

Keywords: colorectal cancer, emodin, macrophages, mouse models
Abstract
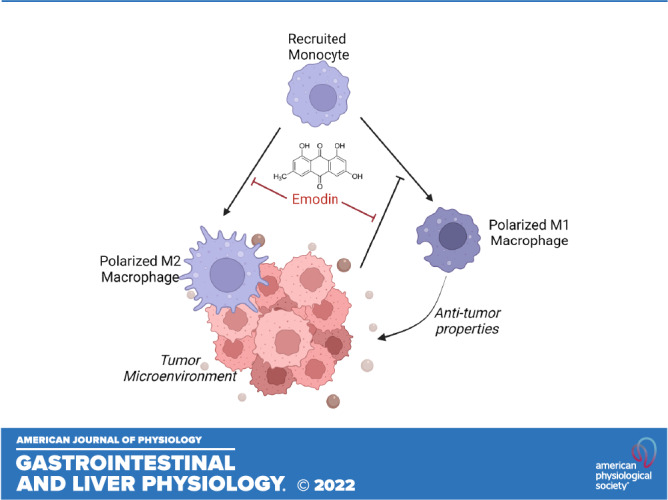
Emodin, a natural anthraquinone, has been shown to have antitumorigenic properties and may be an effective therapy for colorectal cancer (CRC). However, its clinical development has been hampered by a poor understanding of its mechanism of action. The purpose of this study was to 1) evaluate the efficacy of emodin in mouse models of intestinal/colorectal cancer and 2) to examine the impact of emodin on macrophage behavior in the context of CRC. We used a genetic model of intestinal cancer (ApcMin/+) and a chemically induced model of CRC [azoxymethane/dextran sodium sulfate (AOM/DSS)]. Emodin was administered orally (40 or 80 mg/kg in AOM/DSS and 80 mg/kg in ApcMin/+) three times a week to observe its preventative effects. Emodin reduced polyp count and size in both rodent models (P < 0.05). We further analyzed the colon microenvironment of AOM/DSS mice and found that mice treated with emodin exhibited lower protumorigenic M2-like macrophages and a reduced ratio of M2/M1 macrophages within the colon (P < 0.05). Despite this, we did not detect any significant changes in M2-associated cytokines (IL10, IL4, and Tgfb1) nor M1-associated cytokines (IL6, TNFα, IL1β, and IFNγ) within excised polyps. However, there was a significant increase in NOS2 expression (M1 marker) in mice treated with 80 mg/kg emodin (P < 0.05). To confirm emodin’s effects on macrophages, we exposed bone marrow-derived macrophages (BMDMs) to C26 colon cancer cell conditioned media. Supporting our in vivo data, emodin reduced M2-like macrophages. Overall, these data support the development of emodin as a natural compound for prevention of CRC given its ability to target protumor macrophages.
NEW & NOTEWORTHY Our study confirms that emodin is an effective primary therapy against the onset of genetic and chemically induced sporadic colorectal cancer. We established that emodin reduces the M2-like protumorigenic macrophages in the tumor microenvironment. Furthermore, we provide evidence that emodin may be acting to antagonize the P2X7 receptor within the bone tissue and consequently decrease the activation of proinflammatory cells, which may have implications for recruitment of cells to the tumor microenvironment.
INTRODUCTION
Colorectal cancer (CRC) is the third most common cancer worldwide (1, 2). The etiology of CRC is a complex occurrence that encompasses the interaction of genetic and environmental factors. However, only ∼20% of cases can be attributed to genetic factors with the vast majority being ascribed to environmental causes (3). Indeed, diet is thought to be responsible for up to 70% of CRCs (4). As such, bioactive food components offer exciting possibilities for chemoprevention due to their potential to target many factors associated with the development and progression of CRC (5, 6). Furthermore, the ability of bioactive food components to elicit tumoricidal effects without displaying the high toxicity exhibited by standard pharmacological interventions may translate to improved quality of life and survival in patients with cancer. Therefore, there is a critical need for rigorous studies to 1) establish the efficacy and 2) understand the mechanisms of action of bioactive food components so that they can be implemented as dietary chemopreventive strategies.
Emodin (1,3,8-trihydroxy-6-methylanthraquinone) is a natural anthraquinone isolated from several Chinese herbs, including Rheum palmatum, Polygonum cuspidatum, and Polygonum multiflorum. Several preclinical investigations have demonstrated that emodin contains a wide spectrum of pharmacological benefits including anti-inflammatory (7–10) and antitumorigenic (11–16) properties. Indeed, studies by our group have shown that emodin is effective at reducing mammary tumorigenesis in orthotopic mouse models (12, 14) and is safe in mice at 20, 40, and 80 mg/kg dosages (17). In addition, several preclinical studies have documented its benefits in CRC (18, 19). In fact, a recent study using the azoxymethane/dextran sodium sulfate (AOM/DSS) model of CRC reported that emodin reduced the incidence and size of intestinal tumors (18). Consistently, it was reported that emodin reduced the incidence of neoplastic lesions in ApcMin/+ mice treated with dextran sodium sulfate (DSS) (19). Both the National Cancer Institute (NCI) and the National Center for Complementary and Integrative Health (NCCIH) recognize the importance of evidence-based complementary medicine modalities that may be integrated as part of standard care for all patients across the cancer continuum. However, further development of emodin as an effective anticancer agent is hindered by uncertainties surrounding its mechanisms of action.
In some ways, the very broad pleiotropic effects of bioactive food components may have encumbered their clinical development. In actuality, the ability of these bioactive food components to target many factors makes them attractive antitumor strategies. However, without a clear understanding of the mechanisms of action of these compounds, their use in the cancer domain will likely continue to be limited. Preclinical studies have reported that emodin exerts its tumor-suppressive effects in CRC through multiple mechanisms including induction of apoptosis (20–22), inhibition of Wnt signaling (23), inhibition of VEGFR-2 signaling (24), suppression of urokinase secretion (25), and a reduction in inflammatory processes (18). Interestingly, there is now emerging evidence that emodin also can target macrophages (7, 12, 14). Macrophages have been reported to play a major role in CRC; in fact, tumor associated macrophages (TAMs) have been demonstrated to represent the predominant immune cell in the tumor microenvironment (TME) (26–29). It is generally understood that the recruitment of macrophages to the tumor contributes to the maintenance of the TME by exertion of protumoral functions (26, 27, 30). TAMs are characterized as M2-like macrophages and have been shown to promote proliferation, invasion and metastasis, angiogenesis, and tissue remodeling (26, 27, 30). Furthermore, high counts of TAMs have been associated with poor prognosis and therapeutic outcome in CRC (31–33). Therefore, therapeutic interventions targeting TAMs and reducing the M2/M1 ratio within the TME may contribute to a better prognosis. We have reported that emodin is uniquely able to suppress the excessive response of macrophages to both M1 and M2 stimuli and therefore has the potential to restore macrophage homeostasis in various pathologies (7). Indeed, emodin bidirectionally tunes the induction of LPS/IFNγ- and IL4-responsive genes through inhibiting NF-κB/IRF5/STAT1 signaling and IRF4/STAT6 signaling, respectively (7). In breast cancer, we have reported that emodin reduces primary tumor growth (12) as well as pulmonary metastasis (14) through inhibiting macrophage recruitment and M2 polarization in the lungs. However, no studies have examined emodin’s ability to target macrophages in CRC.
In the present study, we aimed to 1) establish the efficacy of emodin as a preventive therapy in two widely used models of CRC and 2) examine its impact on macrophages in CRC as a potential novel mechanism of action. We utilized a genetic model of intestinal cancer (ApcMin/+) and a chemically induced model of CRC (AOM/DSS) for tumorigenesis outcomes; use of two very different models increases translational relevance. For assessment of colonic macrophages, we exclusively used the AOM/DSS model given that these mice develop colon polyps whereas the ApcMin/+ develop mostly small intestinal polyps. Our data highlight a novel mechanism of action of emodin in CRC and suggest that emodin can be further developed as a chemopreventive agent for CRC.
