

Abstract
The kidney plays a critical role in fluid homeostasis, glucose control, and drug excretion. Loss of kidney function due to drug-induced nephrotoxicity affects over 20% of the adult population. The kidney proximal tubule is a complex vascularized structure that is particularly vulnerable to drug-induced nephrotoxicity. Here, we introduce a model of vascularized human kidney spheroids with integrated tissue-embedded microsensors for oxygen, glucose, lactate, and glutamine, providing real-time assessment of cellular metabolism. Our model shows that both the immunosuppressive drug cyclosporine and the anticancer drug cisplatin disrupt proximal tubule polarity at subtoxic concentrations, leading to glucose accumulation and lipotoxicity. Impeding glucose reabsorption using glucose transport inhibitors blocked cyclosporine and cisplatin toxicity by 1000- to 3-fold, respectively. Retrospective study of 247 patients who were diagnosed with kidney damage receiving cyclosporine or cisplatin in combination with the sodium-glucose cotransporter-2 (SGLT2) inhibitor empagliflozin showed significant (P < 0.001) improvement of kidney function, as well as reduction in creatinine and uric acid, markers of kidney damage. These results demonstrate the potential of sensor-integrated kidney-on-chip platforms to elucidate mechanisms of action and rapidly reformulate effective therapeutic solutions, increasing drug safety and reducing the cost of clinical and commercial failures.
INTRODUCTION
Drug-induced nephrotoxicity causes over 1.5 million adverse events annually in the United States alone, affecting about 20% of the adult population. Incidence of drug-induced nephrotoxicity among the elderly may be as high as 66% due to prescription medications related to diabetes and cardiovascular disease. Although renal impairment is often reversible, its management was estimated to cost about $3.5 billion annually (1). Drug-induced nephrotoxicity is thought to be driven by multiple mechanisms including altered intraglomerular hemodynamics (2), tubular cell toxicity, inflammation (2), crystal nephropathy (3), and thrombotic microangiopathy (4, 5). The ability to elucidate particular mechanisms of renal injury is critical for recognizing and preventing drug-induced renal impairment. This is particularly important for drugs such as cyclosporine and cisplatin designated as essential medicines by the World Health Organization (6), as drug-induced renal injury still limits their utility.
Cyclosporine A (CsA) is an immunosuppressive drug naturally produced by the fermentation of Tolypocladium inflatum (7). It is prescribed after solid organ transplantation to prevent rejection (8) and for the treatment of rheumatoid arthritis, psoriasis, and Crohn’s disease (8). However, cyclosporine use is associated with elevated concentrations of serum creatinine and uric acid and the eventual development of renal failure, limiting its usefulness (9). The pathogenesis of cyclosporine-induced renal injury is thought to be mediated by vasculopathy that restricts blood flow to the kidney (8, 10). Cyclosporine has also been shown to interact with cell membranes at low concentration, inducing proximal tubule damage through oxidative stress (11), endoplasmic reticulum stress (12), and autophagy (13). Abnormal microvacuolization is a pathological hallmark of cyclosporine-induced injury, but its mechanistic relevance is unclear (14).
Cisplatin is a chemotherapeutic drug used in the treatment of bladder, breast, lung, ovarian, cervical, and testicular cancers (15). Cisplatin-induced renal insufficiency and acute renal failure are complications that can accompany even a single dose of the drug and are noted in 30% of patients receiving therapy (16). Elevated amount of blood urea nitrogen, serum creatinine, and uric acid occurs during the week after administration, with severe nephrotoxicity appearing after repeated courses, limiting its effectiveness (15). The mechanism of cisplatin-induced renal injury is unclear but appears to be associated with abnormal vacuolization and damage of the proximal tubule (17).
Recent years have seen growing interest in kidney-on-a-chip technology (18, 19). Jang and colleagues (20) demonstrated physiological proximal tubule function on a cross-membrane chip, whereas Musah et al. (21) showed that a glomerulus chip platform could enhance the differentiation of human induced pluripotent stem cell–derived podocytes. More recently, high-throughput models of perfused proximal tubule on chip were demonstrated by Vormann et al. (22), whereas Homan et al. (23) showed that human pluripotent stem cell–derived kidney organoids showed enhanced vascularization, polarized tubular compartments, and mature gene expression under flow compared to static controls. Our work on tissue-embedded sensors promised the ability to elucidate fundamental mechanisms of toxicity: We used tissue-embedded oxygen sensors to demonstrate an N-acetyl-p-benzoquinone imine (NAPQI)–independent mechanism of acetaminophen damage on human liver-on-chip (24), and combined it with glucose and lactate sensors to show that a previously unknown metabolic shift induced by troglitazone may be responsible for its postmarket withdrawal (25). More recently, our sensor-driven metabolic flux balance analysis demonstrated that valproate causes glucose-driven lipogenesis in human liver-on-chip rather than the inhibition of β-oxidation seen in mice (26).
Here, we used a combination of vascularized proximal tubule spheroids-on-a-chip and tissue-embedded sensors to elucidate and reverse a mechanism of drug-induced nephrotoxicity. We showed that both cyclosporine and cisplatin disrupt proximal tubule polarization at concentrations 1000 times lower than the onset of damage. Real-time metabolic analysis revealed that loss of polarization leads to glucose accumulation and subsequent lipid buildup and toxicity. Blocking glucose reabsorption by empagliflozin, a sodium-glucose cotransporter-2 inhibitor (SGLT2i), blocked cyclosporine and cisplatin toxicity. Results were clinically validated in a retrospective study showing significant increase (P < 0.01) in estimated glomerular filtration rates (eGFRs) up to physiological rates and reduction in serum creatinine and uric acid (markers of kidney damage) in patients taking both drug and inhibitor (P < 0.001). Our work demonstrates the potential of sensor-integrated microphysiological platforms to elucidate mechanisms of action and rapidly reformulate effective therapeutic solutions.
RESULTS
Cyclosporine and cisplatin disrupt proximal tubule polarization
To study the effects of cyclosporine and cisplatin, we used human primary proximal tubule cells (hPTCs) in two-dimensional (2D) culture for rapid screening. Cyclosporine and cisplatin cause direct cell damage with toxic concentration-50% (TC50) values of 66 and 117 μM, respectively, concentrations 20-fold higher than their maximal physiological plasma concentration (Cmax) (Fig. 1, A and B). However, exposure to either drug at a clinically relevant concentration of 100 nM caused elevated expression of renal injury marker, kidney injury molecule-1 (KIM-1) (Fig. 1C), suggesting that underlying physiological stress is induced by low-concentration drug exposure.
Fig. 1. Physiological doses of cyclosporine and cisplatin disrupt proximal tubule polarization.
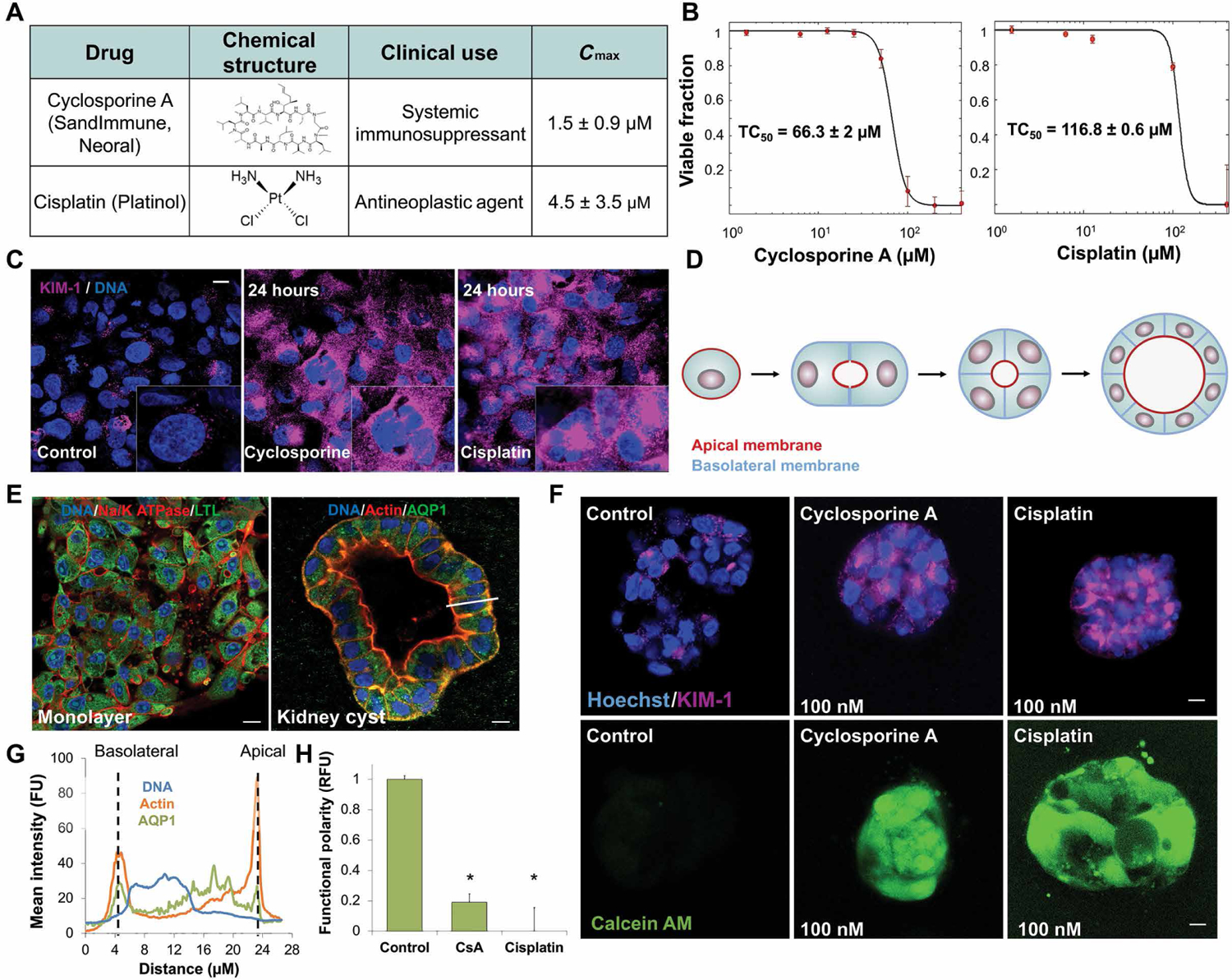
(A) Table of structure, clinical use indications, and maximal physiological concentration (Cmax) of cyclosporine and cisplatin. (B) Dose-dependent toxicity curves of primary human proximal tubule cells (hPTCs) exposed for 24 hours to cyclosporine (TC50 = 66 μM) or cisplatin (TC50 = 95 μM) in 2D cell culture. (C) Immunofluorescence staining of acute kidney injury marker KIM-1 in hPTCs after 24-hour exposure to physiological doses of cyclosporine or cisplatin. (D) Schematic of 3D cyst formation, modeling proximal tubule polarity in vitro from single- and multicell suspensions in matrix. (E) Immunofluorescence staining of hPTCs in monolayer and 3D cyst formation. White bar indicates subsequent analyzed cross section in (G). (F) Functional polarity assay in cysts exposed to minute doses of cyclosporine or cisplatin for 24 hours. Fluorescent calcein is excreted through MDR1 (P-gp) in polarized cells. (G) Mean fluorescence intensity of cross section indicated by the white bar in (E), demonstrating polarized protein expression in hPTC cysts. FU, fluorescent unit; RFU, relative fluorescent unit. (H) Quantification of the loss of functional polarity. Scale bars, 10 μm. n = 4. N = 3. *P < 0.05, Student’s t test. Error bars indicate ± SE.
