

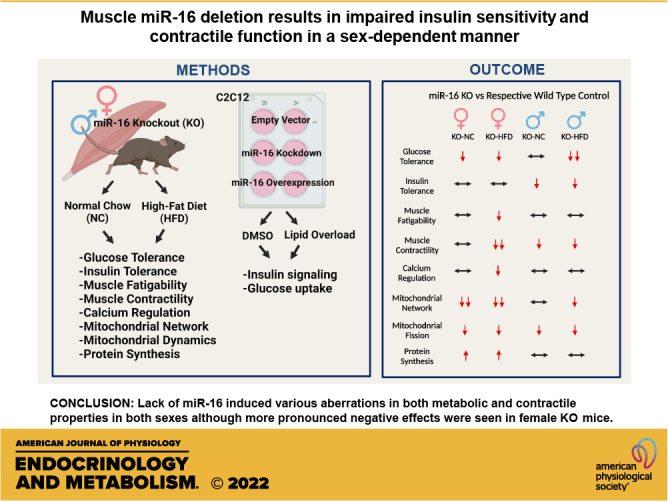
Keywords: insulin resistance, microRNA, mitochondrial quality, protein turnover, type 2 diabetes mellitus
Abstract
microRNAs (miRs) are linked to various human diseases including type 2 diabetes mellitus (T2DM) and emerging evidence suggests that miRs may serve as potential therapeutic targets. Lower miR-16 content is consistent across different models of T2DM; however, the role of miR-16 in muscle metabolic health is still elusive. Therefore, the purpose of this study was to investigate how deletion of miR-16 in mice affects skeletal muscle metabolic health and contractile function in both sexes. This study was conducted using both 1) in vitro and 2) in vivo experiments. In in vitro experiments, we used C2C12 myoblasts to test if inhibition or overexpression of miR-16 affected insulin-mediated glucose handling. In in vivo experiments, we generated muscle-specific miR-16 knockout (KO) mice fed a high-fat diet (HFD) to assess how miR-16 content impacts metabolic and contractile properties including glucose tolerance, insulin sensitivity, muscle contractile function, protein anabolism, and mitochondrial network health. In in vitro experiments, although inhibition of miR-16 induced impaired insulin signaling (P = 0.002) and glucose uptake (P = 0.014), overexpression of miR-16 did not attenuate lipid overload-induced insulin resistance using the diacylglycerol analog 1‐oleoyl‐2‐acetyl‐sn‐glycerol. In in vivo experiments, miR-16 deletion induced both impaired muscle contractility (P = 0.031–0.033), and mitochondrial network health (P = 0.008–0.018) in both sexes. However, although males specifically exhibited impaired insulin sensitivity following miR-16 deletion (P = 0.030), female KO mice showed pronounced glucose intolerance (P = 0.046), corresponding with lower muscle weights (P = 0.015), and protein hyperanabolism (P = 0.023). Our findings suggest distinct sex differences in muscle adaptation in response to miR-16 deletion and miR-16 may serve as a key regulator for metabolic dysregulation in T2DM.
NEW & NOTEWORTHY We set to investigate the role of miR-16 in skeletal muscle during diet-induced insulin resistance. Our data provide novel evidence that the lack of miR-16 induced multiple aberrations in insulin sensitivity, muscle contractility, mitochondrial network health, and protein turnover in a sex-dependent manner. Interestingly, miR-16 deletion leads to insulin resistance in males and exacerbated glucose intolerance in females, suggesting different mechanisms of metabolic dysregulation with a lack of miR-16 between sexes.
INTRODUCTION
Type 2 diabetes mellitus (T2DM) is a fast-growing metabolic disease in Western society. Specifically, 34.2 million individuals, 10.5% of the US population, have T2DM and more than 88 million US adults who are currently prediabetic could lead to T2DM within 5 years unless properly treated (1). Interruptions in skeletal muscle metabolism are often responsible for various diseases such as T2DM (2). Because skeletal muscle is the major site for glucose disposal, insulin resistance within the skeletal muscle is considered the onset point of T2DM (3). To manage this metabolic disease, a myriad of studies have provided useful therapeutic strategies including antidiabetic drugs (4), nutritional management (5, 6), and exercise interventions (7). However, the molecular mechanisms of T2DM are still incompletely understood, resulting in not fully effective therapeutic treatments to resolve this metabolic disease.
In the beginning of the new millennium, emerging evidence of microRNAs (miRs) led to a number of investigations on their multiple roles in biological processes in different disease states and their possible therapeutic potential (8, 9). miRs are short single-stranded (∼22 nucleotides long), noncoding RNA molecules that regulate post-transcriptions of target messenger RNA (mRNA) via inhibiting mRNA translation of protein-coding genes or promoting degradation of the target mRNA (10). miRs are enriched in skeletal muscle and recent studies have examined their role in muscle development (11), atrophy (12, 13), mitochondrial biology (14), and insulin resistance (15). Specifically, we and others (16, 17) have demonstrated lower miR-16 in insulin-resistant skeletal muscle, which is consistently noted in human, rodent, and in vitro models of T2DM. In addition, knockdown of the miR-15 family (miR-15b and miR-16) in myocytes altered cellular signaling cascades responsible for glucose uptake and induced glucose intolerance in vitro (16). In our prior study, we identified that altered miR-16 content was related to aberrations in protein synthesis via upregulation of mechanistic target of rapamycin (mTOR) and ribosomal protein S6 kinase β-1 (p70S6K1) and autophagy via activation of B cell lymphoma-2 (Bcl-2), Beclin-1, and microtubule-associated proteins 1 A/1B light chain 3B (LC3), resulting in altered protein turnover (17). Based on prior evidence, it is plausible that diminished miR-16 in muscle may be sufficient to concomitantly impair insulin sensitivity and muscle functions; however, this hypothesis has yet to be tested.
Therefore, the purpose of this study was to investigate the role of miR-16 in skeletal muscle during diet-induced insulin resistance. Specifically, our aim was twofold: 1) to assess if skeletal muscle miR-16 deletion is sufficient to induce insulin resistance and 2) to determine how loss of miR-16 content affects metabolic and contractile properties including glucose handling, insulin sensitivity, muscle contractility, protein anabolism, and muscle mitochondrial quality using skeletal muscle-specific knockout (KO) of miR-16 in both biological sexes. Here, we provide evidence that reduced miR-16 content corresponds to impaired insulin sensitivity, muscle contractility, mitochondrial network health, and abnormal protein turnover in a sex-dependent manner.
METHODS
Cell Culture Experiments
C2C12 murine myoblasts (CRL-1772, ATCC, Manassas, VA) were plated in six-well plates (5 × 104 cells/well) with 2 mL Dulbecco’s modified Eagle medium (DMEM) combined with 20% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S). Cells were incubated at 37°C with 5% CO2 and media was changed every 48 h. Upon ∼80% confluence, growth media was replaced with DMEM supplemented with 2% horse serum, 1% P/S, 5% HEPES, 0.75% transferrin, and 0.75% insulin for cell differentiation. After 5 days of differentiation, mature C2C12 cells were then treated with 1-oleoyl-2-acetyl-sn-glycerol, a diacylglycerol/DAG analog (OAG; 62600, Cayman, Ann Arbor, MI) at a concentration of 20 μM in either differentiation media or in serum starvation media and incubated overnight to induce insulin resistance (17).
Transfection
Plasmids encoding either an empty vector control (pCMV-miR; PCMVMIR, Origene, Rockville, MD) or miR-16 (pCMV-miR-16; SC400199, Origene) were transferred into DH5-α Escherichia coli as we have previously described (18). Plasmid DNA was amplified and isolated from bacteria using PureLink HiPure Plasmid Filter Maxiprep kit (K211017, Life Technologies, Carlsbad, CA). We used anti-miR inhibitor for miR-16 (AM17000, Ambion, Austin, TX), miR-16-1-5p-clone (AM10339, Ambion), and anti-miR negative control (AM17010, Life Technologies). Plasmid DNA (1 μg) or 30 nM anti-miR was diluted in 50 μL of Opti-mem reduced serum media (31985088, Life Technologies), combined with 4 μL Lipofectamine 2000 (11668019, Life Technologies) diluted in 50 μL Opti-mem and incubated for 20 min to allow lipid/DNA complexes to form. Media was replaced with Opti-mem and lipid/DNA complexes were added and incubated for 5 h at 37°C with 5% CO2 before returning to growth media. For serum starvation or insulin stimulation, cells were incubated overnight in either DMEM with 20% FBS or serum-free DMEM. Overnight fasted cells were treated with either serum-free media or 100 mM insulin supplemented media 9 min before the cell harvest, as described previously (19). In paired experiments, pCMV-miR-16 was used to overexpress miR-16 and compared with pCMV-miR as empty vector control, or anti-16 was transfected to inhibit miR-16 and compared with negative control anti-miR. Insulin-stimulated glucose uptake level was assessed via using a 2-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-2-deoxyglucose (2-NBDG) assay per the manufacturer’s instruction (N13195, Thermo Fisher Scientific, Waltham, MA). The efficiency of the overexpression and inhibition of miR-16 was tested and validated previously (17).
Animals
All animal procedures were approved by the Institutional Animal Care and Use Committees of the University of Arkansas, Fayetteville. miR-15a/16-1 floxed (017641) and MCK Cre Recombinase (006475) mouse breeding pairs were obtained from Jackson Laboratories (Bar Harbor, ME) and crossbred for studies at the University of Arkansas Central Laboratory Animal Facility (CLAF) by PI’s laboratory staff. Breeding was done such that all experimental animals contained the miR-15a/16-1 floxed transgene, and MCK-Cre-positive mice were used as miR-16 knockout (KO) and MCK-Cre-negative animals served as wild-type (WT) control. This design allowed for MCK driven expression of Cre-recombinase to drive specific deletion of miR-15a/16-1 in skeletal muscle. Animals were genotyped through PCR using primer sequences as provided by Jackson Laboratory for each transgene (miR-15a/16-1 floxed and MCK Cre Recombinase) and confirmed by laboratory staff. Genotype confirmation for miR-16 KO and WT animals was provided in Supplemental Fig. S1 (all Supplemental Figures are available at https://doi.org/10.6084/m9.figshare.16615906). Animals were kept on a 12:12-h light-dark cycle, housed 72°F. Experimental animals were housed 4–8 per cage in 16 in. × 10.5 in. × 6 in. cages. Commercial bedding was provided for the animals. A total of 89 (45 males and 44 females) mice were fed either normal chow (NC) or 45% high-fat diet (HFD) from Research Diets (124521, New Brunswick, NJ) beginning at 9 wk of age for 4 wk. Animals were given ad libitum access to food and water for the duration of the study. Upon the start of HFD feeding, food intake was closely monitored by laboratory staff. There were four experimental groups (WT NC, WT HFD, KO NC, and KO HFD) in each sex and 9–12 mice were randomly selected to each group.
Plasmid DNA Amplification and Electroporation
The plasmid containing DH5-α E. coli (20) for MitoTimer was amplified and plasmid DNA was isolated using PureLink HiPure Plasmid Filter Maxiprep kit (K211017, Life Technologies). At 6 wk of age, plasmid transfection into the right flexor digitorum brevis (FDB) muscle was performed as described previously (20, 21). Briefly, while mice were anesthetized using 4% isoflurane, 10 μL of 0.36 mg/mL of hyaluronidase in saline was injected using an insulin syringe subcutaneously under footpad. The needle penetrates the skin at a point close to the heel of the foot and advances the needle subcutaneously toward the base of the toe for ¼ which is above the FDB muscle. After an hour, second anesthesia was applied to inject a total of 20 μg of the MitoTimer plasmid DNA with the same procedure described for the hyaluronidase injection. Anesthesia was disconnected after the plasmid DNA injection for 10–15 min and reconnected for electroporation. Two gold-plated acupuncture needles were placed under the skin at the heel and a second one at the base of the toes. Electrodes were perpendicular to the long axis of the foot and parallel to each other. Ten electrical pulses with 20 ms in duration/each, at 1 Hz and 75 V used an S88 electrical stimulator (Grass Instrument, West Warwick, RI).
