

Abstract
Tuberculosis (TB) is a severe infectious disease worldwide. The increasing emergence of drug-resistant Mycobacterium tuberculosis (Mtb) has markedly hampered TB control. Therefore, there is an urgent need to develop new anti-TB drugs to treat drug-resistant TB and shorten the standard therapy. The discovery of targets of drug action will lay a theoretical foundation for new drug development. With the development of molecular biology and the success of Mtb genome sequencing, great progress has been made in the discovery of new targets and their relevant inhibitors. In this review, we summarized 45 important drug targets and 15 new drugs that are currently being tested in clinical stages and several prospective molecules that are still at the level of preclinical studies. A comprehensive understanding of the drug targets of Mtb can provide extensive insights into the development of safer and more efficient drugs and may contribute new ideas for TB control and treatment.
1. Introduction
Tuberculosis (TB) is a respiratory infectious disease caused by Mycobacterium tuberculosis (Mtb). The World Health Organization (WHO) reported approximately 9.9 million incident cases and 1.28 million deaths related to TB in 2020 [1]. Although a declining trend in the incidence and mortality of TB has been observed since 2010, the global TB burden remains a challenge. In addition, multidrug-resistant (MDR)-TB poses a threat to TB control. For more than 10 years, approximately 3-4% of new TB cases and 18-21% of patients with TB with retreatment had MDR-TB or rifampicin (RFP)-resistant TB (RR-TB) [1]. Therefore, the importance of preventing Mtb transmission and identifying treatments for MDR-TB and XDR-TB must be recognized and addressed.
Effective anti-TB regimens kill Mtb, improve the clinical symptoms of TB, and prevent the malignant development of the disease. The current standard treatment for drug-sensitive TB is a combination of a short-course chemotherapy regimen under direct supervision recommended by the WHO, which uses four first-line drugs [isoniazid (INH), RFP, ethambutol (EMB), and pyrazinamide (PZA)] in the first two months of development, followed by INH and RFP in the last four months of consolidation. The WHO TB guidelines in 2021 showed that this “short-course chemotherapy” has successfully cured 66 million patients with TB since 2000 [1]. However, the treatment of MDR-TB and XDR-TB is difficult and may have no significant effect on the persistent Mtb, because they need to be treated with more toxic and costlier second- and third-line drugs for a longer time, even up to two years, and usually with limited success. The primary causes of drug-resistant TB in clinical cases include gene mutations in drug targets or drug-activating enzymes, compensatory evolution, and the activation of efflux pumps [2, 3]. At present, there are limited varieties of anti-TB drugs in clinics, leaving clinicians with limited options. Therefore, there is an urgent need to develop new drugs with novel mechanisms to cure TB or shorten the treatment time for MDR-TB and provide effective support for TB control [4]. Here, we reviewed the 45 targets of drug action on Mtb and reported the corresponding newly developed drugs in recent years. These new drugs showed excellent anti-TB activity due to their action on different new targets, improved the cure rate of patients with MDR/XDR-TB, and significantly reduced the total mortality; however, some new drugs were more toxic than existing anti-TB drugs. Clearly, the rational and effective implementation of new drug regimens will overcome the “barrier” of drug-resistant TB and provide strong support for the goal of ending TB globally. Table 1 shows the specific anti-TB targets, while Figure 1(a) presents the clinical trial stages of the new anti-TB drugs.
Table 1.
Summary of targets of anti-TB drugs.
| Action targets | Gene Bank | Human homologue | New drugs/lead compounds | Structure | Mechanism of action | NCT number | Phase | Notes | References | |
|---|---|---|---|---|---|---|---|---|---|---|
| Cell wall biosynthesis | InhA | rv1484 | None | Diaryl ether compounds | — | — | — | — | Undesirable bioavailability, limited solubility, and certain cytotoxicity | [6, 8, 9, 11, 12, 168] |
| FabH | rv0533c | None | Alkaloids and small molecule compounds | — | — | — | — | In vitro experiments | [15–17] | |
| Methoxy-keto-mycolic acid | — | None | Delamanid |
|
Inhibit the mycolic acid biosynthesis of Mtb cell wall | NCT03828201 | II | Not yet recruiting | [18–24, 169] | |
| NCT03959566 | II | Recruiting | ||||||||
| NCT04550832 | II | Recruiting | ||||||||
| NCT05007821 | II | Recruiting | ||||||||
| NCT04518228 | IV | Recruiting | ||||||||
| Pretomanid (PA-824) |
|
NCT04179500 | II | Recruiting | ||||||
| NCT05040126 | III | Recruiting | ||||||||
| NCT04207112 | II, III | Recruiting | ||||||||
| TBA-354 |
|
NCT02606214 | I | Terminated (mild reversible neurotoxicity) | ||||||
| DprE1 | rv3790 | None | BTZ-043 |
|
Inhibit the arabinogalactan biosynthesis of Mtb cell wall | NCT04874948 | I | Completed | [4, 25–29, 170] | |
| NCT04044001 | I, II | Recruiting | ||||||||
| PBTZ-169 |
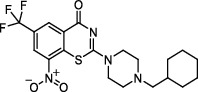
|
NCT03334734 | II | Terminated (very slow enrollment) | ||||||
| NCT03776500 | I | Completed | ||||||||
| OPC-167832 |
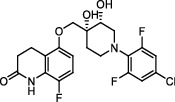
|
NCT03678688 | I, II | Recruiting | ||||||
| TBA-7371 |
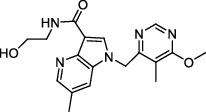
|
NCT03199339 | I | Completed | ||||||
| NCT04176250 | II | Recruiting | ||||||||
| WecA | rv1302 | None | CPZEN-45 |

|
— | Preclinical study | Poor solubility and potentially low bioavailability | [30] | ||
| RmlC | rv3465 | None | Benzimidazolone derivatives |
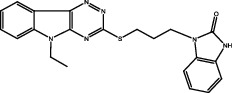
|
— | — | Mild antitubercular activity | [31, 32] | ||
| MurX | rv2156c | None | SQ641 |
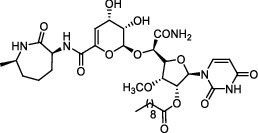
|
Inhibit the peptidoglycan biosynthesis of Mtb cell wall | — | Preclinical study | Low solubility and low bioavailability | [33–35] | |
| MbtI | rv2386c | None | Natural products and small molecule compounds | — | — | — | Carbonyl and 7-hydroxyl groups are the necessary structures of inhibitors | [36–38, 40, 171] | ||
| MbtA | rv2384 | Nucleic acid analogues | Short half-life and low bioavailability | |||||||
| Alr and DdlA |
rv3423c
rv2981c |
None | — | — | — | — | Research needs to be further | [41, 42] | ||
| GlmU | rv1018c | None | Natural products and small molecule compounds | — | — | — | In vitro experiments | [43–46, 172] | ||
| Ino1 | rv0046c | Yes | — | — | — | — | — | Inhibitors rarely reported so far | [48, 49, 100] | |
| MEP pathway | — | None | Alkaloids and small molecule compounds | — | — | — | — | Research needs to be further | [50, 51] | |
| Protein biosynthesis and breakdown | 50s ribosomal subunit | — | None | AZD-5847 |
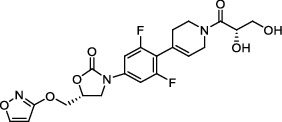
|
Inhibit the proteins biosynthesis of Mtb | NCT01516203 | II | Completed | [54, 55] |
| 23s rRNA | — | None | Sutezolid |
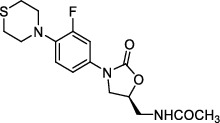
|
NCT03959566 | II | Recruiting | [57–63] | ||
| NCT03237182 | IV | Recruiting | ||||||||
| Delpazolid |
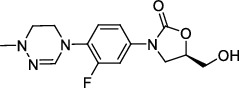
|
NCT04550832 | II | Recruiting | ||||||
| NCT04939779 | I | Completed | ||||||||
| Contezolid |
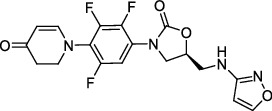
|
NCT03747497 | II | Completed | ||||||
| LeuRS | — | Yes | GSK-3036656 |
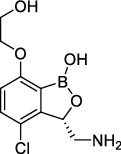
|
NCT03557281 | II | Completed | [64] | ||
| ClpP1 ClpP2 |
rv2461c
rv2460c |
Yes | Acyldepsipeptide antibiotics (ADEPs) and cyclomarinA | — | — | — | — | Research needs to be further | [69] | |
| DNA related enzymes | GyrA rv0006 |
None | DC-159a |
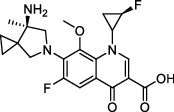
|
Inhibit the activity of DNA gyrase, blocks DNA replication and transcription to lead Mtb death | — | Preclinical study | Mtb is more resistant to DC-159a | [77] | |
| GyrB rv0005 |
None | SPR720 |
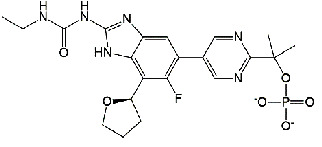
|
I | Completed | [78] | ||||
| topA | rv3646c | Yes | Boldine-derived alkaloids |
|
— | — | Screening and structural optimization | Inhibition activity at μM level | [82–84] | |
| Energy metabolism | ATP synthase | — | Yes | Bedaquiline |
|
Inhibit the ATP synthase of Mtb to block the energy supply | NCT04630145 | II and III | Recruiting | [85, 86] |
| NCT04207112 | II and III | Recruiting | ||||||||
| NADH-2 | rv1854c | None | Phenothiazines, quinoline pyrimidines, quinazolones, and DPI analogues | — | — | — | — | More serious side effects | [90–94, 173] | |
| Respiratory cytochrome bc1 complex | — | Yes | Q203 |
|
Inhibit the synthesis of ATP by acting on the respiratory cytochrome bc1 complex | NCT03563599 | II | Completed | https://www.clinicaltrials.gov/ct2/show/NCT03563599?cond = Q203&draw =2&rank =1 | |
| MenB | rv0548c | None | Succinyl benzoate derivatives and benzoxazine derivatives | — | — | — | — | In vitro experiments | [96–98] | |
| Lipid metabolism | Fatty acid desaturase (DesA3) | rv3229c | Yes | Thiocarlide |
|
Inhibit desA3 to decrease oleic acid and stearic acid synthesis of Mtb | — | — | Require structure optimization | [99, 101, 174] [100] |
| Pantothenate synthetase (PanC) | rv3602c | None | Small molecule compounds | — | — | — | — | In vitro experiments | [102] | |
| Signal transduction pathway | Shikimate pathway | rv2539c | None | Natural products and small molecule compounds | — | — | — | — | In vitro experiments | [103, 104, 175] |
| Other targets | FtsZ | rv2150c | None | Natural products and small molecule compounds | — | — | — | — | In vitro experiments | [106–109, 111] |
| MmpL | rv0206c | None | BM212 |
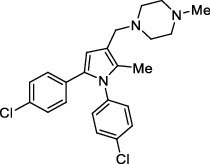
|
Inhibit membrane proteins mmpL of Mtb | — | Preclinical study | Poor pharmacokinetics and more severe toxic reactions | [112–118, 176] | |
| SQ109 |
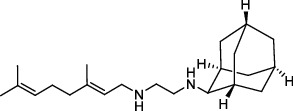
|
NCT01785186 | II | Completed | ||||||
| STPK | — | Yes | — | — | — | — | — | Antituberculosis activity of inhibitors is not significant | [119–122] | |
| AhpD | rv2429 | None | — | — | — | — | — | Inhibitors rarely reported so far | [123] | |
| NATs | rv3566c | Yes | — | — | — | — | — | Inhibitors rarely reported so far | [124] | |
| Diterpene cyclase | rv3377c | None | Diterpenoid and triterpenoid acids | — | — | — | — | Research needs to be further | [125–128] | |
| Tuberculosinol phosphatase | rv3378c | |||||||||
| ICL | rv0467 | None | Natural products and synthetic compounds | — | — | — | — | In laboratory research | [129–131] | |
| Redox-related enzymes (cytochrome P450 enzymes) | — | Yes | Azole antifungals | — | — | — | — | Prevent the synthesis of ergosterol from fungal cell membrane | [132] | |
| MshA | rv0486 | None | Adenosine derivatives and synthetic compounds | — | — | — | — | Research needs to be further | [133–136] | |
| MshC | rv2130c | — | ||||||||
| PptT | rv2794c | None | 8918 |
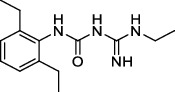
|
Inhibit PptT to block the activation of Pks13 enzyme and various type-І Pks | — | — | Short half-life | [137, 138] | |
| MptpA | rv2234 | None | Natural products and synthetic compounds | — | — | — | — | The best inhibition activity at nM level | [139–142, 177] | |
| MptpB | rv0153c | |||||||||
| Biofilm | — | None | C10 |
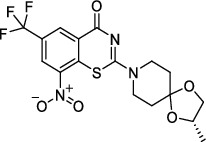
|
Prevent the formation of biofilm to make Mtb more susceptible to antibiotics | — | — | Research needs to be further | [145] | |
Figure 1.
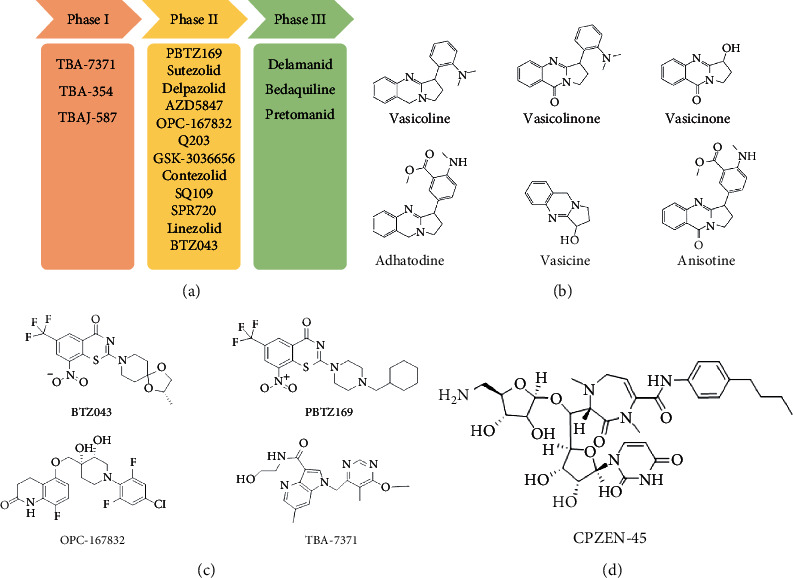
New anti-TB drugs in clinical trials (a), the chemical structures of vasicoline, vasicolinone, vasicinone, vasicine, adhatodine, and anisotine (b), the chemical structures of BTZ-043, PBTZ-169, OPC-167832, and TBA-7371 (c), and the chemical structure of CPZEN-45 (d).
