

Abstract
Alzheimer's disease (AD), the most prominent form of dementia in the elderly, has no cure. Strategies focused on the reduction of amyloid beta or hyperphosphorylated Tau protein have largely failed in clinical trials. Novel therapeutic targets and strategies are urgently needed. Emerging data suggest that in response to environmental stress, mitochondria initiate an integrated stress response (ISR) shown to be beneficial for healthy aging and neuroprotection. Here, we review data that implicate mitochondrial electron transport complexes involved in oxidative phosphorylation as a hub for small molecule-targeted therapeutics that could induce beneficial mitochondrial ISR. Specifically, partial inhibition of mitochondrial complex I has been exploited as a novel strategy for multiple human conditions, including AD, with several small molecules being tested in clinical trials. We discuss current understanding of the molecular mechanisms involved in this counterintuitive approach. Since this strategy has also been shown to enhance health and life span, the development of safe and efficacious complex I inhibitors could promote healthy aging, delaying the onset of age-related neurodegenerative diseases.
KEY WORDS: Mitochondria, Mitochondria signaling, Complex I inhibitors, Alzheimer's disease, Integrated stress response, Neuroprotection, Mitochondria targeted therapeutics, Healthy aging
Abbreviations: AD, Alzheimer's disease; ADP, adenosine diphosphate; AIDS, acquired immunodeficiency syndrome; AMP, adenosine monophosphate; AMPK, AMP-activated protein kinase; APP/PS1, amyloid precursor protein/presenilin 1; ATP, adenosine triphosphate; Aβ, amyloid beta; BBB, blood‒brain barrier; BDNF, brain-derived neurotrophic factor; CP2, tricyclic pyrone compound two; ER, endoplasmic reticulum; ETC, electron transport chain; FADH2, flavin adenine dinucleotide; FDG-PET, fluorodeoxyglucose-positron emission tomography; GWAS, genome-wide association study; HD, Huntington's disease; HIF-1α, hypoxia induced factor 1 α; ISR, integrated stress response; LTP, long term potentiation; MCI, mild cognitive impairment; MPTP, 1-methyl 4-phenyl-1,2,3,6-tetrahydropyridine; mtDNA, mitochondrial DNA; mtUPR, mitochondrial unfolded protein response; NAD+ and NADH, nicotinamide adenine dinucleotide; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NRF2, nuclear factor E2-related factor 2; OXPHOS, oxidative phosphorylation; PD, Parkinson's disease; PGC1α, peroxisome proliferator-activated receptor gamma coactivator 1 alpha; PMF, proton-motive force; pTau, hyper-phosphorylated Tau protein; RNAi, RNA interference; ROS, reactive oxygen species; T2DM, type II diabetes mellitus; TCA, the tricarboxylic acid cycle; ΔpH, proton gradient; Δψm, mitochondrial membrane potential
Graphical abstract
Mild energetic stress via partial inhibition of complex I could induce mitochondria-mediated multifaceted integrated stress response, leading to health and life extension and neuroprotection.

1. Introduction
Alzheimer's disease (AD) is a common neurodegenerative disorder in the elderly without a cure1. Common hallmarks of AD include extracellular plaques formed by amyloid beta (Aβ) peptides, neurofibrillary tangles comprised of hyper-phosphorylated Tau protein (pTau), reactive microgliosis, dystrophic neuritis, and loss of neurons and synapses2. However, failure of recent clinical trials focused on the prevention of Aβ and pTau production or their clearance questions the amyloid cascade hypothesis and the role of Aβ and pTau in the underlying disease mechanism. The outcomes of the most recent studies conducted using advanced biochemistry and multi omics systems biology approaches in well characterized cohorts of AD patients, patient-derived cells and tissues suggest a multifactorial nature of AD where several mechanisms on a whole organismal level become individually or synergistically affected in a disease-stage- and sex-specific manner3, 4, 5. These pathways include reduced glucose uptake and utilization, insulin resistance, altered autophagy and proteostasis, increased inflammation and oxidative stress, and mitochondrial dysfunction6, 7, 8. Conclusive evidence demonstrating that brain hypometabolism precedes clinical presentation and the development of Aβ aggregates provided a justification for interventions focused on metabolism and mitochondrial function as potential disease-modifying strategies that could block the disease progression9. However, most treatments aimed at boosting mitochondrial function or reducing the pathology associated with increased production of reactive oxygen species (ROS) have failed clinical trials10,11. Unexpectedly, partial reduction of the activity of the complexes involved in the oxidative phosphorylation (OXPHOS) and electron transport chain (ETC) machinery using genetic or pharmacological down modulation approaches has been shown to provide significant health benefits, improving mitochondrial function and cellular energetics in multiple model systems in vitro and in vivo. In particular, partial inhibition of mitochondrial complex I using small molecules has emerged as a therapeutic strategy for multiple human conditions, including cancer and neurodegenerative diseases. This counterintuitive strategy has been shown to increase longevity and health span, which ultimately could delay the onset of neurodegenerative diseases of aging, such as AD9. Indeed, the induction of mild energetic stress via partial complex I inhibition with subsequent mitochondria-mediated stress response may increase resilience to the greater stress associated with aging and ensure that mechanisms found instrumental for protection against AD, including inflammation, synaptic function, proteostasis, mitochondrial dynamics and function, and oxidative stress, remain in control12,13. Below, we discuss the current understanding of the neuroprotective mechanisms behind complex I inhibition with respect to mitochondrial signaling via integrated stress response (ISR) and progress in the development of safe and efficacious partial complex I inhibitors.
2. Mitochondria function in energy production and as signaling organelles
Most of the energy required for cellular functions is produced by mitochondria (Fig. 1). These organelles are abundant, occupying up to 25% of the cytoplasmic volume. The mitochondrion is the only cellular organelle other than the nucleus that has its own DNA (mtDNA) and transcriptional and translational machinery (Fig. 1A). These features, together with the unusual dynamics of mitochondrial division and fusion (reminiscent of bacteria), have led to the theory of an ancient endosymbiosis of a nucleated cell and an aerobic prokaryote14. Such cooperation provides the host with significantly increased energy supply, making mitochondria a “power plant” of the cell, while mitochondria enjoy the protection and resources of the host, including the outsourcing of the most of protein synthesis essential for their function. Successful symbiotic integration required the development of a robust communication system. The extensive arsenal of signaling molecules utilized by mitochondria for intracellular communication is discussed below.
Figure 1.

Mitochondria structure and components of the OPXHOS machinery involved in mitochondrial intracellular signaling. (A) Electron micrograph (left) and cartoon (right) show a mitochondrion and its constituents. The organelle has an outer membrane and an inner membrane that folds into cristae that accommodate complexes of the OXPHOS machinery. The TCA cycle and mitochondrial DNA are located in the matrix. Scale bar, 500 nm. (B) The OXPHOS machinery. The series of protein complexes create a flow of electrons via redox reactions. The NADH and FADH2 are converted to NAD+ (complex I) and FAD (complex II), respectively, with H2O formed (complex IV) as a biproduct. This electron transfer causes protons to flow from mitochondrial matrix to intermembrane space, creating an outward gradient of positively charged protons. The inner mitochondrial membrane bound F0 subunit of complex V (ATP synthase) uses this electrochemical gradient to rotate causing conformational changes to F1 subunits that convert ADP to ATP. Changes in the concentrations of all these metabolites could be used for intracellular communication. ADP indicates adenosine diphosphate; ATP, adenosine triphosphate; FADH2, flavin adenine dinucleotide; NAD+, nicotinamide adenine dinucleotide; NADH, nicotinamide adenine dinucleotide; OXPHOS, oxidative phosphorylation; TCA, tricarboxylic acid.
2.1. OXPHOS machinery
Cell populations in the brain are diverse, and each cell type has distinctive energy demands and metabolic profiles. The conventional view is that neurons depend on energy produced by mitochondria via OXPHOS15. In OXPHOS, a series of metabolic reactions leads to the oxidation of glucose or its metabolites, such as pyruvate and lactate, to produce energy in the form of adenosine triphosphate (ATP) (Fig. 1B)16. OXPHOS is the most efficient metabolic pathway, producing approximately 36 molecules of ATP per one molecule of glucose compared to 2 ATP molecules produced during glycolysis, a cytoplasmic process that also uses glucose but does not require mitochondria17. Mitochondrial morphology is essential to maintaining OXPHOS. The organelle has two membrane compartments (Fig. 1A). The outer membrane delimits the organelle and allows the passage of small molecules and ions to maintain mitochondrial homeostasis. An inner membrane consists of multiple folds called crista and defines the mitochondrial matrix as a closed compartment. OXPHOS machinery is located at the inner mitochondrial membrane, while the tricarboxylic acid (TCA) cycle that produces essential components to power OXPHOS takes place in the matrix.