Grip Strength Test
At 8 wk of age, mouse grip strength was tested using a rodent grip strength meter (Harvard Apparatus, Holliston, MA). Briefly, each mouse was allowed to grab a metal grid connected to a force transducer and then pulled backward in the horizontal plane by the tail off the grasping grid. Each mouse underwent three sets of five consecutive repetitions with a 2- to 3-min rest period between each set, which were averaged to determine grip strength for each mouse as previously described (22). Grip strength data were normalized to body weight to account for different body sizes.
Graded Exercise Test
A graded exercise test (GXT) was performed as described previously (23, 24). Briefly, at 8 wk of age, mice performed GXT on a treadmill as follows: 1) The mice were acclimatized to the GXT by walking on the treadmill for 10 min each day for 3 days before the GXT at a speed of 0.5 m/h. 2) The test started with mice running on the treadmill with 0% inclination and 0.5 m/h speed for 10 min. The grade was then increased to 5% and speed to 0.7 m/h and the speed was increased by 0.1 m/h every 30 min until exhaustion (not exceeding 1.2 m/h), which is defined as when mice refuse to run when encouraged by brushing the tail for ∼10–20 s. The total time to exhaustion and total distance were recorded.
Glucose Tolerance Tests
Glucose tolerance tests (GTTs) were performed as described by in our laboratory (23). Briefly, at 9 wk of age, food was removed 6 h before the test. At the start of the test, the tail was cut at the very tip and one drop of blood was used to assess blood glucose level as a baseline using an Ultima diabetic monitor (ReliOn, Spokane alley, WA). Scissors were cleaned using 70% EtOH between animals. Animals’ body weight was then weighed and sterilized d-glucose in saline was given via an intraperitoneal (IP) injection at a dose of 1.5 g glucose/kg body wt. At 30-, 60-, and 120-min postinjection blood glucose level was measured by gently massaging the tail to obtain one drop of blood each time. At the end of 120 min, mice were transferred to a clean cage and food and water were returned.
Insulin Tolerance Test
Insulin tolerance test (ITT) was performed ∼48 h after GTT as previously described (25). Baseline glucose level was measured using the same procedure described for the GTT. Mice were given an IP injection of insulin (1 U/kg body wt), and tail vein blood samples were collected at 0-, 15-, 30-, and 60 min following insulin administration. Data were calculated as the slope from 0- to 15-min post-insulin administration to assess acute insulin responsiveness. Grip strength test, GXT, GTT, and ITT were repeated at 13 wk of age.
In Vivo Muscle Contractility Test
Approximately 48 h before tissue harvest, in vivo skeletal muscle contractility (1/2 relaxation time, time to maximum contraction, and fatigability) of the anterior crural (shin) muscles were performed as previously described (26, 27). Briefly, mice were anesthetized using 2.5% isoflurane, and left hindlimb was shaved and ethanol was applied to clean the skin. The foot was placed in a foot plate attached to a servomotor (Model 300B-LR, Aurora Scientific, Aurora, Ontario, Canada) using surgical tape while their body temperature was maintained at 37°C using a heating pad. Two platinum electrodes (Model E2-12, Grass Instrument, West Warwick, RI) were then inserted percutaneously on either side of the peroneal nerve. Consistent instant stimulation was tested and monitored to ensure the electrodes were neither too deep nor too proximal to cause recruitment of the posterior crural muscles. Muscle contractions were induced via electrical stimulator and stimulus isolation unit (Model S48 and SIU5, respectively; Grass Technologies, West Warwick, RI). After resting the hindlimb for 1 min, 1/2 relaxation time and time to maximum contraction were assessed at 150-Hz torque stimulation (28) followed by resting the hindlimb for 4 min. After measurement of submaximal and maximal isometric torques, we performed a fatigability test via applying 120 isometric contractions at 40 Hz for a total protocol time of 2 min followed by 10-min rest. During the recovery following the fatigability test, the normal movement of experimental mice was closely monitored to determine any injuries during the in vivo muscle contractility test. To account for differences in body size among experimental mice, torque (mN⋅m) was normalized by their body weight per prior reports (29).
Deuterium Injection and 24-h Protein Fractional Synthesis Rate
To determine 24-h fractional synthesis rate (FSR), mice were given an IP injection of deuterium oxide (D2O), heavy labeled water, at 20 μL/g body wt to achieve an ∼2% enrichment of total body water with deuterium 24 h before tissue harvest. Drinking water was thereafter supplemented with 4% D2O to maintain the plasma pool of D2O until tissue harvest (30, 31). A detailed method for FSR has previously been reported (32). Briefly, 15 mg of gastrocnemius muscle was powdered and then homogenized in a 10% trichloroacetic acid (TCA) solution for mixed FSR. The mixed fractions were then washed three times with 10% TCA solution by centrifugation to eliminate cytosolic amino acids. Proteins were then placed in a tube with 200 μL of 6 M HCL and heated at 100°C for 24 h to hydrolyze proteins into amino acids. An aliquot of the hydrolysate was dried down and derivatized with a 3:2:1 vol/vol solution of methyl-8, methanol, and acetonitrile to determine 2H labeling of alanine on its methyl-8 derivative. A 1 μL aliquot was injected into an HP-5ms capillary column in the gas chromatography (GC; 7890 A, Agilent, Santa Clara, CA) as a split injection at a ratio of 20:1. Gas chromatography-mass spectrometry (GC-MS) settings have previously been described (32). Briefly, the initial temperature of the column was set at 90°C and held for 5 min and increased by 5°C/min to 130°C, which was further increased at a rate of 40°C/min to 240°C, with all steps performed at a constant helium flow of 1 mL/min. Peak abundances of ions 99 and 100 were extracted from chromatograms, and M + 1/M ratios were used to calculate percent enrichment of protein-bound alanine using a regression formula generated by [2H]alanine standards (r2 = 0.999). A deuterated alanine/alanine ratio was used to assess protein synthesis. The precursory pool of 2H2O in the plasma was reacted with 10 M NaOH and a 5% solution of acetone in acetonitrile for 24 h to conjugate the free 2H2O to acetone. The solution was extracted by adding Na2SO4 and chloroform and placed in capillary columns to be analyzed on the GC-MS to detect acetone at a 20:1 split. FSR of mixed proteins were calculated using the equation:
where EA represents the amount of protein-bound [2H] alanine (mol% excess), EBW is the quantity of 2H2O in body water (mol% excess), 3.7 represents the exchange of 2H between body water and alanine (3.7 of 4 carbon-bound hydrogens of alanine exchange with water), and t (h) represents the time the label was present in hours.
Insulin Administration
An additional IP injection of insulin (5 U/kg) at 10 min before tissue collection was used to determine insulin-stimulated insulin signaling.
Fluorescence Microscopy for pMitoTimer
At the time of tissue harvesting, freshly collected FDB muscles were fixed with 4% paraformaldehyde (PFA) for 20 min followed by 5-min incubation with PBS at room temperature. Muscles were then spread out on a gelatin-coated glass slide using two forceps and mounted with a drop of 50% glycerol in PBS as mounting media. A coverslip was added on the top of the mounted sample and four drops of nail polish were applied at the corners to anchor the coverslip followed by ∼2-min mild pressure with a thumb. pMitoTimer images were acquired at ×100 magnification using the fluorescein isothiocyanate (FITC) (green, excitation/emission; 488/518 nm) and tetramethylrhodamine (TRITC) (red, excitation/emission; 543/572 nm) fluorescent channels on a Nikon Eclipse Ti-s inverted epifluorescent microscope (Melville, NY) with LED-based light source. Acquisition parameters were followed as described by Laker et al. (20) and a specially generated MATLAB program, a generous gift from Dr. Zhen Yan (University of Virginia), was used to analyze pMitoTimer red to green ratio and number of pure red puncta. Since the assay is optimally performed on fresh tissue, the whole process was performed within 30 min of the tissue collection.
Protein Preparation and Immunoblotting
Frozen gastrocnemius muscle samples were powdered and 20 mg of gastrocnemius muscle was homogenized in protein loading buffer [50 mM Tris-HCl, pH 6.8, 1% sodium dodecyl sulfate (SDS), 10% glycerol, 20 mM dithiothreitol, 127 mM 2-mercaptoethanol, and 0.01% bromophenol blue combined with protease inhibitors, and phosphatase inhibitors] (24). Total protein (40 μg) in protein sample buffer were loaded into 10% gel and resolved by SDS-PAGE at 100 V for 2 h, and transferred to a PVDF membrane at 20 V for 1.5 h. The membranes were then blocked with 5% of milk in Tris-buffered saline (TBS) for an hour at room temperature followed by three times washes with TBS containing 0.2% Tween 20 (TBST), then the primary antibodies were added to the membrane in LI-COR blocking buffer with 0.2% Tween 20. The membranes were incubated overnight for primary antibodies specific for p-Akt (Ser473; 9271, Cell Signaling, Danvers, MA), Akt (9272, Cell Signaling), p-AS160 (Thr642; 8881, Cell Signaling), AS160 (07-741, Millipore Sigma), sarcoplasmic reticulum Ca2+-ATPase (SERCA) (4219, Cell Signaling), p-4EBP1 (Thr37/46; Cell Signaling, 2855), 4EBP1 (Cell Signaling, 9644), Deptor (ABS222, Millipore Sigma), p70S6K1 (9202, Cell signaling), voltage-dependent anion channel (VDAC) (4866S, Cell Signaling), fission protein 1 (Fis1) (Novus, NB100-56646), Dynamin-related protein 1 (Drp1) (Cell Signaling, 14647), and Bcl2 interacting protein 3 (Bnip3) (Cell Signaling, 44060). Primary antibodies were diluted 1:1,000 in TBS blocking buffer with 0.2% Tween 20 and all primary antibodies used are commercially available and validated by the vendors (Cell Signaling, Novous, and Millipore Sigma), and molecular weights were confirmed via protein ladder. Following overnight incubation with primary antibodies, membranes were washed with TBST and probed with IRDye secondary antibodies (LI-COR Biosciences, Lincoln, NE). Membranes were washed again after secondary incubation (LI-COR, IRDye 800CW/680RD) with TBST (1:10,000) + 0.01% SDS and imaged on LI-COR Odyssey FC using IR detection. All bands were normalized to the 45-kDa actin band of Ponceau S stain as a loading control and all data were normalized to same-sex WT-CON and expressed as fold-difference as previously described (21, 33).
RNA Isolation, cDNA Synthesis, and Quantitative Real-Time PCR
Frozen gastrocnemius muscle samples were powdered, and 20 mg of pulverized gastrocnemius samples were suspended in 1 mL of ice-cold TRIzol reagent and homogenized using Polytron for ∼5 s for three times before being transferred into 1.5-mL microtube and placed on ice. After 15 min, 200 μL of chloroform was added, and samples were centrifuged for 25 min. The clear portion of the solution from the top was removed and transferred into a new tube. An equal amount of 70% of diethyl pyrocarbonate (DEPC)-treated ethanol was added, and sample was loaded into a Rneasy column. RNA isolation was then processed using an RNA isolation kit (K145002; Invitrogen, Carlsbad, CA). DNase-treated RNA (30 μL) was collected and RNA concentration and purity were determined by fluorometry using 260/280 nm ratios read on a Bio-Tek Power Wave XC microplate reader (BioTek Instruments, Winooski, VT) with Take3 microvolume plate and Gen5 software. Subsequently, 1 μg of RNA was reverse transcribed into cDNA using VILO Superscript reagent (11755050; Invitrogen); cDNA was diluted to 1:100 (10 ng/mL). Real-time polymerase chain reaction (PCR) was performed using TaqMan probe reagent and Step-One real-time (RT)-qPCR instrumentation (Applied Biosystems, Foster City, CA), and Ct values were analyzed according to the manufacturer’s instructions. PCR was as follows: 4 min of incubations, 45 cycles of denaturation, annealing, and extension at 95°C, 60°C, and 72°C, respectively. Target TaqMan probes for 18 s (Mm03928990_g1), Redd1 (Mm00512504_g1), Deptor (Mm01195339_m1), Ryr receptor (Mm01175211_m1), and Bnip3 (Mm01275600_m1) were purchased from Applied Biosystems (Life Technologies). Primer information for Fis1 and Drp1 has previously been reported (24). Final quantification of mRNA level was run using a QuantSudio3 PCR instrument (Thermo Fisher Scientific, Waltham, MA) and calculated using the ΔΔCT method as described previously (34, 35). All targets were normalized to the 18 s Ct value, which did not differ between groups.
