

SUMMARY
Chromatin misfolding has been implicated in cancer pathogenesis, yet its role in therapy resistance remains unclear. Here, we systematically integrated sequencing and imaging data to examine the spatial and linear chromatin structures in targeted therapy sensitive and resistant human T cell acute lymphoblastic leukemia (T-ALL). We found widespread alterations in successive layers of chromatin organization including spatial compartments, contact domain boundaries and enhancer positioning upon the emergence of targeted therapy resistance. Reorganization of genome folding structures closely coincides with restructuring of chromatin activity and redistribution of architectural proteins. Mechanistically, derepression and repositioning of the B-lineage-determining transcription factor EBF1 from the heterochromatic nuclear envelope to the euchromatic interior instructs widespread genome refolding, and promotes therapy resistance in leukemic T cells. Together, our findings suggest that lineage-determining transcription factors can instruct changes in genome topology as a driving force for epigenetic adaptations in targeted therapy resistance.
Graphical Abstract
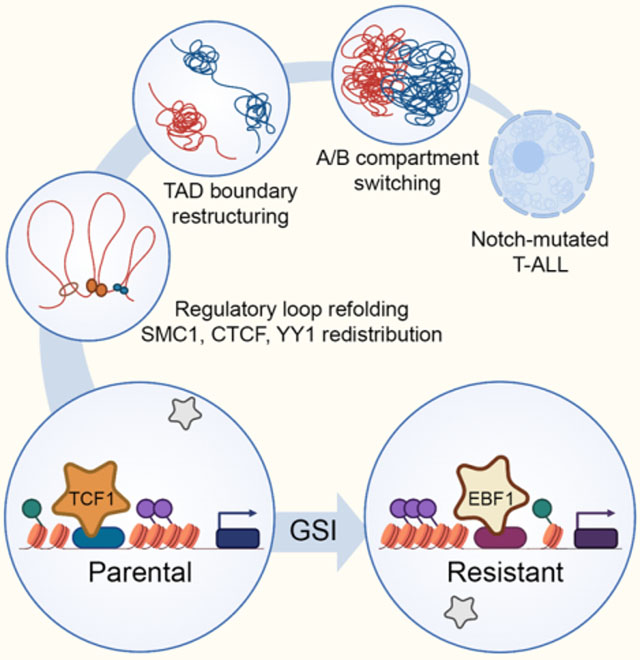
Zhou et al. identifies concerted and widespread restructuring of genome topology during targeted therapy resistance and reveals that derepression and repositioning of the B-lineage-determining transcription factor EBF1 from the nuclear envelope to the interior drive genome folding reorganization and confer Notch-inhibitor resistance in leukemic T cells.
INTRODUCTION
Epigenetic adaptations are emerging as important contributors to anticancer therapy resistance. Recent evidence supports the role of licensing alternative enhancers in anticancer therapy evasion (Marine et al., 2020). Yet, the contribution of chromatin folding reorganization to treatment resistance is largely unknown.
Compaction of chromatin into the nuclear space is made possible through the assembly of structures at successive length scales (Dekker and Misteli, 2015). At the megabase scale, the genome is partitioned into transcriptionally permissive A and repressive B compartments, within which preferentially interacting regions form topologically associating domains (TADs) (Dixon et al., 2012; Lieberman-Aiden et al., 2009). TAD boundaries generally restrain the contacts among promoters and enhancers to achieve accurate spatiotemporal regulation of gene expression (Hnisz et al., 2016). Architectural proteins including the ring-shaped cohesin complex, the insulator protein CCCTC-binding factor (CTCF), and the zinc finger transcription factor YY1 are crucial for connecting higher-order chromatin structures (Stadhouders et al., 2019). We and others revealed that in addition to dysregulating architectural proteins, oncogenic subversion of non-architectural proteins can lead to genome topology changes in treatment-naive cancer cells (Corces and Corces, 2016; Petrovic et al., 2019). Nevertheless, few studies have investigated potential regulatory roles of chromatin folding in therapy resistance. In this study, we identified chromatin folding changes and their underpinning molecular mechanisms in Notch-mutated T-ALL.
Notch gain-of-function mutations are observed in ~ 60% of T-ALL patients, and correlate with poor prognosis (Litzow and Ferrando, 2015). These observations provide a compelling rationale for developing Notch antagonists, such as gamma-secretase inhibitors (GSIs), as a targeted therapy for Notch-mutated T-ALL. Activating Notch mutations often allow ligand-independent release of the intracellular Notch (ICN) domain from the cell membrane by gamma-secretase (Weng et al., 2004). Excessive ICN in the nucleus forms oncogenic Notch transcription complexes (NTCs) with the DNA-binding RBPJ and mastermind-like (MAML) co-factors, resulting in aberrant expression of Notch target genes, such as the proto-oncogene MYC. Besides the role of Notch in activating MYC enhancers (Herranz et al., 2014), we and others showed that GSI-mediated short-term Notch inhibition alters lineage-restricted DNA loops among enhancers and promoters at and beyond the MYC locus, but does not impact chromatin compartment and TAD structures (Kloetgen et al., 2020; Petrovic et al., 2019). However, it is unknown whether and how chromatin folding contributes to GSI-resistance in Notch-mutated T-ALL.
To investigate the contribution of genome refolding to GSI-resistance in T-ALL, we used multi-pronged sequencing, imaging and functional genomic approaches to identify precise changes in gene expression, chromatin activity, and genome topology. Our studies revealed widespread alterations in tandem layers of genome folding, including compartment shifting, TAD boundary disruption and enhancer repositioning. The alterations in successive levels of genome folding structures coincided with chromatin activity and transcriptional changes, promoting GSI-resistance. We further elucidated the mechanisms underpinning genome refolding and showed that GSI-resistant cells repress the T cell developmental transcription factors TCF1 and LEF1, and instead activate the B cell lineage-determining transcription factor EBF1 to connect higher-order chromatin structures and overcome addiction to oncogenic Notch signals. Together, these observations support chromatin refolding as an epigenetic mechanism capable of conferring resistance to targeted therapy, and implicate the differential activity of lineage-determining transcription factors as a driver of chromatin folding reorganization underlying cancer treatment responses.
RESULTS
Generating a GSI-resistant NOTCH1-mutated T-ALL model
T-ALL DND41 cells harbor prototypical activating NOTCH1 mutations and are sensitive to GSI treatment (Figure S1A). To gain insights into mechanisms of targeted therapy resistance in T-ALL, we modeled GSI-resistance and investigated molecular characteristics of resistant DND41. GSI-resistant DND41 cells were derived by prolonged treatment of “parental” cells with 125 nM GSI. In contrast to parental cells, GSI treatment did not induce apoptosis, death, and growth inhibition in resistant cells (Figures S1A–C). But like parental cells, GSI treatment markedly reduced expression of ICN1 and known NOTCH1 targets in the resistant cells (Figures S1C and S1D), where no additional mutation was observed in NOTCH1 transmembrane and gamma-secretase cleavage site sequences (Table S1). Thus, GSI-resistance in DND41 is not due to reactivation of Notch signals.
Chromatin compartmentalization is reorganized in GSI-resistant T-ALL
To examine whether epigenetic adaptation and chromatin restructuring enable GSI-resistance, we mapped genome folding structures using in situ HiC and cohesin subunit SMC1 HiChIP, and used the complementary features of these two assays (Yu and Ren, 2017) to identify reproducible changes in successive layers of chromatin folding structures. Given the high correlation of SMC1 HiChIP- and HiC-defined compartments (Figure S1E), we first marked differential compartments as the ones reproducibly identified by both assays. Although most compartments remained invariant, 115 and 171 loci shifted from A to B or from B to A compartment in resistant cells, respectively (Table S2).
To examine how changes in compartment and chromatin activity align, we compared the abundance of active and repressive histone marks within each compartment. In the resistant cells, A to B compartment shift coincided with repression of active chromatin, as measured by ChIP sequencing (ChIP-seq) of active H3K27ac and H3K4me1 and Cut&Run of polycomb-associated H3K27me3 histone marks (Figure 1A). Conversely, loci that changed from B to A compartment gained active and lost repressive histone marks (Figure 1A). To test the functional impacts of compartment changes, we then compared gene expression in parental and resistant cells. Loci that moved to the A compartment in resistant cells were enriched for upregulated genes (Figure 1A), including the T cell regulator KLF7 and the T-ALL-associated factor ZEB2; where active and repressive histone marks were respectively increased and decreased (Figures S1F and S1G). In contrast, regions that shifted to the B compartment predominantly held genes that were downregulated and assumed repressive chromatin states in resistant cells (Figure 1A), as exemplified by the T-cell-restricted Ikaros factor IKZF2 (Figures 1B and 1C).
Figure 1: Chromatin compartments and TADs are reorganized in GSI-resistant T-ALL.
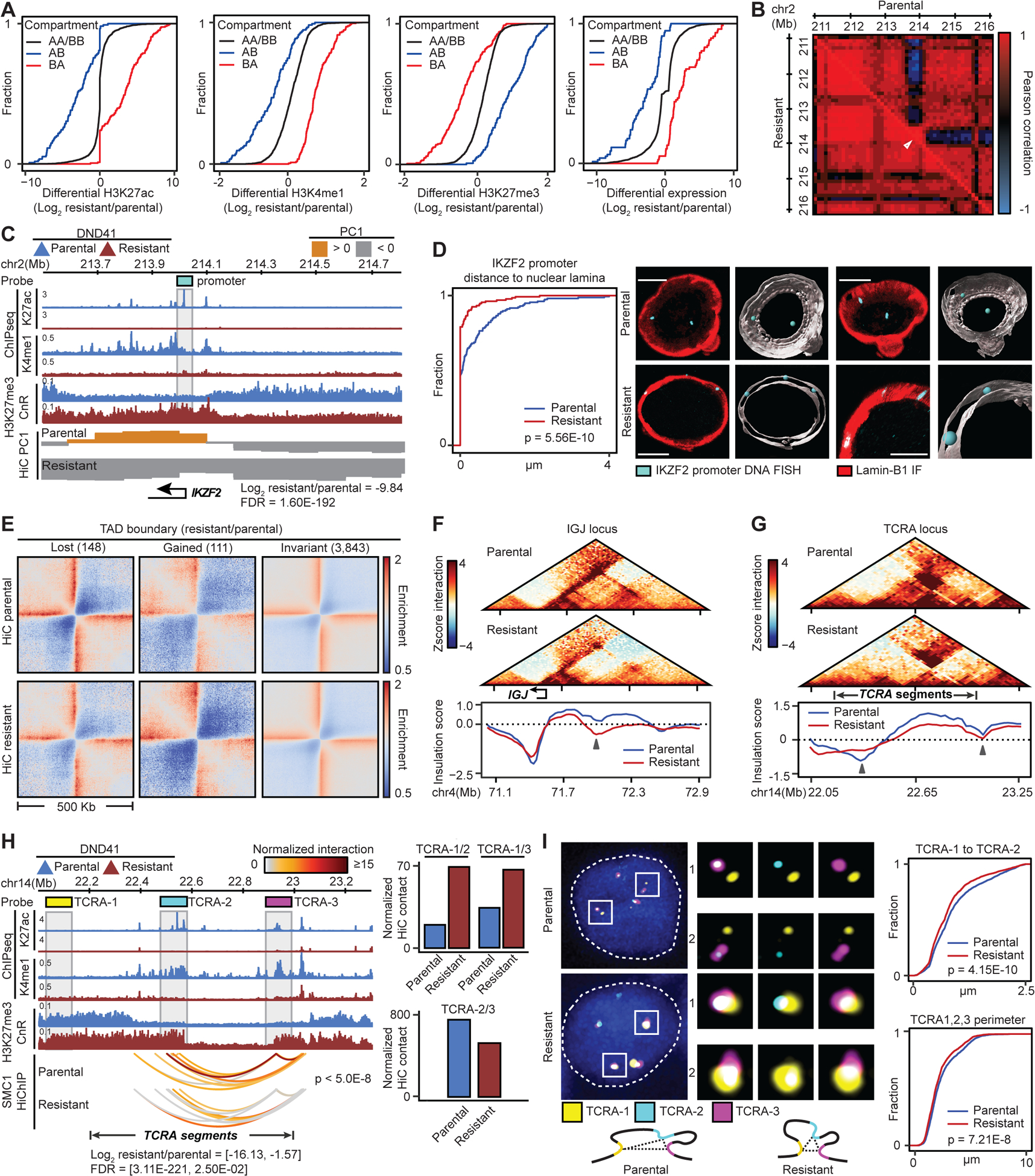
(A) Histone marks and gene expression changes at loci with compartment switching in GSI-resistant DND41.
(B, C) IKZF2 contact frequency Pearson correlation matrix (B), its PC1, expression and histone marks (C) in parental and resistant cells. Gray box: Oligopaint probes.
(D) Left: Distributions of distance between IKZF2 and nuclear envelope in resistant vs parental cells (KS test) with mean (+/− SD) of 0.087 (+/− 0.27) and 0.36 (+/− 0.64) μm, respectively. Right: representative and Imaris-modeled cells.
(E) Pileup HiC maps at TAD boundaries per insulation score change.
(F, G) Examples of TAD boundary insulation gain (F) or loss (G) in resistant cells marked by arrows.
(H) Changes in histone marks, looping (paired t-test), and expression of TCRA gene segments. Gray boxes: Oligopaint probes. Right: HiC signal between the noted probes.
(I) Distributions of TCRA-1 to TCRA-2 distance (right, top) and TCRA-1, TCRA-2 and TCRA-3 spatial perimeter (right, bottom) (model: left, bottom) in resistant vs parental cells (KS test). Parental (resistant) mean (+/− SD) distance and perimeter: 0.92 (+/− 0.57) (0.81 (+/− 0.53)) and 2.46 (+/− 1.40) (2.20 (+/− 1.25)) μm, respectively. Left: representative cells. Blue: DAPI.
A and B compartments are predominantly positioned at the nuclear interior and envelope, respectively (Falk et al., 2019). We thus used high-resolution Oligopaint DNA fluorescence in situ hybridization (FISH) together with Lamin-B1 immunofluorescence (IF) and 3D confocal imaging to visualize whether IKZF2 compartment change relates to its nuclear repositioning in individual cells. Notably, IKZF2 was mainly sequestered at the nuclear envelope in resistant (106/126 alleles) compared to parental cells (72/157 alleles, Kolmogorov–Smirnov (KS) test p = 5.56E-10) (Figures 1B–D). Thus, our data suggest that genomic loci are repositioned in the nuclear space of GSI-resistant cells, and these repositioning events coincide with large scale changes in chromatin and transcriptional activities.
GSI-resistance alters T-cell receptor alpha TAD boundaries
We next investigated whether long-term GSI treatment impacts TAD boundaries, which are invariant to short-term Notch inhibition (Petrovic et al., 2019). Comparison of 4102 TAD boundaries defined by both HiC and SMC1 HiChIP identified 111 boundaries with significant gain of insulation potential in the resistant cells (Figures 1E, S1H and S1I, Table S3). Insulation gain correlated with increased chromatin and transcriptional activities, as exemplified by the immunoglobulin J chain gene IGJ and T cell receptor CD69 loci (Figures 1F, S1J and S1K). Our data further revealed decreased insulation at 148 boundaries (Figures 1E, S1H and S1I, Table S3). For example, T-cell receptor alpha (TCRA) TAD boundaries were disrupted in the resistant cells (Figure 1G). Concordantly, H3K27me3 spread across the boundaries and TCRA gene segments were significantly downregulated in GSI-resistance (Figure 1H).
To visualize the disrupted TCRA TAD boundaries in individual cells, we designed Oligopaint DNA FISH probes spanning three 100 Kbp regions (Figure 1H). TCRA-1 probe visualized a region 5’ of the TCRA TAD, marked with H3K27me3 in both parental and resistant cells, while TCRA-2 and TCRA-3 probes marked regions flanking the disrupted boundaries. SMC1 HiChIP and HiC showed increased looping levels between TCRA-1 and both TCRA-2 and TCRA-3, suggesting that inter-TAD interactions increased across the disrupted boundaries (Figure 1H). Concordant with the genomic data, TCRA-1 and TCRA-2 3D distances were markedly closer in 3418 resistant compared to 2515 parental cells (KS p = 4.15E-10) (Figure 1I). Measurement of the TCRA-1, TCRA-2 and TCRA-3 spatial perimeter further suggested that the TCRA TAD was more compact in the resistant cells, confirming HiC data (Figures 1I and 1H). Together, these data suggest the presence of TAD restructuring during resistance development that largely corresponds to changes in chromatin activity and gene expression.
GSI-resistance reorganizes enhancer-promoter loops
To determine whether changes in contact frequency among regulatory elements, such as enhancers, promoters, and CTCF-bound insulators, contribute to GSI-resistance, we next used HiC and SMC1 HiChIP to identify significant and reproducible high-resolution loops (~5 Kbp).
While most loops remained invariant, looping levels between 484 and 441 loci pairs were significantly decreased or increased in the resistant cells, respectively (Figures 2A, S2A, Table S4). Our data showed that 39% of the significantly decreased loops connected gene promoters to distal enhancers in the parental cells. On the other hand, 65% of increased loops were among enhancers in the resistant cells. Notably, only a small number of structural loops (5%), connecting two CTCF-bound elements, were altered (Figure 2B). Thus, the differential loops observed during GSI-resistance development are mostly among enhancers and promoters.
Figure 2: GSI-resistance restructures enhancer-promoter loops.
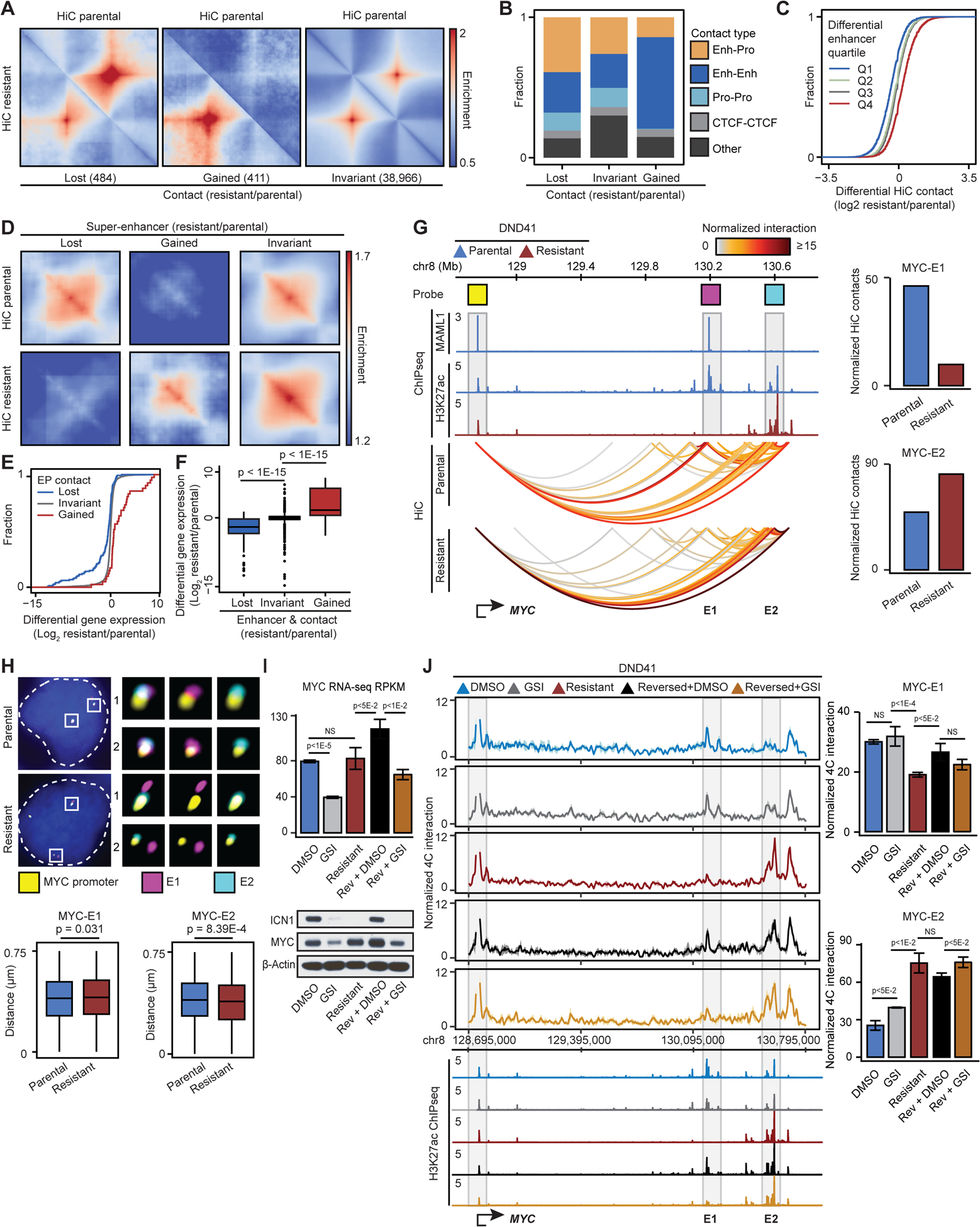
(A) Pileup HiC maps at loops per interaction frequency change.