During OXPHOS, substrates such as nicotinamide adenine dinucleotide (NADH) or flavin adenine dinucleotide (FADH2) produced in the TCA cycle donate electrons that are transferred through the mitochondrial ETC via a series of redox reactions coupled to a final phosphorylation of adenosine diphosphate (ADP) to produce ATP, CO2 and water. This process requires oxygen, and oxygen consumption could be measured using oxygen electrodes or a Seahorse Extracellular Flux Analyzer to inform on the OXPHOS activity18. The ETC (Fig. 1B) includes four protein complexes: NADH dehydrogenase (complex I), succinate dehydrogenase (complex II), cytochrome c oxidoreductase (complex III), and cytochrome c oxidase (complex IV)19. Electrons moving through the ETC promote the translocation of protons (H+) from the matrix to the intermembrane space, establishing an electrochemical gradient or proton-motive force (PMF). The energy generated from the PMF is used to phosphorylate ADP to ATP via ATP synthase (complex V) (Fig. 1B). The PMF is regulated by the electrical potential difference established between the cytoplasm and the matrix, known as mitochondrial membrane potential (Δψm), and the proton gradient (ΔpH) across the inner mitochondrial membrane20. Under normal physiological conditions, PMF is dominated by Δψm, which accounts for over 70% of its potential21,22. However, maintaining the PMF at high potential can lead to a breakdown of the membrane with subsequent formation of ROS, including hydrogen peroxide (H2O2) and superoxide (O2–)23. Hence, the PMF buildup during OXPHOS is counterbalanced by ATP synthesis, during which protons re-enter the matrix diminishing the PMF. Under steady state conditions, the rate of electron transport equilibrates with proton translocation resulting in sufficient energy production and minimal generation of ROS19. During OXPHOS, H2O2 and O2– are produced as byproducts in mitochondria, primarily by complexes I and III, and are sequestered by the antioxidant enzymes, including superoxide dismutases, glutathione peroxidase, glutaredoxins, thioredoxins and catalases7. Under disease conditions, this balance may be altered, leading to excessive ROS production and cellular damage. The balance amongst mitochondrial function, ROS production and the antioxidant defense is essential for normal function, since neuronal cells comprise most (80%–90%) of the energy demand of the brain and primarily depend on OXPHOS24. Thus, levels of ROS, energy metabolites, metabolites of the TCA cycle, and Δψm are tightly linked to mitochondrial function, so changes in concentrations of all these metabolites could be used for intracellular signaling. In essence mitochondria serve as signaling organelles.
2.2. Mitochondria as signaling organelles
Mitochondria have long been recognized as central to ATP production, calcium buffering, and initiation of the apoptosis. In recent years, it became apparent that mitochondrial involvement in regulating cellular fate is more complex25,26. Mitochondria communicate with the rest of the cell by releasing metabolites, mtDNA, and ROS, by changing their size and motility and by interacting with other subcellular organelles (Fig. 2). For example, fluctuations in the TCA cycle metabolites (citrate, α-ketoglutarate, succinate and fumarate) induce epigenetic modifications, including nuclear DNA methylation, histone acetylation, and protein hydroxylation and acetylation27,28. Changes in the levels of nicotinamide adenine dinucleotide (NAD+), a direct product of mitochondrial complex I function, affect the activity of sirtuins, the essential regulators of multiple cellular functions linked to improved mitochondrial function, increased health span and longevity29, 30, 31, 32. Alterations in ROS levels control hypoxic responses, immunity, and stem cell function33. Recent findings demonstrate that mitochondrial ROS is an essential component of signaling that mediates antioxidant (redox) balance and stabilizes hypoxia-induced factor 1 α (HIF-1α)34, an important mediator of life-span extension linked to mitochondrial ISR35. Recent studies also identified mitochondrial ROS signaling as a mitohormetic process where an increase in sublethal levels of ROS could predispose cells to a better response to increased oxidative stress in the future36. Furthermore, mitochondria regulate immune response by releasing mtDNA and through the peptides (e.g., humanin and mitochondrial open-reading-frame of the twelve S rRNA-C, MOTS-c) encoded by mtDNA37,38. Changes in mitochondrial dynamics (fission, fusion, axonal trafficking, biogenesis and mitophagy) are also important determinants of mitochondrial function and quality control39,40. Multiple mechanisms are in place to respond to abnormal mitochondrial dynamics to ensure organelle preservation, including mitochondrial unfolded protein response (mtUPR, a process to maintain monoconidial proteostasis)41, enhanced biogenesis (a mechanism to produce new mitochondria), and mitophagy (a process that removes damaged organelles)42. Changes in Δψm play a key role in mitochondrial homeostasis signaling for selective elimination of damaged organelles through mitophagy by recruiting PTEN-induced kinase 1 (PINK1) and Parkin proteins to the mitochondrial membrane43. It is also a driving force for the translocation of ions and proteins essential for mitochondrial function. Mitochondria interact with other organelles, including the endoplasmic reticulum (ER), to modulate lipid homeostasis, immune response, and cell death44,45. Finally, changes in ATP levels associated with either increased energy utilization or reduced mitochondrial capacity led to an increase in the cellular adenosine monophosphate (AMP)/ATP ratio, which activates AMP-activated protein kinase (AMPK), a master regulator of cellular energy homeostasis46, 47, 48, 49. Active AMPK initiates a robust signaling cascade to restore energy balance. This dynamic process involves changes in lipid and glucose metabolism, mitochondrial dynamics and biogenesis, autophagy, and protein synthesis. Directly relevant to aging and neurodegenerative diseases is the AMPK-dependent reduction of inflammation and increase in levels of sirtuins, signaling molecules that regulate vast networks of metabolic and non-metabolic enzymes essential for healthy aging50, 51, 52. Numerous pathways affected by AMPK have been shown to be neuroprotective brining attention to AMPK as a drug target for neurodegenerative diseases46. However, the development of direct AMPK activators has been proven difficult given the delicate balance required for cellular energy homeostasis46. Nevertheless, it is now broadly accepted that indirect AMPK activation via exercise or caloric restriction is associated with increased health span and slowing down the progression of age-related neurodegenerative diseases53. The availability of such a robust signaling arsenal allows mitochondria to successfully adapt to environmental changes, ensuring sustained energy production and cell survival.
Figure 2.

Mitochondrial arsenal for intracellular signaling. Δψm, mitochondrial membrane potential; NAD+, nicotinamide adenine dinucleotide; TCA, tricarboxylic acid cycle; ROS, reactive oxygen species; AMP, adenosine monophosphate; ATP, adenosine triphosphate; AMPK, AMP-activated protein kinase; mtDNA, mitochondrial DNA.
2.3. Beneficial consequences of mitochondrial stress response
The most common mitochondrial stressors that induce mtUPR and ISR include fluctuations in energy sources, mtDNA mutations, changes in Δψm, Ca2+ and other ions, increased ROS production, and inhibition of the OXPHOS complexes26,42,54. The mechanisms of the mtUPR and the ISR studied in Caenorhabditis elegans and mammalian cells converged on changes in gene expression of chaperones, proteases, detoxification enzymes, and the engagement of mediators of metabolic and epigenetic reprograming, including activation of AMPK (Fig. 2). In C. elegans, initiation of this signaling cascade extended life span55 with epigenetic modification transmitted over four generations through histone H3K4 methylation56. Data in mammalian cells suggest that activation of ISR depends both on the nature of the mitochondrial stressor and the metabolic state of the cell57. Multiple studies conducted to date indicate that mild mitochondrial stress associated with the inhibition of OXPHOS complexes could induce an adaptive stress response that promotes health and longevity and delays the development of neurodegenerative diseases. Early evidence from studies in model organisms have demonstrated that mutations that decrease the activity of the mitochondrial respiratory chain resulted in a 20%–300% increase in the mean adult life span in C. elegans58, 59, 60, 61, 62, 63. Similar effects on longevity were achieved with RNA interference (RNAi) reduction in expression of the ETC components. The complete ablation of major ETC subunits resulted in severe phenotype and shorter lifespan indicating that only mild decrease in ETC activity was beneficial64. In flies, the RNAi of five genes encoding components of mitochondrial respiratory complexes I, III, IV, and V resulted in increased life span65. This phenomenon was not associated with altered assembly of respiratory complexes or reduced ATP production. Targeted RNAi of two complex I genes in adult tissues or in neurons alone was sufficient to extend Drosophila melanogaster life span65.
In mice, decreased expression of proteins involved in the ETC, especially the matrix arm subunits of complex I, increased longevity by 30% and was associated with improved complex I assembly, higher complex I-linked state 3 respiration and decreased ROS production66,67. Partial inhibition of complex IV and cytochrome c oxidase activity not only increased longevity in mice but also protected from neurodegeneration68. The severe deficiency in complex IV or mild deficiency in complex III expression in neurons resulted in a reduction of ROS and Aβ plaques in the APP/PS1 mouse model of AD69. Similarly, the depletion of mtDNA also led to a decrease in plaque accumulation in the same AD mouse model70. Inhibition of complex V has been linked to mitohormetic signaling, which increased neuronal survival in response to toxic agents in vitro and in vivo where mechanistic pathways converged on AMPK and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)71. Furthermore, the uncoupling of OXPHOS using molecules that depolarize mitochondria causing an ATP decrease and activation of AMPK has been considered as a therapeutic strategy for aging, obesity, neurodegeneration, and cancer72, 73, 74, 75, 76, 77. Despite showing an improvement of mitochondrial function and oxidative metabolism via adaptive stress response, the OXPHOS uncouplers had multiple off-target effects and toxicity, which limited their clinical use.
Longitudinal RNA sequencing analysis identified mitochondrial complex I as a hub in a module of genes whose expression was negatively correlated with lifespan in Nothobranchius furzeri, an African turquoise killifish78. Partial pharmacological inhibition of complex I with picomolar concentrations of the small molecule rotenone reversed aging-related regulation of gene expression rejuvenating the transcriptome and increasing life span in N. furzeri by 15%78. This is particularly interesting since high concentrations of rotenone are devastating for the organism due to a high ROS production79. In humans, data generated in a cohort of 2200 ultranonagenarians (and an equal number of controls) have shown that mutations in subunits of complex I that resulted in partial loss of its activity had a beneficial effect on longevity, while the simultaneous presence of mutations in complexes I and III or in complexes I and V appeared to be detrimental80. Thus, the beneficial adaptive stress response could be induced by multiple mitochondria-targeted stressors, including inhibition of OXPHOS complexes, mtDNA depletion, and mitochondrial uncoupling, among others. However, clinical translation in most cases is impeded by the lack of selectivity, specificity, and deleterious side effects.
Molecular mechanisms linked to life-extending interventions associated with mild inhibition of OXPHOS across species included adaptive response to energetic stress via activation of AMPK81. Additional important outcomes involved protection against oxidative stress that was attributed to the decreased rate of OXPHOS leading to overall lower production of ROS, AMPK-induced activation of the nuclear factor E2-related factor 2 (NRF2) signaling pathway, and mitohormetic response where sublethal ROS production associated with the ETC inhibition increased antioxidant defense via retrograde ROS signaling36,82, 83, 84. Furthermore, AMPK activation enhanced autophagy mediating the removal of damaged organelles and misfolded proteins to improve cellular proteostasis85, while increasing the production of “young” mitochondria via biogenesis. Importantly, these mechanisms overlap with the outcomes of non-pharmacological interventions, such as exercise and caloric restriction, known to reduce oxidative damage and inflammation and improve health, life span, and cognitive function86, 87, 88.
