

Abstract
Herein, we define the role of ferroptosis in the pathogenesis of diabetic cardiomyopathy (DCM) by examining the expression of key regulators of ferroptosis in mice with DCM and a new ex vivo DCM model. Advanced glycation end-products (AGEs), an important pathogenic factor of DCM, were found to induce ferroptosis in engineered cardiac tissues (ECTs), as reflected through increased levels of Ptgs2 and lipid peroxides and decreased ferritin and SLC7A11 levels. Typical morphological changes of ferroptosis in cardiomyocytes were observed using transmission electron microscopy. Inhibition of ferroptosis with ferrostatin-1 and deferoxamine prevented AGE-induced ECT remodeling and dysfunction. Ferroptosis was also evidenced in the heart of type 2 diabetic mice with DCM. Inhibition of ferroptosis by liproxstatin-1 prevented the development of diastolic dysfunction at 3 months after the onset of diabetes. Nuclear factor erythroid 2-related factor 2 (NRF2) activated by sulforaphane inhibited cardiac cell ferroptosis in both AGE-treated ECTs and hearts of DCM mice by upregulating ferritin and SLC7A11 levels. The protective effect of sulforaphane on ferroptosis was AMP-activated protein kinase (AMPK)-dependent. These findings suggest that ferroptosis plays an essential role in the pathogenesis of DCM; sulforaphane prevents ferroptosis and associated pathogenesis via AMPK-mediated NRF2 activation. This suggests a feasible therapeutic approach with sulforaphane to clinically prevent ferroptosis and DCM.
KEY WORDS: Advanced glycation end-products, AMPK, Cell death, Diabetic cardiomyopathy, Engineered cardiac tissue, Ferroptosis, Lipid peroxidation, NRF2
Graphical abstract
This study suggests that ferroptosis plays an essential role in the pathogenesis of diabetic cardiomyopathy. Sulforaphane prevents ferroptosis and associated pathogenesis via AMPK-mediated NRF2 activation.
1. Introduction
The global prevalence of diabetes in 2019 was 9.3% (463 million) and is estimated to rise to 10.9% (700 million) by 20451. Diabetic cardiomyopathy (DCM), first described nearly 50 years ago2, remains the main cause of heart failure in individuals with diabetes3. Despite the exponential increase in the number of preclinical and clinical studies on DCM in the past decades, the pathogenesis of this condition has not been fully elucidated.
With the onset of diabetes, systemically increased glucose levels increase the production of advanced glycation end-products (AGEs) that accumulate in the cardiac extracellular matrix4, and metabolic shift in cardiomyocytes increases fatty acid utilization, leading to intracellular lipid accumulation and the associated lipotoxicity5. Both AGE accumulation and intracellular lipid accumulation cause the excessive generation of reactive oxygen and nitrogen species to initiate the pathogenesis of DCM6,7. On the other hand, cell death plays a key role in DCM pathogenesis8. Although four types of cell death (apoptosis, autophagy, necrosis, and other types of uncertain cell death such as ferroptosis) are potentially involved in the development of DCM, as summarized recently9, apoptotic cell death is the main type of cell death observed at the early stage of DCM pathogenesis10,11. However, whether other non-apoptotic and necrotic forms of regulated cell death also play an essential role in the development of DCM, particularly at the late stage, is largely unknown.
Ferroptosis is a non-apoptotic form of regulated cell death that is induced by overproduction of phospholipid hydroperoxides in an iron-dependent manner. It is morphologically, biochemically, and genetically distinct from apoptosis and necroptosis12,13. Ferroptosis has been implicated in the pathological processes associated with cardiac ischemia/reperfusion injury and doxorubicin-induced cardiomyopathy14,15. However, while these changes are observed in acute models of the disease, it is not known whether ferroptosis also plays a role in chronic disease-associated cardiac dysfunction, such as DCM. Increasing evidence suggests that iron overload in individuals with diabetes not only increases the risk of insulin resistance and diabetes progression but also aggravates cardiovascular complications via the Fenton reaction16, 17, 18. Therefore, we aimed to define the role of ferroptosis in DCM pathogenesis.
Transcription factor nuclear factor erythroid 2-related factor 2 (NRF2) is a crucial regulator of the antioxidant response. We have previously shown that sulforaphane (SFN), an NRF2 activator, prevents diabetes-induced oxidative stress and cardiac dysfunction19,20. AMP-activated protein kinase (AMPK), a heterotrimeric enzyme comprising one catalytic subunit (α1 or α2) and two regulatory subunits, plays a pivotal role in the regulation of various physiological and pathological cellular events, including energy metabolism and oxidative stress21. The AMPKα1 isoform is mainly expressed in non-muscle cell types, whereas the AMPKα2 isoform is dominant in the skeletal muscle and heart21,22. Therefore, the AMPKα2 subunit plays a leading role in controlling cardiac physiology22. Intriguingly, AMPK activation facilitates the nuclear accumulation of NRF223,24. We further showed that AMPKα2 is indispensable for the SFN-associated prevention of cardiomyopathy in diabetic mice, acting via the AMPK/AKT/GSK3β/NRF2 signaling pathway25,26. Of note, some of the key regulators of ferroptosis are downstream targets of NRF2, such as SLC7A11, which transports cystine, the precursor of glutathione (GSH), into the cytosol, and ferritin, which plays a central role in the iron metabolism by storing excess cellular iron and alleviating the Fenton reaction15,27, 28, 29. However, the potential mechanism of the AMPK/NRF2 pathway in regulating ferroptosis in DCM has not been explored to date.
Considering the above, this study aimed to determine whether ferroptosis participates in the pathogenesis of DCM, and if so, whether NRF2 activation could protect the heart from ferroptosis ex vivo and in vivo. We also asked whether AMPK plays a role in activating NRF2-mediated cardiac protection against ferroptosis ex vivo and in vivo. We addressed these important questions using the recently established ex vivo model of DCM induced by AGE in engineered cardiac tissue (ECT) and type 2 diabetes (T2D) mouse models. We verified the crucial role of ferroptosis in DCM. NRF2 activation by SFN inhibited ferroptosis, and AMPK was indispensable for SFN-induced prevention of ferroptosis via NRF2 activation. We believe that this study provides important clues for developing novel DCM treatments targeting ferroptosis by regulating iron hemostasis and lipid peroxidation.
2. Materials and methods
2.1. Isolation of neonatal mouse ventricular cardiac cells and ECT construction
As recently described30, heart cells were isolated from the heart of FVB pups within 3 days of birth, and ECTs were constructed30,31. ECTs were maintained in culture medium for 3–15 days, and the medium was changed every 3 days30. On Day 4, they were treated with AGEs (150 μg/mL, Sigma–Aldrich, Huntington, MA, USA) or bovine serum albumin (BSA 150 μg/mL, as a control for AGEs; Sigma–Aldrich). The AGE-containing medium was changed every 3 days, until Day 12 after the beginning of treatment. ECTs were exposed to dimethyl sulfoxide (DMSO, as a control for the following treatments) or ferrostatin-1 (Fer-1, 10 μmol/L), the iron chelator deferoxamine (DFO, 100 μmol/L), necrostatin-1s (Nec-1s, 20 μmol/L), Z-VAD-FMK (25 μmol/L), SFN (10 μmol/L), or compound C (10 μmol/L) for 2 h before AGE treatment, as required. AGEs and the other compounds were added each time the medium was changed.
2.2. ECT function classification
ECT function was digitally recorded for 15 s every day after ECT construction and then qualitatively classified, as previously described30,31. Briefly, it was classified as follows: normal function (N); increased ECT deformation during shortening with regional dyssynchrony (A); increased ECT deformation during shortening with global dyssynchrony (B); decreased beating rate and deformation with global dyssynchrony (C); or arrested beating (D).
2.3. Cardiomyocyte staining
ECTs were fixed in situ with 4% (w/v) paraformaldehyde, removed from the Tissue Train (FLEXCELL International Corporation, Burlington, MA, USA) plate, stored in phosphate-buffered saline (PBS) at 4 °C, dehydrated in a graded alcohol series, cleared with xylene, embedded in paraffin, and sectioned to 5 μm thickness, as described elsewhere30. For troponin T (TnT) staining, after antigen retrieval and a PBS wash, ECT sections were incubated with mouse anti-cTnT (a cardiomyocyte [CM] marker) for 1 h (1:500 dilution; Thermo Fisher Scientific, Carlsbad, CA, USA) and then secondary antibody for 1 h (1:1000, Abcam, Cambridge, MA, USA), and nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen). Ten fields per ECT were randomly selected, and double-stained cTnT (red) and DAPI (blue)-positive cells were counted (as cardiomyocytes) to determine the percentage of cardiomyocytes in ECTs.