(B) Fraction of classes of genomic elements connected to differential loops. Enh: Enhancer, Pro: Promoter.
(C) HiC changes per quartile of differential H3K27ac on connected enhancers.
(D) Pileup HiC maps at SEs per H3K27ac change.
(E) Differential expression distribution per connected enhancer-promoter (EP) loop change.
(F) Differential expression per enhancer activity and EP contact change (Wilcoxon test).
(G) Left: Notch-bound (MAML1) E1 SE (magenta), Notch-unbound E2 SE (cyan) and MYC promoter (yellow) Oligopaint probes. Right: HiC signal between the noted probes.
(H) Bottom: distributions of distance between the noted elements in parental vs resistant cells (Wilcoxon test). Parental (resistant) MYC-E1 and MYC-E2 mean (+/− SD) distance: 0.399 (+/− 0.176) (0.407 (+/− 0.176)) and 0.401 (+/− 0.173) (0.389 (+/− 0.177)) μm. Top: representative cells. Blue: DAPI.
(I) MYC transcript (top), ICN1 and MYC protein (bottom) levels in DMSO- and GSI-treated parental, GSI-resistant, DMSO-treated reversed, and GSI-treated reversed DND41. Control: β-Actin.
(J) MYC-promoter-viewpoint UMI 4C-seq track (left), quantification (right, mean ± SD of 3 replicates) and H3K27ac per condition in (I) (t-test). NS: p>0.05.
Changes in enhancer activity and positioning coincide with transcriptional changes in GSI-resistant cells
To investigate how differential transcriptional outputs relate to chromatin activity and loop alterations, we first linked the changes in enhancer activity and positioning. Our data revealed that in general, enhancer activity changes closely related to enhancer-promoter looping alterations (Figures 2C, S2B and S2C). More specifically, hyperacetylation or gain of super-enhancers (SEs) in the resistant cells coincided with marked increases in looping levels at these regions and within the SE loci (Figures 2D and S2D). Conversely, looping levels were significantly decreased at the loci with loss of SE activity.
We next asked how changes in chromatin activity and looping relate to gene expression. Differential enhancer-promoter looping led to significant and concordant gene expression changes (Figure 2E). Furthermore, gain and loss of SE activity, respectively, led to upregulation and downregulation of genes in the resistant cells (Figure S2E). Notably, the most significant differential gene expression occurred when enhancer activity and position concomitantly changed (Figure 2F). Together, these data indicate concordant changes in enhancer activity and positioning, which in addition to TAD and compartment reorganization, coincide with differential transcriptional outputs of GSI-resistant cells.
MYC super-enhancers are repositioned in GSI-resistant cells
Notch-dependent long-range control of MYC expression is critical for T-ALL pathogenesis (Yashiro-Ohtani et al., 2014). We thus captured enhancer activity and looping at the MYC locus to elucidate their contribution to GSI-resistance. TAD boundaries at ~4 Mbp sequences flanking MYC remained unchanged (Figure S2F). Comparing the activity of enhancers contacting MYC showed that Notch-bound super-enhancer (E1), located ~1.35 Mbp 3’ of the promoter in parental cells, was inactivated in the resistant cells (Figure 2G). Conversely, the Notch-unbound super-enhancer (E2), located further ~0.5 Mbp 3’ of E1, markedly gained activity in these cells (Figure 2G). Knowing that Notch relies on different combinations of enhancer activation and positioning to control its targets (Petrovic et al., 2019), we next asked whether MYC enhancers are repositioned in resistant cells. Our sequencing data revealed that the contact frequency between the Notch-bound E1 SE and the MYC promoter decreased in the resistant cells (Figure 2G); in contrast, the looping level between the Notch-unbound E2 SE and the MYC promoter increased (Figure 2G).
We next sought to establish how the differential MYC promoter-enhancer contact frequencies observed by HiC and SMC1 HiChIP relate to physical distances between the interacting elements in individual T-ALL cells. We thus measured 3D distances between the MYC promoter, E1 and E2 SEs in 1652 DMSO-treated parental, 1323 GSI-treated parental, and 1694 GSI-resistant cells with 50 Kbp Oligopaint DNA FISH probes (Figure 2G). Unlike Notch-mutated triple-negative breast cancer (TNBC) and mantle cell lymphoma (MCL) (Petrovic et al., 2019), short-term treatment did not reposition E1 and E2 in Notch-mutated T-ALL (Figure S2G). Yet in the resistant cells, the MYC promoter and E1 were significantly separated while E2 became markedly closer to the promoter (Figure 2H). Together, our optical mapping supports sequencing data and showed the repositioning of both MYC E1 and E2 SEs in individual GSI-resistant T-ALL cells.
To investigate the functional consequences of chromatin restructuring, we assessed changes in MYC expression. Notably, changes in the activity and positioning of the E1 and E2 SEs coincided with reactivation of MYC and its known targets in the resistant cells (Figures 2I, S2H, and Table S6). Thus, despite the lack of new genomic alterations and detectable NOTCH1 activity, the resistant cells bypass their addiction to oncogenic Notch signals and re-express MYC, a key survival gene required for both parental and GSI-resistant T-ALL (Figures 2I, S2I and Table S1).
GSI withdrawal reverses MYC locus chromatin activity and folding in resistant cells
To test the hypothesis that epigenetic adaptation leads to MYC recovery in GSI-resistance, we generated ‘reversed’ DND41 cells by withdrawing GSI for 6 weeks from the resistant cells. While E2 SE did not change and remained GSI-insensitive, E1 regained activity and was sensitive to short-term treatment in the reversed cells (Figure 2J). We next performed MYC promoter-centric 4C-seq to investigate plasticity of MYC locus folding. These data confirmed our 3D DNA FISH and showed that contact frequencies between E1, E2 and the MYC promoter were invariant to short-term Notch inhibition (Figures 2J and S2G). In line with HiC and 3D DNA FISH studies, the 4C-seq data further showed significant repositioning of E1 and E2 SEs in GSI-resistance (Figure 2J). While E2 still contacted MYC, E1 recovered its interaction with the MYC promoter in the reversed cells (Figure 2J). In concordance with E1 reactivation and repositioning, MYC expression increased in the reversed cells, and was sensitive to short-term GSI treatment (Figure 2I). Thus, the resistant cells can be resensitized to GSI due to highly dynamic chromatin structure at the MYC locus.
Differential architectural protein loading coincides with refolded loops in GSI-resistance
Folding of chromatin into structural and regulatory loops is facilitated by architectural proteins, including the cohesin complex, CTCF and YY1 (Rowley and Corces, 2018). To elucidate the underlying mechanisms of widespread chromatin refolding in GSI-resistance, we investigated the relationship between the changes in architectural protein binding and chromatin looping. Although the expression levels of these proteins were unchanged (Figure S3A), we identified 4143, 2272 and 5376 loci with SMC1, CTCF and YY1 differential occupancy, respectively. Among the 503 elements connected by the intensified DNA loops, more than 80% gained SMC1 in the resistant cells (Figures 2A, 3A and S3B). Conversely, 64% of the 689 elements connected to attenuated DNA loops lost SMC1 (Figures 3A and S3B). As expected, chromatin loops remained invariant at the sites similarly bound to SMC1 in parental and resistant cells (Figures 3A and S3B). Comparison of CTCF and YY1 binding events also showed that loss and gain of their chromatin occupancy coincided with decreased and increased looping in GSI-resistance, respectively (Figures 3B, 3C, S3C, and S3D).
Figure 3: Differential SMC1, CTCF, or YY1 loading coincides with refolded loops.
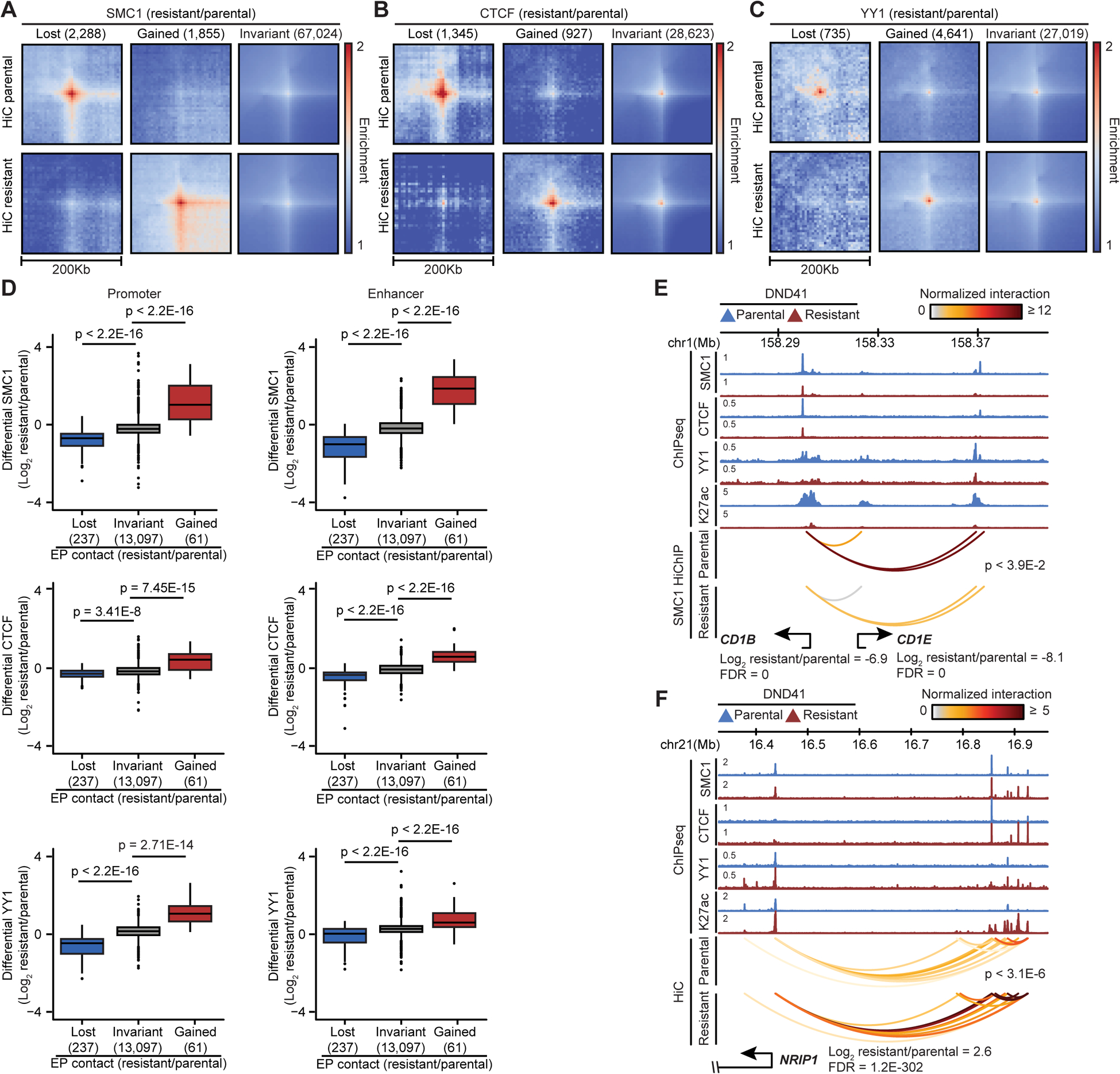
(A, B, C) Pileup HiC maps at SMC1 (A), CTCF (B) and YY1 (C) peaks per differential chromatin binding (number of events).
(D) SMC1 (top), CTCF (middle) and YY1 (bottom) loading at the connected promoters (left) and enhancers (right) per looping change (Wilcoxon test).
(E, F) Concordant changes in expression, SMC1, CTCF, YY1, H3K27ac levels, and EP loops (paired t-test).
To further quantitate how changes in architectural protein chromatin occupancy relate to looping reorganization, we evaluated the differential SMC1, CTCF and YY1 binding at the sites of altered looping. SMC1, CTCF and YY1 occupancy was significantly decreased at loci with reduced looping in resistant cells (Figure S3E). By contrast, binding of these proteins was markedly increased at the loci with enhanced looping (Figure S3E). Concordant with these observations, higher changes in SMC1, CTCF, and YY1 loading were associated with more differential looping between their bound loci (Figure S3F). Thus, the redistribution of CTCF, YY1 and SMC1 chromatin binding facilitates DNA loop reorganization in GSI-resistance.
We next investigated the differential propensity of architectural proteins for altering structural and regulatory loops. Concomitant differential loading of SMC1, CTCF, and YY1 had the highest impact on differential looping, while differential CTCF and YY1 binding events were mutually exclusive in the absence of differential SMC1 (Figure S3G). Given that GSI-resistance only altered a small number of structural loops (Figure 2B), we specifically investigated the impact of differential SMC1, CTCF and YY1 binding on enhancer-promoter (EP) loops. The occupancy of all three proteins was altered at differential EP loops (Figure 3D). However, CTCF, which is not employed by most EP loops (Weintraub et al., 2017), showed the least differential loading, while SMC1 exhibited the highest changes at the elements connected to differential EP loops (Figure 3D). Compared to the promoters, YY1 was preferentially recruited to distal enhancers that gained looping in GSI-resistance, as exemplified by the enhancers connected to the early cortical thymocyte marker CD1B and transcriptional coregulator NRIP1 (Figures 3D–F). Concomitant with significant reduction in YY1, CTCF, and SMC1 loading, drug resistance markedly decreased expression and looping of CD1B to an enhancer located 65 Kbp away (Figure 3E). Conversely, NRIP1 upregulation in the resistant cells coincided with recruitment of these architectural proteins and repositioning of resistance-restricted enhancers located 400 Kbp 5’ of the gene promoter (Figure 3F). Hence, differential chromatin recruitment of YY1, SMC1 and, to a lesser extent, CTCF contributes to repositioning of enhancers and promoters in GSI-resistant T-ALL.
TCF1/LEF1 and EBF1 deregulation links to differential accessibility in GSI-resistance
We found that chromatin occupancy of SMC1, YY1 and, to some extent, CTCF was significantly altered at the repositioned elements in GSI-resistance. Yet, these architectural proteins are equally expressed in the parental and resistant cells (Figure S3A). This observation led us to investigate the molecular underpinnings of CTCF, YY1 or SMC1 differential recruitment and enhancer repositioning. We first leveraged ATAC-seq to map differential chromatin opening. Although short-term Notch inhibition had no significant effect on chromatin opening, long-term Notch inhibition markedly altered the accessibility of more than 7000 elements (Figures 4A, S4A, and Table S5). In line with reduced accessibility, we observed substantial H3K27ac and H3K4me1 loss, and H3K27me3 gain at ~3000 parental-cell-restricted accessible elements (Figures 4B and S4B). Conversely, concomitant with H3K27me3 loss, H3K27ac and H3K4me1 were deposited at ~4000 resistance-restricted accessible elements (Figures 4B and S4B).
Figure 4: TCF1/LEF1 and EBF1 differential activity underlies GSI-resistant T-ALL enhancer positioning.
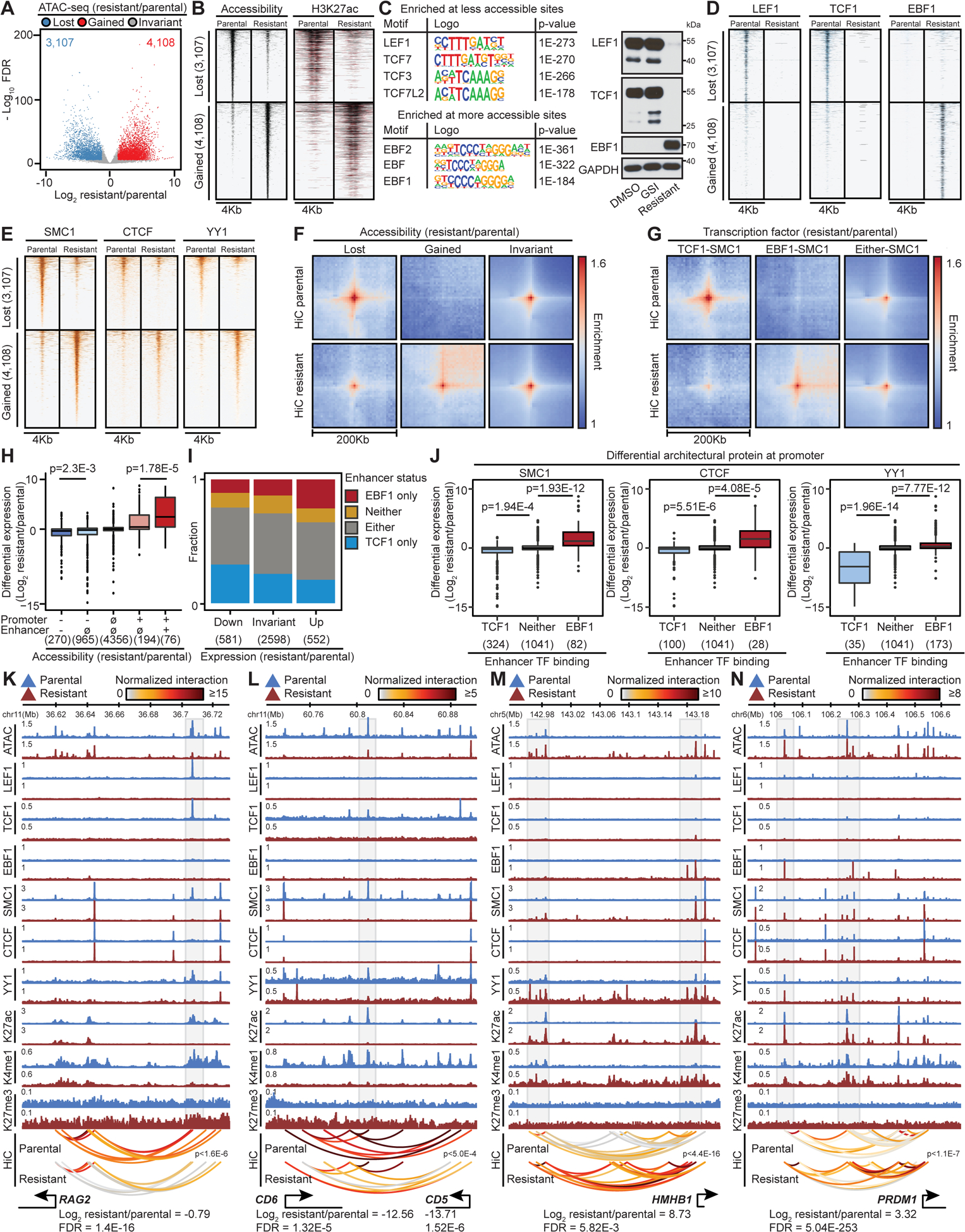
(A, B, C, D, E) Differential accessible elements (A), as well as their ATAC-seq and H3K27ac levels (B), the most enriched motifs (C, left), LEF1, TCF1, EBF1 (D), SMC1, CTCF and YY1 (E) binding in resistant vs parental cells. LEF1, TCF1 and EBF1 expression in DMSO- and GSI-treated parental and resistant DND41 (C, Right). Control: GAPDH.
(F) Pileup HiC maps at differential accessible sites per (A).
(G) Pileup HiC maps at pairs of accessible sites per EBF1/TCF1 and SMC1 change. Either: EBF1 replaced TCF1 (STAR Methods).
(H) Differential expression (gene count) per combinatorial lost (−), invariant (∅) or gained (+) opening of promoter and connected enhancer (Wilcoxon test).
(I) Fraction of enhancers bound only to EBF1 or TCF1, neither, or TCF1 in parental and EBF1 in resistant cells (Either) as a function of connected gene expression change (count).
(J) Differential expression (gene count) per change in binding of SMC1, CTCF and YY1 at promoter and TCF1/EBF1 at connected enhancers (Wilcoxon test).