2. Cell Wall Biosynthesis
Cell wall biosynthesis offers several molecular targets because biosynthetic enzymes do not have homologs in the mammalian system. Traditional anti-TB first-line drugs (INH and EMB) and second-line drugs (cycloserine [CS] and ethionamide [ETH]) inhibit cell wall synthesis by acting on different targets.
2.1. Targeting Mycolic Acid Biosynthesis
The cell wall of Mtb contains a large amount of mycolic acid, which surrounds the peptidoglycan layer. Mycolic acids are considered the main virulence factor of Mtb because they make Mtb naturally resistant to most antibiotics [5]. The synthesis of the Mtb cell wall is mainly regulated by fatty acid synthases (FAS). FAS-I is also involved in the synthesis of fatty acids in eukaryotes, while FAS-II is unique to Mtb cells and is a target of anti-TB drugs. The biological enzymes enoyl-acyl carrier protein reductase (inhA) and β-ketoacyl-acyl carrier protein synthase III (FabH) are important targets of anti-TB drugs.
2.1.1. Enoyl-acyl Carrier Protein Reductase
InhA is closely related to the extension of the fatty acid chain in the cell wall [6]. InhA is the only target of ETH and protionamide (PTH) and is one of the targets of INH. Mutations in inhA (rv1484) could induce Mtb resistance to ETH, PTH, and INH [7]. It has been confirmed that <10% INH-resistant Mtb clinical isolates are associated with mutations in inhA [7]. Several compounds with a diaryl ether structure and their derivatives have been identified as inhibitors of inhA, some of which showed activity against both drug-sensitive and drug-resistant Mtb [8, 9]. Triclosan, a diaryl ether derivative, is a potential anti-TB drug candidate because its polychlorinated molecular structure does not require any biological activation in vivo and directly targets inhA. However, these compounds have several limitations, such as undesirable bioavailability, limited solubility, and cytotoxicity [8]. Mtb has been reported to be resistant to triclosan, but the specific drug resistance mechanism is still unclear [10]. In addition, 3-nitropropanoic acid, gallic acid derivatives, pyrrolidone derivatives, tetrahydrofuran derivatives, 4-hydroxy-2-pyridone analogs, aryl amides, and other inhA inhibitors have been reported [9, 11–14].
2.1.2. β-Ketoacyl-acyl Carrier Protein Synthase III
β-Ketoacyl-acyl carrier protein synthase III (FabH) is an important link between type I and type II FAS, and it catalyzes the condensation of malonyl-ACP with acyl-CoA to form β-ketoacyl-ACP [15]. Mtb fabH has no homologous proteins in humans. The six alkaloids (vasicoline, vasicolinone, vasicinone, vasicine, adhatodine, and anisotine, Figure 1(b)) extracted from the leaves of Justicia adhatoda and the selected synthetic small molecule compounds were found to have moderate activity against fabH [16, 17].
2.1.3. Methoxy-mycolic Acid and Keto-mycolic Acid
Mycolic acid synthesized by Mtb is classified into three types according to positional modification: α-, keto-, and methoxy-mycolic acid [18]. Nitroimidazole compounds exert anti-TB activity by mainly targeting methoxy-mycolic acid.
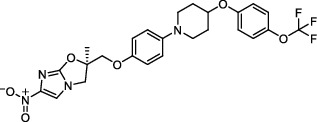
Delamanid, a nitro-dihydro-imidazooxazole derivative, inhibits keto- and methoxy-mycolic acid synthesis and displays higher activity against Mtb than RFP, INH, EMB, and streptomycin (SM), without obvious cross-resistance with first-line anti-TB drugs [19]. However, delamanid monotherapy can rapidly produce delamanid-resistant Mtb strains.
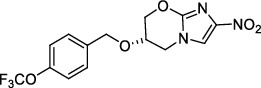
In addition, PA-824 also acts on deazaflavin-dependent nitroreductase (Ddn). Ddn is mainly responsible for catalyzing the formation of lethal nitrogen atoms in cells by nitroimidazoles to cause bacterial cell lysis [20]. Phase II clinical trials showed that PA-824 disrupted the formation of mycolic acids and demonstrated potent activity against drug-sensitive and drug-resistant Mtb, including nonreplicating strains (https://www.croiconference.org/abstract/efficacy-bedaquiline-pretomanid-moxifloxacin-pza-bpamz-against-ds-mdr-tb/). In an anoxic environment, Ddn activates PA-824 and further generates lethal reactive nitrogen in Mtb cells, leading to nitric oxide (NO) release. In addition, as an NO donor, PA-824 can cause respiratory toxicity in bacterial cells and enhance the inherent killing mechanism of the innate immune system, resulting in bacterial death [21]. Under aerobic conditions, the methoxy and keto groups of mycolic acids in the cell wall of Mtb could be effectively inhibited by PA-824 to exert significant antibacterial activity [22].
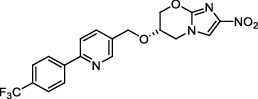
TBA-354 has a narrow-spectrum bactericidal effect in vitro against replicating and nonreplicating Mtb. Its potency is similar to that of delamanid and greater than that of PA-824. TBA-354 has a higher bioavailability and a longer elimination half-life than other nitroimidazole drugs [23]. Unfortunately, the findings from the clinical trials showed that TBA-354 could result in mild signs of reversible neurotoxicity; therefore, Global TB Alliance discontinued this research [24].
2.2. Targeting Arabinogalactan Biosynthesis
2.2.1. Decaprenylphosphoryl-β-D-ribose-2′-epimerase
Decaprenylphosphoryl-β-D-ribose-2′-epimerase (DprE) is a heterodimeric enzyme comprising DprE1 and DprE2 proteins. DprE1, a key enzyme in the cell wall biosynthesis of Mtb, was initially discovered as a target of benzothiazinones, which show potent activity against Mtb [25, 26]. Recently, four new drugs, namely, BTZ-043, PBTZ-169, OPC-167832, and TBA-7371 (Figure 1(c)), have been formally tested in the clinical trials (http://www.newtbdrugs.org/pipelines/clinical).
BTZ-043, the first discovered DprE1 inhibitor, exhibited more significant anti-TB activity than INH and EMB in vitro (minimum inhibitory concentration [MIC] 2.3 nM) and in vivo [27]. A phase I clinical trial of BTZ-043 has been completed (ClinicalTrials.gov identifier: NCT04874948), and a phase III clinical trial is recruiting now (ClinicalTrials.gov identifier: NCT04044001). Meanwhile, PBTZ-169 had lower cytotoxicity and better efficacy in a murine model than BTZ-043. The protonation of piperazine nitrogen makes it more soluble and improves its potency in vivo [28]. The phase II clinical trial of PBTZ-169 has been suspended owing to slow enrollment (ClinicalTrials.gov Identifier: NCT03334734).
OPC-167832, a newly synthesized carbostyril derivative, exerts potent inhibitory effects on both growing and intracellular Mtb. The MICs of OPC-167832 against Mtb, including MDR/XDR-Mtb, ranged from 0.24 to 2 ng/mL (https://www.newtbdrugs.org/pipeline/compound/opc-167832). In vivo experiments in mice showed that optimized regimens of OPC-167832 combined with delamanid have the potential to shorten the therapy course and significantly improve the prognosis of drug-sensitive TB and MDR-TB.
TBA-7371 is a noncovalent DprE1 inhibitor that has completed a phase I clinical trial. It inhibits DprE1 with an IC50 value of 10 nM and is active against Mtb with a MIC range of 0.78–3.12 μM (https://www.newtbdrugs.org/). TBA-7371 could shorten the standard therapy course and has no cross-resistance to the current anti-TB drugs [29]. At present, a phase II clinical trial to assess the safety, early bactericidal activity, and pharmacokinetics of TBA-7371 is recruiting participants (ClinicalTrials.gov Identifier: NCT04176250).
2.2.2. GlcNAc-1-P Transferase
GlcNAc-1-P transferase (WecA) is a phosphotransferase that catalyzes uridine diphosphate (UDP)-GlcNAc and decaprenyl-P to form decaprenyl-P-P-GlcNAc, which is further extended by rhamnosyl transferase to form decaprenyl-P-P-GlcNAc-rhamnose. WecA has been proposed as a target of the caprazamycin derivative CPZEN-45 (a Streptomyces-derived product, Figure 1(d)). CPZEN-45 showed excellent activity against both replicating and nonreplicating Mtb strains, with a MIC of 1.56 μg/mL and 6.25 μg/mL for Mtb H37Rv strain and MDR-Mtb strain, respectively. However, the development of an oral formulation for CPZEN-45 may not be feasible owing to its poor solubility and low bioavailability [30].
2.2.3. TDP-6-deoxy-D-xylo-4-hexulose 3,5-Epimerase
TDP-6-deoxy-D-xylo-4-hexulose 3,5-epimerase (RmlC), the rate-limiting enzyme in the production of thymidine diphospho-L-rhamnose, is responsible for converting dTDP-4-keto-6-deoxy-glucose into dTDP-4-keto-rhamnose [31]. Potential RmlC inhibitors with benzimidazolone structures were identified using high-throughput screening of 201,368 compounds. Nevertheless, in vitro experiments showed that most of these compounds displayed no significant anti-TB activities, except for compound SID 7975595 (Figure 2(a)), which exhibited an IC50 of 0.398 μM on RmlC [32].
Figure 2.
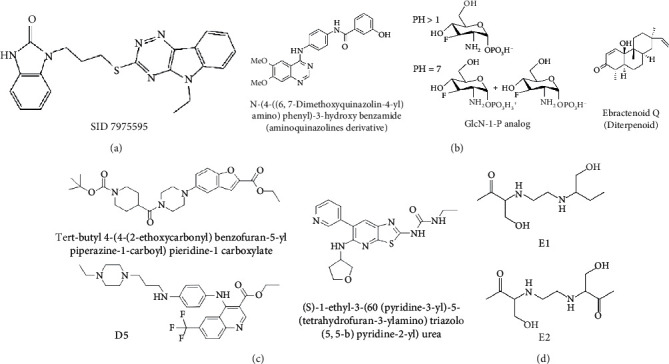
The chemical structures of SID 7975595 (a), the inhibitors of GlmU (b), the inhibitors of gyrB (c), and the inhibitors of E1 and E2 (d).
2.3. Targeting Peptidoglycan Biosynthesis
2.3.1. Translocase I
Translocase I (murX), an essential enzyme for the growth of Mtb, converts UDP-N-acetylmuramyl-pentapeptide into prenyl-N-acetylmuramyl-pentapeptide (lipid I) in peptidoglycan biosynthesis [33]. MurX inhibitors (e.g., lead compound SQ641) rapidly killed Mtb during the growing stage. SQ641, a chemically modified nucleoside analog, killed Mtb faster than any existing anti-TB drugs and had a pronounced postantibiotic effect up to 55 h. However, a lipophilic decanoyl side chain in SQ641 likely contributes to its low solubility and poor oral absorption, resulting in modest intracellular activity against Mtb and poor in vivo activity [34]. Scientists have developed an SQ641 phospholipid nanoemulsion structure (SQ641-NE), which accommodates higher drug loading and is compatible with human use. SQ641-NE demonstrates better intracellular and in vivo activity and has synergistic interactions with the three first-line anti-TB drugs, INH, EMB, and PZA [35].
2.3.2. Mycobactin
Mtb releases high-affinity siderophores, namely, mycobactin, which binds to iron more efficiently than host proteins [36]. MbtI, the product of rv2386c in Mtb, is the presumed isochorismate synthase that catalyzes the transformation of chorismate into salicylate and pyruvate in the first step of the mycobactin biosynthesis pathway. Being a chorismite-utilizing enzyme, mbtI does not exist in the host; therefore, mbtI inhibitors, including chromane-based derivatives and synthetic compounds, such as benzodihydropyranones, furans, and benzimidazole derivatives, are likely to be good therapeutic candidates for humans [37–39]. In addition, carbonyl and 7-hydroxyl groups are the necessary structures for these inhibitors.
MbtA, an adenylating enzyme encoded by mbtA, the initiating gene of mycobactin biosynthesis, catalyzes the synthesis of salicylic acid and ATP into salicyl adenylate. Nucleoside analogs, reported as mbtA inhibitors, exhibited potent anti-TB activity under iron-deficient conditions and had no obvious toxicity to mammals. However, these inhibitors possess unsatisfactory pharmacokinetic properties, including poor oral bioavailability, low oral exposure, and rapid clearance [40].
2.3.3. Alanine Racemase and D-Ala-D-Ala Ligase
Among the existing second-line anti-TB drugs, CS is known to inhibit alanine racemase (alr) and D-Ala-D-Ala ligase (ddlA) with MIC values in the micromolar range. Alr is an essential enzyme for the racemization of L-alanine to D-alanine in most bacteria [41]. DdlA is a multidomain protein that binds two D-Ala fragments in the presence of ATP to form the precursor of the peptidoglycan D-Ala-D-Ala, which acts as a dipeptide donor in pentapeptide peptidoglycan biosynthesis [42]. Alr and ddlA are considered attractive drug targets owing to their essentiality in Mtb and the absence of a homolog in humans.
2.3.4. N-Acetylglucosamine-1-phosphate Uridyltransferase
UDP-GlcNAc, an essential precursor of peptidoglycan and lipopolysaccharide in Mtb, is produced by a four-step biosynthetic pathway in the cytoplasm catalyzed by three enzymes [D-fructose-6-phosphate aminotransferase, phosphoglucosamine mutase, and N-acetylglucosamine-1-phosphate uridyltransferase (GlmU)] [43]. Deletion of GlmU in Mtb can lead to extensive perturbation of bacterial morphology and substantial reduction in cell wall thickness under normal and hypoxic conditions [44]. Three classes of compounds were identified as promising inhibitors of GlmU, namely, aminoquinazolines, GlcN-1-P analogs, and some diterpenoids extracted from the traditional Chinese medicines (Figure 2(b)) [45–47].
2.4. Targeting Other Targets in Cell Wall Biosynthesis
2.4.1. Inositol-1-phosphate Synthase
Inositol is vital for the biogenesis of mycothiol, phosphatidylinositol, and glycosylphosphatidylinositol anchors linked to complex carbohydrates in Mtb. In the inositol biosynthesis pathway, glucose-6-phosphate is converted to inositol-1-phosphate through ino1. Ino1 is further dephosphorylated by inositol monophosphatase to produce inositol [48]. Ino1, encoded by rv0046c, is essential for Mtb growth. A previous study has shown that ino1 mutation in Mtb strains attenuated growth and reproduction in the normal medium [49]. Therefore, ino1 may be a rational target for the development of anti-TB drugs. However, it is necessary to consider the possible side effects of ino1 inhibitors due to the ubiquity of ino1 in eukaryotic cells.