Statistics
For cell experiments, pre-planned t tests were used to compare controls and treatments (miR-16 inhibition) using GraphPad Prism (La Jolla, CA; Fig. 1, A and B) and a two-way ANOVA was used as the global analysis for each dependent variable. Independent variables were gene condition; pCMV versus miR-16 and media condition; dimethyl sulfoxide (DMSO) versus OAG. For animal experiments, a two-way ANOVA was utilized. Independent variables within each sex included genotype (WT vs. KO) and diet (NC vs. HFD). Sex was not a statistical factor due to the excessive comparisons that would result in a 2 × 2 × 2 ANOVA that would not provide meaningful information. When significant F ratios were found, statistical differences among means were determined by Tukey’s post hoc test. The comparison-wise error rate, α, was set at P < 0.05 for all statistical tests. All two-way ANOVA data were analyzed using the Statistical Analysis System (SAS), version 9.3 (Cary, NC). Figures were compiled using GraphPad Prism and data were expressed as means ± standard error (SE).
Figure 1.
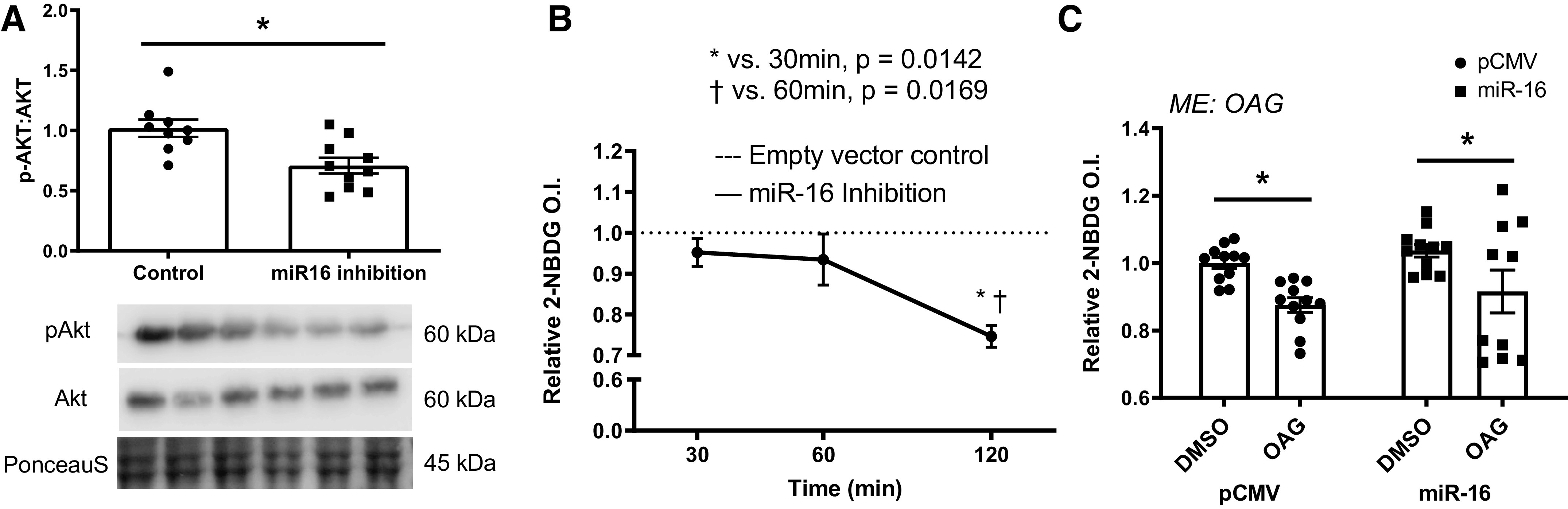
In vitro glucose handling. A: protein content of phosphorylation of Akt (p-Akt)/Akt via immunoblotting in response to miR-16 inhibition. B: insulin-stimulated glucose uptake level (black line) via 2-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-2-deoxyglucose (2-NBDG) following miR-16 inhibition in a time-dependent manner (at 30, 60, and 120 min following the miR-16 inhibition). The dotted line is the basal state of 2-NBDG level (empty vector control) and 2-NBDG level of miR-16 inhibition was normalized to empty vector control (dotted line). C: insulin-stimulated glucose uptake via 2-NBDG via immunoblotting following miR-16 overexpression in lipid overload-induced insulin resistant myoblasts. Data described as means ± SE. Statistical differences are denoted as *P < 0.05 (Student’s t test) Panel C analyzed by two-way ANOVA. An n of ∼9 replicates (wells) per condition was used and each condition was tested in three independent experiments (three replicates per each experiment). ME, main effect of indicated factor.
RESULTS
Inhibition of miR-16 Induces Insulin Resistance, but Overexpression Is Not Sufficient to Ameliorate Glucose Disposal In Vitro
To determine if loss of miR-16 is sufficient to induce insulin resistance, we assessed insulin sensitivity in vitro. Insulin sensitivity via insulin-stimulated phosphorylation of Akt (Ser473)/Akt was ∼30% lower in myotubes following miR-16 inhibition compared with control (P = 0.002; Fig. 1A). Glucose uptake via assessment of 2-NBDG was ∼22% lower following miR-16 inhibition compared with vector control (P = 0.014–0.017; Fig. 1B). To examine if miR-16 overexpression is sufficient to attenuate insulin resistance induced via OAG in vitro, we applied overexpression of miR-16 in myotubes. Although glucose uptake via 2-NBDG was lower in OAG-treated myotubes compared with control cells (P = 0.009), overexpression of miR-16 did not significantly attenuate the suppressed glucose uptake or insulin signaling (P > 0.05; Fig. 1C).
HFD Results in Higher Body and Fat Weights in Both Sexes, but KO Induces Smaller Muscles in Females
Considering that in vitro inhibition of miR-16 was sufficient to impair insulin sensitivity, we next moved to an in vivo model utilizing miR-15a/16-1 floxed mice (36) bred on muscle-specific MCK Cre Recombinase to specifically delete this miR family in skeletal muscle. In males, there was a main effect of diet, with HFD mice having greater body weight (∼20%; P < 0.0001), soleus muscle weight (∼13%; P = 0.012), and gonadal fat (∼216%; P < 0.0001) compared with animals fed with NC (Table 1). There were no additional statistical differences in other tissue weights (gastrocnemius, tibialis anterior (TA), plantaris, extensor digitorum longus (EDL), heart, lungs, spleen, and tibia length; Table 1). In females, there was a main effect of diet, with HFD mice having greater body weight (∼13%; P = 0.0067), heart (∼13%; P = 0.0009), and gonadal fat (∼135%; P < 0.0001) compared with animals fed with NC (Table 2). In addition, female miR-16 KO mice had ∼8% lower plantaris (P = 0.015) and ∼6% lower TA weights (P = 0.06) compared with WT animals (Table 2). There was an interaction effect on liver weight. Specifically, female WT HFD had ∼13% lower liver weight than KO HFD animals (P = 0.013; Table 2). Similarly, female KO NC had ∼18% lower spleen weight compared with WT NC mice (P = 0.006; Table 2). There were no additional statistical differences in other tissues (soleus, gastrocnemius, TA, EDL, lungs, and tibia length; Table 2).
Table 1.
Male characteristics
| Male | WT NC, n = 12 | WT HFD, n = 12 | KO NC, n = 10 | KO HFD, n = 11 | Main Effect |
|---|---|---|---|---|---|
| Body weight, g | 26.62 ± 2.11 | 32.48 ± 5.12* | 26.06 ± 2.28 | 30.69 ± 2.53* | Diet (P < 0.001) |
| Soleus, mg | 12.35 ± 1.2 | 14.55 ± 2.74* | 12.61 ± 1.82 | 13.63 ± 1.85* | Diet (P = 0.012) |
| Gastrocnemius, mg | 146.36 ± 13.15 | 145.29 ± 6.93 | 137.37 ± 6.12 | 147.71 ± 9.22 | N/A |
| TA, mg | 53.86 ± 4.97 | 55.36 ± 4.00 | 53.65 ± 2.81 | 51.86 ± 5.84 | N/A |
| Plantaris, mg | 20.91 ± 2.82 | 21.37 ± 2.51 | 21.12 ± 2.82 | 21.01 ± 2.47 | N/A |
| EDL, mg | 10.88 ± 1.70 | 10.98 ± 1.87 | 11.4 ± 1.49 | 11.17 ± 1.52 | N/A |
| Heart, mg | 139.15 ± 18.99 | 143.83 ± 15.20 | 134.60 ± 10.15 | 148.15 ± 19.91 | N/A |
| Liver, mg | 1,076.68 ± 180.05 | 1,187.78 ± 230.98 | 1,175.72 ± 120.83 | 1,109.00 ± 139.76 | N/A |
| Lungs, mg | 170.53 ± 25.24 | 168.48 ± 28.49 | 170.00 ± 22.05 | 187.38 ± 18.25 | N/A |
| Spleen, mg | 91.04 ± 14.55 | 89.30 ± 14.55 | 76.53 ± 12.22 | 91.60 ± 16.64 | N/A |
| Fat, mg | 369.42 ± 198.52 | 1,573.46 ± 806.23* | 504.43 ± 194.24 | 1,186.51 ± 615.77* | Diet (P < 0.001) |
| Tibia length, mm | 17.14 ± 0.07 | 14.24 ± 0.07 | 17.14 ± 0.07 | 17.15 ± 0.07 | N/A |
Tissue weights are recorded as wet weights. All values are represented as means ± SE. An n of 10–12 animals per group was used. EDL, extensor digitorum longus; HFD, high-fat diet; KO, knockout; NC, normal chow; TA, tibialis anterior; WT, wild type. Data analyzed by two-way ANOVA with Tukey’s post hoc test where appropriate. *Statistical significance for main effect of genotype or diet with α value set at P < 0.05.
Table 2.