(K-N) Coordinated changes in gene expression, looping (paired t-test), and opening of connected enhancers in resistant cells.
We next performed motif analyses to identify the transcription factors contributing to differential chromatin opening. Sequences of parental-cell-restricted accessible elements were most enriched with motifs of TCF (Figure 4C), a family of HMG proteins (Yui and Rothenberg, 2014). RNA-seq and immunoblotting showed significant downregulation of LEF1 and TCF1 (encoded by TCF7) in the resistant cells (Figure S4C and 4C). Conversely, EBF recognition sequences were the most enriched motifs in the resistance-restricted accessible elements (Figure 4C). Among the EBF protein family, EBF1 was undetectable in parental cells but was highly expressed in resistant ones (Figures 4C and S4C). Notably, EBF1 and TCF1 can create de novo chromatin opening during early B and T-lineage differentiation, respectively (Boller et al., 2016; Johnson et al., 2018).
To further substantiate the effect of TCF1, LEF1 and EBF1 on differential accessibility observed in GSI-resistance, we measured their chromatin binding. As motif analysis predicted, TCF1 and LEF1 only bound to parental-cell-restricted accessible elements (Figures 4C and 4D). Conversely, resistance-restricted accessible elements bound to EBF1 after its derepression (Figures 4C and 4D). Hence, differential TCF1/LEF1 and EBF1 expression and binding coincide with the differential chromatin opening and may contribute to GSI-resistance development.
Differential TCF1/LEF1 and EBF1 binding directs SMC1, CTCF, or YY1 to repositioned elements in GSI-resistance
As removal of nucleosomes might be a key step to allow architectural protein recruitment, we considered the possibility that differential TCF1/LEF1 and EBF1 activities might serve a crucial role in CTCF, SMC1 and YY1 redistribution and enhancer repositioning in GSI-resistance. Indeed, ChIP-seq analysis revealed significant loss of SMC1, YY1, and CTCF at 81%, 52% and 30% of TCF1-bound parental-cell-restricted elements, respectively. On the other hand, 95%, 65% and 27% of the EBF1-bound resistance-restricted accessible elements exhibited significant gain of SMC1, YY1, and CTCF, respectively (Figures 4B, 4D and 4E).
We next assessed the impact of differential TCF1/LEF1 and EBF1 binding on DNA looping to substantiate the role of TCF1/LEF1 and EBF1-mediated architectural protein redistribution. Contact frequencies were greatly reduced among the parental elements that lost TCF1/LEF1 binding and accessibility, but markedly enhanced among the ones that bound EBF1 and gained opening in GSI-resistance (Figures 4F and S4D). Analysis of paired loci revealed that concordant loss of TCF1/LEF1 on one anchor and SMC1, CTCF, or YY1 at the other anchor similarly diminished DNA looping (Figures 4G, S4E, S4F and S4G). Conversely, concordant gain of EBF1 and these architectural proteins at either anchor intensified looping in GSI-resistance. Together, these data further support the potential role of TCF1/LEF1 and EBF1 in selective recruitment of architectural proteins and genome refolding that confers differential GSI response in Notch-mutated T-ALL.
Coordinated differential recruitment of TCF1/LEF1, EBF1, and architectural proteins mediates GSI-resistant transcriptional program
To assess how changes in TCF1/LEF1- and EBF1-mediated chromatin opening and folding relate to the GSI-resistant transcriptional program, we linked H3K27ac-marked enhancers to gene promoters using our high resolution in situ HiC and SMC1 HiChIP, and quantified the impact of accessibility changes on gene expression. 1460 downregulated genes were generally associated with T-cell-related pathways, particularly the ones differentiating T from B lymphocytes (Figure S4H, Table S6). As expected, changes in opening at promoters and distal enhancers greatly altered gene expression (Figure 4H). Notably, changes in opening of both promoters and enhancers, compared to promoters alone, led to significantly higher differential gene expression (Figure 4H), further supporting that distal element changes contribute to GSI-resistance.
After observing the impact of chromatin opening changes on gene expression and the link between TCF1/LEF1 and EBF1 and opening (Figures 4B, 4D, and 4H), we next assessed the direct contribution of these transcription factors to differential gene expression. Genes markedly upregulated in the resistant cells were preferentially connected to enhancers gaining EBF1 (Figure 4I, proportion test p = 0.01). TCF1/LEF1 was significantly depleted at enhancers connected to downregulated genes (proportion test p < 1E-15). Notably, only 11% of differentially expressed genes did not connect to enhancers with EBF1 gain or TCF1/LEF1 loss (Figure 4I). Together, these data support the importance of differential activity and binding of TCF1 and EBF1 in orchestrating GSI-resistant gene expression.
Given the association between differential TCF1/LEF1 and EBF1 binding and redistribution of architectural proteins (Figures 4D and 4E), we next investigated how TCF1/LEF1- and EBF1-mediated recruitment of SMC1, YY1, and CTCF impacts gene expression. Concordant loss of TCF1/LEF1 and architectural proteins led to markedly higher gene repression (Figure 4J). Examining 540 TCF1/LEF1 direct targets showed that TCF1/LEF1 loss led to repression of genes encoding components of T cell activation, differentiation, and signaling (Figures S4I and S4J, Tables S6 and S7). For example, concomitant loss of TCF1/LEF1, SMC1 and YY1 coincided with reduced looping frequency between parental-cell-specific elements and RAG2, CD6 and CD5 promoters (Figures 4K and 4L).
Besides downregulating TCF1/LEF1 targets, GSI-resistance significantly upregulated 1128 genes, including 400 direct EBF1 targets (Table S6). Notably, concomitant gain of EBF1 and SMC1, CTCF, or YY1 led to significantly higher gene activation (Figure 4J). Pathway analysis revealed that EBF1 binding directly upregulates genes associated with B cell differentiation, activation, and lineage choice (Figure S4K, Table S7), as exemplified by the B cell receptor component CD79A, plasma cell master regulator PRDM1, and B-cell leukemia-associated antigen HB-encoding gene HMHB1 (Figures 4M, 4N, and S4L). Examination of these loci revealed that EBF1 binding led to de novo opening and recruitment of architectural proteins to regulatory elements, which in turn increased looping to target genes. Together, these results indicate that TCF1/LEF1 repression and EBF1 derepression impact chromatin activity and folding to shift the GSI-resistant T-ALL transcriptional program away from a T cell-associated towards a B cell-associated one.
In addition to directing architectural proteins to the elements connected to differentially expressed genes, EBF1 derepression also guided SMC1, CTCF, or YY1 to MYC-connected enhancers, whose reactivation was crucial for GSI-resistant cell survival (Figures 2I and S2I). Notably, MYC reactivation coincided with the replacement of TCF1/LEF1 with EBF1 at both E1 and E2 SEs (Figures 2J, S4M and S4N). Loss of TCF1/LEF1 in the resistant cells led to architectural protein depletion, H3K27me3 deposition, and closing of several accessible elements within the Notch-dependent E1 (Figures 2J and S4M). Conversely, resistant cells concomitantly gained EBF1, SMC1, YY1, as well as chromatin opening and activity at two elements within the Notch-independent E2 (Figure S4N). Together, these data demonstrate the underlying mechanisms of enhancer switching that maintain MYC expression in GSI-resistance.
EBF1 and TCF1 but not LEF1 can create de novo chromatin accessibility and looping in Notch-mutated TNBC
Although our data in GSI-resistance established a strong link between differential TCF1, LEF1 and EBF1 binding and chromatin restructuring, it was unclear whether these factors were equally able to create de novo chromatin opening and looping. We thus examined their direct impact on these chromatin structures in gain-of-function models. We reasoned that Notch-mutated breast cancer cells could serve as an ideal model since their chromatin is distinct from Notch-mutated hematolymphoid cells, and they lack endogenous expression of lymphocyte lineage-determining transcription factors, including TCF1, LEF1, and EBF1 (Figure S5A). We thus transduced Notch-mutated TNBC MB157 with doxycycline-inducible LEF1, TCF1 or EBF1, and assessed changes in chromatin opening, architectural protein loading, and DNA looping 3 days post induction (Figure S5A). Ectopic LEF1, TCF1, or EBF1 markedly increased opening of 411, 1691, and 8550 elements, respectively (Figures 5A, Table S5). Notably, the elements that significantly gained opening after expression of TCF1 and EBF1, but not LEF1, were enriched for their recognition sequences and mostly bound TCF1 and EBF1, respectively (Figures 5A and 5B). Concordantly, H3K27ac was deposited at 23% of the EBF1-bound and 12% of the TCF1-bound elements (Figure 5B). Thus, EBF1 and TCF1, but not LEF1, can create de novo chromatin opening in MB157, in line with their pioneering potential during lymphocyte differentiation (Boller et al., 2016; Johnson et al., 2018).
Figure 5: EBF1 and TCF1 can instruct looping in Notch-mutated TNBC.
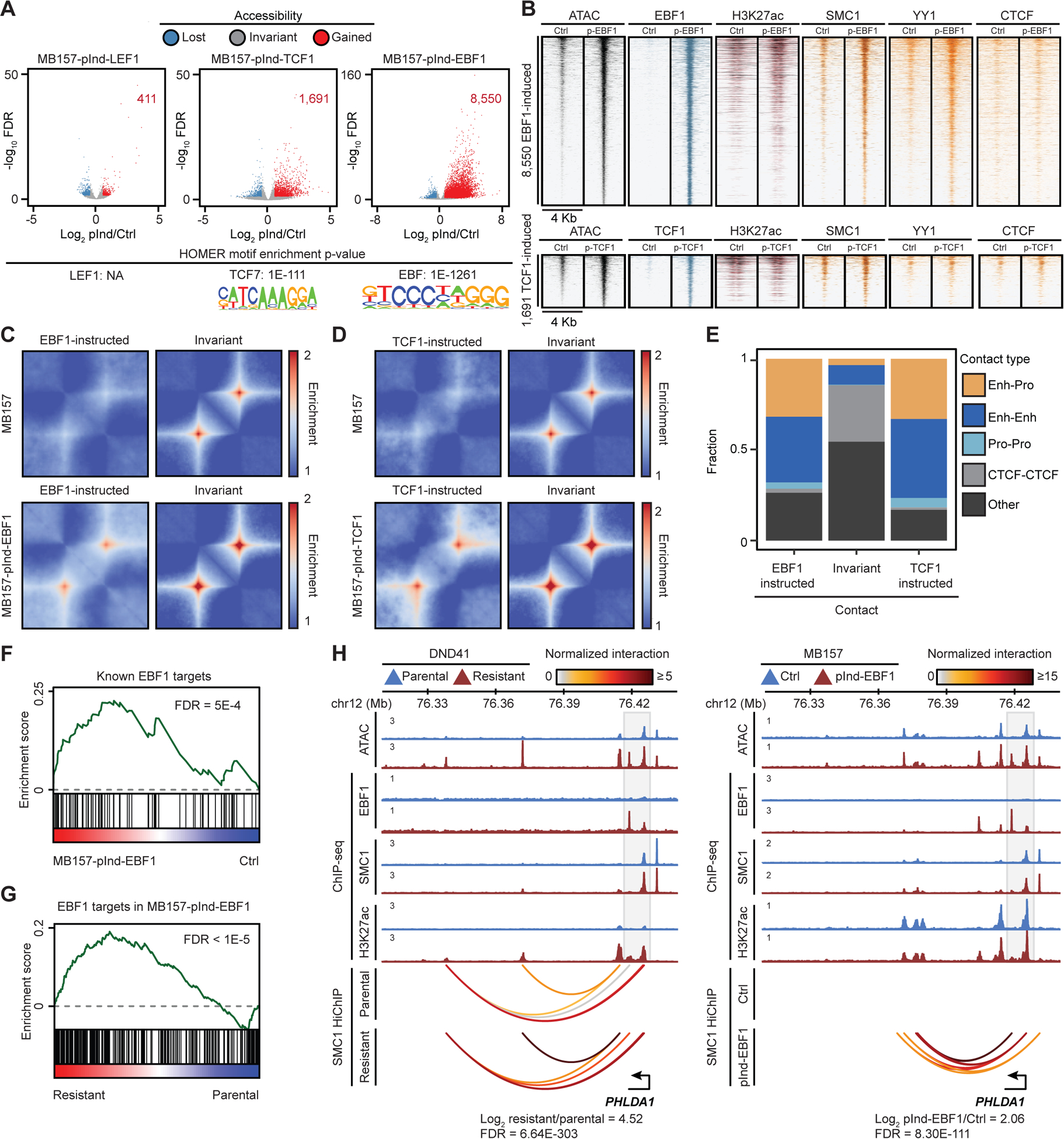
(A, B) Differential opening (A) and binding of noted proteins (B) in MB157 (MB157-pInd-) after LEF1, TCF1, or EBF1 induction. Bottom: enriched motifs.
(C, D) Pileup SMC1 HiChIP maps per accessibility change in MB157-pInd-EBF1 (C) and MB157-pInd-TCF1 (D).
(E) Fraction of classes of genomic elements connected to differential loops. Enh: Enhancer, Pro: Promoter.
(F, G) EBF1 induces overlapping genes in normal B cells, MB157 (F), and GSI-resistant DND41 (G) (permutation test).
(H) Concordant increase of EBF1, SMC1 and H3K27ac loading, accessibility and looping in GSI-resistant DND41 and MB157-pInd-EBF1.
In addition to changes in chromatin opening, our data also showed concomitant differential TCF1 and EBF1 binding, SMC1 and YY1 recruitment, and enhancer repositioning in GSI-resistant T-ALL (Figures 4B–G). Concordantly, ectopic EBF1 and TCF1 chromatin binding in TNBC greatly intensified 1354 and 562 DNA loops, respectively (Figures 5C and 5D, Table S4). This result was illustrated by HEY2 and IL15RA-connected loops (Figures S5B and S5C). Notably, EBF1- and TCF1-instructed loops in TNBC mainly connected to enhancers and promoters and only marginally affected structural CTCF-CTCF loops (Figure 5E). Furthermore, SMC1 and/or YY1, but not CTCF, markedly increased at the loci connected to EBF1- or TCF1-instructed loops (Figures S5D and S5E). Together, these data suggest that similar to T-ALL (Figures 4G and S4E–G), EBF1 and, to a lesser extent, TCF1 can instruct DNA looping in MB157, potentially via recruitment of architectural proteins.
Ectopic EBF1 induces GSI-resistant T-ALL-associated genes in TNBC
To evaluate how widespread EBF1 and TCF1-induced chromatin restructuring controls MB157 gene expression, we next measured transcriptional changes 3 days post EBF1 or TCF1 induction. Ectopic EBF1 or TCF1 markedly upregulated 1463 and 898 genes, respectively (Figure S5F, Table S6). Linking gene promoters to distal enhancers revealed that the concomitant EBF1- or TCF1-induced enhancer and promoter opening led to significantly higher increases in gene expression compared to opening of only one of these elements (Figure S5G), supporting that EBF1 and TCF1 activate both proximal and distal regulatory elements. Notably, the EBF1- or TCF1-bound enhancers that were both activated and repositioned induced higher expression compared to only activated or repositioned EBF1- or TCF1-bound enhancers (Figure S5H), further supporting the role of these transcription factors in enhancer positioning.
Examining EBF1 direct targets in TNBC MB157 revealed that they were overrepresented in genes involved in adaptive immunity and associated signaling (Figure S5I, Tables S6 and S7), consistent with the role of EBF1 in activating key genes during early B cell development. More specifically, several known EBF1 targets in early B cell differentiation (Nechanitzky et al., 2013; Treiber et al., 2010) were upregulated in EBF1-expressing TNBC MB157 (Figure 5F). Similarly, TCF1 targets in MB157 were enriched in the components of known TCF-dependent pathways (Figure S5J, Tables S6 and S7). Remarkably, EBF1 directly induced an overlapping set of genes in epigenetically disparate TNBC and GSI-resistant T-ALL (Figures 5G and S5K), as exemplified by CD79A, TCF3, and the therapy resistance-associated gene PHLDA1 (Fearon et al., 2018) (Figures 5H and S5L). Notably, EBF1 binding within the 13 Kbp region flanking PHLDA1 instructed DNA looping in addition to increasing chromatin activity and gene expression in both GSI-resistant T-ALL and EBF1-expressing TNBC (Figure 5H). Thus, EBF1 can create de novo chromatin opening and looping to control transcription in hematopoietic and non-hematopoietic cancer cells.
TCF1 or LEF1, but not both, are dispensable for T-ALL chromatin opening maintenance
TCF1/LEF1 loss coincided with reduced chromatin opening in GSI-treated T-ALL and ectopic TCF1 (although less than EBF1) created de novo chromatin opening in TNBC (Figures 4B, 4D, 5A and 5B). These observations led us to test the requirement of TCF1 and LEF1 for viability and chromatin opening in drug-naive T-ALL. LEF1 and TCF1 double knockout resulted in a mild but significant decrease in proliferation of Cas9-expressing DND41 (Figures S6A and S6B). While loss of TCF1 or LEF1 alone had negligible effect on DND41 chromatin opening, deletion of both factors markedly reduced opening of 80, 158 and 263 elements with TCF consensus binding sequences 3, 6 and 9 days post sgRNA co-transduction, respectively (Figures 6A and 6B). By contrast, regions that gained accessibility upon TCF1 and LEF1 deletion were not enriched for TCF motifs, and were opened potentially due to indirect effects of their loss. Similar to Notch-dependent DND41, loss of TCF1 and LEF1 together, but not alone, significantly reduced chromatin opening at the sites with TCF binding motifs in T-ALL Jurkat cells (Figures S6C and S6D) that express NOTCH1 but exhibited Notch-independent growth (O’Neil et al., 2007). Together, our data suggest that although TCF1 and LEF1 have highly similar protein structures and binding motifs (Cadigan and Waterman, 2012), and both can maintain accessibility, only TCF1 can induce de novo chromatin opening.
Figure 6: EBF1, but not TCF1 or LEF1 alone, maintains chromatin opening.
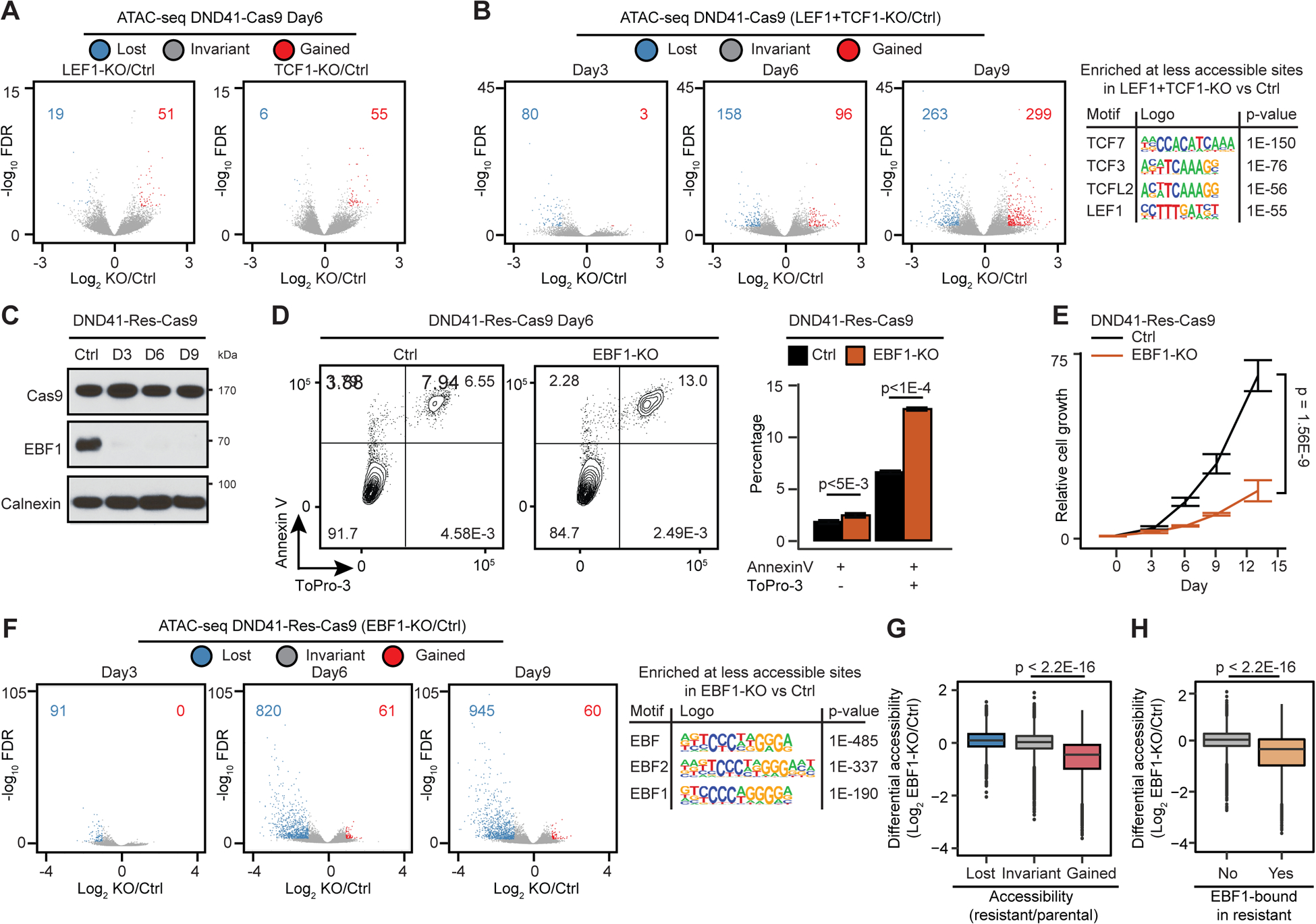
(A, B) Differential opening post deletion of LEF1 or TCF1 (A), or both (B) in Cas9-expressing DND41 (DND41-Cas9) at the noted dates. (B, left): the most enriched motifs.
