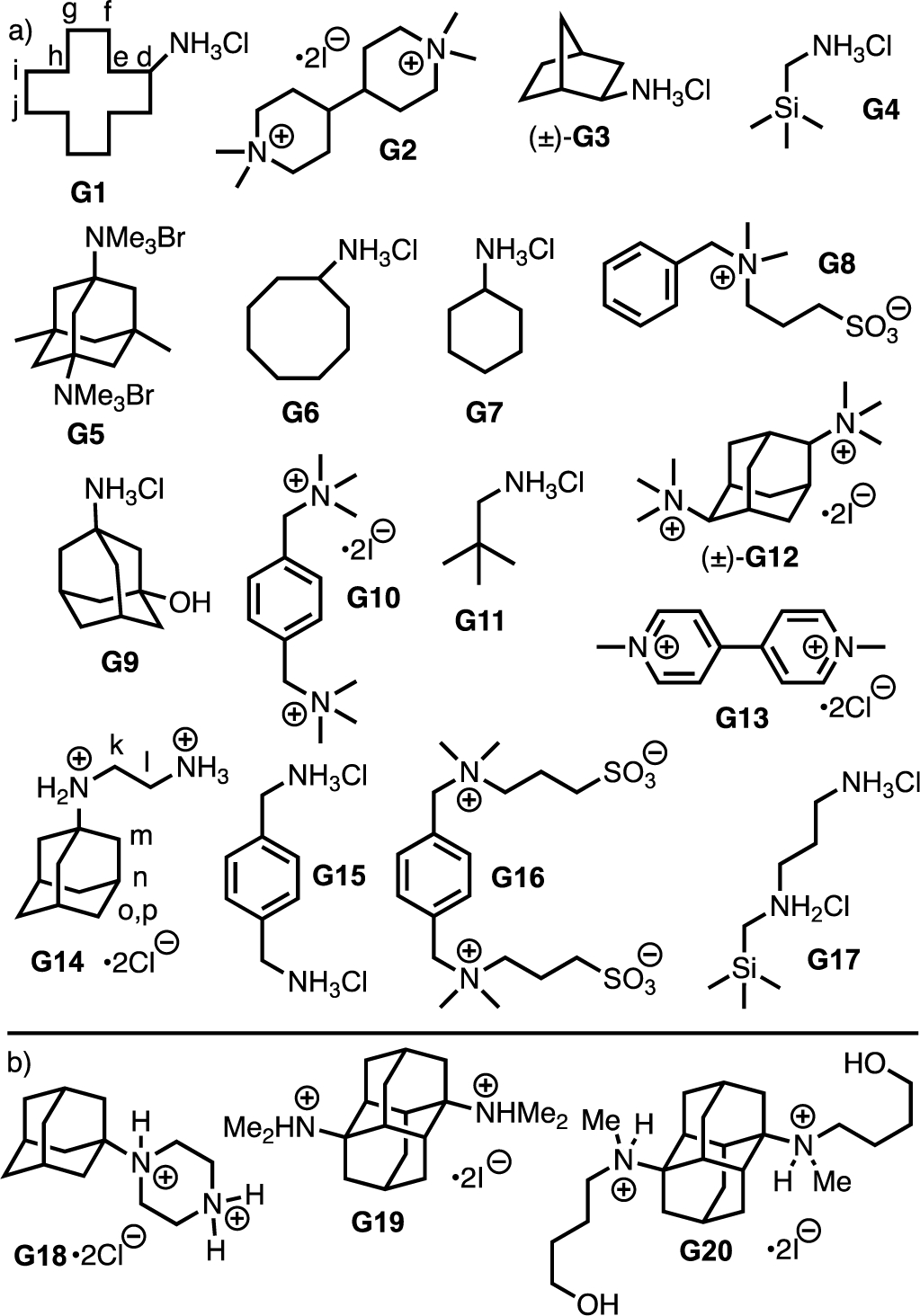
Structures of guests G1 – G20 used in this study. Panel a) guests studied by 1H NMR and ITC, b) guests studied only by 1H NMR.
Qualitative 1H NMR Host•Guest Recognition Study
As drawn in Figure 1, WP6 features a C6-axis and overall D6-symmetry and is therefore chiral.13b However, because the OCH2CO2Na substituents can rotate through the annulus of the macrocycle WP6 is isolated as a racemic mixture of planar chiral macrocycles (e.g. Rp and Sp).17 Figure 3a shows the 1H NMR spectrum recorded for WP6 which features a single sharp resonance for Ha, Hb, and Hc on the chemical shift timescale. This observation strongly suggests that rotation through the annulus is fast on the chemical shift timescale. Initially, we studied the binding of WP6 toward the panel of guests (G1 – G20) in D2O by 1H NMR stoichiometry at 1:1 and 1:2 WP6:guest stoichiometry (Supporting Information). The 1H NMR spectra recorded for WP6, G1, and 1:1 and 1:2 mixtures of WP6 and G1 (Figure 3) illustrate the spectral changes that are commonly observed. For example, at a 1:1 WP6:G1 stoichiometry (Figure 3c), the resonances for G1 within the WP6•G1 complex undergo substantial upfield shifts due to their location in the magnetically shielding environment of the macrocyclic cavity defined by the aromatic walls. Conversely, host resonance Ha undergoes a smaller downfield shift which can be explained by changes in the orientation of the aromatic walls with respect to each other. More interestingly, the Hc resonance of the OCH2CO2Na groups with the WP6•G1 complex shift downfield and split into an AB quartet (Hc, Hc’) for the diastereotopic methylene groups. In combination, this indicates that rotation through the annulus is slow on the chemical shift timescale for WP6•G1 but that exchange of guest G1 is fast on the chemical shift timescale which renders the top and bottom portals of WP6 equivalent. Figure 3d shows the 1H NMR spectrum recorded at a 1:2 WP6:G1 stoichiometry. Compared to Figure 3c, the resonances for G1 shift back toward their locations for uncomplexed G1 which further confirms the fast exchange of G1 on the chemical shift timescale. The D6-symmetric conformation of uncomplexed WP6 in dominant in aqueous solution. However, pillararenes are capable of conformational diastereoisomerism when one or more of the aromatic rings flips. For example, in the case of the WP6•G13 complex we observe a dramatic increase in complexity in the 5.5 – 7.5 ppm region of the spectrum which is consistent with reduced symmetry of the complexes (Supporting Information, Figure S12).
Figure 3.