METHODS
Animals
Male WT C57BL/6 and ApcMin/+ mice were purchased from Jackson Laboratories (Bar Harbor, ME) and were cared for in the Department of Laboratory Animal Resources at the University of South Carolina. Mice were randomized on arrival to the animal facility and were housed 4 to 5 per cage and maintained on a 12:12-h light-dark cycle in a low-stress environment (22°C, 50% humidity, low noise). Mice were kept in a room isolated from all other ongoing animal experiments and were only handled by the primary investigators. All mice were habituated to the AIN-76A diet before any interventions and were given food and water ad libitum through the course of the study. All methods were in accordance with the American Association for Laboratory Animal Science, and the Institutional Animal Care and Usage Committee of the University of South Carolina approved all experiments.
ApcMin/+ Protocol
Figure 1A illustrates the protocol used to treat ApcMin/+ mice with emodin. Briefly, 16 male ApcMin/+ mice were randomly divided into two groups: ApcMin/+ + vehicle (n = 8) or ApcMin/+ + 80 mg/kg emodin (n = 8). This dose based on body weight corresponds to ∼2 mg of emodin per mouse (assuming a 25-g mouse).
Figure 1.

Emodin (80 mg/kg) reduces polyp count in ApcMin/+ mice. A: experimental design. B: absolute body weight measurements. C: symptom score. D: total polyp count. E: polyp size. *Statistical significance (P < 0.05) from Student’s t test, n = 6 or 7/group.
AOM/DSS Protocol
Figure 2A illustrates the protocol used to chemically induce CRC using the AOM/DSS protocol and concurrent treatment with emodin. Briefly, 65 mice were randomly divided into four groups: control + vehicle (n = 5), AOM/DSS + vehicle (n = 20), AOM/DSS + 40 mg/kg emodin (n = 20), and AOM/DSS + 80 mg/kg emodin (n = 20). These doses based on body weight correspond to ∼1 and 2 mg of emodin per mouse (assuming a 25-g mouse), respectively. At 12 wk of age (baseline week 0) mice received either an intraperitoneal injection of the carcinogen (34), AOM (10 mg/kg; Sigma, St. Louis, MO), diluted in PBS (AOM/DSS) or PBS alone (control). Mice receiving the AOM injection were subjected to two cycles of DSS-supplemented water (36–50 kDa; MP Biomedical, Solon, OH) at final concentrations of 2% and 1% at weeks 1 and 4, respectively. Each DSS cycle lasted for a 1-wk period.
Figure 2.

Emodin reduces polyp count in AOM/DSS mice. A: experimental design. B: percent (%) change in body weight from start of experiment (W1). C: symptom score. #Statistical significance (P < 0.05) between 40 mg/kg and vehicle-treated mice, $statistical significance (P < 0.05) between 80 mg/kg and vehicle-treated mice, %statistical significance (P < 0.05) between 40 and 80 mg/kg-treated mice from one-way ANOVA, n = 5–20/group. D: survival curve. E: total polyp count. F: polyp size. G: polyp location. Groups not containing the same letter (a and b) indicates statistical significance between groups (P < 0.05) from one-way ANOVA, n = 5–20/group. AOM/DSS, azoxymethane/dextran sodium sulfate.
Emodin Preparation
Emodin was purchased from Nanjing Zelang Medical Technology Co., Ltd, (Nanjing, China). Emodin was independently analyzed by the Mass Spectrometry Center at the University of South Carolina before the initiation of the experimental study; LC-UV-MS and NMR were performed to confirm the purity of emodin (>98%) and the molecular structure. Emodin was delivered to mice three times a week via oral gavage using flexible plastic tubing gavage needles (Instech, no. FTP-20). It was prepared fresh on the day of treatment in a 24 mg/mL stock solution using pure propylene glycol (VWR) and mixed for 6–8 h at room temperature and protected from light. The 24 mg/mL solution was used for 80 mg/kg dosing (AOM/DSS and ApcMin/+ models) and further diluted to a 12 mg/mL dilution for delivering the 40 mg/kg dose (AOM/DSS). Pure propylene glycol was used as a vehicle in all experiments.
Symptom Monitoring and Score Calculation
Both ApcMin/+ and AOM/DSS mice underwent the same symptom monitoring and score calculation. Body weights and symptom scores were determined twice a week along with food and water intake measurements. Calculation of symptom score was performed as previously described (35), taking into account percent body weight loss, stool consistency, and rectal bleeding. Briefly, fresh colonic evacuates were smeared onto “Hemoccult” tape to assess severity of diarrhea and were tested with developer (Beckman Coulter, Brea, CA) to assess rectal bleeding. Bleeding was scored as follows: no positive detection of blood (0), detection of blood but not grossly visible (2), and gross visibility of blood (4). Diarrhea was scored as the following: solid cylinder (0), soft cylinder and easily spreadable (2), and noncylindrical or runny (4). Body weight was calculated as percent body weight loss: 0%–5% (0), 6%–10% (1), 11%–15% (2), 15%–20% (3), 20%–25% (4), and >25% (5). Scores of all three categories were summed to obtain an overall symptom score for each mouse.
Tissue Collection
Mice were euthanized by isoflurane overdose 24 h postfinal treatment with emodin or vehicle. Blood was collected from the inferior vena cava for a complete blood count described below. Tissues (small intestine, colon, liver, spleen, epididymal fat, and mesenteric fat) were removed and weighed. The colon and small intestine from ApcMin/+ mice were carefully dissected and separated into five equal segments, the fifth being the colon-distal to the cecum and proximal to the anus. Intestines were opened longitudinally, and polyps/tumors were counted and measured using a dissecting microscope. Polyps in the ApcMin/+ mouse are largely found in segments 2–4. The colon from AOM/DSS mice was carefully dissected distal to the cecum and proximal to the anus, opened longitudinally, and polyps/tumors were counted and measured using a dissecting microscope. Furthermore, colon length was measured using a caliper. A subset of AOM/DSS mice (n = 4) were used for flow cytometry analysis in which the colon was placed in ice-cold PBS until digestion as described below. The remaining colons (n = 16) were snap frozen in liquid nitrogen and stored at −80°C until qPCR analysis. Femoral shafts were removed for isolation of bone marrow.
Blood Panel Analysis
A complete blood panel analysis was performed using the VetScan HMT (Abaxis, Union City, CA) for determination of white blood cells (WBC), lymphocytes (LYM), monocytes (MON), neutrophils (NEU), red blood cells (RBC), hematocrit (Hct), and hemoglobin (Hb). The neutrophil/lymphocyte ratio (NLR) was calculated from obtained values. Briefly, whole blood was placed in an EDTA-coated microtube and analyzed on the VetScan HMT according to the manufacturer’s instructions.
Leukocyte Isolation and Flow Cytometry
Colons were removed and digested to obtain single cell suspensions using the Miltenyi Biotech Lamina Propria Dissociation Kit (no. 130-097-410). Bone marrow was taken from both femoral shafts by flushing with saline. Red blood cell lysis was performed with 20-s hypotonic solution (0.2% NaCl) treatment followed by hypertonic (1.6% NaCl) cessation. All isolates were passed through a 70-μm strainer before antibody staining. Bone marrow and lamina propria isolates were stained with a macrophage panel as follows: CD11b– FITC, F4/80– PE, CD206− APC, CD11c– APC/Cy7, Ly6c– PerCP/Cy5.5, and CD45– PE/Cy7. All antibodies were purchased from Biolegend (San Diego, CA). Cell suspensions were blocked with CD16/32 FC Block and staining was performed for 1 h at 4°C. Leukocytes were identified by gating for CD45+ cells. Active monocytes were identified as Ly6C+ cells. Data were acquired using a BD FACSAria II cell sorter and analyzed by FlowJo v10.6.2.