To elucidate this effect, we embedded hPTCs without endothelial cells in soft basement membrane, allowing them to form polarized 3D cysts, hollow cavities with a single lumen lined with hPTCs and filled with fluid (Fig. 1, D to F), displaying apical-basal polarity (Fig. 1G). Up-regulation of acute renal injury marker KIM-1 and loss of polarized organization were detected in proximal cysts exposed to either cyclosporine or cisplatin at concentrations as low as 100 nM (Fig. 1F). To measure disruption of tubule polarization, we exposed the cysts to calcein AM, which is secreted out of polarized epithelium by multidrug resistance transporter 1 (MDR1) (fig. S1) (27). Both cyclosporine and cisplatin caused a disruption of epithelial polarity at concentrations far below Cmax (Fig. 1, F to H).
Characterization of nephrotoxicity in sensor-embedded vascularized kidney spheroids
Vascularized kidney organoids were recently shown to develop polarized tubular compartments and mature gene expression under perfusion (23). As the endothelium is thought to be important in renal toxicity (28), we formed 3D spheroids by seeding rat microvascular endothelial cells with hPTCs in microwells (Fig. 2A). Fluid shear stress in humans was estimated to range from 0.7 to 1.2 dyne/cm2 (29, 30). To mimic this environment, spheroids were exposed to shear forces of 0.75 dyne/cm2 at perfusion rates of 5 μl/min. Under physiological flow conditions, enhanced green fluorescent protein (EGFP)–labeled endothelial microvascular structures interlaced between proximal tubule-like structures, showing distinct lumen in confocal microscopy (Fig. 2A). Confocal microscopy also showed Lotus tetragonolobus lectin (LTL)–positive apical membranes indicating polarization. 3D confocal reconstruction showed unidirectional tubule structures 240 ± 40 μm in length mimicking cortical cross section of the human kidney (Fig. 2B and movie S1).
Fig. 2. Characterization of nephrotoxicity in sensor-embedded vascularized kidney spheroids.
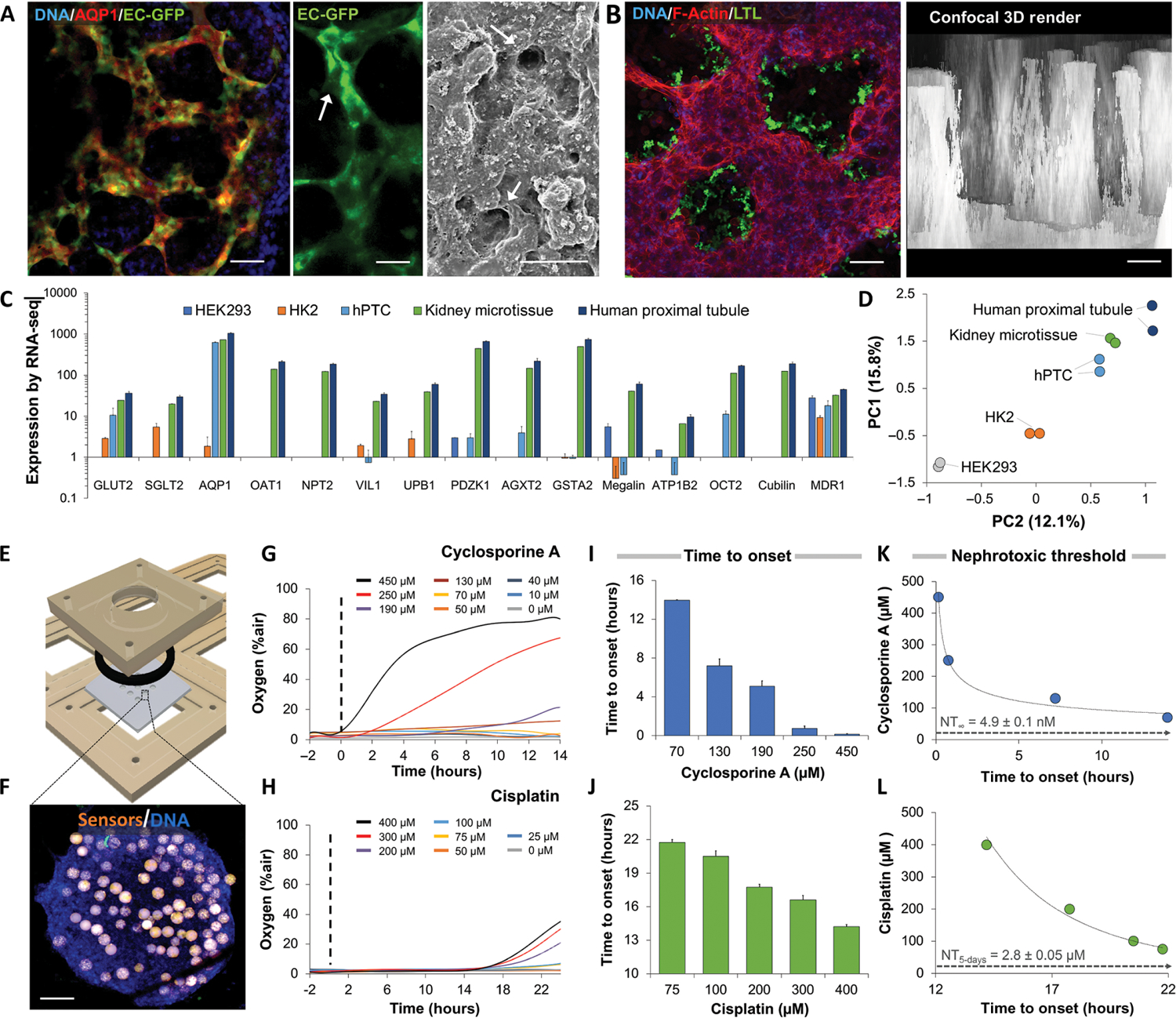
(A) Confocal microscopy of kidney spheroids composed of EGFP-labeled microvascular endothelial cells and aquaporin 1–positive (AQP1+) proximal tubule cells after 4 days in culture. Scanning electron micrograph of vascularized kidney spheroid shown on right. Arrows indicate patent endothelial lumens. Scale bars, 10 μm. (B) Confocal microscopy of proximal tubules expressing apical surface LTL in a spheroid. 3D reconstruction shows parallel longitudinal tubule-like structures 240 ± 40 μm in length. (C) Gene expression of RNA-seq data from human embryonic kidney (HEK) 293 cells, human kidney 2 (HK2) cells, and primary proximal tubule cells (hPTCs) in 2D culture compared to vascularized kidney spheroids and human proximal tubule tissue. (D) Principal component analysis (PCA) of gene expression patterns of vascularized kidney spheroids compared to HEK293, HK2, hPTCs, and proximal tubule tissue in vivo. (E) 3D design of a computer numerical control–fabricated nine-microwell bioreactor. Laser-cut disposable microwell chips containing nine spheroids are seeded in an open configuration and perfused until metabolic stabilization is achieved. (F) Fluorescent microscopy shows a human kidney spheroid with embedded oxygen sensors (orange). Scale bar, 100 μm. (G) Representative oxygen uptake over time response of kidney spheroids exposed to increasing concentrations of cyclosporine or (H) cisplatin. Dotted line notes onset of drug exposure. (I) Time to onset of response of kidney spheroids exposed to cyclosporine or (J) cisplatin. (K) Analytical derivation of nephrotoxic threshold (NT) using the time-to-onset–dependent flux accumulation equation. NT∞ was defined as the horizontal asymptote, concentration for which the onset of damage will only occur at infinite time. Cyclosporine and (L) cisplatin showed NT of 4.9 ± 0.1 nM, a 1000-fold lower than clinically reported Cmax, and 2.8 ± 0.05 μM, respectively. Scale bars, 50 μm. N = 9. N = 3. Error bars indicate ± SE. For RNA-seq analysis, P values from a negative binomial Wald test are reported.