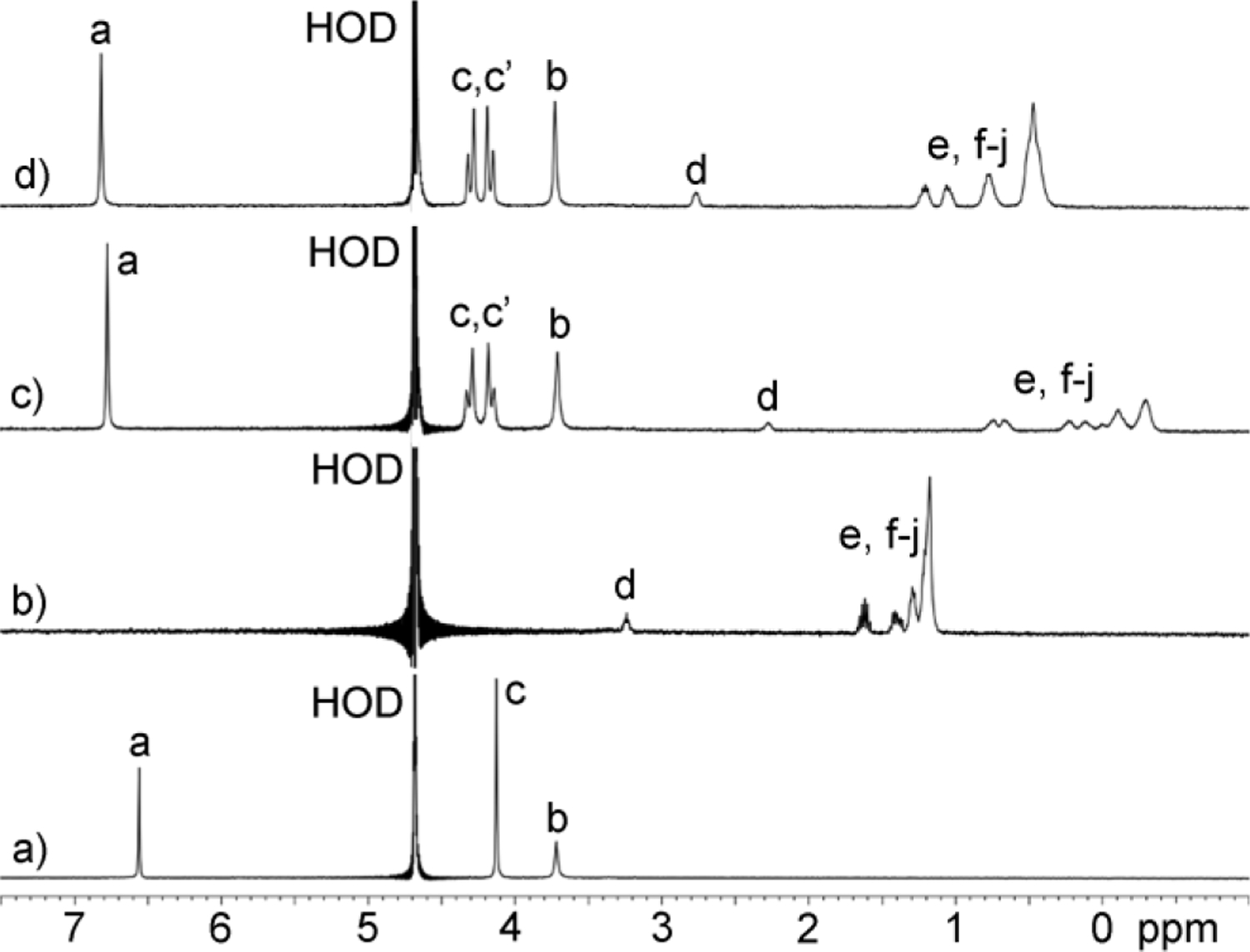
1H NMR spectra recorded (400 MHz, RT, D2O) for: a) WP6 (1 mM), b) G1 (1 mM), c) a mixture of WP6 (1 mM) and G1 (1 mM), and d) a mixture of host WP6 (1 mM) and G1 (2 mM).
The 1H NMR spectra recorded for mixtures of WP6 and guest G14 (Figure 4) provide a beautiful example of stereochemistry and chemical exchange in host•guest chemistry. For example, the observation of a sharp singlets for WP6 (Figure 4a) indicates that the top and bottom portals of WP6 are equivalent due to the presence of a C6-axis and six perpendicular C2-axes resulting in D6 point group symmetry. Similarly, the adamantane residue of guest G14 has a C3-axis and three mirror planes which results in single resonances for Hm and Hn, whereas Ho and Hp are part of the diastereotopic CH2-group (Figure 4b). The protons on the N-CH2CH2-N group (Hk and Hl) appear as coupled triplets as expected. The situation changes completely within the WP6•G14 complex (Figure 4c). As can be seen (Figure 4a,c), the aromatic resonance Ha splits into two singlets (Ha, Ha’). Apparently, the WP6•G14 complex undergoes slow guest exchange which renders the top and bottom portals of the complex chemically distinct with different chemical shifts; complexation maintains the C6-axis but eliminates the six perpendicular C2-axes. The presence of four doublets for Hc (Hc – Hc’’’) for WP6•G14 reflects the top-bottom dissymmetry and that this CH2-group is diastereotopic within the overall chiral and racemic complex. Figure 5 shows an MMFF minimized molecular model of WP6•G14 which illustrates these symmetry considerations. Even more interesting is the appearance of the resonances for guest G14 within the WP6•G14 complex. For example, Hk and Hl split into four resonances Hl, Hl’, Hk, Hk’ because the chiral WP6•G14 complex renders these CH2-groups diastereotopic and all four protons are chemically distinct. Protons Hn still appear as a single resonance in WP6•G14 because the C3-axis present in G14 is maintained in the WP6•G14 complex. Even more interesting is that the six protons Hm that appear as a single resonance in G14 split into a pair of coupled doublets Hm and Hm’ within WP6•G14. The three mirror planes that are present in the adamantane skeleton of G14 are destroyed upon complexation to form the chiral WP6•G14 complex which renders these three CH2-groups diastereotopic. All of the protons of guest G14 experience a large upfield shift upon complexation which reflects their complexation inside the hydrophobic magnetically shielding environment of the WP6 cavity. At a 1:2 WP6:G14 stoichiometry the guest exchange rate increases which results in averaged NMR where the ethylene diammonium ion tail can point out of either portal which results in a merging of the Ha and Ha’ resonances as well as the Hc – Hc’’’ resonances as expected based on symmetry considerations. The guest resonances also merge and shift back toward the chemical shift for uncomplexed G14 as expected.
Figure 4.
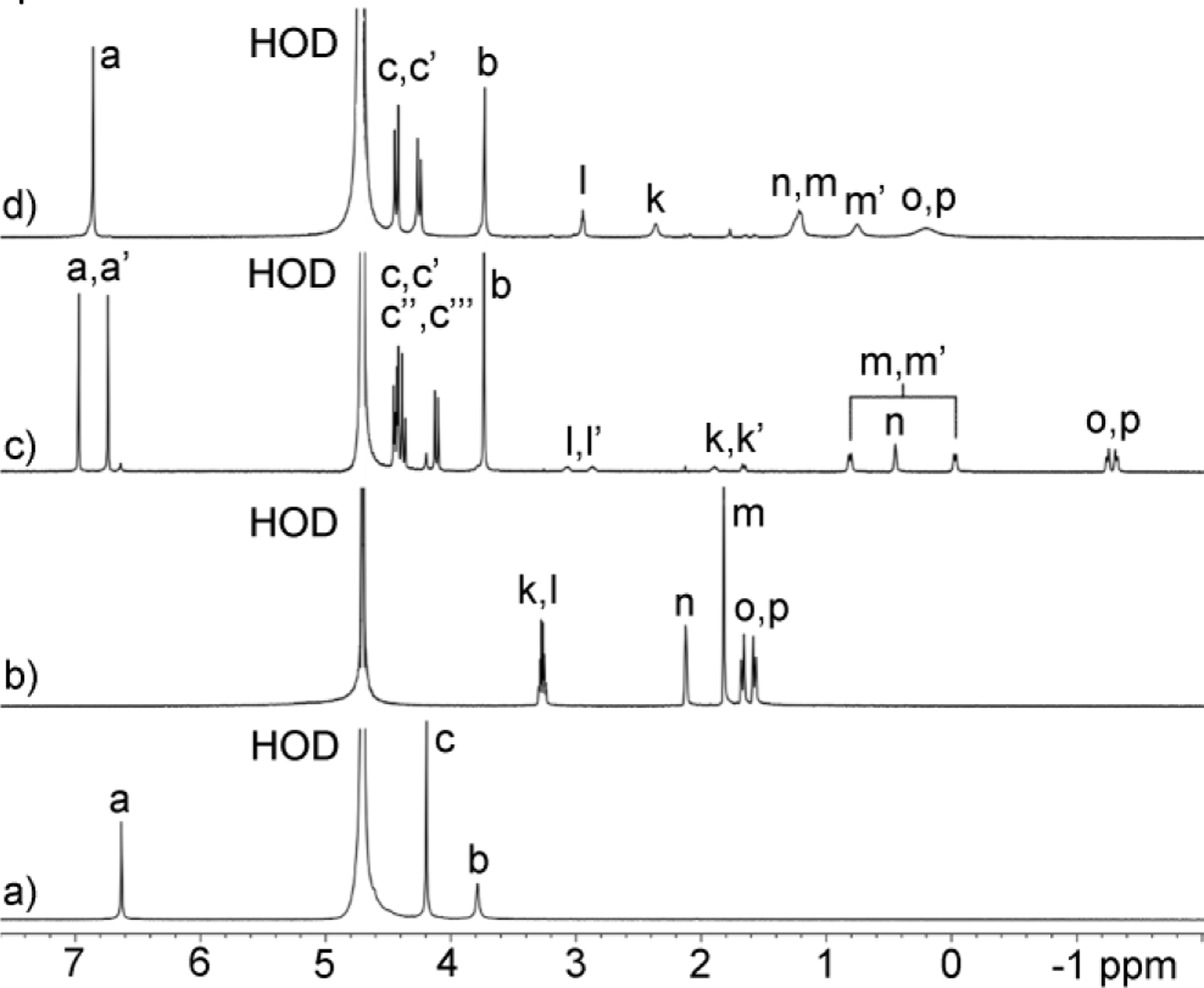
1H NMR spectra recorded (600 MHz, RT, D2O) for: a) WP6 (1 mM), b) G14 (1 mM), c) a mixture of WP6 (1 mM) and G14 (1 mM), and d) a mixture of host WP6 (1 mM) and G14 (2 mM).