Gene Expression
Quantification of colonic expression of the chemokine monocyte chemoattractant protein 1 (MCP-1), M1-associated cytokines interleukin 6 (IL-6), tumor necrosis factor-α (TNF-α), IL-1β, interferon γ (IFNγ) and nitric oxide synthase 2 (NOS2), and M2-associated cytokines IL-10, IL4, and transforming growth factor β (TGF-β) were performed as previously described (33). Briefly, RNA was extracted using TRIzol reagent (Life Technologies, Gibco-BRL, Carlsbad, CA) and chloroform procedures. RNA sample quality and quantities were verified using an Agilent Bioanalyzer and determined to be of good quality based on A260/A280 values (>1.8) before cDNA synthesis using QuantiTect Reverse Transcription kit (Qiagen 205313). Quantitative RT-PCR analysis was carried out as per the manufacturer’s instructions (Applied Biosystems) using Taq-Man Gene Expression Assays. Data were normalized to vehicle-treated controls and compared with five reference targets (B2M, TBP, HPRT, HMBS, and H2AFV), which were evaluated for expression stability using the GeNorm algorithm.
Cell Culture
Bone marrow-derived macrophages (BMDM) were isolated and assessed as previously described (36). Briefly, healthy C57BL/6 mice were euthanized by isoflurane overdose and femurs were cleared of muscle and debris. The epiphyses of the femurs were cut, and the medullary cavity flushed with DMEM with 10% FBS and passed through a 70-µm strainer. Isolates were incubated in 10-cm dish at 37°C in DMEM for 4 h to allow for mature monocytes and dendritic cells to adhere and be discarded. Floating, immature undifferentiated monocytes were collected and subjected to RBC lysis and incubated in bone marrow maintenance media (20% L929-conditioned media, 10% FBS, 1% Penn/Strep DMEM) for 6 days. After 6 days of maturation, macrophages were given either serum-free media + dimethylsulfoxide (DMSO), C26-conditioned media + DMSO, serum-free media with 50 µM emodin, or C26 condition media with 50-µM emodin. Cells were incubated in their respective conditions for 48 h. After 48 h cells were washed twice with PBS before removal by incubation in 5 mM EDTA PBS at 4°C for 20 min. Cells were gently scraped and washed in PBS again before antibody staining. Cells were stained and analyzed via flow cytometry similar to described above.
Western Blotting
Bone marrow cell isolates were homogenized in Mueller buffer containing a protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO). Total protein concentration was determined by the Bradford method. Equal amounts of crude protein homogenates were separated by SDS-PAGE and transferred to a PVDF membrane. Membranes were stained with Ponceau S solution to verify equal protein loading and transfer efficiency. Subsequently, membranes were blocked for 1 h in 5% nonfat milk in Tris-buffered saline-0.1% Tween-20 (TBST). Primary antibodies for P2X7 (Alomone Labs; Cat. No. 136–152) and total (Cat. No. 12640) and phosphorylated Tyr705 (Cat. No. 9145) STAT3 from Cell Signaling (Danvers, MA) were diluted 1:1,000 in 5% nonfat milk in TBST and incubated on membranes for 1 h at room temperature or overnight at 4°C. Anti-rabbit IgG horseradish peroxidase-conjugated secondary antibody (Cell Signaling) was incubated with the membranes at 1:2,000 dilution in 5% nonfat milk in TBST for 1 h at room temperature. An enhanced chemiluminescent substrate for detection of horseradish peroxidase (Thermo Scientific, Waltham, MA) was used to visualize the antibody-antigen interaction. Autoradiography films were scanned, and blots were quantified using scientific imaging software (Image J). All samples were run on the same gel for each protein and scanned together; images were cropped (dotted line) for representation. After completion of the Western blot, all membranes were stained with Amido black solution, and the densitometry of each lane was calculated and utilized for total protein normalization. This method of normalization has been shown to be an acceptable alternative to typical loading controls.
Statistical Analyses
All data were analyzed using commercial software (SigmaStat V3.5, SPSS, Chicago, IL). All in vivo outcomes were analyzed using a Student’s t test or one-way ANOVA where appropriate. If a significant difference was observed, a multiple comparisons (least significant difference, LSD) test was performed to further define group differences. In vitro studies were analyzed using a two-way ANOVA. A Student-Newman-Keuls test was used for all post hoc analyses. Survival curve analysis was conducted by log-rank (Mantel-Cox) test. Any data that were not normally distributed or did not display equal variance were logarithmically transformed so that those criteria were met. Statistical significance was set with an α value of P < 0.05. Data are presented as means ± SE. All figures were generated using GraphPad Prism v9.0.
RESULTS
Emodin Reduced Polyp Load in ApcMin/+ Mice
After 11 wk of emodin or vehicle treatment, mice (∼16 wk of age) were euthanized, the entire small intestines were stained with methylene blue, and polyps were counted. We found that although no differences in body weight or calculated symptom score were observed (Fig. 1, B and C), 11 wk of emodin treatment reduced total polyps in ApcMin/+ mice (Fig. 1D; P < 0.05). Further analysis confirmed that emodin reduced the number of large (>2 mm) and medium (1–2 mm) sized polyps in the small intestine compared with vehicle-treated controls (Fig. 1E; P < 0.05). However, there was no difference in small polyp count (<1 mm; Fig. 1E) among the groups.
Emodin Did Not Alter Organ Weight Nor Circulating Blood Cell Counts in ApcMin/+ Mice
Splenomegaly, anemia/cytopenia, and cachexia are hallmarks of advanced colorectal/intestinal cancer. At the conclusion of the study, there was no detectable difference in liver, spleen, epididymal fat, or mesenteric fat pad weight (Table 1) between vehicle and emodin-treated mice. However, it is important to note that splenomegaly was observed at this stage with both groups having average spleen weights greater than 200 mg (splenomegaly in mice is described as spleen weight >100 mg). We also observed no difference in small intestine or colon length (Table 1). We performed a comprehensive blood panel analysis at the conclusion of the study and found no significant differences among all groups in all major parameters (Table 2).
Table 1.
Emodin does not alter organ weight in Apcmin/+ mice
| Group | Liver, mg | Spleen, mg | Epididymal Fat, mg | Mesenteric Fat, mg | Small Intestine, cm | Colon, cm |
|---|---|---|---|---|---|---|
| Apcmin + vehicle | 1,237.5 ± 115.1 | 233.0 ± 44.4 | 625.8 ± 80.6 | 224.0 ± 36.9 | 32.4 ± 0.5 | 8.2 ± 0.2 |
| Apcmin + 80 mg/kg | 1,328.0 ± 47.1 | 277.1 ± 65.6 | 613.7 ± 106.5 | 201.2 ± 20.7 | 32.9 ± 0.9 | 8.2 ± 0.1 |
Liver, spleen, epididymal fat, mesenteric fat, small intestine, and colon length weights at the conclusion of the study.
Table 2.
Emodin does not alter hematological perturbations in ApcMin/+ mice
| Group | WBC (109) | LYM (109) | MON (109) | NEU (109) | NLR (A.U.) | RBC (1012) | HGB, g/dL | HCT, % |
|---|---|---|---|---|---|---|---|---|
| Apcmin + vehicle | 3.83 ± 0.71 | 2.27 ± 0.65 | 0.120 ± 0.048 | 1.46 ± 0.19 | 0.779 ± 0.204 | 6.32 ± 1.12 | 9.75 ± 1.59 | 27.26 ± 4.60 |
| Apcmin + 80 mg/kg | 5.42 ± 0.64 | 3.63 ± 0.64 | 0.301 ± 0.078 | 1.48 ± 0.17 | 0.642 ± 0.265 | 5.52 ± 0.95 | 9.00 ± 1.21 | 24.60 ± 3.68 |
White blood cells (WBC), lymphocytes (LYM), monocytes (MON), neutrophils (NEU), neutrophil-to-lymphocyte ratio (NLR), red blood cells (RBC), hemoglobin (HGB), and hematocrit (HCT) were measured using a VetScan HMT (Abaxis, Union City, CA). n = 6 or 7/group.