Female characteristics
| Female | WT NC, n = 12 | WT HFD, n = 12 | KO NC, n = 11 | KO HFD, n = 9 | Main Effect |
|---|---|---|---|---|---|
| Body weight, g | 21.44 ± 1.37 | 23.73 ± 3.53* | 21.60 ± 1.40 | 24.82 ± 5.32* | Diet (P = 0.006) |
| Soleus, mg | 10.57 ± 1.30 | 10.64 ± 0.91 | 9.50 ± 1.55 | 10.43 ± 2.60 | N/A |
| Gastrocnemius, mg | 106.88 ± 4.74 | 112.82 ± 7.72 | 108.82 ± 11.02 | 112.87 ± 11.28 | N/A |
| TA, mg | 42.10 ± 3.50 | 42.27 ± 4.56 | 41.23 ± 4.04 | 38.48 ± 2.86 | Geno (P = 0.06) |
| Plantaris, mg | 15.85 ± 1.64 | 17.05 ± 1.45 | 15.14 ± 1.57* | 15.26 ± 1.34* | Geno (P = 0.013) |
| EDL, mg | 9.35 ± 0.74 | 9.56 ± 1.13 | 9.21 ± 0.89 | 9.74 ± 1.27 | N/A |
| Heart, mg | 112.53 ± 12.00 | 127.63 ± 15.57* | 112.34 ± 7.29 | 125.92 ± 12.82* | Diet (P = 0.001) |
| Liver, mg | 1,001.07 ± 71.93ab | 878.14 ± 107.19a | 910.36 ± 131.36ab | 1,009.52 ± 228.70b | Interaction (P = 0.013) |
| Lungs, mg | 152.38 ± 17.15 | 151.83 ± 23.26 | 153.90 ± 17.60 | 165.72 ± 13.47 | N/A |
| Spleen, mg | 100.91 ± 12.00a | 93.39 ± 13.87ab | 82.93 ± 9.43b | 98.73 ± 16.01a | Interaction (P < 0.006) |
| Fat, mg | 167.43 ± 78.15 | 443.30 ± 244.46* | 226.06 ± 97.87 | 481.73 ± 302.75* | Diet (P < 0.001) |
| Tibia length, mm | 17.04 ± 0.1 | 17.10 ± 0.09 | 17.03 ± 0.09 | 17.10 ± 0.08 | N/A |
Tissue weights are recorded as wet weights. All values are represented as means ± SE. An n of 10–12 animals per group was used. EDL, extensor digitorum longus; HFD, high-fat diet; KO, knockout; NC, normal chow; TA, tibialis anterior; WT, wild type. Superscript “different letters” denote an interaction effect with α value set at P < 0.05.
Data analyzed by two-way ANOVA with Tukey’s post hoc test where appropriate. *: Statistical significance for main effect of genotype or diet with α value set at P < 0.05.
miR-16 KO Differentially Affects Glucose and Insulin Handling by Biological Sex
We examined both glucose tolerance and insulin sensitivity under the conditions of miR-16 KO and HFD. We observed a main effect of diet on glucose tolerance for both sexes, with HFD mice having ∼14%–32% (males) or ∼14%–19% (females) greater blood glucose levels after 30, 60, and 120 min (males; P = 0.001–0.050; Fig. 2A) or after 30 and 120 min (females; P = 0.017–0.026; Fig. 2B) following glucose challenge compared with o NC animals. Integrative area under the curve (IAUC) indicates ∼81% greater IAUC in male HFD animals compared with NC animals (P = 0.001; Fig. 2C) whereas the IAUC was ∼15% greater in female KO mice compared with WT animals (P = 0.046; Fig. 2D). In males, a main effect of genotype was noted in insulin-mediated blood glucose level, with KO animals having ∼14% higher compared with WT animals at 15 min following insulin challenge (P = 0.048; Fig. 2E) although there were no statistical significances in ITT curve in females (Fig. 2F). There was a main effect of genotype on ITT slope during the first 15 min in males, where male KO mice had ∼81% blunted ITT slope compared with WT animals (P = 0.03; Fig. 2G) whereas no statistical differences in ITT slope were observed in females (Fig. 2H). Although there were no statistical differences in insulin-mediated phosphorylation of Akt (p-Akt; Ser473)/Akt level in males, a main effect of genotype was noted in female p-Akt/Akt content, with female KO mice having ∼46% lower p-Akt/Akt level compared with WT animals (P = 0.007; Fig. 2, I and J). An interaction was noted in male p-AS160 (Thr642)/AS160 level, with male WT HFD mice having ∼71% lower p-AS160/AS160 level compared with WT NC animals (P = 0.017) although there were no statistical significances observed in females (Fig. 2, K and L). The basal levels of GTT, ITT, GTT IAUC, and ITT slope did not differ between WT and KO before diet intervention within each sex (Supplemental Fig. S3, A–D).
Figure 2.
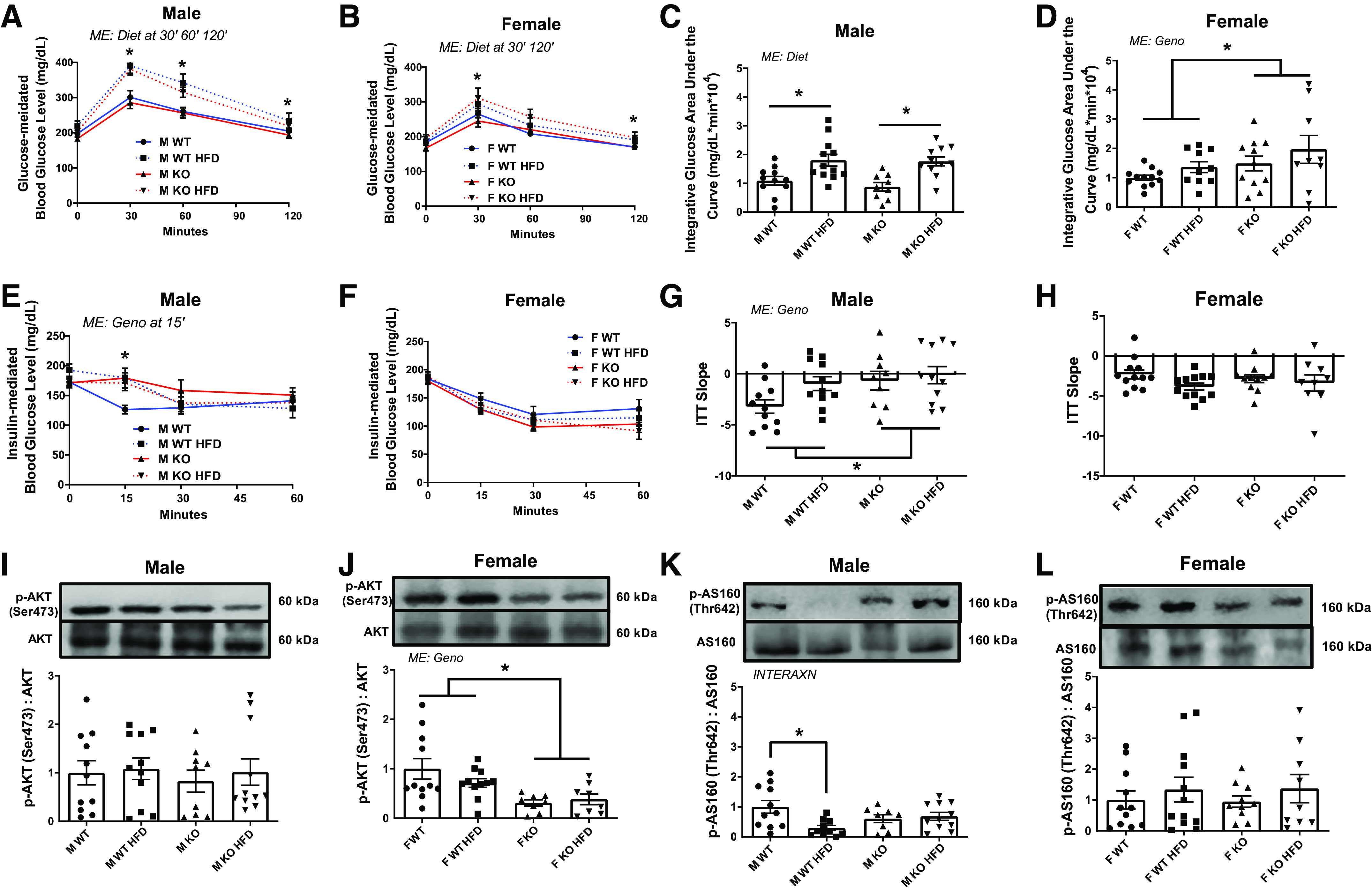
In vivo glucose handling. A and B: blood glucose level before (0 min) and after glucose administration (30, 60, and 120 min). C and D: integrative area under the curve (IAUC) for A and B. E and F: blood glucose level before (0 min) and after insulin administration (15, 30, and 60 min). G and H: insulin tolerance test (ITT) slope for first 15 min of insulin-mediated blood glucose level. I and J: insulin-mediated phosphorylation of Akt (p-Akt) (Ser473) level via immunoblotting. K and L: p-AS160 (Thr642) level via immunoblotting. Data analyzed by two-way ANOVA with Tukey’s post hoc test where appropriate. Data described as means ± SE. Statistical differences are denoted as *P < 0.05 (An n of 9–12 animals per group was used). INTERAXN, interaction effect; KO, knockout; ME, main effect of indicated factor; WT, wild type.
miR-16 KO Results in Impaired Muscle Contractility in Males While Both KO and HFD Impair Muscle Contractility in Females
Insulin resistance is often associated with lower skeletal muscle strength (37). To determine muscle contractile function in response to miR-16 KO, in vivo muscle contractility tests were performed. Muscle fatigability did not show any statistical significance in males (Fig. 3A). However, an interaction effect in fatigability was noted in females. Specifically, KO HFD female mice had greater fatigability (lower % peak torque) during 50–110 electrical stimulations compared with WT control (P = 0.039–0.009; Fig. 3B). Although not statistically significant, KO animals had ∼28% (males; P = 0.058) and ∼20% (females; P = 0.063) lower time to maximal contraction compared with WT animals (Fig. 3, C and D). There was a main effect of genotype on half relaxation time, with male KO animals having ∼20% delayed half relaxation time compared with WT mice (P = 0.033; Fig. 3E). In females, there was an interaction in half relaxation time, with female KO HFD mice having ∼24% delayed half relaxation time compared with all other groups (P = 0.031; Fig. 3F). To determine if impaired muscle contractility was associated with altered regulation of calcium handling, we assessed calcium regulation via SERCA and ryanodine receptor (Ryr) levels. In males, there was a main effect of genotype on SERCA protein content, with male KO mice having ∼35% greater SERCA compared with WT animals (P = 0.002) although Ryr receptor mRNA content did not change (Fig. 3, G and I). In females, although SERCA protein content did not change between experimental groups, there was an interaction in Ryr receptor mRNA content, with female KO HFD mice having ∼42% lower Ryr receptor mRNA levels compared with KO WT mice (P = 0.007; Fig. 3, H and J). There was a main effect of diet on grip strength on both sexes, with HFD mice having ∼16% (male; P = 0.020) and ∼15% (female; P = 0.023) lower compared with NC animals although initial grip strength level did not differ among experimental mice before the diet intervention (Supplemental Fig. S2, A, C, and D). There was a main effect of diet on male GXT, where male HFD mice were ∼23% lower on GXT total distance compared with WT (P = 0.035) although the initial GXT did not differ between WT and KO mice before the diet intervention (Supplemental Fig. S2, B and E). In females, there were no statistical differences among experimental groups before and after the diet intervention (Supplemental Fig. S2, B and F).
Figure 3.