(C) EBF1 levels post its deletion in Cas9-expressing resistant DND41 (DND41-Res-Cas9). D: day; control: Calnexin.
(D, E) Effect of EBF1 loss on apoptosis, death (D) and relative growth (E) in DND41-Res-Cas9 cells. 3 biological with mean ± SD of 5 technical replicates (t-test).
(F) Differential opening and enriched motifs post EBF1 deletion in DND41-Res-Cas9.
(G, H) Differential ATAC-seq post EBF1 deletion in DND41-Res-Cas9 per differential opening (G) or EBF1 binding (H) during resistance development (Wilcoxon test).
EBF1 is required for accessibility of GSI-resistance-restricted elements
We next used CRISPR-Cas9 to delete EBF1 in GSI-resistant DND41 to directly test its requirement for chromatin opening (Figure 6C). EBF1 deletion induced apoptosis and suppressed proliferation of Cas9-expressing resistant DND41 cells (DND41-Res-Cas9) (Figures 6D and 6E). Moreover, EBF1 loss markedly reduced opening of at least 90, 600, and 900 elements with EBF recognition sequences 3, 6, and 9 days post sgRNA transduction in two independent lines (Figures 6F and S6E), suggesting their direct dependency on EBF1. Notably, EBF1 loss preferentially reduced the opening of the sites that bound EBF1 and/or gained accessibility in GSI-resistance (Figures 6G and 6H), suggesting that EBF1 loss specifically alters opening of elements promoting drug resistance.
To further substantiate the role of EBF1 in chromatin restructuring during GSI-resistance development, we assessed accessibility 4 weeks into treatment. Time-course study of cell proliferation showed an intermediate GSI sensitivity at week 4 (Figure S6F). More refined time-course studies revealed that while TCF1 and LEF1 rapidly reduced upon persistent GSI treatment, EBF1 gradually increased, and its kinetics correlated with MYC expression (Figures S6G and S6H). Concomitant with TCF1/LEF1 loss in 4-week-GSI-treated cells, chromatin opening was markedly reduced at sequences with TCF binding motifs, suggesting their direct dependency on TCF1/LEF1 (Figure S6I). In line with a rapid TCF/LEF1 repression, the number of sites with reduced opening was comparable between the resistant and 4-week-GSI-treated cells (Figures S6I and S6J). Conversely, two-fold more elements gained opening in resistant compared to 4-week-GSI-treated cells, where EBF1 expression was markedly lower (Figures S6I, S6J and S6G). The MYC locus exemplified these dynamics, where opening of E1 and E2 SEs four weeks into treatment was halfway between the parental and resistant cells (Figure S6K). Together, these data further support the role of TCF1 and EBF1 in regulation of chromatin accessibility.
Intrigued by DND41 data, we next examined EBF1 function in other Notch-dependent cells. Like GSI-resistant T-ALL, loss of EBF1 markedly decreased growth, viability and chromatin opening in EBF1-expressing MCL Granta-519 (Figures S6L–P). Importantly, gain of EBF1 decreased GSI sensitivity in Notch-addicted HPB-ALL and KOPTK1 T-ALL (Figures S6Q and S6R). Thus, the widespread effect of EBF1 on chromatin promotes survival of EBF1-expressing cells including Notch-addicted T-ALL.
EBF1 is required for positioning of GSI-resistance-restricted accessible elements
In addition to opening chromatin, EBF1 recruited SMC1, YY1, and instructed DNA looping in TNBC (Figures 5B and 5C). Moreover, EBF1 binding coincided with the repositioned elements in GSI-resistant T-ALL (Figures 4G and S4E–G). These observations led us to examine whether EBF1 chromatin binding is required for GSI-resistant genome folding. We thus focused on early EBF1 genomic targets to overcome the significant time-scale difference between GSI-resistance development (> 12 weeks) and EBF1 genetic deletion (6 days). We reasoned that “early” EBF1 targets were the elements that bound EBF1 and gained accessibility in GSI-resistant cells, and concordantly lost opening 6 days post EBF1 sgRNA transduction (EBF1-KO) (Figure 7A). Notably, early EBF1 target elements mostly gained opening 4 weeks into treatment (Figure 7A). SMC1, YY1 and, to a lesser extent, CTCF had the highest increases at the early EBF1 target elements (Figure 7B). If EBF1 dynamically regulates the level of DNA loops connected to early EBF1 target elements, then these loops should be attenuated following EBF1 knockout. Indeed, EBF1 deletion markedly reduced interaction frequency of loops connected to 83% of the early EBF1 target elements in two independent GSI-resistant DND41 lines (Figures 7C, 7D, S7A, and S7B p-value < 2.2E-16). Importantly, EBF1 deletion significantly reduced MYC as well as EBF1 target genes activated upon GSI-resistance (Figures 7E and S7C). In addition to DNA loops, time-course analysis further supported the involvement of EBF1 in regulating the integrity of TAD boundaries, as exemplified by TCRA and IGJ loci. In line with an intermediate EBF1 expression, a parental-cell-restricted TCRA TAD boundary was partially disrupted 4 weeks into treatment (Figures S6E and S7D). Conversely, an IGJ TAD boundary gradually formed, and its insulation 4 weeks into treatment was midway between parental and resistant cells (Figure S7E). The time-course data further supported the role of EBF1 in controlling A/B compartmentalization. This result was illustrated by the IKZF2 locus, where PC1 and gene expression were partially attenuated 4 weeks into treatment; however, the reduction was much greater in resistant cells with markedly higher EBF1 expression (Figures S7F and S6G).
Figure 7: EBF1 derepression confers GSI-resistance in T-ALL.
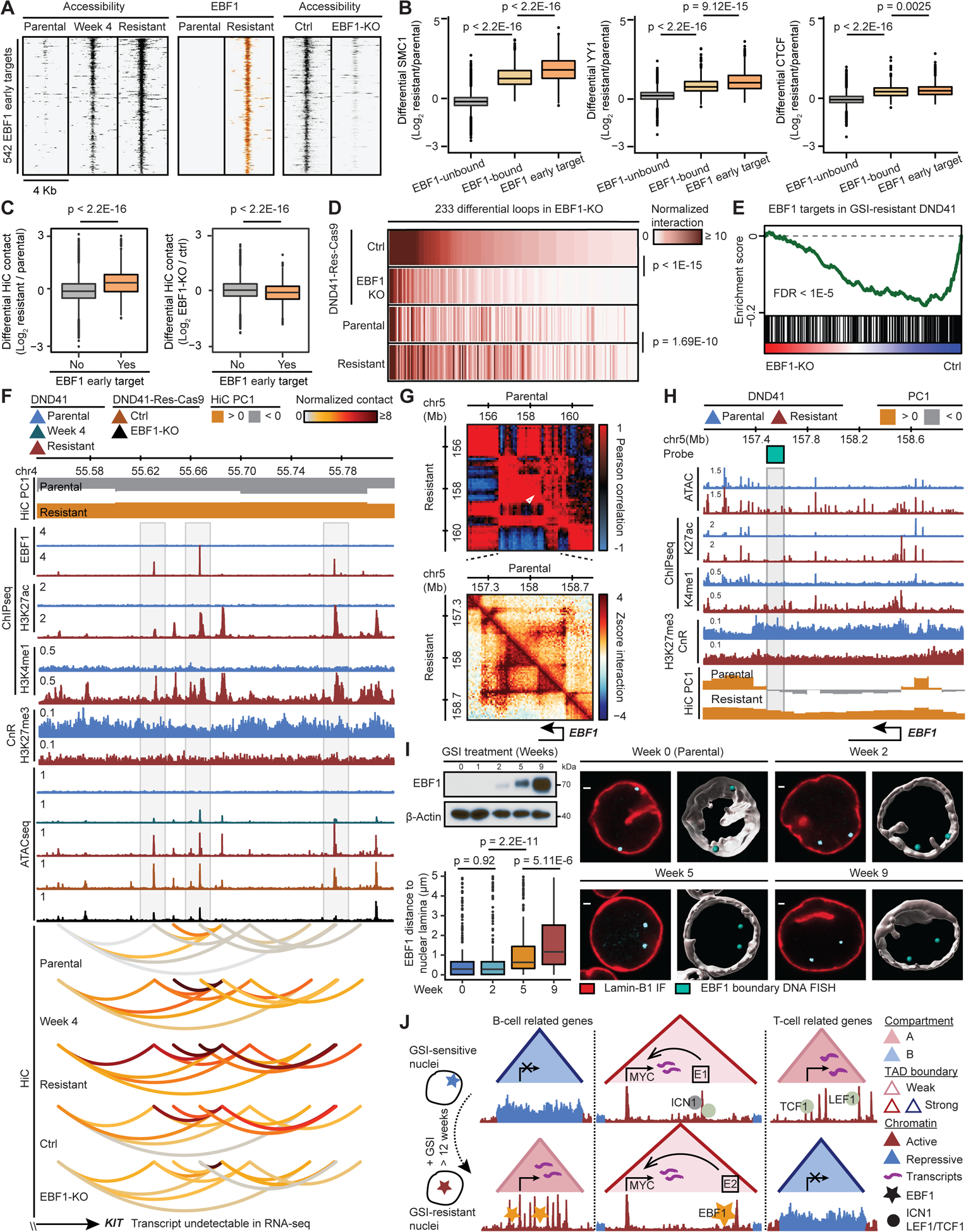
(A) Gradual gain and loss of opening during resistance acquisition and EBF1 deletion (EBF1-KO), respectively, at EBF1 early target elements.
(B) Architectural proteins increase the most at the EBF1 early target elements. EBF1-unbound: EBF1-unbound accessible elements in resistant DND41; EBF1-bound: resistant EBF1-bound elements without change of accessibility in EBF1-KO; EBF1 early target elements per (A) (Wilcoxon test).
(C) Differential HiC signal during resistance development (left) and post EBF1 deletion (right) per loop connected to EBF1 early target element (Wilcoxon test).
(D) HiC signal at EBF1-dependent loops connected to EBF1 early target elements (paired t-test).
(E) Repression of GSI-resistant EBF1 targets (Figure S4K) upon EBF1 deletion in DND41-Res-Cas9.
(F) KIT-connected EBF1-bound elements with EBF1-dependent opening and looping (gray boxes) showing chromatin restructuring can precede expression (STAR Methods).
(G) EBF1 locus Pearson correlation (top) and zoomed-in HiC contact frequency (bottom) matrices.
(H) EBF1 acquired active chromatin state and compartmentalization in resistant cells. Gray box: Oligopaint probe at EBF1-containing TAD boundary.
(I) Gradual EBF1 activation (left, top) and increase of distance between EBF1 and nuclear envelope (left, bottom) during resistance acquisition. Mean (+/− SD) distance at week 0 to 9: 0.74 (+/− 1.19), 0.72 (+/− 1.20), 1.18 (+/− 1.35) and 1.67 (+/− 1.44) μm. Right: representative and Imaris-modeled cells.
(J) Dissociation of B-lineage determining transcription factor EBF1 from nuclear lamina during GSI-resistance development instructs widespread and concerted chromatin refolding that regulates key genes including MYC to circumvent Notch-addiction in T-ALL.
Although our data established a strong link between differential chromatin activity, looping and gene expression in GSI-resistance (Figure 4H, 4J, S4I and S4J), it was still unclear whether transcription precedes or follows chromatin restructuring (Stadhouders et al., 2018). We thus interrogated loci to identify genes for which changes in the chromatin structures preceded activation. Hematopoietic stem cell marker KIT was inactive in parental, 4-week-GSI-treated, EBF1-wildtype GSI-resistant, and EBF1-KO GSI-resistant cells (Figure 7F). EBF1 bound to three silent sites that had been inaccessible in the parental cells and gradually opened. Concomitant with activation of EBF1-bound elements, the KIT locus topology was restructured. Notably, looping to the EBF1-bound elements was markedly increased, as they moved from B to A compartment during resistance development. EBF1 deletion in the resistant cells significantly reduced opening and looping levels of the three EBF1-induced de novo accessible elements. Thus, the EBF1 requirement for activation and positioning of genomic elements can occur independent of transcriptional activation during GSI-resistance acquisition.
EBF1 derepression confers GSI-resistance in T-ALL
We next sought to elucidate mechanisms of EBF1 derepression during resistance development (Figures 4C and S4C). In addition to loss of NOTCH1 signals in resistant cells, we observed concurrent, but gradual, downregulation of GATA3, a known EBF1 repressor (Banerjee et al., 2013) (Figures S1C and S7G). TCF3, a known EBF1 activator (Rothenberg, 2010), was also markedly upregulated in these cells (Figure S7G). Moreover, EBF1 deletion did not change GATA3 and TCF3 in resistant cells (Figure S7G), consistent with their roles upstream of EBF1.
Intrigued by these data (Figures S7G and 4C), we next examined whether EBF1 repositioning to the euchromatic nuclear compartment activates its expression. Sequencing data revealed that EBF1, which was located at the B compartment and marked by H3K27me3 in GSI-sensitive cells, moved to the A compartment and concomitantly gained substantial chromatin activity and opening in the resistant cells (Figures 7G and 7H). Consistent with sequencing data, time-course optical mapping of 3D distance between the EBF1 and the Lamin-B1-marked nuclear envelope visualized gradual relocalization of EBF1 away from the nuclear periphery to the interior (Figures 7I and S7H), and further confirmed EBF1 repositioning from heterochromatic to euchromatic compartment during GSI-resistance development. Notably, EBF1 deletion did not revert EBF1 locus back to the nuclear envelope in 6 days (Figure S7I). Together, these data suggest that loss of EBF1 repressors and gain of EBF1 activators repositioned EBF1 from the nuclear envelope to the interior, thus resulting in its activation that is required for chromatin reorganization and GSI-resistance (Figures 7J).
DISCUSSION
The roles and mechanisms of chromatin folding adaptation in cancer therapy resistance remain enigmatic (Debruyne et al., 2019). Using high-resolution sequencing and optical mapping of genome topology, we revealed that GSI-resistance affects chromatin folding structures at various length scales, allowing GSI-treated NOTCH1-mutated cells to replace T-lineage with B-lineage regulatory programs to bypass their addiction to oncogenic NOTCH1, a key driver of T cell fate choice (Taghon et al., 2005).
Here, we showed changes in chromatin compartments during GSI-resistance development, in contrast to their insensitivity to short-term treatment (Petrovic et al., 2019). This observation resembles the compartment switching during lymphocyte differentiation. Lack of Notch signals promotes B-lineage specification, where Ebf1 nuclear positioning and activation orchestrates cell fate choice (Lin et al., 2010). During the pre-B to pro-B cell transition, Ebf1 moves from the nuclear envelope to the euchromatic interior (Lin et al., 2012). Conversely, during terminal differentiation to plasma cells, Ebf1 moves to heterochromatic nuclear space (Bortnick et al., 2020). A similar role for compartment switching was reported in T cell development. During differentiation to DN2 T cells, Bcl11b is activated and repositioned from the nuclear lamina to the interior (Isoda et al., 2017). Here, we demonstrated the role of spatial compartmentalization in anticancer therapy resistance (Figures 7J). Our detailed studies of T-lineage-specific IKZF2 and B-lineage-specific EBF1 loci exemplify widespread compartment repositioning during GSI-resistance acquisition. The capability of Notch1-mutated DND41 leukemic cells to repress T-lineage and derepress B-lineage programs suggest their distinct plastic developmental state. Notably, we found elevated EBF1 and reduced TCF1 expression in the immature compared to the mature primary T-ALL (Homminga et al., 2011; Zhang et al., 2012) (Figure S7J). In summary, our data reveal that chromatin refolding engendered by lineage-determining transcription factors can result in epigenetic adaptation and cell fate plasticity conferring T-ALL therapy resistance (Figure 7J), an observation which may extend to other cancers.
LIMITATIONS OF THE STUDY
This study employs in vitro cell line systems for in-depth mechanistic study of GSI-resistance and examines EBF1 and TCF1 roles in genome folding regulation. Cell culture studies lack many cancer hallmarks. Thus, further in vivo models could better define the clinical relevance of EBF1 derepression mechanism and precisely reveal how and when it contributes to T-ALL progression and treatment response.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and request for reagents may be directed to and will be fulfilled by the corresponding author, Robert B. Faryabi (faryabi@pennmedicine.upenn.edu).
Materials availability
Plasmids generated in this study will be available upon request.
Data and code availability
Next-generation sequencing data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Original western blot images have been deposited at Mendeley and are publicly available as of the date of publication. Microscopy data reported in this paper will be shared by the lead contact upon request.