Emodin Reduced Polyp Number and Tumor Burden in AOM/DSS Mice
As expected, AOM/DSS mice had significantly lower body weight compared with control mice. However, emodin-treated mice had improved body weight maintenance compared with vehicle-treated mice; 40 mg/kg of emodin yielded a sparing of body weight loss at week 4.5 and 80 mg/kg of emodin yielded a sparing of body weight loss at weeks 2, 4.5, and 5 (Fig. 2, A and B; P < 0.05). Furthermore, within AOM/DSS mice treated with emodin there was a significant difference in body weight loss between 40 mg/kg treatment and 80 mg/kg treatment at week 2 with the 80 mg/kg group losing less weight (Fig. 2, A and B; P < 0.05). These findings were consistent with a significantly lower symptom score calculation in 40 and 80 mg/kg mice compared with vehicle-treated mice at weeks 5, 8, 9, and 10 (Fig. 2, A and C; P < 0.05). Survival was monitored throughout the study. Although not statistically significant, through the course of study there were three deaths in the AOM/DSS + vehicle group (17/20 survival), two deaths in the AOM/DSS + 40 mg/kg group (18/20 survival), and no deaths in the AOM/DSS + 80 mg/kg group (20/20 survival; Fig. 2, A and D).
At the conclusion of the study, colons were excised and polyps were counted under a dissecting microscope. There was a statistically significant difference in colonic polyp number between vehicle-treated mice and mice treated with 40 and 80 mg/kg emodin (Fig. 2E; P < 0.05); indeed, both doses of emodin reduced overall colonic polyp number. Similar to the ApcMin/+ study, we categorized polyp counts by size and revealed that both 40 mg/kg and 80 mg/kg emodin treatment significantly reduced medium-sized (1–2 mm; P < 0.05) polyps but not small (<1 mm) nor large (>2 mm) polyps (Fig. 2F). The location of polyps within the colon has been shown to be important in the prognosis of CRC (37). With this understanding, we analyzed colon polyps in distal, middle, and proximal sections of the colon. We found that both 40 and 80 mg/kg emodin does not change the presence of distal or proximal polyps but does significantly reduce the presence of polyps in the middle portion of the colon (Fig. 2G; P < 0.05).
Emodin Improves Liver Weight in AOM/DSS Mice but Does Not Impact Other Tissues
We observed splenomegaly in all AOM/DSS mice (P < 0.05), which was not changed with emodin treatment (Table 3). Advanced CRC causes organ wasting, which is observed experimentally through decreased major organ weight. We observe decreased epididymal fat and mesenteric fat weight in all AOM/DSS mice (Table 3; P < 0.05). Interestingly, we found that mice treated with 80 mg/kg emodin had lower epididymal fat pad weight compared with mice treated with 40 mg/kg emodin (P < 0.05) but not vehicle-treated controls (Table 3). There was no difference in mesenteric fat pad weight with emodin treatment (Table 3). We show that vehicle-treated AOM/DSS mice had significantly lower liver weights compared with noncancer control (Table 3; P < 0.05). Interestingly, we found that both 40 mg/kg and 80 mg/kg had increased liver weight compared with vehicle-treated AOM/DSS mice, but this did not reach statistical significance (Table 3).
Table 3.
Emodin treatment does not alter organ weight in AOM/DSS mice
| Group | Liver, mg | Spleen, mg | Epididymal Fat, mg | Mesenteric Fat, mg | Tibia, mm | Small Intestine, cm | Colon, cm |
|---|---|---|---|---|---|---|---|
| Control | 1,478.2 ± 43.1a | 82.2 ± 1.4a | 1,426.0 ± 127.0a | 586.0 ± 52.3a | 18.6 ± 0.2 | 32.6 ± 0.4 | 8.0 ± 0.2 |
| AOM/DSS + vehicle | 1,245.2 ± 46.7b,c | 152.5 ± 20.0b | 714.8 ± 77.4b,c | 310.9 ± 28.5b | 18.3 ± 0.1 | 32.3 ± 0.3 | 7.5 ± 0.1 |
| AOM/DSS + 40 mg/kg | 1,336.3 ± 41.8a,c | 139.7 ± 12.6b | 844.8 ± 59..4b | 347.3 ± 25.0b | 18.0 ± 0.1 | 32.6 ± 0.4 | 7.6 ± 0.1 |
| AOM/DSS + 80 mg/kg | 1,314.6 ± 25.1a,c | 144.7 ± 13.4b | 626.9 ± 35.0c | 317.2 ± 13.8b | 18.3 ± 0.1 | 33.8 ± 0.6 | 7.6 ± 0.1 |
Liver, spleen, epididymal fat, mesenteric fat, small intestine, and colon length weights at the conclusion of the study. AOM/DSS, azoxymethane/dextran sodium sulfate. Groups not containing the same letter (a–c) indicates statistical significance between groups (P < 0.05) from one-way ANOVA.
Emodin Reduced Protumoral M2-Like Macrophage Count in Colon Lamina Propria of AOM/DSS Mice
TAMs are understood to play an essential role in the prognosis and treatment outcome of CRC. Using flow cytometry, we show here that AOM/DSS increased total lamina propria macrophage (CD45+ CD11b+Ly6C+ F4/80+) cells. Compared with healthy controls, total macrophages were significantly greater in AOM/DSS mice treated with vehicle (Fig. 3, A and B; P < 0.05). However, both 40 and 80 mg/kg emodin-treated mice were not different from healthy controls nor were they different from vehicle-treated AOM/DSS mice (Fig. 3, A and B).
Figure 3.

Emodin reduces protumorigenic M2-like macrophages within the AOM/DSS mouse colon lamina propria. A: flow plots identifying mature macrophages (CD45+CD11b+Ly6C+F480+ cells). B: quantification of mature macrophages. C: flow plots identifying M1-like (CD11c+) vs. M2-like (CD206+) macrophages. CD11c/CD206+ cells are considered transitioning cells and CD11c-CD206− cells are considered M0 macrophages. D: quantification of M1, transition, M2, and M0 macrophages. E: ratio of M1/M2 macrophage counts. Groups not containing the same letter (a and b) indicates statistical significance between groups (P < 0.05) from one-way ANOVA, n = 2 control and n = 4 AOM/DSS/group. AOM/DSS, azoxymethane/dextran sodium sulfate. AOM/DSS, azoxymethane/dextran sodium sulfate.
We defined M1-like macrophages as CD45+ CD11b+F4/80+ CD11c+ CD206− cells and M2-like macrophages as CD45+ CD11b+F4/80+ CD11c−CD206+. CD11c+ CD206+ double-positive macrophages are characterized as cells undergoing transition and not completely polarized M1- or M2-like macrophages. Furthermore, CD45+ CD11b+F4/80+ macrophages expressing neither CD11c nor CD206 are characterized as M0 (i.e., naïve) macrophages. As expected, we observed a significant increase in M2-like macrophages in the lamina propria of AOM/DSS mice treated with vehicle (Fig. 3, C and D; P < 0.05). Excitingly, and consistent with our breast cancer studies on emodin (12, 14), both 40 and 80 mg/kg emodin treatment decreased the abundance of CD206+ M2-like macrophages in the lamina propria (Fig. 3, C and D; P < 0.05). There was a similar trend in CD11c−CD206− M0 macrophages (Fig. 3, C and D; P < 0.05); however, this did not reach statistical significance (Fig. 3, C and D). There was no significant difference between groups in the presence of M1 or transitioning macrophages (Fig. 3, C and D).
M1 macrophages in the tumor are described as having antitumor properties and M2-like macrophages are described as having protumor properties (29). To observe the general pro- versus antitumor niche, we show the ratio of M2 versus M1 macrophages within the lamina propria. We confirm the presence of a more protumoral niche by reporting an increase in the ratio of M2/M1 macrophages in AOM/DSS vehicle-treated mice demonstrating an increase in relative M2 macrophages, which is ameliorated with 40 and 80 mg/kg emodin treatment (Fig. 3E).
High Dose Emodin Increased Intrapolyp NOS2
Polyps were removed and RNA was extracted for inflammatory cytokine analysis via qRT-PCR. There was a significant increase in NOS2, an M1 and tumoricidal marker, expression in mice treated with 80 mg/kg emodin compared with vehicle-treated controls and mice treated with 40 mg/kg emodin (Fig. 4; P < 0.05). However, despite reduced polyp load, mice treated with 40 and 80 mg/kg emodin did not exhibit significant differences in MCP1, IL6, TNFα, IL1β, IFNγ, IL10, IL4, or Tgfb1 (Fig. 4) when compared with vehicle-treated AOM/DSS mice.
Figure 4.