2.4. Isolation of RNA from ECTs and quantitative real-time polymerase chain reaction (RT-qPCR)
Ptgs2, Nqo1, and Gapdh expression were determined using RT-qPCR, as described previously30,31. All primers were purchased from Thermo Fisher Scientific. RT-qPCR was performed in a 20 μL volume (10 μL of TaqMan Universal PCR Master Mix, 1 μL of primer, and 9 μL of cDNA) using the ABI 7300 Real-Time PCR system. Fold differences in expression between different samples were determined based on the comparative cycle time (CT), and the CT value was normalized to that of the endogenous reference gene (Gapdh).
2.5. Transmission electron microscopy
ECT samples (1 mm × 1 mm × 2 mm) were quickly removed from the culture plate and immediately fixed in 3% phosphate-glutaraldehyde. They were post-fixed, embedded, cut, and mounted at the Electron Microscopy Core Facility (Department of Anatomy, University of Louisville, KY, USA). Ultrathin sections were analyzed using a Hitachi H-7500 transmission electron microscope (Hitachi, Tokyo, Japan).
2.6. Animals
Mated FVB male and female mice, male mice with global AMPKα2 knockout (AMPKα2-KO), and the corresponding wild-type (WT) (C57BL/6J) were housed in the University of Louisville Research Resources Center (University of Louisville, KY, USA). All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Louisville and performed according to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH). Mice were kept at 22 °C with a 12-h light/dark cycle and provided free access to standard rodent chow and tap water.
T2D mice with DCM were established as characterized elsewhere25. Briefly, eight-week-old WT and AMPKα2-KO mice were fed a high-fat diet (HFD) for three months to induce insulin resistance, followed by a single intraperitoneal injection of streptozotocin (STZ) (in 0.1 mol/L of citrate acid buffer, 100 mg/kg) to induce partial insulin deficiency25,32 and hyperglycemia (fasting blood glucose ≥220 mg/dL), seven days after STZ injection. Age-matched mice were fed a normal diet (ND) for three months and used as a control. Next, T2D and control mice were injected subcutaneously with SFN (Sigma–Aldrich; 0.5 mg/kg, except for the control mice) five days a week for three months and exposed to continual HFD or ND. When SFN treatment was completed, mice were fed an HFD or ND without SFN injection for an additional three months, until sacrifice, six months after T2D onset.
For the above experiments, the mice were randomly allocated into seven groups: WT-ND + vehicle (WT-Ctrl, n = 5); WT-ND + SFN (WT-Ctrl + SFN, n = 5); WT-HFD/STZ + vehicle (WT-T2D, n = 5); WT-HFD/STZ + SFN (WT-T2D + SFN, n = 5); KO-ND + vehicle (KO-Ctrl, n = 4); KO-HFD/STZ + vehicle (KO-T2D, n = 7); or KO-HFD/STZ + SFN (KO-T2D + SFN, n = 6).
To study the effect of the ferroptosis inhibitor (liproxstatin-1) on the diabetic heart, C57BL/6J mice were housed in the Institute of Translational Medicine, the First Hospital of Jilin University (University of Jilin, Jilin, China). All animal procedures were approved by the Ethics Committee of the First Hospital of Jilin University. Mice with diabetes were established as above. Liproxstatin-1 treatment was started (10 mg/kg, i.p. injection) along with the vehicle control (1% DMSO in PBS) once a day until the end of the experiment.
2.7. Echocardiography
Transthoracic echocardiography was performed to evaluate cardiac function. In brief, mice were anesthetized with 2,2,2-tribromoethanol, placed supinely on a temperature-controlled platform, and analyzed through a high-resolution imaging system (Vevo 770; Visual Sonics, Toronto, Canada) equipped with a high-frequency ultrasound probe (RMV-707B). Ejection fraction and fractional shortening were calculated using Vevo 770 software. The E/A ratio, E/e′ ratio, and deceleration time were calculated using the Vevo 1100 system. The data were averaged over ten cardiac cycles.
2.8. Protein extraction and Western blotting
Protein extraction from ECTs or cardiac tissue and Western blotting were performed as previously described30,31. Nuclear protein was extracted using a nuclei isolation kit (NUC201, Sigma–Aldrich, Huntington, MA, USA). Antibodies against ferritin heavy chain (1:1000), KEAP1 (1:1000), nuclear NRF-2 (dilution at 1:1000), and histone H3 (1:1000) were purchased from Abcam (Cambridge, MA, USA); against β-actin (1:3000) from Santa Cruz Biotechnology (Dallas, TX, USA); and against phosphorylated (p) AMPK (Thr172, 1:1000), AMPK (1:2000), p-AKT (Ser473, Thr308, 1:1000), AKT (1:2000), p-GSK3β (Ser9, 1:500), GSK3β (1:2000), and SLC7A11 (1:500) from Cell Signaling (Danvers, MA, USA). Anti-rabbit/anti-mouse horseradish peroxidase-linked secondary antibody (1:5000) was also purchased from Cell Signaling.
2.9. Determination of lipid peroxidation
ECT and cardiac lipid peroxidation levels were examined by measuring the formation of MDA using a Lipid Peroxidation (MDA) Assay Kit (Cat#ab233471, Abcam), following the manufacturer's instructions.
2.10. Determination of labile iron levels
Labile iron levels in ECTs and the cardiac tissue were measured using an Iron Colorimetric Assay Kit (Cat#K390-100, BioVision, Milpitas, CA, USA), following the manufacturer's instructions.
2.11. Determination of GSH and oxidized GSH (GSSG) levels
ECT and cardiac GSH and GSSG levels were measured using the Glutathione Fluorometric Assay Kit (Cat#K264-100, BioVision), following the manufacturer's instructions.
2.12. Statistical analysis
Data were analyzed using GraphPad Prism 8 software (San Diego, CA, USA). Data are presented as the normalized mean ± standard deviation (SD). Statistical differences were determined using two-sided, unpaired Student's t-test or two-way analysis of variance followed by Tukey's multiple comparisons test. P-values <0.05 were considered statistically significant.
3. Results
3.1. AGEs induce ferroptosis in ECTs ex vivo
AGEs are important pathogenic factors that initiate DCM pathogenesis mainly by increasing oxidative stress levels. Therefore, we used AGE-exposed ECTs to establish a unique ex vivo DCM model to reflect specific molecular alterations, structural remodeling, and contractible dysfunction of DCM30. The concentration of AGEs used in this study was 150 μg/mL, which does not induce acute toxicity (apoptosis assessed by cleaved caspase 3 and TUNEL staining or necrosis assessed by lactate dehydrogenase in the medium), but can induce ECT remodeling and dysfunction according to our previous study30. Staining ECTs with cTnT revealed that a long-term AGE exposure led to the reduction in CM numbers, which was not rescued by the necroptosis inhibitor Nec-1s or the apoptosis pan-caspase inhibitor Z-VAD-FMK (Fig. 1A and B). However, lipid peroxidation, a hallmark of ferroptosis13,15, was elevated in AGE-treated ECTs, as reflected by the assessment of malondialdehyde (MDA) levels (Fig. 1E). AGEs also increased the expression of the ferroptosis marker gene Ptgs213,15 in ECTs, as shown in Fig. 1F. Furthermore, AGE-induced cell loss and ferroptosis marker levels (MDA and Ptgs2) were attenuated by the ferroptosis-specific inhibitor ferrostatin-1 (Fer-1) (Fig. 1C–F) and DFO (Fig. 1G–J). Of note, inhibition of ferroptosis by Fer-1 and DFO also prevented ECT dysfunction (Table 1). Finally, transmission electron microscopy analysis revealed the presence of shrunken mitochondria with enhanced membrane density, a morphologic feature of ferroptosis, in AGE-treated cardiomyocytes (Fig. 1K). Collectively, these observations suggest that ferroptosis was predominantly responsible for AGE-induced cell death under the experimental conditions tested and was an essential contributor to AGE-induced ECT dysfunction.
Figure 1.