2.4.2. Methylerythritol Phosphate Pathway
Isopentenyl diphosphate (IPP) and its isomer dimethylallyl diphosphate (DMAPP) are crucial for the synthesis of arabinogalactan, peptidoglycan, and mycolic acid in the cell wall of Mtb [50]. It has been found that the mevalonate pathway and methylerythritol phosphate (MEP) pathway can be used in the biosynthesis of IPP and DMAPP. Most bacterial pathogens utilize the MEP pathway, whereas the mevalonate pathway is present in humans. A series of potent inhibitors of several enzymes in the MEP pathway, including fosmidomycin, thiazolyl pyrimidines, and natural products such as alkaloids, have been evaluated in the preliminary stage; therefore, further studies are needed on their pharmacological activities [50, 51].
3. Targeting Protein Biosynthesis and Breakdown
3.1. Targeting Protein Biosynthesis
In Mtb, the functional ribosomes are composed of 50S subunits (including 23S rRNA, 5S rRNA, and 35 proteins) and 30S subunits (including 16S rRNA and 22 proteins) [52]. Preliminary studies have demonstrated that most oxazolidinone compounds inhibit bacterial protein synthesis at the initial stage of transformation by binding to the V domain of 23S rRNA [53]. Certain other compounds act on the 50S ribosome subunit or leuRS to exhibit anti-TB activity.
3.1.1. 50S Ribosome Subunits
AZD5847, an oxazolidinone derivative, blocks translation by binding with 50S ribosome subunits to inhibit protein synthesis. AZD5847 is a prodrug that is mainly activated into disodium phosphonate of AZD5847 in vivo to play an anti-TB role [54]. With regard to safety, AZD5847 showed adverse reactions of thrombocytopenia and hyperbilirubinemia when administered at high doses in phase II clinical studies [55].
3.1.2. 23S rRNA
Sutezolid (PUN-100480), a novel oxazolidinone and a thiomorpholinyl analog of linezolid, significantly reduces the number of colony-forming units and acts by binding to the 23S ribosome [56]. Preliminary evidence showed that the activity of prodrug sutezolid in killing intracellular Mtb in patients was 10 times higher than that of its metabolite [57]. Phase I studies revealed that sutezolid was well tolerated at all dosages, was more active against MDR-Mtb isolates, and was less toxic than linezolid [58]. Phase II clinical trials showed that 14% of patients had a slight elevation in alanine aminotransferase levels [59]. At present, phase II and phase IV clinical trials are ongoing (ClinicalTrials.gov Identifiers: NCT03959566 and NCT03237182, respectively).
Delpazolid (LCB01-0371) is a second-generation oxazolidinone compound that contains a cyclic amidrazone. The compound had high aqueous solubility, good absorption, distribution, metabolism, excretion, and pharmacokinetic profiles, and low toxicity. In clinical trials, no obvious serious side effects were observed in any subjects, but mild diarrhea, dyspepsia, headache, nausea, and common adverse reactions were observed [60]. Currently, a phase II clinical trial was performed to evaluate the safety, efficacy, tolerability, and pharmacokinetics of delpazolid (combined with bedaquiline, delamanid, and moxifloxacin) in patients with pulmonary TB. This clinical trial is recruiting, and the result is not published (ClinicalTrials.gov Identifier: NCT04550832).
Contezolid (MRX-I), a new class of oxazolidinone protein inhibitors, is mainly used for the treatment of gram-positive bacteria. Eckburg et al. [61] reported that MRX-I had high bioavailability when taken with food and low drug accumulation after multidose administration. When a single dose was increased, the subjects showed good tolerance to MRX-I. Other studies have demonstrated that the anti-TB activity of MRX-I is comparable to that of linezolid, while MRX-I is less toxic in inhibiting bone marrow and monoamine oxidase [62, 63].
3.1.3. Leucyl-tRNA Synthase
Leucyl-tRNA synthase (LeuRS) belongs to the class I aminoacyl-tRNA synthase subgroup and plays an important role in intracellular transport. GSK-3036656, a benzoxazole compound, exerts anti-TB effects by inhibiting Mtb LeuRS to block protein synthesis. The results of a phase I clinical trial showed that GSK3036656 was generally well tolerated after single and multiple doses; no serious adverse events were reported. In addition, GSK3036656 showed 2- to 3-fold accumulation when administered repeatedly, and the pharmacokinetic parameter experiments were not altered in the presence of food [64]. Recently, to evaluate the early bactericidal activity, safety, and tolerability of GSK3036656 in TB patients, a phase II clinical trial was conducted by GlaxoSmithKline in drug-sensitive pulmonary TB patients (ClinicalTrials.gov Identifier: NCT03557281); this clinical trial has been completed but the results are not yet available. Moreover, the results from another randomized, double-blind, placebo-controlled FTIH study showed that GSK3036656 is safe and generally well tolerated after single and multiple doses in healthy subjects (ClinicalTrials.gov Identifier: NCT03075410) [64].
3.2. Targeting Clp Proteases
Clp proteases, which are responsible for the degradation of abnormal proteins and nonfunctional proteins, are highly conserved intracellular proteolytic enzymes in prokaryotes and eukaryotes [65, 66]. The clp protease complex consists of a protease subunit (clpP) and an ATPase subunit (clpC or clpX) [67]. In contrast to the clp system of Escherichia coli with only one subunit, Mtb contains two coexpressed clpP subunits (clpP1 and clpP2) that function together with clpC1 and clpX, but both subunits have different substrate specificities [68]. It has been demonstrated that clp protease is a promising drug target [69]. To date, several compounds have been shown to target clp systems. For example, acyldepsipeptide antibiotics (ADEPs) and peptide boronates target clpP [70, 71], and four compounds, including lassomycin, cylomarin A, ecumicin, and rufomycin, targeting clpC1 ATPase showed potent activity against Mtb [72–75].
4. DNA-Related Enzymes
4.1. DNA Gyrase
DNA gyrase, which binds to DNA as a tetramer comprising two subunits A (gyrA) and two subunits B (gyrB), is the only type II topoisomerase in Mtb. The inhibition of the gene encoding DNA gyrase results in significant cell death because there are no viable alternative mechanisms for performing this function.
Fluoroquinolones (FQs), a class of antibiotics showing anti-TB activity, inhibit the supercoiling action of DNA gyrase by binding to the gyrA subunit and trapping the gyrase-DNA covalent complex. DC-159a is a newly synthesized broad-spectrum 8-methoxyfluoroquinolone that exhibits considerable inhibitory activity on the gyrA subunit of DNA gyrase in Mtb [76]. The MIC90 of DC-159a against drug-susceptible Mtb was 4 and 8 times higher than that of moxifloxacin and levofloxacin, respectively, with an MIC90 of 0.5 μg/mL against MDR-Mtb isolates resistant to other FQs [77].
A novel GyrB inhibitor, SPR720 (a phosphate prodrug of SPR719), is currently in clinical development for the treatment of PTB. A randomized, double-blind, placebo-controlled, phase I clinical trial evaluated the safety, drug resistance, and pharmacokinetics of SPR720. The results showed that SPR720 was well tolerated orally, and the most common adverse events were mild to moderate gastrointestinal reactions and headache, which were shown to be dose-dependent (ClinicalTrials.gov Identifier: NCT03796910) [78]. Besides, active compounds with aromatic skeleton structures were identified through high-throughput screening, and they showed desirable gyrB inhibitory and anti-Mtb activities in vitro (Figure 2(c)) [79–81].
4.2. DNA Topoisomerase I
DNA topoisomerase I, encoded by topA, catalyzes the cleavage and binding of DNA strands by cleaving phosphodiester bonds on one strand of DNA and then religating the seal. It is a basic enzyme that maintains the biological growth activity of Mtb without the need for energy factors such as ATP [82]. Depletion of intracellular protein levels upon downregulation of topA expression led to the loss of Mtb viability.
Several inhibitors of DNA topoisomerase I have been identified. For example, boldine-derived alkaloids, dihydrobenzofuranyl urea, and amsacrine derivatives inhibited DNA topoisomerase I and showed moderate inhibitory effects on Mtb [82–84].
5. Energy Metabolism
5.1. ATP Synthase
ATP synthase provides the energy needed by Mtb during its life cycle (aerobic and hypoxic dormant stages). Bedaquiline (TMC207), sold under the brand name Sirturo, acts as an active substance against Mtb by inhibiting ATP synthase responsible for generating energy to Mtb cells.
Bedaquiline inhibits mycobacterial ATP synthase by binding to the C subunit and depletes cellular energy stores. It is highly selective for Mtb and has no obvious cytotoxicity to host cells. In addition, it has no cross-resistance with other anti-TB drugs and may be an important treatment option for patients with MDR-TB [85]. Bedaquiline has shown potent activity against drug-sensitive and MDR-Mtb strains with a MIC value of 0.03 μg/mL, thus shortening the duration of treatment to 2–4 months [86]. Bedaquiline was approved for marketing by the Food and Drug Administration and was officially approved in China. In clinical trials, combining bedaquiline with other anti-TB drugs (e.g., FQs, clofazimine, PA-824, delamanid, and azithromycin) increased the risk of QTc prolongation in patients. As an indispensable drug in short-term MDR-TB regimens, the safety and efficacy of bedaquiline administrated with PA-824 and linezolid are being validated in phase III clinical trials (ClinicalTrials.gov Identifier: NCT 03086486). In addition, several clinical trials of bedaquiline are currently underway in different countries (https://www.clinicaltrials.gov/ct2/results?term=bedaquiline&cond=tuberculosis&Search=Apply&recrs=b&recrs=a&recrs=f&recrs=d&age_v=&gndr=&type=&rslt=).
At present, bedaquiline resistance has appeared. The drug resistance mechanisms can be summarized into three types: (1) as the target of bedaquiline, the mutation of aptE gene encoding the C subunit of ATP synthase weakened the binding force between bedaquiline and C subunit [87]; (2) the mutation of Rv0678 gene encoding transcription suppressor of efferent pump MmpS5/MmpL5 will lead to upregulation of MmpS5/MmpL5 expression in efferent pump system and decrease intracellular drug concentration [88]; and (3) mutations in the pepQ gene may enhance efflux effect of the drug. Besides, bedaquiline and clofazimine showed complete cross-resistance to Rv0678 and pepQ mutations [88, 89]. Therefore, the emergence of bedaquiline resistance will bring a new challenge to this novel drug treatment of TB.
5.2. Type-II NADH Dehydrogenase
Type-II NADH dehydrogenase (NDH-2) is a key enzyme in the respiratory chain for the growth and metabolism of Mtb. The absence of NDH-2 in mammalian mitochondria renders this enzyme an attractive target for antibiotic development [90]. Several compounds, such as phenothiazines, quinoline pyrimidines, quinazolones, and diphenyl iodine (DPI) analogs, have been found to be potent inhibitors of this enzyme [91–94].
5.3. Respiratory Cytochrome bc1 Complex
The respiratory cytochrome bc1 complex is responsible for catalyzing the transmission of electrons from hydroquinone to cytochrome, allowing protons to cross the plasma membrane in the respiratory chain of Mtb. Q203, an imidazopyridine amide, inhibits ATP synthesis by acting on the respiratory cytochrome bc1 complex. It inhibited MDR- and XDR-Mtb isolates at the nanomolar level and exhibited satisfactory anti-TB activity in mice infected with Mtb at a dose of <1 mg/kg. Q203 is effective against drug-resistant, MDR- and XDR-Mtb strains [95]. The result of phase І clinical trials demonstrated that Q203 was safe and well tolerated at different doses, and no significant adverse reactions were observed in the subjects (http://www.qurient.com/bbs/content.php?co_id=q203).
5.4. 1,4-Dihydroxy-2-naphthoyl CoA Synthase
Prokaryotes utilize menaquinone (vitamin K2) as a lipid-soluble redox cofactor in the electron transport chain. Menaquinone is mainly produced from mycolic acid, and the reaction is catalyzed by 1,4-dihydroxy-2-naphthoyl CoA synthase (menB); this biosynthesis pathway is absent in humans [96]. Therefore, mutation of the gene encoding menB hinders the transmission of electrons among membrane-bound protein complexes, and thus, it serves as a potential target for anti-TB drugs. It has been reported that 2-amino-4-oxo-4-phenylbutanoate derivatives containing a succinylbenzoate skeleton and some compounds with a benzoxazine skeleton structure exhibited good inhibitory effects on both replicating and nonreplicating Mtb by inhibiting menB [97, 98].
6. Lipid Metabolism
6.1. Fatty Acid Desaturase
Fatty acid desaturase in Mtb catalyzes the oxidation of alkyl-saturated fatty acids to produce alkyl-unsaturated fatty acids by introducing two cis-double bonds at the proximal or distal end [99]. Three potential aerobic desaturases (encoded by desA1, desA2, and desA3) were identified through the whole genome analysis of Mtb [100]. DesA3 (Rv3229c) is a membrane enzyme with stearyl-CoA9-desaturase (Δ9-stearyl desaturase) activity that produces oleic acid, which is an essential component of mycobacterial membrane phospholipids and triglycerides. Thiocarlide, an effective anti-TB drug developed in the 1970s against a range of MDR-TB strains, causes a decrease in oleic acid synthesis by inhibiting desA3 and in stearic acid synthesis of Mtb, eventually resulting in cell death [101].
6.2. Pantothenate Synthetase
Pantothenic acid, also known as vitamin B5, is an important precursor of coenzyme A (CoA) and ACP and plays a decisive role in the energy exchange of Mtb. In the process of pantothenic acid biosynthesis, four pantothenate synthetases (pan) (B, C, D, and E) are involved. Of these, panC is the rate-limiting enzyme in the synthesis process, which catalyzes the final step of the reaction. Some compounds containing aromatic heterocyclic skeletons have been identified as inhibitors of panC of Mtb [102]. However, further modifications need to be considered to improve potency and selectivity based on the structural characteristics of the active site of the enzyme.
7. Signal Transduction Pathways
7.1. Shikimic Acid Pathway
In the shikimate pathway, erythrose-4-phosphate is converted to the final product chorismate through seven enzymatic steps (Figure 3) [103]. It is thought to inhibit the growth of Mtb by inhibiting the activities of seven related enzymes involved. Shikimate kinase (SK), encoded by arok, is a key enzyme that converts shikimic acid to shikimic acid-3-phosphate catalyzed by ATP.
Figure 3.
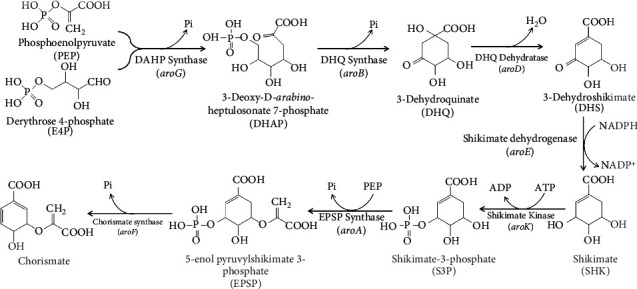
The shikimate pathway of Mycobacterium tuberculosis.
Dicyclic sesquiterpenes, which are natural products linked to quinones and synthetic small molecular compounds 3-nitrobenzyl derivatives, are promising SK inhibitors. The sesquiterpene derivative ilimaquinone (IQ) showed time-dependent inhibitory activity against Mtb shikimate kinase (MSK). Besides, 3-nitrobenzyl derivatives and pyrazole derivatives compounds also showed strong inhibitory activity against MSK [104, 105].