Emodin (80 mg/kg) increases NOS2 expression within polyps of AOM/DSS mice. qPCR analysis of MCP-1, IL6, TNFα, IL1b, IFNg, NOS2, IL10, IL4, and Tgfb1 genes. Data were normalized to vehicle-treated controls and compared with five reference targets (B2M, TBP, HPRT, HMBS, and H2AFV), which were evaluated for expression stability using GeNorm. Groups not containing the same letters (a and b) indicates statistical significance between groups (P < 0.05) from one-way ANOVA, n = 12–15/group. AOM/DSS, azoxymethane/dextran sodium sulfate.
Emodin Reduced Bone Marrow-Associated Inflammation but Did Not Ameliorate Cancer-Associated Anemia
Hematological perturbations are a significant hallmark of advanced CRC. We found that all AOM/DSS mice had significantly greater monocyte counts compared with healthy controls (Table 4; P < 0.05). Although not statistically significant, there was a trending decrease in monocyte counts (P = 0.121) in 80 mg/kg mice compared with vehicle-treated mice. Furthermore, there was a significant decrease in RBC counts, HGB, and HCT in all AOM/DSS mice (P < 0.05), but no difference was detected in mice treated with emodin compared with vehicle controls (Table 4). There were no significant differences detected in WBC, LYM, NEU, or NLR detected in any experimental groups (Table 4).
Table 4.
Emodin does not significantly alter hematological parameters within AOM/DSS mice
| Group | WBC (109) | LYM (109) | MON (109) | NEU (109) | NLR (A.U.) | RBC (1012) | HGB, g/dL | HCT, % |
|---|---|---|---|---|---|---|---|---|
| Control | 5.38 ± 0.48 | 4.27 ± 0.40 | 0.040 ± 0.004a | 1.04 ± 0.11 | 0.24 ± 0.01 | 10.28 ± 0.27a | 16.00 ± 0.26a | 41.96 ± 1.18a |
| AOM/DSS + vehicle | 4.57 ± 0.27 | 3.22 ± 0.26 | 0.215 ± 0.029b | 1.13 ± 0.12 | 0.43 ± 0.09 | 8.19 ± 0.32b | 13.11 ± 0.46b | 33.86 ± 1.19b |
| AOM/DSS + 40 mg/kg | 4.83 ± 0.43 | 3.48 ± 0.36 | 0.194 ± 0.038b | 1.16 ± 0.09 | 0.36 ± 0.02 | 8.77 ± 0.29b | 13.76 ± 0.31b | 35.75 ± 1.00b |
| AOM/DSS + 80 mg/kg | 5.73 ± 0.26 | 4.15 ± 0.23 | 0.166 ± 0.031b | 1.41 ± 0.11 | 0.36 ± 0.03 | 8.38 ± 0.24b | 12.88 ± 0.29b | 34.90 ± 0.68b |
White blood cells (WBC), lymphocytes (LYM), monocytes (MON), neutrophils (NEU), neutrophil-to-lymphocyte ratio (NLR), red blood cells (RBC), hemoglobin (HGB), and hematocrit (HCT) were measured using a VetScan HMT (Abaxis, Union City, CA). n = 5 control and n = 17–20. AOM/DSS/group. AOM/DSS, azoxymethane/dextran sodium sulfate. Groups not containing the same letter (a and b) indicates statistical significance between groups (P < 0.05) from one-way ANOVA.
Given that intestinal macrophages are largely derived from the bone marrow, we next assessed the effects of emodin on bone marrow macrophages by flow cytometry in a subset of mice (n = 2–4). Interestingly, mice treated with both 40 and 80 mg/kg emodin had significantly lower total macrophages (CD45+ CD11b+Ly6C+ F4/80+) compared with control and AOM/DSS alone (Fig. 5, A and B; P < 0.05). On further analysis, we found that AOM/DSS mice had increased M1 macrophages (CD45+ CD11b+F480+ CD11c+ CD206−) compared with control mice (Fig. 5, C and D; P < 0.05). Intriguingly, 40 and 80 mg/kg emodin treatment reduced M1 macrophage counts to similar values as noncancer control mice (Fig. 5, C and D; P < 0.05). In all AOM/DSS groups, there was a significant decrease in M2 (CD45+ CD11b+F480+ CD11c−CD206+) and M0 (CD45+ CD11b+F480+ CD11c−CD206−) macrophages compared with noncancer controls (Fig. 5, C and D; P < 0.05). There was no difference in CD45+ CD11b+F480+ CD11c+ CD206+ cells among all groups. We confirm this increase in M1 macrophages by reporting a significant increase in the M1/M2 ratio of AOM/DSS mice compared with noncancer controls (Fig. 5E).
Figure 5.

Increasing dose of emodin reduces proinflammatory macrophage environment within the bone marrow of AOM/DSS mice. A: flow plots identifying mature macrophages (CD45+/CD11b+/Ly6C+/F480+ cells). B: quantification of mature macrophages. C: flow plots identifying M1-like (CD11c+) vs. M2-like (CD206+) macrophages. CD11c/CD206+ cells are considered transitioning cells and CD11c-CD206− cells are considered M0 macrophages. D: quantification of M1, transition, M2, and M0 macrophages. E: ratio of M1/M2 macrophage counts. Groups not containing the same letter (a and b) indicates statistical significance between groups (P < 0.05) from one-way ANOVA, n = 2 control and n = 4 AOM/DSS/group. AOM/DSS, azoxymethane/dextran sodium sulfate.
Emodin Reduced Expression of P2X7 and STAT3 Signaling in Bone Tissue of AOM/DSS Mice
Given the reduction in M1 macrophages in the bone marrow with emodin, we next assessed the presence of P2X7, a receptor present on macrophages that is known to activate inflammatory responses and that can be targeted by emodin (9, 13, 38). We observed a significant increase in P2X7 expression in AOM/DSS mice that was reduced by 40 and 80 mg/kg emodin treatment (Fig. 6, A and B; P < 0.05). As activation of the P2X7 receptor has been reported to promote STAT3 signaling, we assessed STAT3. STAT3 activation by phosphorylation at Y705 was significantly increased in AOM/DSS mice treated with vehicle but consistent with the P2X7 findings was decreased with both 40 and 80 mg/kg emodin treatment (Fig. 6, A and C; P < 0.05). Interestingly, there was a significant increase in total STAT3 protein expression in all AOM/DSS mice which was not changed with emodin treatment but caused a significant decrease in the ratio of phospho-to-total STAT3 protein expression (Fig. 6, A and C; P < 0.05).
Figure 6.

Increasing dose of emodin reduces P2X7 receptor expression and STAT3 phosphorylation/activation within the bone tissue of AOM/DSS mice. A: representative Western blots of P2X7, p-STAT3 (Y705), t-STAT3, and amido black stain. Quantification of relative expression of P2X7 receptor (B) and STAT3 (C). Groups not containing the same letter (a and b) indicate statistical significance between groups (P < 0.05) from one-way ANOVA, n = 2 control and n = 4 AOM/DSS/group. AOM/DSS, azoxymethane/dextran sodium sulfate.
Emodin Prevents the Induction of TAMs In Vitro
Bone marrow-derived macrophages were exposed to C26-conditioned media (CM) and coincubated with 50 µM emodin for 48 h. There was a main effect of C26 CM in decreasing M1 macrophages and a main effect of emodin in increasing M1-like macrophages (Fig. 7, P < 0.05). C26 CM increased M2-like macrophages compared with serum-free control consistent with the expected increase in TAMs. Emodin was able to decrease M2-like macrophages alone and more importantly, emodin blocked the increase in M2-like macrophages with C26 CM (Fig. 7, P < 0.05).
Figure 7.

Emodin prevents C26-induced polarization of macrophages in vitro. A: representative flow plot of bone marrow-derived macrophages (BMDMs). B: quantification of relative abundance of CD11b+CD68+ macrophages in BMDMs treated with C26 conditioned media (CM) and emodin. C: representative flow plot of BMDMs and their relative expression of CD11b and CD68. D: quantification of relative abundance of CD11b+CD68+CD11c+CD206− M1 like macrophages in BMDMs treated with C26-conditioned media (CM) and emodin. E: representative flow plot of BMDMs and their relative expression of CD11c and CD206. F: quantification of relative abundance of CD11b+CD68+CD11c−CD206+ M2-like macrophages in BMDMs treated with C26-conditioned media (CM) and emodin. *Main effect of emodin (P < 0.05); ^main effect of CM (P < 0.05); groups not containing the same letter (a–c) indicates statistical significance between groups (P < 0.05) from two-way ANOVA n = 3/treatment.