Figure 5.
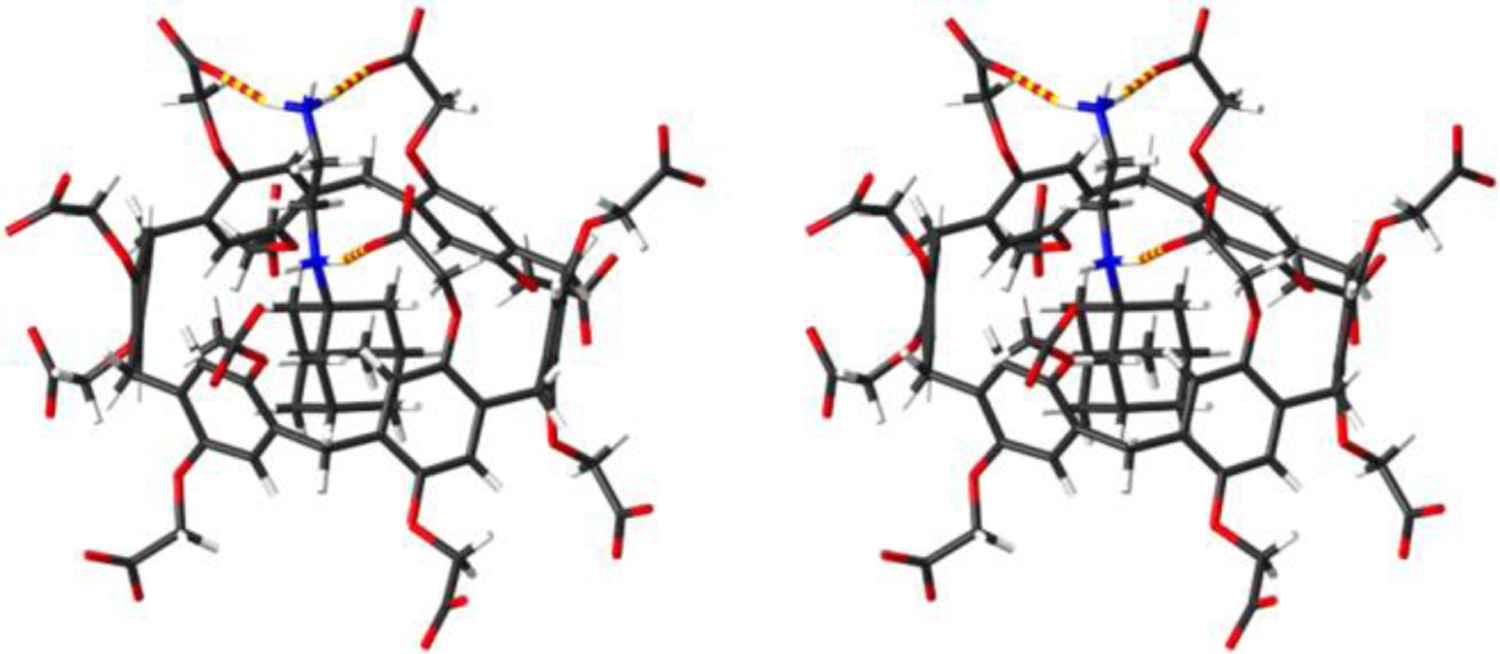
Cross-eyed stereoview of an MMFF minimized model of WP6•G14. Color code: C, gray; H, white; N, blue; O, red; H-bonds, red-yellow striped.
Measurement and Discussion of the Thermodynamic Parameters of Complex Formation.
After having qualitatively assessed the binding properties of WP6 toward the guest panel by 1H NMR spectroscopy we decided to measure the thermodynamic parameters of complexation. Given that WP6 is known to display tight binding and our desire to use a single analytical method across our measurements we turned to isothermal titration calorimetry (ITC) measurements which allows accurate Ka determination over a wide dynamic range.18 Figure 6a shows the thermogram measured when WP6 (200 μM) in the ITC cell was titrated with a solution of G7 (2.0 mM) in the syringe. All ITC experiments were conducted in duplicate. Figure 6b shows the fitting of the integrated heat values to a 1:1 binding model implemented in the PEAQ ITC data analysis software with Ka = 1.31 × 105 M−1 and ΔH = −3.18 kcal mol−1. The Ka and ΔH values for the weaker complexes (Ka ≤ 5 × 106 M−1) were determined in an analogous manner by direct ITC titrations and are presented in Table 1. In these direct titrations, the fixed concentration of WP6 in the cell was manipulated in order to optimize the c-value18c and therefore sample a larger portion of the binding isotherm and therefore deliver more reliable results.
Figure 6.