RNA-sequencing (RNA-seq) analysis showed that whereas the gene expression of hPTCs in 2D culture was more similar to human proximal tubules than that of immortalized cell lines (Fig. 2C), vascularized spheroids more closely resembled the mature gene expression profile of the adult proximal tubule (Fig. 2, C and D). Enrichment analysis showed that both mature proximal tubule tissue and our vascularized spheroids up-regulate similar genes compared to isolated primary proximal tubule cells (table S1). Kidney spheroids embedded with oxygen sensors were placed in bioreactors under continuous, single-pass perfusion (Fig. 2, E and F). Exposure to increasing concentrations of cyclosporine and cisplatin showed a slow accumulative response, defined as an increase in interstitial oxygen, a sign of cellular damage in the spheroids (Fig. 2, G and H). Time from exposure to the onset of damage varied in a dose-dependent manner, from 4 to 14 hours in cyclosporine to 14 to 22 hours in cisplatin (Fig. 2, I and J). Extrapolating the dose-dependent time-to-onset data permits the calculation of nephrotoxic threshold (NT), an in vitro analog of no observed effect level evaluated in animals (26). NT concentrations calculated were 4.9 ± 0.1 nM for cyclosporine in chronic exposure and 2.8 ± 0.05 μM for cisplatin for 5-day exposure—a clinically relevant treatment duration for each (Fig. 2, K and L). These results suggest that the safety margins for both drugs are inadequate as both NT values are 100-fold or more below Cmax, explaining the high incidence of drug-induced nephrotoxicity.
Metabolic analysis reveals drug-induced glucose accumulation and lipogenesis
Changes in fuel utilization are sensitive markers of physiological stress (25) that can be followed in real time through the integration of embedded sensors (Fig. 3A). Here, we used an on-chip potentiostat (PSTAT) to monitor an eight-electrode array integrated in a 1-μl microchip connected to the bioreactor outflow (Fig. 3B and fig. S2). A single central processing unit (CPU) synchronizes optical and electrochemical signals permitting real-time quantification of metabolic fluxes and adenosine triphosphate (ATP) production (Fig. 3C).
Fig. 3. Metabolic analysis reveals drug-induced glucose accumulation and lipogenesis.
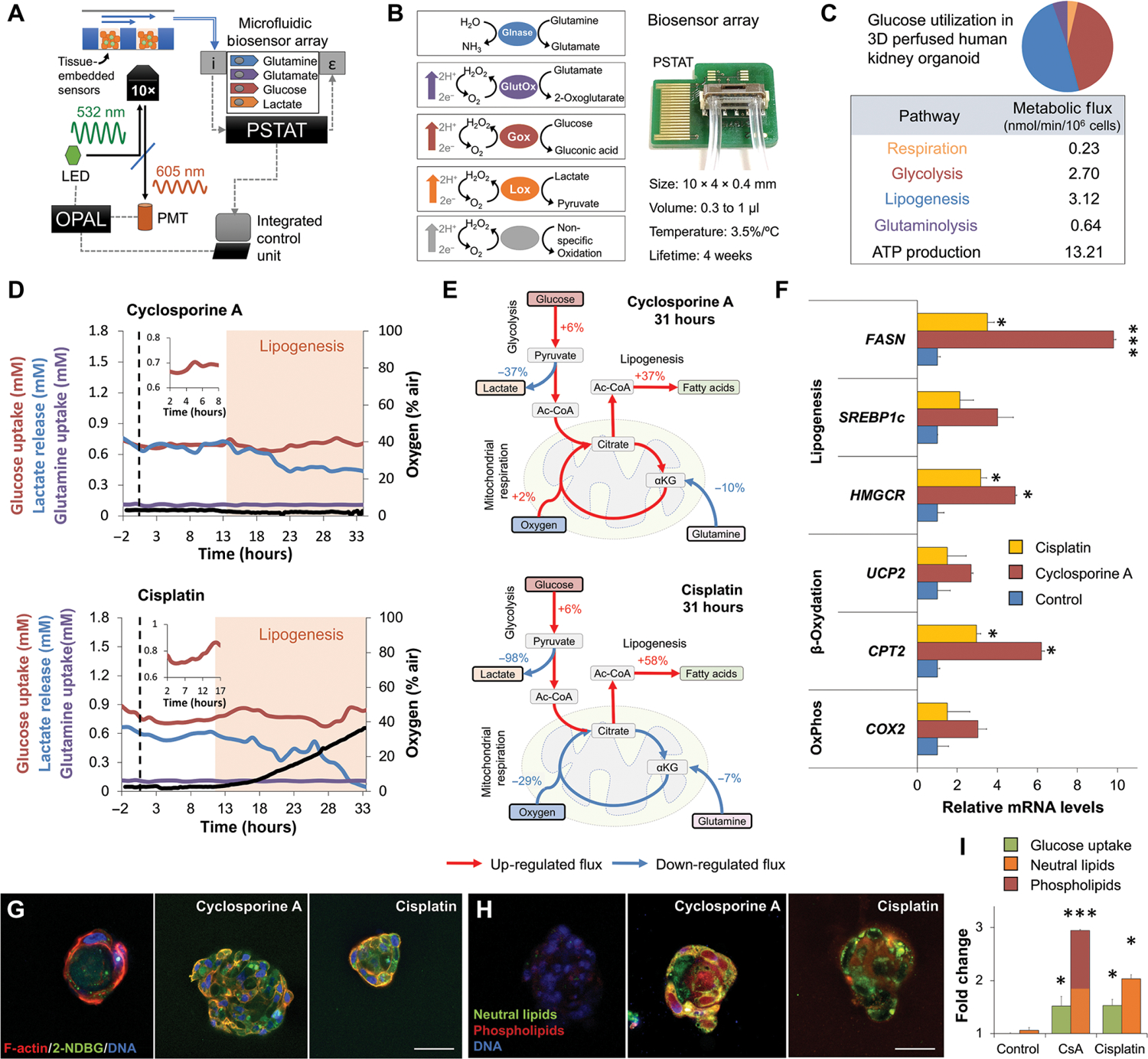
(A) Schematic of sensor-embedded spheroid-on-a-chip platform. The optical O2 measurement system (OPAL)–modulated light emitting diode (LED) signal excites tissue-embedded oxygen sensors. Phase shift is measured through a hardware-filtered photomultiplier (PMT). Chip outflow is connected to a biosensor array containing electrochemical sensors and an integrated potentiostat (PSTAT). Measurements are synchronized in real time by a single microprocessor. The arrows above the spheroids represent the direction of the flow. i, chemical input; ε, electrical output. (B) Photograph (right) and schematic (left) of microfluidic four-analyte biosensor array for glucose, lactate, glutamine, and glutamate. Glnase, glutaminase; GlutOx, l-glutamate oxidase; Gox, glucose oxidase; Lox, lactate oxidase. Array has an internal volume of 0.3 to 1 μl and integrated temperature control. (C) Intracellular metabolic fluxes of vascularized kidney spheroids in steady state. Glucose utilization and calculated ATP production are shown as nmol/min/106 cells. (D) Dynamics of oxygen, glucose, lactate, and glutamine fluxes during continuous exposure of vascularized kidney spheroids to physiological doses of cyclosporine or cisplatin under flow. n = 9. N = 3. (E) Flux balance analysis showing up-regulation of lipogenesis in kidney spheroids exposed to cyclosporine or cisplatin for 31 hours. Red and blue arrows note up- and down-regulated fluxes, respectively. (F) Relative gene expression after 48 hours of drug exposure. (G) Uptake and transport of 2-NBDG glucose analog in 3D hPTC cysts exposed to physiological concentrations of cyclosporine or cisplatin for 30 min. Student’s t test. n = 3. N = 3. (H) Fluorescence staining for neutral lipids and phospholipids in 3D cysts exposed to physiological concentrations of cyclosporine or cisplatin for 48 hours. (I) Quantification of glucose and lipid accumulation from (G) and (H). Scale bars, 25 μm. *P < 0.05, ***P < 0.001, Student’s t test. Error bars indicate ±SE. n = 4. N = 3.