DISCUSSION
There is an abundance of evidence to support the development of natural compounds for the prevention and treatment of CRC. Indeed, natural compounds offer exciting opportunities for chemoprevention due to their potential to target many factors associated with the development and progression of CRC (5, 6). In addition, they can provoke antitumor effects without portraying the high toxicity exhibited by standard pharmacological interventions. However, their clinical development has been hampered by a poor understanding of their mechanism of action as well as concerns surrounding bioavailability and safety. In the current study, we sought to examine the effects of dietary emodin, a natural anthraquinone isolated from several Chinese herbs, on intestinal/colorectal cancer. Furthermore, as a potential mechanism of action, we examined the impact of emodin on macrophages in the context of CRC. We report that emodin successfully reduced tumorigenesis in two widely used mouse models (Apcmin/+ and AOM/DSS) of intestinal/colorectal cancer. These effects of emodin were associated with a decrease in protumorigenic M2-like macrophages and a reduced ratio of M2/M1 macrophages within the colon (AOM/DSS mouse only) and this was confirmed in vitro.
Our findings for a benefit of emodin on tumorigenesis related outcomes are supported by several published studies in rodents and using a variety of CRC models. To our knowledge, the earliest available study reported that emodin effectively suppressed tumor growth in nude mice xenografts bearing LS1034, colorectal carcinoma cell, tumors (20). More recently, it was reported that emodin inhibited tumorigenesis of established solid colon tumors (CC-531) in rats (39). Likewise, and consistent with our findings, emodin inhibited carcinogenesis in an AOM/DSS mouse model of colon tumorigenesis (18). Similarly, in a xenograft model of colon cancer (RKO), emodin suppressed tumor growth (40). Finally, it was reported that aloe-emodin reduced the number of colorectal tumors in ApcMin/+ mice, treated with or without DSS (19). The mechanisms underlying these tumoricidal effects of emodin continue to be unearthed; however, to date studies have implicated an induction of apoptosis (20–22), inhibition of Wnt signaling (23), inhibition of VEGFR-2 signaling (24), suppression of urokinase secretion (25), and a reduction in inflammatory processes (18) as candidate mechanisms. Recently, our group has reported that emodin can bidirectionally modulate macrophages in a context-dependent manner (7); however, the role for macrophages in emodin’s anticolorectal cancer therapy had not yet been evaluated.
We sought to examine whether the antitumor effects of emodin in CRC may be mediated by macrophages. Previous work from our group has documented the importance of macrophages in CRC (31, 41). Indeed, a preceding study from our group documented that deletion of MCP-1, the most important chemokine for monocyte/macrophage recruitment, results in a decrease in polyp number and size in the ApcMin/+ mouse (41). Furthermore, we reported that macrophage depletion using clodronate liposome administration decreased tumor number and specifically large (≥1 mm) tumors in the AOM/DSS model (31). In the current study and as hypothesized, there was an increase in M2-like protumor macrophages in the colon, which was significantly reduced by emodin treatment. This was further confirmed in vitro using C26-conditioned media. To our knowledge, this is the first study to report an emodin-induced decrease in protumor macrophages in the context of CRC. However, previous work by our team has documented emodin’s ability to target macrophages in breast cancer (12, 14, 42). Indeed, in a 4T1 and EO771 orthotopic mouse model of breast cancer, emodin reduced lung metastasis and this was consistent with a reduction in infiltration of F4/80+ macrophages and M2-like macrophages (14). In a follow-up study using the same models, we reported that emodin attenuated breast tumor growth by inhibiting macrophage infiltration and M2-like polarization, which was accompanied by increased T-cell activation and a reduction in angiogenesis (12). Furthermore, we reported that emodin’s actions on TAMs can suppress epithelial mesenchymal-transition (EMT) and cancer stem cell (CSC) formation of breast cancer cells (42). Although emodin was able to reduce the number of M2-like protumor macrophages in the colon, we did not detect any significant difference in M2-associated cytokines in excised polyps. However, we did document an increase in the M1-associated marker, NOS2, with emodin treatment supporting promotion of M1 macrophages, which are considered antitumor. A limitation of our study is that macrophages were not assessed in the colons of the ApcMin/+ model given that this model primarily develops intestinal polyps; therefore, we cannot assume that emodin can decrease M2 macrophages in this model or other models of CRC.
Given that intestinal macrophages are largely considered to be derived from the bone marrow (43), we next examined emodin’s effect on bone marrow macrophages. Interestingly, the bone marrow data appears opposite to the colon data; in the bone marrow, emodin suppressed M1-like macrophages but had no impact on M2-like macrophages. This may not be entirely surprising as we have previously reported that emodin is uniquely able to suppress the excessive response of macrophages to both M1 and M2 stimuli and therefore has the potential to restore macrophage homeostasis in various pathologies (7). Indeed, previous work from our group has reported that emodin bidirectionally tunes the induction of LPS/IFNγ− and IL4-responsive genes through inhibiting NF-κB/IRF5/STAT1 signaling and IRF4/STAT6 signaling, respectively (7). This was further supported by the finding that emodin inhibited the removal of H3K27 trimethylation (H3K27m3) marks and the addition of H3K27 acetylation (H3K27ac) marks on genes required for M1 or M2 polarization of macrophages (7). This phenomenon suggests that emodin may exert very different homeostasis-maintaining effects on macrophages in different locations and thus target two very different pathologies within the same individual as documented in the current study with colon (decrease in M2) and bone marrow (decrease in M1) derived macrophages. We did not perform fate mapping studies and therefore cannot confirm as to whether this effect of emodin on bone marrow macrophages had any direct impact on the tumorigenesis outcomes. However, we can speculate that if emodin can reduce the inflammatory status of cells in the bone marrow, it could potentially limit the recruitment of cells like macrophages, to the tumor microenvironment, which would then be educated to become TAMs. Indeed, our complete blood count data indicates a trend for emodin to decrease circulating monocytes in a dose-dependent fashion in AOM/DSS-treated mice. Additionally, this finding, supports our previous report on the ability of emodin to bidirectionally depolarize macrophages depending on environmental cues (7). Further studies involving fate mapping are necessary to fully understand emodin’s impact on bone marrow cells and subsequent recruitment to and education in the TME.
Given the reduction in M1 macrophages in the bone marrow with emodin, we next assessed the presence of P2X7, a receptor on macrophages that is known to activate inflammatory responses and that can be targeted by emodin (9, 13, 38). Our data indicate that the P2X7 receptor is increased in the bone marrow of AOM/DSS-treated mice, and this effect is rescued by emodin treatment. Previous work has documented that antagonizing the P2X7 receptor can reduce inflammatory signaling (44). Therefore, we examined emodin’s effect on STAT3 signaling in bone marrow homogenates and confirm that STAT3 was indeed reduced by emodin and in an apparent dose-dependent manner. Thus, the culmination of previous investigations on the role of the P2X7 receptor would suggest that emodin is working to antagonize the P2X7 receptor, which is present on activated proinflammatory immune cells and therefore contributes to the decreased expression of STAT3 signaling. However, this mechanism needs further investigation, particularly in the context of CRC.
Overall, our findings indicate the efficacy of emodin as an agent for the prevention of intestinal/colon-based cancers. The exact in vivo mechanisms of emodin have proven to be elusive, at least in the context of CRC. However, here we provide evidence that emodin can decrease protumorigenic M2-like macrophages in the colon of AOM/DSS, which we confirmed in vitro using C26-conditioned media. We further demonstrate that emodin ameliorates the increased systemic monocyte counts and bone marrow M1 macrophages in CRC mice, possibly by inhibiting the activity of the P2X7 receptor. Although these findings do not definitively indicate the mechanism of action of emodin, we are the first to show evidence of emodin’s ability to target macrophages in vivo in a model of CRC. Consistent with our previous findings (7), these data indicate that the benefit of emodin is likely specific to the environment and warrants further investigation to confirm the mechanisms discussed here using additional mouse models of CRC. Importantly, we show these results utilizing a dose of emodin that has demonstrated no apparent toxicities (17).