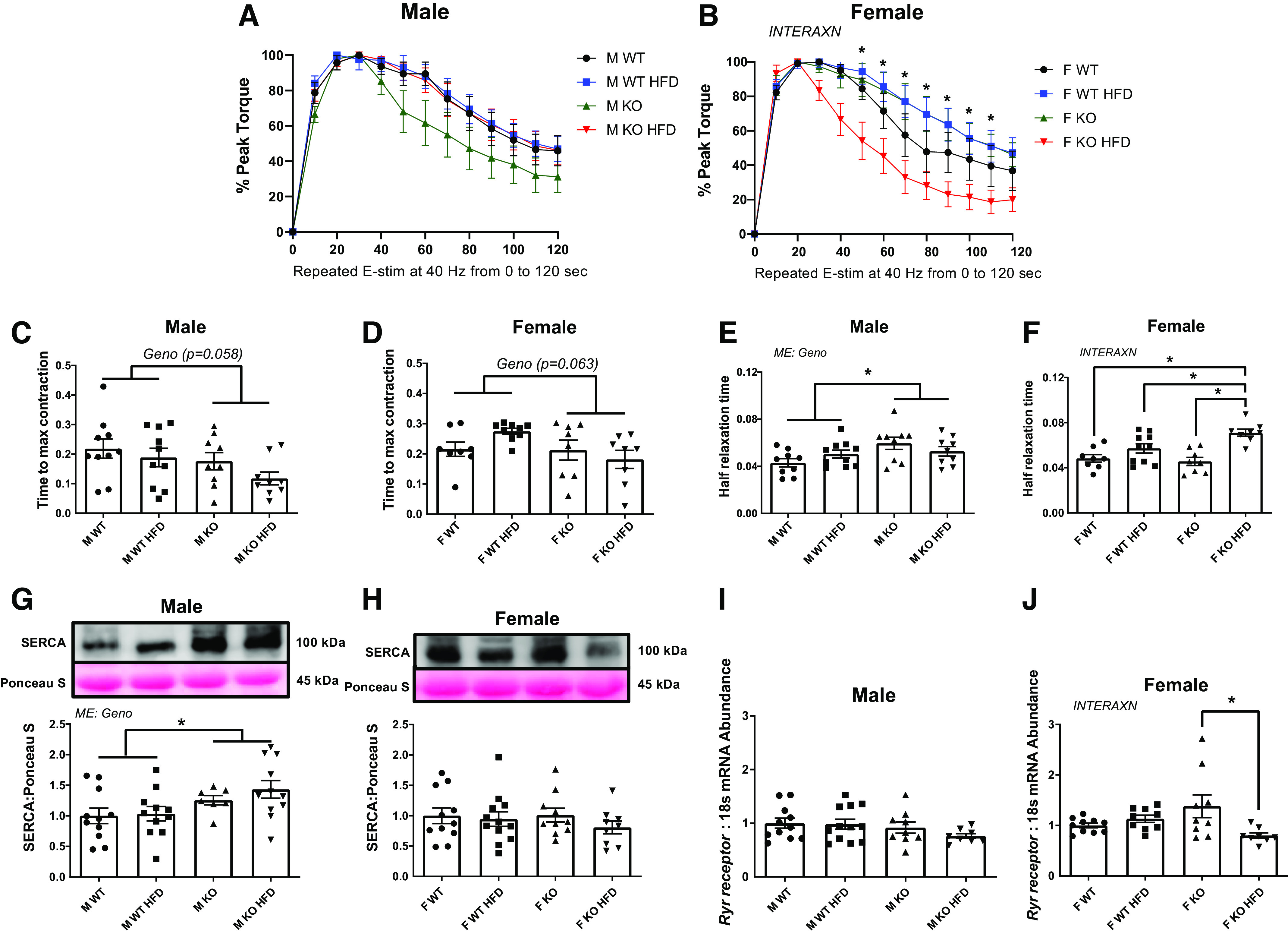
Skeletal muscle contractility. A and B: fatigability data shows % decrease in peak torque following 120 times of electrical stimulation in 2 min at 40 Hz. C and D: time to maximal contraction. E and F: one half relaxation time after the muscle contraction. G and H: protein content of sarcoplasmic reticulum Ca2+-ATPase (SERCA) via immunoblotting. I and J: mRNA content of Ryr receptor. Data analyzed by two-way ANOVA with Tukey’s post hoc test where appropriate. Data described as means ± SE. Statistical significance identified by denoting as * for main effect of genotype or diet, and as “different letters” for an interaction effect with α value set at P < 0.05 (An n of 8–10 animals per group was used). HFD, high-fat diet; INTERAXN, interaction effect; KO, knockout; ME, main effect of indicated factor; WT, wild type.
Biological Sex Differentially Affects Mitochondrial Network Degeneration following miR-16 Deletion
Since mitochondrial dysfunction has been commonly seen in T2DM, we sought to determine the role of miR-16 in mitochondrial network health. Although MitoTimer red to green ratio (a mitochondrial oxidative stress marker) did not reach statistical significances in either sex (data not presented), male HFD mice had approximately fourfold greater pure red puncta (a marker for completely degenerated mitochondria via mitophagy) level compared with animals fed with NC (P = 0.012; Fig. 4, A and E) with no effect of genotype. In females, KO animals had ∼2.5-fold greater pure red puncta compared with WT animals with no effect of diet (P = 0.018; Fig. 4, B and F). Mitochondrial content assessed via voltage-dependent anion channel (VDAC) protein level did not differ between experimental groups in either sex (Fig. 4, C, D, G, and H). In males, there was a main effect of genotype on the mitochondrial fission marker Dynamin-related protein 1 (Drp1), with male KO mice having ∼24% lower mitochondrial fission protein 1 (Fis1) and ∼19% lower Drp1 mRNA content compared with WT animals (P = 0.0150 and P = 0.025, respectively; Fig. 4, I and M). In females, there was a main effect of genotype in Fis1 mRNA and an interaction in Drp1 mRNA content such that female KO mice had ∼25% lower Fis1 compared with WT animals (P = 0.039; Fig. 4J) and female KO HFD mice had ∼32% lower Drp1 compared with WT HFD (P = 0.049; Fig. 4N). There were no statistical differences in protein contents for Fis1 and Drp1 of both sexes (Fig. 4, C, D, K, L, O, and P). Mitochondrial autophagy activity was assessed via Bcl2 interacting protein 3 (Bnip3) mRNA content, where an interaction was noted that male KO HFD mice had ∼43% higher Bnip3 compared with WT HFD and KO NC animals (P = 0.008; Fig. 4Q). An interaction was observed in Bnip3 mRNA of females, with female KO NC mice having ∼88% higher Bnip3 compared with WT NC mice (P = 0.012; Fig. 4R). A main effect of diet was noted in Bnip3 protein content in both sexes, where HFD animals were ∼23% (P = 0.026; males) and ∼35% (P = 0.007; female) higher in Bnip3 protein contents compared with NC animals (Fig. 4, C, D, S, and T).
Figure 4.
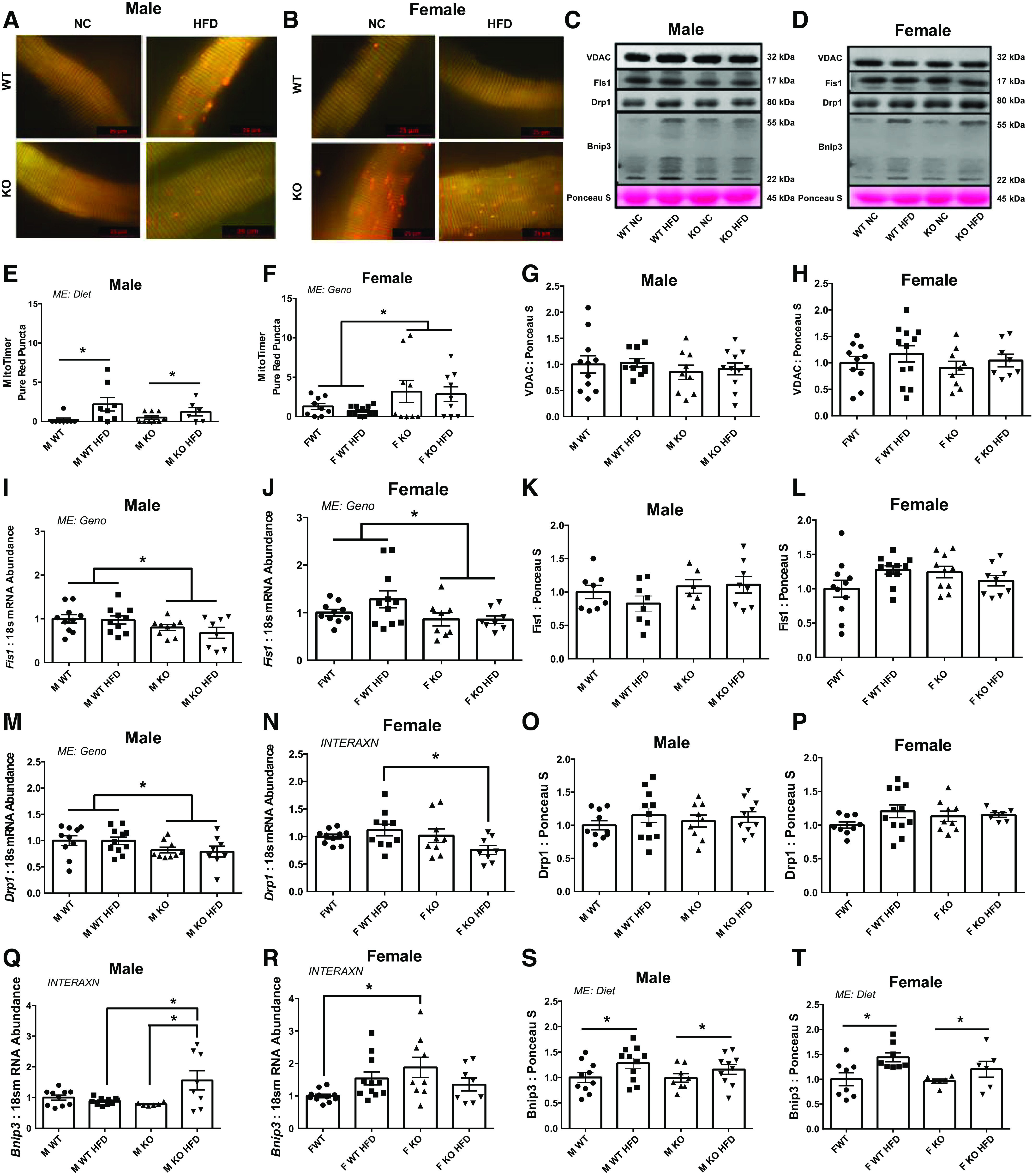
Mitochondrial network health. A and B: representative images of pMitoTimer in single muscle fiber. C and D: representative Western blot images for voltage-dependent anion channel (VDAC), fission protein 1 (Fis1), dynamin-related protein 1 (Drp1), and Bcl2 interacting protein 3 (Bnip3). E and F: pMitoTimer pure red puncta level. G and H: protein content of VDAC via immunoblotting. mRNA (I and J) and Protein content (K and L) for Fis1. mRNA (M and N) and protein content (O and P) for Drp1. mRNA (Q and R) and protein content (S and T) for Bnip3. Data analyzed by two-way ANOVA with Tukey’s post hoc test where appropriate. Data described as means ± SE. Statistical significance identified by denoting as * for main effect of genotype or diet, and as “different letters” for an interaction effect with α value set at P < 0.05 (An n of 7–11 animals per group was used). HFD, high-fat diet; INTERAXN, interaction effect; KO, knockout; ME, main effect of indicated factor; WT, wild type.
miR-16 Deletion Results in Protein Hyperanabolism in Female Mice
Based on our prior in vitro evidence for miR-16 regulation of protein anabolism, we next sought to determine impacts of miR-16 on protein anabolism in vivo, we assessed FSR via 24 h incorporation of amino acids and mRNA/protein markers for mTORC1 downstream and mTORC1 repressors. Although FSR was not statistically different in male mice, we observed a main effect of genotype in females, with female KO mice having ∼33% greater FSR compared with WT animals (P = 0.024; Fig. 5, A and B). Phosphorylation of 4EBP1 (Thr37/42)/4EBP1 did not differ among experimental groups in both sexes (Fig. 5, C and D). In males, a main effect of genotype was noted in Deptor such that KO mice had ∼22% (P = 0.017) lower Deptor mRNA content compared with WT animals (Fig. 5E). In females, there was an interaction in Deptor, with KO HFD animals having ∼33% lower Deptor mRNA content compared with WT HFD and KO NC animals (P = 0.001; Fig. 5F). In males, there was an interaction in Deptor protein, with KO NC animals having ∼23% higher compared with WT NC (P = 0.012; Fig. 5G) although there was no main effect of Deptor protein content in females (Fig. 5H). Although Redd1 mRNA content was not different among experimental groups in males, ∼69% higher Redd1 mRNA content was observed in female HFD compared with NC mice (P = 0.045; Fig. 5, I and J). Protein content of p70S6K1 did not differ among experimental groups in both sexes (Fig. 5, K and L).