3. Mitochondria-targeted therapeutics
While inhibition of mitochondrial ETC to achieve healthy aging and prevent neurodegeneration appears counterintuitive, broad application of metformin (1,1-dimethylbiguanide), an inexpensive U.S. Food and Drug Administration (FDA)-approved drug to treat type II diabetes mellitus (T2DM), supports the feasibility of such an approach in humans. Metformin is a natural product derived from the medicinal herb ‘goat's-rue’, Galega officinalis. It has a robust safety record having been used in herbal medicine since medieval times89. Metformin became the most prescribed antidiabetic drug after the results of a prospective study conducted in overweight patients with T2DM with a median follow up of over 10 years where blood glucose control with metformin reduced the incidence of diabetes-related endpoints and all-cause mortality90. Metformin exerts its glucose-lowering effect by inhibiting hepatic gluconeogenesis and opposing the action of glucagon89,91,92. Among other multiple targets, metformin could inhibit mitochondrial complex I to result in defective cyclic AMP and protein kinase A signaling in response to glucagon and the stimulation of AMPK93. Metformin can cross the blood‒brain barrier (BBB) and have specific effects on the central nervous system. Biological, clinical, and epidemiological data suggest that T2DM increases risk of mild cognitive impairment (MCI), vascular dementia and AD. Clinical trials have found that application of antidiabetic drugs including metformin protected against cognitive decline in patients with MCI and AD, improving executive functioning, learning, memory, and attention94, 95, 96 (Table 1). These antidiabetic drugs positively affected mitochondrial and synaptic function, reduced neuroinflammation, and improved brain metabolism97. Interestingly, a recent systematic review reported that metformin reduced mortality and diseases of aging (cardiovascular disease and cancer) in patients who did not have diabetes, demonstrating that the effect of metformin on health span is independent of its antidiabetic properties98. Thus, metformin appears to mimic mechanisms involved in caloric restriction and exercise shown to slow the aging process, improve memory, and reduce oxidative stress99, 100, 101, 102, 103, 104. However, a few reports based on data generated in experimental animal models and collected in studies in diabetic patients suggest that metformin could increase amyloid accumulation and risk of developing AD105, 106, 107, 108, 109. These effects have been linked to overactivation of AMPK and vitamin B12 deficiency potentiated by metformin, which contribute to cognitive impairment110,111. Furthermore, it remains uncertain to what extent complex I inhibition contributes to the beneficial effect of metformin. Analysis of the literature indicates that plasma protein binding of metformin is negligible, and after oral administration at the recommended doses and dosing schedules, steady-state plasma concentrations are reached within 24–48 h and are generally less than 1 μg/mL (6.04 μmol/L). In controlled clinical trials, maximum metformin plasma levels did not exceed 5 μg/mL even at maximum doses112. The experimental data, however, indicate that metformin does not inhibit complex I at concentrations below 25 μmol/L93. Nevertheless, it was reported that metformin accumulates in mitochondria where it could reach concentrations sufficient for complex I inhibition113. Thus, while increasing evidence supports strong therapeutic potential for metformin as a neuroprotective therapy for neurodegenerative diseases of aging, additional safety and feasibility studies and mechanistic studies aimed at evaluating the contribution of complex I inhibition in different tissues to the drug efficacy are needed to identify potential risk factors, windows of therapeutic opportunity, and regimens114,115.
Table 1.
Mitochondrial complex I inhibitors in clinical trials.
| Complex I inhibitor | Structure | Condition or disease | Clinical trial IDa |
|---|---|---|---|
| Metformin | 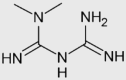 |
AD MCI Aging |
NCT01965756 NCT00620191 NCT02432287 |
| T2DM, obesity, cancer, inflammation, infectious diseases | 1681 trials completed and 2587 trials in total | ||
| Resveratrol (also inhibits complexes III and V) | 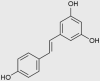 |
AD MCI AD Aging |
NCT02502253 NCT01219244 NCT01504854 NCT02095873 |
| Inflammation, T2DM, metabolic syndrome, mitochondrial myopathies, COVID-19 | 124 trials completed and 185 trials in total | ||
| Berberine |  |
AD, MCI | NCT03221894 |
| Inflammation, T2DM, obesity, metabolic disorder, hypertension, COVID-19 | 39 trials completed and 73 trials in total | ||
| Epigallocatechin-3-gallate (also inhibits complexes II and V) | 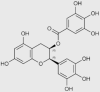 |
AD AD Huntington's disease Multiple sclerosis Down syndrome |
NCT00951834 NCT03978052 NCT01357681 NCT03740295 NCT01699711 |
| T2DM, metabolic syndrome, hypertension, inflammation, cancer | 60 trials completed and 95 trials in total | ||
| Droquinone and tricyclic ortho-carbonyl analogs |
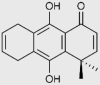 
|
Melasma HI/AIDS COVID-19 |
22 trials completed and 38 trials in total |
| Elesclomol |  |
Cancer |
NCT00888615 6 trials completed and 9 trials in total |
| IACS-10759 |  |
Acute myeloid leukemia Cancer |
NCT02882321 NCT03291938 |
| BAY 87-2243 | 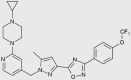 |
Cancer | NCT01297530 terminated |
| Benzophenone | 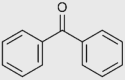 |
Breast cancer Melasma Infertility |
NCT03885648 4 trials completed |
| Capsaicin (also inhibits complex III) | 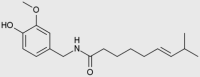 |
Pain, neuropathy | 193 trials completed and 286 trials in total |
| ME-143 | 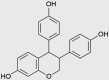 |
Solid tumors | NCT01401868 |
| ME-344 | 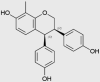 |
Solid tumors Breast cancers |
NCT01544322 NCT02806817 2 trials completed and 3 trials in total |
AD, Alzheimer's disease; MCI, mild cognitive impairment; T2DM, type 2 diabetes; HI, human immunodeficiency; AIDS, acquired immunodeficiency syndrome.
Listed are the most resent representative clinical trials as of September, 2021. Additional trials could be found on https://clinicaltrials.gov/.
Recent studies have identified other ETC inhibitors with a wide range of biological properties, including antioxidant, anticancer, anti-inflammatory, and cardio- and neuroprotective effects116, 117, 118, 119, 120. Resveratrol, a promising therapeutic compound that activates sirtuins121, has been shown to reduce the activity of mitochondrial complexes I, III, and V122, 123, 124. Similar to metformin, resveratrol stimulates key signaling pathways, including antioxidant defenses, reduction of inflammation via inhibiting NF-κB signaling, and AMPK activation, leading to improved mitochondrial function and biogenesis through sirtuin 1/AMPK/peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC1α) pathway and vitagenes, which prevent the deleterious effects triggered by oxidative stress125,126. Results of studies conducted in in vitro and in vivo models of AD provided evidence that resveratrol normalizes cholinergic neurotransmission and brain-derived neurotrophic factor (BDNF) expression, reduces oxidative stress, promotes Aβ peptide clearance and anti-amyloidogenic cleavage of APP, and reduces neuronal apoptosis127. Application of resveratrol was also beneficial in models of metabolic disorders, Huntington's disease (HD), and Parkinson's disease (PD), amyotrophic lateral sclerosis, stroke, and alcohol-induced neurodegenerative disorders128. However, the use of resveratrol in humans has been challenging, with limited bioavailability, pronounced adverse side effects, and inconsistent results were reported in healthy and unhealthy participants of clinical trials129. While data generated to date strongly support the importance of resveratrol for human health, the design of better analogs with greater potency, solubility, and bioavailability are needed122. Taken together, these data demonstrate that mild inhibition of the OXPHOS complexes engages a multifaceted mitochondria-mediated signaling cascade that improves multiple mechanisms of AD pathogenesis, including inflammation, mitochondrial dysfunction, abnormal energy and lipid homeostasis, the ER and oxidative stress making this therapeutic approach appealing7,9,40,130.
4. Partial mitochondrial complex I inhibition as a therapeutic strategy for AD and other diseases
Partial inhibition of complex I with small molecules emerged as a promising strategy to induce beneficial ISR. Table 1 lists complex I inhibitors that are in clinical trials for various human conditions, including T2DM, cancers, metabolic disorder, obesity, inflammatory and infectious diseases. Only metformin, resveratrol, berberine, and epigallocatechin-3-gallate were trialed in a limited number of studies for neurodegenerative diseases, including AD, HD, MCI, multiple sclerosis, and Down syndrome. Metformin improved cognitive function in patients with amnestic MCI, while resveratrol, berberine and epigallocatechin-3-gallate did not show statistically significant improvements in cognitive performance in patients with AD, HD, or MCI. While all four complex I inhibitors penetrate the BBB, the therapeutic effect of resveratrol, berberine and epigallocatechin-3-gallate was limited, probably due to a poor stability, short half-life, and a very low bioavailability (<1%) in contrast to metformin, which is stable and has better bioavailability. Therefore, modifications of current complex I inhibitors or the development of new small molecules with improved drug-like properties and bioavailability are needed to increase therapeutic efficacy for neurodegenerative diseases.