In conclusion, the present study confirms that emodin is an effective primary therapy against the onset of genetic and chemically induced sporadic intestinal/CRC. We established that emodin reduces the M2-like protumorigenic macrophages within the colon of AOM/DSS mice. Furthermore, we provide evidence that emodin may be acting to antagonize the P2X7 receptor within the bone tissue and consequently decrease the activation of proinflammatory cells, which may have implications for recruitment of cells to the TME. Our data support the continuing investigation of emodin as an effective anti-inflammatory and anticancer therapeutic and provide insight into the tissue specific mechanisms in which this novel drug might benefit patients with CRC.
GRANTS
This work was supported by National Institutes of Health Grants R01CA218578 (to D.F. and E.A.M.), R41AT009964 (to E.A.M.), and F31AT009820 (to A.T.S.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.T.S., B.V., M.N., D.F., and E.A.M. conceived and designed research; A.T.S., B.V., and J.L.K. performed experiments; A.T.S., B.V., I.C., J.L.K., and D.F. analyzed data; A.T.S., B.V., I.C., J.L.K., M.N., D.F., and E.A.M. interpreted results of experiments; A.T.S. and B.V. prepared figures; A.T.S. and E.A.M. drafted manuscript; A.T.S., B.V., I.C., J.L.K., M.N., D.F., and E.A.M. edited and revised manuscript; A.T.S., B.V., I.C., J.L.K., M.N., D.F., and E.A.M. approved final version of manuscript.
REFERENCES
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer J Clin 58: 71–96, 2008. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Meissner HI, Breen N, Klabunde CN, Vernon SW. Patterns of colorectal cancer screening uptake among men and women in the United States. Cancer Epidemiol Biomarkers Prev 15: 389–394, 2006. doi: 10.1158/1055-9965.EPI-05-0678. [DOI] [PubMed] [Google Scholar]
- 3.Rustgi AK. The genetics of hereditary colon cancer. Genes Dev 21: 2525–2538, 2007. doi: 10.1101/gad.1593107. [DOI] [PubMed] [Google Scholar]
- 4.Willett WC. Diet and cancer: an evolving picture. JAMA 293: 233–234, 2005. doi: 10.1001/jama.293.2.233. [DOI] [PubMed] [Google Scholar]
- 5.Kim YS, Young MR, Bobe G, Colburn NH, Milner JA. Bioactive food components, inflammatory targets, and cancer prevention. Cancer Prev Res (Phila) 2: 200–208, 2009. doi: 10.1158/1940-6207.CAPR-08-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aggarwal BB, Kunnumakkara AB, Harikumar KB, Tharakan ST, Sung B, Anand P. Potential of spice-derived phytochemicals for cancer prevention. Planta Med 74: 1560–1569, 2008. doi: 10.1055/s-2008-1074578. [DOI] [PubMed] [Google Scholar]
- 7.Iwanowycz S, Wang J, Altomare D, Hui Y, Fan D. Emodin bidirectionally modulates macrophage polarization and epigenetically regulates macrophage memory. J Biol Chem 291: 11491–11503, 2016. doi: 10.1074/jbc.M115.702092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li HL, Chen HL, Li H, Zhang KL, Chen XY, Wang XW, Kong QY, Liu J. Regulatory effects of emodin on NF-κB activation and inflammatory cytokine expression in RAW 264.7 macrophages. Int J Mol Med 16: 41–47, 2005. doi: 10.3892/ijmm.16.1.41. [DOI] [PubMed] [Google Scholar]
- 9.Liu L, Zou J, Liu X, Jiang LH, Li J. Inhibition of ATP-induced macrophage death by emodin via antagonizing P2X7 receptor. Eur J Pharmacol 640: 15–19, 2010. doi: 10.1016/j.ejphar.2010.04.036. [DOI] [PubMed] [Google Scholar]
- 10.Song ZC, Wang ZS, Bai JH, Li Z, Hu J. Emodin, a naturally occurring anthraquinone, ameliorates experimental autoimmune myocarditis in rats. Tohoku J Exp Med 227: 225–230, 2012. doi: 10.1620/tjem.227.225. [DOI] [PubMed] [Google Scholar]
- 11.Guo HC, Bu HQ, Luo J, Wei WT, Liu DL, Chen H, Tong HF, Wang ZH, Wu HY, Li HH, Zuo MM, Li W, Lin SZ. Emodin potentiates the antitumor effects of gemcitabine in PANC-1 pancreatic cancer xenograft model in vivo via inhibition of inhibitors of apoptosis. Int J Oncol 40: 1849–1857, 2012. doi: 10.3892/ijo.2012.1389. [DOI] [PubMed] [Google Scholar]
- 12.Iwanowycz S, Wang J, Hodge J, Wang Y, Yu F, Fan D. Emodin inhibits breast cancer growth by blocking the tumor-promoting feedforward loop between cancer cells and macrophages. Mol Cancer Ther 15: 1931–1942, 2016. doi: 10.1158/1535-7163.MCT-15-0987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jelassi B, Anchelin M, Chamouton J, Cayuela ML, Clarysse L, Li J, Goré J, Jiang LH, Roger S. Anthraquinone emodin inhibits human cancer cell invasiveness by antagonizing P2X7 receptors. Carcinogenesis 34: 1487–1496, 2013. doi: 10.1093/carcin/bgt099. [DOI] [PubMed] [Google Scholar]
- 14.Jia X, Yu F, Wang J, Iwanowycz S, Saaoud F, Wang Y, Hu J, Wang Q, Fan D. Emodin suppresses pulmonary metastasis of breast cancer accompanied with decreased macrophage recruitment and M2 polarization in the lungs. Breast Cancer Res Treat 148: 291–302, 2014. doi: 10.1007/s10549-014-3164-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuo TC, Yang JS, Lin MW, Hsu SC, Lin JJ, Lin HJ, Hsia TC, Liao CL, Yang MD, Fan MJ, Wood WG, Chung JG. Emodin has cytotoxic and protective effects in rat C6 glioma cells: roles of Mdr1a and nuclear factor κB in cell survival. J Pharmacol Exp Ther 330: 736– 7–44., 2009. doi: 10.1124/jpet.109.153007. [DOI] [PubMed] [Google Scholar]
- 16.Lee KH, Lee MS, Cha EY, Sul JY, Lee JS, Kim JS, Park JB, Kim JY. Inhibitory effect of emodin on fatty acid synthase, colon cancer proliferation and apoptosis. Mol Med Rep 15: 2163–2173, 2017. doi: 10.3892/mmr.2017.6254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sougiannis AT, Enos RT, VanderVeen BN, Velazquez KT, Kelly B, McDonald S, Cotham W, Chatzistamou I, Nagarkatti M, Fan D, Murphy EA. Safety of natural anthraquinone emodin: an assessment in mice. BMC Pharmacol Toxicol 22: 9, 2021. doi: 10.1186/s40360-021-00474-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y, Pu W, Bousquenaud M, Cattin S, Zaric J, Sun LK, Rüegg C. Emodin inhibits inflammation, carcinogenesis, and cancer progression in the AOM/DSS model of colitis-associated intestinal tumorigenesis. Front Oncol 10: 564674, 2020. doi: 10.3389/fonc.2020.564674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shimpo K, Chihara T, Kaneko T, Beppu H, Wakamatsu K, Shinzato M, Yukitake J, Sonoda S. Inhibitory effects of low-dose aloe-emodin on the development of colorectal tumors in min mice. Asian Pac J Cancer Prev 15: 5587–5592, 2014. doi: 10.7314/apjcp.2014.15.14.5587. [DOI] [PubMed] [Google Scholar]
- 20.Ma YS, Weng SW, Lin MW, Lu CC, Chiang JH, Yang JS, Lai KC, Lin JP, Tang NY, Lin JG, Chung JG. Antitumor effects of emodin on LS1034 human colon cancer cells in vitro and in vivo: roles of apoptotic cell death and LS1034 tumor xenografts model. Food Chem Toxicol 50: 1271–1278, 2012. doi: 10.1016/j.fct.2012.01.033. [DOI] [PubMed] [Google Scholar]