8. Other Targets
8.1. Filamenting Temperature-Sensitive Protein Z (FtsZ)
FtsZ, a bacterial tubulin homolog, is a significant cell division protein that attaches to the membrane of the bacterial center and forms a cell dynamic ring called the Z ring. Inactivation of ftsZ or alteration of ftsZ protein can inhibit the formation of the Z ring and septum, which in turn affects cell division [106].
Several ftsZ inhibitors have been found to be effective against Mtb, including natural products containing curcumin, coumarins, berberine, and resveratrol, small-molecule compounds comprising bisindole methane derivatives, benzamides, taxanes, and rhodamine derivatives, and some polypeptides and nucleic acids [107–110]. Most of these compounds have been tested in vitro. Some tricyclic substituted benzimidazoles have been evaluated in vivo in mice, and they showed potent anti-Mtb activity [111].
8.2. Mycobacterial Membrane Protein Large Proteins
Resistance-nodulation-division (RND) is a ubiquitous family of efflux pumps that may contribute to the recognition and transport of a great diversity of cationic, anionic, or neutral compounds in bacteria [112, 113]. Among the RND superfamily of transporters, mmpL proteins play an important role in the elaboration of the cell envelope of mycobacteria. The genes encoding for mmpL proteins are associated with gene clusters involved in the synthesis of cell wall-associated glycolipids [114]. The anti-TB lead compound BM212 and the new drug SQ109 target mmpL3 [115].
The potent compound BM212, a 1,5-diarylpyrrole derivative, showed MIC values ranging from 0.7 to 1.5 μg/mL against several Mtb strains. However, owing to its poor bioavailability and severe toxicity, BM212 was not subjected to clinical trials [115].
SQ109, an ethylenediamine compound optimized from a library based on EMB, is currently the only candidate drug for use in clinical research. It was considered a potential candidate owing to its submicromolar MIC values (0.78-2 μM) and low cytotoxicity. SQ109 showed improved activity on intracellular Mtb compared to EMB and at a similar level to INH. Several studies have reported that in addition to inhibiting Mtb cell growth, SQ109 also acts on the growth of other bacteria, fungi, and malarial parasites, all of which lack the common functional target point mmpL3 orthologs [116–118]. Therefore, it can be presumed that mmpL3 is not the only target of SQ109.
8.3. Ser/Thr Protein Kinase
The presence of several STPKs suggests that protein phosphorylation plays a central role in regulating various biological functions, ranging from environmental adaptive responses to bacterial pathogenicity. Among the 11 Mtb STPKs, only protein kinase A (pknA), pknB, pknG, and serine/threonine phosphatase are essential for intracellular survival of Mtb cells [119]. The anti-TB efficacy of pknA inhibitors was not significant. The IC50 values of many compounds that inhibited pknB were at the micromolar level; however, these compounds had no effect on Mtb [120–122].
8.4. Alkyl Hydroperoxidases
Although the alkyl hydroperoxidases ahpD and ahpC of Mtb have no sequence homology, they are regulated by the same promoter. AhpC provides hydrogen to protect Mtb from H2O2 or other oxidants, while ahpD reduces ahpC in an oxidized state, thereby ensuring that ahpC can continue to catalyze the reactions and guarantee recycling. On the other hand, ahpD exhibits alkylhydroperoxidase activity, and the highly oxidative environment in macrophages prevents ahpD from providing antioxidant protection to the internally retained Mtb. Therefore, the catalytic reaction of ahpC can be blocked by inhibiting ahpD activity in the antioxidant system of Mtb. In this case, Mtb is no longer protected by alkylhydroperoxidase, resulting in cell lysis [123]. These features make ahpD a potentially attractive target for anti-TB drugs.
8.5. Arylamine N-Acetyltransferase
NATs constitute a major family of enzymes that acetylate arylamines and hydrazines using acetyl-CoA as an acetyl donor [124]. A NAT isoenzyme in humans (known as human NAT2) is responsible for the acetylation and inactivation of INH. If INH is acetylated by NAT2, the product acetylisoniazid cannot be oxidized to its active form (isonicotinic acid) by katG. Therefore, the bioavailability of INH can be improved by effectively inhibiting the activity of NATs to kill Mtb.
8.6. Diterpene Cyclase and Diterpene Synthase
Diterpene cyclase and diterpene synthase, encoded by rv3377c and rv3378c, respectively, produce diterpenoids of tuberculosinols in the cell membrane of Mtb, which ensures the pathogenicity and virulence of Mtb [125]. Rv3377c cyclizes bicyclization and rearrangement of (E, E, E)-geranylgeranyl diphosphate to halimadienyl-diphosphate (HPP). Meanwhile, Rv3378c hydrolyzes HPP to the novel tricyclic diterpene edaxadiene, which directly inhibits phagosomal maturation in vitro [126–128]. Diterpenoid and triterpenoid acids, including isosteviol and betulinic, oleanolic, and ursolic acids, as well as binuclear isosteviol derivatives, showed moderate anti-TB activity by competitively binding to the active site of diterpene synthase.
8.7. Isocitrate Lyase (ICL)
ICL is a pivotal enzyme that catalyzes the first step of the glyoxylate cycle and has been demonstrated to be essential for Mtb survival against macrophages [129]. Studies have shown that the persistence of Mtb disappears after the deletion of the ICL gene. The 3-nitropropionamide derivatives, which are synthetic compounds, showed potent antimycobacterial activity with an IC50 value of 0.1 μM [130, 131].
8.8. Redox-Related Enzymes
Cytochrome P450 enzymes, belonging to the monooxygenase system, are mainly distributed in the endoplasmic reticulum and mitochondrial inner membrane of eukaryotic cells and are found freely in the cytoplasm of prokaryotic cells. Antifungal azoles, such as fluconazole and itraconazole, inhibit cytochrome P450-dependent enzymes. Studies have shown that econazole and clotrimazole are most effective against Mycobacterium smegmatis (MIC values <0.2 and 0.3 μM, respectively). Moreover, they are superior inhibitors of mycobacterial growth than RFP and INH, suggesting that CYP51 might be a potential target for azole drugs to exert anti-TB activities [132].
8.9. Mycothiol
Mycothiol (MSH) protects Mtb from oxidants and cytotoxins. The biosynthesis of MSH is a multistep process that four enzymatic reactions catalyzed by mshA, mshB, mshC, and mshD. Both mshB and mshD are not essential for the growth of Mtb, while the knockout of mshA and mshC could cause fatal damage to the growth of Mtb [133]. Three classes of compounds comprising the adenosine derivatives 5-O-[N-(L-cysteinyl) sulfamonyl] adenosine, NTF1836, and dequalinium chloride, were identified as promising inhibitors of mshC [134–136].
8.10. 4′-Phosphopantetheinyl Transferases
Phosphopantetheinyl transferases (PptTases) play a major role in activating fatty acids, polyketides, and nonribosomal peptide synthases, which endow Mtb with its unique ability to produce an impressive variety of lipids with unusual structures. PptT, one of two PptTases produced by Mtb, is encoded by rv2794c and is responsible for the activation of Pks13 enzyme and various type-І Pks required for the formation of mycolic acids and lipid virulence factors in mycobacteria [137]. Amidino-urea 1-[(2,6-diethylphenyl)-3-N-ethylcarbamimodoyl]urea, called “8918”, effectively killed Mtb, including drug-resistant clinical strains [138]. The “8918” showed an MIC90 of 3.1 μM and 0.56–3.0 μM against Mtb laboratory strain and 29 Mtb clinical strains, respectively [138]. However, the short half-life of “8918” leads to rapid microsomal metabolism, resulting in insufficient time to eliminate Mtb in vivo.
8.11. MptpA and mptpB
Mtb encodes two protein tyrosine phosphatases, mptpA and mptpB, which act as key virulence factors and are secreted during infection and are important for the entry and survival of Mtb in the host cell. At present, four classes of compounds have been identified as promising inhibitors, namely, chalcones, aryldifluoromethylphosphonic acids, cyclic peptides, and halogen-containing aromatic compounds [139–142]. The IC50 values of compounds with the best anti-mptpA and anti-mptpB activities were 0.16 and 0.038 nM, respectively [142].
8.12. Biofilm
Biofilm is an organized colony of bacteria surrounded by large extracellular molecules of bacteria attached to the surface of living or inanimate objects. Mtb forms biofilms in an anaerobic environment and is highly resistant to anti-TB drugs. Although the mechanism of biofilm formation of Mtb is not clear, the formation process of it can be divided into reversible adhesion and irreversible adhesion [143]. The formation mechanism of Mtb biofilm is similar to that of other bacterial biofilm. The biofilm maturation process of Mtb includes four steps, such as attachment, sessile growth, biofilm maturation, and dispersal [144]. It has been reported that compound C10 could prevent the formation of a pellicle biofilm to a large extent, making Mtb more susceptible to antibiotics [145]. C10 in conjunction with INH may restore the efficacy of INH in patients with INH-resistant TB and increase the efficacy of antibiotics in killing Mtb, including those of INH-sensitive strains.
8.13. Type VII Secretion System
The pathogenicity of Mtb is related to its type VII secretion system, which secretes a large number of effector proteins to resist the host's immune defense and promote Mtb infection. The type VII secretion system of Mtb consists of five members: ESAT-6 secretion systems (ESX) 1 to 5, among which ESX-1 is the most widely studied. The ESX-1 gene cluster is located in the RD1 region, which is not included in the BCG strain, suggesting that ESX-1 plays a critical role in Mtb virulence [146, 147]. The pathogenicity of ESX-1 deficient Mtb mutants is highly attenuated [148, 149]. Several ESX-1 inhibitors have been successfully identified and may be promising anti-TB drugs [150, 151]. Recent studies have shown that IMB-BZ can specifically inhibit the secretion of CFP-10 to reduce virulence, which will significantly reduce the survival of mycobacteria in the intracellular and in vivo [152]. In addition, the inhibitors of protein synthesis (chloramphenicol and kanamycin) and protein degradation (lassomycin and bortezomib) can specifically block ESX-1 secretion activity in Mtb and reduce the growth of Mtb by 50% or less [153]. Moreover, subinhibitory concentrations of chloramphenicol can specifically attenuate ESX-1-mediated Mtb virulence in macrophages [153]. Taken together, targeting ESX-1 may promote the development of new TB drugs, and these potential inhibitors still need to be further studied.
9. Problems and Prospects
At present, steady progress has been made in the research and development of new anti-TB drugs, many promising drug targets have been identified, and many new and different action mechanisms of the leading compounds, candidate drugs, and anti-TB drugs have been developed. However, there are still problems and challenges.
9.1. Elucidate the Biological Characteristics and Pathogenesis of Mtb
The genes, proteins, and pathways involved in the growth and metabolism of Mtb and their pathogenesis are undoubtedly the strategies for discovering new drug targets. Based on the physiological characteristics of bacterial membranes, Chen et al. [154] presumed that Mtb cell membranes could be used as a feasible target for blocking bacterial persistence and sustained survival. All biological organisms, regardless of their replication state, need to rely on functionally and structurally complete cell membranes to survive. Bacterial cell membranes are disrupted when numerous physiological metabolic targets and their transduction pathways are inhibited. However, at present, the selection of targets in the membrane is a major obstacle to the development of cell membrane inhibitors. Lipophilic drugs may cause toxic effects on mammalian cell membranes, resulting in irreversible damage to host cells [154]. These challenges notwithstanding, selective targeting of Mycobacterium cell membranes offers an opportunity to solve the problems of existing anti-TB drugs.
The pathogenicity of Mtb is related to the inflammatory response caused by the mass reproduction of Mtb in tissue cells, the components of the bacteria and cell walls, the toxicity of metabolites, and the anti-TB immune response of the body. Although there is abundant evidence that lipids may be the main pathogenic agent of Mtb, all pathogenic substances and their pathogenic mechanisms have not been fully elucidated. At least five different cell wall glycolipids are released by Mtb into the body's macrophages. The release of these components may affect physiological processes in and between cells. Therefore, lipid studies are essential to elucidate the pathogenesis of Mtb and to identify effective targets.
9.2. Strengthen the Research of Anti-TB Drug Targets
Identifying drug targets is the key to the research and development of new anti-TB drugs, and the ideal anti-TB drug target obtained should meet the following conditions: (1) the target should be the molecule necessary for the growth and metabolism of Mtb; once inactivated, it will lead to death or the lack of retention ability of Mtb; (2) the target does not rapidly produce drug resistance or bypass pathway; (3) there is no homology between the target and human molecules; and (4) the target should be different from other compounds already discovered. Some new action mechanisms and action targets, such as the biological characteristics of retentive Mtb, tissue liquefaction and cavity formation, and the host immune mechanism of latent tuberculosis infection, have also become research hotspots.
If the latent mechanism of Mtb could be elucidated and identify the targets clearing latent Mtb, it will greatly shorten the course of TB treatment and reduce the production of drug-resistant Mtb. For instance, the glyoxylate pathway is involved in the pathogenicity of a variety of pathogens, which can play a broad-spectrum antibacterial role as a drug target.
In addition, we need to expand the research ideas to focus on those proteins related to TB in the host, in which increasing or reducing the expression of these proteins can destroy the survival environment of Mtb and enhance the resistance of the body. The latent mechanism underlying Mtb in macrophages can be elucidated to find a target to break its immune escape in macrophages to promote the clearance of latent Mtb.
One of the traditional methods of drug target discovery is to identify the target through molecular pharmacology studies of drugs with significant pharmacological effects. Although lead compounds with pharmacological effects can be observed through animal experiments, disadvantages including duration of the study, high cost, and low efficiency are noted. Bioinformatics, including proteomics, genomics, gene function prediction, and data mining, have been used in drug development processes in recent years. A study established a characteristic model of cell body-related genes and identified 127 potential cell wall-related genes through bioinformatics analysis of Mtb, which provided strong support for the discovery of drug targets in the future [155]. In addition, a recent study confirmed that arabinosyltransferase C, which is involved in the cell wall synthesis of Mtb, was a potential anti-TB target from the insight of molecular docking and found that E1 and E2 (Figure 2(d)) with binding affinities of −5.77 kal/mol and−5.13 kal/mol, respectively, could be potential inhibitors of arabinosyltransferase C [156]. The combination of biotechnology, such as gene sequencing, connectivity mapping, and molecular docking, has become a new strategy for identifying drug targets for different diseases. Simulated binding with small molecular compounds using a structure simulation technique may quickly screen the inhibitors of the target from a compound library. Moreover, the application of gene manipulation techniques, such as gene knockout and gene transfer, makes the identification of drug targets easier. Therefore, the analyses of drug targets from the perspective of biology and statistics have broken through the bottleneck of traditional cytological or molecular screening methods and have greatly improved the screening speed and efficiency of new anti-TB drug targets.