This paper does not report any original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-MYC [Y69] | Abcam | Cat# ab32072, RRID:AB_731658 |
| Anti-EBF-1 Antibody | Millipore | Cat# AB10523, RRID:AB_2636856 |
| Anti-Early B-cell factor (EBF1) Antibody | Sigma-Aldrich | Cat# ABE1294, RRID:AB_2893472 |
| Rabbit anti-EBF1 | (Boller et al., 2016) | N/A |
| TCF1 (C46C7) Rabbit mAb antibody | CST | Cat# 2206, RRID:AB_2199300 |
| LEF1 (D6J2W) Rabbit mAb | CST | Cat# 76010, RRID:AB_2799877 |
| Maml1 (D3K7B) Rabbit mAb | CST | Cat# 12166, RRID:AB_2797838 |
| Cleaved Notch1 (Val1744) (D3B8) Rabbit mAb | CST | Cat# 4147, RRID:AB_2153348 |
| Caspase-3 Antibody | CST | Cat# 9662, RRID:AB_331439 |
| Annexin V Recom Horizon V450 50 test antibody | BD | Cat# 560506, RRID:AB_2869356 |
| Monoclonal Anti-β-Actin antibody, clone AC-74 | Sigma-Aldrich | Cat# A5316, RRID:AB_476743 |
| Calnexin (C5C9) Rabbit mAb antibody | CST | Cat# 2679, RRID:AB_2228381 |
| GAPDH (D16H11) XP Rabbit mAb antibody | CST | Cat# 5174, RRID:AB_10622025 |
| Rabbit polyclonal anti-H3 acetyl-K27 | Active Motif | Cat# 39133, RRID:AB_2561016 |
| Tri-Methyl-Histone H3 (Lys27) (C36B11) Rabbit mAb | CST | Cat# 9733, RRID:AB_2616029 |
| Rabbit polyclonal anti-H3 mono methyl-K4 | Abcam | Cat# ab8895, RRID:AB_306847 |
| Rabbit anti-SMC1 Antibody, Affinity Purified | Bethyl | Cat# A300–055A, RRID:AB_2192467 |
| Rabbit polyclonal anti-CTCF | Millipore | Cat# 07–729, RRID:AB_441965 |
| YY1 antibody (pAb) | Active Motif | Cat# 61779, RRID:AB_2793763 |
| Cas9 (7A9-3A3) antibody | CST | Cat# 14697, RRID:AB_2750916 |
| Rabbit Anti-Lamin B1 Polyclonal Antibody | Abcam | Cat# ab16048, RRID:AB_443298 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 | Thermo | Cat# A10042, RRID:AB_2534017 |
| Goat Anti-Rabbit IgG, H & L Chain Specific Peroxidase Conjugate antibody | Millipore | Cat# 401315, RRID:AB_437787 |
| Goat Anti-Mouse IgG, H & L Chain Antibody, Peroxidase Conjugated | Millipore | Cat# 401215, RRID:AB_10682749 |
| Bacterial and virus strains | ||
| One Shot Stbl3 Chemically Competent E.coli | Thermo | Cat# C737303 |
| Chemicals, peptides, and recombinant proteins | ||
| Trypsin (2.5%) | Gibco | Cat# 15090-046 |
| gamma-Secretase Inhibitor XXI (compound E) | Calbiochem | Cat# 565790 |
| FuGene HD | Promega | Cat# E2311 |
| Penicillin-Streptomycin | Corning | Cat# 30-002-CI |
| MEM Non-Essential Amino Acids | Gibco | Cat# 11140-050 |
| Sodium Pyruvate | Gibco | Cat# 11360-070 |
| 2-mercaptoethanol | Sigma | Cat# M3148 |
| RPMI 1640 | Corning | Cat# 10-040-CM |
| DMEM | Corning | Cat# 10-013-CV |
| Dulbecco’s Phosphate-Buffered Salt Solution 1X | Corning | Cat# 21031CV |
| HyClone Fetal bovine serum | Thermo Fisher Scientific | Cat# SH30070.03 |
| L-glutamine | Corning | Cat# 25-005-CI |
| Polybrene | Sigma-Aldrich | Cat# H9268 |
| Gibco Puromycin Dihydrochloride (10 mg/mL) | Thermo | Cat# A1113803 |
| Complete Protease Inhibitor Cocktail Tablets | Roche | Cat# 11697498001 |
| Complete Mini EDTA-free Protease Inhibitor Cocktail | Roche | Cat# 11836170001 |
| Recombinant Protein G Agarose | Invitrogen | Cat# 15920-010 |
| Lithium Chloride, 8M | Sigma-Aldrich | Cat# 7447-41-8 |
| Sodium bicarbonate | Sigma-Aldrich | Cat#144-55-8 |
| RNase A | Roche | Cat# 10109169001 |
| Proteinase K | Invitrogen | Cat# 25530-049 |
| Pierce™ 16% Formaldehyde | Thermo | Cat# 28908 |
| EGS | Thermo | Cat# 21565 |
| Glycine | Invitrogen | Cat# 15527-013 |
| MboI | NEB | Cat# R0147 |
| SalI-HF | NEB | Cat# R3138S |
| AscI | NEB | Cat# R0558S |
| NEBNext End Repair Module | NEB | Cat# E6050 |
| Klenow Fragment (3’ to 5’ exo-) | NEB | Cat# M0212 |
| CIP | NEB | Cat# M0290 |
| TO-PRO3 Ready Flow Reagent | Thermo | Cat# R37170 |
| Hygromycin B Gold 1 g (10 × 1 ml) | Invivogen | Cat# ant-hg-1 |
| Biotin-14-dATP 200 μl (1 mM) | Jena Bioscience | Cat# NU-835-BIO14-S |
| Maxima H Minus Reverse Transcriptase (200 U/μL) | Thermo | Cat# EP0752 |
| T4 DNA Ligase | NEB | Cat# M0202L |
| DNA Polymerase I, Large (Klenow) Fragment | NEB | Cat# M0210S |
| Triton X-100 | Roche | Cat# 10789704001 |
| BSA, 50mg/ml | Invitrogen | Cat# AM2616 |
| SurePAGE, Bis-Tris, 10×8, 4–12%, 15 wells | GenScript | Cat# M00654 |
| MES SDS Running Buffer Powder | GenScript | Cat# M00677 |
| PageRuler Prestained Protein Ladder, 10 to 180 kDa | Thermo | Cat# 26616 |
| PageRuler Plus Prestained Protein Ladder, 10 to 250 kDa | Thermo | Cat# PI26619 |
| Tween 20 | BIO-RAD | Cat# 170-6531 |
| Protein A Magnetic Beads | Pierce | Cat# 88846 |
| Dynabeads MyOne Streptavidin C1 | Invitrogen | Cat# 65001 |
| Dynabeads Protein G for Immunoprecipitation | Invitrogen | Cat# 10003D |
| Phenol-chloroform-isoamyl (PCI) | Invitrogen | Cat# 15593049 |
| Dimethylformamide | Sigma-Aldrich | Cat# D4551 |
| Power SYBR™ Green PCR Master Mix | Applied Biosystems | Cat# 4367659 |
| Tris-HCI, pH 7.5, 1M | Invitrogen | Cat# 15567-027 |
| Tris-HCI, pH 8.0, 1M | Invitrogen | Cat# 15568-025 |
| EDTA, pH 8.0, 0.5M | Invitrogen | Cat# 15575-020 |
| Sodium Chloride | Fisher BioReagents | Cat# 7647-14-5 |
| MgCl2, 1M | Thermo | Cat# AM9530G |
| NP-40 (Igepal CA-630) | Sigma | Cat# I8896 |
| Sodium deoxycholate | Sigma-Aldrich | Cat# D6750 |
| SDS solution, 10% | Invitrogen | Cat# 15553-027 |
| Geneticin Selective Antibiotic (G418) (50 mg/mL) | Thermo | Cat# 10131035 |
| Doxycycline | Sigma | Cat# D9891 |
| Gibco Blasticidin S HCl (10 mg/mL) | Thermo | Cat# A1113903 |
| Phusion PCR Master Mix | NEB | Cat# M0531 |
| Phusion Flash High-Fidelity PCR Master Mix | Thermo | Cat# F548S |
| NEBNext High-Fidelity 2X PCR Master Mix | NEB | Cat# M0541 |
| AMPure XP | Beckman Coulter | Cat# A63881 |
| SuperScript III Reverse Transcriptase | Invitrogen | Cat# 18080093 |
| Gateway™ LR Clonase™ II Enzyme mix | Invitrogen | Cat# 11791020 |
| Gateway™ BP Clonase™ II Enzyme mix | Invitrogen | Cat# 11789020 |
| poly-L-lysine-treated glass slides | Thermo | Cat# P4981-001 |
| Polyvinylsulfonic acid (PVSA) | Sigma Aldrich | Cat# 278424 |
| 20x SSC Buffer | Corning | Cat# 46-020-CM |
| Slowfade | Invitrogen | Cat# 336936 |
| Critical commercial assays | ||
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat# G7571 |
| DC Protein Assay Reagents Package | Bio-rad | Cat# 5000116 |
| QIAquick PCR Purification Kit | QIAGEN | Cat# 28106 |
| MinElute Reaction Cleanup Kit | QIAGEN | Cat# 28204 |
| DNeasy Blood & Tissue Kit | QIAGEN | Cat# 69504 |
| RNeasy Plus Mini Kit | QIAGEN | Cat# 74136 |
| RNeasy Plus Micro Kit | QIAGEN | Cat# 74034 |
| SeqCap Adapter Kit A | Roche | Cat# 7141530001 |
| KAPA Library Quant Kit | Roche | Cat# KK4824 |
| ZymoPURE Plasmid Miniprep Kit 100 preps | Zymo | Cat# D4210 |
| Zymoclean Gel DNA Recovery Kit | Zymo | Cat# D4007 |
| NEBuilder HiFi DNA Assembly Cloning Kit | NEB | Cat# E5520S |
| NEBNext Ultra II DNA Library Prep Kit | NEB | Cat# E7645S |
| HiScribe T7 High Yield RNA Synthesis Kit | NEB | Cat# E2040S |
| SMARTer Stranded Total RNA Sample Prep Kit - HI Mammalian | Takara | Cat# 634873 |
| NucleoBond Xtra Maxi EF | Takara | Cat# 740424 |
| ECL Prime Western Blotting Detection Reagents | Cytiva | Cat# RPN2232 |
| D1000 ScreenTape | Agilent | Cat# 5067-5582 |
| D1000 Reagents | Agilent | Cat# 5067-5583 |
| High Sensitivity D1000 ScreenTape | Agilent | Cat# 5067-5584 |
| High Sensitivity D1000 Reagents | Agilent | Cat# 5067-5585 |
| Genomic DNA ScreenTape | Agilent | Cat# 5067-5365 |
| Genomic DNA Reagents | Agilent | Cat# 5067-5366 |
| RNA ScreenTape | Agilent | Cat# 5067-5576 |
| RNA ScreenTape Ladder | Agilent | Cat# 5067-5578 |
| RNA ScreenTape Sample Buffer | Agilent | Cat# 5067-5577 |
| Qubit dsDNA HS Assay Kit | Invitrogen | Cat# Q32851 |
| Qubit ssDNA Assay Kit | Invitrogen | Cat# Q10212 |
| Arima-HiC Kit (8 Reactions) | Arima | Cat# A510008 |
| Accel-NGS 2S Plus DNA Library Kit (24 rxns) | Swift | Cat# 21024 |
| 2S SET A INDEXING KIT (12 INDICES, 48 RXNS) | Swift | Cat# 26148 |
| NEBNext Multiplex Oligos for Illumina (Dual Index Primers Set 1) | NEB | Cat# E7600S |
| NEBNext Multiplex Oligos for Illumina (Dual Index Primers Set 2) | NEB | Cat# E7780S |
| Nextera XT Index Kit v2 Set A (96 indexes, 384 samples) | Illumina | Cat# FC-131-2001 |
| Illumina Tagment DNA Enzyme and Buffer Kit | Illumina | Cat# 20034197 |
| Nextseq 500/550 high output kit v2.5 (75 cycles) | Illumina | Cat# 20024906 |
| NovaSeq 6000 S1 Reagent Kit v1.5 (100 cycles) | Illumina | Cat# 20028319 |
| Deposited data | ||
| Raw and analyzed data | This study | GSE173872 |
| MB157 HiChIP | (Petrovic et al., 2019) | GSE116869 |
| Experimental models: Cell lines | ||
| HPB-ALL | DSMZ | Cat# ACC 483 |
| KOPTK1 | (Wang et al., 2014) | N/A |
| Granta-519 | (Ryan et al., 2017) | N/A |
| DND41 | DSMZ | Cat# ACC 525 |
| MB157 | ATCC | Cat# CRL-7721 |
| HEK293T | ATCC | Cat# CRL-3216 |
| Oligonucleotides | ||
| Primer: EEF1A1 qPCR forward: TTGTCGTCATTGGACACGTAG | (Petrovic et al., 2019) | N/A |
| Primer: EEF1A1 qPCR reverse: TGCCACCGCATTTATAGATCAG | (Petrovic et al., 2019) | N/A |
| Primer: MYC qPCR forward: ACCCTCTCAACGACAGCAGC | (Petrovic et al., 2019) | N/A |
| Primer: MYC qPCR reverse: ACTCCGTCGAGGAGAGCAGA | (Petrovic et al., 2019) | N/A |
| Primer: CD300A qPCR forward: GCGGCCAAGACCTCAACAA | This study | N/A |
| Primer: CD300A qPCR reverse: TGAGTTCACCACCTCCTCAGT | This study | N/A |
| Primer: DTX1 qPCR forward: ATGGAGCGACTGGTCACAGC | This study | N/A |
| Primer: DTX1 qPCR reverse: CCGTAGATGGCCTTGCAGGT | This study | N/A |
| Primer: CR2 qPCR forward: CACACACCGGGAGTTCCTTA | This study | N/A |
| Primer: CR2 qPCR reverse: ACAACGGATGGTGCTCTCTC | This study | N/A |
| Primer: LEF1 qPCR forward: TAATGCACGTGAAGCCTCAGCA | This study | N/A |
| Primer: LEF1 qPCR reverse: CACTCAGCAACGACATTCGCTC | This study | N/A |
| Primer: TCF7 qPCR forward: CCAACCATCAAGAAGCCCCT | This study | N/A |
| Primer: TCF7 qPCR reverse: TGTGCACTCTGCAATGACCT | This study | N/A |
| Primer: CD79A qPCR forward: GCACCAAGAACCGAATCATC | This study | N/A |
| Primer: CD79A qPCR reverse: CTTCTCGTTCTGCCATCGTT | This study | N/A |
| Primer: pDONR221-LEF1 PCR forward: GGGGACAAGTTTGTACAAAAAAGCAGGCTTCATGCCCCAACTCTCCGGAGGA | This study | N/A |
| Primer: pDONR221-LEF1 PCR reverse: GGGGACAAGTTTGTACAAAAAAGCAGGCTTCATGCCCCAACTCTCCGGAGGA | This study | N/A |
| Primer: pDONR221-TCF7 PCR forward: GGGGACAAGTTTGTACAAAAAAGCAGGCTTCAGATCGGCCACCATGTATCC | This study | N/A |
| Primer: pDONR221-TCF7 PCR reverse: GGGGACCACTTTGTACAAGAAAGCTGGGTCGttatctagagcactgtcatcgg | This study | N/A |
| Primer: TRE3G-EBF1-puro PCR forward: TACCCTCGTAAAACCGGTCGATGTTTGGGATTCAGGAAAG | This study | N/A |
| Primer: TRE3G-EBF1-puro PCR reverse: GTACAAGAAAGCTGGGTCGGTCACATAGGAGGAACAATC | This study | N/A |
| sgRNA: MYC targeting sequence E3g5: GCGTGACCAGATCCCGGAGT | This study | N/A |
| sgRNA: LEF1 targeting sequence g3: CCCTCCTACTCGAGTTATTC | This study | N/A |
| sgRNA: TCF7 targeting sequence g5: TGTTCGTAGAGAGAGAGTTG | This study | N/A |
| sgRNA: EBF1 targeting sequence g7: TCTCATGTGTGAGCAAGACT | This study | N/A |
| Secondary Oligopaint probe: /5Alex488N/CACACGCTCTTCCGTTCTATGCGACGTCGGTG/3AlexF488N/ | (Beliveau et al., 2018) | N/A |
| Secondary Oligopaint probe: /5ATTO565N/ACACCCTTGCACGTCGTGGACCTCCTGCGCTA/3ATTO565N/ | (Beliveau et al., 2018) | N/A |
| Secondary Oligopaint probe: /5Alex647N/TGATCGACCACGGCCAAGACGGAGAGCGTGTG/3AlexF647N/ | (Beliveau et al., 2018) | N/A |
| Recombinant DNA | ||
| LentiV_Cas9_puro | (Grevet et al., 2018) | Addgene Cat# 108100 |
| LRG2.1 | (Grevet et al., 2018) | Addgene Cat# 108098 |
| LRCherry2.1 | (Grevet et al., 2018) | Addgene Cat# 108099 |
| Gateway pDONR221 | Invitrogen | Cat# 12536017 |
| pInducer20 | Addgene | Cat# 44012 |
| pINDUCER21 (ORF-EG) | Addgene | Cat# 46948 |
| pENTR221-TCF7 | Addgene | Cat# 79498 |
| pDONR221-EBF1 | DNASU | Cat# HsCD00296820 |
| pDONR221-LEF1 | This study | N/A |
| pDONR221-TCF7 | This study | N/A |
| pInducer20-LEF1 | This study | N/A |
| pInducer20-TCF7 | This study | N/A |
| pInducer20-EBF1 | This study | N/A |
| pInducer21-EBF1 | This study | N/A |
| TRE3G-TRF1-puro | (Dilley et al., 2016) | N/A |
| CMV-rtTA3G-blast | (Dilley et al., 2016) | N/A |
| TRE3G-EBF1-puro | This study | N/A |
| Software and algorithms | ||
| Benchling | N/A | https://www.benchling.com |
| BEDTools v2.25.0 | (Quinlan and Hall, 2010) |
https://bedtools.readthedocs.io/en/stable; RRID:SCR_006646 |
| biomaRt v2.28.0 | (Durinck et al., 2005) | https://www.rdocumentation.org/packages/biomaRt/versions/2.28.0; RRID:SCR_006442 |
| BWA v0.7.13 | (Li and Durbin, 2009) | http://bio-bwa.sourceforge.net; RRID:SCR_010910 |
| DESeq2 v1.10.1 | (Love et al., 2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html; RRID:SCR_015687 |
| FlowJo v9.7.7 | FlowJo LLC | https://www.flowjo.com; RRID:SCR_008520 |
| ggplot2 v2.2.1 | (Wickham, 2009) | https://ggplot2.tidyverse.org; RRID:SCR_014601 |
| HiC-Pro v2.5.0 | (Servant et al., 2015) | https://github.com/nservant/HiC-Pro |
| HOMER v4.8 | (Heinz et al., 2010) | http://homer.ucsd.edu/homer; RRID:SCR_010881 |
| JASPAR | (Sandelin et al., 2004) | http://jaspar.genereg.net; RRID:SCR_003030 |
| Juicer tools v1.7.6 | (Durand et al., 2016b) | https://github.com/aidenlab/juicer |
| Juicebox v1.0 | (Durand et al., 2016a) | https://www.aidenlab.org/juicebox |
| MACS v2.0.9 | (Zhang et al., 2008) | https://github.com/taoliu/MACS; RRID:SCR_013291 |
| Mango v1.2.0 | (Phanstiel et al., 2015) | https://github.com/dphansti/mango |
| MEME v4.11.1 | (Bailey et al., 2015) | http://meme-suite.org/index.html; RRID:SCR_001783 |
| MSigDB v6.1 | (Liberzon et al., 2015) | http://software.broadinstitute.org/gsea/msigdb/index.jsp; RRID:SCR_003199 |
| normR v1.6.0 | (Helmuth and Chung, 2018) | https://bioconductor.org/packages/release/bioc/html/normr.html; RRID:SCR_006442 |
| Picard v2.1.0 | Broad Institute | https://broadinstitute.github.io/picard; RRID:SCR_006525 |
| pheatmap v 1.0.10 | R. Kodle | https://github.com/raivokolde/pheatmap; RRID:SCR_016418 |
| RepeatMasker | AFA Smit, R Hubley, P. Green | http://www.repeatmasker.org; RRID:SCR_012954 |
| SAMTools v1.3 | (Li et al., 2009) | http://samtools.sourceforge.net; RRID:SCR_002105 |
| STAR v2.5 | (Dobin et al., 2013) | https://github.com/alexdobin/STAR; RRID:SCR_015899 |
| Sushi v1.18.0 | (Phanstiel et al., 2014) | https://bioconductor.org/packages/release/bioc/html/Sushi.html; RRID:SCR_006442 |
| Trim Galore v0.4.1 | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore; RRID:SCR_011847 |
| UCSC tools v329 | (Kent et al., 2010) | https://github.com/ucscGenomeBrowser/kent |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture and generation of GSI-resistant DND41
DND41 was purchased from Leibniz-Institute DSMZ-German Collection of Microorganisms and Cell Lines (DSMZ, cat# ACC525). MB157 (CRL-7721), HEK293T (CRL-11268) and Jurkat E6-1 (TIB-152) were purchased from ATCC. Granta519 was from the Broad Novartis Cancer cell line encyclopedia (Ryan et al., 2017). DND41, Jurkat E6-1, HPB-ALL and KOPTK1 were cultured in RPMI 1640 (Corning, cat# 10-040-CM) supplemented with 10% fetal bovine serum (Hyclone, cat# SH30070.03), 2 mM L-glutamine (Corning, cat# 25-005-CI), 100 U/mL and 100 μg/mL penicillin/streptomycin (Corning, cat# 30-002-CI), 100 mM nonessential amino acids (Gibco, cat# 11140-050), 1mM sodium pyruvate (Gibco, cat#11360-070) and 0.1mM of 2-mercaptoethanol (Sigma, cat# M3148). MB157, HEK293T and Granta519 were cultured in DMEM (Corning, cat# 10-013-CV) supplemented with 10% fetal bovine serum (Hyclone, cat# SH30070.03) and 100 U/mL and 100 μg/mL penicillin/streptomycin (Corning, cat# 30-002-CI). To generate GSI-resistant DND41, parental DND41 was treated with 125nM gamma-secretase inhibitor (GSI) compound E (Calbiochem cat# 565790) for more than 12 weeks with regular refreshment of media and maintained in 125nM GSI. To generate GSI-resistant-reversed cells, GSI-resistant DND41 were cultured in media without GSI for 6–8 weeks. The process of GSI-resistant DND41 generation was independently repeated by different personnel ten times and activation of EBF1 was observed in all replicates. All cell lines, including the Cas9-expressing and pInducer20 cell lines described below, were grown at 37°C and 5% CO2 and were used at a low passage number (<12) and subjected to regular mycoplasma tests and short tandem repeat (STR) profiling.