Complex I is the largest (970 kDa) multisubunit complex of the ETC with 14 central subunits involved in the oxidation of NADH to NAD+ at the flavin mononucleotide domain (FMN), transfer of the electrons along eight canonical iron-sulfur clusters to ubiquinone and its reduction, and proton pumping (Fig. 3)131. There are an additional 31 accessory subunits that are not directly associated with energy production131. Structures of bacterial and mammalian complex I have been determined by X-ray crystallography and cryogenic electron microscopy (cryo-EM) at high resolution, providing new insights into its assembly, proton-pumping machinery, the enzyme's catalytic mechanism, and dysfunctions associated with disease-causing mutations131, 132, 133. Complex I contributes significantly to the formation of ROS134. Interestingly, there are more than 60 complex I inhibitors that have a differential effect on the enzyme kinetics or ROS production, where molecules including rotenone, piericidin A, and rolliniastatin 1 and 2 increase ROS, while inhibitors such as stigmatellin, mucidin, capsaicin, and coenzyme Q2 prevent ROS formation134. Similarly, some mutations in complex I could preserve the conversion of NADH to NAD+ and, therefore, complex I activity while completely blocking pathological ROS production135. These data suggest that it is possible to develop safe and efficacious complex I inhibitors that are selective to the target and do not induce mitochondrial dysfunction associated with increased ROS production. These observations help to address concerns associated with the development of complex I inhibitors for chronic use in the elderly population to treat/delay the development of AD. For example, it is well established that mitochondrial complex I inhibitors such as 1-methyl 4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) or 3-nitropropionic acid could be used to mimic PD and HD, respectively. Indeed, the metabolic product of MPTP, MPP+, binds complex I at two sites and induces significant ROS formation. However, the pathophysiology of PD involves other mechanisms that affect mitochondrial function, including altered mitophagy and biogenesis, where the involvement of complex I inhibition has been called into question136,137. Moreover, a recent study demonstrated that α-synuclein aggregation was less common in complex I deficient neurons in the substantia nigra, implying that partial complex I inhibition in PD may be a beneficial compensatory mechanism for reducing increased ROS production138. Similarly, the involvement of complex I inhibition in HD pathogenesis may not be a primary mechanism underlying the disease pathogenesis139,140. Our data generated in primary mouse neurons from HD mice118 and in a bacterial artificial chromosome (BAC)-mediated transgenic mouse model of HD (unpublished observations) demonstrate that partial reduction of complex I activity improves multiple mechanisms affected by the expression of mutant huntingtin protein. Thus, a mounting body of evidence supports the feasibility of targeting complex I as a therapeutic strategy for neurodegenerative diseases. However, to develop safe and efficacious complex I inhibitors, it is imperative to determine the binding site, extent of inhibition of complex I activity, selectivity, and levels of ROS production.
Figure 3.

Redox-linked proton translocation by complex I. Electrons are transferred from the nicotinamide adenine dinucleotide (NADH) oxidation site (the flavin mononucleotide domain, FMN) to the ubiquinone reduction site via a chain of iron‒sulfur clusters (in gold); selected critical residues of the ubiquinone reduction site are shown in green (Tyr144, His95, His91). The FMN and ubiquinone are the main sites of reactive oxygen species (ROS) production. The membrane arm comprises three antiporter type subunits with discontinuous helices (ND5, marine; ND4, cyan; ND2, pink) corresponding to three potential proton translocation sites (black arrows). In the proximal part of the membrane arm (PP module) the π-bulge helix of ND6 (orange) and the discontinuous helix of ND1 (red) are highlighted. Residues constituting a fourth putative proton pathway (dashed arrow) are found in subunits ND2 and ND4L. In the center of the membrane arm a series of protonable residues (basic, blue; acidic, red) extends from subunit ND5 to subunit ND1 and terminates below the ubiquinone reduction site with a loop comprising a cluster of highly conserved acidic residues. Conformational changes linked to the redox chemistry of ubiquinone are proposed to induce an electric pulse that ultimately triggers proton translocation events in the membrane arm. Reprinted from Ref. 131 with the permission from the Elsevier.
We recently identified a small molecule tricyclic pyrone compound (CP2) that penetrates the BBB and accumulates in mitochondria where it mildly inhibits the activity of complex I117,141,142. CP2 is bioavailable, has low toxicity in vitro and in vivo, and has good drug-like properties and safety profile, demonstrating the lack of off-target activity against human receptors and ion channels141, 142, 143, 144. The genome-wide associations study using 196 human lymphoblastoid cell lines from healthy individuals with diverse age, sex and racial background demonstrated the safety of CP2 application at therapeutic doses144. The effect of CP2 on the activity of each of the respiratory complexes examined using enzymatic assays and mitochondria isolated from the mouse or postmortem human brain confirmed selective and specific inhibition of complex I117,142. The bioenergetics studies conducted in mouse primary cortical neurons using a Seahorse Extracellular Flux Analyzer (Agilent Technologies, Inc.) demonstrated that CP2 improved cellular bioenergetics augmenting spare respiratory capacity, an indicator of mitochondrial ability to produce energy under conditions of increased workload or stress, which is essential for long-term survival and function145. Similarly, CP2 increased mitochondrial respiratory control ratio and reduced proton leak, suggesting better coupling efficiency of the neuronal ETC, greater bioenergetic reserve, and enhanced ability to withstand stress. In vivo efficacy of chronic CP2 administration was examined in independent cohorts of male and female mice that express mutant human amyloid precursor protein (APP), mutant human presenilin 1 protein (PS1), mutant APP and PS1 (APP/PS1) or mutant APP, PS1 and human Tau protein (3xTgAD) starting in utero for 14 months, at pre- or symptomatic stages of the disease141, 142, 143. In all studies, chronic CP2 treatment did not induce toxicity or affect development. In all treatment paradigms, animals were allowed to have CP2 in drinking water ad lib. Remarkably, in all treatment groups, CP2 improved energy homeostasis in the brain and periphery (glucose uptake and utilization, glucose tolerance, and insulin resistance), synaptic activity, long-term potentiation, dendritic spine maturation, cognitive function and proteostasis (reduced Aβ and pTau levels), and reduced oxidative stress and inflammation in the brain and periphery, ultimately blocking the ongoing neurodegeneration (Fig. 4)142,143. We observed increased levels of ATP consistent with improvement of brain energy homeostasis and reduced levels of ceramides, indicative of the release of the ER stress prominent in patients with AD142. Therapeutic efficacy was monitored using translational in vivo biomarker fluorodeoxyglucose-positron emission tomography (FDG-PET), phosphorus-31 magnetic resonance imaging (31P MRI), and blood-based metabolomics. Interestingly, this treatment augmented mitochondrial dynamics and function, including restoration of axonal trafficking in neurons from CP2-treated PS1 and APP/PS1 mice117. While CP2 was demonstrated to be selective and specific complex I inhibitor that lacks the off-target activities142,144, it was shown to interfere with the formation of Aβ aggregates146, 147, 148, which could also contribute to its beneficial properties.
Figure 4.

Partial inhibition of mitochondria complex I with small molecule compound CP2 activates multiple AMP-activated protein kinase-dependent mechanisms leading to neuroprotection in mouse models of Alzheimer's disease.
Further translational support for this therapeutic strategy was provided by the cross-validation of transcriptomic data generated in CP2-treated AD mice with the human brain transcriptome data available through the co-expression meta-analysis in the Accelerating Medicines Partnership Program for Alzheimer's disease database (ampadportal.org). Beneficial changes in gene expression associated with CP2 treatment in APP/PS1 mice overlap with signatures established in patients with AD, female patients in particular, supporting high translational potential of this approach142. Major translational targets included the immune system response and multiple pathways involved in synaptic function and neurotransmission, which underlie early pathology in patients with AD149. Since CP2 improved axonogenesis and dendritic spine morphology and function, it is feasible that this treatment could also induce neuronal regeneration.
Molecular mechanisms of neuroprotection converged on the AMPK activation and the downstream signaling that resulted in increased resistance to oxidative stress, augmented mitochondrial bioenergetics, improved glucose uptake and utilization, increased production of sirtuins 1 and 3, reduction of glycogen synthase kinase 3 beta (GSK3β) activity, significant reduction in levels of pTau and Aβ, and increased autophagy and levels of BDNF and synaptic proteins in vivo117,142,143. With CP2-inhibited complex I activity, the overall energy levels in the brain measured using 31P MRI were not decreased, which could be attributed to enhanced mitochondrial biogenesis and bioenergetics and improved brain energy homeostasis142. The translational relevance of this approach is emphasized by the fact that the intervention was started after the onset of Aβ neuropathology150, cognitive symptoms151, bioenergetic dysfunction152, and progressive neurodegeneration153. These data provide further support for brain energy rescue as a novel concept for treatment of neurodegenerative diseases of aging9,142,143. Furthermore, similar to metformin and resveratrol, CP2 also enhanced health and life span in chronologically aged wild-type mice and mice fed with a high-fat diet (our unpublished observations), implying that the activation of mitochondria-induced ISR using complex I as a small molecule druggable target could delay the onset or block the progression of age-related neurodegenerative diseases.
5. Conclusions
We summarized here evidence for a novel therapeutic approach to exploit the incredible ability of mitochondria to engage multifaceted neuroprotective stress response triggered by partial complex I inhibition. This approach promises relief for multiple human conditions, including, but not limited to mitochondrial diseases, HD, PD, and amyotrophic lateral sclerosis, and to promote healthy aging to delay the onset of neurogenerative diseases, AD in particular, where age is the greatest risk factor. There is a mounting body of evidence generated in model organisms and humans in support of the safety of chronic application of complex I inhibitors. However, a better understanding of the molecular mechanisms is required to establish safety in translation to humans, including the development of biomarkers that inform on mitochondrial function and the capacity to induce the beneficial stress response. Further therapeutic developments should produce selective and specific complex I inhibitors capable of penetrating the BBB with excellent safety profile.
Acknowledgments
We thank Drs. Thomas Chung and Graham Johnson for constructive suggestions, and Ms. Shelly Gochnauer for help with the manuscript. This research was supported by grants from the National Institutes of Health [grant numbers RF1AG55549, R01NS107265, RO1AG062135, AG59093, AG072899, UG3/UH3NS 113776, all to Eugenia Trushina, USA]. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of the NIH. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences
Author contributions
Eugenia Trushina conceptualized the manuscript, Eugenia Trushina, Sergey Trushin, and Md Fayad Hasan wrote the manuscript, Md Fayad Hasan developed figures, all authors approved the manuscript.