Perfused spheroids were exposed to a nontoxic concentration of cyclosporine (1 μM), and their metabolism was tracked in real time (Fig. 3D and figs. S3 and S4). Metabolism was unchanged for 13 hours followed by a 6% increase in glucose uptake and 37% decrease in lactate production. Flux balance analysis demonstrated a 37% increase in lipogenesis (Fig. 3E). Metabolic analysis showed consistent 34% increase in intracellular acetyl–coenzyme A (CoA) after cyclosporine exposure, validating our flux balance (fig. S3). Exposure to a nontoxic concentration of cisplatin (1 μM) showed a similar increase in glucose uptake and a sharp 98% decline in lactate production (Fig. 3D). Flux balance analysis demonstrated a 58% increase in lipogenesis (Fig. 3E). Metabolic analysis showed a similar 15% increase in intracellular acetyl-CoA after cisplatin exposure, validating our flux balance (fig. S4). Gene expression analysis 24 hours after exposure showed the up-regulation of lipogenesis-associated genes including fatty acid synthase (FASN) and 3-hydroxy-3-methylglutaryl (HMG)–CoA reductase (HMGCR) (Fig. 3F).
To further validate the flux balance analysis calculations, we exposed proximal tubule cells to 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl) amino]-2-deoxy-d-glucose (2-NBDG), a fluorescent analog of glucose, for 30 min (Fig. 3G). Proximal cysts showed a twofold increase in glucose uptake during cyclosporine or cisplatin exposure (Fig. 3I and figs. S3 and S4). To demonstrate lipid accumulation, we stained proximal tubule cells for neutral lipid and phospholipids after cyclosporine or cisplatin exposure (Fig. 3H). Quantification showed twofold increase in neutral lipids accumulating in cisplatin-treated cells but a significant increase (P < 0.001) in phospholipids accumulating in cyclosporine-treated cells (Fig. 3I and figs. S3 and S4). Activation of β-oxidation using fenofibrate reversed this metabolic effect (figs. S3 and S4).
Cyclosporine and cisplatin toxicity reversal by SGLT2 inhibition
Empagliflozin (Jardiance) is an SGLT2 inhibitor approved for the treatment of type 2 diabetes (Fig. 4A). Exposure of kidney spheroids to increasing concentrations of cyclosporine or cisplatin in the presence of empagliflozin or a cocktail of empagliflozin and phloretin, a GLUT2 inhibitor, showed a protracted response (fig. S5). The calculated NT of cisplatin predicted not to cause damage increased by 3-fold when combined with empagliflozin above its observed Cmax. The calculated NT of cyclosporine predicted not to cause damage also increased by 1000-fold to 7.6 μM (Fig. 4B), which is higher than its observed Cmax (Fig. 1A). The proximal tubule reabsorbs glucose from the filtrate through the apical SGLT2 transporter and releases glucose to the bloodstream through the basal facilitative glucose transporter-2 (GLUT2) (Fig. 4C) (31). We have recently shown that GLUT2 is actively translocated to the basal membrane (32), suggesting that disruption of polarization would prevent proximal tubule cells from releasing intracellular glucose to the blood. Exposing proximal cysts to cyclosporine or cisplatin in the presence of 5 μM empagliflozin markedly reduced glucose accumulation (Figs. 3I and 4D and) followed by a significant decrease (P < 0.05) in lipid accumulation (Figs. 3I and 4E). These results suggest that the reformulation of cyclosporine or cisplatin with empagliflozin could markedly lower the toxicity of either compound.
Fig. 4. Cyclosporine and cisplatin toxicity are reversed by SGLT2 inhibition.
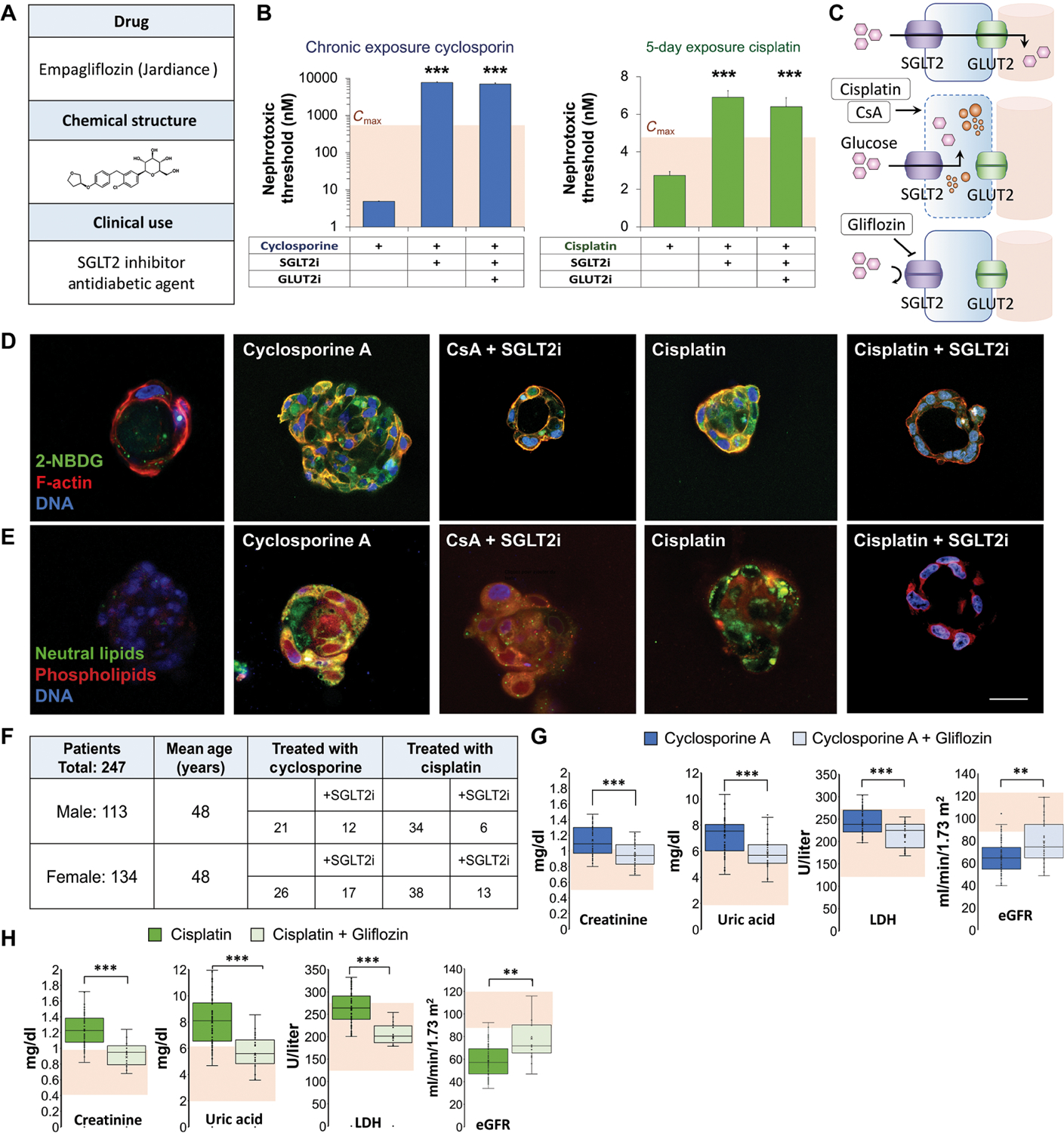
(A) Structure and clinical indication of U.S. Food and Drug Administration–approved SGLT2 inhibitor (SGLT2i) empagliflozin. (B) Quantification of NTs in vascular kidney spheroids exposed to cyclosporin or cisplatin at various concentrations (400, 200, 100, 50, 25, 12, 6.2, 3.1, and 1.5 μM) in the presence of empagliflozin (SGLT2i) or empagliflozin and phloretin, glucose transporter-2 inhibitor (GLUT2i). Student’s t test. n = 9. N = 3. (C) Schematic of glucose transport in proximal tubule cells and mechanism of nephroprotective effect of empagliflozin (gliflozin). (D) Fluorescent glucose analog (2-NBDG) accumulation in 3D cysts exposed to low concentrations (100 nM) of cyclosporine or cisplatin for 30 min in the presence or absence of empagliflozin (SGLT2i). Control, cyclosporine A, and cisplatin images are reproduced from Fig. 3G for comparison. (E) Lipid accumulation in 3D cysts exposed to cyclosporine or cisplatin for 48 hours in the presence or absence of empagliflozin. Control, cyclosporine A, and cisplatin images are reproduced from Fig. 3H for comparison. (F) Table summarizing the number of patients with kidney damage in each group of a retrospective clinical study according to their treatment. (G) Box plots showing serum creatinine, uric acid, lactate dehydrogenase (LDH) concentrations, and estimated glomerular filtration rate (eGFR) in patients treated with cyclosporine compared to those treated with both cyclosporine and empagliflozin. Shaded area indicates normal values. (H) Box plots showing serum creatinine, uric acid, LDH concentrations, and eGFR in patients treated with cisplatin compared to those treated with both cisplatin and empagliflozin. Shaded area indicates normal values. Scale bars, 25 μm. Error bars indicate ±SE. Box plot center line indicates median; the cross indicates mean; box upper and lower limits indicate third and first quartiles, respectively; whiskers indicate 1.5× interquartile range; and points are data points. **P < 0.01, ***P < 0.001, Student’s t test.