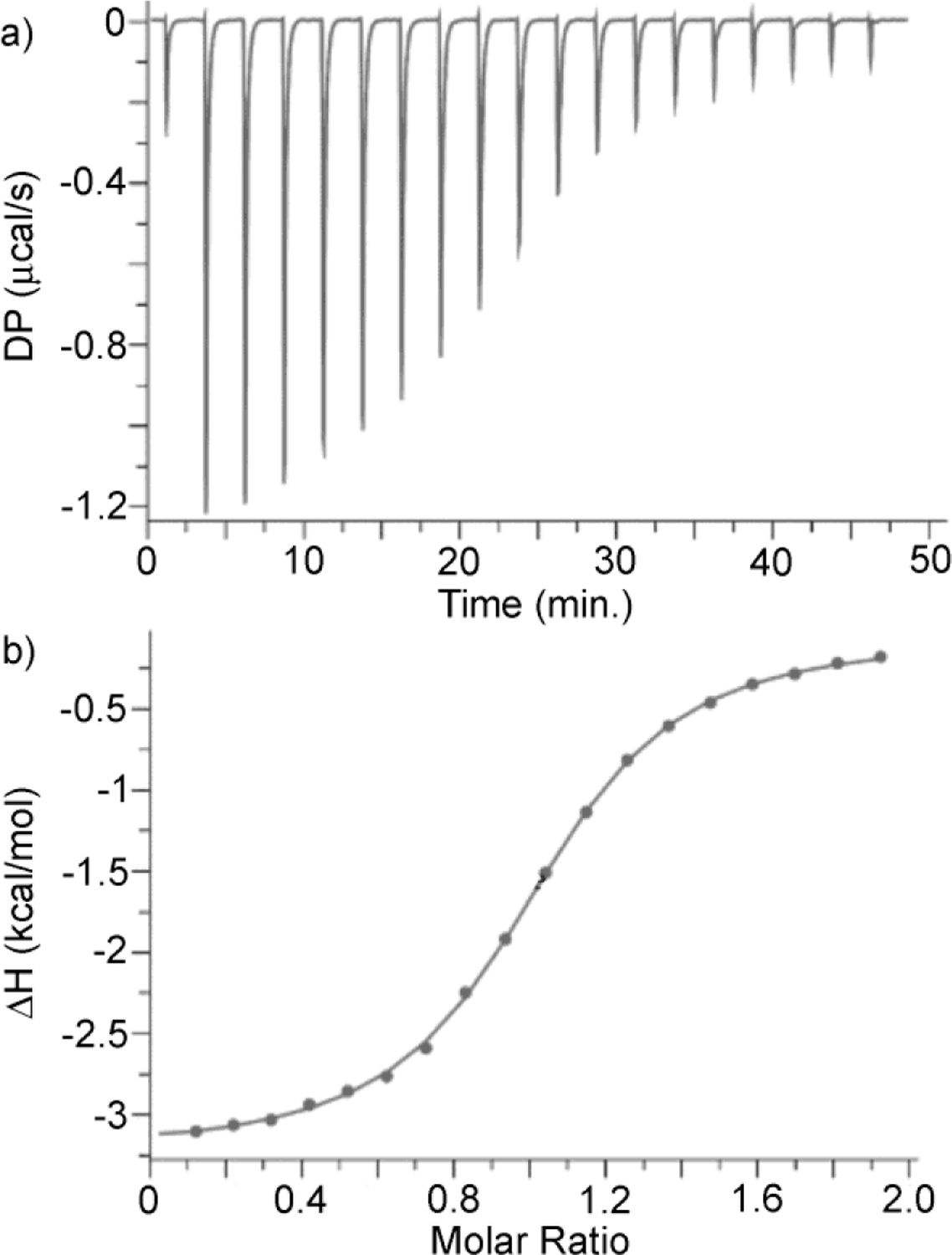
a) ITC thermogram recorded during the direct titration of WP6 (200 μM) in the cell with G7 (2.0 mM) in the syringe, b) Fitting of the data to a 1:1 binding model with Ka = 1.31 × 105 M−1.
Table 1.
Binding constants (Ka, M−1) and enthalpies (ΔH, kcal mol−1) measured for WP6•guest complexes. Conditions: 1x PBS buffer, pH 7.4, 298.15 K.
| Guest | Ka (M−1) | ΔH (kcal mol−1) |
|---|---|---|
| G1 a | (5.29 ± 0.07) × 104 | −8.08 ± 0.02 |
| G2 b | (4.59 ± 0.35) × 107 | −6.10 ± 0.02 |
| (±)-G3 a | (6.45 ± 0.18) × 105 | −4.75 ± 0.02 |
| G4 e | (5.08 ± 0.11) × 104 | −4.15 ± 0.02 |
| G5 f | (9.01 ± 0.23) × 103 | −3.95 ± 0.03 |
| G6 c | (7.09 ± 0.44) × 105 | −6.90 ± 0.07 |
| G7 d | (1.31 ± 0.05) × 105 | −3.18 ± 0.02 |
| G8 e | (2.35 ± 0.04) × 104 | −9.55 ± 0.05 |
| G9 f | (3.75 ± 0.31) × 104 | −5.31 ± 0.08 |
| G10 b | (1.61 ± 0.08) × 107 | −6.23 ± 0.02 |
| G11 f | (3.37 ± 0.05) × 104 | −5.61 ± 0.02 |
| (±)-G12 g | (9.43 ± 0.31) × 107 | −7.45 ± 0.02 |
| G13 c | (1.63 ± 0.11) × 106 | −4.98 ± 0.04 |
| G14 i | (4.69 ± 0.09) × 109 | −16.4 ± 0.02 |
| G15 h | (1.76 ± 0.06) × 107 | −7.03 ± 0.03 |
| G16 e | (1.32 ± 0.03) × 104 | −7.49 ± 0.06 |
| G17 a | (2.29 ± 0.06) × 105 | −4.15 ± 0.02 |
Measured by direct ITC titration of WP6 in the cell with guest in the syringe
[WP6] = 0.1 mM, [guest] = 1.0 mM
[WP6] = 0.05 mM, [guest] = 0.5 mM
[WP6] = 0.2 mM, [guest] = 2.0 mM
[WP6] = 0.5 mM, [guest] = 5.0 mM
[WP6] = 1.0 mM, [guest] = 10 mM. Measured by competitive ITC titration of a mixture of WP6 (0.1 mM) and G7 in the cell with guest (1 mM) in the syringe
[G7] = 0.2 mM
[G7] = 0.5 mM
[G7] = 1.0 mM.
Measured by competitive ITC titration of a mixture of WP6 (0.1 mM) and G15 (0.5 mM) in the cell with guest (1 mM) in the syringe.
For the tighter binding complexes WP6•G2 and WP6•G12 with Ka > 107 M−1 we could not optimize the c-values by reducing the fixed concentration of WP6 in the cell and therefore turned to competitive ITC titrations.18b In competitive ITC titrations the cell contains a solution of WP6 and an excess of a weaker binding guest into which a solution of the tighter binding guest is titrated. The integrated heat data from the competitive ITC titration is fitted to the competitive binding model implemented in the PEAQ ITC data analysis software using the known concentrations of host and weak binding guest along with the known Ka and ΔH values for the host•weak guest complexes as inputs to extract the Ka and ΔH values for the host•tight guest complex. Experimentally, it is important that the host•weak guest and host•tight guest complexes have significantly different ΔH values otherwise the titration will not produce sufficient heat to allow a proper fitting of the data. Experimentally, we selected G7 as the weak binding complex because its Ka toward WP6 is large enough to make it a reasonable competitor and the ΔH for the WP6•G7 complex is significantly smaller than those of the other complexes. Figure 7a shows the thermogram recorded during the titration of a solution of WP6 (100 μM) and G7 (0.2 mM) in the cell with G2 (1 mM) in the syringe. Figure 7b shows the fitting of the integrated heat versus WP6:G7 molar ratio to the competitive binding model that allowed us to determine Ka = 4.59 × 107 M−1 and ΔH = −6.10 kcal mol−1 for the tighter WP6•G2 complex. Please note that the limiting ΔH value at low molar ratio (≈ −3.2 kcal mol−1; Figure 7b) corresponds to the difference between the ΔH values for the WP6•G7 and WP6•G2 complexes. The Ka and ΔH values for the WP6•G12, WP6•G14, WP6•G15 complexes were determined by an analogous competitive ITC titration (Supporting Information).
Figure 7.