Conflicts of interest
Eugenia Trushina is an inventor on the patent US20180044295A1 (“Compounds for modulating mitochondrial function”). The authors declare no conflict of interest.
References
- 1.Alzheimer's Association 2017 Alzheimer's disease facts and figures. Alzheimers Dement. 2017;13:325–373. [Google Scholar]
- 2.Mayeux R., Stern Y. Epidemiology of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006239. doi: 10.1101/cshperspect.a006239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Talwar P., Sinha J., Grover S., Rawat C., Kushwaha S., Agarwal R., et al. Dissecting complex and multifactorial nature of Alzheimer's disease pathogenesis: a clinical, genomic, and systems biology perspective. Mol Neurobiol. 2016;53:4833–4864. doi: 10.1007/s12035-015-9390-0. [DOI] [PubMed] [Google Scholar]
- 4.Deming Y., Dumitrescu L., Barnes L.L., Thambisetty M., Kunkle B., Gifford K.A., et al. Sex-specific genetic predictors of Alzheimer's disease biomarkers. Acta Neuropathol. 2018;136:857–872. doi: 10.1007/s00401-018-1881-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Toledo J.B., Arnold M., Kastenmüller G., Chang R., Baillie R.A., Han X., et al. Metabolic network failures in Alzheimer's disease: a biochemical road map. Alzheimers Dement. 2017;13:965–984. doi: 10.1016/j.jalz.2017.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swerdlow R.H. Mitochondria and mitochondrial cascades in Alzheimer's disease. J Alzheimers Dis. 2018;62:1403–1416. doi: 10.3233/JAD-170585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tönnies E., Trushina E. Oxidative stress, synaptic dysfunction, and Alzheimer's disease. J Alzheimers Dis. 2017;57:1105–1121. doi: 10.3233/JAD-161088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mosconi L., Pupi A., De Leon M.J. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer's disease. Ann N Y Acad Sci. 2008;1147:180–195. doi: 10.1196/annals.1427.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cunnane S.C., Trushina E., Morland C., Prigione A., Casadesus G., Andrews Z.B., et al. Brain energy rescue: an emerging therapeutic concept for neurodegenerative disorders of ageing. Nat Rev Drug Discov. 2020;19:609–633. doi: 10.1038/s41573-020-0072-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Steinhubl S.R. Why have antioxidants failed in clinical trials?. Am J Cardiol. 2008;101:S14D–S19D. doi: 10.1016/j.amjcard.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 11.Wang W., Karamanlidis G., Tian R. Novel targets for mitochondrial medicine. Sci Transl Med. 2016;8:326rv3. doi: 10.1126/scitranslmed.aac7410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andersen S.L. Centenarians as models of resistance and resilience to Alzheimer's disease and related dementias. Adv Geriatr Med Res. 2020;2 doi: 10.20900/agmr20200018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seto M., Weiner R.L., Dumitrescu L., Hohman T.J. Protective genes and pathways in Alzheimer's disease: moving towards precision interventions. Mol Neurodegener. 2021;16:29. doi: 10.1186/s13024-021-00452-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dyall S.D., Brown M.T., Johnson P.J. Ancient invasions: from endosymbionts to organelles. Science. 2004;304:253–257. doi: 10.1126/science.1094884. [DOI] [PubMed] [Google Scholar]
- 15.Bélanger M., Allaman I., Magistretti P.J. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011;14:724–738. doi: 10.1016/j.cmet.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 16.Vacanti N.M., Divakaruni A.S., Green C.R., Parker S.J., Henry R.R., Ciaraldi T.P., et al. Regulation of substrate utilization by the mitochondrial pyruvate carrier. Mol Cell. 2014;56:425–435. doi: 10.1016/j.molcel.2014.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mookerjee S.A., Gerencser A.A., Nicholls D.G., Brand M.D. Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J Biol Chem. 2017;292:7189–7207. doi: 10.1074/jbc.M116.774471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang L., Trushina E. In: Neuromethods techniques to investigate mitochondrial function in neurons. Strack S., Usachev Y, editors., editors. vol. 123. Springer Nature; New York: 2017. Chapter 5; pp. 95–113. [Google Scholar]
- 19.Hüttemann M., Lee I., Pecinova A., Pecina P., Przyklenk K., Doan J.W. Regulation of oxidative phosphorylation, the mitochondrial membrane potential, and their role in human disease. J Bioenerg Biomembr. 2008;40:445–456. doi: 10.1007/s10863-008-9169-3. [DOI] [PubMed] [Google Scholar]
- 20.Brand M.D., Nicholls D.G. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mitchell P., Moyle J. Estimation of membrane potential and pH difference across the cristae membrane of rat liver mitochondria. Eur J Biochem. 1969;7:471–484. doi: 10.1111/j.1432-1033.1969.tb19633.x. [DOI] [PubMed] [Google Scholar]
- 22.Nicholls D.G. The influence of respiration and ATP hydrolysis on the proton-electrochemical gradient across the inner membrane of rat-liver mitochondria as determined by ion distribution. Eur J Biochem. 1974;50:305–315. doi: 10.1111/j.1432-1033.1974.tb03899.x. [DOI] [PubMed] [Google Scholar]
- 23.Brand M.D., Chien L.F., Ainscow E.K., Rolfe D.F., Porter R.K. The causes and functions of mitochondrial proton leak. Biochim Biophys Acta. 1994;1187:132–139. doi: 10.1016/0005-2728(94)90099-x. [DOI] [PubMed] [Google Scholar]
- 24.Yellen G. Fueling thought: management of glycolysis and oxidative phosphorylation in neuronal metabolism. J Cell Biol. 2018;217:2235–2246. doi: 10.1083/jcb.201803152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chandel N.S. Evolution of mitochondria as signaling organelles. Cell Metab. 2015;22:204–206. doi: 10.1016/j.cmet.2015.05.013. [DOI] [PubMed] [Google Scholar]
- 26.Mottis A., Herzig S., Auwerx J. Mitocellular communication: shaping health and disease. Science. 2019;366:827–832. doi: 10.1126/science.aax3768. [DOI] [PubMed] [Google Scholar]
- 27.Campbell S.L., Wellen K.E. Metabolic signaling to the nucleus in cancer. Mol Cell. 2018;71:398–408. doi: 10.1016/j.molcel.2018.07.015. [DOI] [PubMed] [Google Scholar]
- 28.Weinberg S.E., Singer B.D., Steinert E.M., Martinez C.A., Mehta M.M., Martínez-Reyes I., et al. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature. 2019;565:495–499. doi: 10.1038/s41586-018-0846-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Verdin E. NAD+ in aging, metabolism, and neurodegeneration. Science. 2015;350:1208–1213. doi: 10.1126/science.aac4854. [DOI] [PubMed] [Google Scholar]
- 30.Katsyuba E., Auwerx J. Modulating NAD+ metabolism, from bench to bedside. EMBO J. 2017;36:2670–2683. doi: 10.15252/embj.201797135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Samant S.A., Zhang H.J., Hong Z., Pillai V.B., Sundaresan N.R., Wolfgeher D., et al. SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics during stress. Mol Cell Biol. 2014;34:807–819. doi: 10.1128/MCB.01483-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng A., Yang Y., Zhou Y., Maharana C., Lu D., Peng W., et al. Mitochondrial SIRT3 mediates adaptive responses of neurons to exercise and metabolic and excitatory challenges. Cell Metab. 2016;23:128–142. doi: 10.1016/j.cmet.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Diebold L., Chandel N.S. Mitochondrial ROS regulation of proliferating cells. Free Radic Biol Med. 2016;100:86–93. doi: 10.1016/j.freeradbiomed.2016.04.198. [DOI] [PubMed] [Google Scholar]
- 34.Chandel N.S., Maltepe E., Goldwasser E., Mathieu C.E., Simon M.C., Schumacker P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A. 1998;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schieber M., Chandel N.S. ROS function in redox signaling and oxidative stress. Curr Biol. 2014;24:R453–R462. doi: 10.1016/j.cub.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ristow M. Unraveling the truth about antioxidants: mitohormesis explains ROS-induced health benefits. Nat Med. 2014;20:709–711. doi: 10.1038/nm.3624. [DOI] [PubMed] [Google Scholar]
- 37.Kim J., Gupta R., Blanco L.P., Yang S., Shteinfer-Kuzmine A., Wang K., et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science. 2019;366:1531–1536. doi: 10.1126/science.aav4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.West A.P., Khoury-Hanold W., Staron M., Tal M.C., Pineda C.M., Lang S.M., et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520:553–557. doi: 10.1038/nature14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Labbé K., Murley A., Nunnari J. Determinants and functions of mitochondrial behavior. Annu Rev Cell Dev Biol. 2014;30:357–391. doi: 10.1146/annurev-cellbio-101011-155756. [DOI] [PubMed] [Google Scholar]
- 40.Flannery P.J., Trushina E. Mitochondrial dynamics and transport in Alzheimer's disease. Mol Cell Neurosci. 2019;98:109–120. doi: 10.1016/j.mcn.2019.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu L., Zhou Q., He L., Chen L. Mitochondrial unfolded protein response: an emerging pathway in human diseases. Free Radic Biol Med. 2021;163:125–134. doi: 10.1016/j.freeradbiomed.2020.12.013. [DOI] [PubMed] [Google Scholar]
- 42.Eisner V., Picard M., Hajnóczky G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat Cell Biol. 2018;20:755–765. doi: 10.1038/s41556-018-0133-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zorova L.D., Popkov V.A., Plotnikov E.Y., Silachev D.N., Pevzner I.B., Jankauskas S.S., et al. Mitochondrial membrane potential. Anal Biochem. 2018;552:50–59. doi: 10.1016/j.ab.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.López-Crisosto C., Bravo-Sagua R., Rodriguez-Peña M., Mera C., Castro P.F., Quest A.F., et al. ER-to-mitochondria miscommunication and metabolic diseases. Biochim Biophys Acta. 2015;1852:2096–2105. doi: 10.1016/j.bbadis.2015.07.011. [DOI] [PubMed] [Google Scholar]