We validated our approach through a retrospective clinical study at the Hadassah Medical Center. Blood and urine analysis was conducted for 247 patients who were diagnosed with kidney damage, 113 males and 134 females (Fig. 4F). Average patient age was 48. Patients with hepatic or renal diseases were excluded from the study. Patients took either cyclosporine or cisplatin alone or in combination with an SGLT2 inhibitor. In the cyclosporine-treated patients, serum creatinine, uric acid, and lactate dehydrogenase (LDH) were elevated. eGFRs were decreased, indicating kidney damage (Fig. 4G). However, patients taking both cyclosporine and empagliflozin showed significant down-regulation (P < 0.001) of serum creatinine, uric acid, and LDH, with eGFR rising above 90 ml/min per 1.73 m2, suggesting an improvement in renal function (Fig. 4G and fig. S6). Patients taking cisplatin showed similar elevation of serum creatinine, uric acid, LDH, and lower eGFRs, whereas patients taking both cisplatin and the SGLT2 inhibitor showed much lower amounts of serum creatinine, uric acid, LDH, and higher eGFR, demonstrating an improvement in renal function (Fig. 4H and fig. S6). These results validate our kidney-on-a-chip results, strongly supporting a glucose-driven mechanism of drug-induced nephrotoxicity.
DISCUSSION
Self-assembled spheroids are an important tool for the study of human physiology (28, 33) that present critical aspects of tissue organization and function. Recent work showed that human pluripotent stem cell–derived kidney organoids show enhanced vascularization and epithelial polarization under flow (23), possibly due to endothelial-secreted hepatocyte growth factor (28, 34). Our model, composed of primary cells, demonstrates similar endothelial capillaries forming under physiological shear forces of 0.8 dyne/cm2. The vessels are interwoven around parallel tubule-like structure about 240 ± 40 μm in length, reminiscent of mature kidney organization. Genome-wide expression analysis indicated high similarity of our kidney spheroids to human proximal tubules.
The capacity of human microtissues and organ-on-chip models to predict human-specific drug response has been previously described (18, 35). However, it is the embedding of sensors in human tissue spheroids that offers a unique ability to obtain a mechanistic understanding that is not possible in animal models. Sensors enable kinetic measurements that can be used to distinguish between direct chemical effect occurring in minutes (25), transcriptional changes occurring in hours, and slow, accumulative response developing over days (26). The integration of multiple metabolic sensors allows us to balance the intracellular fluxes of central carbon metabolism, detecting minute changes in cellular energetics induced due to chemical, mechanical, or electrical damage. This metabolic analysis revealed the dysregulation of glucose absorption within 30 min of drug exposure, followed by lipogenesis over the ensuing hours.
The link between lipid accumulation and kidney injury was suggested over 160 years ago by Virchow (36) and has since been shown definitively in animal models of diabetes, metabolic syndrome (37, 38), and drug-induced injury (12, 39). Renal lipid accumulation was also demonstrated in human diabetic nephropathy (40) and is thought to be mediated by excessive lipid uptake and the suppression of peroxisome proliferator–activated receptor α (PPARα) and β-oxidation (41). Cisplatin toxicity was shown to increase lipid accumulation in the kidney due to PPARα suppression in mice (39), while Lhoták and colleagues (12) suggested that cyclosporine-induced endoplasmic reticulum stress led to lipid accumulation due to sterol regulatory element–binding protein-2 (SREBP-2) activation in mice.
Our results show dysregulation of glucose absorption occurring within 30 min of drug exposure due to disruption of epithelial polarity. Although human kidney spheroids exposed to physiological doses of cyclosporine and cisplatin do not show the down-regulation of β-oxidation observed in diabetic nephropathy, we did observe a transient induction of endoplasmic reticulum stress several hours after exposure (fig. S7). The observed dynamics suggest that glucose-driven lipogenesis is the cause of lipid accumulation in human kidneys, rather than increased lipid uptake observed in mouse models. These results underscore a fundamental difference in lipid metabolism between species. Lipid synthesis is driven primarily by glucose in human metabolism, but rodents use short-chain fatty acids instead. These results support data coming out of empagliflozin cardiovascular outcome event trial in type 2 diabetes mellitus patients (EMPA-REG OUTCOME) and canagliflozin cardiovascular assessment study (CANVAS) clinical studies, suggesting that SGLT2is slow the progression of diabetic nephropathy independently of their systemic effects (42, 43).
This previously unknown mechanistic insight of injury allowed us to suggest the combination therapy of either cyclosporine or cisplatin with SGLT2is and to validate this approach in a clinical setting. Reformulation with SGLT2is increased the nephrotoxicity threshold, defined as the lowest concentration extrapolated to cause injury for standard treatment period, of both drugs. Threshold for the new cyclosporine formulation increased to 7.6 μM, sevenfold higher than its maximal plasma concentration (Cmax). Patients receiving cyclosporine and SGLT2i showed no significant markers of kidney injury. Threshold for the new cisplatin formulation increased by 2.5-fold to 6.9 μM in a 5-day exposure regiment. Thus, patients receiving cisplatin and SGLT2i were similarly protected.
Although the results of our study are promising, our analysis was limited to the proximal tubule and clinical evidence has not address the pharmacokinetics of the proposed drug combinations or possible changes in drug efficacy. Thus, the combination of empagliflozin with cyclosporine or cisplatin should be carefully studied in a properly randomized placebo-controlled clinical study. Further work must also provide histological evidence for the reduction in kidney damage, specifically loss of vacuolization, as well as detailed analysis of the molecular mechanism of drug-induced loss of polarity (44). Our results demonstrate the potential of sensor-integrated microphysiological platforms to model human physiology for drug development applications, unraveling mechanisms of action that shed critical light on new therapeutic strategies.
MATERIALS AND METHODS
Study design
This study aimed to design a physiologically relevant human kidney-on-chip system focusing on proximal tubules to follow real-time measurement of metabolism through multiple sensor–embedded spheroids. We observed differences between a control group of cultured renal spheroids in normal conditions versus exposed to subtoxic concentrations of known nephrotoxic drugs (cyclosporine and cisplatin). We found that these drugs induced glucose-led lipotoxicity and hypothesized that reducing glucose accumulation would prevent these effects. On the basis of in vitro results, we performed a retrospective clinical study to compare markers of kidney damage in blood and urine from patients taking either cyclosporine or cisplatin alone (control groups) with patients taking either cyclosporine or cisplatin in combination with an SGLT2 inhibitor like empagliflozin. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. This study was approved by the local institutional review board (IRB) of Hadassah Medical Center Helsinki Committee (IRB approval number no. HMO 0171–20).
Renal cultures or spheroids in kidney-on-chip were randomly assigned for the various treatment groups in all the assays. The investigators were not blinded on any of the reported experiments. No sample size calculations were performed. Statistical significance was assessed using Student’s t test unless stated otherwise. No data were excluded unless obvious failure was observed from the kidney-on-chip system, bacterial infection, or hardware and software crashes that halted measurements during the experiments. Experiments involving proximal tubule cells in cultures, renal spheroids, or the kidney-on-chip system had minimum n ≥ 3 biological replicates for each group. Technical replicates were not considered as independent samples; therefore, multiple technical replicates of the same biological sample were counted as N = 1. Experiments were repeated multiple times in multiple occasions. For RNA-seq data, renal spheroid transcriptomics were derived from three samples, with each sample containing RNA from a minimum of nine spheroids.
Cell culture
Primary hPTCs were purchased from Lonza (Basel, Switzerland) and Upcyte Technologies GmbH (Hamburg, Germany). Cells were cultured in molecular, cellular, and developmental biology (MCDB) 153 basal medium (Sigma-Aldrich, USA) supplemented with 0.5% fetal bovine serum (Biological Industries, Israel), insulin-transferrin-selenium supplement (Gibco, USA), 0.1 μM dexamethasone (Sigma-Aldrich, USA), epidermal growth factor (10 ng/ml; PeproTech, USA), 5 pM triiodothyronine (Sigma-Aldrich, USA), epinephrine (0.5 μg/ml) (Sigma-Aldrich, USA), and penicillin and streptomycin (100 U/ml). Primary cells were cultured up to eight population doublings. Rat primary microvascular endothelial cells (Vec Technologies, USA) were cultured on gelatin-coated flasks using Endothelial Cell Growth Medium-2MV (Lonza, Switzerland) according to the manufacturer’s directions. All cells were cultured under standard conditions in a humidified incubator at 37°C under 5% CO2.
Quantitative RT-PCR
RNA was isolated and purified using an RNeasy Mini kit (Qiagen, Germany) according to the manufacturer’s instructions. RNA concentration and purity were determined using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific). Complementary DNA (cDNA) was synthesized from 1 μg of RNA sample using qScript cDNA SuperMix (Quanta BioSciences, USA) according to the manufacturer’s protocol. Gene expression analysis was carried out using KAPA SYBR FAST Universal 2× qPCR Master Mix (Kapa Biosystems, USA) on Applied Biosystems QuantStudio 5 System for a 384-well plate format (Applied Biosystems, USA). Gene transcription was evaluated using the ΔΔCt method normalized to 60S ribosomal protein L32 (RPL32), ubiquitin C (UBC), or β-actin. Primers are listed in table S2.