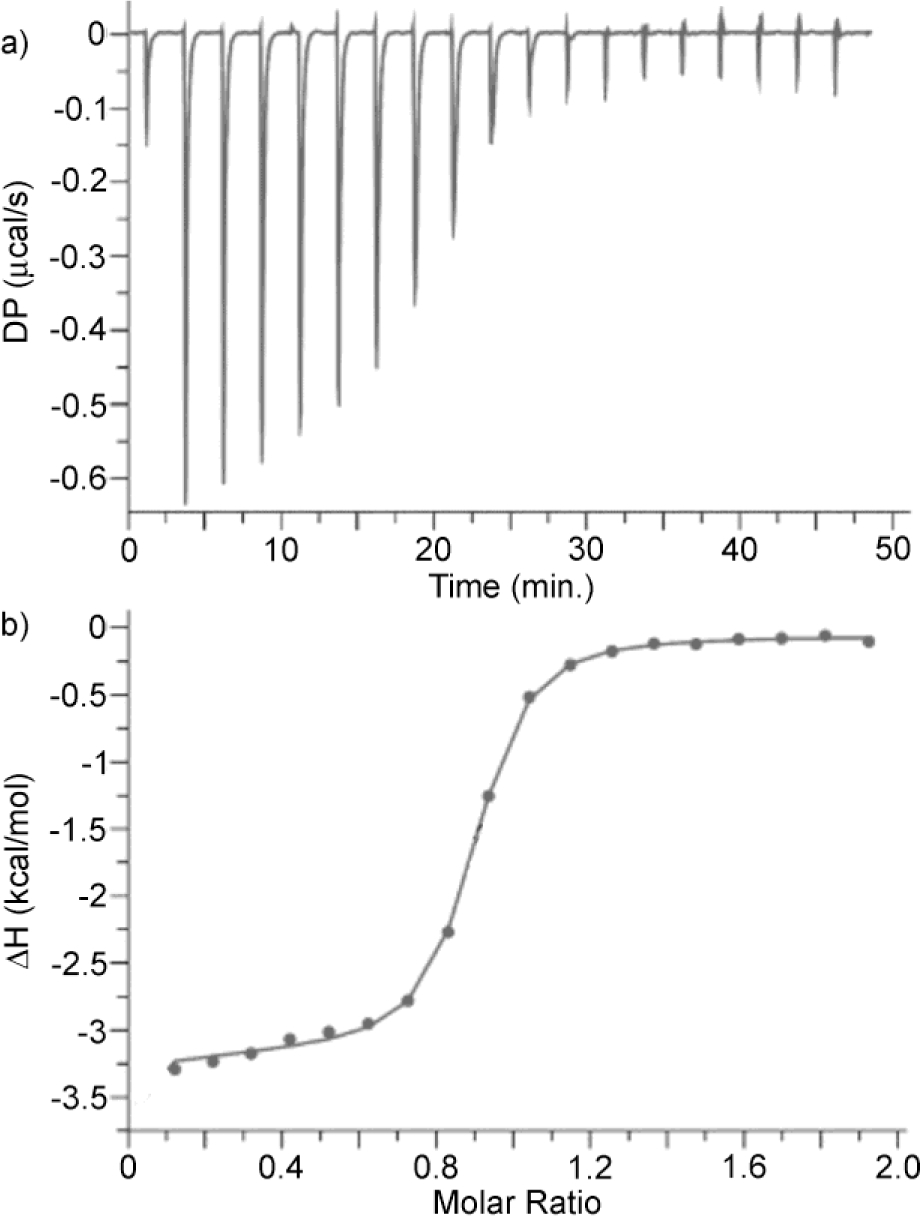
a) ITC thermogram recorded during the competitive titration of a mixture of WP6 (100 μM) and G7 (0.2 mM) in the cell with G2 (1.00 mM) in the syringe, b) Fitting of the data to a competitive binding model with Ka = 4.59 × 107 M−1 and ΔH = −6.10 kcal mol−1.
Measurement of the pKa values for WP6.
Given the importance of electrostatic interactions on the measured WP6•guest Ka values and the complications likely to be encountered by the computationalists in determining the average charge state of WP6 at neutral pH, we decided to measure the pKa values for WP6. Previously, the Silveira group reported the pKa values for WP5 obtained by pH metric titrations.19 The authors assume that each portal acts independently and report a total of five pKa values: 4.35, 4.49, 4.89. 5.30 and 6.34. Similar pH metric titrations were performed by a contract research organization (Pion, Supporting Information) in three different THF/water mixtures and the pKa values for WP6 were determined as 3.62 ± 0.01, 4.16 ± 0.01, 4.41 ± 0.03, 4.80 ± 0.07, and 5.66 ± 0.01 after extrapolation to pure water using the Yasuda-Shedlovsky equation. Accordingly, WP6 is predominantly present in the dodeca anionic form at pH 7.4
X-ray Crystal Structure of G2.
We attempted to grow single crystals of different host•guest complexes of WP6 but were unsuccessful. In one attempt, we obtained single crystals of G2•2I− and performed x-ray diffraction measurements and solved the crystal structure of G2 (Figure 8, CCDC 2114714).20 In brief, guest G2 adopts a linear geometry in the crystal with both dimethyl piperidine rings in the chair conformation. The dihedral angle of the central HC-CH unit of G2 is 180˚ which minimizes unfavorable gauche butane type interactions.
Figure 8.
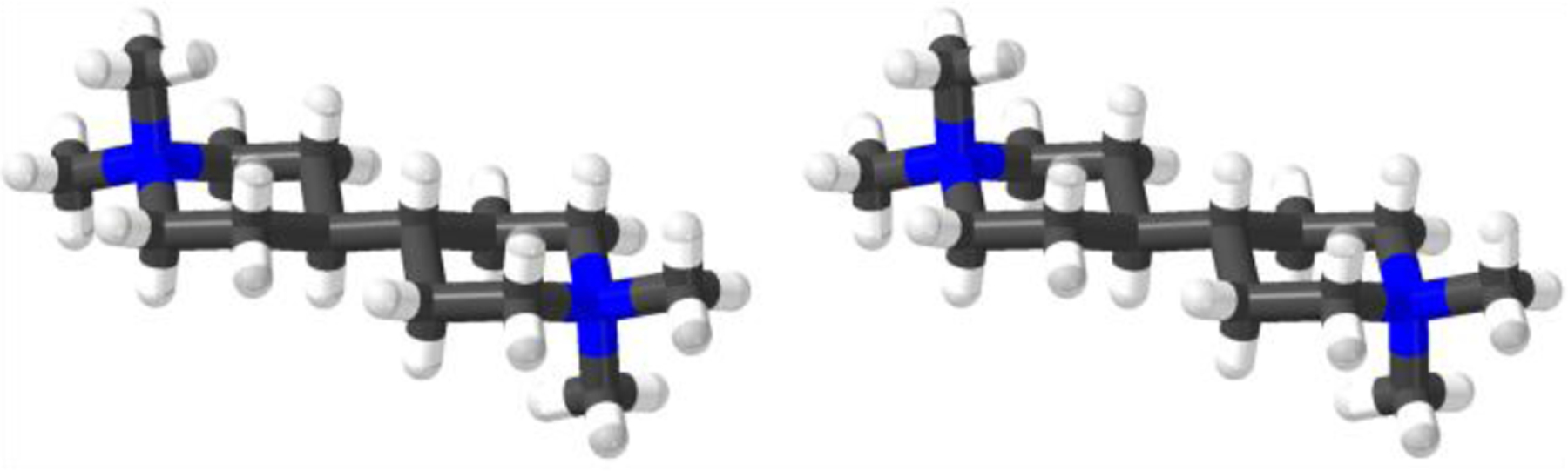
Cross-eyed stereoview of the x-ray crystal structure of G2. Color code: C, gray; H, white; N, blue; I, purple.
Discussion of the Trends in Binding Affinity.
The binding constants measured for the complexation between WP6 and G1 – G17 differ by over five orders of magnitude from 9010 M−1 to 4.69 × 109 M−1 (Table 1). The WP6•G1 – WP6•G17 complexes all uniformly driven by favorable changes in enthalpy with ΔH values ranging from −3.18 kcal mol−1 for WP6•G7 to −16.4 kcal mol−1 for WP6•G14. Most of the complexes are also driven by energetically favorable entropic changes with -TΔS values (Supporting Information) ranging from −0.57 kcal mol−1 for WP6•G11 to −4.35 kcal mol−1 for WP6•G2 and WP6•G10; the WP6•G1 (+1.63 kcal mol−1), WP6•G8 (+3.58 kcal mol−1), WP6•G14 (+3.25 kcal mol−1), WP6•G16 (+1.87 kcal mol−1) complexes are exceptions with positive -TΔS values. These thermodynamic signatures for WP6•guest binding are consistent with the non-classical hydrophobic effect that was established in cyclophane chemistry by Diederich21 and documented in other systems most notably cucurbiturils.9 Some trends are discernible within this limited dataset and are discussed below.