- 45.Raeisossadati R., Ferrari M.F.R. Mitochondria–ER tethering in neurodegenerative diseases. Cell Mol Neurobiol. 2020 doi: 10.1007/s10571-020-01008-9. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Steinberg G.R., Carling D. AMP-activated protein kinase: the current landscape for drug development. Nat Rev Drug Discov. 2019;18:527–551. doi: 10.1038/s41573-019-0019-2. [DOI] [PubMed] [Google Scholar]
- 47.Hardie D.G. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- 48.Hardie D.G., Ross F.A., Hawley S.A. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Herzig S., Shaw R.J. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2018;19:121–135. doi: 10.1038/nrm.2017.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peixoto C.A., Oliveira W.H., Araújo S.M.D.R., Nunes A.K.S. AMPK activation: role in the signaling pathways of neuroinflammation and neurodegeneration. Exp Neurol. 2017;298:31–41. doi: 10.1016/j.expneurol.2017.08.013. [DOI] [PubMed] [Google Scholar]
- 51.Ruderman N.B., Xu X.J., Nelson L., Cacicedo J.M., Saha A.K., Lan F., et al. AMPK and SIRT1: a long-standing partnership?. Am J Physiol Endocrinol Metab. 2010;298:E751–E760. doi: 10.1152/ajpendo.00745.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van de Ven R.A.H., Santos D., Haigis M.C. Mitochondrial sirtuins and molecular mechanisms of aging. Trends Mol Med. 2017;23:320–331. doi: 10.1016/j.molmed.2017.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Flannery P.J., Trushina E. Mitochondrial dysfunction in Alzheimer's disease and progress in mitochondria-targeted therapeutics. Curr Behav Neurosci Rep. 2019;6:88–102. [Google Scholar]
- 54.Nargund A.M., Pellegrino M.W., Fiorese C.J., Baker B.M., Haynes C.M. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science. 2012;337:587–590. doi: 10.1126/science.1223560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Merkwirth C., Jovaisaite V., Durieux J., Matilainen O., Jordan S.D., Quiros P.M., et al. Two conserved histone demethylases regulate mitochondrial stress-induced longevity. Cell. 2016;165:1209–1223. doi: 10.1016/j.cell.2016.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ma C., Niu R., Huang T., Shao L.W., Peng Y., Ding W., et al. N6-Methyldeoxyadenine is a transgenerational epigenetic signal for mitochondrial stress adaptation. Nat Cell Biol. 2019;21:319–327. doi: 10.1038/s41556-018-0238-5. [DOI] [PubMed] [Google Scholar]
- 57.Mick E., Titov D.V., Skinner O.S., Sharma R., Jourdain A.A., Mootha V.K. Distinct mitochondrial defects trigger the integrated stress response depending on the metabolic state of the cell. Elife. 2020;9:e49178. doi: 10.7554/eLife.49178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee S.S., Lee R.Y., Fraser A.G., Kamath R.S., Ahringer J., Ruvkun G. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat Genet. 2003;33:40–48. doi: 10.1038/ng1056. [DOI] [PubMed] [Google Scholar]
- 59.Dillin A., Hsu A.L., Arantes-Oliveira N., Lehrer-Graiwer J., Hsin H., Fraser A.G., et al. Rates of behavior and aging specified by mitochondrial function during development. Science. 2002;298:2398–2401. doi: 10.1126/science.1077780. [DOI] [PubMed] [Google Scholar]
- 60.Feng J., Bussière F., Hekimi S. Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev Cell. 2001;1:633–644. doi: 10.1016/s1534-5807(01)00071-5. [DOI] [PubMed] [Google Scholar]
- 61.Hamilton B., Dong Y., Shindo M., Liu W., Odell I., Ruvkun G., et al. A systematic RNAi screen for longevity genes in C. elegans. Genes Dev. 2005;19:1544–1555. doi: 10.1101/gad.1308205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miyadera H., Amino H., Hiraishi A., Taka H., Murayama K., Miyoshi H., et al. Altered quinone biosynthesis in the long-lived clk-1 mutants of Caenorhabditis elegans. J Biol Chem. 2001;276:7713–7716. doi: 10.1074/jbc.C000889200. [DOI] [PubMed] [Google Scholar]
- 63.Kayser E.B., Sedensky M.M., Morgan P.G., Hoppel C.L. Mitochondrial oxidative phosphorylation is defective in the long-lived mutant clk-1. J Biol Chem. 2004;279:54479–54486. doi: 10.1074/jbc.M403066200. [DOI] [PubMed] [Google Scholar]
- 64.Rea S.L., Ventura N., Johnson T.E. Relationship between mitochondrial electron transport chain dysfunction, development, and life extension in Caenorhabditis elegans. PLoS Biol. 2007;5:e259. doi: 10.1371/journal.pbio.0050259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Copeland J.M., Cho J., Lo T., Jr., Hur J.H., Bahadorani S., Arabyan T., et al. Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr Biol. 2009;19:1591–1598. doi: 10.1016/j.cub.2009.08.016. [DOI] [PubMed] [Google Scholar]
- 66.Miwa S., Jow H., Baty K., Johnson A., Czapiewski R., Saretzki G., et al. Low abundance of the matrix arm of complex I in mitochondria predicts longevity in mice. Nat Commun. 2014;5:3837. doi: 10.1038/ncomms4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lapointe J., Hekimi S. Early mitochondrial dysfunction in long-lived Mclk1+/– mice. J Biol Chem. 2008;283:26217–26227. doi: 10.1074/jbc.M803287200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dell'agnello C., Leo S., Agostino A., Szabadkai G., Tiveron C., Zulian A., et al. Increased longevity and refractoriness to Ca2+-dependent neurodegeneration in Surf1 knockout mice. Hum Mol Genet. 2007;16:431–444. doi: 10.1093/hmg/ddl477. [DOI] [PubMed] [Google Scholar]
- 69.Fukui H., Diaz F., Garcia S., Moraes C.T. Cytochrome c oxidase deficiency in neurons decreases both oxidative stress and amyloid formation in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2007;104:14163–14168. doi: 10.1073/pnas.0705738104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pinto M., Pickrell A.M., Fukui H., Moraes C.T. Mitochondrial DNA damage in a mouse model of Alzheimer's disease decreases amyloid beta plaque formation. Neurobiol Aging. 2013;34:2399–2407. doi: 10.1016/j.neurobiolaging.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.García-Aguilar A., Cuezva J.M. A review of the inhibition of the mitochondrial ATP synthase by IF1 in vivo: reprogramming energy metabolism and inducing mitohormesis. Front Physiol. 2018;9:1322. doi: 10.3389/fphys.2018.01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ross E., Ata R., Thavarajah T., Medvedev S., Bowden P., Marshall J.G., et al. AMP-activated protein kinase regulates the cell surface proteome and integrin membrane traffic. PLoS One. 2015;10:e0128013. doi: 10.1371/journal.pone.0128013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rossmeisl M., Barbatelli G., Flachs P., Brauner P., Zingaretti M.C., Marelli M., et al. Expression of the uncoupling protein 1 from the aP2 gene promoter stimulates mitochondrial biogenesis in unilocular adipocytes in vivo. Eur J Biochem. 2002;269:19–28. doi: 10.1046/j.0014-2956.2002.02627.x. [DOI] [PubMed] [Google Scholar]
- 74.Birk A.V., Chao W.M., Bracken C., Warren J.D., Szeto H.H. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br J Pharmacol. 2014;171:2017–2028. doi: 10.1111/bph.12468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Desquiret V., Loiseau D., Jacques C., Douay O., Malthièry Y., Ritz P., et al. Dinitrophenol-induced mitochondrial uncoupling in vivo triggers respiratory adaptation in HepG2 cells. Biochim Biophys Acta. 2006;1757:21–30. doi: 10.1016/j.bbabio.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 76.Urra F.A., Muñoz F., Córdova-Delgado M., Ramírez M.P., Peña-Ahumada B., Rios M., et al. FR58P1a; a new uncoupler of OXPHOS that inhibits migration in triple-negative breast cancer cells via Sirt1/AMPK/β1-integrin pathway. Sci Rep. 2018;8:13190. doi: 10.1038/s41598-018-31367-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vaughan R.A., Garcia-Smith R., Bisoffi M., Trujillo K.A., Conn C.A. Effects of caffeine on metabolism and mitochondria biogenesis in rhabdomyosarcoma cells compared with 2,4-dinitrophenol. Nutr Metab Insights. 2012;5:59–70. doi: 10.4137/NMI.S10233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Baumgart M., Priebe S., Groth M., Hartmann N., Menzel U., Pandolfini L., et al. Longitudinal RNA-Seq analysis of vertebrate aging identifies mitochondrial complex I as a small-molecule-sensitive modifier of lifespan. Cell Syst. 2016;2:122–132. doi: 10.1016/j.cels.2016.01.014. [DOI] [PubMed] [Google Scholar]
- 79.Heinz S., Freyberger A., Lawrenz B., Schladt L., Schmuck G., Ellinger-Ziegelbauer H. Mechanistic investigations of the mitochondrial complex I inhibitor rotenone in the context of pharmacological and safety evaluation. Sci Rep. 2017;7:45465. doi: 10.1038/srep45465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Raule N., Sevini F., Li S., Barbieri A., Tallaro F., Lomartire L., et al. The co-occurrence of mtDNA mutations on different oxidative phosphorylation subunits, not detected by haplogroup analysis, affects human longevity and is population specific. Aging Cell. 2014;13:401–407. doi: 10.1111/acel.12186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Salminen A., Kaarniranta K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res Rev. 2012;11:230–241. doi: 10.1016/j.arr.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 82.Chen A., Raule N., Chomyn A., Attardi G. Decreased reactive oxygen species production in cells with mitochondrial haplogroups associated with longevity. PLoS One. 2012;7:e46473. doi: 10.1371/journal.pone.0046473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.D'Aquila P., Bellizzi D., Passarino G. Mitochondria in health, aging and diseases: the epigenetic perspective. Biogerontology. 2015;16:569–585. doi: 10.1007/s10522-015-9562-3. [DOI] [PubMed] [Google Scholar]