Figure 5.
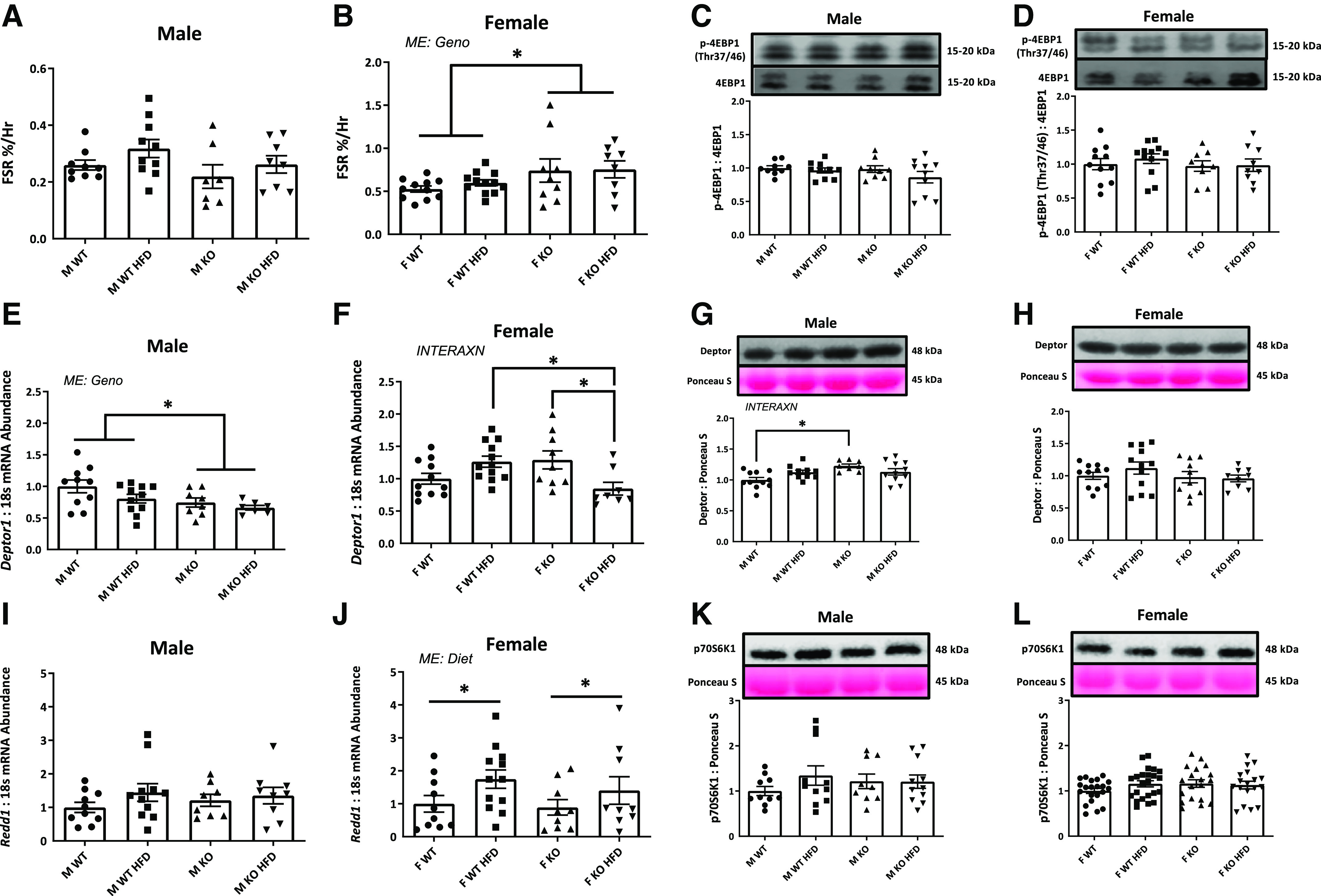
Protein anabolism. A and B: mixed muscle fractional synthesis rate (FSR) via 24 h of amino acid incorporation. C and D: protein content for p-4EBP1/4EBP1. mRNA (E and F) and protein content (G and H) of Deptor. I and J: mRNA content of Redd1. K and L: protein content for p70S6K1. Data analyzed by two-way ANOVA with Tukey’s post hoc test where appropriate. Data described as means ± SE. Statistical significance identified by denoting as * for main effect of genotype or diet, and as “different letters” for an interaction effect with α value set at P < 0.05 (An n of 7–12 animals per group was used). HFD, high-fat diet; INTERAXN, interaction effect; KO, knockout; ME, main effect of indicated factor; WT, wild type.
Primary miR-16 Content Is Not Lower in Either In Vivo or In Vitro Models of Insulin Resistance
Finally, since lower mature miR-16 content in muscle has been consistently noted in human, rodent, and in vitro models of T2DM (16, 17), we asked if primary miR-16 content is similarly lower in insulin-resistant condition in both in vitro and in vivo to better understand the mechanism of lowered mature miR-16 in insulin-resistant states. Primary miR-16 content in OAG-induced myotubes was not different from control vehicle in vitro (Fig. 6A) and was ∼31% higher in HFD-induced obese mice (P = 0.033; Fig. 6B).
Figure 6.

Pri-miR16 level in both in vitro and in vivo. A: pri-miR16 content between control and 1-oleoyl-2-acetyl-sn-glycerol (OAG)-induced insulin resistance. B: pri-miR16 content between control and diet-induced obese mice. Data described as means ± SE. Statistical differences are denoted as *P < 0.05 (Student’s t test) (An n of 6–10 replicates/animals as appropriate per condition was used).
DISCUSSION
In the present study, we initially sought to determine if lowered miR-16 content in skeletal muscle is sufficient to induce insulin resistance, we found knockdown of miR-16 is sufficient to induce insulin resistance in vitro; however, overexpression is not able to protect from insulin resistance in vitro. We then investigated how deletion of miR-16 affects metabolic and contractile properties in vivo using skeletal muscle-specific deletion of miR-16 with concurrent HFD-induced insulin resistance in both biological sexes. We observed that miR-16 KO induced impaired metabolic and contractile properties including insulin sensitivity, muscle contractility, calcium signaling, protein anabolism, and mitochondrial network health in a biological sex-dependent manner.
First, in vitro data revealed impaired insulin sensitivity in myotubes following miR-16 inhibition, consistent with lowered muscle miR-16 in insulin-resistant models (16, 17, 38). However, when we overexpressed miR-16, it did not rescue the dampened insulin action induced via lipid overload. A prior study demonstrated improved glucose uptake level in response to the overexpression of miR-16 in myocytes (38). The disparity between the two studies may be due to the usage of different cell lines and different lipid overload method. In fact, while we tested glucose uptake in response to miR-16 overexpression in C2C12 derived from mouse muscle and used OAG to induce insulin resistance in vitro, Talari et al. (38) assessed insulin-stimulated glucose uptake in response to miR-16 overexpression in L6 myocytes derived from rat muscle and used palmitate. Therefore, based on our in vitro findings reduced muscle miR-16 may be sufficient to induce insulin resistance and thus a likely mechanism of this condition; however, rescue of miR-16 may not be a directly viable target to treat this condition.
HFD has been used commonly in preclinical animal studies to induce obesity and diabetes (23, 39) and here we also found increased body and fat tissue weights in both sexes (Tables 1 and 2). Although miR-16 deletion did not affect changes in tissue weights in males, female KO mice revealed lower muscle weights compared with WT counterparts. Moreover, we observed compromised glucose tolerance in female KO mice while HFD induced glucose intolerance in males. These glucose intolerance effects corresponded to insulin-stimulated signaling. Although male HFD mice exhibited impaired insulin signaling, this impairment was observed in female KO mice. We then sought to determine insulin response via insulin-stimulated blood glucose clearance. Interestingly, male KO mice revealed impaired insulin-stimulated glucose clearance while females were unaffected (Fig. 2). Our data suggest male mice are more vulnerable to HFD-induced glucose intolerance and insulin signaling compared with female counterparts, in line with prior evidence that females are generally protected from diet-induced metabolic disorders (40) and more sensitive to insulin-mediated insulin signaling than males (41, 42). Moreover, females exhibited unchanged insulin-mediated glucose disposal while insulin signaling (p-Akt/Akt) was impaired in female KO mice. However, signaling downstream of Akt (p-AS160/AS160) was unchanged in females, suggesting impairment of p-Akt per se may not be responsible for insulin-mediated glucose clearance in female KO mice. Overall, there was a dichotomy whereby miR-16 deletion induces insulin resistance in male mice but glucose intolerance in female mice.
Impaired muscle and physical function in combination with glucose intolerance are commonly observed in T2DM (43), often leading to impaired quality of life and increased mortality (44). Here, we observed compromised muscle contractility in both sexes following miR-16 deletion. In fact, although KO induced delayed one-half relaxation times in males, miR-16 deletion induced greater fatigability and slower one-half relaxation time specifically following HFD in female KO mice. Overall muscle contractility may be more compromised in female KO mice compared with males. This may be due to lower muscle mass in female KO mice. Indeed, female KO mice had ∼6%–8% smaller TA and plantaris muscle weights compared with WT. Especially, TA muscle is the major muscle to contribute to peak-isometric torque of the ankle dorsiflexors (as measured through our approach) (45), therefore, lower muscle weight may partially account for compromised muscle contractility in female KO mice. Moreover, delayed one-half relaxation times in both sexes could be partially explained by altered calcium handling during muscle contraction. Calcium is essential during muscle contraction triggering it by allowing the interaction between actin and myosin (46). Normally, calcium is stored in the sarcoplasmic reticulum (SR) at rest and released during muscle contraction through Ryanodine receptor, and restored through SR calcium-pump or SR Ca2+-ATPase (SERCA) pump (47). In the current study, higher SERCA protein levels in male KO mice and lower Ryr receptor mRNA content was observed in female KO HFD compared with KO NC. Elevated SERCA protein in muscle of male KO mice is likely a compensation for impaired calcium reuptake in these mice. However, this interpretation is speculative and should be further tested directly. Since both calcium transport and the cross-bridge muscle contraction cycle require ATP hydrolysis (48), it is plausible that impaired muscle contractility may partially be due to the alteration of the ATP energy system in miR-16 KO mice.
Considering female miR-16 KO mice displayed greater fatigability and that mitochondrial dysfunction has been well established in skeletal muscle during T2DM (49), we next examined muscle mitochondrial network health (23, 25). Previously, Laker et al. (20) observed greater mitochondrial degeneration in mice fed with HFD. Here, we observed increased pure red puncta (an indicator for mitochondria-targeted for removal from the cell via mitophagy) induced by HFD in male mice, whereas female mice exhibited this increase specifically in miR16 KO mice. Considering the connection of pMitoTimer pure red puncta with mitophagy and mitochondrial fission (20), we assessed mRNA and protein contents of mitophagy marker Bnip3 and mitochondrial fission markers Fis1, and Drp1. Consistent with other literature (50), BNIP3 protein suggested the indication of high-fat feeding induced mitophagy though this effect was not impacted by miR16 deletion regardless of biological sex. Taken together, although these data suggest HFD was responsible for elevated mitophagy in protein levels of both sexes, mRNA levels of mitochondrial fission and mitophagy were disrupted by miR-16 KO in both sexes. It is feasible that these disrupted mRNA targets by miR-16 KO may account for additional aberrance in mitochondrial quality control. Though it is worth noting that both males and females have a drop in markers of mitochondrial fission and no change in mitochondrial content (VDAC) was observed. These results appear to imply a discoordination of mitochondrial turnover with miR16 KO regardless of sex. Collectively, these data suggest miR-16 may in part function in regulation of mitochondrial network health that is likely mediated through impacts on mitophagy and mitochondrial fission. However, these data should be interpreted with caution and require further validation to fully define the role of miR-16 in the regulation of mitochondrial quality.