9.3. In-Depth Study and Modification of Lead Compounds with Anti-TB Activity
It is well known that target inhibitors are identified mainly through high-throughput screening, which often ignores the inhibitory activity of compounds against Mtb and the adverse reactions to the host itself, leading to the slow discovery of effective lead compounds. A large number of novel lead compounds with anti-TB activity have been identified using high-throughput screening methods, but the mechanism of action of a few new drugs remains unclear. Studying the active molecular groups and mechanism of action of the lead compounds can modify the lead compounds to obtain more active anti-TB drugs. In recent years, after an in-depth study of the structure and anti-TB mechanism of bedaquiline, which still has safety concerns including QTc prolongation and hepatotoxicity, scientists have found that replacing the naphthalene C units with different heterocycles could confirm its efficacy and reduce its side effects, among which the most classical structural modification compound is TBAJ-587 [157, 158]. Recent animal studies have shown that TBAJ-587, with additional improved properties, has better anti-TB activity, safety, and pharmacokinetic properties and a lower predicted clinical dose than bedaquiline (https://www.newtbdrugs.org/pipeline/compound/tbaj-587-diarylquinoline, https://www.tballiance.org/portfolio/compound/tbaj-587-diarylquinoline). Currently, a randomized, double-blind, placebo-controlled phase I clinical trial is recruiting healthy adults to evaluate the safety, tolerability, and PK of TBAJ-587 (NCT04890535).
9.4. Identify Anti-TB Activity from Existing Antibiotics or Natural Products
There is no doubt that the discovery of new chemical scaffolds with novel mechanisms of action is risky. Therefore, it is an effective strategy to identify the anti-TB activity of existing antibiotics or optimize their structure to find new anti-TB drugs. Linezolid is a synthetic oxazolidinone antibiotic approved by the United States Food and Drug Administration in 2000 to treat infections caused by gram-positive cocci. In recent years, linezolid has been shown to be effective in the clinical treatment of MDR-TB [159]. Due to the lack of drugs for treating drug-resistant TB, a combination therapy strategy containing linezolid has been reported to be a promising option for MDR-TB and XDR-TB treatment [160]. At present, owing to the serious side effects of longer-term use, including bone marrow suppression and optic and peripheral neuropathy, limits the clinical application of linezolid [161]. If this concern is amended through structural modification, linezolid analogs could be developed as new anti-TB drugs in the future. Metronidazole, a class of nitroimidazole antibiotics, has been widely used to treat local infections caused by anaerobic bacteria in clinical settings and has entered the clinical trial stage of anti-TB in recent years (https://www.clinicaltrials.gov/ct2/results?cond=tuberculosis&term=metronidazole&cntry=&state=&city=&dist=). The approved delamanid and PA-824 were derived from the metronidazole scaffold through different chemical structure modifications. The advantage of this strategy is that there is a large amount of pharmacodynamics and human safety data available that can be quickly entered into clinical trials, but the number of antibiotics that can be reevaluated is limited.
Although a large number of natural products have emerged with anti-TB activity, few natural products or derivatives have been developed into drugs since the discovery of RFP in 1968. There were few unmodified natural products that could exert the drug activity directly, all of which need to be modified by introducing chemical groups. CPZEN-45 and capuramycins, natural product derivatives with detailed synthetic routes discovered in recent years, were all suitable for the treatment of TB. Nevertheless, it is still necessary to develop more unknown natural products to expand the pipeline of anti-TB drugs. Scientists have isolated more than 170 compounds with anti-TB properties from marine organisms, of which 10 had strong activities and potential for further development [162]. In addition, traditional Chinese medicine is a promising source for the development of anti-TB lead compounds. For example, cordycepin, an efficient component of Cordyceps spp., kills Mtb by hijacking bacterial adenosine kinase [163]. An active compound isolated from Arisaema sinii Krause could inhibit biofilm formation of Mtb [164]. However, these studies are limited to in vitro experimental studies, and there are few reports on further animal experiments and clinical trials. Therefore, the development of traditional Chinese medicine against TB is relatively slow.
9.5. Computational Technologies for Screening and Developing Novel Anti-TB Drugs
In recent years, with the development of bioinformatics, a growing number of computational technologies have been used for drug design and development. Compared with the traditional drug screening methods, the emerging computational approaches for drug screening greatly improve the chance of determining effective drug molecules and save time and effort. For example, Puhl et al. developed several computational screening approaches based on the identified crystal structure of DG167 in KasA protein which is involved in mycolic acids synthesis and identified several drug molecules that bind effectively to KasA protein [165]. Almeleebia et al. selected 224,205 compounds and screened the catalytic site of Mtb proteasome by the computational approach and then docked the top hit compounds with the Mtb proteasome molecule, discovering the interaction mechanism between these compounds and the proteasome and successfully identifying several Mtb proteasome inhibitors [166]. The effective combination of machine learning, artificial intelligence, CRIS, and other technologies will greatly save time and reduce the cost, providing a powerful weapon in the fight against TB in the future [167].
10. Conclusion
In summary, there are currently more than 15 kinds of anti-TB drugs in clinical trials, but only bedaquiline and delamanid have been listed with a new target and structure as of yet. Screening more anti-TB candidates is urgently needed to develop more effective and safer anti-TB drugs, especially against MDR-TB and XDR-TB. Cell wall synthesis, ATP synthesis, protein synthesis, DNA synthesis, and other signal transduction pathways of Mtb mentioned in this review have been considered the research hotspots of anti-TB drug development in recent years. Targeting persistent Mtb in infected host cells has become a focus of research in this field. In the future, we should find more reasonable and effective targets to develop new drug candidates with high activity and fewer adverse reactions, and the research and development of anti-TB drugs still have a long way to go.
Acknowledgments
This work was supported by the Beijing Science and Technology Committee Research Project Foundation of China [grant number Z171100001717010] and General Subject Foundation of the 8th Medical Center of PLA General Hospital [grant number 2018MS-010].
Abbreviations
- 8918:
Amidino-urea 1-[(2,6-diethylphenyl)-3-N-ethylcarbamimodoyl]urea
- Alr:
Alanine racemase
- DprE:
Decaprenylphosphoryl-β-D-ribose-2′-epimerase
- DdlA:
D-Ala-D-Ala ligase
- Ddn:
Deazaflavin-dependent nitroreductase
- FabH:
β-Ketoacyl-acyl carrier protein synthase III
- Ftsz:
Filamenting temperature-sensitive protein Z
- InhA:
Enoyl-acyl carrier protein reductase
- Ino1:
Inositol-1-phosphate synthase
- ICL:
Isocitrate lyase
- LCB01-0371:
Delpazolid
- LeuRS:
Leucyl-tRNA synthase
- MDR:
Multidrug-resistant
- Mtb:
Mycobacterium tuberculosis
- MurX:
Translocase І
- MRX-І:
Contezolid
- MenB:
1,4-Dihydroxy-2-naphthoyl CoA synthase
- SK:
Shikimate kinase
- MmpL:
Mycobacterial membrane protein large
- MSH:
Mycothiol
- MIC:
Minimum inhibitory concentration
- NO:
Nitric oxide
- NDH-2:
Type-II NADH dehydrogenase
- NATs:
Arylamine N-acetyltransferase
- PA-824:
Pretomanid
- PUN-100480:
Sutezolid
- PptT:
4′-Phosphopantetheinyl transferase
- STPK:
Ser/Thr protein kinase
- TB:
Tuberculosis
- WecA:
GlcNAc-1-P transferase
- WHO:
Word Health Organization
- XDR:
Extensively drug-resistant.
Data Availability
The data used to support the findings of this study are included in this article.
Conflicts of Interest
The authors declare no conflicts of interest.
Authors' Contributions
XQW was responsible for the conception of the manuscript, and JM and WPG contributed to the assessment of the literature content and writing of the manuscript. All authors read and approved the final manuscript. Jie Mi and Wenping Gong contributed equally to this work.
References
- 1.WHO. Global Tuberculosis Report . Vol. 2021. World Health Organization; 2021. [Google Scholar]
- 2.Miotto P., Zhang Y., Cirillo D. M., Yam W. C. Drug resistance mechanisms and drug susceptibility testing for tuberculosis. Respirology . 2018;23(12):1098–1113. doi: 10.1111/resp.13393. [DOI] [PubMed] [Google Scholar]
- 3.Pi Rui L. Q., Qian G. Fitness cost and compensatory evolution of drug-resistant mycobacterium tuberculosis. Journal of Microbes and Infections. . 2017;12:362–368. [Google Scholar]
- 4.Campaniço A., Moreira R., Lopes F. Drug discovery in tuberculosis. New drug targets and antimycobacterial agents. European Journal of Medicinal Chemistry . 2018;150:525–545. doi: 10.1016/j.ejmech.2018.03.020. [DOI] [PubMed] [Google Scholar]
- 5.Alahari A., Trivelli X., Guérardel Y., et al. Thiacetazone, an antitubercular drug that inhibits cyclopropanation of cell wall mycolic acids in mycobacteria. PloS One . 2007;2(12, article e1343) doi: 10.1371/journal.pone.0001343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quemard A., Sacchettini J. C., Dessen A., et al. Enzymic characterization of the target for isoniazid in mycobacterium tuberculosis. Biochemistry . 1995;34(26):8235–8241. doi: 10.1021/bi00026a004. [DOI] [PubMed] [Google Scholar]
- 7.Ho Y. M., Sun Y.-J., Wong S.-Y., Lee A. S. Contribution ofdfrAandinhAMutations to the detection of Isoniazid-ResistantMycobacterium tuberculosisIsolates. Antimicrobial Agents and Chemotherapy . 2009;53(9):4010–4012. doi: 10.1128/AAC.00433-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vosátka R., Krátký M., Vinšová J. Triclosan and its derivatives as antimycobacterial active agents. European Journal of Pharmaceutical Sciences . 2018;114:318–331. doi: 10.1016/j.ejps.2017.12.013. [DOI] [PubMed] [Google Scholar]
- 9.AlMatar M., Makky E. A., Var I., Kayar B., Koksal F. Novel compounds targeting InhA for TB therapy. Pharmacological Reports . 2018;70(2):217–226. doi: 10.1016/j.pharep.2017.09.001. [DOI] [PubMed] [Google Scholar]
- 10.Gomez A., Andreu N., Ferrer-Navarro M., Yero D., Gibert I. Triclosan-induced genes Rv1686c - Rv1687c and Rv3161c are not involved in triclosan resistance in Mycobacterium tuberculosis. Scientific Reports . 2016;6(1, article 26221) doi: 10.1038/srep26221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Inturi B., Pujar G. V., Purohit M. N. Recent advances and structural features of enoyl-ACP reductase inhibitors of mycobacterium tuberculosis. Arch Pharm (Weinheim) . 2016;349(11):817–826. doi: 10.1002/ardp.201600186. [DOI] [PubMed] [Google Scholar]
- 12.Pan P., Knudson S. E., Bommineni G. R., et al. Time-dependent diaryl ether inhibitors of InhA: structure-activity relationship studies of enzyme inhibition, antibacterial activity, and invivo efficacy. ChemMedChem . 2014;9(4):776–791. doi: 10.1002/cmdc.201300429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Songsiriritthigul C., Hanwarinroj C., Pakamwong B., et al. Inhibition of Mycobacterium tuberculosisInhAby 3-nitropropanoic acid. Proteins: Structure, Function, and Bioinformatics . 2022;90(3):898–904. doi: 10.1002/prot.26268. [DOI] [PubMed] [Google Scholar]
- 14.Prasad M. S., Bhole R. P., Khedekar P. B., Chikhale R. V. Mycobacterium enoyl acyl carrier protein reductase (InhA): A key target for antitubercular drug discovery. Bioorganic Chemistry . 2021;115, article 105242 doi: 10.1016/j.bioorg.2021.105242. [DOI] [PubMed] [Google Scholar]
- 15.Zhang H. J., Li Z. L., Zhu H. Advances in the research of β-Ketoacyl-ACP synthase III (FabH) inhibitors. Current Medicinal Chemistry . 2012;19(8):1225–1237. doi: 10.2174/092986712799320484. [DOI] [PubMed] [Google Scholar]
- 16.Jha D. K., Panda L., Lavanya P., Ramaiah S., Anbarasu A. Detection and confirmation of alkaloids in leaves of Justicia adhatoda and bioinformatics approach to elicit its anti-tuberculosis activity. Applied Biochemistry and Biotechnology . 2012;168(5):980–990. doi: 10.1007/s12010-012-9834-1. [DOI] [PubMed] [Google Scholar]
- 17.Liu Y., Zhong W., Li R. J., Li S. Synthesis of potent inhibitors of β-ketoacyl-acyl carrier protein synthase III as potential antimicrobial agents. Molecules . 2012;17(5):4770–4781. doi: 10.3390/molecules17054770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang C.-c., Smith C. V., Glickman M. S., Jacobs W. R., Sacchettini J. C. Crystal Structures of Mycolic Acid Cyclopropane Synthases from Mycobacterium tuberculosis. The Journal of Biological Chemistry . 2002;277(13):11559–11569. doi: 10.1074/jbc.M111698200. [DOI] [PubMed] [Google Scholar]
- 19.Matsumoto M., Hashizume H., Tomishige T., et al. OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. Plos Medicine . 2006;3(11, article e466) doi: 10.1371/journal.pmed.0030466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cellitti S. E., Shaffer J., Jones D. H., et al. Structure of Ddn, the Deazaflavin-Dependent Nitroreductase from Mycobacterium tuberculosis Involved in Bioreductive Activation of PA-824. Structure . 2012;20(1):101–112. doi: 10.1016/j.str.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh R., Manjunatha U., Boshoff H. I. M., et al. PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science . 2008;322(5906):1392–1395. doi: 10.1126/science.1164571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feuerriegel S., Köser C. U., Baù D., et al. Impact of fgd 1 and ddn diversity in Mycobacterium tuberculosis complex on in vitro susceptibility to PA-824. Antimicrobial Agents and Chemotherapy . 2011;55(12):5718–5722. doi: 10.1128/AAC.05500-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Upton A. M., Cho S., Yang T. J., et al. In vitro and in vivo activities of the nitroimidazole TBA-354 against Mycobacterium tuberculosis. Antimicrobial Agents and Chemotherapy . 2015;59(1):136–144. doi: 10.1128/AAC.03823-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alliance T. B. Phase 1 clinical trial of TB drug candidate TBA-354 discontinued . TB Alliance; 2018. [Google Scholar]