- 84.Weir H.J., Yao P., Huynh F.K., Escoubas C.C., Goncalves R.L., Burkewitz K., et al. Dietary restriction and AMPK increase lifespan via mitochondrial network and peroxisome remodeling. Cell Metab. 2017;26:884–896.e5. doi: 10.1016/j.cmet.2017.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kim J., Kundu M., Viollet B., Guan K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ma L., Wang R., Dong W., Zhao Z. Caloric restriction can improve learning and memory in C57/BL mice probably via regulation of the AMPK signaling pathway. Exp Gerontol. 2018;102:28–35. doi: 10.1016/j.exger.2017.11.013. [DOI] [PubMed] [Google Scholar]
- 87.Il'yasova D., Fontana L., Bhapkar M., Pieper C.F., Spasojevic I., Redman L.M., et al. Effects of 2 years of caloric restriction on oxidative status assessed by urinary F2-isoprostanes: the CALERIE 2 randomized clinical trial. Aging Cell. 2018;17:e12719. doi: 10.1111/acel.12719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Escobar K.A., Cole N.H., Mermier C.M., VanDusseldorp T.A. Autophagy and aging: maintaining the proteome through exercise and caloric restriction. Aging Cell. 2019;18:e12876. doi: 10.1111/acel.12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rena G., Hardie D.G., Pearson E.R. The mechanisms of action of metformin. Diabetologia. 2017;60:1577–1585. doi: 10.1007/s00125-017-4342-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.UK Prospective Diabetes Study (UKPDS) Group Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34) Lancet. 1998;352:854–865. Erratum in: Lancet 1998;352:1558. [PubMed] [Google Scholar]
- 91.Cameron A.R., Logie L., Patel K., Erhardt S., Bacon S., Middleton P., et al. Metformin selectively targets redox control of complex I energy transduction. Redox Biol. 2018;14:187–197. doi: 10.1016/j.redox.2017.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang C.S., Li M., Ma T., Zong Y., Cui J., Feng J.W., et al. Metformin activates AMPK through the lysosomal pathway. Cell Metab. 2016;24:521–522. doi: 10.1016/j.cmet.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 93.Wang Y., An H., Liu T., Qin C., Sesaki H., Guo S., et al. Metformin improves mitochondrial respiratory activity through activation of AMPK. Cell Rep. 2019;29:1511–1523.e5. doi: 10.1016/j.celrep.2019.09.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cao B., Rosenblat J.D., Brietzke E., Park C., Lee Y., Musial N., et al. Comparative efficacy and acceptability of antidiabetic agents for Alzheimer's disease and mild cognitive impairment: a systematic review and network meta-analysis. Diabetes Obes Metab. 2018;20:2467–2471. doi: 10.1111/dom.13373. [DOI] [PubMed] [Google Scholar]
- 95.Koenig A.M., Mechanic-Hamilton D., Xie S.X., Combs M.F., Cappola A.R., Xie L., et al. Effects of the insulin sensitizer metformin in Alzheimer disease: pilot data from a randomized placebo-controlled crossover study. Alzheimer Dis Assoc Disord. 2017;31:107–113. doi: 10.1097/WAD.0000000000000202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Luchsinger J.A., Perez T., Chang H., Mehta P., Steffener J., Pradabhan G., et al. Metformin in amnestic mild cognitive impairment: results of a pilot randomized placebo controlled clinical trial. J Alzheimers Dis. 2016;51:501–514. doi: 10.3233/JAD-150493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Herath P.M., Cherbuin N., Eramudugolla R., Anstey K.J. The effect of diabetes medication on cognitive function: evidence from the PATH through life study. BioMed Res Int. 2016;2016:7208429. doi: 10.1155/2016/7208429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Campbell J.M., Bellman S.M., Stephenson M.D., Lisy K. Metformin reduces all-cause mortality and diseases of ageing independent of its effect on diabetes control: a systematic review and meta-analysis. Ageing Res Rev. 2017;40:31–44. doi: 10.1016/j.arr.2017.08.003. [DOI] [PubMed] [Google Scholar]
- 99.Cameron A.R., Morrison V.L., Levin D., Mohan M., Forteath C., Beall C., et al. Anti-inflammatory effects of metformin irrespective of diabetes status. Circ Res. 2016;119:652–665. doi: 10.1161/CIRCRESAHA.116.308445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Handschin C. Caloric restriction and exercise “mimetics’’: ready for prime time?. Pharmacol Res. 2016;103:158–166. doi: 10.1016/j.phrs.2015.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Anisimov V.N., Berstein L.M., Egormin P.A., Piskunova T.S., Popovich I.G., Zabezhinski M.A., et al. Effect of metformin on life span and on the development of spontaneous mammary tumors in HER-2/neu transgenic mice. Exp Gerontol. 2005;40:685–693. doi: 10.1016/j.exger.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 102.Anderson R.M., Shanmuganayagam D., Weindruch R. Caloric restriction and aging: studies in mice and monkeys. Toxicol Pathol. 2009;37:47–51. doi: 10.1177/0192623308329476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Witte A.V., Fobker M., Gellner R., Knecht S., Flöel A. Caloric restriction improves memory in elderly humans. Proc Natl Acad Sci U S A. 2009;106:1255–1260. doi: 10.1073/pnas.0808587106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Campbell J.M., Stephenson M.D., de Courten B., Chapman I., Bellman S.M., Aromataris E. Metformin use associated with reduced risk of dementia in patients with diabetes: a systematic review and meta-analysis. J Alzheimers Dis. 2018;65:1225–1236. doi: 10.3233/JAD-180263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chen Y., Zhou K., Wang R., Liu Y., Kwak Y.D., Ma T., et al. Antidiabetic drug metformin (GlucophageR) increases biogenesis of Alzheimer’s amyloid peptides via up-regulating BACE1 transcription. Proc Natl Acad Sci U S A. 2009;106:3907–3912. doi: 10.1073/pnas.0807991106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Imfeld P., Bodmer M., Jick S.S., Meier C.R. Metformin, other antidiabetic drugs, and risk of Alzheimer's disease: a population-based case-control study. J Am Geriatr Soc. 2012;60:916–921. doi: 10.1111/j.1532-5415.2012.03916.x. [DOI] [PubMed] [Google Scholar]
- 107.Moore E.M., Mander A.G., Ames D., Kotowicz M.A., Carne R.P., Brodaty H., et al. Increased risk of cognitive impairment in patients with diabetes is associated with metformin. Diabetes Care. 2013;36:2981–2987. doi: 10.2337/dc13-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mairet-Coello G., Courchet J., Pieraut S., Courchet V., Maximov A., Polleux F. The CAMKK2-AMPK kinase pathway mediates the synaptotoxic effects of Aβ oligomers through Tau phosphorylation. Neuron. 2013;78:94–108. doi: 10.1016/j.neuron.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cai Z., Yan L.J., Li K., Quazi S.H., Zhao B. Roles of AMP-activated protein kinase in Alzheimer's disease. NeuroMolecular Med. 2012;14:1–14. doi: 10.1007/s12017-012-8173-2. [DOI] [PubMed] [Google Scholar]
- 110.Chapman L.E., Darling A.L., Brown J.E. Association between metformin and vitamin B12 deficiency in patients with type 2 diabetes: a systematic review and meta-analysis. Diabetes Metab. 2016;42:316–327. doi: 10.1016/j.diabet.2016.03.008. [DOI] [PubMed] [Google Scholar]
- 111.Moore E., Mander A., Ames D., Carne R., Sanders K., Watters D. Cognitive impairment and vitamin B12: a review. Int Psychogeriatr. 2012;24:541–556. doi: 10.1017/S1041610211002511. [DOI] [PubMed] [Google Scholar]
- 112.Bristol-Myers Squibb Company . 2020. GLUCOPHAGE® (metformin hydrochloride extended-release tablets) Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/020357s031,021202s016lbl.pdf. [Google Scholar]
- 113.Hu D., Xie F., Xiao Y., Lu C., Zhong J., Huang D., et al. Metformin: a potential candidate for targeting aging mechanisms. Aging Dis. 2021;12:480–493. doi: 10.14336/AD.2020.0702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Arnold S.E., Arvanitakis Z., Macauley-Rambach S.L., Koenig A.M., Wang H.Y., Ahima R.S., et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat Rev Neurol. 2018;14:168–181. doi: 10.1038/nrneurol.2017.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rotermund C., Machetanz G., Fitzgerald J.C. The therapeutic potential of metformin in neurodegenerative diseases. Front Endocrinol (Lausanne) 2018;9:400. doi: 10.3389/fendo.2018.00400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Vingtdeux V., Dreses-Werringloer U., Zhao H., Davies P., Marambaud P. Therapeutic potential of resveratrol in Alzheimer’s disease. BMC Neurosci. 2008;9 Suppl 2:S6. doi: 10.1186/1471-2202-9-S2-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhang L., Zhang S., Maezawa I., Trushin S., Minhas P., Pinto M., et al. Modulation of mitochondrial complex I activity averts cognitive decline in multiple animal models of familial Alzheimer's disease. EBioMedicine. 2015;2:294–305. doi: 10.1016/j.ebiom.2015.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Trushina E., Rana S., McMurray C.T., Hua D.H. Tricyclic pyrone compounds prevent aggregation and reverse cellular phenotypes caused by expression of mutant huntingtin protein in striatal neurons. BMC Neurosci. 2009;10:73. doi: 10.1186/1471-2202-10-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Leifert W.R., Abeywardena M.Y. Cardioprotective actions of grape polyphenols. Nutr Res. 2008;28:729–737. doi: 10.1016/j.nutres.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 120.Salehi B., Mishra A.P., Nigam M., Sener B., Kilic M., Sharifi-Rad M., et al. Resveratrol: a double-edged sword in health benefits. Biomedicines. 2018;6:91. doi: 10.3390/biomedicines6030091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hubbard B.P., Sinclair D.A. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol Sci. 2014;35:146–154. doi: 10.1016/j.tips.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gueguen N., Desquiret-Dumas V., Leman G., Chupin S., Baron S., Nivet-Antoine V., et al. Resveratrol directly binds to mitochondrial complex I and increases oxidative stress in brain mitochondria of aged mice. PLoS One. 2015;10:e0144290. doi: 10.1371/journal.pone.0144290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zini R., Morin C., Bertelli A., Bertelli A.A., Tillement J.P. Effects of resveratrol on the rat brain respiratory chain. Drugs Exp Clin Res. 1999;25:87–97. [PubMed] [Google Scholar]