Mitochondrial degeneration has been associated with altered protein anabolism (51, 52), often seen in T2DM (53, 54). Specifically, the obese Zucker rat, a common model of genetic obesity and T2DM, has an inherently greater basal protein anabolism (32, 55). While concurrently, we have observed lowered muscle miR-16 content in the obese Zucker rat (17). Furthermore, in that same study we noted direct regulation of muscle protein turnover by miR-16 in vitro (17). Specifically, inhibition of miR-16 was sufficient to elevate protein anabolism and its signaling markers including mTOR and p70S6K1 while inhibiting autophagy. Based on our prior in vitro data suggesting miR-16 regulation of protein anabolism (17) and to further determine protein anabolic regulation in vivo, we set to determine protein anabolism in miR-16 KO mice. In the current study, female KO mice exhibited elevated protein anabolism measured via D2O labeling-based FSR (24 h amino acid labeling method allows measure of protein synthesis in a freely fed state over a complete light/dark cycle), though no differences were noted in males. To further provide insight into the mechanism of elevated protein anabolism, we assessed downstream targets for mTORC1 and its suppressors. However, higher protein FSR in females did not correspond with protein contents of mTORC1 downstream (total p70S6K or phosphorylation of 4EBP1) nor suppressor targets (Deptor and Redd1). We should note that transient phosphorylations of the mTOR signaling cascade may not be reflective of the “activity” of basal anabolic systems that can be directly measured over a full light/dark cycle and in freely fed state when using longer labeling approaches with D2O. Elevated protein anabolism in the female KO mouse corresponds to our observation of exacerbated glucose intolerance in females following miR-16 deletion. However, the mechanism of protein hyperanabolism in the female miR-16 KO mouse remains elusive. Interestingly, despite elevated basal protein anabolism, many muscles appear smaller in female mice following miR-16 deletion. These observations of basal hyperanabolism with smaller muscle weights correspond to prior observations in the obese Zucker rat (32, 55). These data suggest: 1) protein hyperanabolism mediated by reduced miR-16 may be a direct mechanism of glucose intolerance and 2) this hyperanabolism is likely counteracted by hypercatabolic muscle protein metabolism leading to lowered or unchanged muscle size.
Finally, we (17) and others (16, 38) previously observed consistent downregulation of mature miR-16 in skeletal muscle across cell culture, animal, and human models. Therefore, to gain some insight as to how mature miR-16 is lowered in these models, we assessed primary miR-16 (pri-miR16). Accordingly, we examined the pri-miR16 in both insulin resistant myotubes and HFD-induced obese animals, using the same samples as in Lee et al. (17) to determine if they express lower pri-miR16 content in insulin resistance. By doing so, this investigation allowed us to examine whether the reduction of miR-16 was due to lower production of miR-16. Our data indicate that pri-miR16 content was not lowered in either insulin-resistant muscle cells or muscle samples of HFD-induced obese mice compared with healthy controls, suggesting that lowered mature miR-16 in insulin-resistant muscle is likely the product of either export or degradation as we do not see any suggestion of altered transcription of the pri-miR form. However, pri-miR16 content from HFD-induced obese animals was derived from male animals and this aspect has yet to be tested in females. Further studies are warranted to assess these two potential fates of mature miR-16 in both sexes as the mechanism of miR-16 reduction in T2DM.
Interestingly, the present data clearly present a dichotomy in biological sex. miR-16 KO resulted in impaired muscle contractility and regulation of calcium handling, and upregulated mitophagy in both sexes. However, although male mice specifically exhibited impaired insulin sensitivity following miR-16 deletion, female KO mice demonstrate impaired glucose tolerance that corresponded with lower muscle weight, and muscle protein hyperanabolism. So, while females are relatively protected from HFD-induced metabolic dysregulation, females become more susceptible to metabolic dysregulation and contractile dysfunction when mature muscle miR-16 is reduced. Although further investigations are required to correspond to clinical application, these outcomes may indicate that changes in miR-16 in muscle could serve as a mechanism of metabolic dysregulation in a sex-dependent manner and support the use of miR-16 as a therapeutic target for metabolic disease.
We acknowledge a limitation herein is that all animals were insulin-stimulated to assess maximal insulin-stimulated signaling, this does limit interpretation for insulin response. However, our main focus was to assess insulin-mediated insulin signaling in HFD or/and KO compared with WT NC mice. In addition, this study was not statistically designed to measure sex differences for each measurement, instead we demonstrated group differences within each sex.
In conclusion, the present study demonstrated that lack of miR-16, observed in multiple models of T2DM, induced various aberrations in both metabolic and contractile properties in both sexes although more pronounced negative effects were seen in female KO mice. Interestingly, although miR-16 deletion appears to lead to insulin resistance in males (failed response during ITT), in females exacerbated glucose intolerance is observed. Furthermore, while miR-16 deletion led to various impairments in contractile function and mitochondrial regulation, it is of interest to note basal protein hyperanabolism in female miR-16 KO mice. This specific effect is similar to our prior observations in vitro regarding miR-16 function in muscle and parallels observations in the obese Zucker rat. Although questions on the role of miR-16 in metabolic disorders remain, the current study establishes the role of this miRNA in muscle metabolic and contractile function.
SUPPLEMENTAL DATA
Supplemental Figs. S1–S3: https://doi.org/10.6084/m9.figshare.16615906.
GRANTS
This study was funded by the American College of Sports Medicine Research Endowment Grant, University of Arkansas Graduate-Professional Student Congress Research Council, the Arkansas Bioscience Institute, and by the National Institute of General Medical Sciences of the National Institutes of Health under Award No. 1P20GM125503.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
D.E.L., J.D.F., and N.P.G. conceived and designed research; S.L., J.W.D., M.E.R.-C., D.E.L., F.M.d.S., A.R.C., E.R.S., and L.W.S. performed experiments; S.L., J.W.D., M.E.R.-C., D.E.L., F.M.d.S., A.R.C., E.R.S., and L.W.S. analyzed data; S.L., M.E.R.-C., T.A.W., J.D.F., and N.P.G. interpreted results of experiments; S.L. and D.E.L. prepared figures; S.L. drafted manuscript; S.L., J.W.D., M.E.R.-C., D.E.L., F.M.d.S., A.R.C., E.R.S., L.W.S., T.A.W., J.D.F., and N.P.G. edited and revised manuscript; N.P.G. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank all the various faculty, staff, and students of the Exercise Science Research Center at the University of Arkansas for their support and contributions herein. The authors specifically thank Dr. Jarrod Call for consultation in the measure of muscle contractile function and Michael Davis for contribution in tissue preparation. They also thank Dr. Kevin Murach who developed the graphical abstract for the authors using BioRender.
REFERENCES
- 1.Centers for Disease Control and Prevention. National Diabetes Statistics Report, 2020. Atlanta, GA: Centers for Disease Control and Prevention, US Department of Health and Human Services, 2020, p. 12–15. https://www.cdc.gov/diabetes/pdfs/data/statistics/national-diabetes-statistics-report.pdf. [Google Scholar]
- 2.Musi N, Fujii N, Hirshman MF, Ekberg I, Fröberg S, Ljungqvist O, Thorell A, Goodyear LJ. AMP-activated protein kinase (AMPK) is activated in muscle of subjects with type 2 diabetes during exercise. Diabetes 50: 921–927, 2001. doi: 10.2337/diabetes.50.5.921. [DOI] [PubMed] [Google Scholar]
- 3.DeFronzo RA, Tripathy D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes care 32: S157–S163, 2009. doi: 10.2337/dc09-S302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Inzucchi SE, Maggs DG, Spollett GR, Page SL, Rife FS, Walton V, Shulman GI. Efficacy and metabolic effects of metformin and troglitazone in type II diabetes mellitus. N Engl J Med 338: 867–873, 1998. doi: 10.1056/NEJM199803263381303. [DOI] [PubMed] [Google Scholar]
- 5.Joslin EP. The treatment of diabetes mellitus. Can Med Assoc J 6: 673–684, 1916. [PMC free article] [PubMed] [Google Scholar]
- 6.Bantle JP, Wylie-Rosett J, Albright AL, Apovian CM, Clark NG, Franz MJ, Hoogwerf BJ, Lichtenstein AH, Mayer-Davis E, Mooradian AD, Wheeler ML; American Diabetes Association. Nutrition recommendations and interventions for diabetes: a position statement of the American Diabetes Association. Diabetes Care 31, Suppl 1: S61–S78, 2008. doi: 10.2337/dc08-S061. [DOI] [PubMed] [Google Scholar]
- 7.Goodyear LJ, Kahn BB. Exercise, glucose transport, and insulin sensitivity. Annu Rev Med 49: 235–261, 1998. doi: 10.1146/annurev.med.49.1.235. [DOI] [PubMed] [Google Scholar]
- 8.Poy MN, Eliasson L, Krutzfeldt J, Kuwajima S, Ma X, Macdonald PE, Pfeffer S, Tuschl T, Rajewsky N, Rorsman P, Stoffel M. A pancreatic islet-specific microRNA regulates insulin secretion. Nature 432: 226–230, 2004. doi: 10.1038/nature03076. [DOI] [PubMed] [Google Scholar]
- 9.Trang P, Weidhaas JB, Slack FJ. MicroRNAs as potential cancer therapeutics. Oncogene 27: S52–S57, 2008. doi: 10.1038/onc.2009.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75: 843–854, 1993. doi: 10.1016/0092-8674(93)90529-Y. [DOI] [PubMed] [Google Scholar]
- 11.Chen J-F, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, Conlon FL, Wang D-Z. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet 38: 228–233, 2006. doi: 10.1038/ng1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu J, Li R, Workeneh B, Dong Y, Wang X, Hu Z. Transcription factor FoxO1, the dominant mediator of muscle wasting in chronic kidney disease, is inhibited by microRNA-486. Kidney Int 82: 401–411, 2012. doi: 10.1038/ki.2012.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen H, Liu T, Fu L, Zhao S, Fan B, Cao J, Li X. Identification of microRNAs involved in dexamethasone-induced muscle atrophy. Mol Cell Biochem 381: 105–113, 2013. doi: 10.1007/s11010-013-1692-9. [DOI] [PubMed] [Google Scholar]
- 14.Das S, Bedja D, Campbell N, Dunkerly B, Chenna V, Maitra A, Steenbergen C. miR-181c regulates the mitochondrial genome, bioenergetics, and propensity for heart failure in vivo. PLoS One 9: e96820, 2014. doi: 10.1371/journal.pone.0096820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lovis P, Roggli E, Laybutt DR, Gattesco S, Yang J-Y, Widmann C, Abderrahmani A, Regazzi R. Alterations in microRNA expression contribute to fatty acid-induced pancreatic β-cell dysfunction. Diabetes 57: 2728–2736, 2008. doi: 10.2337/db07-1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bork-Jensen J, Scheele C, Christophersen DV, Nilsson E, Friedrichsen M, Fernandez-Twinn DS, Grunnet LG, Litman T, Holmstrøm K, Vind B, Højlund K, Beck-Nielsen H, Wojtaszewski J, Ozanne SE, Pedersen BK, Poulsen P, Vaag A. Glucose tolerance is associated with differential expression of microRNAs in skeletal muscle: results from studies of twins with and without type 2 diabetes. Diabetologia 58: 363–373, 2015. doi: 10.1007/s00125-014-3434-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee DE, Brown JL, Rosa ME, Brown LA, Perry RA, Wiggs MP, Nilsson MI, Crouse SF, Fluckey JD, Washington TA, Greene NP. microRNA‐16 is downregulated during insulin resistance and controls skeletal muscle protein accretion. J Cell Biochem 117: 1775–1787, 2016. doi: 10.1002/jcb.25476. [DOI] [PubMed] [Google Scholar]
