

Abstract
This manuscript was sent to Joyce Bischoff, Guest Editor, for review by expert referees, editorial decision, and final disposition. Final decisions were approved by Jane Leopold, Guest Editor-in-Chief.
Subject Terms: Inflammation, Metabolism
A prevailing notion is that increased glucose uptake in response to hyperglycemia in cells involved in atherosclerosis contributes to the increased risk of cardiovascular complications of diabetes. Because macrophages play critical roles in atherosclerosis, numerous studies have focused on how hyperglycemia affects these cells. It is unknown, however, if diabetes indeed increases glucose uptake in macrophages. We therefore asked whether diabetes leads to increased glucose uptake and glycolysis in macrophages, using a model of diabetes-accelerated atherosclerosis; LDL receptor-deficient (Ldlr−/−) mice in which diabetes was induced by streptozotocin.
Diabetic hyperglycemic mice and non-diabetic littermates were fed a low-fat semipurified diet1 for 4 weeks (A). Peritoneal macrophages were isolated and sorted by negative selection 4 days after thioglycollate elicitation.2 Measurements were done after an additional 1 h adhesion purification. Flow cytometry was performed immediately after isolation, identifying macrophages as live CD11B+ F4/80+ cells.
Streptozotocin had no detectable toxic effects prior to development of diabetes (A).
Rather than increased glucose uptake, CD11B+ F4/80+ macrophages from diabetic mice (B) exhibited reduced expression of GLUT1, the main glucose transporter in these cells, concomitant with reduced glucose uptake and reduced glycolysis estimated by extracellular acidification rate (ECAR). The enzymatic activity of GAPDH (Abcam; ab204732) was not reduced by diabetes (1.10±0.04 fold of ND; mean±SD; n=5–9), as has been reported in endothelial cells.3 However, phosphofructokinase, platelet type (Pfkp) exhibited reduced gene expression (B). The reduced ECAR in macrophages from diabetic mice was not due to the downregulation of GLUT1 because mice with myeloid cell-targeted knockdown of GLUT1 matching the diabetes-induced GLUT1 reduction did not exhibit reduced ECAR (C).
To investigate if the reduced macrophage ECAR in diabetes is associated with reduced mitochondrial activity, mitochondrial membrane potential was measured by flow cytometry, using tetramethylrhodamine (TMRM). MitoTracker Green (MTG) assessed total mitochondrial mass. Macrophages from diabetic mice exhibited reduced mitochondrial membrane potential, without a reduction in mitochondrial mass (D).
Functionally, macrophages from diabetic mice showed hampered CCL2 release in response to IFNβ (D), perhaps due to desensitization.
Next, we used global proteomics and pathway analyses to interrogate the overall effect of diabetes on macrophages (E). Macrophages from diabetic mice were enriched in processes related to mRNA metabolism, complement and coagulation cascades, and immune and inflammatory responses. In contrast, macrophages from non-diabetic mice were enriched in proteins involved in insulin receptor signaling and recycling, iron uptake, and regulation of intracellular pH via vacuolar ATPases. Thus, diabetes profoundly alters the global macrophage proteome in the absence of increased glucose uptake.
We used two strategies described previously1 to investigate if the effects of diabetes on macrophage metabolism are mediated by hyperglycemia or by insulin deficiency. Diabetic mice were treated with an intensive insulin regimen or a sodium-glucose co-transporter 2 inhibitor (SGLT2i; dapagliflozin), which suppresses blood glucose levels through insulin-independent urinary glucose excretion. Intensive insulin treatment using subcutaneous insulin pellets and long-acting insulin injections reduced blood glucose levels in diabetic mice over the 4-week study (F). Administration of the SGLT2i (25 mg/kg/day; 4 weeks) also suppressed blood glucose levels. At the end of the study, thioglycollate-elicited peritoneal macrophages were isolated as in A. Oxygen consumption rate (OCR) analysis by Seahorse revealed that diabetes suppressed basal OCR by 48±5% (P=0.0004) and maximal OCR by 53±6% (P=0.0005), consistent with the reduced mitochondrial membrane potential (F). The SGLT2i was unable to normalize macrophage OCR whereas OCR in insulin-treated diabetic mice was not significantly different from that in non-diabetic mice (F). A similar patten was observed for ATP-linked respiration. Likewise, diabetes suppressed ECAR in macrophages through a mechanism dependent on insulin deficiency rather than hyperglycemia, because the SGLT2i failed to restore ECAR in macrophages from diabetic mice.