AGEs induce ferroptosis in murine-derived ECTs. (A) and (B) Representative immunofluorescent staining of the cardiomyocytes (CMs; troponin, red) and nuclei (DAPI, blue) in ECTs following AGE treatment (40× magnification). Necroptosis inhibitor necrostatin-1s (Nec-1s) or apoptosis pan-caspase inhibitor Z-VAD-FMK was added 2 h before AGE treatment (n = 4 per group). (C) and (D) Representative immunofluorescent staining of CMs (troponin, red) and nuclei (DAPI, blue) in ECTs following AGE treatment (40× magnification). Ferrostatin-1 (Fer-1) was added 2 h before Ctrl/AGE treatment (n = 4 per group). (E) Effect of Fer-1 (added 2 h before AGE treatment) on lipid peroxidation in ECTs, assessed by observing the changes in MDA levels (n = 3 or 4 per group). (F) Effect of Fer-1 (added 2 h before AGE treatment) on Ptgs2 expression in ECTs, assessed by RT-qPCR (n = 3–4 per group). (G) and (H) Representative immunofluorescent staining of CMs (troponin, red) and nuclei (DAPI, blue) in ECTs following AGE treatment (40× magnification). Deferoxamine (DFO) was added 2 h before AGE treatment (n = 4 per group). (I) Effect of DFO (added 2 h before AGE treatment) on Ptgs2 expression in ECTs, assessed using RT-qPCR (n = 3–4 per group). (J) Effect of DFO (added 2 h before AGE treatment) on lipid peroxidation in ECTs, assessed by changes in MDA levels (n = 3–4 per group). (K) Representative transmission electron microscopy images of mitochondria in ECTs after AGE treatment. Data are presented as the normalized mean ± SD (to control) or mean ± SD. AGEs, advanced glycation end-products; ECT, engineered cardiac tissue; MDA, malondialdehyde. ns, no significant.
Table 1.
Fer-1 and DFO rescues the global function of ECTs treated with AGEs.
| ECT function | Ctrl (n = 6) |
BSA (n = 6) |
AGEs (n = 8) |
Fer-1 (n = 6) |
AGEs + Fer-1 (n = 10) |
DFO (n = 6) |
AGEs + DFO (n = 10) |
|---|---|---|---|---|---|---|---|
| N | 6 | 6 | 0 | 6 | 8 | 6 | 7 |
| A | 0 | 0 | 0 | 0 | 1 | 0 | 1 |
| B | 0 | 0 | 3 | 0 | 1 | 0 | 2 |
| C | 0 | 0 | 5 | 0 | 0 | 0 | 0 |
| D | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Rate of dysfunction | 0 | 0 | 100% | 0 | 20% | 0 | 30% |
AGEs, advanced glycation end-products; Fer-1, ferrostatin 1, inhibitor of ferroptosis; DFO, deferoxamine, iron chelator and inhibitor of ferroptosis.
Next, we sought to investigate the mechanism by which AGEs induce ferroptosis. We examined the expression of key inhibitors of ferroptosis. In terms of iron metabolism, AGE treatment transiently increased ferritin expression, which was attenuated in a time-dependent manner from 24 h to the end of culture (12 days) (Fig. 2A and B), with concomitantly increased levels of labile iron in AGE-treated ECTs (Fig. 2C). In addition to iron overload, loss of GSH is another key mechanism of ferroptosis. Accordingly, SLC7A11, required for the transport of cystine to generate intracellular GSH, was downregulated in AGE-treated ECTs (Fig. 2D). This suggests that the downregulation of SLC7A11 levels contributes to the decreased GSH levels and GSH/GSSG ratio in AGE-treated ECTs (Fig. 2E and F).
Figure 2.

AGEs induce alterations of key ferroptosis-associated molecules. (A) AGE-induced time-dependent ferritin expression, assessed using Western blotting (n = 3 per group). (B) AGE-induced expression of ferritin, assessed using Western blotting (n = 4 per group). (C) Labile iron levels in AGE-treated ECTs, assessed using an Iron Colorimetric Assay Kit (n = 4 per group). (D) AGE-induced expression of SLC7A11, assessed using Western blotting (n = 4 per group). (E) and (F) Alterations of GSH and GSH/GSSG levels, assessed using the Glutathione Fluorometric Assay Kit (n = 5 per group). Data are presented as the normalized mean ± SD (to control) or mean ± SD. AGEs, advanced glycation end-products; ECT, engineered cardiac tissue; GSH, glutathione; GSSG, oxidized glutathione; SLC7A11, solute carrier family 7 member 11.
3.2. Ferroptosis is essential for the pathogenesis of DCM in vivo
Next, we asked whether ferroptosis is involved in the pathogenesis of DCM. DCM was induced in T2D mice due to HFD and intraperitoneal injection of STZ, with reduced cardiac function, as confirmed by echocardiography25. Levels of MDA (a lipid peroxidation marker) and the expression of Ptgs2 (a ferroptosis marker) were increased in mice with DCM (Fig. 3A and B). We observed that the expression of SLC7A11, GSH levels, and GSH/GSSG ratio were reduced in mice with DCM (Fig. 3C, D and E). As described above, the ex vivo study demonstrated a transient elevation of intracellular ferritin levels, followed a decrease after 24 h in a time-dependent manner upon AGE treatment. In vivo experiments revealed that ferritin levels in diabetic heart homogenates were significantly lower than those in the hearts of healthy controls (Fig. 3C). In line with the observations for AGE-treated ECTs, labile iron levels were significantly increased in heart homogenates from diabetic mice compared with those in healthy controls (Fig. 3F). We then treated diabetic mice with a ferroptosis inhibitor (liproxstatin-1) and found that after 3 months with diabetes, the diastolic function decreased (Fig. 3G–J). The systolic function (not shown) was not significantly decreased, which is consistent with the pathogenic process of DCM. Of note, liproxstatin-1 alleviated the decrease in diastolic function at 3 months after the onset of diabetes (Fig. 3G–I). Hence, ferroptosis was involved in the pathogenesis of DCM.
Figure 3.

Ferroptosis is involved in the pathogenesis of DCM. (A) Lipid peroxidation in DCM mice, assessed by observing the changes in MDA levels (n = 5 per group). (B) Expression of Ptgs2 in mice with DCM, assessed using RT-qPCR (n = 5 per group). (C) Expression of SLC7A11 and ferritin in mice with DCM, assessed using Western blotting (n = 5 per group). (D) and (E) Alterations of GSH and GSH/GSSG levels, assessed using the Glutathione Fluorometric Assay Kit (n = 5 per group). (F) Labile iron levels in DCM mice, assessed using the Iron Colorimetric Assay Kit (n = 5 per group). (G)–(I) Diastolic function in DCM mice, assessed using ultrasonography (n = 5 per group). Data are presented as the normalized mean ± SD (to control) or mean ± SD. DCM, diabetic cardiomyopathy; GSH, glutathione; GSSG, oxidized glutathione; MDA, malondialdehyde; SLC7A11, solute carrier family 7 member 11.
3.3. NRF2 inhibits AGE-induced ferroptosis in ECTs
We have previously shown that NRF2 activation plays a pivotal role in preventing DCM by enhancing the expression of antioxidants and alleviating oxidative stress20 and that both ferritin and SLC7A11 are NRF2 targets28. Thus, we determined whether NRF2 activation rescues AGE-induced ferroptosis in ECTs. Nuclear NRF2 expression decreased in AGE-treated ECTs and mice with DCM (Supporting Information Fig. S1A and S1C), consistent with its downstream target Nqo1 mRNA (Fig. S1B and S1D). SFN, a potent agonist of NRF2, increased nuclear accumulation of NRF2, thus rescuing the decrease in nuclear NRF2 expression induced by long-term AGE exposure (Fig. 4A and B). As expected, NRF2 activation attenuated the increase in Ptgs2 expression, decreased MDA levels, allowed CM survival (Fig. 4C–F), and improved ECT function in the AGE-treated group (Table 2). We also observed that NRF2 activation upregulated levels of Nqo1 mRNA (Fig. S1E), ferritin (Fig. 4G and H), and SLC7A11 (Fig. 4G and I), attenuated the increase in labile iron levels (Fig. 4J), and increased GSH levels and the GSH/GSSG ratio (Fig. 4K and L). Taken together, these observations indicate that NRF2 activation inhibited AGE-induced ferroptosis in ECTs.
Figure 4.