Influence of the Number of Carbons Among Primary Mono Ammonium Ions.
Guests G11 (5 C-atoms), G7 (6 C-atoms), G3 (7 C-atoms), G6 (8 C-atoms), and G1 (12 C-atoms) are all primary mono-ammonium ions that differ in the number of C-atoms in the hydrophobic residue. The Ka values increase as the number of carbon atoms increases from G11 to G6 which can be explained by the increasing hydrophobicity of the scaffold as CH2 units are incrementally added; we have seen related trends previously with P6AS and CB[n]-type receptors.14a, 16d, 22 Cyclododecylammonium ion G1 binds more weakly (Ka = 5.29 × 104 M−1) which suggests that G1 may be too large for the cavity of WP6. Alternatively, the -TΔS value for WP6•G1 is +1.63 kcal mol−1 which suggests that confinement of the conformationally flexible G1 imposes a large entropic penalty which reduces Ka. Other primary mono ammoniums whose Ka values were measured include G4 and G9. Guest G4 (Ka = 5.08 × 104 M−1) which contains one silicon atom was found to bind somewhat more strongly than G11 (Ka = 3.37 × 104 M−1) which can be attributed to the slightly larger volume of G4 due to the longer C-Si bonds. Adamantane guest G9 (Ka = 3.75 × 104 M−1) contains 10 C-atoms but binds even more weakly than G1 presumably due to the need for the hydrophilic OH functional group of G9 to remain solvated within the WP6•G9 complex.
Influence of Guest Charge on Binding Affinity.
Diammonium ion guests G13 (Ka = 1.63 × 106 M−1), G10 (Ka = 1.61 × 107 M−1), G2 (Ka = 4.59 × 107 M−1), G12 (Ka = 9.43 × 107 M−1), and G14 (Ka = 4.69 × 109 M−1) are the tighter binders within this dataset. The central hydrophobic cores of G2, G12, and G13 each contain 10 carbon atoms which suggests that the lower Ka measured for G13 is most likely due to the more hydrophilic viologen skeleton. The Ka for WP6•G13 was previously measured by Huang in less competitive unbuffered water where Ka = 1.02 × 108 M−1.15 Guest G10 which contains only 8 C-atoms in its central hydrophobic core binds somewhat stronger than G13 but weaker than G2 and (±)-G12. The ability of guests G2, G10, G12, and G13 to engage in favorable ammonium ion•••carboxylate interactions at both portals of WP6 is likely the source of their high binding affinity. Complex WP6•G14 which is the tightest complex in the dataset two ammonium•••carboxylate interactions at a single portal (Figure 5).
Cavity size effects.
Interestingly, bis quaternary ammonium ion G5 binds very poorly to WP6 (Ka = 9010 M−1) despite its dimethyl adamantane core and its 2+ charge. Figure 9 shows an MMFF minimized molecular model for WP6•G5 which shows that WP6 is too narrow to engulf the hydrophobic core of G5 and instead simply binds to one of the pendant NMe3+ groups which explains the especially poor affinity. Related trends have been observed previously by us with CB[n]-type receptors.8b
Figure 9.
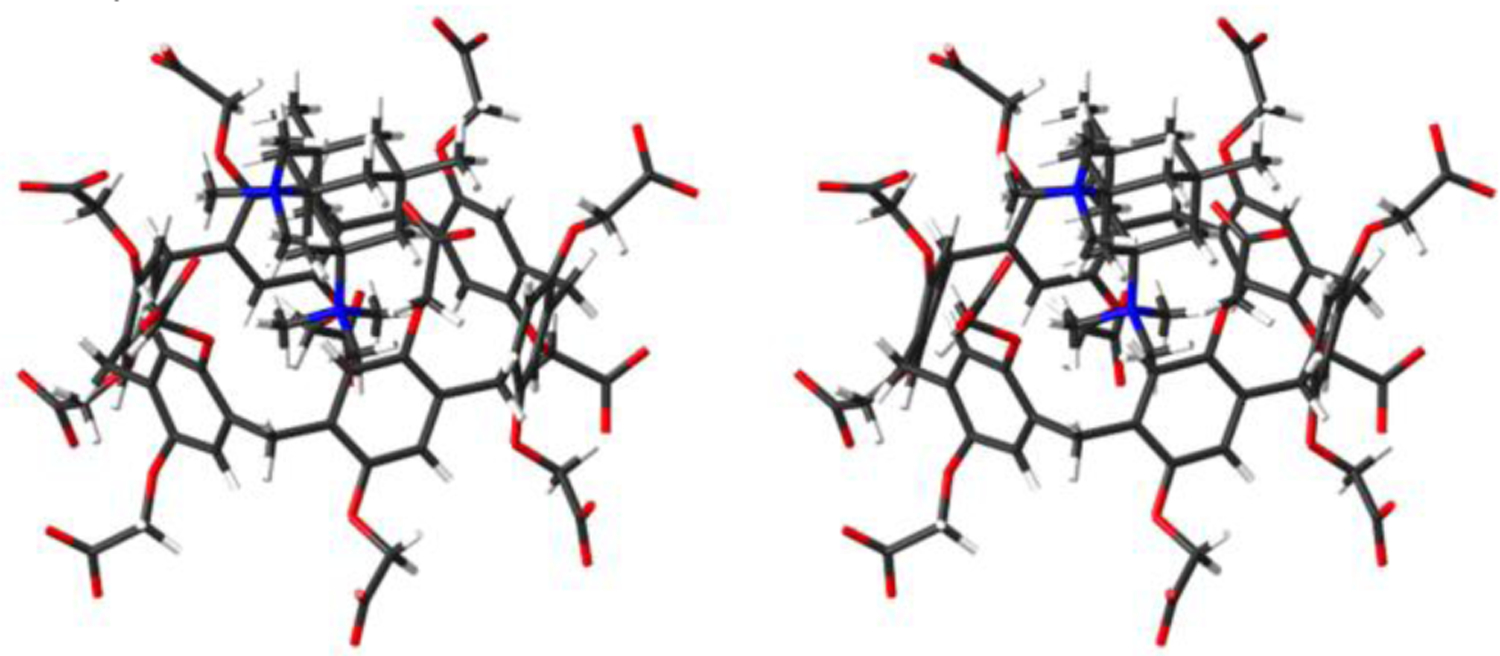
Cross-eyed stereoview of an MMFF minimized model of WP6•G5. Color code: C, gray; H, white; N, blue; O, red.
Influence of Secondary Electrostatic Interactions.
The diammonium ion guests G2, G10, and G12 locate their cationic centers near the anionic portals of WP6. We wondered about the influence of pendant charged functionality on the observed Ka values. For example, G17 is an analogue of G4 that features a cationic (CH2)3NH3+ sidearm that would be expected to engage in attractive secondary electrostatic interactions with anionic WP6. We find that the WP6•G17 complex is 4.5-fold tighter than the WP6•G4 complex which corresponds to a difference of −0.89 kcal mol−1. Similarly, G16 is an analogue of G10 that features two anionic (CH2)3SO3− sidearms that would be expected to engage in repulsive secondary electrostatic interactions with anionic WP6. Complex WP6•G16 is 1220-fold weaker than WP6•G10 which corresponds to a difference of +4.2 kcal mol−1 (or +2.1 kcal mol−1 per sidearm). Apparently, repulsive secondary electrostatic interactions exert a larger influence on Ka that attractive secondary electrostatic interactions.
Influence of Guest Methylation State.