- 124.Zheng J., Ramirez V.D. Inhibition of mitochondrial proton F0F1-ATPase/ATP synthase by polyphenolic phytochemicals. Br J Pharmacol. 2000;130:1115–1123. doi: 10.1038/sj.bjp.0703397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tellone E., Galtieri A., Russo A., Giardina B., Ficarra S. Resveratrol: a focus on several neurodegenerative diseases. Oxid Med Cell Longev. 2015;2015:392169. doi: 10.1155/2015/392169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bastianetto S., Ménard C., Quirion R. Neuroprotective action of resveratrol. Biochim Biophys Acta. 2015;1852:1195–1201. doi: 10.1016/j.bbadis.2014.09.011. [DOI] [PubMed] [Google Scholar]
- 127.Rege S.D., Geetha T., Griffin G.D., Broderick T.L., Babu J.R. Neuroprotective effects of resveratrol in Alzheimer disease pathology. Front Aging Neurosci. 2014;6:218. doi: 10.3389/fnagi.2014.00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sun A.Y., Wang Q., Simonyi A., Sun G.Y. Resveratrol as a therapeutic agent for neurodegenerative diseases. Mol Neurobiol. 2010;41:375–383. doi: 10.1007/s12035-010-8111-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wahab A., Gao K., Jia C., Zhang F., Tian G., Murtaza G., et al. Significance of resveratrol in clinical management of chronic diseases. Molecules. 2017;22:1329. doi: 10.3390/molecules22081329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Guo T., Zhang D., Zeng Y., Huang T.Y., Xu H., Zhao Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer's disease. Mol Neurodegener. 2020;15:40. doi: 10.1186/s13024-020-00391-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wirth C., Brandt U., Hunte C., Zickermann V. Structure and function of mitochondrial complex I. Biochim Biophys Acta. 2016;1857:902–914. doi: 10.1016/j.bbabio.2016.02.013. [DOI] [PubMed] [Google Scholar]
- 132.Fiedorczuk K., Letts J.A., Degliesposti G., Kaszuba K., Skehel M., Sazanov L.A. Atomic structure of the entire mammalian mitochondrial complex I. Nature. 2016;538:406–410. doi: 10.1038/nature19794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Parey K., Haapanen O., Sharma V., Köfeler H., Züllig T., Prinz S., et al. High-resolution cryo-EM structures of respiratory complex I: mechanism, assembly, and disease. Sci Adv. 2019;5:eaax9484. doi: 10.1126/sciadv.aax9484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Fato R., Bergamini C., Bortolus M., Maniero A.L., Leoni S., Ohnishi T., et al. Differential effects of mitochondrial complex I inhibitors on production of reactive oxygen species. Biochim Biophys Acta. 2009;1787:384–392. doi: 10.1016/j.bbabio.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yin Z., Burger N., Kula-Alwar D., Aksentijević D., Bridges H.R., Prag H.A., et al. Structural basis for a complex I mutation that blocks pathological ROS production. Nat Commun. 2021;12:707. doi: 10.1038/s41467-021-20942-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Borsche M., Pereira S.L., Klein C., Grünewald A. Mitochondria and Parkinson's disease: clinical, molecular, and translational aspects. J Parkinsons Dis. 2021;11:45–60. doi: 10.3233/JPD-201981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Choi W.S., Kruse S.E., Palmiter R.D., Xia Z. Mitochondrial complex I inhibition is not required for dopaminergic neuron death induced by rotenone, MPP+, or paraquat. Proc Natl Acad Sci U S A. 2008;105:15136–15141. doi: 10.1073/pnas.0807581105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Flønes I.H., Fernandez-Vizarra E., Lykouri M., Brakedal B., Skeie G.O., Miletic H., et al. Neuronal complex I deficiency occurs throughout the Parkinson's disease brain, but is not associated with neurodegeneration or mitochondrial DNA damage. Acta Neuropathol. 2018;135:409–425. doi: 10.1007/s00401-017-1794-7. [DOI] [PubMed] [Google Scholar]
- 139.Intihar T.A., Martinez E.A., Gomez-Pastor R. Mitochondrial dysfunction in Huntington's disease; interplay between HSF1, p53 and PGC-1α transcription factors. Front Cell Neurosci. 2019;13:103. doi: 10.3389/fncel.2019.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Trushina E., Dyer R.B., Badger J.D., 2nd, Ure D., Eide L., Tran D.D., et al. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol Cell Biol. 2004;24:8195–8209. doi: 10.1128/MCB.24.18.8195-8209.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zhang L., Zhang S., Maezawa I., Trushin S., Minhas P., Pinto M., et al. Corrigendum to "Modulation of mitochondrial complex I activity averts cognitive decline in multiple animal models of familial Alzheimer's disease" [EBioMedicine 2 (2015) 294-305] EBioMedicine. 2019;42:532. doi: 10.1016/j.ebiom.2015.03.009. Erratum for: EBioMedicine 2015;2:294-305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Stojakovic A., Trushin S., Sheu A., Khalili L., Chang S.Y., Li X., et al. Partial inhibition of mitochondrial complex I ameliorates Alzheimer's disease pathology and cognition in APP/PS1 female mice. Commun Biol. 2021;4:61. doi: 10.1038/s42003-020-01584-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Stojakovic A., Chang S.Y., Nesbitt J., Pichurin N.P., Ostroot M.A., Aikawa T., et al. Partial inhibition of mitochondrial complex I reduces Tau pathology and improves energy homeostasis and synaptic function in 3xTg-AD mice. J Alzheimers Dis. 2021;79:335–353. doi: 10.3233/JAD-201015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gao H., Tripathi U., Trushin S., Okromelidze L., Pichurin N.P., Wei L., et al. A genome-wide association study in human lymphoblastoid cells supports safety of mitochondrial complex I inhibitor. Mitochondrion. 2021;58:83–94. doi: 10.1016/j.mito.2021.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Choi S.W., Gerencser A.A., Nicholls D.G. Bioenergetic analysis of isolated cerebrocortical nerve terminals on a microgram scale: spare respiratory capacity and stochastic mitochondrial failure. J Neurochem. 2009;109:1179–1191. doi: 10.1111/j.1471-4159.2009.06055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hong H.S., Rana S., Barrigan L., Shi A., Zhang Y., Zhou F., et al. Inhibition of Alzheimer's amyloid toxicity with a tricyclic pyrone molecule in vitro and in vivo. J Neurochem. 2009;108:1097–1108. doi: 10.1111/j.1471-4159.2008.05866.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Jin L.W., Hua D.H., Shie F.S., Maezawa I., Sopher B., Martin G.M. Novel tricyclic pyrone compounds prevent intracellular APP C99-induced cell death. J Mol Neurosci. 2002;19:57–61. doi: 10.1007/s12031-002-0011-9. [DOI] [PubMed] [Google Scholar]
- 148.Maezawa I., Hong H.S., Wu H.C., Battina S.K., Rana S., Iwamoto T., et al. A novel tricyclic pyrone compound ameliorates cell death associated with intracellular amyloid-beta oligomeric complexes. J Neurochem. 2006;98:57–67. doi: 10.1111/j.1471-4159.2006.03862.x. [DOI] [PubMed] [Google Scholar]
- 149.Johnson E.C.B., Dammer E.B., Duong D.M., Ping L., Zhou M., Yin L., et al. Large-scale proteomic analysis of Alzheimer's disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat Med. 2020;26:769–780. doi: 10.1038/s41591-020-0815-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wengenack T.M., Whelan S., Curran G.L., Duff K.E., Poduslo J.F. Quantitative histological analysis of amyloid deposition in Alzheimer's double transgenic mouse brain. Neuroscience. 2000;101:939–944. doi: 10.1016/s0306-4522(00)00388-2. [DOI] [PubMed] [Google Scholar]
- 151.Holcomb L.A., Gordon M.N., Jantzen P., Hsiao K., Duff K., Morgan D. Behavioral changes in transgenic mice expressing both amyloid precursor protein and presenilin-1 mutations: lack of association with amyloid deposits. Behav Genet. 1999;29:177–185. doi: 10.1023/a:1021691918517. [DOI] [PubMed] [Google Scholar]
- 152.Trushina E., Nemutlu E., Zhang S., Christensen T., Camp J., Mesa J., et al. Defects in mitochondrial dynamics and metabolomic signatures of evolving energetic stress in mouse models of familial Alzheimer’s disease. PLoS One. 2012;7:e32737. doi: 10.1371/journal.pone.0032737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Matchett B.J., Grinberg L.T., Theofilas P., Murray M.E. The mechanistic link between selective vulnerability of the locus coeruleus and neurodegeneration in Alzheimer's disease. Acta Neuropathol. 2021;141:631–650. doi: 10.1007/s00401-020-02248-1. [DOI] [PMC free article] [PubMed] [Google Scholar]