METHOD DETAILS
Lentiviral packaging
Lentivirus was produced in HEK293T cells as previously described (Petrovic et al., 2019). Briefly, 4.5×106 HEK293T cells were plated in 8 mL DMEM media in 10 cm dishes 12–16 hours before transfection. The lentiviral constructs, packaging plasmid (pCMVdelta) and envelope plasmid (VSV-G) were co-transfected using FuGene HD (Promega, cat# E2311). The cells were returned to the incubator for 6–8 hours before replacement with 6 mL media. Lentiviral supernatants were harvested 48 hours post-transfection, subjected to 0.45 μm filtration and stored at −80°C.
CRISPR-Cas9 Editing
CRISPR/Cas9 system was used for knocking out MYC, LEF1, TCF1 and EBF1. Codon-optimized version of Cas9 carrying puromycin resistance gene (Cas9-puro) and sgRNA vectors carrying GFP (LRG2.1) or mCherry (LRmCherry2.1) (Grevet et al., 2018) were used. Parental DND41, GSI-resistant DND41, Jurkat E6-1 and Granta519 cells were transduced with Cas9-puro lentivirus by spinfection at 2000 rpm for 90 min at 22°C in the presence of 6 μg/mL polybrene (Sigma-Aldrich, cat# H9268). Transduced DND41 cells were selected 3 days after spinfection with incremental 0.1–1 μg/mL puromycin until most cells are viable and maintained in 1 μg/mL puromycin. Transduced Jurkat E6-1 and Granta519 cells were selected and maintained with 1–4 μg/mL puromycin. Expression of Cas9 was confirmed with western blot (CST cat# 14697S).
sgRNA targeting MYC, LEF1, TCF7 or EBF1 exons were designed with UCSC genome browser CRISPR Targets Track and Benchling (https://www.benchling.com/). A total of 5, 8, 16 and 8 sgRNAs were designed for MYC, LEF1, TCF7 and EBF1, respectively. MYC and LEF1 targeting sgRNAs were cloned into LRG2.1 while TCF7 and EBF1 sgRNAs were cloned into LRmCherry2.1. The efficacy of each sgRNA was determined by TIDE assay and western blot with antibodies against MYC Y69 (Abcam cat# ab32072), LEF1 (D6J2W) (CST cat# 76010S), TCF1 (C46C7) (CST cat# 2206S) and EBF1 (EMD Millipore cat# ABE1294 or AB10523). MYC-E3g5, LEF1-g3, TCF7-g5 and EBF1-g7 were used in the subsequent experiments.
For targeting MYC, Cas9-expressing parental DND41 (DND41-Cas9) and GSI-resistant DND41 cells (DND41-Res-Cas9) were transduced with lentivirus produced with LRG2.1-MYC-E3g5 using the spinfection method above. For targeting LEF1 and TCF7 individually and simultaneously, DND41-Cas9 or Jurkat-E6-1-Cas9 cells were transduced with lentivirus produced with LRG2.1-LEF1-g3 and LRmCherry2.1-TCF7-g5 individually or with a mixture of 1:1 ratio using the spinfection method above. For targeting EBF1, DND41-Res-Cas9 or Granta519-Cas9 cells were transduced with lentivirus produced with LRmCherry2.1-EBF1-g7 using the spinfection method above. In control condition, Cas9 expressing cells were transduced with LRG2.1 and/or LRmCherry2.1.
Ectopic induction of LEF1, TCF1 and EBF1
LEF1 CDS with attB1/2 overhang was produced by PCR from cDNA of parental DND41. TCF7 CDS with attB1/2 overhang was produced by PCR from pENTR221-TCF7 (addgene cat# 79498). These two CDS were then cloned into pDONR221 with attP1/2 using Gateway cloning BP reactions (Invitrogen cat# 11789020) to produce pDONR221-LEF1 and pDONR221-TCF7 plasmids. pDONR221-EBF1 was purchased from DNASU (cat# HsCD00296820). pInducer20 (Addgene cat# 44012) or pInducer21 (Addgene cat# 46948) was then used with pDONR221-LEF1, -TCF1 and -EBF1 in LR reactions (Invitrogen cat# 11791020) to produce pInducer20-LEF1, pInducer20-TCF1, pInducer20-EBF1 and pInducer21-EBF1. TRE3G-TRF1-puro and CMV-rtTA3G-blast plasmids were generous gifts from the Greenberg lab. 5 μg TRE3G-TRF1-puro was digested with SalI-HF (NEB cat# R3138S) and AscI (NEB cat# R0558S) at 37 °C for 2 hours to produce TRE3G-puro backbone. EBF1 CDS with proper overhang was amplified from pInducer20-EBF1 with primers designed by NEBuilder (https://nebuilder.neb.com/#!/). TRE3G-puro backbone and EBF1 CDS were assembled with NEBuilder HiFi DNA Assembly Cloning Kit (cat# E5520S) to produce TRE3G-EBF1-puro. All plasmids were subject to Sanger sequencing validation using Genewiz (https://www.genewiz.com/). MB157 cells were transduced with lentivirus produced with pInducer20-LEF1, pInducer20-TCF1 and pInducer20-EBF1 and selected under 1.2 mg/mL G418 (Gibco cat# 10131027) for 7 days. To ectopically express LEF1, TCF1 and EBF1, 10 μg/mL doxycycline was added for 72 hours and MB157-pInducer20 cells were harvested to perform western blot, ATAC-seq, RNA-seq, ChIP-seq and SMC1 HiChIP. HBP-ALL cells were transduced with lentivirus produced with TRE3G-EBF1-puro and CMV-rtTA3G-blast and selected under 1 μg/mL puromycin (cat# A1113803) and 10 μg/mL blasticidin (cat# A1113903) for 14 days. KOPTK1 cells were transduced with lentivirus produced with pInducer21-EBF1 and sorted for top 10% GFP positive cells three days post transduction. To ectopically express EBF1 in HPB-ALL-TRE3G-EBF1 and KOPTK1-pInd21-EBF1, 0.5 μg/mL doxycycline was added for 72 hours, and then cells were harvested to perform western blot in two independent biological replicates.
Assay for Transposase-Accessible Chromatin (ATAC-seq)
ATAC-seq was performed as previously described (Buenrostro et al., 2013) and three replicates were performed for each condition. Briefly, 50,000 cells were pelleted at 800 × g and washed with 50 μl of ice cold 1 × PBS (Corning cat# 21031CV), followed by 2min treatment with 50 μl lysis buffer (10 mM Tris-HCl, pH 7.4, 3 mM MgCl2, 10 mM NaCl, 0.1% Igepal cat# CA-630). Pelleted nuclei were resuspended in 50 μl of transposition buffer (25 μl of 2 × TD buffer, 22.5 μl of molecular biology grade water and 2.5 μl Tn5 transposase (Illumina cat# FC-121-1030) to tag the accessible chromatin for 45min at 37°C. Tagmented DNA was purified with MinElute Reaction Cleanup kit (Qiagen cat# 28204) and amplified with 5 PCR cycles. Additional number of PCR cycles was determined from the side reaction and ranged from 10–12 total cycles of PCR. Libraries were purified using QiaQuick PCR purification kit (Qiagen cat# 28106) and eluted in 20 μl EB buffer. Indexed libraries were assessed for nucleosome patterning on TapeStation 4150 (Agilent) and paired-end sequenced (38bp+38bp) on Illumina NextSeq 550.
For comparison of chromatin accessibility in parental, GSI-resistant and four-week-GSI-treated DND41 cells, parental DND41 was treated with DMSO or 125 nM GSI for 24 hours and GSI-resistant cells were refreshed with media containing 125 nM GSI for 24 hours, and DAPI-negative single live cells in four-week-GSI-treated DND41 were sorted on BD FACSAria Fusion with 100 μm nozzle and immediately proceeded to ATAC-seq. For assessing the impact of LEF1 or TCF1 loss on parental DND41 genome, DND41-Cas9 cells carrying LRG2.1-LEF1-g3 or LRmCherry2.1-TCF7-g5 were sorted 6 days post transduction. To assess the impact of LEF1 and TCF1 loss on parental DND41 genome, DND41-Cas9 carrying LRG2.1-LEF1-g3 and LRmCherry2.1-TCF7-g5 were sorted 3, 6 and 9 days post transduction. DND41-Cas9 carrying LRG2.1 and LRmCherry2.1 were sorted 3 days post transduction as control. Jurkat-E-6-1-Cas9 with LRG2.1-LEF1-g3 and/or LRmCherry2.1-TCF7-g5 and control cells were sorted 6 days post transduction. To assess the impact of EBF1 loss on GSI-resistant DND41 genome, DND41-Res-Cas9 with LRmCherry2.1-EBF1-g7 were sorted 3, 6 and 9 days post transduction and control cells were sorted 3 days post transduction. Granta519-Cas9 with LRmCherry2.1-EBF1-g7 and control cells were sorted 3 days post transduction. Cell sorting was done on BD FACSAria Fusion with 100 μm nozzle gating for single live cells with DAPI staining and top 50% GFP and/or mCherry expression. To assess the impact of ectopic LEF1, TCF1 and EBF1 expression on MB157 genome accessibility, MB157 cells and doxycycline induced MB157pInducer20-LEF1, TCF1 and EBF1 cells were sorted on BD FACSJazz with 100 μm nozzle gating for single live cells with DAPI staining.
RNA sequencing
Strand-specific RNA-seq was performed in the following cells with three replicates per condition: 1) parental and GSI-resistant-reversed DND41 treated with DMSO or 125 nM GSI for 24 hours, GSI-resistant DND41 refreshed with media containing 125 nM GSI for 24 hours, and FACS-sorted single live cells in four-week-GSI-treated DND41; 2) MB157 and induced MB157pInducer20-TCF1 and -EBF1; 3) DND41-Res-Cas9 control and LRmCherry2.1-EBF1-g7 cells sorted 6 days post transduction. In all experiments, 3–5×10^5 cells were washed with 1 × PBS and lysed with 350 μl RLT Plus buffer (Qiagen) supplemented with 2-mercaptoethanol, vortexed briefly and immediately proceeded to RNA isolation with RNeasy Plus Micro Kit (Qiagen #74034). RNA integrity numbers were determined using TapeStation 4150 (Agilent), and all samples used for RNA-seq library preparation had RIN numbers greater than 9.5. 300–800 ng of total RNA was used, and libraries were prepared using the SMARTer Stranded Total RNA Sample Prep Kit-HI Mammalian (Clontech cat# 634873). Libraries were paired-end sequenced (38bp+38bp) on Illumina NextSeq 550.
Chromatin Immunoprecipitation Sequencing (ChIP-seq)
ChIP-seq for histone marks was performed as previously described (Petrovic et al., 2019). Briefly, 1 × 10^7 cells were crosslinked with 1% formaldehyde and lysed. Nuclei were extracted and resuspended in 1% SDS and sonicated using Brandson 450 sonicator with 25% amplitude, 0.5s on, 1s off for a total of 4min 30s on. Solubilized chromatin was then diluted and cleared with IgG (CST cat# 2729S) and recombinant protein G–conjugated Agarose beads (Invitrogen #15920–010) for 1 hour at 4°C. The cleared supernatant was subsequently immunoprecipitated with antibodies recognizing H3K27ac (Active Motif cat# 39133) or H3K4me1 (Abcam cat# ab8895) overnight at 4°C. Buffers in all steps above were supplemented with protease inhibitors (Roche cat# 11697498001). Antibody-chromatin complexes were captured with recombinant protein G–conjugated Agarose beads, washed with low salt wash buffer, high salt wash buffer, LiCl wash buffer and TE buffer with 50mM NaCl and eluted. Input sample was prepared by the same approach without immunoprecipitation.
ChIP-seq for transcription factors was performed as previously described (Bossen et al., 2015). Briefly, Dynabeads Protein G (ThermoFisherScientific cat# 10003D) was incubated with antibodies recognizing MAML1 (D3K7B) (CST cat# 12166S), TCF1/TCF7 (C46C7) (CST cat# c2206S), LEF1 (D6J2W) (CST cat# 76010S), EBF1 (Boller et al., 2016), SMC1a (Bethyl, cat# A300-055A), YY1 (Active motif cat# 61779) and CTCF (EMD Millipore cat# 07-729) for 8–12 hours in PBS+0.5% BSA at 4°C. 4 × 10^7 cells were crosslinked with 1% formaldehyde and 1.5mM EGS (ThermoFisherScientific cat# 21565) and sonicated using Brandson 450 sonicator with 17% amplitude, 10s on, 1min off for 10 times. Lysate was then cleared by centrifuging for 5min at 16 Kg, 4°C and incubated with antibody-bound beads and 1% Triton Tx-100 (Roche cat# 10789704001) overnight at 4°C. Buffers in all steps above were supplemented with protease inhibitors (Roche cat# 11697498001). Antibody-chromatin complexes captured on beads were then separated on magnet and washed with wash buffer 1, 2, 3, LiCl wash Buffer and TE buffer and eluted.
Following elution of histone mark and transcription factor samples, RNase (Roche cat# 10109169001) and Proteinase K (Invitrogen cat# 25530-049) treatments were performed, and reverse crosslinked at 65°C overnight. DNA was purified with QiaQuick PCR Purification Kit (Qiagen, cat# 28106). Libraries were then prepared using the NEBNext Ultra II DNA library Prep Kit for Illumina (NEB cat# E7645S) with single (NEB cat# E7335, cat# E7710) or dual (NEB cat# E7600, cat# E7780) indexing. Two replicates were performed for each condition. Indexed libraries were validated for quality and size distribution using a TapeStation 4150 (Agilent), quantified by KAPA Library Quantification Kit (Roche cat# KK4824) and paired-end sequenced (38 bp+38 bp) on Illumina NextSeq 550.
CUT & RUN
CUT & RUN was performed as described (Skene and Henikoff, 2017). Briefly, 5×10^5 cells were immobilized on Concanavalin-coated magnetic beads (Bangs Laboratories cat# cBP531) and incubated with Tri-Methyl-Histone H3 (Lys27) (C36B11) antibody (CST cat# 9733S) overnight at 4°C. Beads were washed with digitonin wash buffer and mixed in pA-MNase (gift from Henikoff lab) at 4°C for 1 hour. After activation by Ca2+ on wet ice for 30min, 2XSTOP buffer was added and incubated 37°C for 10min to release CUT & RUN fragments from the insoluble nuclear chromatin. DNA was then extracted from the supernatant using Phenol-chloroform-isoamyl alcohol 25:24:1 (PCI; Invitrogen cat# 15593049) method. Library construction, quantification and sequencing were done as described above for ChIP-seq.
In situ HiC
In situ HiC was performed in parental, GSI-resistant and FACS-sorted single live four-week-GSI-treated DND41 cells, and DND41-Res-Cas9 control and LRmCherry2.1-EBF1-g7 cells sorted 6 days post transduction. Briefly, 1–2×10^6 cells were washed with 1×PBS containing 3% BSA and fixed with 2% formaldehyde and proceeded with Arima-HiC Kit per manufacturer’s instructions. Samples passing Arima-QC1 were used for library construction with Accel-NGS 2S Plus DNA Library Kit (Swift cat# 21024) and 2S SET A INDEXING KIT (Swift cat# 26148) and assessed with Arima-QC2. Samples passing Arima-QC2 were subsequently PCR amplified based on Arima-QC2 calculations, quantified with TapeStation 4150 (Agilent) and KAPA Library Quantification Kit (Roche cat# KK4824) and paired-end (38bp+38bp) sequenced on Illumina NextSeq 550.
SMC1 HiChIP
SMC1 HiChIP was performed in parental and GSI-resistant DND41 (2 replicates each) and induced MB157pInducer20-TCF1 and EBF1 cells (1 replicate each) as previously described (Petrovic et al., 2019). DMSO and GSI-washout treated MB157 HiChIP samples were previously published (Petrovic et al., 2019). Briefly, 2 × 107 cells were crosslinked with 1% formaldehyde (Thermo Fisher Scientific, cat# 28908) for 10 min and subsequently quenched with 0.125M glycine (Invitrogen, cat# 15527-013). Chromatin was digested using MboI restriction enzyme (NEB, cat# R0147), followed by biotin incorporation with Biotin-14-dATP (Invitrogen, cat# 19524-016 or Jena Bioscience cat# NU-835-BIO14-S). DNA was then ligated with T4 ligase (NEB cat# M0202L) and sonicated on Covaris LE220. Sheared chromatin was 4-fold diluted with ChIP dilution buffer (16.7mM Tris pH 7.5, 167mM NaCl, 1.2mM EDTA, 0.01% SDS, 1.1% Triton X-100), cleared with IgG (CST cat# 2729S) and then incubated with anti-SMC1 antibody (Bethyl, cat# A300-055A) at 4°C overnight. Chromatin-antibody complexes were captured by Protein-A magnetic beads (Pierce, cat# 88846) and subsequently washed with Low Salt Wash Buffer, High Salt Wash Buffer, LiCl Wash Buffer and eluted. DNA was purified with MinElute PCR Purification Kit (Qiagen, cat# 28004) and quantified using Qubit dsDNA HS Assay Kit (Invitrogen, cat# Q32851). 50–150ng was used for capture with Dynabeads MyOne Streptavidin C-1 (Invitrogen, cat# 65001) and an appropriate amount of Tn5 enzyme (Illumina, cat# FC-121-1030) was added to captured DNA to generate sequencing library. Paired-end sequencing (38 bp+38 bp) was performed on Illumina NextSeq 550.
UMI 4C-seq
UMI 4C-seq was performed for parental and GSI-resistant-reversed DND41 treated with DMSO or 125 nM GSI for 24 hours and GSI-resistant DND41 refreshed with media containing 125 nM GSI for 24 hours. Method was previously described (Cho et al., 2018) with minor modifications. 5–10 × 10^6 cells were crosslinked with 1% formaldehyde and lysed with ice-cold Hi-C Lysis buffer (10 mM Tris-HCl pH 8.0, 10 mM NaCl, 0.2% NP-40, 1X protease inhibitors) at 4°C for 30 minutes. Pelleted nuclei were resuspended in 0.5% SDS at 62°C for 10min and then quenched with Triton X-100 at 37°C for 15 min. Chromatin was then digested with MboI (NEB cat# R0147) at 37°C for 2 hours and ligated with T4 DNA Ligase (NEB cat# M0202) at room temperature for 4 hours with rotation. Pelleted nuclei were resuspended in 200 μL of Proteinase K Buffer (10 mM Tris pH 7.0, 100 mM NaCl, 1mM EDTA, 0.5% SDS) at 55°C for 1hr, reverse crosslinked at 67°C overnight, and treated with RNase A before DNA precipitation by ethanol and NaAc. 10 μg of eluted DNA was sonicated on Bioruptor for 5 cycles of 30sec on, 60sec off followed by sequential incubation with end repair mix (NEB cat# E6050), Klenow exo- (NEB cat# M0212) and CIP (NEB cat# M0290). DNA was ligated with Illumina forked indexed adapters (Roche cat# 7141530001) with Quick Ligase (NEB cat# M2200L) at room temperature for 30min and denatured at 95°C for 2min to release the non-ligated strand of the adapter. Single-stranded DNA was quantified by Qubit ssDNA Assay Kit (Invitrogen cat# Q10212) and 200ng of 4C DNA template was amplified by nested PCR using primer sets targeting MYC promoter. 3–5 replicates were performed for each condition. Indexed libraries were validated for quality and size distribution using a TapeStation 4150 (Agilent), quantified by KAPA Library Quantification Kit (Roche #KK4824) and paired-end sequenced (38 bp+38 bp) on Illumina NextSeq 550.
Cancer-associated gene panel sequencing
Ultra-deep genomic sequencing of parental and GSI-resistant DND41 cells was performed using CLIA-approved clinical assay used for sequencing of cancer patients at the Hospital of the University of Pennsylvania. Briefly, genomic DNA of parental and GSI-resistant DND41 cells was extracted (296.6 and 293.5 ng respectively) using DNeasy Blood & Tissue Kits (Qiagen cat# 69506) and sheared with Covaris LE220-plus ultrasonicator to achieve an average length of 200 bp. Whole genome libraries were generated with a Kapa Hyper Prep kit (Roche cat #07962363001) targeting the full-coding regions of 505 cancer-associated genes listed in Table S1. The target-enriched libraries were then PCR amplified and paired-end sequenced (150bp + 150bp) on Illumina NovaSeq 6000.