In our recent study of P6AS we found that higher degrees of guest methylation (e.g. 1˚ < 2˚< 3˚ < 4˚) resulted in significantly higher Ka values.14a In the present dataset, p-xylenediamine derived guests G10 and G15 differ only in the degree of methylation. We find that the Ka values for WP6 toward G10 (1.61 × 107 M−1) and G15 (1.76 × 107 M−1) are quite similar which establishes that methylation state changes do not play a major role in the host•guest trends of WP6.
Conclusions.
In summary, we have reported an investigation of the binding of WP6 toward a panel of cationic hydrophobic guests G1 – G20 by a combination of 1H NMR spectroscopy and ITC. The 1H NMR measurements establish that the hydrophobic binding domains of the guest are located in the hydrophobic cavity of WP6 which constitutes an anisotropic shielding region. The 1H NMR spectra of WP6•guest complexes may appear simple when guest exchange is fast (e.g. WP6•G1, Figure 3), present a workshop on symmetry considerations when guest exchange is slower (e.g. WP6•G14, Figure 4), or be uninterpretable when WP6 assumes an unsymmetrical conformation (e.g. WP6•G13, Supporting Information). The thermodynamic parameters of binding (Ka, ΔH) were measured by direct or competitive ITC and span from a low of 9010 M−1 for WP6•G5 to 4.69 × 109 M−1 for WP6•G14. The WP6•guest complexes are generally driven by favorable ΔH and less favorable -TΔS values which means that the non-classical hydrophobic effect governs the molecular recognition of WP6. The overall guest charge, the number of C-atoms in the hydrophobic binding domain, the presence of secondary electrostatic interactions, and cavity size effects all play a significant role in determining WP6•guest binding affinity. Perhaps most significantly, the thermodynamic data presented in Table 1 serves as a blinded dataset for the SAMPL9 challenge to allow to validate and improve their methods to compute binding free energies in aqueous solution. When those methods reach maturity it will significantly advance wide areas of supramolecular and medicinal chemistry.
Experimental.
WP6 was synthesized according to the reported procedure.15 Guests were available from previous studies.8, 11b, 11d, 16 1H NMR spectra were measured on Bruker spectrometers operating at 400 or 600 MHz using D2O as solvent. Chemical shifts (δ) are referenced relative to the residual resonances for HOD (4.80 ppm). ITC experiments were conducted in the 200 μL working volume of the sample cell of a PEAQ ITC instrument (Malvern) using a 40 μL injection syringe. Host and guest solutions were prepared in phosphate buffered saline (PBS) at pH 7.4. The sample cell was filled to capacity (200 μL) with the host solution and the guest solution was titrated in (first injection = 0.4 μL, subsequent 18 injections = 2 μL). In select cases, competitive titrations were required where host and an excess of weaker binding guest were included in the cell and the tighter binding guest was titrated into the cell. For direct titrations, the binding data was fitted using the 1:1 binding model implemented in the PEAQ-ITC analysis software whereas for competitive titrations the competition binding model was used.
Supplementary Material
Acknowledgements.
We thank the National Institutes of Health (GM-124270) and the National Science Foundation (CHE-2105857) for financial support. We thank Profs. Kata Majerski, Marina Sekutor, and Robert Glaser for the samples of G5, G12, G19, and G20.
Footnotes
Conflicts of Interest.
The authors have no conflicts of interest in relation to the work contained in the paper.
Electronic Supplementary Information (ESI) available: Details of synthesis, NMR, and ITC experiments. See DOI: 10.1039/D1NJ05209H
Notes and References
- 1.Mohs RC and Greig NH, Alzheimers Dement, 2017, 3, 651–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Isaacs L, SAMPL Challenge, https://en.wikipedia.org/wiki/SAMPL_Challenge, (accessed June 11, 2021); [Google Scholar]; (b) Muddana HS, Daniel Varnado C, Bielawski CW, Urbach AR, Isaacs L, Geballe MT and Gilson MK, J. Comput.-Aided Mol. Des, 2012, 26, 475–487; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Muddana HS, Fenley AT, Mobley DL and Gilson MK, J. Comput.-Aided Mol. Des, 2014, 28, 305–317; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Yin J, Henriksen NM, Slochower DR, Shirts MR, Chiu MW, Mobley DL and Gilson MK, J. Comput.-Aided Mol. Des, 2017, 31, 1–19; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Rizzi A, Murkli S, McNeill JN, Yao W, Sullivan M, Gilson MK, Chiu MW, Isaacs L, Gibb BC, Mobley DL and Chodera JD, J. Comput.-Aided Mol. Des, 2018, 32, 937–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Isik M, Levorse D, Rustenburg AS, Ndukwe IE, Wang H, Wang X, Reibarkh M, Martin GE, Makarov AA, Mobley DL, Rhodes T and Chodera JD, J. Comput.-Aided Mol. Des, 2018, 32, 1117–1138; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Isik M, Levorse D, Mobley DL, Rhodes T and Chodera JD, J. Comput.-Aided Mol. Des, 2020, 34, 405–420; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mobley DL, Liu S, Lim NM, Wymer KL, Perryman AL, Forli S, Deng N, Su J, Branson K and Olson AJ, J. Comput.-Aided Mol. Des, 2014, 28, 327–345; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Guthrie JP, J. Phys. Chem. B, 2009, 113, 4501–4507. [DOI] [PubMed] [Google Scholar]
- 4.Lowe AJ, Pfeffer FM and Thordarson P, Supramol. Chem, 2012, 24, 585–594. [Google Scholar]
- 5.(a) Dempsey JM, Zhai C, McGarraugh HH, Schreiber CL, Stoffel SE, Johnson A and Smith BD, Chem. Commun, 2019, 55, 12793–12796; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Assaf KI and Nau WM, Chem. Soc. Rev, 2015, 44, 394–418; [DOI] [PubMed] [Google Scholar]; (c) Liu Y, Zhou F, Yang F and Ma D, Org. Biomol. Chem, 2019, 17, 5106–5111; [DOI] [PubMed] [Google Scholar]; (d) Jordan JH and Gibb BC, Chem. Soc. Rev., 2015, 44, 547–585; [DOI] [PubMed] [Google Scholar]; (e) Bom A, Bradley M, Cameron K, Clark JK, Van Egmond J, Feilden H, MacLean EJ, Muir AW, Palin R, Rees DC and Zhang M-Q, Angew. Chem., Int. Ed, 2002, 41, 265–270; [DOI] [PubMed] [Google Scholar]; (f) Yin H, Zhang X, Wei J, Lu S, Bardelang D and Wang R, Theranostics, 2021, 11, 1513–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Freeman WA, Mock WL and Shih N-Y, J. Am. Chem. Soc, 1981, 103, 7367–7368; [Google Scholar]; (b) Kim J, Jung I-S, Kim S-Y, Lee E, Kang J-K, Sakamoto S, Yamaguchi K and Kim K, J. Am. Chem. Soc, 2000, 122, 540–541; [Google Scholar]; (c) Day AI, Arnold AP, Blanch RJ and Snushall B, J. Org. Chem, 2001, 66, 8094–8100; [DOI] [PubMed] [Google Scholar]; (d) Day AI, Blanch RJ, Arnold AP, Lorenzo S, Lewis GR and Dance I, Angew. Chem. Int. Ed, 2002, 41, 275–277; [DOI] [PubMed] [Google Scholar]; (e) Liu S, Zavalij PY and Isaacs L, J. Am. Chem. Soc, 2005, 127, 16798–16799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Ganapati S and Isaacs L, Isr. J. Chem, 2018, 58, 250–263; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Isaacs L, Chem. Commun, 2009, DOI: 10.1039/b814897j, 619–629. [DOI] [PubMed] [Google Scholar]