NRF2 activation inhibits AGE-induced ferroptosis in ECTs. (A) and (B) Effect of sulforaphane (SFN) (added 2 h before AGE treatment) on the expression of NRF2, assessed using Western blotting (n = 4 per group). (C) Effect of SFN (added 2 h before AGE treatment) on the expression of Ptgs2 in AGE-treated ECTs, assessed using RT-qPCR (n = 3–4 per group). (D) Effect of SFN (added 2 h before AGE treatment) on the level of lipid peroxidation in AGE-treated ECTs, assessed by observing the changes in MDA levels (n = 3–4 per group). (E) and (F) Representative immunofluorescent staining of the cardiomyocytes (CMs) (troponin, red) and nuclei (DAPI, blue) in ECTs following AGE treatment (40× magnification). SFN was added 2 h before AGE treatment (n = 4 per group). (G) and (H) Effect of SFN (added 2 h before AGE treatment) on the expression of ferritin, assessed using Western blotting (n = 4 per group). (G) and (I) Effect of SFN (added 2 h before AGE treatment) on the expression of SLC7A11, assessed using Western blotting (n = 4 per group). (J) Effect of SFN (added 2 h before AGE treatment) on labile iron levels, assessed using the Iron Colorimetric Assay Kit (n = 4 per group). (K) and (L) Effect of SFN (added 2 h before AGE treatment) on GSH and GSH/GSSG levels, assessed using the Glutathione Fluorometric Assay Kit (n = 5 per group). Data are presented as the normalized mean ± SD (to control) or mean ± SD. AGEs, advanced glycation end-products; ECT, engineered cardiac tissue; GSH, glutathione; GSSG, oxidized glutathione; MDA, malondialdehyde; SLC7A11, solute carrier family 7 member 11.
Table 2.
SFN rescues the global function of ECTs treated with AGEs and the effect is AMPK-dependent.
| ECT function | Ctrl (n = 6) |
AGEs (n = 6) |
SFN (n = 6) |
AGEs + SFN (n = 8) |
Compound C (n = 6) |
AGEs + SFN + Compound C (n = 8) |
|---|---|---|---|---|---|---|
| N | 6 | 0 | 6 | 5 | 6 | 0 |
| A | 0 | 1 | 0 | 2 | 0 | 1 |
| B | 0 | 1 | 0 | 1 | 0 | 3 |
| C | 0 | 4 | 0 | 0 | 0 | 4 |
| D | 0 | 0 | 0 | 0 | 0 | 0 |
| Rate of dysfunction | 0 | 100% | 0 | 37.5% | 0 | 100% |
AGEs, advanced glycation end-products; SFN, sulforaphane, a potent agonist of NRF2; compound C, a potent inhibitor of AMPK phosphorylation.
3.4. Protective effects of SFN on AGE-induced ferroptosis in ECTs are AMPK-dependent
Emerging evidence, including our previous work, indicates that AMPK phosphorylation upregulates AKT-mediated inactivation of GSK3β and, ultimately, the activation of NRF223, 24, 25. Therefore, we next examined the role of AMPK in the protective effects of NRF2 in AGE-induced ferroptosis. We found that AGE-induced downregulation of p-AMPK was rescued by SFN (Fig. 5A and B). The addition of compound C to inhibit AMPK activation (Fig. 5C and D) abolished AKT/GSK3β-mediated NRF2 activation by SFN (Fig. 5E, F and Supporting Information Fig. S2A–S2C). The expression of KEAP1, a regulator of NRF2 activity33, however, remained unchanged in this model (Fig. S2D). Consequently, AMPK inactivation also abolished the protective effects of SFN in attenuating ferroptosis markers (MDA and Ptgs2) (Fig. 5G and H), increasing the expression of Nqo1 mRNA (Fig. S1F), SLC7A11, and ferritin, GSH levels, and the GSH/GSSG ratio, while attenuating the increase in free iron levels and rescuing the CM survival and ECT function in AGE-treated ECTs (Fig. 6 and Table 2). Therefore, the protective effects of SFN, exerted via NRF2 activation, on AGE-induced ferroptosis ex vivo were AMPK-dependent.
Figure 5.

Protective effects of SFN against AGE-induced ferroptosis in ECTs are AMPK-dependent. (A) and (B) Effect of sulforaphane (SFN, added 2 h before AGE treatment) on the expression of p-AMPK and AMPK, assessed using Western blotting (n = 4 per group). (C) and (D) Effect of SFN and compound C (added 2 h before AGE treatment) on the expression of p-AMPK and AMPK, assessed using Western blotting (n = 4 per group). (E) and (F) Effect of SFN and compound C (added 2 h before AGE treatment) on the expression of NRF2, assessed using Western blotting (n = 4 per group). (G) Effect of SFN and compound C (added 2 h before AGE treatment) on the level of lipid peroxidation, assessed by changes in MDA levels (n = 3–4 per group). (H) Effect of SFN and compound C (added 2 h before AGE treatment) on the expression of Ptgs2, assessed using RT-qPCR (n = 3–4 per group). Data are presented as the normalized mean ± SD (to control) or mean ± SD. AGEs, advanced glycation end-products; ECT, engineered cardiac tissue; MDA, malondialdehyde.
Figure 6.

SFN activation of key regulators against AGE-induced ferroptosis is AMPK-dependent. (A) and (B) Effect of sulforaphane (SFN) and compound C (added 2 h before AGE treatment) on the expression of SLC7A11, assessed using Western blotting (n = 4 per group). (A) and (C) Effect of SFN and compound C (added 2 h before AGE treatment) on ferritin expression, assessed using Western blotting (n = 4 per group). (D) Effect of SFN and compound C (added 2 h before AGE treatment) on labile iron levels, assessed using the Iron Colorimetric Assay Kit (n = 4 per group). (E) and (F) Effect of SFN and compound C (added 2 h before AGE treatment) on GSH and GSH/GSSG levels, assessed using the Glutathione Fluorometric Assay Kit (n = 5 per group). (G) and (H) Representative immunofluorescent staining of the cardiomyocytes (CMs) (troponin, red) and nuclei (DAPI, blue) in ECTs following AGE treatment (40× magnification). SFN and compound C were added 2 h before AGE treatment (n = 4 per group). Data are presented as the normalized mean ± SD (to control) or mean ± SD. AGEs, advanced glycation end-products; ECT, engineered cardiac tissue; GSH, glutathione; GSSG, oxidized glutathione; MDA, malondialdehyde; SLC7A11, solute carrier family 7 member 11. ns, no significant.
3.5. Protective effects of SFN on cardiac ferroptosis and DCM are AMPK-dependent
We further examined the role of AMPKα2-KO in the SFN-induced prevention of cardiac ferroptosis in mice with DCM. As we have shown recently, SFN prevents T2D-induced oxidative damage, remodeling (hypertrophy and fibrosis), and progression of cardiac dysfunction in WT (control) diabetic mice, but not in AMPKα2-KO mice25. Using cardiac samples from these animals, we confirmed that NRF2 expression decreased in both WT and AMPKα2-KO diabetic mice. Of note, SFN increased the expression of nuclear NRF2 in the heart of diabetic WT mice via AKT/GSK3β (not KEAP1; Supporting Information Fig. S3A–S3D), but not in AMPKα2-KO mice (Fig. 7A and B).
Figure 7.

Protective effects of the AMPK/NRF2 pathway against ferroptosis in mice with DCM. (A) and (B) Expression of NRF2 in WT and AMPKα2 KO mice, assessed using Western blotting (n = 4–7 per group). (C) Expression of Ptgs2 in WT and AMPKα2 KO mice, assessed by RT-qPCR (n = 4–7 per group). (D) and (E) Expression of SLC7A11 in WT and AMPKα2 KO mice, assessed using Western blotting (n = 4–7 per group). (D) and (F) Expression of ferritin in WT and AMPKα′2 KO mice, assessed using Western blotting (n = 4–7 per group). (G) and (H) Alterations of GSH and GSH/GSSG levels in WT and AMPKα2 KO mice, assessed using the Glutathione Fluorometric Assay Kit (n = 4–7 per group). (I) Labile iron levels in WT and AMPKα2 KO mice, assessed using the Iron Colorimetric Assay Kit (n = 4–7 per group). (J) Level of lipid peroxidation in WT and AMPKα2 KO mice, assessed by changes in MDA levels (n = 4–7 per group). Data are presented as the normalized mean ± SD (to control) or mean ± SD. GSH, glutathione; GSSG, oxidized glutathione; MDA, malondialdehyde; SLC7A11, solute carrier family 7 member 11. ns, no significant.
Finally, we examined the key regulators of ferroptosis-related pathways. SFN attenuated the expression of Ptgs2 in WT diabetic mice but not in AMPKα2-KO diabetic mice (Fig. 7C). The downregulation of Nqo1 mRNA (Fig. S3E), SLC7A11, ferritin, and GSH levels and the increased levels of free iron were rescued by SFN treatment in WT mice but not in AMPKα2-KO diabetic mice (Fig. 7D–I). The analysis of MDA levels confirmed that lipid peroxidation was not attenuated in AMPKα2-KO mice after SFN treatment (Fig. 7J). Therefore, the protective effects of SFN, exerted via NRF2 activation, on ferroptosis in mouse models of DCM were AMPK-dependent.