Cyst formation
Kidney cysts were formed as previously described (32). Briefly, 30 μl of growth factor–reduced Matrigel (Corning, CA) was coated on each well of black 96-well glass bottom plate (Greiner Bio-one, Austria). Primary hPTCs were mixed with ice-cold Matrigel solution at a density of 15 × 104 cells/ml and added as 70-μl cell suspension to each precoated well. Plate was incubated for 60 min at 37°C to complete polymerization before media addition. Final cell concentration was 10,500 cells per well. Cysts develop a single central lumen within 2 to 6 weeks of seeding.
Immunofluorescence microscopy
Spheroids and cysts were fixed with 4% paraformaldehyde (PFA) for 1 hour on ice and washed three times with Dulbecco’s phosphate-buffered saline (DPBS) with calcium and magnesium (Sigma-Aldrich, USA). Samples were incubated for 1 hour at room temperature with 100 mM glycine and washed for 30 min with DPBS. Permeabilization was carried out overnight at 4°C with 0.5% Triton X-100 in DPBS. Blocking buffer was composed of 3.8 g of NaCl, 0.94 g of NaHPO4, 0.2 g of NaH2PO4, 5 g of Bovine Serum Albumin Fraction V (MP Biomedicals, USA), 0.5% Triton X-100, and 0.25 ml of Tween 20 in 50 ml of distilled water. Samples were incubated in blocking buffer for 48 hours at 4°C, washed, and incubated with primary antibodies diluted in blocking buffer for additional 48 hours at 4°C. Samples were washed 24 hours at room temperature before addition of secondary antibodies diluted in blocking buffer and 48-hour incubation at 4°C. Nuclei were counterstained with Hoechst 33258 (Sigma-Aldrich, USA) at 1:1000 concentration. Samples were finally washed for 24 hours before microscopy. Confocal microscopy was performed on an LSM-700 Zeiss microscope. Antibodies and probes are listed in table S3.
Functional polarity assay
To measure epithelial polarity, cysts were incubated with 100 nM CsA (Merck, Germany), 100 nM cisplatin (Merck, Germany), and 100 μM verapamil (Merck, Germany) for positive control overnight. Cysts were incubated with 2 μM calcein AM (Molecular Probes, USA) for 1 hour at 37°C before imaging. Living cells metabolize calcein AM to green fluorescent calcein, which is excreted through apical MDR1 (P-gp) in polarized cells to the central lumen, resulting in weak central fluorescence. If the polarity is disturbed, cells will accumulate calcein and show high green fluorescence in their cytoplasm.
Bioreactor fabrication and assembly
Bioreactor manifold and disposable polydimethylsiloxane (PDMS) microwell inserts were fabricated as previously described (26). Briefly, manifold was machined from biocompatible polyetherimide blocks using Haas VF-2SSYT (Hass Automations, USA), whereas PDMS microwell inserts were cut using 355-nm pulsed Nd-YAG laser (3D-Micromac, Germany). Microwells were cut to 1.5 mm diameter and a center-to-center distance of 3 mm, followed by bonding to 0.5-mm-thick glass coverslips using oxygen plasma activation. Inserts were sterilized with 70% ethanol (EtOH) and 30-min exposure to ultraviolet light before cell seeding.
Bioreactor manifolds were housed with preseeded hPTCs and endothelial cells mixed in Matrigel at a ratio of 2:1 in microwell inserts, in which spheroids were protected from the negative effect of shear as previously described (26). Each bioreactor was perfused via 0.762-mm Tygon low-adhesion tubing (Saint-Gobain, France) at a flow rate of 5 μl/min modeled to reach shear forces of 0.75 dyne/cm2 as previously described (25).
Spheroid seeding and perfusion
Proximal tubule cells and microvascular endothelial cells were mixed at 2:1 ratio and centrifuged at 300g for 5 min. Cell pellet was mixed with ruthenium-phenanthroline–based phosphorescence dye 50-μm-diameter microbeads (CPOx-50-RuP) (Colibri Photonics, Germany) and resuspended in 100 μl of ice-cold Matrigel (Corning, CA). PDMS microwells were loaded with 1.35 μl of Matrigel suspension containing 7.5 × 104 cells and oxygen-sensing beads and incubated for 30 min at 37°C for polymerization. Culture medium was added for overnight incubation at 37°C, allowing the cells to self-assemble into spheroids 750 ± 100 μm in diameter.
Microtissue inserts were closed in the bioreactor after assembly and set in a climate control chamber of a DynamiX bioanalyzer (Tissue Dynamics, Israel). Bioreactors were continuously perfused with medium supplemented with vascular endothelial growth factor (VEGF; 5 ng/ml; PeproTech, USA) and 10 mM Hepes (Biological Industries, Israel) at a flow rate of 5 μl/min. Spheroids were monitored by continuous measurements of oxygen and metabolites, stabilizing their metabolic activity and 3D organization by day 4 of perfusion.
Real-time oxygen measurement
Real-time oxygen measurement was performed as previously described (26). Briefly, CPOx-50-RuP sensors show a decrease in phosphorescence decay time as a function of oxygen concentration. Sensors are excited by a sinusoidal amplitude–modulated 532-nm light emitting diode (LED) source and emit a sinusoidal amplitude–modulated light at 605 nm that is shifted in phase due to oxygen quenching. Background interference was filtered using 53.5- and 31.3-kHz two-frequency phase modulation. Measurements were carried out by averaging five consecutive 4-s exposures. Measurements were taken every 15 min. Under similar conditions, 28 days of measurement of spheroids was carried out with no phototoxicity, signal drift, or loss of signal intensity. Raw data can be found in data file S1.
Assessment of cellular toxicity and time to onset
Bioreactors were perfused with different concentrations of compounds in a single pass (either with CsA at concentrations of 450, 250, 190, 130, 70, 50, 40, 10, and 0 μM or with cisplatin at concentrations of 400, 300, 200, 100, 75, 50, 25, and 0 μM), saturating non-specific absorption and reaching constant exposure as previously described (25). Cell viability was determined by oxygen uptake after 24 hours of exposure unless otherwise noted. TC50 concentrations were determined using MATLAB by sigmoidal curve fitting. All error bars indicate ±95% confidence range. Time to onset was analyzed by MATLAB based on low-pass filter and trend assessment.
Real-time glucose, lactate, glutamine, and glutamate measurements
Amperometric four-analyte sensors (B.LV5) were purchased from Innovative Sensor Technology (IST; Switzerland). The sensors are based on the enzymatic reactions of glucose oxidase (with a linear range of 0.05 to 30 mM), lactate oxidase (with a linear range of 0.05 to 20 mM), and glutamate oxidase (with a linear range of 0.04 to 10 mM), and glutamine is indirectly measured by a first enzymatic reaction converting glutamine to glutamate with glutaminase (with a linear range of 0.04 to 20 mM). Sensors produce H2O2 in amounts proportional to the measured metabolite, which is detected with platinum electrodes under polarized conditions. Measurements were made 8 to 24 hours before drug exposure and until respiratory response was complete and synchronized and integrated on a single CPU. Measurements were carried out and calibrated to sensitivity decrease by on-chip potentiostat (IST, Switzerland).
Metabolic pathways and ATP production
Oxygen, glucose, lactate, and glutamine fluxes were measured by calculating the change in metabolite concentration between the inand outflow as a function of perfusion rate and cell density as previously described (25). Here, direct glutamine measurement allowed us to calculate lipid metabolism fluxes. Oxidative phosphorylation flux was calculated by dividing the oxygen uptake rate by six. We estimated 32 ATP molecules generated by complete oxidation of glucose. Glycolysis flux was calculated by dividing lactate production rate by two, with maximal rate defined by glucose uptake rate minus the oxidative phosphorylation flux. ATP production in glycolysis was estimated to be two molecules per molecule of glucose. Glutaminolysis was calculated directly by glutamine uptake. ATP production in glutaminolysis was estimated to be three molecules per molecule of lactate generated. Excess glucose was thus directed toward citrate production and lipogenesis, because the contribution of pentose phosphate pathway in nonproliferating cells is minor (45).
Glucose accumulation assay
To monitor glucose accumulation, we used a fluorescent analog of glucose, 2-NBDG (Invitrogen, USA). After 30 min of drug exposure in DPBS containing 3 mM 2-NBDG at 37°C, cysts were fixed in 4% PFA for 20 min at room temperature. After fixation, cysts were stained with phalloidin rhodamine for actin stain and Hoechst 33258 (Sigma-Aldrich, USA) for DNA stain. Cysts were washed with PBST (DPBS + 0.1% Triton X-100) four times for 15 min before confocal microscopy. 2-NBDG is transported like glucose in proximal tubule cysts using SGLT2 and GLUT2 transporters. If transport is disturbed, 2-NBDG will accumulate in the cells, showing higher green fluorescence.