- 21.Ma L, Li W. Emodin inhibits LOVO colorectal cancer cell proliferation via the regulation of the Bcl-2/Bax ratio and cytochrome c. Exp Ther Med 8: 1225–1228, 2014. doi: 10.3892/etm.2014.1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng C, Dong W. Aloe-emodin induces endoplasmic reticulum stress-dependent apoptosis in colorectal cancer cells. Med Sci Monit 24: 6331–6339, 2018. doi: 10.12659/MSM.908400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bienz M, Clevers H. Linking colorectal cancer to Wnt signaling. Cell 103: 311– 3–20., 2000. doi: 10.1016/S0092-8674(00)00122-7. [DOI] [PubMed] [Google Scholar]
- 24.Dai G, Ding K, Cao Q, Xu T, He F, Liu S, Ju W. Emodin suppresses growth and invasion of colorectal cancer cells by inhibiting VEGFR2. Eur J Pharmacol 859: 172525, 2019. doi: 10.1016/j.ejphar.2019.172525. [DOI] [PubMed] [Google Scholar]
- 25.Schörkhuber M, Richter M, Dutter A, Sontag G, Marian B. Effect of anthraquinone-laxatives on the proliferation and urokinase secretion of normal, premalignant and malignant colonic epithelial cells. Eur J Cancer 34: 1091–1098, 1998. doi: 10.1016/s0959-8049(98)00037-9. [DOI] [PubMed] [Google Scholar]
- 26.Bain CC, Schridde A. Origin, differentiation, and function of intestinal macrophages. Front Immunol 9: 2733, 2018. doi: 10.3389/fimmu.2018.02733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jedinak A, Dudhgaonkar S, Sliva D. Activated macrophages induce metastatic behavior of colon cancer cells. Immunobiology 215: 242–249, 2010. doi: 10.1016/j.imbio.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 28.Kwak Y, Koh J, Kim DW, Kang SB, Kim WH, Lee H S. Immunoscore encompassing CD3+ and CD8+ T cell densities in distant metastasis is a robust prognostic marker for advanced colorectal cancer. Oncotarget 7: 81778–81790, 2016. doi: 10.18632/oncotarget.13207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li J, Li L, Li Y, Long Y, Zhao Q, Ouyang Y, Bao W, Gong K. Tumor-associated macrophage infiltration and prognosis in colorectal cancer: systematic review and meta-analysis. Int J Colorectal Dis 35: 1203–1210, 2020. doi: 10.1007/s00384-020-03593-z. [DOI] [PubMed] [Google Scholar]
- 30.Cranford TL, Enos RT, Velázquez KT, McClellan JL, Davis JM, Singh UP, Nagarkatti M, Nagarkatti PS, Robinson CM, Murphy EA. Role of MCP-1 on inflammatory processes and metabolic dysfunction following high-fat feedings in the FVB/N strain. Int J Obes 40: 844–851, 2016. doi: 10.1038/ijo.2015.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bader JE, Enos RT, Velázquez KT, Carson MS, Nagarkatti M, Nagarkatti PS, Chatzistamou I, Davis JM, Carson JA, Robinson CM, Murphy EA. Macrophage depletion using clodronate liposomes decreases tumorigenesis and alters gut microbiota in the AOM/DSS mouse model of colon cancer. Am J Physiol Gastrointest Liver Physiol 314: G22–G31, 2018. doi: 10.1152/ajpgi.00229.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kang JC, Chen JS, Lee CH, Chang JJ, Shieh YS. Intratumoral macrophage counts correlate with tumor progression in colorectal cancer. J Surg Oncol 102: 242–248, 2010. doi: 10.1002/jso.21617. [DOI] [PubMed] [Google Scholar]
- 33.Sougiannis AT, VanderVeen BN, Enos RT, Velazquez KT, Bader JE, Carson M, Chatzistamou I, Walla M, Pena MM, Kubinak JL, Nagarkatti M, Carson JA, Murphy EA. Impact of 5 fluorouracil chemotherapy on gut inflammation, functional parameters, and gut microbiota. Brain Behav Immun 80: 44–55, 2019. doi: 10.1016/j.bbi.2019.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Enos RT, Velázquez KT, McClellan JL, Cranford TL, Nagarkatti M, Nagarkatti PS, Davis JM, Murphy EA. High-fat diets rich in saturated fat protect against azoxymethane/dextran sulfate sodium-induced colon cancer. Am J Physiol Gastrointest Liver Physiol 310: G906–G919, 2016. doi: 10.1152/ajpgi.00345.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaur K, Saxena A, Debnath I, O’Brien JL, Ajami NJ, Auchtung TA, Petrosino JF, Sougiannis AJ, Depaep S, Chumanevich A, Gummadidala PM, Omebeyinje MH, Banerjee S, Chatzistamou I, Chakraborty P, Fayad R, Berger FG, Carson JA, Chanda A. Antibiotic-mediated bacteriome depletion in Apc. Cancer Med 7: 2003–2012, 2018. doi: 10.1002/cam4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ying W, Cheruku PS, Bazer FW, Safe SH, Zhou B. Investigation of macrophage polarization using bone marrow derived macrophages. J Vis Exp 76: 50323, 2013. doi: 10.3791/50323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Phipps AI, Lindor NM, Jenkins MA, Baron JA, Win AK, Gallinger S, Gryfe R, Newcomb PA. Colon and rectal cancer survival by tumor location and microsatellite instability: the Colon Cancer Family Registry. Dis Colon Rectum 56: 937–944, 2013. doi: 10.1097/DCR.0b013e31828f9a57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oliveira SD, Nanini HF, Savio LEB, Waghabi MC, Silva CLM, Coutinho-Silva R. Macrophage P2X7 receptor function is reduced during schistosomiasis: putative role of TGF-β1. Mediators Inflamm 2014: 134974, 2014. doi: 10.1155/2014/134974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Höhn P, Braumann C, Freiburger M, Koplin G, Dubiel W, Luu AM. Anti-tumorigenic effects of emodin and its' homologue BTB14431 on vascularized colonic cancer in a rat model. Asian Pac J Cancer Prev 21: 205–210, 2020. doi: 10.31557/APJCP.2020.21.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gu J, Cui CF, Yang L, Wang L, Jiang XH. Emodin inhibits colon cancer cell invasion and migration by suppressing epithelial-mesenchymal transition via the Wnt/β-catenin pathway. Oncol Res 27: 193–202, 2019. [Erratum in Oncol Res 28: 681–682, 2021]. doi: 10.3727/096504018X15150662230295. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 41.McClellan JL, Davis JM, Steiner JL, Enos RT, Jung SH, Carson JA, Pena MM, Carnevale KA, Berger FG, Murphy EA. Linking tumor-associated macrophages, inflammation, and intestinal tumorigenesis: role of MCP-1. Am J Physiol Gastrointest Liver Physiol 303: G1087–G1095, 2012. doi: 10.1152/ajpgi.00252.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Q, Hodge J, Wang J, Wang Y, Wang L, Singh U, Li Y, Yao Y, Wang D, Ai W, Nagarkatti P, Chen H, Xu P, Murphy EA, Fan D. Emodin reduces breast cancer lung metastasis by suppressing macrophage-induced breast cancer cell epithelial-mesenchymal transition and cancer stem cell formation. Theranostics 10: 8365–8381, 2020. doi: 10.7150/thno.45395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laviron M, Boissonnas A. Ontogeny of tumor-associated macrophages. Front Immunol 10: 1799, 2019. doi: 10.3389/fimmu.2019.01799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu Y, Wu Y, Gu S, Yin Q, Li H, Wang J, Geng D, Xu Y. The P2X7 receptor (P2X7R)-specific antagonist A804598 inhibits inflammatory reaction in human fibroblast-like synoviocytes. Am J Transl Res 12: 45–53, 2020. [PMC free article] [PubMed] [Google Scholar]