- 8.(a) Cao L, Sekutor M, Zavalij PY, Mlinaric-Majerski K, Glaser R and Isaacs L, Angew. Chem. Int. Ed, 2014, 53, 988–993; [DOI] [PubMed] [Google Scholar]; (b) Liu S, Ruspic C, Mukhopadhyay P, Chakrabarti S, Zavalij PY and Isaacs L, J. Am. Chem. Soc, 2005, 127, 15959–15967. [DOI] [PubMed] [Google Scholar]
- 9.(a) Nau WM, Florea M and Assaf KI, Isr. J. Chem, 2011, 51, 559–577; [Google Scholar]; (b) Biedermann F, Uzunova VD, Scherman OA, Nau WM and De Simone A, J. Am. Chem. Soc, 2012, 134, 15318–15323; [DOI] [PubMed] [Google Scholar]; (c) Biedermann F, Nau WM and Schneider H-J, Angew. Chem. Int. Ed, 2014, 53, 11158–11171. [DOI] [PubMed] [Google Scholar]
- 10.(a) Deng C-L, Murkli SL and Isaacs LD, Chem. Soc. Rev, 2020, 49, 7516–7532; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jung H, Park KM, Yang J-A, Oh EJ, Lee D-W, Park K, Ryu SH, Hahn SK and Kim K, Biomaterials, 2011, 32, 7687–7694; [DOI] [PubMed] [Google Scholar]; (c) Sun C, Zhang H, Li S, Zhang X, Cheng Q, Ding Y, Wang L-H and Wang R, ACS Appl. Mater. Interfaces, 2018, 10, 25090–25098; [DOI] [PubMed] [Google Scholar]; (d) Wheate NJ and Limantoro C, Supramol. Chem, 2016, 28, 849–856. [Google Scholar]
- 11.(a) Ma D, Glassenberg R, Ghosh S, Zavalij PY and Isaacs L, Supramol. Chem, 2012, 24, 325–332; [Google Scholar]; (b) Cao L and Isaacs L, Supramol. Chem, 2014, 26, 251–258; [Google Scholar]; (c) She NF, Moncelet D, Gilberg L, Lu XY, Sindelar V, Briken V and Isaacs L, Chem. Eur. J, 2016, 22, 15270–15279; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Murkli S, McNeill JN and Isaacs L, Supramol. Chem, 2019, 31, 150–158; [Google Scholar]; (e) Ndendjio SZ, Liu W, Yvanez N, Meng Z, Zavalij PY and Isaacs L, New J. Chem, 2020, 44, 338–345; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Murkli S, Klemm J, Brockett AT, Shuster M, Briken V, Roesch MR and Isaacs L, Chem. - Eur. J, 2021, 27, 3098–3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Sullivan MR, Yao W and Gibb BC, Supramol. Chem, 2019, 31, 184–189; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Suating P, Nguyen TT, Ernst NE, Wang Y, Jordan JH, Gibb CLD, Ashbaugh HS and Gibb BC, Chem. Sci, 2020, 11, 3656–3663; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kellett K, Slochower DR, Schauperl M, Duggan BM and Gilson MK, J. Comput.-Aided Mol. Des, 2021, 35, 95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Xue M, Yang Y, Chi X, Zhang Z and Huang F, Acc. Chem. Res, 2012, 45, 1294–1308; [DOI] [PubMed] [Google Scholar]; (b) Ogoshi T, Yamagishi T-A and Nakamoto Y, Chem. Rev, 2016, 116, 7937–8002; [DOI] [PubMed] [Google Scholar]; (c) Wu J-R and Yang Y-W, Chem. Commun, 2019, 55, 1533–1543; [DOI] [PubMed] [Google Scholar]; (d) Li Z and Yang Y-W, Acc. Mater. Res., 2021, 2, 292–305; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Lou X-Y and Yang Y-W, Adv. Mater, 2020, 32, 2003263; [DOI] [PubMed] [Google Scholar]; (f) Song N, Lou X-Y, Ma L, Gao H and Yang Y-W, Theranostics, 2019, 9, 3075–3093; [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Song N, Kakuta T, Yamagishi T-A, Yang Y-W and Ogoshi T, Chem, 2018, 4, 2029–2053. [Google Scholar]
- 14.(a) Xue W, Zavalij PY and Isaacs L, Angew. Chem., Int. Ed, 2020, 59, 13313–13319; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yu G, Xue M, Zhang Z, Li J, Han C and Huang F, J. Am. Chem. Soc, 2012, 134, 13248–13251. [DOI] [PubMed] [Google Scholar]
- 15.Yu G, Zhou X, Zhang Z, Han C, Mao Z, Gao C and Huang F, J. Am. Chem. Soc, 2012, 134, 19489–19497. [DOI] [PubMed] [Google Scholar]
- 16.(a) Cao L, Hettiarachchi G, Briken V and Isaacs L, Angew. Chem. Int. Ed, 2013, 52, 12033–12037; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cao L, Skalamera D, Zavalij PY, Hostas J, Hobza P, Mlinaric-Majerski K, Glaser R and Isaacs L, Org. Biomol. Chem, 2015, 13, 6249–6254; [DOI] [PubMed] [Google Scholar]; (c) Sigwalt D, Sekutor M, Cao L, Zavalij PY, Hostas J, Ajani H, Hobza P, Mlinaric-Majerski K, Glaser R and Isaacs L, J. Am. Chem. Soc, 2017, 139, 3249–3258; [DOI] [PubMed] [Google Scholar]; (d) Liu W, Lu X, Meng Z and Isaacs L, Org. Biomol. Chem, 2018, 16, 6499–6506. [DOI] [PubMed] [Google Scholar]
- 17.Zhu H, Li Q, Gao Z, Wang H, Shi B, Wu Y, Shangguan L, Hong X, Wang F and Huang F, Angew. Chem., Int. Ed, 2020, 59, 10868–10872. [DOI] [PubMed] [Google Scholar]
- 18.(a) Wiseman T, Williston S, Brandts JF and Lin L-N, Anal. Biochem, 1989, 179, 131–137; [DOI] [PubMed] [Google Scholar]; (b) Velazquez-Campoy A and Freire E, Nat. Protocols, 2006, 1, 186–191; [DOI] [PubMed] [Google Scholar]; (c) Broecker J, Vargas C and Keller S, Anal. Biochem, 2011, 418, 307–309. [DOI] [PubMed] [Google Scholar]
- 19.Silveira EV, Wanderlind EH, Masson AK, Cordeiro PS, Nascimento V, Affeldt RF and Micke GA, New J. Chem, 2020, 44, 2701–2704. [Google Scholar]
- 20.Crystal Data for C14H30N2I2 (M =480.20 g/mol): monoclinic, space group P21/c (no. 14), a = 6.5208(2) Å, b = 14.5116(5) Å, c = 10.1283(3) Å, β = 104.2182(5)°, V = 929.05(5) Å3, Z = 2, T = 296(2) K, μ(MoKα) = 3.375 mm-1, Dcalc = 1.717 g/cm3, 16743 reflections measured (5.01° ≤ 2Θ ≤ 62.496°), 3037 unique (Rint = 0.0262, Rsig = 0.0147) which were used in all calculations. The final R1 was 0.0240 (I > 2σ(I)) and wR2 was 0.0553 (all data). Refinement details: H atoms were positioned from the geometric considerations and refined as riding on the attached atoms with Uiso constrained to be 20% (50% for methyl group) larger than Ueqv of the attached group. Orientation of methyl groups was optimized.
- 21.(a) Meyer EA, Castellano RK and Diederich F, Angew. Chem., Int. Ed, 2003, 42, 1210–1250; [DOI] [PubMed] [Google Scholar]; (b) Persch E, Dumele O and Diederich F, Angew. Chem., Int. Ed, 2015, 54, 3290–3327. [DOI] [PubMed] [Google Scholar]
- 22.Murkli S, Klemm J, King D, Zavalij PY and Isaacs L, Chem. - Eur. J, 2020, 26, 15249–15258. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.