Oligopaint DNA FISH and Lamin-B1 IF
Oligopaint libraries for DND41 were designed using the OligoMiner pipeline (Beliveau et al., 2018) using the default parameters and synthesized with CustomArray (GenScript). 42 nucleotide sequences with homology to the regions of interest were mined from the hg19 genome. Each probe library targeted a 50Kbp, 75Kbp or 100Kbp region for EBF1 and MYC, IKZF2 and TCRA loci, respectively. DNA secondary oligos conjugated with Alexa-488, Atto-565 and Alexa-647 were purchased from IDT with high-performance liquid chromatography (HPLC) purification. Probe amplification primers and bridges were purchased from IDT with standard desalting.
Parental, GSI-resistant, FACS-sorted single live cells in two-, five- and nine-week-GSI-treated DND41 cells, and DND41-Res-Cas9 control and LRmCherry2.1-EBF1-g7 cells sorted 6 days post transduction were settled on poly-L-lysine-treated glass slides (ThermoScientific cat# P4981-001) for 30min in a humidified chamber and fixed for 10min in 4% formaldehyde at room temperature, followed by permeabilization in 0.5% Triton X-100 for 15min. For combined Lamin-B1 immunofluorescence, slides were block with 10% BSA in 1× PBS for 1 hour and incubated with 100 μl PBS + 5% donkey serum + 1μl Lamin-B1 antibody (Abcam cat# ab16048) at room temperature for 2 hours. After washing with 1 × PBS 3 × 5min, slides were incubated in 100 μl PBS + 5% donkey serum + 1μl secondary antibody (Invitrogen cat# A10042) at room temperature for 2 hours, washed with 1 × PBS 3 × 10min, and fixed again with 4% formaldehyde. For DNA FISH, slides were washed with 70%, 90%, and 100% ethanol for 2min each and slightly dried before treated with SSCT (2 × SSC, 0.1% Tween) + 50% formamide for 5min at room temperature, 2.5min at 92°C and 20min at 60°C. 10–50 pmol of primary Oligopaint probes in hybridization buffer (10% dextran sulfate, 2xSSCT, 50% formamide, 4% PVSA, 5.6mM dNTPs, 10μg RNase A) were then added to the cells, covered with a coverslip, and sealed with rubber cement. Cells were denatured at 92°C for 2.5min and incubated at 37°C overnight in a humidified chamber. Approximately 16–18 hours later, coverslips were removed with a razor blade, and slides were washed in 2×SSCT at 60°C for 15min and room temperature for 10min, and 0.2×SSC at room temperature for 10min. 10pmol of secondary probes containing fluorophores (Alexa-488, Atto-565, and Alexa-647) and 2pmol bridges were added to slides in secondary hybridization buffer (10% dextran sulfate, 10% formamide, and 4% PVSA), and covered with a coverslip sealed with rubber cement. Slides were incubated at room temperature for 2 hours in a humidified chamber, followed by washes in 2×SSCT at 60°C for 15 minutes, 2×SSCT at RT for 10 minutes, and 0.2×SSC with 1:4,000 DAPI at RT for 10 minutes and mounted in SlowFade Gold Antifade Mountant (Invitrogen cat# S36936).
Cell proliferation
Cell proliferation was measured with CellTiter Glo Luminescent Cell Viability Assay (Promega cat# G7571) according to the manufacturer’s instructions. 1) Parental and GSI-resistant DND41 cells were plated with 0, 1nM, 10nM, 100nM, 1μM, 10μM and 100μM GSI in 5 replicates with 2,000 cells per well on 96-well plates and luminescence was measured on day 6. 2) Parental, GSI-resistant, FACS-sorted single live cells in four- and eight-week GSI-treated DND41 cells were plated with 0, 10nM, 100nM, 1μM, 10μM and 100μM GSI in 5 replicates with 2,000 cells per well on 96-well plates and luminescence was measured on day 6. 3) HPB-ALL-TRE3G-EBF1 and KOPTK1-pInducer21-EBF1 were plated with 0, 10nM, 100nM, 1μM, 10μM and 100μM GSI and 0 or 0.5 μg/mL doxycycline in 5 replicates with 2,000 cells per well on 96-well plates and luminescence was measured on day 6. 4) Control and EBF1 targeted DND41-Res-Cas9 cells were sorted 3 days post gRNA infection and plated in 5 replicates with 2,000 cells per well on 96-well plates. Luminescence was measured 24 hours after plating (day 0) and every 3 days for a total of 15 days. The same procedure was applied to control and LEF1+TCF1 targeted DND41-Cas9 cells. Control and MYC-targeted DND41-Cas9 or DND41-Res-Cas9 cells were measured for 6 days. For control and EBF1 targeted Granta519, cells were plated at 1,000 per well and measured for a total of 12 days. Statistics for cell growth changes were calculated using Student’s t-test.
Cell apoptosis
Parental and GSI-resistant DND41 cells were treated with 125nM GSI for 6 days. Control and EBF1 targeted DND41-Res-Cas9 or Granta519-Cas9 cells were sorted 3 days post transduction and cultured for 3 days (day6). For each replicate, 5 × 10^5 cells were washed in 1 × PBS and resuspended in 100 μl 1 × Annexin V binding buffer (BD cat# 556454). 5 μl V450-Annexin V (BD cat #560506) was added and incubated in the dark at room temperature for 15min. 400 μl of 1 × Annexin V binding buffer containing 1:1000 TO-PRO3 (ThermoFisherScientific cat# R37170) was added and immediately proceeded to Flow cytometry analysis on BD LSR II. Experiments were repeated three times with 3–5 replicates per condition.
Quantitative RT-PCR
Primers were designed using Primer-BLAST software. RNA was extracted with RNeasy Plus Micro Kit (Qiagen cat# 74034) and synthesized to cDNA with SuperScript III (Invitrogen cat# 18080093). Quantitative PCR was performed on an Applied Biosystem ViiA 7 real-time PCR System using Power SYBR Green PCR Master Mix (Applied Biosystems cat# 4367659). Relative expression level was calculated by the 2^−(ΔΔCt) method using EEF1A1 as internal control. Statistics was calculated using a Student’s t test.
Western blot
0.2–1 × 10^6 cells were washed in ice cold 1×PBS and lysed with whole lysis buffer (2% SDS, 60mM Tris) supplemented with proteinase inhibitors. Protein concentration was determined with DC Protein Assay Reagents Package (Bio-rad cat# 5000116) and 5–20 μg proteins were used for SDS-PAGE electrophoresis in tris-mes-sds running buffer (Genescript cat# M00654, cat# M00677). Gels were then transferred to methanol activated PVDF membrane (Cytive cat# 10600023), blocked with 5% skim milk in 1×TBST at room temperature for 1 hour, and incubated with primary antibodies at 1:1,000 to 1:20,000 dilutions in 5% skim milk at 4°C overnight. After washing with 1×TBST 3×10min, secondary antibodies (Millipore cat# 401315-2ml, cat# 401215-2ml) were added at 1:3,000 or 1:6,000 dilution in 5% skim milk and incubated at room temperature for 1 hour. Following 3×15min wash in 1×TBST, imaging was done with ECL Prime Western Blotting Detection Reagents (Cytiva cat# RPN2232) and autoradiography films.
QUANTIFICATION AND STATISTICAL ANALYSIS
The statistical significance of differences between measurements was determined by Wilcoxon rank sum test using R (version 3.6.1) wilcox.test function, unless otherwise stated. Statistical details of experiments can be found in figure legends. Visualizations were done with R.
Definition of regulatory elements
The following definitions of regulatory elements were used throughout the manuscript. Promoters: promoters were defined as ± 2.5 kilobases (Kbp) from the transcription start site (TSS) of each expressed gene. Enhancers: Enhancers were defined as H3K27ac peaks excluding the ones overlapping with promoters.
Gene annotation
A total of 2,828,317 Ensembl transcripts in GRCh37.75 assembly were downloaded in gtf format. For each Ensembl gene id (ENSG), the longest transcript (ENST) was used to assign a unique transcriptional start site and gene position. After exclusion of genes annotated as rRNA or on chromosome M, 57,209 gene annotations were used in RNA-seq analysis.
ATAC-seq data analysis
Alignment
Reads from ATAC-seq experiments were trimmed with Trim Galore (version 0.4.1) with parameters -q 15 --phred33 --gzip --stringency 5 -e 0.1 --length 20. Trimmed reads were aligned to the Ensembl GRCh37.75 primary assembly including chromosome 1–22, chrX, chrY, chrM and contigs using BWA (version 0.7.13) (Li and Durbin, 2009) with parameters bwa aln -q 5 -l 32 -k 2 -t 6. Paired-end reads were group with bwa sampe -P -o 1000000. Reads mapped to contigs, ENCODE blacklist and marked as duplicates by Picard (version 2.1.0) were discarded and the remaining reads were used in downstream analyses and visualization.
Peak calling and Differential accessibility analysis
Peaks in each aligned replicate were identified using MACS2 (version 2.0.9) (Zhang et al., 2008) with parameters --nomodel --nolambda --format=BAM -g hs -p 1E-5 --bw=300 --keep-dup=1. All peaks from replicates of each experiment were combined using bedtools ‘merge’ function and the union of peaks was quantified over each aligned bam file using bedtools ‘coverage’ and normalized to RPKM. Differential accessibility analysis was performed using DEseq2 (Love et al., 2014) with parameters test = “Wald”, betaPrior = F, fitType = “parametric”. For comparison of parental and GSI-resistant DND41, significance cutoff was log2 fold change > 1 or < −1 and FDR < 1E-5. For MB157, MB157pInducer20-LEF1, TCF1 and EBF1, significance cutoff was log2 fold change > 0.5 or < −0.5 and FDR < 0.05. For control and LEF1/TCF1/EBF1 targeted Cas9 expressing cells, significance cutoff was log2 fold change > 1 or < −1 and FDR < 1E-3.
Motif Analysis
Motif enrichment was performed with HOMER (version 4.8) (Heinz et al., 2010) findMotifsGenome.pl with parameters ‘hg19 -mask -size given -len 8,9,10,11,12’. For parental/GSI-resistant DND41, regions significantly gained accessibility were used as foreground and regions significantly lost accessibility were used as background and vice versa. For MB157pInducer20-LEF1, -TCF1 and -EBF1, regions significantly gained accessibility were used as foreground and equal number of randomly sampled regions without significantly differential accessibility were used as background. For LEF1/TCF1/EBF1 targeted Cas9 expressing cells, regions with significant loss of accessibility were used as foreground and regions significantly gained accessibility or did not change accessibility were used as background. Motif logos and associated p-values determined by HOMER were reported.
ChIP-seq and CUT&RUN data analysis
Reads from ChIP-seq and CUT&RUN experiments were aligned with the same procedure as ATAC-seq above. Aligned bam files of the two replicates of each condition were merged using samtools (version 1.3) (Li et al., 2009) ‘cat’ command for peak calling and quantification. For each merged library, fragment length was estimated with HOMER ‘makeTagDirectory’. Peaks were identified using MACS with parameters -p 1E-3 -g hs --nomodel --shiftsize=0.5*fragment_length --format=BAM --bw=300 --keep-dup=1 and with corresponding input control. Log2 fold change of SMC1, CTCF and YY1 load was calculated as log2 RPKM of GSI-resistant vs parental. Significance of change was determined by using ‘enrichR’ and ‘getQvalues’ functions from normr R package (Helmuth and Chung, 2018) using parameters minP = 1, eps = 0.00001, iterations = 10, procs = 12. The criteria for differential SMC1 peaks were log2 fold change > 1 or < −1 and FDR < 1E-5; differential YY1 and CTCF peaks were log2 fold change > 0.5 or < −0.5 and FDR < 1E-5. Super-enhancers were defined by implementing previously described methods(Whyte et al., 2013) in R and applying it to H3K27ac peaks.
For comparing protein loading at accessible regions, the union of ATAC-seq peaks were quantified on merged ChIP-seq bam files using bedtools ‘coverage’ and normalized to RPKM. For tag density plots, HOMER ‘annotatePeaks.pl’ was used on merged libraries and visualized with R function ‘pheatmap’. Bin size of 50bp was used in Figures 4B, 4D, 4E, 5B and 7A. Bin size of 250bp was used in Figure S4B. For merged libraries genome tracks, bedgraph of reads normalized to reads per million (RPM) were generated with bedtools genomecov. Selected genomic loci were visualized with R package Sushi (version 1.18.0) (Phanstiel et al., 2014) function ‘plotBedgraph’. Genome-wide uploadable bigWig files were generated with UCSC tools (version 329) (Kent et al., 2010) ‘bedGraphToBigWig’.
HiC and SMC1 HiChIP analysis
Alignment and significant interaction calling
Raw reads for each HiC sample were processed with HiC-Pro (version v2.5.0) (Servant et al., 2015) to obtain putative interactions with default parameters except LIGATION_SITE and GENOME_FRAGMENT provided by Arima. Raw reads for each SMC1 HiChIP sample were processed with HiC-Pro to obtain putative interactions with default parameters except LIGATION_SITE = GATCGATC and GENOME_FRAGMENT generated for MboI restriction enzyme. Valid pairs (VI), self-circle (SC) and dangling-end (DE) interactions in cis were used as input for significant interaction calling in ‘.bedpe’ format. Mango (version 1.2.0) (Phanstiel et al., 2015) step 4 identified putative significant interaction anchors by MACS peak calling with MACS_qvalue = 0.05 and MACS_shiftsize = 75. Mango step 5 identified significant interactions with default parameters except maxinteractingdist = 2000000 and MHT = found. Only significant interactions with PETs >= 4 were used in the following analyses.
Long-range regulatory loops
Long-range regulatory loops were defined using SMC1 HiChIP significant interactions. For parental and GSI-resistant DND41, significant interactions with SMC1 ChIP-seq and ATAC-seq peaks on both loop anchors and are present in at least two out of four HiChIP samples are considered reproducible. Reproducible loops were classified into enhancer-enhancer (EE), enhancer-promoter (EP) promoter-promoter (PP), CTCF-CTCF and other interactions based on the presence of enhancers, promoters and CTCF peaks at the summit of the two anchors +/− 5 Kbp. To identify differential long-range interactions between parental and GSI-resistant DND41, annotated reproducible loops were quantified on .bedpe files of HiC and HiChIP experiments and normalized to contacts per million, i.e. divided by the number of reproducible interactions timed 1E6. Log2 fold change was calculated as log2 GSI-resistant vs parental of normalized interactions. Significance of fold change was determined by using ‘enrichR’ and ‘getQvalues’ functions from normr R package (Helmuth and Chung, 2018) using parameters minP = 1, eps = 0.00001, iterations = 10, procs = 12. The criteria for differential contact were log2 fold change > 0.5 or < −0.5 and FDR < 0.05. Only contacts significantly increased or decreased in both HiC and HiChIP are considered reproducibly differential. For DMSO-treated and GSI-washout MB157 (Petrovic et al., 2019), MB157pInducer20-TCF1 and MB157pInducer20-EBF1, reproducible long-range contacts were defined as significant contacts that are present in at least two out of the four HiChIP samples. The same annotation and normalization process was applied for these contacts. Fold change and significance were calculated as MB157pInducer20-TCF1 or MB157pInducer20-EBF1 vs GSI-washout MB157. The criteria for differential contact in MB157 were log2 fold change > 1 or < −1 and FDR < 1E-5. The criterion for differential contact in EBF1-KO vs Ctrl DND41-Res-Cas9 was log2 (EBF1-KO / Ctrl) < −0.5. Rows in Figure 7D were sorted in descending order of contact frequencies in Ctrl DND41-Res-Cas9. Pileup plots of differential or invariant long-range contacts were calculated using coolpup.py with --rescale --local on .cool files (described below) and visualized with plotpup.py (Flyamer et al., 2020). 100 bin scaling was used in Figures 2A, 2D. Normalized interactions at selected loci were visualized with R package Sushi (version 1.18.0) (Phanstiel et al., 2014) function ‘plotBedpe’.
Pileup plots of normalized HiC contact maps in Figure 4G depict pairwise contact between accessible elements with loss of TCF1 and SMC1 (left); gain of EBF1 and SMC1 (middle); loss of TCF1 and gain of EBF1 and invariant SMC1 (right) in GSI-resistant cells. Each heatmap showing +/− 100 Kbp flanking the center with 5 Kbp resolution.
TAD boundary identification
For DND41, the valid pairs (allValidPairs files) of both HiC and SMC1 HiChIP from HiC-Pro (Servant et al., 2015) were used to generate .cool files using hicpro2higlass.sh in HiC-Pro. Genome-wide insulation scores and boundary scores were calculated using balanced interaction matrices with 5 Kbp bin size and 100 Kbp window size using cooltools ‘diamond-insulation’ function (https://cooltools.readthedocs.io/en/latest/). Bins with boundary score > 0.3 in at least half of the HiC and HiChIP samples were considered valid boundaries. Boundaries with (parental – GSI-resistant) boundary score >= 0.4 and (parental – GSI-resistant) insulation score <= −0.1 are considered less insulated in GSI-resistant cells, and bins with (parental – GSI-resistant) boundary score <= −0.4 and (parental – GSI-resistant) insulation score > 0.1 are considered more insulated in GSI-resistant cells. Adjacent differential boundaries are merged. Only differential boundaries identified in both HiC and HiChIP were considered reproducibly differential. Pileup plots of differential or invariant boundaries were calculated using coolpup.py (Flyamer et al., 2020) with --pad 500 --local on .cool files and visualized with plotpup.py.
Z-scored contact map
To emphasize the boundaries identified by insulation score, z-score transformation on 25 Kbp-resolution square root vanilla coverage (VC_SQRT) normalized contact map was applied per chromosome per sample as described(Crane et al., 2015) using R function loessFit with parameters iter = 100, span = 0.02. Selected loci on transformed maps were visualized using R package Sushi (version 1.18.0)(Phanstiel et al., 2014) function ‘plotHic’.
Differential Compartment calling
Resolutions from 50 Kbp to 200 Kbp for A/B compartment detection were tested. Given the consistency of results at this resolution range, we used HOMER (Heinz et al., 2010) to identify differential A/B compartments at 100 Kbp resolution. Briefly, HOMER ‘analyzeHiC’ function was used to create distance-normalized pairwise contact correlation matrix, and calculate its first principal component (PC1) for each experiment. H3K27ac ChIP-seq signal was used to guide HOMER and avoid arbitrary PC1 sign assignment. After PC1 calculation, getHiCcorrDiff.pl was used to directly calculate the difference in contact correlation profile of each loci between parental and GSI-resistant conditions as measured by HiC and HiChIP separately. Then findHiCCompartments.pl with stringent criteria of absolute differential correlation less than 0.4 and absolute differential PC1 greater than 0.4 was used to determine if a region shifted its compartment according to both HiC and HiChIP measurements. To ensure reproducibility, only regions that were designated as changed compartment by both HiC and HiChIP were considered for subsequent analysis.
UMI 4C-seq analysis
UMI 4C-seq data was processed with Juicer (version 1.5) (Durand et al., 2016b) with parameters -g hg19 -s MboI to generate .hic files. Unnormalized contact matrices of 5 Kbp resolution were extracted from .hic files using Juicer tools ‘dump’ function. The bin containing the TSS of MYC was determined as “viewpoint”. To visualize interactions surrounding the viewpoint, the row of the viewpoint in the contact matrix with the columns within 10 bins upstream and 410 bins downstream of the viewpoint were extracted and contact frequencies were normalized by the total number of contacts in this range. Mean +/− S.D. normalized contacts in each condition were plotted with R package ggplot2.
RNA-seq data analysis
RNA-seq data was aligned to Ensembl GRCh37.75 primary assembly including chromosome 1–22, chrX, chrY, chrM and contigs using STAR (version 2.5) (Dobin et al., 2013) with parameters -outFilterIntronMotifs RemoveNoncanonicalUnannotated --alignIntronMax 100000 -- outSAMstrandField intronMotif --outSAMunmapped Within --chimSegmentMin 25 --chimJunctionOverhangMin 25. Strand-specific read counts were quantified using Subread (version 1.5.1) featureCounts with parameters -t exon -g gene_id -s 1 -T 6 and used as input to differential gene expression analysis. Read counts were normalized to reads per million per kilobase (RPKM) for each gene. Expressed genes were determined as genes with > 1 RPKM in at least half of the samples in each experiment.