- 18.Froger A, Hall JE. Transformation of plasmid DNA into E. coli using the heat shock method. J Vis Exp 6: 253, 2007. doi: 10.3791/253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goodman CA, Mabrey DM, Frey JW, Miu MH, Schmidt EK, Pierre P, Hornberger TA. Novel insights into the regulation of skeletal muscle protein synthesis as revealed by a new nonradioactive in vivo technique. FASEB J 25: 1028–1039, 2011. doi: 10.1096/fj.10-168799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laker RC, Xu P, Ryall KA, Sujkowski A, Kenwood BM, Chain KH, Zhang M, Royal MA, Hoehn KL, Driscoll M, Adler PN, Wessells RJ, Saucerman JJ, Yan Z. A novel MitoTimer reporter gene for mitochondrial content, structure, stress, and damage in vivo. J Biol Chem 289: 12005–12015, 2014. doi: 10.1074/jbc.M113.530527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosa-Caldwell ME, Lim S, Haynie WS, Jansen LT, Westervelt LC, Amos MG, Washington TA, Greene NP. Altering aspects of mitochondrial quality to improve musculoskeletal outcomes in disuse atrophy. J Appl Physiol (1985) 129: 1290–1303, 2020. doi: 10.1152/japplphysiol.00407.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.White JP, Gao S, Puppa MJ, Sato S, Welle SL, Carson JA. Testosterone regulation of Akt/mTORC1/FoxO3a signaling in skeletal muscle. Mol Cell Endocrinol 365: 174–186, 2013. doi: 10.1016/j.mce.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosa-Caldwell ME, Jansen LT, Lim S, Dunlap KR, Haynie WS, Washington TA, Greene NP. Neither autophagy nor exercise training mode affect exercise-induced beneficial adaptations in high fat-fed mice. Sports Med Health Sci 2: 44–53, 2020. doi: 10.1016/j.smhs.2020.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greene NP, Lee DE, Brown JL, Rosa ME, Brown LA, Perry RA, Henry JN, Washington TA. Mitochondrial quality control, promoted by PGC‐1α, is dysregulated by Western diet‐induced obesity and partially restored by moderate physical activity in mice. Physiol Rep 3: e12470, 2015. doi: 10.14814/phy2.12470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laker RC, Lillard TS, Okutsu M, Zhang M, Hoehn KL, Connelly JJ, Yan Z. Exercise prevents maternal high-fat diet-induced hypermethylation of the Pgc-1α gene and age-dependent metabolic dysfunction in the offspring. Diabetes 63: 1605–1611, 2014. doi: 10.2337/db13-1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baltgalvis KA, Call JA, Nikas JB, Lowe DA. Effects of prednisolone on skeletal muscle contractility in mdx mice. Muscle Nerve 40: 443–454, 2009. doi: 10.1002/mus.21327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Call JA, McKeehen JN, Novotny SA, Lowe DA. Progressive resistance voluntary wheel running in the mdx mouse. Muscle Nerve 42: 871–880, 2010. doi: 10.1002/mus.21764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones LA, Hunter IW. Force sensation in isometric contractions: a relative force effect. Brain Res 244: 186–189, 1982. doi: 10.1016/0006-8993(82)90919-2. [DOI] [PubMed] [Google Scholar]
- 29.Nichenko AS, Sorensen JR, Southern WM, Qualls AE, Schifino AG, McFaline-Figueroa J, Blum JE, Tehrani KF, Yin H, Mortensen LJ, Thalacker-Mercer AE, Greising SM, Call JA. Lifelong Ulk1-mediated autophagy deficiency in muscle induces mitochondrial dysfunction and contractile weakness. Int J Mol Sci 22: 1937, 2021. doi: 10.3390/ijms22041937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gasier HG, Riechman SE, Wiggs MP, Previs SF, Fluckey JD. A comparison of 2H2O and phenylalanine flooding dose to investigate muscle protein synthesis with acute exercise in rats. Am J Physiol Endocrinol Metab 297: E252–E259, 2009. doi: 10.1152/ajpendo.90872.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hudson MB, Smuder AJ, Nelson WB, Wiggs MP, Shimkus KL, Fluckey JD, Szeto HH, Powers SK. Partial support ventilation and mitochondrial-targeted antioxidants protect against ventilator-induced decreases in diaphragm muscle protein synthesis. PLoS One 10: e0137693, 2015. doi: 10.1371/journal.pone.0137693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nilsson MI, Greene NP, Dobson JP, Wiggs MP, Gasier HG, Macias BR, Shimkus KL, Fluckey JD. Insulin resistance syndrome blunts the mitochondrial anabolic response following resistance exercise. Am J Physiol Endocrinol Metab 299: E466–E474, 2010. doi: 10.1152/ajpendo.00118.2010. [DOI] [PubMed] [Google Scholar]
- 33.Rosa-Caldwell ME, Lim S, Haynie WA, Brown JL, Deaver JW, Morena Da Silva F, Jansen LT, Lee DE, Wiggs MP, Washington TA, Greene NP. Female mice may have exacerbated catabolic signalling response compared to male mice during development and progression of disuse atrophy. J Cachexia Sarcopenia Muscle 12: 717–730, 2021. doi: 10.1002/jcsm.12693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lim S, Dunlap KR, Rosa‐Caldwell ME, Haynie WS, Jansen LT, Washington TA, Greene NP. Comparative plasma proteomics in muscle atrophy during cancer‐cachexia and disuse: the search for atrokines. Physiol Rep 8: e14608, 2020. doi: 10.14814/phy2.14608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown JL, Rosa-Caldwell ME, Lee DE, Blackwell TA, Brown LA, Perry RA, Haynie WS, Hardee JP, Carson JA, Wiggs MP, Washington TA, Greene NP. Mitochondrial degeneration precedes the development of muscle atrophy in progression of cancer cachexia in tumour‐bearing mice. J Cachexia Sarcopenia Muscle 8: 926–938, 2017. doi: 10.1002/jcsm.12232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klein U, Lia M, Crespo M, Siegel R, Shen Q, Mo T, Ambesi-Impiombato A, Califano A, Migliazza A, Bhagat G, Dalla-Favera R. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell 17: 28–40, 2010. doi: 10.1016/j.ccr.2009.11.019. [DOI] [PubMed] [Google Scholar]
- 37.Srikanthan P, Karlamangla AS. Relative muscle mass is inversely associated with insulin resistance and prediabetes. Findings from the third National Health and Nutrition Examination Survey. J Clin Endocrinol Metab 96: 2898–2903, 2011. [Erratum in J Clin Endocrinol Metab 97: 2203, 2012]. doi: 10.1210/jc.2011-0435. [DOI] [PubMed] [Google Scholar]
- 38.Talari M, Kapadia B, Kain V, Seshadri S, Prajapati B, Rajput P, Misra P, Parsa KV. MicroRNA-16 modulates macrophage polarization leading to improved insulin sensitivity in myoblasts. Biochimie 119: 16–26, 2015. doi: 10.1016/j.biochi.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 39.Hancock CR, Han D-H, Chen M, Terada S, Yasuda T, Wright DC, Holloszy JO. High-fat diets cause insulin resistance despite an increase in muscle mitochondria. Proc Natl Acad Sci USA 105: 7815–7820, 2008. doi: 10.1073/pnas.0802057105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rich-Edwards JW, Kaiser UB, Chen GL, Manson JE, Goldstein JM. Sex and gender differences research design for basic, clinical, and population studies: essentials for investigators. Endocr Rev 39: 424–439, 2018. doi: 10.1210/er.2017-00246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nuutila P, Knuuti MJ, Mäki M, Laine H, Ruotsalainen U, Teräs M, Haaparanta M, Solin O, Yki-Järvinen H. Gender and insulin sensitivity in the heart and in skeletal muscles: studies using positron emission tomography. Diabetes 44: 31–36, 1995. doi: 10.2337/diab.44.1.31. [DOI] [PubMed] [Google Scholar]
- 42.Lundsgaard A-M, Kiens B. Gender differences in skeletal muscle substrate metabolism–molecular mechanisms and insulin sensitivity. Front Endocrinol (Lausanne) 5: 195, 2014. doi: 10.3389/fendo.2014.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sayer AA, Dennison EM, Syddall HE, Gilbody HJ, Phillips DIW, Cooper C. Type 2 diabetes, muscle strength, and impaired physical function: the tip of the iceberg? Diabetes care 28: 2541–2542, 2005. doi: 10.2337/diacare.28.10.2541. [DOI] [PubMed] [Google Scholar]
- 44.Hamasaki H, Kawashima Y, Katsuyama H, Sako A, Goto A, Yanai H. Association of handgrip strength with hospitalization, cardiovascular events, and mortality in Japanese patients with type 2 diabetes. Sci Rep 7: 1–9, 2017. doi: 10.1038/s41598-017-07438-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Juneja P, Hubbard JB. Anatomy, bony pelvis and lower limb, tibialis anterior muscles. StatPearls [Internet], 2018. https://www.ncbi.nlm.nih.gov/books/NBK513304/. [PubMed] [Google Scholar]
- 46.Szent-Györgyi AG. Calcium regulation of muscle contraction. Biophys J 15: 707–723, 1975. doi: 10.1016/S0006-3495(75)85849-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Santulli G, R Marks A. Essential roles of intracellular calcium release channels in muscle, brain, metabolism, and aging. Curr Mol Pharmacol 8: 206–222, 2015. doi: 10.2174/1874467208666150507105105. [DOI] [PubMed] [Google Scholar]
- 48.Geeves MA. The dynamics of actin and myosin association and the crossbridge model of muscle contraction. Biochem J 274: 1–14, 1991. doi: 10.1042/bj2740001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 51: 2944–2950, 2002. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- 50.Ehrlicher SE, Stierwalt HD, Newsom SA, Robinson MM. Short-term high-fat feeding does not alter mitochondrial lipid respiratory capacity but triggers mitophagy response in skeletal muscle of mice. Front Endocrinol 12: 284, 2021. doi: 10.3389/fendo.2021.651211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hsu C-C, Wang C-H, Wu L-C, Hsia C-Y, Chi C-W, Yin P-H, Chang C-J, Sung M-T, Wei Y-H, Lu S-H, Lee H-C. Mitochondrial dysfunction represses HIF-1α protein synthesis through AMPK activation in human hepatoma HepG2 cells. Biochim Biophys Acta 1830: 4743–4751, 2013. doi: 10.1016/j.bbagen.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 52.Cheung M, Verity MA. Methyl mercury inhibition of synaptosome protein synthesis: role of mitochondrial dysfunction. Environ Res 24: 286–298, 1981. doi: 10.1016/0013-9351(81)90158-4. [DOI] [Google Scholar]
- 53.Flaim KE, Copenhaver ME, Jefferson LS. Effects of diabetes on protein synthesis in fast-and slow-twitch rat skeletal muscle. Am J Physiol Endocrinol Physiol 239: E88–E95, 1980. doi: 10.1152/ajpendo.1980.239.1.E88. [DOI] [PubMed] [Google Scholar]
- 54.Fedele MJ, Hernandez JM, Lang CH, Vary TC, Kimball SR, Jefferson LS, Farrell PA. Severe diabetes prohibits elevations in muscle protein synthesis after acute resistance exercise in rats. J Appl Physiol 88: 102–108, 2000. doi: 10.1152/jappl.2000.88.1.102. [DOI] [PubMed] [Google Scholar]
- 55.Nilsson MI, Dobson JP, Greene NP, Wiggs MP, Shimkus KL, Wudeck EV, Davis AR, Laureano ML, Fluckey JD. Abnormal protein turnover and anabolic resistance to exercise in sarcopenic obesity. FASEB J 27: 3905–3916, 2013. doi: 10.1096/fj.12-224006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figs. S1–S3: https://doi.org/10.6084/m9.figshare.16615906.