4. Discussion
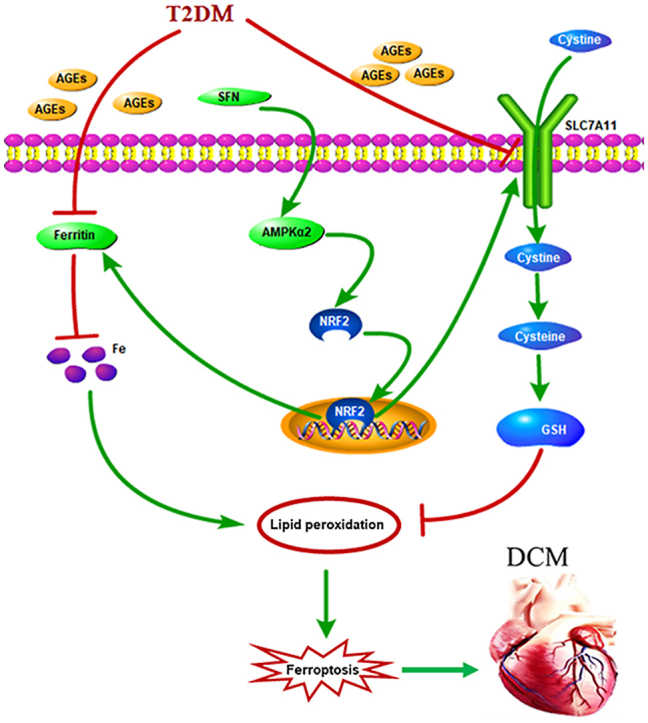
In this study, we showed that ferroptosis was essential for the pathogenesis of DCM. NRF2 activation by SFN inhibited lipid peroxidation and, consequently, ferroptosis. Furthermore, AMPK was indispensable for SFN-induced prevention of ferroptosis in DCM, via NRF2 activation (Fig. 8). These observations provided new insight into the novel pathogenic mechanism underlying DCM and new theoretical evidence for the application of SFN to prevent and treat DCM.
Figure 8.

Schematic representation of AGE-induced ferroptosis in diabetic cardiomyopathy (DCM). AGEs, as pivotal pathogenic factors of DCM, inhibit the expression of SLC7A11 and ferritin, which in turn decrease the GSH levels and increase the labile iron levels, respectively. These alterations lead to increased lipid peroxidation and cardiomyocyte ferroptosis, which then initiates cardiac inflammation, remodeling (cardiomyocyte hypertrophy, pro-fibrotic responses, and fibrosis), and eventually, cardiac dysfunction, that is, DCM. Sulforaphane (SFN) promotes the nuclear accumulation of NRF2 and stimulates the downstream expression of SLC7A11 and ferritin via AMPK activation, which shields the cardiomyocytes from ferroptosis. AGEs, advanced glycation end-products; AMPK, AMP-activated protein kinase; DCM, diabetic cardiomyopathy; GSH, glutathione; SLC7A11, solute carrier family 7 member 11.
Myocardial cell death, such as apoptosis, has been considered key for the pathogenesis of DCM8,9; however, it predominantly occurs at the early stage of diabetes10,34, 35, 36. Ferroptosis is morphologically, biochemically, and genetically distinct from apoptosis and necroptosis and is recognized as an iron-dependent cell death because it is attenuated by ferroptosis-specific inhibitor Fer-1 and iron chelation12,15. Previous studies have documented the association of metal toxicity with diabetic complications, specifically how iron chelation attenuates cardiac oxidative stress, inflammation, and remodeling37,38. However, it remains unclear whether this association is mediated by ferroptosis. Importantly, in this study, we identified the following specific mechanisms in AGE-treated ECTs ex vivo and the heart of diabetic mice in vivo: ferroptosis significantly increased CM loss and ECT dysfunction; inhibition of ferroptosis by ferroptosis-specific inhibitors or iron chelation prevented AGE-induced CM loss and ECT dysfunction; in diabetic mice, key regulators of ferroptosis were altered concomitantly; and inhibition of ferroptosis by liproxstatin-1 prevented the development of diastolic dysfunction at 3 months after the onset of diabetes, which further proved the role of ferroptosis in the pathogenesis of DCM. This suggested the essential role of ferroptosis in the development of DCM. This is a particularly important finding because, while early cardiac apoptosis is crucial for the development of DCM, relatively low numbers of apoptotic cells are observed in the heart of diabetic animals at the late stage of the disease, three and six months post-diabetes11,34,39. In this study, we detected ferroptosis in the heart of diabetic mice even six months after diabetes onset.
AGEs are glycated proteins or lipids formed in response to clinical hyperglycemia, a typical marker of diabetes. AGE levels are increased in patients with diabetes and mice with DCM, and AGEs are considered to be the most important pathogenic initiators of DCM through the interactions with the receptor for AGEs (RAGE) and promotion of oxidative stress40, 41, 42, 43, 44. Studies45 have found that serum AGEs were 10.9 ± 3.5 μg/mg of serum protein in patients with diabetes and 14.8 ± 5.5 μg/mg in patients with diabetic peripheral artery disease, very similar to the concentration we used in this study. In previous work, we determined that AGE-induced ECT remodeling and dysfunction could be rescued by treating ECTs with either N-acetylcysteine or inhibitors of RAGE30. Herein, we further found that AGEs induced ferroptosis in murine ECTs. Consistent with previous studies with cultured cells, AGEs increased the levels of COX2 (Ptgs2)46,47, a newly identified ferroptosis marker, and induced ECT dysfunction. Intracellular ferritin levels were transiently elevated in AGE-treated ECTs; however, 24 h after treatment, they started to decrease in a time-dependent manner. Labile iron levels in both AGE-treated ECTs and diabetic hearts were significantly increased, consistent with previous studies on ferroptosis48,49. The decreased ferritin levels and increased labile iron levels were in accordance with the pathological alterations in mice with DCM. Of note, increased oxidative stress generated by iron overload, via the Fenton reaction, enhances AGE formation50, sustaining a vicious cycle. Therefore, iron overload contributes to lipid peroxidation, an important mechanism underlying DCM51,52. Furthermore, GSH, the formation of which is SLC7A11-dependent15,29, is an important molecule that alleviates lipid peroxidation and ferroptosis. In this study, we showed that SLC7A11 and GSH levels were significantly downregulated in AGE-treated ECTs and hearts of mice with DCM. These pathophysiological alterations might disrupt normal cell function and potentiate lipid peroxidation, resulting in ferroptosis, as an important pathogenic factor for DCM development.
NRF2 is a key regulator of the cellular antioxidant response, controlling the expression of genes that counteract oxidative and electrophilic stresses20. Previous studies revealed that AGEs and hyperglycemia inhibit NRF2 either via promoting ERK phosphorylation53, 54, 55 or reducing the expression of the NRF2-activating mediator Sirt156,57. Therefore, overexpression of cardiac NRF2 or activation of NRF2 by SFN is able to attenuate oxidative stress and prevents DCM20,25. SFN could also reduce AGE-induced oxidative stress and inflammation through suppressing RAGE expression58,59. Of note, SLC7A11 and ferritin, as key inhibitors of ferroptosis, are downstream targets of NRF228,60,61. Herein, we confirmed that SFN, as a potent NRF2 activator, facilitates the nuclear translocation of NRF2 to upregulate ferritin and SLC7A11 levels, attenuating lipid peroxidation and subsequent ferroptosis, and finally, rescuing ECT function after AGE treatment and cardiac function in T2D. Therefore, SFN treatment inhibited ferroptosis, mainly by activating NRF2-regulated antioxidants that counteract lipid peroxidation. This is the first study to reveal the role of SFN in inhibiting ferroptosis.