Lipid accumulation assay
Quantification of lipid accumulation was performed using the HCS LipidTOX Phospholipidosis and Steatosis Detection Kit (Thermo Fisher Scientific, USA). Briefly, proximal tubule cells were incubated with different concentrations of drugs dissolved in culture medium and 1× phospholipidosis detection reagent for 48 hours. Cells were subsequently fixed in 4% PFA and stained with 1× neutral lipid detected reagent for 45 min and counterstained with Hoechst 33258 (1 μg/ml). Staining intensity was normalized to the number of Hoechst 33258–positive nuclei.
Acetyl-CoA detection assay
Quantification of intracellular acetyl-CoA was performed using the PicoProbe Acetyl-CoA Fluorometric Assay Kit (catalog no. K317–100, BioVision, USA) according to the manufacturer’s instructions. hPTCs were incubated for 48 hours with 100 nM CsA, 100 nM cisplatin, and vehicle for control. Cells were lysed on ice, and fluorescence was measured with a plate reader (PerkinElmer, USA).
SGLT2 and GLUT2 inhibition studies
Empagliflozin (AdipoGen Life Sciences, USA), an SGLT2 inhibitor, was added to cysts or spheroids 1 hour before drug exposure at a concentration of 5 μM. Because spheroids in our setup are perfused on both apical and basal surfaces, we included an inhibitor cocktail composed of 5 μM empagliflozin and 1 mM phloretin, a GLUT2 inhibitor, as a positive control. No major difference was seen between the activity of empagliflozin and the complete inhibitor cocktail.
RNA-seq analysis
RNA-seq data for adult human renal proximal tubule were downloaded from gene set enrichment analysis (GSEA) series GSE95425 and GSE19090 along with accompanying metadata. RNA-seq data for human embryonic kidney (HEK) 293 cells were downloaded from GSEA series GSE19090 along with accompanying metadata. RNA extraction from cell cultures was performed using the RNeasy Micro kit (Qiagen, USA) according to the manufacturer’s directions. Library preparation and RNA sequencing were performed by the Hebrew University Center for Genomic Technologies. Briefly, library construction was conducted using Illumina TruSeq RNA Library Prep V2 Kit (Illumina, USA) and sequenced on Illumina NextSeq500 with single-end, 86–base pair reads using the High Output V2 Kit. Sequencing reads were mapped to the University of California Santa Cruz (UCSC) human transcriptome (genome build hg19) using Bowtie2 (46). Expression of all genes was quantified using RNA-seq by expectation-maximization (RSEM), yielding an expression matrix of inferred gene counts (47). Differential expression analysis was performed using R package DESeq2 using default parameters (48). P values from a negative binomial Wald test are reported.
Processing, analysis, and graphic display of genomic data
R studio (https://rstudio.com/) was used to perform principle components analysis (prcomp package) (49), scatter plots, and volcano plots (ggplot package) (50). Hierarchical clustering, heatmaps, correlation plots, and similarity matrices were created in Morpheus. Gene ontology enrichment analyses (fig. S1) and clustering were performed using DAVID Informatics Resources 6.7 (51). Network maps were with the McGill’s Network Analyst Tool using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (52).
Analysis of canonical splice variants
Reads were downloaded from Sequence Read Archive (SRA) (GSE147735) and filtered and trimmed to remove low-quality reads and sequencing artifacts with fastp v20 (https://github.com/OpenGene/fastp.git). Reads were pseudoaligned to the GRCh38 genecode human transcriptome (GRCh38.p13, version 32) using Kallisto version 0.46.1 (https://github.com/pachterlab/kallisto) run with the default k-mer length of 31, in single-read, single-overhang mode, with fragment mean length of 400 and 100 SDs. Differentially expressed transcripts/genes were identified using Sleuth based on a likelihood ratio test comparing the condition of interest and 100 Kallisto bootstrap samples.
Scanning electron microscopy
Spheroid samples were precoated with an Au-Pd nanolayer using a SC7640 Sputter coater. Scanning electron microscopy (SEM) imaging was performed using the FEI Sirion High Resolution Scanning Electron Microscope (Holland). Images were taken at secondary electron detection with an accelerating voltage of 5 kV, a spot size of 4.0, and at a 5.3-mm working distance using high-resolution mode. Tex SEM Laboratories-Energy Dispersive X-ray Analysis (TSL-EDAX) (EDAX, USA) system was mounted for electron backscattered diffraction (EBSD).
Retrospective clinical study
A retrospective case-control study of patients who were diagnosed with kidney damage between January 2010 and June 2019 in the Hadassah Medical Center was carried out after ethics approval. This tertiary hospital serves greater Jerusalem and comprises a heterogeneous, multicultural, and multinational population from a heavily populated urban center and from the surrounding rural areas. The study group consisted of a registry of patients between the ages of 25 and 70, treated with either cisplatin (Platinol) or cyclosporine (Neoral, Sandimmune), that were suspected or diagnosed with kidney injury. The group was divided on the basis of treatment and subcategorized by the presence or absence of treatment with SGLT2 inhibitors (gliflozins) due to a background illness. Patients with Crohn’s disease, nephrotic syndrome, or underlying liver injury were excluded from the cohort.
Statistical analysis
Experiments were repeated two or three times with triplicate samples for each experimental condition, unless stated otherwise. Data from representative experiments are presented, and similar trends were seen in multiple trials. A parametric two-tailed Student’s t test was used for calculating significant differences between two groups. All error bars represent ±SE, unless otherwise noted.
For RNA-seq analysis, P values from a negative binomial Wald test are reported. For statistical evaluation of clinical data, we used the Statistical Package for the Social Sciences (SPSS Inc., Chicago, USA) 11.0.0 program. Continuous variables were calculated using Mann-Whitney rank-sum or Student’s t test, and categorical variables were calculated using χ2 and analysis of variance (ANOVA) tests.
Supplementary Material
Acknowledgments:
We thank N. Rainy, O. Leitersdorf, P. Mednikova, and H. Ornstein.
Funding:
This work was supported by European Research Council Consolidator Grant OCLD (project no. 681870) and the gifts of the Nikoh Foundation and the Sam and Rina Frankel Foundation.
Footnotes
SUPPLEMENTARY MATERIALS
stm.sciencemag.org/cgi/content/full/13/582/eabd6299/DC1
Fig. S1. Inhibition of P-gp retains calcein AM in proximal tubule cysts.
Fig. S2. Design and sensitivity of in-line metabolic sensors.
Fig. S3. CsA induces a shift to lipogenesis.
Fig. S4. Cisplatin induces a shift to lipogenesis.
Fig. S5. Glucose inhibition shifts the onset of cellular damage.
Fig. S6. Clinical validation in human patients segmented by sex.
Fig. S7. Expression of endoplasmic reticulum stress markers after drug exposure.
Table S1. Expression analysis of vascularized kidney spheroids.
Table S2. List of qRT-PCR primers.
Table S3. List of antibodies and probes.
Data file S1. Raw data.
Movie S1. 3D reconstruction from LSM700 confocal Z-stack of a vascularized kidney spheroid labeled with AQP1.
Competing interests: Y.N. holds equity in Tissue Dynamics Ltd. Y.N., A.E., and A.C. are coinventors on international patent application number PCT/IL2020/050173, “A method for reduction of drug-induced nephrotoxicity,” associated with this study. The other authors declare that they have no competing interests.
Data and materials availability:
All data associated with this study are in the paper or the Supplementary Materials. The RNA-seq data are available in the NCBI GEO repository (accession GSE147735).