We have previously shown that increased glucose uptake in myeloid cells in non-diabetic mice does not phenocopy the effect of diabetes on atherosclerosis.4 The present study demonstrates that macrophages in diabetic mice exhibit reduced - rather than increased - glucose uptake and glycolysis relative to those from non-diabetic mice.
These findings challenge dogma by suggesting that hyperglycemia associated with diabetes does not directly increase macrophage glucose uptake. Instead, our findings support the proposal that lack of insulin associates with dampened macrophage metabolism. We cannot, however, rule out the possibility of accumulation of specific glucose metabolites, or that the macrophages we used might not accurately reflect lesion macrophages.
Our results cast doubt on increased glucose uptake as a direct contributor to diabetes-induced macrophage phenotypic changes associated with atherosclerosis. This interpretation is consistent with clinical data, which largely implicate risk factors other than hyperglycemia in promoting cardiovascular disease risk in diabetes.5
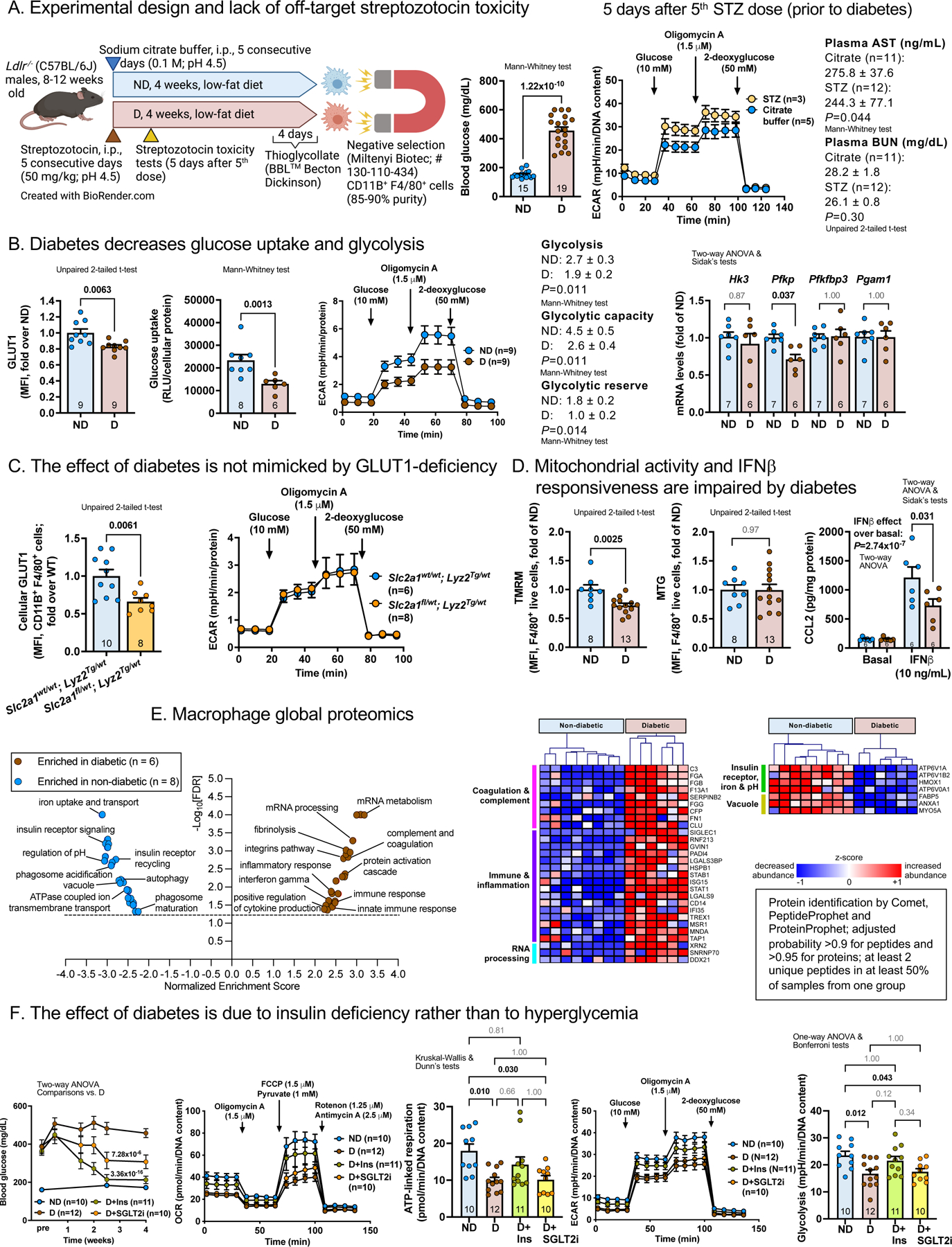
(A) Diabetes was induced in fasted mice by streptozotocin. Non-diabetic littermates received vehicle. Thioglycollate-elicited CD11B+F4/80+ macrophages were purified by negative selection. Streptozotocin showed no toxicity prior to diabetes induction in macrophages by ECAR or in plasma by aspartate aminotransferase (AST; Abcam, ab263882) and blood urea nitrogen (BUN; Arbor Assays, K024). (B) Macrophage GLUT1 levels measured by flow cytometry (Alexa Fluor 647, #ab195020; 1 μg/mL) and glucose uptake by Glucose Uptake-Glo™ Assay at 60 min (Promega). ECAR analyzed by Seahorse (Agilent) in XF base medium with 2 mM glutamine. Gene expression by real-time PCR. (C) GLUT1 (Slc2a1)-flox mice (10 generations C57BL/6J) were crossed with Lyz2-Cre mice. Mice heterozygous for Slc2a1 and Lyz2-Cre exhibited reduced GLUT1 but not reduced ECAR. (D) TMRM and MTG (both 100 nM; ThermoFisher) assessed by flow cytometry. CCL2 release by ELISA (Invitrogen, # 88–7391) 6 h after IFNβ stimulation (E) Shotgun proteomics (Orbitrap Lumos; ThermoFisher)1 and pathway analyses identify enriched processes in diabetic vs. non-diabetic mice (FDR<0.05; volcano plot; http://www.webgestalt.org); heatmaps showing differentially abundant proteins nominally significant (P<0.05) calculated by a negative binomial model (http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html) within these pathways (https://uwmdi.org/bornfeldt-data-sharing-portal). (F) Diabetic mice treated with an intensive insulin regimen or SGLT2i.1 Blood glucose, OCR and ECAR in peritoneal macrophages measured as above. Statistics (GraphPad Prism 9.0.2): two-tailed unpaired t-tests (two groups, normally distributed data; D’Agostino-Pearson normality tests), Mann-Whitney (two groups; non-parametric data or small groups), ANOVA (parametric) or Kruskal-Wallis (non-parametric) tests, as indicated. ND, non-diabetic mice; D, diabetic mice. Mean ± SEM; n=mice/group as indicated. FCCP, carbonyl cyanide-p-trifluoromethoxyphenylhydrazone.
Sources of funding
NIH grants R35HL150754, P01HL151328, R01DK121756, P30DK017047, American Diabetes Association #9-18-CVD1-002, American Heart Association #828090, Grants-in-Aid for Scientific Research in Japan (20K17121).
Footnotes
Disclosures
None.
References
- 1.Kanter JE, Shao B, Kramer F, Barnhart S, Shimizu-Albergine M, Vaisar T, Graham MJ, Crooke RM, Manuel CR, Haeusler RA, et al. Increased apolipoprotein C3 drives cardiovascular risk in type 1 diabetes. J Clin Invest. 2019;129:4165–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kothari V, Tang J, He Y, Kramer F, Kanter JE, Bornfeldt KE. ADAM17 boosts cholesterol efflux and downstream effects of high-density lipoprotein on inflammatory pathways in macrophages. Arterioscler Thromb Vasc Biol. 2021;41:1854–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brownlee M Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–20. [DOI] [PubMed] [Google Scholar]
- 4.Nishizawa T, Kanter JE, Kramer F, Barnhart S, Shen X, Vivekanandan-Giri A, Wall VZ, Kowitz J, Devaraj S, O’Brien KD, et al. Testing the role of myeloid cell glucose flux in inflammation and atherosclerosis. Cell Rep. 2014;7:356–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eckel RH, Bornfeldt KE, Goldberg IJ. Cardiovascular disease in diabetes, beyond glucose. Cell Metab. 2021;33:1519–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]