- 25.Brecik M., Centárová I., Mukherjee R., et al. DprE1 is a vulnerable tuberculosis drug target due to its cell wall localization. ACS Chemical Biology . 2015;10(7):1631–1636. doi: 10.1021/acschembio.5b00237. [DOI] [PubMed] [Google Scholar]
- 26.Lv K., You X., Wang B., et al. Identification of better pharmacokinetic benzothiazinone derivatives as new antitubercular agents. ACS Medicinal Chemistry Letters . 2017;8(6):636–641. doi: 10.1021/acsmedchemlett.7b00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Makarov V. V., Manina G., Mikusova K., et al. Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science . 2009;324(5928):801–804. doi: 10.1126/science.1171583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Makarov V., Lechartier B., Zhang M., et al. Towards a new combination therapy for tuberculosis with next generation benzothiazinones. Embo Molecular Medicine . 2014;6(3):372–383. doi: 10.1002/emmm.201303575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tiberi S., du Plessis N., Walzl G., et al. Tuberculosis: progress and advances in development of new drugs, treatment regimens, and host-directed therapies. The Lancet Infectious diseases . 2018;18(7):e183–e198. doi: 10.1016/S1473-3099(18)30110-5. [DOI] [PubMed] [Google Scholar]
- 30.Salomon J. J., Galeron P., Schulte N., et al. Biopharmaceutical in vitro characterization of CPZEN-45, a drug candidate for inhalation therapy of tuberculosis. Therapeutic Delivery . 2013;4(8):915–923. doi: 10.4155/tde.13.62. [DOI] [PubMed] [Google Scholar]
- 31.Kantardjieff K. A., Chang Y. K., Naranjo C., Waldo G. S., Rupp B. Mycobacterium tuberculosis RmlC epimerase (Rv3465): a promising drug-target structure in the rhamnose pathway. Acta Crystallographica Section D: Biological Crystallography . 2004;60(5):895–902. doi: 10.1107/S0907444904005323. [DOI] [PubMed] [Google Scholar]
- 32.Sivendran S., Jones V., Sun D., et al. Identification of triazinoindol-benzimidazolones as nanomolar inhibitors of the Mycobacterium tuberculosis enzyme TDP-6-deoxy- d- xylo -4-hexopyranosid-4-ulose 3,5-epimerase (RmlC) Bioorganic & Medicinal Chemistry . 2010;18(2):896–908. doi: 10.1016/j.bmc.2009.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siricilla S., Mitachi K., Skorupinska-Tudek K., Swiezewska E., Kurosu M. Biosynthesis of a water-soluble lipid I analogue and a convenient assay for translocase I. Analytical Biochemistry . 2014;461:36–45. doi: 10.1016/j.ab.2014.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bogatcheva E., Dubuisson T., Protopopova M., Einck L., Nacy C. A., Reddy V. M. Chemical modification of capuramycins to enhance antibacterial activity. The Journal of Antimicrobial Chemotherapy . 2011;66(3):578–587. doi: 10.1093/jac/dkq495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nikonenko B., Reddy V. M., Bogatcheva E., Protopopova M., Einck L., Nacy C. A. Therapeutic efficacy of SQ641-NE against Mycobacterium tuberculosis. Antimicrobial Agents and Chemotherapy . 2014;58(1):587–589. doi: 10.1128/AAC.01254-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meneghetti F., Villa S., Gelain A., et al. Iron acquisition pathways as targets for antitubercular drugs. Current Medicinal Chemistry . 2016;23(35):4009–4026. doi: 10.2174/0929867323666160607223747. [DOI] [PubMed] [Google Scholar]
- 37.Reddy P. V., Puri R. V., Chauhan P., et al. Disruption of mycobactin biosynthesis leads to attenuation of Mycobacterium tuberculosis for growth and virulence. The Journal of Infectious Diseases . 2013;208(8):1255–1265. doi: 10.1093/infdis/jit250. [DOI] [PubMed] [Google Scholar]
- 38.Chiarelli L. R., Mori M., Barlocco D., et al. Discovery and development of novel salicylate synthase (MbtI) furanic inhibitors as antitubercular agents. European Journal of Medicinal Chemistry . 2018;155:754–763. doi: 10.1016/j.ejmech.2018.06.033. [DOI] [PubMed] [Google Scholar]
- 39.Mori M., Stelitano G., Chiarelli L. R., et al. Pharmaceuticals . 2. Vol. 14. Pharmaceuticals (Basel, Switzerland): 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dawadi S., Boshoff H. I. M., Park S. W., Schnappinger D., Aldrich C. C. Conformationally constrained cinnolinone nucleoside analogues as siderophore biosynthesis inhibitors for tuberculosis. ACS Medicinal Chemistry Letters . 2018;9(4):386–391. doi: 10.1021/acsmedchemlett.8b00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dong H., Hu T., He G., et al. Structural features and kinetic characterization of alanine racemase from _Bacillus pseudofirmus_ OF4. Biochemical and Biophysical Research Communications . 2018;497(1):139–145. doi: 10.1016/j.bbrc.2018.02.041. [DOI] [PubMed] [Google Scholar]
- 42.Yang S., Xu Y., Wang Y., et al. The biological properties and potential interacting proteins of d-alanyl-d-alanine ligase a from Mycobacterium tuberculosis. Molecules . 2018;23(2):324–339. doi: 10.3390/molecules23020324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Milewski S., Gabriel I., Olchowy J. Enzymes of UDP-GlcNAc biosynthesis in yeast. Yeast (Chichester, England) . 2006;23(1):1–14. doi: 10.1002/yea.1337. [DOI] [PubMed] [Google Scholar]
- 44.Soni V., Upadhayay S., Suryadevara P., et al. Depletion of M. tuberculosis GlmU from infected murine lungs effects the clearance of the pathogen. PLoS Pathogens . 2015;11(10, article e1005235) doi: 10.1371/journal.ppat.1005235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Y., Zhou Y., Ma Y., Li X. Design and synthesis of novel cell wall inhibitors of Mycobacterium tuberculosis GlmM and GlmU. Carbohydrate Research . 2011;346(13):1714–1720. doi: 10.1016/j.carres.2011.05.024. [DOI] [PubMed] [Google Scholar]
- 46.Yu Z., Wei Y., Tian X., et al. Diterpenoids from the roots of _Euphorbia ebracteolata_ and their anti- tuberculosis effects. Bioorganic Chemistry . 2018;77:471–477. doi: 10.1016/j.bioorg.2018.02.007. [DOI] [PubMed] [Google Scholar]
- 47.Patel H. M., Palkar M., Karpoormath R. Exploring MDR-TB inhibitory potential of 4-aminoquinazolines as Mycobacterium tuberculosis N-acetylglucosamine-1-phosphate uridyltransferase (GlmUMTB) inhibitors. Chemistry & Biodiversity . 2020;17(8, article e2000237) doi: 10.1002/cbdv.202000237. [DOI] [PubMed] [Google Scholar]
- 48.Bachhawat N., Mande S. C. Identification of the INO1 gene of Mycobacterium tuberculosis H37Rv reveals a novel class of inositol-1-phosphate synthase enzyme. Journal of Molecular Biology . 1999;291(3):531–536. doi: 10.1006/jmbi.1999.2980. [DOI] [PubMed] [Google Scholar]
- 49.Movahedzadeh F., Smith D. A., Norman R. A., et al. The Mycobacterium tuberculosis ino1 gene is essential for growth and virulence. Molecular Microbiology . 2004;51(4):1003–1014. doi: 10.1046/j.1365-2958.2003.03900.x. [DOI] [PubMed] [Google Scholar]
- 50.Wang X., Dowd C. S. The methylerythritol phosphate pathway: promising drug targets in the fight against Tuberculosis. Infectious Diseases . 2018;4(3):278–290. doi: 10.1021/acsinfecdis.7b00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haymond A., Dowdy T., Johny C., et al. A high-throughput screening campaign to identify inhibitors of DXP reductoisomerase (IspC) and MEP cytidylyltransferase (IspD) Analytical Biochemistry . 2018;542:63–75. doi: 10.1016/j.ab.2017.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li X., Sun Q., Jiang C., et al. Structure of Ribosomal Silencing Factor Bound to Mycobacterium tuberculosis Ribosome. Structure . 2015;23(10):1858–1865. doi: 10.1016/j.str.2015.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Z., Pang Y., Wang Y., Liu C., Zhao Y. Beijing genotype of _Mycobacterium tuberculosis_ is significantly associated with linezolid resistance in multidrug-resistant and extensively drug- resistant tuberculosis in China. International Journal of Antimicrobial Agents . 2014;43(3):231–235. doi: 10.1016/j.ijantimicag.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 54.Pstragowski M., Zbrzezna M., Bujalska-Zadrozny M. Advances in pharmacotherapy of tuberculosis. Acta Poloniae Pharmaceutica . 2017;74(1):3–11. [PubMed] [Google Scholar]
- 55.Furin J. J., Du Bois J., van Brakel E., et al. Early bactericidal activity of AZD5847 in patients with pulmonary tuberculosis. Antimicrobial Agents and Chemotherapy . 2016;60(11):6591–6599. doi: 10.1128/AAC.01163-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.WHO. Global Tuberculosis Report 2020. Genevapp: World Health Organization . Geneva: 2020. [Google Scholar]
- 57.Zhu T., Friedrich S. O., Diacon A., Wallis R. S. Population pharmacokinetic/pharmacodynamic analysis of the bactericidal activities of sutezolid (PNU-100480) and its major metabolite against intracellular Mycobacterium tuberculosis in ex vivo whole-blood cultures of patients with pulmonary tuberculosis. Antimicrobial Agents and Chemotherapy . 2014;58(6):3306–3311. doi: 10.1128/AAC.01920-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wallis R. S., Jakubiec W. M., Kumar V., et al. Pharmacokinetics and whole-blood bactericidal activity against Mycobacterium tuberculosis of single doses of PNU-100480 in healthy volunteers. The Journal of Infectious Diseases . 2010;202(5):745–751. doi: 10.1086/655471. [DOI] [PubMed] [Google Scholar]
- 59.Wallis R. S., Dawson R., Friedrich S. O., et al. Mycobactericidal activity of sutezolid (PNU-100480) in sputum (EBA) and blood (WBA) of patients with pulmonary tuberculosis. Plo S one . 2014;9(4, article e94462) doi: 10.1371/journal.pone.0094462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Choi Y., Lee S. W., Kim A., et al. Safety, tolerability and pharmacokinetics of 21 day multiple oral administration of a new oxazolidinone antibiotic, LCB01-0371, in healthy male subjects. The Journal of Antimicrobial Chemotherapy . 2018;73:183–190. doi: 10.1093/jac/dkx367. [DOI] [PubMed] [Google Scholar]
- 61.Eckburg P. B., Ge Y., Hafkin B. Single- and multiple-dose study to determine the safety, tolerability, pharmacokinetics, and food effect of oral MRX-I versus linezolid in healthy adult subjects. Antimicrobial Agents and Chemotherapy . 2017;61(4, article e02181-16) doi: 10.1128/AAC.02181-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gordeev M. F., Yuan Z. Y. New potent antibacterial oxazolidinone (MRX-I) with an improved class safety profile. Journal of Medicinal Chemistry . 2014;57(11):4487–4497. doi: 10.1021/jm401931e. [DOI] [PubMed] [Google Scholar]
- 63.Shoen C., DeStefano M., Hafkin B., Cynamon M. In VitroandIn VivoActivities of contezolid (MRX-I) against Mycobacterium tuberculosis. Antimicrobial Agents and Chemotherapy . 2018;62(8, article e00493-18) doi: 10.1128/AAC.00493-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tenero D., Derimanov G., Carlton A., et al. First-time-in-human study and prediction of early bactericidal activity for GSK3036656, a potent leucyl-tRNA synthetase inhibitor for tuberculosis treatment. Antimicrobial Agents and Chemotherapy . 2019;63(8, article e00240-19) doi: 10.1128/AAC.00240-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Porankiewicz J., Wang J., Clarke A. K. New insights into the ATP-dependent Clp protease: Escherichia coli and beyond. Molecular Microbiology . 1999;32(3):449–458. doi: 10.1046/j.1365-2958.1999.01357.x. [DOI] [PubMed] [Google Scholar]
- 66.Mdluli K., Kaneko T., Upton A. Cold Spring Harbor Perspectives in Medicine . 2015;5(6) doi: 10.1101/cshperspect.a021154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Parish T. Targeting mycobacterial proteolytic complexes with natural products. Chemistry & Biology . 2014;21(4):437–438. doi: 10.1016/j.chembiol.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 68.Personne Y., Brown A. C., Schuessler D. L., Parish T. Mycobacterium tuberculosis ClpP proteases are co-transcribed but exhibit different substrate specificities. Plo S one . 2013;8(4, article e60228) doi: 10.1371/journal.pone.0060228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ollinger J., O'Malley T., Kesicki E. A., Odingo J., Parish T. Validation of the essential ClpP protease in Mycobacterium tuberculosis as a novel drug target. Journal of Bacteriology . 2012;194(3):663–668. doi: 10.1128/JB.06142-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Famulla K., Sass P., Malik I., et al. Acyldepsipeptide antibiotics kill mycobacteria by preventing the physiological functions of the ClpP1P2 protease. Molecular Microbiology . 2016;101(2):194–209. doi: 10.1111/mmi.13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Akopian T., Kandror O., Tsu C., et al. Cleavage Specificity of Mycobacterium tuberculosis ClpP1P2 Protease and Identification of Novel Peptide Substrates and Boronate Inhibitors with Anti- bacterial Activity. The Journal of Biological Chemistry . 2015;290(17):11008–11020. doi: 10.1074/jbc.M114.625640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gavrish E., Sit C. S., Cao S., et al. Lassomycin, a ribosomally synthesized cyclic peptide, kills Mycobacterium tuberculosis by targeting the ATP-dependent protease ClpC1P1P2. Chemistry & Biology . 2014;21(4):509–518. doi: 10.1016/j.chembiol.2014.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bürstner N., Roggo S., Ostermann N., et al. Gift from nature: cyclomarin a kills mycobacteria and malaria parasites by distinct modes of action. ChemBioChem . 2015;16(17):2433–2436. doi: 10.1002/cbic.201500472. [DOI] [PubMed] [Google Scholar]