REFERENCES AND NOTES
- 1.Davis-Ajami ML, Fink JC, Wu J, Nephrotoxic medication exposure in U.S. Adults with predialysis chronic kidney disease: Health services utilization and cost outcomes. J. Manag. Care Spec. Pharm 22, 959–968 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schetz M, Dasta J, Goldstein S, Golper T, Drug-induced acute kidney injury. Curr. Opin. Crit. Care 11, 555–565 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Perazella MA, Crystal-induced acute renal failure. Am. J. Med 106, 459–465 (1999). [DOI] [PubMed] [Google Scholar]
- 4.Pisoni R, Ruggenenti P, Remuzzi G, Drug-induced thrombotic microangiopathy: Incidence, prevention and management. Drug Saf. 24, 491–501 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Medina PJ, Sipols JM, George JN, Drug-associated thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Curr. Opin. Hematol 8, 286–293 (2001). [DOI] [PubMed] [Google Scholar]
- 6.World Health Organization, WHO Model Lists of Essential Medicines (World Health Organization, 2019); http://www.who.int/medicines/publications/essentialmedicines/en/. [Google Scholar]
- 7.Yang X, Feng P, Yin Y, Bushley K, Spatafora JW, Wang C, Cyclosporine biosynthesis in Tolypocladium inflatum benefits fungal adaptation to the environment. MBio 9, e01211–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Busauschina A, Schnuelle P, van der Woude FJ, Cyclosporine nephrotoxicity. Transplant. Proc 36, S229–S233 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Johnson RJ, Nakagawa T, Jalal D, Sánchez-Lozada LG, Kang D-H, Ritz E, Uric acid and chronic kidney disease: Which is chasing which? Nephrol. Dial. Transplant 28, 2221–2228 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Humes HD, Jackson NM, O’Connor RP, Hunt DA, White MD, Pathogenetic mechanisms of nephrotoxicity: Insights into cyclosporine nephrotoxicity. Transplant. Proc 17, 51–62 (1985). [PubMed] [Google Scholar]
- 11.Moon D, Kim J, Cyclosporin A aggravates hydrogen peroxide-induced cell death in kidney proximal tubule epithelial cells. Anat. Cell Biol 52, 312–323 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lhoták S, Sood S, Brimble E, Carlisle RE, Colgan SM, Mazzetti A, Dickhout JG, Ingram AJ, Austin RC, ER stress contributes to renal proximal tubule injury by increasing SREBP-2-mediated lipid accumulation and apoptotic cell death. Am. J. Physiol. Renal Physiol 303, F266–F278 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Havasi A, Dong Z, Autophagy and tubular cell death in the kidney. Semin. Nephrol 36, 174–188 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng C-H, Shu K-H, Chang H-R, Chou M-C, Cyclosporine-induced tubular vacuolization: The role of Bip/Grp78. Nephron Exp. Nephrol 122, 1–12 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Dasari S, Tchounwou PB, Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol 740, 364–378 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller RP, Tadagavadi RK, Ramesh G, Reeves WB, Mechanisms of cisplatin nephrotoxicity. Toxins 2, 2490–2518 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Landau SI, Guo X, Velazquez H, Torres R, Olson E, Garcia-Milian R, Moeckel GW, Desir GV, Safirstein R, Regulated necrosis and failed repair in cisplatin-induced chronic kidney disease. Kidney Int. 95, 797–814 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soo JY-C, Jansen J, Masereeuw R, Little MH, Advances in predictive in vitro models of drug-induced nephrotoxicity. Nat. Rev. Nephrol 14, 378–393 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilmer MJ, Ng CP, Lanz HL, Vulto P, Suter-Dick L, Masereeuw R, Kidney-on-a-chip technology for drug-induced nephrotoxicity screening. Trends Biotechnol. 34, 156–170 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Jang K-J, Mehr AP, Hamilton GA, McPartlin LA, Chung S, Suh K-Y, Ingber DE, Human kidney proximal tubule-on-a-chip for drug transport and nephrotoxicity assessment. Integr. Biol 5, 1119–1129 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Musah S, Dimitrakakis N, Camacho DM, Church GM, Ingber DE, Directed differentiation of human induced pluripotent stem cells into mature kidney podocytes and establishment of a glomerulus chip. Nat. Protoc 13, 1662–1685 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vormann MK, Gijzen L, Hutter S, Boot L, Nicolas A, van den Heuvel A, Vriend J, Ng CP, Nieskens TTG, van Duinen V, de Wagenaar B, Masereeuw R, Suter-Dick L, Trietsch SJ, Wilmer M, Joore J, Vulto P, Lanz HL, Nephrotoxicity and kidney transport assessment on 3D perfused proximal tubules. AAPS J. 20, 90 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Homan KA, Gupta N, Kroll KT, Kolesky DB, Skylar-Scott M, Miyoshi T, Mau D, Valerius MT, Ferrante T, Bonventre JV, Lewis JA, Morizane R, Flow-enhanced vascularization and maturation of kidney organoids in vitro. Nat. Methods 16, 255–262 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prill S, Bavli D, Levy G, Ezra E, Schmälzlin E, Jaeger MS, Schwarz M, Duschl C, Cohen M, Nahmias Y, Real-time monitoring of oxygen uptake in hepatic bioreactor shows CYP450-independent mitochondrial toxicity of acetaminophen and amiodarone. Arch. Toxicol 90, 1181–1191 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Bavli D, Prill S, Ezra E, Levy G, Cohen M, Vinken M, Vanfleteren J, Jaeger M, Nahmias Y, Real-time monitoring of metabolic function in liver-on-chip microdevices tracks the dynamics of mitochondrial dysfunction. Proc. Natl. Acad. Sci. U.S.A 113, E2231–E2240 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ehrlich A, Tsytkin-Kirschenzweig S, Ioannidis K, Ayyash M, Riu A, Note R, Ouedraogo G, Vanfleteren J, Cohen M, Nahmias Y, Microphysiological flux balance platform unravels the dynamics of drug induced steatosis. Lab Chip 18, 2510–2522 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vellonen K-S, Mannermaa E, Turner H, Häkli M, Wolosin JM, Tervo T, Honkakoski P, Urtti A, Effluxing ABC transporters in human corneal epithelium. J. Pharm. Sci 99, 1087–1098 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nahmias Y, Schwartz RE, Hu W-S, Verfaillie CM, Odde DJ, Endothelium-mediated hepatocyte recruitment in the establishment of liver-like tissue in vitro. Tissue Eng. 12, 1627–1638 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Essig SM, Mittenberg W, Petersen RS, Strauman S, Cooper JT, Practices in forensic neuropsychology: Perspectives of neuropsychologists and trial attorneys. Arch. Clin. Neuropsychol 16, 271–291 (2001). [PubMed] [Google Scholar]
- 30.Hoeng J, Bovard D, Peitsch MC, Organ-on-a-Chip: Engineered Microenvironments for Safety and Efficacy Testing (Academic Press, 2019). [Google Scholar]
- 31.Ghezzi C, Loo DDF, Wright EM, Physiology of renal glucose handling via SGLT1, SGLT2 and GLUT2. Diabetologia 61, 2087–2097 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cohen M, Kitsberg D, Tsytkin S, Shulman M, Aroeti B, Nahmias Y, Live imaging of GLUT2 glucose-dependent trafficking and its inhibition in polarized epithelial cysts. Open Biol. 4, 140091 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, Clevers H, Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Santos OF, Barros EJ, Yang XM, Matsumoto K, Nakamura T, Park M, Nigam SK, Involvement of hepatocyte growth factor in kidney development. Dev. Biol 163, 525–529 (1994). [DOI] [PubMed] [Google Scholar]
- 35.Kang HM, Lim JH, Noh KH, Park D, Cho H-S, Susztak K, Jung C-R, Effective reconstruction of functional organotypic kidney spheroid for in vitro nephrotoxicity studies. Sci. Rep 9, 17610 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Virchow R, Cellular Pathology as Based Upon Physiological and Pathological Histology (J. B. Lippincott, 1863). [DOI] [PubMed] [Google Scholar]
- 37.Jaikumkao K, Pongchaidecha A, Chueakula N, Thongnak L, Wanchai K, Chatsudthipong V, Chattipakorn N, Lungkaphin A, Dapagliflozin, a sodium-glucose co-transporter-2 inhibitor, slows the progression of renal complications through the suppression of renal inflammation, endoplasmic reticulum stress and apoptosis in prediabetic rats. Diabetes Obes. Metab 20, 2617–2626 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Wang D, Luo Y, Wang X, Orlicky DJ, Myakala K, Yang P, Levi M, The sodium-glucose cotransporter 2 inhibitor dapagliflozin prevents renal and liver disease in western diet induced obesity mice. Int. J. Mol. Sci 19, 137 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li S, Nagothu K, Ranganathan G, Ali SM, Shank B, Gokden N, Ayyadevara S, Megyesi J, Olivecrona G, Chugh SS, Kersten S, Portilla D, Reduced kidney lipoprotein lipase and renal tubule triglyceride accumulation in cisplatin-mediated acute kidney injury. Am. J. Physiol. Renal. Physiol 303, F437–F448 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Herman-Edelstein M, Scherzer P, Tobar A, Levi M, Gafter U, Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J. Lipid Res 55, 561–572 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Izquierdo-Lahuerta A, Martínez-García C, Medina-Gómez G, Lipotoxicity as a trigger factor of renal disease. J. Nephrol 29, 603–610 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, Shaw W, Law G, Desai M, Matthews DR, Canagliflozin and cardiovascular and renal events in type 2 diabetes. N. Engl. J. Med 377, 644–657 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Wanner C, Inzucchi SE, Lachin JM, Fitchett D, von Eynatten M, Mattheus M, Johansen OE, Woerle HJ, Broedl UC, Zinman B, Empagliflozin and progression of kidney disease in type 2 diabetes. N. Engl. J. Med 375, 323–334 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Gu N, Park S-I, Chung H, Jin X, Lee S, Kim T-E, Possibility of pharmacokinetic drug interaction between a DPP-4 inhibitor and a SGLT2 inhibitor. Transl. Clin. Pharmacol 28, 17–33 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Levy G, Habib N, Guzzardi MA, Kitsberg D, Bomze D, Ezra E, Uygun BE, Uygun K, Trippler M, Schlaak JF, Shibolet O, Sklan EH, Cohen M, Timm J, Friedman N, Nahmias Y, Nuclear receptors control pro-viral and antiviral metabolic responses to hepatitis C virus infection. Nat. Chem. Biol 12, 1037–1045 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Langmead B, Salzberg SL, Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li B, Dewey CN, RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sigg CD, Buhmann JM, in 25th International Conference on Machine Learning (ICML) (ACM, 2008). [Google Scholar]
- 50.Wickham H, ggplot2: Elegant Graphics for Data Analysis (Springer International Publishing, ed. 2, 2016); https://www.springer.com/gp/book/9783319242750. [Google Scholar]
- 51.Huang DW, Sherman BT, Lempicki RA, Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
- 52.Xia J, Gill EE, Hancock REW, NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat. Protoc 10, 823–844 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are in the paper or the Supplementary Materials. The RNA-seq data are available in the NCBI GEO repository (accession GSE147735).