Furthermore, we show here that the protective effects of SFN, exerted via NRF2 activation, against ferroptosis were AMPK-dependent in DCM mice. AMPK, which functions as a central regulator of cell survival in response to stressful stimuli, was downregulated in AGE-treated cells62 and in mice with DCM25,63. Herein, we confirmed decreased levels of AMPK phosphorylation in AGE-treated ECTs and in the heart of mice with DCM. AMPK also acts as an upstream regulator of the anti-oxidative response64,65. AGEs decrease cell viability by increasing reactive oxygen species levels, and metformin rescues cell viability via AMPK-dependent inhibition of reactive oxygen species production, and the increase of glutathione peroxidase and GSH levels66. We also showed that SFN treatment activated AMPK and prevented DCM in T2D mice25,63. Our finding is consistent with studies that showed the important role of NRF2 in upregulating downstream antioxidants in AMPK-mediated anti-oxidative effects25,67, 68, 69. Intriguingly, we found that the baseline expression of NRF2 was not affected by inhibiting AMPK phosphorylation with compound C70, which might be associated with the multiple regulators, other than AMPK, that affect the expression of NRF271,72. It was previously demonstrated that active GSK3β stimulates Fyn translocation to the nucleus, where Fyn exports nuclear NRF2 to the cytosol and is degraded via ubiquitination. However, AMPK increases AKT-mediated GSK3β phosphorylation to inactivate GSK3β and ultimately preserves the nuclear function of NRF219,23,25. Herein, we further show that SFN increased AMPK phosphorylation that promoted NRF2 nuclear translocation along with the prevention of ferroptosis. These effects were abolished by the inhibition of AMPK with compound C in AGE-treated ECTs or the heart of AMPKα2-KO mice, even after SFN treatment. Therefore, we proved ferroptosis as an important mechanism for the development of DCM, which could be prevented by SFN treatment via NRF2 activation in an AMPK-dependent manner.
In summary, we reported a mechanism by which ferroptosis mediates the pathogenesis of DCM. We also showed that SFN potentiates AMPK-dependent and NRF2-mediated protection of ECTs ex vivo during AGE-induced ECT dysfunction and of the heart during diabetes-induced cardiac remodeling and dysfunction. These findings suggest that the combination of SFN and selective removal of excess iron to inhibit ferroptosis, together with the prevention of chronic oxidative stress, may represent a feasible therapeutic approach for the prevention of DCM in individuals with diabetes. From a clinical perspective, studies are needed to test the potential clinical implications of metformin (or SFN) in combination with low-dose iron chelators that are used in the clinic, such as deferasirox and deferiprone, for the prevention of DCM.
5. Conclusions
These findings suggest that ferroptosis plays an essential role in the pathogenesis of DCM; SFN prevents ferroptosis and associated pathogenesis via AMPK-mediated NRF2 activation.
Acknowledgments
We would like to express our great appreciation to Dr. Jason Xu for his valuable and constructive suggestions during the research. We would like to thank Editage (www.editage.cn) for English language editing. This work was supported in part by American Diabetes Association (1-18-IBS-082 to LC, USA); the National Key R&D Program of China (2016YFC0900903 to YZ, China); and University of Louisville–China Pediatric Research Exchange Program (to BBK and LC, USA, without salary payment). All personnel expenses and some research-related expenses for Xiang Wang, Wenqian Zhou, Hongbo Men, Terigen Bao, and Yike Sun were covered by the First Hospital of Jilin University (Changchun, China). The sponsors did not play any role in study design; in the collection, analysis, and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2021.10.005.
Contributor Information
Qian Tong, Email: tongqian@jlu.edu.cn.
Yang Zheng, Email: zhengyanghappy07@sina.com.cn.
Lu Cai, Email: lu.cai@louisville.edu.
Author contribution
Conception or design of the study: Xiang Wang, Xinxin Chen, Bradley B. Keller, Qian Tong, Yang Zheng, and Lu Cai; data collection: Xiang Wang, Xinxin Chen, Wenqian Zhou, Hongbo Men, Terigen Bao, and Quanwei Wang; data analysis and interpretation: Xiang Wang, Xinxin Chen, Wenqian Zhou, Hongbo Men, Terigen Bao, Yike Sun, and Quanwei Wang; drafting the article: Xiang Wang, Xinxin Chen, Yi Tan, Bradley B. Keller, Qian Tong, Yang Zheng, and Lu Cai; critical revision of the article: Xiang Wang, Xinxin Chen, Yi Tan, Bradley B. Keller, Qian Tong, Yang Zheng, and Lu Cai. All authors approved the final version of the manuscript.
Conflicts of interest
The authors declare no conflict of interest.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Saeedi P., Petersohn I., Salpea P., Malanda B., Karuranga S., Unwin N., et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res Clin Pract. 2019;157:107843. doi: 10.1016/j.diabres.2019.107843. [DOI] [PubMed] [Google Scholar]
- 2.Rubler S., Yuceoglu Y.Z., Kumral T., Branwood A.W., Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol. 1972;30:595–602. doi: 10.1016/0002-9149(72)90595-4. [DOI] [PubMed] [Google Scholar]
- 3.Boudina S., Abel E.D. Diabetic cardiomyopathy, causes and effects. Rev Endocr Metab Disord. 2010;11:31–39. doi: 10.1007/s11154-010-9131-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brunvand L., Heier M., Brunborg C., Hanssen K.F., Fugelseth D., Stensaeth K.H., et al. Advanced glycation end products in children with type 1 diabetes and early reduced diastolic heart function. BMC Cardiovasc Disord. 2017;17:133. doi: 10.1186/s12872-017-0551-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palomer X., Salvado L., Barroso E., Vazquez-Carrera M. An overview of the crosstalk between inflammatory processes and metabolic dysregulation during diabetic cardiomyopathy. Int J Cardiol. 2013;168:3160–3172. doi: 10.1016/j.ijcard.2013.07.150. [DOI] [PubMed] [Google Scholar]
- 6.Cai L., Kang Y.J. Oxidative stress and diabetic cardiomyopathy: a brief review. Cardiovasc Toxicol. 2001;1:181–193. doi: 10.1385/ct:1:3:181. [DOI] [PubMed] [Google Scholar]
- 7.Wilson A.J., Gill E.K., Abudalo R.A., Edgar K.S., Watson C.J., Grieve D.J. Reactive oxygen species signalling in the diabetic heart: emerging prospect for therapeutic targeting. Heart. 2018;104:293–299. doi: 10.1136/heartjnl-2017-311448. [DOI] [PubMed] [Google Scholar]
- 8.Cai L., Kang Y.J. Cell death and diabetic cardiomyopathy. Cardiovasc Toxicol. 2003;3:219–228. doi: 10.1385/ct:3:3:219. [DOI] [PubMed] [Google Scholar]
- 9.Chen Y., Hua Y., Li X., Arslan I.M., Zhang W., Meng G. Distinct types of cell death and the implication in diabetic cardiomyopathy. Front Pharmacol. 2020;11:42. doi: 10.3389/fphar.2020.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cai L., Liu W., Wang G., Guo L., Jiang Y., Kang Y.J. Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome C-mediated caspase-3 activation pathway. Diabetes. 2002;51:1938–1948. doi: 10.2337/diabetes.51.6.1938. [DOI] [PubMed] [Google Scholar]
- 11.Cai L., Wang Y., Zhou G., Chen T., Song Y., Li X., et al. Attenuation by metallothionein of early cardiac cell death via suppression of mitochondrial oxidative stress results in a prevention of diabetic cardiomyopathy. J Am Coll Cardiol. 2006;48:1688–1697. doi: 10.1016/j.jacc.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 12.Dixon S.J., Lemberg K.M., Lamprecht M.R., Skouta R., Zaitsev E.M., Gleason C.E., et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stockwell B.R., Friedmann Angeli J.P., Bayir H., Bush A.I., Conrad M., Dixon S.J., et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–285. doi: 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fang X., Wang H., Han D., Xie E., Yang X., Wei J., et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. 2019;116:2672–2680. doi: 10.1073/pnas.1821022116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fang X., Cai Z., Wang H., Han D., Cheng Q., Zhang P., et al. Loss of cardiac ferritin H facilitates cardiomyopathy via Slc7a11-mediated ferroptosis. Circ Res. 2020;127:486–501. doi: 10.1161/CIRCRESAHA.120.316509. [DOI] [PubMed] [Google Scholar]