- 74.Gao W., Kim J. Y., Anderson J. R., et al. The cyclic peptide ecumicin targeting ClpC1 is active against Mycobacterium tuberculosis in vivo. Antimicrobial Agents and Chemotherapy . 2015;59(2):880–889. doi: 10.1128/AAC.04054-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Choules M. P., Wolf N. M., Lee H., et al. Rufomycin targets ClpC1 proteolysis in Mycobacterium tuberculosis and M. abscessus. Antimicrobial Agents and Chemotherapy . 2019;63(3, article e02204-18) doi: 10.1128/AAC.02204-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ahmad Z., Minkowski A., Peloquin C. A., et al. Activity of the fluoroquinolone DC-159a in the initial and continuation phases of treatment of murine tuberculosis. Antimicrobial Agents and Chemotherapy . 2011;55(4):1781–1783. doi: 10.1128/AAC.01514-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Disratthakit A., Doi N. In vitro activities of DC-159a, a novel fluoroquinolone, against Mycobacterium species. Antimicrobial Agents and Chemotherapy . 2010;54(6):2684–2686. doi: 10.1128/AAC.01545-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Talley A. K., Thurston A., Moore G., et al. First-in-human evaluation of the safety, tolerability, and pharmacokinetics of SPR720, a novel oral bacterial DNA gyrase (GyrB) inhibitor for mycobacterial infections. Antimicrobial Agents and Chemotherapy . 2021;65(11, article e0120821) doi: 10.1128/AAC.01208-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Renuka J., Reddy K. I., Srihari K., et al. Design, synthesis, biological evaluation of substituted benzofurans as DNA gyraseB inhibitors of _Mycobacterium tuberculosis. Bioorganic & Medicinal Chemistry . 2014;22(17):4924–4934. doi: 10.1016/j.bmc.2014.06.041. [DOI] [PubMed] [Google Scholar]
- 80.Kale M. G., Raichurkar A., Waterson D., et al. Thiazolopyridine ureas as novel antitubercular agents acting through inhibition of DNA gyrase B. Journal of Medicinal Chemistry . 2013;56(21):8834–8848. doi: 10.1021/jm401268f. [DOI] [PubMed] [Google Scholar]
- 81.Islam M. A., Pillay T. S. Identification of promising DNA GyrB inhibitors for tuberculosis using pharmacophore-based virtual screening, molecular docking and molecular dynamics studies. Chemical Biology & Drug Design . 2017;90(2):282–296. doi: 10.1111/cbdd.12949. [DOI] [PubMed] [Google Scholar]
- 82.Ravishankar S., Ambady A., Awasthy D., et al. Genetic and chemical validation identifies Mycobacterium tuberculosis topoisomerase I as an attractive anti-tubercular target. Tuberculosis . 2015;95(5):589–598. doi: 10.1016/j.tube.2015.05.004. [DOI] [PubMed] [Google Scholar]
- 83.García M. T., Carreño D., Tirado-Vélez J. M., et al. Boldine-derived alkaloids inhibit the activity of DNA topoisomerase I and growth of Mycobacterium tuberculosis. Frontiers in Microbiology . 2018;9:1659–1668. doi: 10.3389/fmicb.2018.01659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Szafran M. J., Kołodziej M., Skut P., et al. Amsacrine derivatives selectively inhibit mycobacterial topoisomerase I (top a), impair M. smegmatis growth and disturb chromosome replication. Frontiers in Microbiology . 2018;9:1592–1605. doi: 10.3389/fmicb.2018.01592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Huitric E., Verhasselt P., Andries K., Hoffner S. E. In vitro antimycobacterial spectrum of a diarylquinoline ATP synthase inhibitor. Antimicrobial Agents and Chemotherapy . 2007;51(11):4202–4204. doi: 10.1128/AAC.00181-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dhillon J., Andries K., Phillips P. P. J., Mitchison D. A. Bactericidal activity of the diarylquinoline TMC207 against Mycobacterium tuberculosis outside and within cells. Tuberculosis . 2010;90(5):301–305. doi: 10.1016/j.tube.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 87.Huitric E., Verhasselt P., Koul A., Andries K., Hoffner S., Andersson D. I. Rates and mechanisms of resistance development in Mycobacterium tuberculosis to a novel diarylquinoline ATP synthase inhibitor. Antimicrobial Agents and Chemotherapy . 2010;54(3):1022–1028. doi: 10.1128/AAC.01611-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kadura S., King N., Nakhoul M., et al. Systematic review of mutations associated with resistance to the new and repurposed Mycobacterium tuberculosis drugs bedaquiline, clofazimine, linezolid, delamanid and pretomanid. The Journal of Antimicrobial Chemotherapy . 2020;75(8):2031–2043. doi: 10.1093/jac/dkaa136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chesov E., Chesov D., Maurer F. P., et al. Emergence of bedaquiline-resistance in a high-burden country of tuberculosis. European respiratory journal . 2021;59(article 2100621) doi: 10.1183/13993003.00621-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bald D., Villellas C., Lu P., Koul A. Targeting energy metabolism inMycobacterium tuberculosis, a new paradigm in antimycobacterial drug discovery. MBio . 2017;8(2):p. e00272-17. doi: 10.1128/mBio.00272-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yano T., Li L.-S., Weinstein E., Teh J.-S., Rubin H. Steady-state Kinetics and Inhibitory Action of Antitubercular Phenothiazines on Mycobacterium tuberculosis Type-II NADH-Menaquinone Oxidoreductase (NDH-2) The Journal of Biological Chemistry . 2006;281(17):11456–11463. doi: 10.1074/jbc.M508844200. [DOI] [PubMed] [Google Scholar]
- 92.Shirude P. S., Paul B., Roy Choudhury N., Kedari C., Bandodkar B., Ugarkar B. G. Quinolinyl pyrimidines: potent inhibitors of NDH-2 as a novel class of anti-TB agents. ACS Medicinal Chemistry Letters . 2012;3(9):736–740. doi: 10.1021/ml300134b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Murugesan D., Ray P. C., Bayliss T., et al. 2-Mercapto-Quinazolinones as Inhibitors of Type II NADH Dehydrogenase andMycobacterium tuberculosis: Structure–Activity Relationships, Mechanism of Action and Absorption, Distribution, Metabolism, and Excretion Characterization. ACS infectious diseases . 2018;4(6):954–969. doi: 10.1021/acsinfecdis.7b00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nguyen N., Wilson D. W., Nagalingam G., et al. Broad activity of diphenyleneiodonium analogues against _Mycobacterium tuberculosis_ , malaria parasites and bacterial pathogens. European Journal of Medicinal Chemistry . 2018;148:507–518. doi: 10.1016/j.ejmech.2017.10.010. [DOI] [PubMed] [Google Scholar]
- 95.Malík I., Čižmárik J., Kováč G., Pecháčová M., Hudecova L. Telacebec (Q203): is there a novel effective and safe anti-tuberculosis drug on the horizon. Ceska a Slovenska farmacie: casopis Ceske farmaceuticke spolecnosti a Slovenske farmaceuticke spolecnosti . 2021;70(5):164–171. doi: 10.5817/CSF2021-5-164. [DOI] [PubMed] [Google Scholar]
- 96.Johnston J. M., Arcus V. L., Baker E. N. Structure of naphthoate synthase (MenB) from Mycobacterium tuberculosis in both native and product-bound forms. Acta Crystallographica . 2005;61(9):1199–1206. doi: 10.1107/S0907444905017531. [DOI] [PubMed] [Google Scholar]
- 97.Li X., Liu N., Zhang H., et al. CoA adducts of 4-Oxo-4-phenylbut-2-enoates: inhibitors of MenB from the M. tuberculosis menaquinone biosynthesis pathway. CoA adducts of 4-oxo-4-phenylbut-2-enoates: Inhibitors of Men B from the M. tuberculosis menaquinone biosynthesis pathway. ACS medicinal chemistry letters . 2011;2(11):818–823. doi: 10.1021/ml200141e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li X., Liu N., Zhang H., Knudson S. E., Slayden R. A., Tonge P. J. Synthesis and SAR studies of 1,4-benzoxazine MenB inhibitors: Novel antibacterial agents against Mycobacterium tuberculosis. Bioorganic & Medicinal Chemistry Letters . 2010;20(21):6306–6309. doi: 10.1016/j.bmcl.2010.08.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yeruva V. C., Savanagouder M., Khandelwal R., Kulkarni A., Sharma Y., Raghunand T. R. The Mycobacterium tuberculosis desaturase DesA1 (Rv0824c) is a Ca2+ binding protein. Biochemical and Biophysical Research Communications . 2016;480(1):29–35. doi: 10.1016/j.bbrc.2016.10.014. [DOI] [PubMed] [Google Scholar]
- 100.Cole S. T., Brosch R., Parkhill J., et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature . 1998;393(6685):537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 101.Phetsuksiri B., Jackson M., Scherman H., et al. Unique Mechanism of Action of the Thiourea Drug Isoxyl on Mycobacterium tuberculosis. The Journal of Biological Chemistry . 2003;278(52):53123–53130. doi: 10.1074/jbc.M311209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Velaparthi S., Brunsteiner M., Uddin R., Wan B., Franzblau S. G., Petukhov P. A. 5-tert-Butyl-N-pyrazol-4-yl-4,5,6,7-tetrahydrobenzo[d]isoxazole-3-carboxamide derivatives as novel potent inhibitors ofMycobacterium tuberculosisPantothenate synthetase: initiating a quest for new antitubercular drugs. Journal of Medicinal Chemistry . 2008;51(7):1999–2002. doi: 10.1021/jm701372r. [DOI] [PubMed] [Google Scholar]
- 103.Parish T., Stoker N. G. The common aromatic amino acid biosynthesis pathway is essential in Mycobacterium tuberculosis. Microbiology (Reading, England) . 2002;148(10):3069–3077. doi: 10.1099/00221287-148-10-3069. [DOI] [PubMed] [Google Scholar]
- 104.Simithy J., Fuanta N. R., Hobrath J. V., et al. Mechanism of irreversible inhibition of _Mycobacterium tuberculosis_ shikimate kinase by ilimaquinone. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics . 2018;1866(5-6):731–739. doi: 10.1016/j.bbapap.2018.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rahul Reddy M. B., Krishnasamy S. K., Kathiravan M. K. Identification of novel scaffold using ligand and structure based approach targeting shikimate kinase. Bioorganic Chemistry . 2020;102, article 104083 doi: 10.1016/j.bioorg.2020.104083. [DOI] [PubMed] [Google Scholar]
- 106.Erickson P. H. Modeling the physics of FtsZ assembly and force generation. Proceedings of the National Academy of Sciences of the United States of America . 2009;106(23):9238–9243. doi: 10.1073/pnas.0902258106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Haranahalli K., Tong S., Ojima I. Recent advances in the discovery and development of antibacterial agents targeting the cell-division protein FtsZ. Bioorganic & Medicinal Chemistry . 2016;24(24):6354–6369. doi: 10.1016/j.bmc.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Singh D., Bhattacharya A., Rai A., et al. SB-RA-2001 inhibits bacterial proliferation by targeting FtsZ assembly. Biochemistry . 2014;53(18):2979–2992. doi: 10.1021/bi401356y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Khadkikar P., Goud N. S., Mohammed A., et al. An efficient and facile green synthesis of bisindole methanes as potentialMtbFtsZ inhibitors. Chemical Biology & Drug Design . 2018;92(6):1933–1939. doi: 10.1111/cbdd.13363. [DOI] [PubMed] [Google Scholar]
- 110.Akinpelu O. I., Kumalo H. M., Mhlongo S. I., Mhlongo N. N. Identifying the analogues of berberine as promising antitubercular drugs targetingMtb‐FtsZpolymerisation through ligand‐based virtual screening and molecular dynamics simulations. Journal of Molecular Recognition . 2022;35(2, article e2940) doi: 10.1002/jmr.2940. [DOI] [PubMed] [Google Scholar]
- 111.Knudson S. E., Awasthi D., Kumar K., et al. Cell division inhibitors with efficacy equivalent to isoniazid in the acute murine Mycobacterium tuberculosis infection model. The Journal of Antimicrobial Chemotherapy . 2015;70(11):3070–3073. doi: 10.1093/jac/dkv226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Daury L., Orange F., Taveau J. C., et al. Tripartite assembly of RND multidrug efflux pumps. Nature Communications . 2016;7(1):1–8. doi: 10.1038/ncomms10731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Delmar J. A., Su C. C., Yu E. W. Bacterial multidrug efflux transporters. Annual Review of Biophysics . 2014;43(1):93–117. doi: 10.1146/annurev-biophys-051013-022855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Geetha V. R. Mmp L3 a potential new target for development of novel anti-tuberculosis drugs. Expert Opinion on Therapeutic Targets . 2013;18(3):247–256. doi: 10.1517/14728222.2014.859677. [DOI] [PubMed] [Google Scholar]
- 115.La Rosa V., Poce G., Canseco J. O., et al. MmpL3 is the cellular target of the antitubercular pyrrole derivative BM212. Antimicrobial Agents and Chemotherapy . 2012;56(1):324–331. doi: 10.1128/AAC.05270-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tahlan K., Wilson R., Kastrinsky D. B., et al. SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrobial Agents and Chemotherapy . 2012;56(4):1797–1809. doi: 10.1128/AAC.05708-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Makobongo M. O., Einck L., Peek R. M., Jr., Merrell D. S. In vitro characterization of the anti-bacterial activity of SQ109 against Helicobacter pylori. Plo S one . 2013;8(7, article e68917) doi: 10.1371/journal.pone.0068917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Veiga-Santos P., Li K., Lameira L., et al. SQ109, a new drug lead for Chagas disease. Antimicrobial Agents and Chemotherapy . 2015;59(4):1950–1961. doi: 10.1128/AAC.03972-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Khan M. Z., Kaur P., Nandicoori V. K. Targeting the messengers: serine/threonine protein kinases as potential targets for antimycobacterial drug development. IUBMB Life . 2018;70(9):889–904. doi: 10.1002/iub.1871. [DOI] [PubMed] [Google Scholar]
- 120.Magnet S., Hartkoorn R. C., Székely R., et al. Leads for antitubercular compounds from kinase inhibitor library screens. Tuberculosis . 2010;90(6):354–360. doi: 10.1016/j.tube.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 121.Székely R., Wáczek F., Szabadkai I., et al. A novel drug discovery concept for tuberculosis: inhibition of bacterial and host cell signalling. Immunology Letters . 2008;116(2):225–231. doi: 10.1016/j.imlet.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 122.Walburger A., Koul A., Ferrari G., et al. Protein kinase G from pathogenic mycobacteria promotes survival within macrophages. Science . 2004;304(5678):1800–1804. doi: 10.1126/science.1099384. [DOI] [PubMed] [Google Scholar]
- 123.Koshkin A., Nunn C. M., Djordjevic S., Ortiz de Montellano P. R. The Mechanism of Mycobacterium tuberculosis Alkylhydroperoxidase AhpD as Defined by Mutagenesis, Crystallography, and Kinetics. The Journal of Biological Chemistry . 2003;278(32):29502–29508. doi: 10.1074/jbc.M303747200. [DOI] [PubMed] [Google Scholar]
- 124.Sikora A. L., Frankel B. A., Blanchard J. S. Kinetic and chemical mechanism of arylamine N-acetyltransferase from Mycobacterium tuberculosis. Biochemistry . 2008;47(40):10781–10789. doi: 10.1021/bi800398c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kataev V. E., Khaybullin R. N., Garifullin B. F., Sharipova R. R. New targets for growth inhibition of Mycobacterium tuberculosis: why do natural terpenoids exhibit antitubercular activity? Bioorganic Chemistry . 2018;44(4):438–452. doi: 10.1134/S1068162018040106. [DOI] [Google Scholar]
- 126.Mann F. M., Xu M., Chen X., Fulton D. B., Russell D. G., Peters R. J. Edaxadiene: a new bioactive diterpene from Mycobacterium tuberculosis. Journal of the American Chemical Society . 2009;131(48):17526–17527. doi: 10.1021/ja9019287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Copp B. R., Pearce A. N. Natural product growth inhibitors of Mycobacterium tuberculosis. Natural Product Reports . 2007;24(2):278–297. doi: 10.1039/B513520F. [DOI] [PubMed] [Google Scholar]
- 128.Garifullin B. F., Strobykina I. Y., Mordovskoi G. G., Mironov V. F., Kataev V. E. Synthesis and antituberculosis activity of derivatives of the diterpenoid isosteviol with azine, hydrazide, and hydrazone moieties. Chemistry of Natural Compounds . 2011;47(1):55–58. doi: 10.1007/s10600-011-9829-0. [DOI] [Google Scholar]
- 129.Sharma A. K., Damle S. G., Das O., Sharma R. Isocitrate lyase: a potential target for anti-tubercular drugs. Recent Patents on Inflammation & Allergy Drug Discovery . 2013;7(2):114–123. doi: 10.2174/1872213X11307020003. [DOI] [PubMed] [Google Scholar]
- 130.Liu Y., Zhou S., Deng Q., et al. Identification of a novel inhibitor of isocitrate lyase as a potent antitubercular agent against both active and non-replicating Mycobacterium tuberculosis. Tuberculosis . 2016;97:38–46. doi: 10.1016/j.tube.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 131.Lee Y. V., Wahab H. A., Choong Y. S. Potential inhibitors for isocitrate lyase of Mycobacterium tuberculosis and non-M. tuberculosis: a summary. Biomed research. BioMed Research International . 2015;2015:20. doi: 10.1155/2015/895453.895453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.McLean K. J., Marshall K. R., Richmond A., et al. Azole antifungals are potent inhibitors of cytochrome P 450 mono-oxygenases and bacterial growth in mycobacteria and streptomycetes. Microbiology . 2002;148(10):2937–2949. doi: 10.1099/00221287-148-10-2937. [DOI] [PubMed] [Google Scholar]
- 133.Buchmeier N., Fahey R. C. ThemshAgene encoding the glycosyltransferase of mycothiol biosynthesis is essential inMycobacterium tuberculosisErdman. FEMS Microbiology Letters . 2006;264(1):74–79. doi: 10.1111/j.1574-6968.2006.00441.x. [DOI] [PubMed] [Google Scholar]
- 134.Fan F., Luxenburger A., Painter G. F., Blanchard J. S. Steady-state and pre-steady-state kinetic analysis ofMycobacterium smegmatisCysteine ligase (MshC) Biochemistry . 2007;46(40):11421–11429. doi: 10.1021/bi7011492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Newton G. L., Buchmeier N., Clair J. J. L., Fahey R. C. Evaluation of NTF1836 as an inhibitor of the mycothiol biosynthetic enzyme MshC in growing and non-replicating Mycobacterium tuberculosis. Bioorganic & Medicinal Chemistry . 2011;19(13):3956–3964. doi: 10.1016/j.bmc.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gutierrez-Lugo M.-T., Baker H., Shiloach J., Boshoff H., Bewley C. A. Dequalinium, a new inhibitor of Mycobacterium tuberculosis mycothiol ligase identified by high-throughput screening. Journal of Biomolecular Screening . 2009;14(6):643–652. doi: 10.1177/1087057109335743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Leblanc C., Prudhomme T., Tabouret G., et al. 4′-Phosphopantetheinyl transferase PptT, a new drug target required for Mycobacterium tuberculosis growth and persistence in vivo. PLoS Pathogens . 2012;8(12, article e1003097) doi: 10.1371/journal.ppat.1003097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ballinger E., Mosior J., Hartman T., et al. Opposing reactions in coenzyme A metabolism sensitizeMycobacterium tuberculosisto enzyme inhibition. Science . 2019;363(6426, article eaau8959) doi: 10.1126/science.aau8959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chiaradia L. D., Mascarello A., Purificação M., et al. Synthetic chalcones as efficient inhibitors of Mycobacterium tuberculosis protein tyrosine phosphatase PtpA. Bioorganic & Medicinal Chemistry Letters . 2008;18(23):6227–6230. doi: 10.1016/j.bmcl.2008.09.105. [DOI] [PubMed] [Google Scholar]
- 140.Rawls K. A., Lang P. T., Takeuchi J., et al. Fragment-based discovery of selective inhibitors of the Mycobacterium tuberculosis protein tyrosine phosphatase PtpA. Bioorganic & Medicinal Chemistry Letters . 2009;19(24):6851–6854. doi: 10.1016/j.bmcl.2009.10.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chandra K., Dutta D., Das A. K., Basak A. Design, synthesis and inhibition activity of novel cyclic peptides against protein tyrosine phosphatase A from Mycobacterium tuberculosis. Bioorganic & Medicinal Chemistry . 2010;18(23):8365–8373. doi: 10.1016/j.bmc.2010.09.052. [DOI] [PubMed] [Google Scholar]
- 142.Dutta N. K., He R., Pinn M. L., et al. Mycobacterial protein tyrosine phosphatases A and B inhibitors augment the bactericidal activity of the standard anti-tuberculosis regimen. Acs Infectious Diseases . 2016;2(3):231–239. doi: 10.1021/acsinfecdis.5b00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kumar A., Alam A., Rani M., Ehtesham N. Z., Hasnain S. E. Biofilms: survival and defense strategy for pathogens. International Journal of Medical Microbiology . 2017;307(8):481–489. doi: 10.1016/j.ijmm.2017.09.016. [DOI] [PubMed] [Google Scholar]
- 144.Islam M. S., Richards J. P., Ojha A. K. Targeting drug tolerance in mycobacteria: a perspective from mycobacterial biofilms. Expert Review of Anti-Infective Therapy . 2012;10(9):1055–1066. doi: 10.1586/eri.12.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Flentie K., Harrison G. A., Tükenmez H., et al. Chemical disarming of isoniazid resistance inMycobacterium tuberculosis. Proceedings of the National Academy of Sciences . 2019;116(21):10510–10517. doi: 10.1073/pnas.1818009116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mahairas G. G., Sabo P. J., Hickey M. J., Singh D. C., Stover C. K. Molecular analysis of genetic differences between Mycobacterium bovis BCG and virulent M. bovis. Journal of Bacteriology . 1996;178(5):1274–1282. doi: 10.1128/jb.178.5.1274-1282.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Gröschel M. I., Sayes F., Simeone R., Majlessi L., Brosch R. ESX secretion systems: mycobacterial evolution to counter host immunity. Nature Reviews. Microbiology . 2016;14(11):677–691. doi: 10.1038/nrmicro.2016.131. [DOI] [PubMed] [Google Scholar]
- 148.Falkow S. Molecular Koch's postulates applied to bacterial pathogenicity--a personal recollection 15 years later. Nature Reviews. Microbiology . 2004;2(1):67–72. doi: 10.1038/nrmicro799. [DOI] [PubMed] [Google Scholar]
- 149.Tiwari S., Casey R., Goulding C. W., Hingley-Wilson S., Jacobs W. R., Jr. Infect and inject: HowMycobacterium tuberculosisExploits its major virulence-associated type VII secretion system, ESX-1. Microbiol Spectrum . 2019;7(3) doi: 10.1128/microbiolspec.BAI-0024-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Rybniker J., Chen J. M., Sala C., et al. Anticytolytic screen identifies inhibitors of mycobacterial virulence protein secretion. Cell Host & Microbe . 2014;16(4):538–548. doi: 10.1016/j.chom.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 151.Johnson B. K., Colvin C. J., Needle D. B., Mba Medie F., Champion P. A., Abramovitch R. B. The carbonic anhydrase inhibitor ethoxzolamide inhibits the Mycobacterium tuberculosis PhoPR regulon and Esx-1 secretion and attenuates virulence. Antimicrobial Agents and Chemotherapy . 2015;59(8):4436–4445. doi: 10.1128/AAC.00719-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Jia P., Zhang Y., Xu J., et al. IMB-BZ as an inhibitor targeting ESX-1 secretion system to control mycobacterial infection. The Journal of infectious diseases . 2022;225(4) doi: 10.1093/infdis/jiab486. [DOI] [PubMed] [Google Scholar]
- 153.Drever K., Lim Z. L., Zriba S., Chen J. M. Protein synthesis and degradation inhibitors potently BlockMycobacterium tuberculosistype-7 secretion system ESX-1 activity. ACS Infectious Diseases . 2021;7(2):273–280. doi: 10.1021/acsinfecdis.0c00741. [DOI] [PubMed] [Google Scholar]
- 154.Chen H., Nyantakyi S. A., Li M., et al. The mycobacterial membrane: a novel target space for anti-tubercular drugs. Frontiers in Microbiology . 2018;9:p. 1627. doi: 10.3389/fmicb.2018.01627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Huaidong W. Analysis of Potential Drug Target of Drug Resistant Mycobacterium tuberculosis Based on Bioinformatics . JiLin University; 2016. [Google Scholar]
- 156.Das N., Jena P. K., Pradhan S. K. Arabinosyltransferase C enzyme of Mycobacterium tuberculosis , a potential drug target: An insight from molecular docking study. Heliyon . 2020;6(2, article e02693) doi: 10.1016/j.heliyon.2019.e02693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Sutherland H. S., Tong A. S. T., Choi P. J., et al. Structure-activity relationships for analogs of the tuberculosis drug bedaquiline with the naphthalene unit replaced by bicyclic heterocycles. Bioorganic & Medicinal Chemistry . 2018;26(8):1797–1809. doi: 10.1016/j.bmc.2018.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Choi P. J., Sutherland H. S., Tong A. S. T., et al. Synthesis and evaluation of analogues of the tuberculosis drug bedaquiline containing heterocyclic B-ring units. Bioorganic & Medicinal Chemistry Letters . 2017;27(23):5190–5196. doi: 10.1016/j.bmcl.2017.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lee M., Lee J., Carroll M. W., et al. Linezolid for treatment of chronic extensively drug-resistant tuberculosis. New England Journal of Medicine . 2012;367(16):1508–1518. doi: 10.1056/NEJMoa1201964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hashemian S. M. R., Farhadi T., Ganjparvar M. Linezolid: a review of its properties, function, and use in critical care. Drug Design, Development and Therapy . 2018;12:1759–1767. doi: 10.2147/DDDT.S164515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Von der Lippe B., Sandven P., Brubakk O. Efficacy and safety of linezolid in multidrug resistant tuberculosis (MDR- TB)--a report of ten cases. Journal of Infection . 2006;52(2):92–96. doi: 10.1016/j.jinf.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 162.Hou X. M., Wang C. Y., Gerwick W. H., Shao C. L. Marine natural products as potential anti-tubercular agents. European Journal of Medicinal Chemistry . 2019;165:273–292. doi: 10.1016/j.ejmech.2019.01.026. [DOI] [PubMed] [Google Scholar]
- 163.Huang F., Li W., Xu H., Qin H., He Z. G. Cordycepin kills Mycobacterium tuberculosis through hijacking the bacterial adenosine kinase. Plo S one . 2019;14(6, article e0218449) doi: 10.1371/journal.pone.0218449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Jiang C. H., Gan M. L., An T. T., Yang Z. C. Bioassay-guided isolation of a Mycobacterium tuberculosis bioflim inhibitor from Arisaema sinii Krause. Microbial Pathogenesis . 2019;126:351–356. doi: 10.1016/j.micpath.2018.11.022. [DOI] [PubMed] [Google Scholar]
- 165.Puhl A. C., Lane T. R., Vignaux P. A., et al. Computational approaches to identify molecules binding toMycobacterium tuberculosisKasA. ACS Omega . 2020;5(46):29935–29942. doi: 10.1021/acsomega.0c04271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Almeleebia T. M., Shahrani M. A., Alshahrani M. Y., et al. Identification of new Mycobacterium tuberculosis proteasome inhibitors using a knowledge-based computational screening approach. Molecules . 2021;26(8):p. 2326. doi: 10.3390/molecules26082326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ejalonibu M. A., Ogundare S. A., Elrashedy A. A., et al. Drug discovery for Mycobacterium tuberculosis using structure-based computer-aided drug design approach. International Journal of Molecular Sciences . 2021;22(24, article 13259) doi: 10.3390/ijms222413259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Borsari C., Ferrari S., Venturelli A., Costi M. P. Target-based approaches for the discovery of new antimycobacterial drugs. Drug Discovery Today . 2017;22(3):576–584. doi: 10.1016/j.drudis.2016.11.014. [DOI] [PubMed] [Google Scholar]
- 169.Blair H. A., Scott L. J. Delamanid: a review of its use in patients with multidrug-resistant tuberculosis. Drugs . 2015;75(1):91–100. doi: 10.1007/s40265-014-0331-4. [DOI] [PubMed] [Google Scholar]
- 170.Lechartier B., Rybniker J., Zumla A., Cole S. T. Tuberculosis drug discovery in the post-post-genomic era. Embo Molecular Medicine . 2014;6(2):158–168. doi: 10.1002/emmm.201201772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Harrison A. J., Yu M., Gardenborg T., et al. The structure of MbtI fromMycobacterium tuberculosis, the first enzyme in the biosynthesis of the siderophore mycobactin, reveals it to be a salicylate synthase. Journal of Bacteriology . 2006;188(17):6081–6091. doi: 10.1128/JB.00338-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Tran A. T., Wen D., West N. P., Baker E. N., Britton W. J., Payne R. J. Inhibition studies on Mycobacterium tuberculosis N-acetylglucosamine-1-phosphate uridyltransferase (GlmU) Organic & Biomolecular Chemistry . 2013;11(46):8113–8126. doi: 10.1039/c3ob41896k. [DOI] [PubMed] [Google Scholar]
- 173.Amaral L., Kristiansen J. E. Phenothiazines: an alternative to conventional therapy for the initial management of suspected multidrug resistant tuberculosis. A call for studies. International Journal of Antimicrobial Agents . 2000;14(3):173–176. doi: 10.1016/S0924-8579(99)00153-3. [DOI] [PubMed] [Google Scholar]
- 174.Okuyama H., Kankura T., Nojima S. Positional distribution of fatty acids in phospholipids from mycobacteria. Journal of Biochemistry . 1967;61(6):732–737. doi: 10.1093/oxfordjournals.jbchem.a128607. [DOI] [PubMed] [Google Scholar]
- 175.Prado V., Lence E., Maneiro M., et al. Targeting the motion of shikimate kinase: development of competitive inhibitors that stabilize an inactive open conformation of the enzyme. Journal of Medicinal Chemistry . 2016;59(11):5471–5487. doi: 10.1021/acs.jmedchem.6b00483. [DOI] [PubMed] [Google Scholar]
- 176.Belardinelli J. M., Yazidi A., Yang L., et al. Structure-function profile of MmpL3, the essential mycolic acid transporter fromMycobacterium tuberculosis. ACS Infectious Diseases . 2016;2(10):702–713. doi: 10.1021/acsinfecdis.6b00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Nören‐Müller A., Wilk W., Saxena K., Schwalbe H., Kaiser M., Waldmann H. Discovery of a new cass of inhibitors of Mycobacterium tuberculosis protein tyrosine phosphatase B by biology-oriented synthesis. Angewandte Chemie International Edition . 2010;47:5973–5977. doi: 10.1002/anie.200801566. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are included in this article.