- 16.Swaminathan S., Fonseca V.A., Alam M.G., Shah S.V. The role of iron in diabetes and its complications. Diabetes Care. 2007;30:1926–1933. doi: 10.2337/dc06-2625. [DOI] [PubMed] [Google Scholar]
- 17.Liu Q., Sun L., Tan Y., Wang G., Lin X., Cai L. Role of iron deficiency and overload in the pathogenesis of diabetes and diabetic complications. Curr Med Chem. 2009;16:113–129. doi: 10.2174/092986709787002862. [DOI] [PubMed] [Google Scholar]
- 18.White D.L., Collinson A. Red meat, dietary heme iron, and risk of type 2 diabetes: the involvement of advanced lipoxidation endproducts. Adv Nutr. 2013;4:403–411. doi: 10.3945/an.113.003681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xin Y., Bai Y., Jiang X., Zhou S., Wang Y., Wintergerst K.A., et al. Sulforaphane prevents angiotensin II-induced cardiomyopathy by activation of Nrf2 via stimulating the Akt/GSK-3β/Fyn pathway. Redox Biol. 2018;15:405–417. doi: 10.1016/j.redox.2017.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gu J., Cheng Y., Wu H., Kong L., Wang S., Xu Z., Zhang Z., Tan Y., Keller B., Zhou H., Wang Y., Xu Z., Cai L. Metallothionein is downstream of Nrf2 and partially mediates Sulforaphane prevention of diabetic cardiomyopathy. Diabetes. 2017;66:529–542. doi: 10.2337/db15-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Evangelos P., Beauloye Christophe, Bertrand Luc, Horman Sandrine. AMPK in cardiovascular diseases. Exp Suppl. 2016;107:179–201. doi: 10.1007/978-3-319-43589-3_8. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y., Gao E., Tao L., Lau W.B., Yuan Y., Goldstein B.J., et al. AMP-activated protein kinase deficiency enhances myocardial ischemia/reperfusion injury but has minimal effect on the antioxidant/antinitrative protection of adiponectin. Circulation. 2009;119:835–844. doi: 10.1161/CIRCULATIONAHA.108.815043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi S.H., Kim Y.W., Kim S.G. AMPK-mediated GSK3β inhibition by isoliquiritigenin contributes to protecting mitochondria against iron-catalyzed oxidative stress. Biochem Pharmacol. 2010;79:1352–1362. doi: 10.1016/j.bcp.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 24.Salazar M., Rojo A.I., Velasco D., de Sagarra R.M., Cuadrado A. Glycogen synthase kinase-3β inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J Biol Chem. 2006;281:14841–14851. doi: 10.1074/jbc.M513737200. [DOI] [PubMed] [Google Scholar]
- 25.Sun Y., Zhou S., Guo H., Zhang J., Ma T., Zheng Y., et al. Protective effects of sulforaphane on type 2 diabetes-induced cardiomyopathy via AMPK-mediated activation of lipid metabolic pathways and NRF2 function. Metabolism. 2020;102:154002. doi: 10.1016/j.metabol.2019.154002. [DOI] [PubMed] [Google Scholar]
- 26.Rizvi F., Shukla S., Kakkar P. Essential role of PH domain and leucine-rich repeat protein phosphatase 2 in Nrf2 suppression via modulation of Akt/GSK3β/Fyn kinase axis during oxidative hepatocellular toxicity. Cell Death Dis. 2014;5 doi: 10.1038/cddis.2014.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song X., Long D. Nrf2 and Ferroptosis: a new research direction for neurodegenerative diseases. Front Neurosci. 2020;14:267. doi: 10.3389/fnins.2020.00267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dodson M., Castro-Portuguez R., Zhang D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019;23:101107. doi: 10.1016/j.redox.2019.101107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen D., Tavana O., Chu B., Erber L., Chen Y., Baer R., et al. NRF2 is a major target of ARF in p53-independent tumor suppression. Mol Cell. 2017;68:224–232. doi: 10.1016/j.molcel.2017.09.009. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang X., Chen X., Yu H., Tan Y., Lin Q., Keller B.B., Zheng Y., Cai L. Engineered cardiac tissues: a novel in vitro model to investigate the pathophysiology of mouse diabetic cardiomyopathy. Acta Pharmacol Sin. 2021;42:932–941. doi: 10.1038/s41401-020-00538-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu H., Ye F., Yuan F., Cai L., Ji H., Keller B.B. Neonatal murine engineered cardiac tissue toxicology model: impact of metallothionein overexpression on cadmium-induced injury. Toxicol Sci. 2018;165:499–511. doi: 10.1093/toxsci/kfy177. [DOI] [PubMed] [Google Scholar]
- 32.Taylor R., Al-Mrabeh A., Sattar N. Understanding the mechanisms of reversal of type 2 diabetes. Lancet Diabetes Endocrinol. 2019;7:726–736. doi: 10.1016/S2213-8587(19)30076-2. [DOI] [PubMed] [Google Scholar]
- 33.Lu M.C., Ji J.A., Jiang Z.Y., You Q.D. The Keap1–Nrf2–ARE pathway as a potential preventive and therapeutic target: an update. Med Res Rev. 2016;36:924–963. doi: 10.1002/med.21396. [DOI] [PubMed] [Google Scholar]
- 34.Zhang M., Zhang L., Hu J., Lin J., Wang T., Duan Y., et al. MST1 coordinately regulates autophagy and apoptosis in diabetic cardiomyopathy in mice. Diabetologia. 2016;59:2435–2447. doi: 10.1007/s00125-016-4070-9. [DOI] [PubMed] [Google Scholar]
- 35.Guo R., Liu W., Liu B., Zhang B., Li W., Xu Y. SIRT1 suppresses cardiomyocyte apoptosis in diabetic cardiomyopathy: an insight into endoplasmic reticulum stress response mechanism. Int J Cardiol. 2015;191:36–45. doi: 10.1016/j.ijcard.2015.04.245. [DOI] [PubMed] [Google Scholar]
- 36.Liu Z., Zhu H., Chen K., Dong X., Wei J., Qiu C., et al. Protein kinase RNA-like endoplasmic reticulum kinase (PERK) signaling pathway plays a major role in reactive oxygen species (ROS)-mediated endoplasmic reticulum stress-induced apoptosis in diabetic cardiomyopathy. Cardiovasc Diabetol. 2013;12:158. doi: 10.1186/1475-2840-12-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng Y., Li X., Wang Y., Cai L. The role of zinc, copper and iron in the pathogenesis of diabetes and diabetic complications: therapeutic effects by chelators. Hemoglobin. 2008;32:135–145. doi: 10.1080/03630260701727077. [DOI] [PubMed] [Google Scholar]
- 38.Zou C., Liu X., Xie R., Bao Y., Jin Q., Jia X., et al. Deferiprone attenuates inflammation and myocardial fibrosis in diabetic cardiomyopathy rats. Biochem Biophys Res Commun. 2017;486:930–936. doi: 10.1016/j.bbrc.2017.03.127. [DOI] [PubMed] [Google Scholar]
- 39.Zhou G., Li X., Hein D.W., Xiang X., Marshall J.P., Prabhu S.D., et al. Metallothionein suppresses angiotensin II-induced nicotinamide adenine dinucleotide phosphate oxidase activation, nitrosative stress, apoptosis, and pathological remodeling in the diabetic heart. J Am Coll Cardiol. 2008;52:655–666. doi: 10.1016/j.jacc.2008.05.019. [DOI] [PubMed] [Google Scholar]
- 40.Cheng G., Wang L., Qu W., Long L., Cui H., Liu H., et al. C16, a novel advanced glycation endproduct breaker, restores cardiovascular dysfunction in experimental diabetic rats. Acta Pharmacol Sin. 2005;26:1460–1466. doi: 10.1111/j.1745-7254.2005.00240.x. [DOI] [PubMed] [Google Scholar]
- 41.Bodiga V.L., Eda S.R., Bodiga S. Advanced glycation end products: role in pathology of diabetic cardiomyopathy. Heart Fail Rev. 2014;19:49–63. doi: 10.1007/s10741-013-9374-y. [DOI] [PubMed] [Google Scholar]
- 42.Hou J., Zheng D., Fung G., Deng H., Chen L., Liang J., et al. Mangiferin suppressed advanced glycation end products (AGEs) through NF-kappaB deactivation and displayed anti-inflammatory effects in streptozotocin and high fat diet-diabetic cardiomyopathy rats. Can J Physiol Pharmacol. 2016;94:332–340. doi: 10.1139/cjpp-2015-0073. [DOI] [PubMed] [Google Scholar]
- 43.Wang Y., Luo W., Han J., Khan Z.A., Fang Q., Jin Y., et al. MD2 activation by direct AGE interaction drives inflammatory diabetic cardiomyopathy. Nat Commun. 2020;11:2148. doi: 10.1038/s41467-020-15978-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tan Y., Zhang Z., Zheng C., Wintergerst K.A., Keller B.B., Cai L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: preclinical and clinical evidence. Nat Rev Cardiol. 2020;17:585–607. doi: 10.1038/s41569-020-0339-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lapolla A., Piarulli F., Sartore G., Ceriello A., Ragazzi E., Reitano R., et al. Advanced glycation end products and antioxidant status in type 2 diabetic patients with and without peripheral artery disease. Diabetes Care. 2007;30:670–676. doi: 10.2337/dc06-1508. [DOI] [PubMed] [Google Scholar]
- 46.Sun M., Li Y., Bu W., Zhao J., Zhu J., Gu L., et al. DJC suppresses advanced glycation end products-induced JAK–STAT signaling and ROS in mesangial cells. Evid Based Complement Alternat Med. 2017;2017:2942830. doi: 10.1155/2017/2942830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen J., Sun Z., Jin M., Tu Y., Wang S., Yang X., et al. Inhibition of AGEs/RAGE/Rho/ROCK pathway suppresses non-specific neuroinflammation by regulating BV2 microglial M1/M2 polarization through the NF-kappaB pathway. J Neuroimmunol. 2017;305:108–114. doi: 10.1016/j.jneuroim.2017.02.010. [DOI] [PubMed] [Google Scholar]
- 48.Mayo J.K.P., Zhang D., Winzerling J. Effects of sham air and cigarette smoke on A549 lung cells: implications for iron-mediated oxidative damage. Am J Physiol Lung Cell Mol Physiol. 2004;286:L866–L876. doi: 10.1152/ajplung.00268.2003. [DOI] [PubMed] [Google Scholar]
- 49.Yoshida M., Minagawa S., Araya J., Sakamoto T., Hara H., Tsubouchi K., et al. Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat Commun. 2019;10:3145. doi: 10.1038/s41467-019-10991-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mirlohi M.S., Yaghooti H., Shirali S., Aminasnafi A., Olapour S. Increased levels of advanced glycation end products positively correlate with iron overload and oxidative stress markers in patients with beta-thalassemia major. Ann Hematol. 2018;97:679–684. doi: 10.1007/s00277-017-3223-3. [DOI] [PubMed] [Google Scholar]
- 51.Katunga L.A., Gudimella P., Efird J.T., Abernathy S., Mattox T.A., Beatty C., et al. Obesity in a model of gpx4 haploinsufficiency uncovers a causal role for lipid-derived aldehydes in human metabolic disease and cardiomyopathy. Mol Metab. 2015;4:493–506. doi: 10.1016/j.molmet.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu M., Li Y., Liang B., Li Z., Jiang Z., Chu C., et al. Hydrogen sulfide attenuates myocardial fibrosis in diabetic rats through the JAK/STAT signaling pathway. Int J Mol Med. 2018;41:1867–1876. doi: 10.3892/ijmm.2018.3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tan Y., Ichikawa T., Li J., Si Q., Yang H., Chen X., et al. Diabetic downregulation of Nrf2 activity via ERK contributes to oxidative stress-induced insulin resistance in cardiac cells in vitro and in vivo. Diabetes. 2011;60:625–633. doi: 10.2337/db10-1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ko S., Chang S., Lin I., Chen H. Suppression of antioxidant Nrf-2 and downstream pathway in H9c2 cells by advanced glycation end products (AGEs) via ERK phosphorylation. Biochimie. 2015;118:8–14. doi: 10.1016/j.biochi.2015.07.019. [DOI] [PubMed] [Google Scholar]
- 55.Ko S., Ko H., Shieh T., Chi T., Chen H., Chen Y., et al. Advanced glycation end products influence oral cancer cell survival via Bcl-xl and Nrf-2 regulation in vitro. Oncol Lett. 2017;13:3328–3334. doi: 10.3892/ol.2017.5809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang K., Huang J., Xie X., Wang S., Chen C., Shen X., et al. Sirt1 resists advanced glycation end products-induced expressions of fibronectin and TGF-β1 by activating the Nrf2/ARE pathway in glomerular mesangial cells. Free Radic Biol Med. 2013;65:528–540. doi: 10.1016/j.freeradbiomed.2013.07.029. [DOI] [PubMed] [Google Scholar]
- 57.Chen X., Gong X., Jie J., Yu W., Chen X., Du X., et al. Receptor for advanced glycation end products reveals a mechanism regulating thyroid hormone secretion through the SIRT1/Nrf2 pathway. J Cell Biochem. 2019;120:4582–4598. doi: 10.1002/jcb.27747. [DOI] [PubMed] [Google Scholar]
- 58.Maeda S., Matsui T., Ojima A., Takeuchi M., Yamagishi S. Sulforaphane inhibits advanced glycation end product-induced pericyte damage by reducing expression of receptor for advanced glycation end products. Nutr Res. 2014;34:807–813. doi: 10.1016/j.nutres.2014.08.010. [DOI] [PubMed] [Google Scholar]
- 59.Matsui T., Nakamura N., Ojima A., Nishino Y., Yamagishi S.I. Sulforaphane reduces advanced glycation end products (AGEs)-induced inflammation in endothelial cells and rat aorta. Nutr Metabol Cardiovasc Dis. 2016;26:797–807. doi: 10.1016/j.numecd.2016.04.008. [DOI] [PubMed] [Google Scholar]
- 60.Lei G., Zhang Y., Koppula P., Liu X., Zhang J., Lin S.H., et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020;30:146–162. doi: 10.1038/s41422-019-0263-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sun X., Chen R., Niu X., Chen D., Kang R., Tang D.L. Activation of the p62–Keap1–NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63:173–184. doi: 10.1002/hep.28251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chung M., Chen Y., Pei D., Cheng Y., Sun B., Nicol C., et al. The neuroprotective role of metformin in advanced glycation end product treated human neural stem cells is AMPK-dependent. Biochim Biophys Acta. 2015;1852:720–731. doi: 10.1016/j.bbadis.2015.01.006. [DOI] [PubMed] [Google Scholar]
- 63.Choi K.M., Lee Y.S., Kim W., Kim S.J., Shin K.O., Yu J.Y., et al. Sulforaphane attenuates obesity by inhibiting adipogenesis and activating the AMPK pathway in obese mice. J Nutr Biochem. 2014;25:201–207. doi: 10.1016/j.jnutbio.2013.10.007. [DOI] [PubMed] [Google Scholar]
- 64.Zhang J., Zhang S., Wang P., Guo N., Wang W., Yao L., et al. Pinolenic acid ameliorates oleic acid-induced lipogenesis and oxidative stress via AMPK/SIRT1 signaling pathway in HepG2 cells. Eur J Pharmacol. 2019;861:172618. doi: 10.1016/j.ejphar.2019.172618. [DOI] [PubMed] [Google Scholar]
- 65.Saito Y., Chapple R.H., Lin A., Kitano A., Nakada D. AMPK protects leukemia-initiating cells in myeloid leukemias from metabolic stress in the bone marrow. Cell Stem Cell. 2015;17:585–596. doi: 10.1016/j.stem.2015.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lin C., Cheng Y., Nicol C., Lin K., Yen C., Chiang M. Activation of AMPK is neuroprotective in the oxidative stress by advanced glycosylation end products in human neural stem cells. Exp Cell Res. 2017;359:367–373. doi: 10.1016/j.yexcr.2017.08.019. [DOI] [PubMed] [Google Scholar]
- 67.Lee E.H., Baek S.Y., Park J.Y., Kim Y.W. Rifampicin activates AMPK and alleviates oxidative stress in the liver as mediated with Nrf2 signaling. Chem Biol Interact. 2020;315:108889. doi: 10.1016/j.cbi.2019.108889. [DOI] [PubMed] [Google Scholar]
- 68.Xu W., Zhao T., Xiao H. The Implication of oxidative stress and AMPK–Nrf2 antioxidative signaling in pneumonia pathogenesis. Front Endocrinol (Lausanne) 2020;11:400. doi: 10.3389/fendo.2020.00400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zimmermann K., Baldinger J., Mayerhofer B., Atanasov A.G., Dirsch V.M., Heiss E.H. Activated AMPK boosts the Nrf2/HO-1 signaling axis—a role for the unfolded protein response. Free Radic Biol Med. 2015;88:417–426. doi: 10.1016/j.freeradbiomed.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu X., Chhipa R.R., Nakano I., Dasgupta B. The AMPK inhibitor compound C is a potent AMPK-independent antiglioma agent. Mol Cancer Therapeut. 2014;13:596–605. doi: 10.1158/1535-7163.MCT-13-0579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tonelli C., Chio I.I.C., Tuveson D.A. Transcriptional regulation by Nrf2. Antioxidants Redox Signal. 2018;29:1727–1745. doi: 10.1089/ars.2017.7342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Niture S.K., Khatri R., Jaiswal A.K. Regulation of Nrf2—an update. Free Radic Biol Med. 2014;66:36–44. doi: 10.1016/j.freeradbiomed.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.