Pairwise differential gene expression analysis was performed using DEseq2 (Love et al., 2014) with parameters test = “Wald”, betaPrior = F, fitType = “parametric”. For DND41 cells, significance cutoff was log2 fold change > 0.5 or < −0.5 with FDR < 0.05. In comparison to DMSO-treated parental cells, genes that did not change significantly in 24hr GSI treatment but significantly decreased or increased in GSI-resistant cells were determined as downregulated or upregulated in GSI-resistant cells, respectively. Downregulated genes with TCF1-bound promoter or connected enhancer in parental cells were considered TCF1 target genes. Upregulated genes with EBF1-bound promoter or connected enhancer in GSI-resistant cells were considered EBF1 target genes. Genes that significantly decreased expression in 24hr GSI versus DMSO parental cells but significantly increased in GSI-resistant cells compared to 24hr GSI were determined as recovered. For differential analyses between MB157 and MB157pInducer20-TCF1 or MB157pInducer20-EBF1 cells, genes that showed log2 fold change > 0.5 or < −0.5 with FDR < 0.01 were considered differentially expressed. Genes upregulated in MB157pInducer20-TCF1 with TCF1-bound promoter or connected enhancer were considered TCF1 targets in MB157. Genes upregulated in MB157pInducer20-EBF1 with EBF1-bound promoter or connected enhancer were considered EBF1 targets in MB157.
Expressed genes in GSI-resistant versus parental DND41 were ranked by log2 fold change and used to conduct gene-set enrichment analysis (GSEA) (Subramanian et al., 2005) with 100,000 permutations against the GO and C7 gene sets from Molecular Signatures Database (MSigDB, version 6.1) (Liberzon et al., 2015), and EBF1 targets defined in MB157pInducer20-EBF1. Expressed genes in MB157pInducer20-EBF1 versus MB157 were ranked by log2 fold change and used to conduct GSEA against EBF1 targets defined in pro-B cells (Nechanitzky et al., 2013) and EBF1 targets defined in DND41 cells. Expressed genes in EBF1-targeted versus control DND41-resistant-Cas9 were ranked by log2 fold change and used to conduct GSEA against EBF1 targets defined in DND41. Genes that recovered in GSI-resistant DND41 (Fig S2H), TCF1 targets in DND41 (Fig S4I), EBF1 targets in DND41 (Fig S4K), EBF1 targets in MB157pInducer20-EBF1 (Fig S5I) and TCF1 targets in MB157pInducer20-TCF1 (Fig S5J) were used for pathway and gene-ontology enrichment analyses (http://www.gsea-msigdb.org/gsea/msigdb/annotate.jsp). Selected pathways and gene-ontology sets with FDR < 1E-3 were visualized in R.
Data in Figure 7F
Figure 7F depicts chromatin restructuring preceding KIT expression. From top to bottom: the PC1 values of HiC contact matrices in parental and GSI-resistant cells showing shift from B (<0) to A (>0) compartment. ChIP-seq and Cut&Run tracks showing gain of active histone marks H3K27ac and H3K4me1 and loss of repressive mark H3K27me3. Gray boxes mark three EBF1-bound elements with gradual increase in accessibility in four-week-GSI-treated and GSI-resistant compared to parental cells, which decreased in EBF1-KO compared to Ctrl DND41-Res-Cas9 cells. HiC arcs showing normalized contacts among accessible elements, highlighting gradual increase in HiC contact frequencies in four-week-GSI-treated and GSI-resistant compared to parental cells (paired t-test p-value = 2.48E-4), which decreased in EBF1-KO compared to Ctrl DND41-Res-Cas9 cells (paired t-test p-value = 0.03). Bottom track indicating KIT gene body location, whose expression was undetectable in parental, four-week-GSI-treated, GSI-resistant, and DND41-Res-Cas9 cells. ATAC-seq, HiC and RNA-seq of EBF1-KO were conducted 6 days post transduction of control or EBF1-targeting sgRNAs.
Single nucleotide variant and Copy number alteration analyses
Single nucleotide variants and copy number alteration analyses were performed using CLIA-approved clinical pipeline used for analysis of cancer patients at the Hospital of the University of Pennsylvania. Very briefly, reads from cancer gene panel sequencing were aligned to GRCh38 using BWA (Li and Durbin, 2009) and subjected to GATK (McKenna et al., 2010) base quality score recalibration and duplicate read removal. Samples achieving an average of 500x mean coverage across the 505 targeted cancer associated genes were used in subsequent analyses. Small variant discovery and read filtration were performed according to GATK best practice recommendations (DePristo et al., 2011; Van der Auwera et al., 2013) and copy number segments were called using CNVKit (Talevich et al., 2016). Single nucleotide variations, insertions, deletions and copy number variations were then annotated using Variant Effect Predictor (McLaren et al., 2016). Resulting variations were further filtered with internally validated quality control metrics evaluating coding alterations greater than 50x coverage at a given loci and variant allele frequency higher than 4%, as well as any copy number alterations. Lastly, the variant call set was evaluated for clinical significance based on standards and guidelines by the Association for Molecular Pathology (Li et al., 2017), and manually assessed by a variant reviewer.
Immunofluorescence and DNA FISH imaging and analysis
MYC and TCRA three-color DNA FISH slides were imaged on a Leica SP8 using a 40x oil immersion objective with pixels of 135 × 135 nm and z spacing of 350 nm. We obtained stacks representing 10μm in total axial thickness. Analysis was carried out on the raw images in a semi-automated manner on a cell-by-cell basis as previously described (Fasolino et al., 2020; Raj et al., 2008). Briefly, the DAPI signal was used for manual nuclei segmentation. The exact numbers of nuclei analyzed per cell type and locus are as follows: 2,515 nuclei for the TCRA in parental DND41, 3,418 nuclei for the TCRA in GSI-resistant DND41, 1,652 nuclei for the MYC locus in DMSO-treated parental DND41, 1,323 nuclei for the MYC locus in short-term GSI-treated parental DND41, 1,694 nuclei the MYC locus in GSI-resistant DND41. Spots for each of the 3 channels (Alexa-488, Atto-565, and Alexa-647) were individually detected using a linear filter approximately conforming to a Laplacian convolved with a Gaussian. A plot of the number of spots as a function of the threshold value enabled human-directed thresholding of spots for each of the 3 channels individually. For each spot, the brightest z slice was used as the z coordinate. Centroid positions for each spot in xy were found by fitting a Gaussian. X, Y, and Z coordinates were extracted, and pairwise Euclidean distances between nearest neighbors were calculated. The Kolmogorov-Smirnov test was used to compare differences in the cumulative distribution functions, and the Wilcoxon rank sum test was used to compare differences in the medians. Representative images were carried out on a Leica SP8 using a 63x oil immersion objective and individually adjusted brightness, contrast, minimum, maximum and smoothing in Fiji (https://imagej.net/Fiji).
IKZF2 (Alexa-488) and EBF1 (Alexa-647) DNA FISH combined with Lamin-B1 IF (Alexa-568) were imaged on a Leica SP8 using a 63x oil immersion objective with pixels of 85 × 85 nm and z spacing of 350 nm in a total of 10 μm. Analysis was performed with Imaris (https://imaris.oxinst.com/). Briefly, the DAPI signal was used for automatic nuclei segmentation and relatively round shaped nuclei were selected. For each nucleus, Lamin-B1 signal was automatically detected with mild pruning and masked from the inner nuclear space. DNA FISH signal was automatically detected and the shortest Euclidean distances from the centroid of DNA FISH spot to the masked Lamin-B1 signal were calculated. Negative values were corrected as zero as they represented that DNA FISH targeted sequences were buried within nuclear lamina. The Kolmogorov-Smirnov test (IKZF2) or Wilcoxon rank sum test (EBF1) was used to compare differences in the cumulative distribution functions or medians, respectively. The exact numbers of alleles or nuclei analyzed per cell type and locus are as follows: 126 and 157 alleles for the IKZF2 locus in parental and GSI-resistant DND41; 98, 98, 97 and 99 nuclei for EBF1 in parental, two-week-, five-week- and nine-week-GSI-treated DND41; 95 and 99 nuclei in Ctrl and EBF1-KO DND41-Res-Cas9 cells.
Supplementary Material
Table S1: Ultra-deep genomic sequencing of parental and GSI-resistant DND41 cells. Related to Figure 1.
Table S2: Differential compartments in GSI-resistant and parental cells. Related to Figure 1.
Table S3: Differential TAD boundaries in GSI-resistant and parental cells. Related to Figure 1.
Table S4: Annotation of loci connected by statistically significant long-range interactions in DND41 and MB157. Related to Figures 2 and 5.
Table S5: Differential chromatin accessibility in T-ALL and TNBC cell lines. Related to Figures 4, 5, and 6.
Table S6: Differential gene expression in T-ALL and TNBC cell lines. Related to Figures 2, 4, 5, 6, and 7.
Table S7: Pathways and ontologies associate with GSI-resistance, as well as TCF1- and EBF1-dependent genes in DND41 and MB157 cells. Related to Figures 2, 4, and 5.
Genome refolding drives epigenetic adaptation and confers drug resistance in T-ALL
Drug resistant T-ALL switches from TCF1-driven to EBF1-driven epigenetic program
Lineage-determining transcription factors EBF1 and TCF1 can instruct genome folding
EBF1 dynamically repositions within individual nuclei during resistance acquisition
Acknowledgments
The authors thank Drs Vahedi, Shi, Greenberg, Jain, Grosschedl, Meijerink, Henikoff, as well as Penn Microscopy Core for helpful discussion and technical support. This work was supported by Shanghai Rising-Star 19QA1407800 (W.Z.), R01CA215518 (W.S.P.); Susan G Komen CCR185472448 and R01CA230800 (R.B.F.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
REFERENCES
- Bailey TL, Johnson J, Grant CE, and Noble WS (2015). The MEME Suite. Nucleic Acids Res 43, W39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A, Northrup D, Boukarabila H, Jacobsen SE, and Allman D (2013). Transcriptional repression of Gata3 is essential for early B cell commitment. Immunity 38, 930–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beliveau BJ, Kishi JY, Nir G, Sasaki HM, Saka SK, Nguyen SC, Wu CT, and Yin P (2018). OligoMiner provides a rapid, flexible environment for the design of genome-scale oligonucleotide in situ hybridization probes. Proc Natl Acad Sci U S A 115, E2183–E2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boller S, Ramamoorthy S, Akbas D, Nechanitzky R, Burger L, Murr R, Schubeler D, and Grosschedl R (2016). Pioneering Activity of the C-Terminal Domain of EBF1 Shapes the Chromatin Landscape for B Cell Programming. Immunity 44, 527–541. [DOI] [PubMed] [Google Scholar]
- Bortnick A, He Z, Aubrey M, Chandra V, Denholtz M, Chen K, Lin YC, and Murre C (2020). Plasma Cell Fate Is Orchestrated by Elaborate Changes in Genome Compartmentalization and Inter-chromosomal Hubs. Cell Rep 31, 107470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossen C, Murre CS, Chang AN, Mansson R, Rodewald HR, and Murre C (2015). The chromatin remodeler Brg1 activates enhancer repertoires to establish B cell identity and modulate cell growth. Nat Immunol 16, 775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadigan KM, and Waterman ML (2012). TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb Perspect Biol 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SW, Xu J, Sun R, Mumbach MR, Carter AC, Chen YG, Yost KE, Kim J, He J, Nevins SA, et al. (2018). Promoter of lncRNA Gene PVT1 Is a Tumor-Suppressor DNA Boundary Element. Cell 173, 1398–1412 e1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, and Corces VG (2016). The three-dimensional cancer genome. Curr Opin Genet Dev 36, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane E, Bian Q, McCord RP, Lajoie BR, Wheeler BS, Ralston EJ, Uzawa S, Dekker J, and Meyer BJ (2015). Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature 523, 240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Coninck S, Berx G, Taghon T, Van Vlierberghe P, and Goossens S (2019). ZEB2 in T-cells and T-ALL. Adv Biol Regul 74, 100639. [DOI] [PubMed] [Google Scholar]
- Debruyne DN, Dries R, Sengupta S, Seruggia D, Gao Y, Sharma B, Huang H, Moreau L, McLane M, Day DS, et al. (2019). BORIS promotes chromatin regulatory interactions in treatment-resistant cancer cells. Nature 572, 676–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J, and Misteli T (2015). Long-Range Chromatin Interactions. Cold Spring Harb Perspect Biol 7, a019356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilley RL, Verma P, Cho NW, Winters HD, Wondisford AR, and Greenberg RA (2016). Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 539, 54–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, and Ren B (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, and Aiden EL (2016a). Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst 3, 99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Shamim MS, Machol I, Rao SS, Huntley MH, Lander ES, and Aiden EL (2016b). Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst 3, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S, Moreau Y, Kasprzyk A, Davis S, De Moor B, Brazma A, and Huber W (2005). BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 21, 3439–3440. [DOI] [PubMed] [Google Scholar]
- Falk M, Feodorova Y, Naumova N, Imakaev M, Lajoie BR, Leonhardt H, Joffe B, Dekker J, Fudenberg G, Solovei I, et al. (2019). Heterochromatin drives compartmentalization of inverted and conventional nuclei. Nature 570, 395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasolino M, Goldman N, Wang W, Cattau B, Zhou Y, Petrovic J, Link VM, Cote A, Chandra A, Silverman M, et al. (2020). Genetic Variation in Type 1 Diabetes Reconfigures the 3D Chromatin Organization of T Cells and Alters Gene Expression. Immunity 52, 257–274 e211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon AE, Carter EP, Clayton NS, Wilkes EH, Baker AM, Kapitonova E, Bakhouche BA, Tanner Y, Wang J, Gadaleta E, et al. (2018). PHLDA1 Mediates Drug Resistance in Receptor Tyrosine Kinase-Driven Cancer. Cell Rep 22, 2469–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flotho C, Coustan-Smith E, Pei D, Cheng C, Song G, Pui CH, Downing JR, and Campana D (2007). A set of genes that regulate cell proliferation predicts treatment outcome in childhood acute lymphoblastic leukemia. Blood 110, 1271–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flyamer IM, Illingworth RS, and Bickmore WA (2020). Coolpup.py: versatile pile-up analysis of Hi-C data. Bioinformatics 36, 2980–2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grevet JD, Lan X, Hamagami N, Edwards CR, Sankaranarayanan L, Ji X, Bhardwaj SK, Face CJ, Posocco DF, Abdulmalik O, et al. (2018). Domain-focused CRISPR screen identifies HRI as a fetal hemoglobin regulator in human erythroid cells. Science 361, 285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmuth J, and Chung H (2018). normr: Normalization and difference calling in ChIP-seq data.
- Herranz D, Ambesi-Impiombato A, Palomero T, Schnell SA, Belver L, Wendorff AA, Xu L, Castillo-Martin M, Llobet-Navas D, Cordon-Cardo C, et al. (2014). A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat Med 20, 1130–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Day DS, and Young RA (2016). Insulated Neighborhoods: Structural and Functional Units of Mammalian Gene Control. Cell 167, 1188–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homminga I, Pieters R, Langerak AW, de Rooi JJ, Stubbs A, Verstegen M, Vuerhard M, Buijs-Gladdines J, Kooi C, Klous P, et al. (2011). Integrated transcript and genome analyses reveal NKX2-1 and MEF2C as potential oncogenes in T cell acute lymphoblastic leukemia. Cancer Cell 19, 484–497. [DOI] [PubMed] [Google Scholar]
- Isoda T, Moore AJ, He Z, Chandra V, Aida M, Denholtz M, Piet van Hamburg J, Fisch KM, Chang AN, Fahl SP, et al. (2017). Non-coding Transcription Instructs Chromatin Folding and Compartmentalization to Dictate Enhancer-Promoter Communication and T Cell Fate. Cell 171, 103–119 e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JL, Georgakilas G, Petrovic J, Kurachi M, Cai S, Harly C, Pear WS, Bhandoola A, Wherry EJ, and Vahedi G (2018). Lineage-Determining Transcription Factor TCF-1 Initiates the Epigenetic Identity of T Cells. Immunity 48, 243–257 e210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Zweig AS, Barber G, Hinrichs AS, and Karolchik D (2010). BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics 26, 2204–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloetgen A, Thandapani P, Ntziachristos P, Ghebrechristos Y, Nomikou S, Lazaris C, Chen X, Hu H, Bakogianni S, Wang J, et al. (2020). Three-dimensional chromatin landscapes in T cell acute lymphoblastic leukemia. Nat Genet 52, 388–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, Tsimberidou AM, Vnencak-Jones CL, Wolff DJ, Younes A, et al. (2017). Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn 19, 4–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Benner C, Mansson R, Heinz S, Miyazaki K, Miyazaki M, Chandra V, Bossen C, Glass CK, and Murre C (2012). Global changes in the nuclear positioning of genes and intra- and interdomain genomic interactions that orchestrate B cell fate. Nat Immunol 13, 1196–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Jhunjhunwala S, Benner C, Heinz S, Welinder E, Mansson R, Sigvardsson M, Hagman J, Espinoza CA, Dutkowski J, et al. (2010). A global network of transcription factors, involving E2A, EBF1 and Foxo1, that orchestrates B cell fate. Nat Immunol 11, 635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litzow MR, and Ferrando AA (2015). How I treat T-cell acute lymphoblastic leukemia in adults. Blood 126, 833–841. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marine JC, Dawson SJ, and Dawson MA (2020). Non-genetic mechanisms of therapeutic resistance in cancer. Nat Rev Cancer 20, 743–756. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, and Cunningham F (2016). The Ensembl Variant Effect Predictor. Genome Biol 17, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechanitzky R, Akbas D, Scherer S, Gyory I, Hoyler T, Ramamoorthy S, Diefenbach A, and Grosschedl R (2013). Transcription factor EBF1 is essential for the maintenance of B cell identity and prevention of alternative fates in committed cells. Nat Immunol 14, 867–875. [DOI] [PubMed] [Google Scholar]
- O’Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, Hardwick J, Welcker M, Meijerink JP, Pieters R, et al. (2007). FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med 204, 1813–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovic J, Zhou Y, Fasolino M, Goldman N, Schwartz GW, Mumbach MR, Nguyen SC, Rome KS, Sela Y, Zapataro Z, et al. (2019). Oncogenic Notch Promotes Long-Range Regulatory Interactions within Hyperconnected 3D Cliques. Mol Cell 73, 1174–1190 e1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phanstiel DH, Boyle AP, Araya CL, and Snyder MP (2014). Sushi.R: flexible, quantitative and integrative genomic visualizations for publication-quality multi-panel figures. Bioinformatics 30, 2808–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phanstiel DH, Boyle AP, Heidari N, and Snyder MP (2015). Mango: a bias-correcting ChIA-PET analysis pipeline. Bioinformatics 31, 3092–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, and Tyagi S (2008). Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods 5, 877–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenberg EV (2010). B cell specification from the genome up. Nat Immunol 11, 572–574. [DOI] [PubMed] [Google Scholar]
- Rowley MJ, and Corces VG (2018). Organizational principles of 3D genome architecture. Nat Rev Genet 19, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan RJH, Petrovic J, Rausch DM, Zhou Y, Lareau CA, Kluk MJ, Christie AL, Lee WY, Tarjan DR, Guo B, et al. (2017). A B Cell Regulome Links Notch to Downstream Oncogenic Pathways in Small B Cell Lymphomas. Cell Rep 21, 784–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandelin A, Alkema W, Engstrom P, Wasserman WW, and Lenhard B (2004). JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res 32, D91–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuettpelz LG, Gopalan PK, Giuste FO, Romine MP, van Os R, and Link DC (2012). Kruppel-like factor 7 overexpression suppresses hematopoietic stem and progenitor cell function. Blood 120, 2981–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant N, Varoquaux N, Lajoie BR, Viara E, Chen CJ, Vert JP, Heard E, Dekker J, and Barillot E (2015). HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol 16, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, and Henikoff S (2017). An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadhouders R, Filion GJ, and Graf T (2019). Transcription factors and 3D genome conformation in cell-fate decisions. Nature 569, 345–354. [DOI] [PubMed] [Google Scholar]
- Stadhouders R, Vidal E, Serra F, Di Stefano B, Le Dily F, Quilez J, Gomez A, Collombet S, Berenguer C, Cuartero Y, et al. (2018). Transcription factors orchestrate dynamic interplay between genome topology and gene regulation during cell reprogramming. Nat Genet 50, 238–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taghon TN, David ES, Zuniga-Pflucker JC, and Rothenberg EV (2005). Delayed, asynchronous, and reversible T-lineage specification induced by Notch/Delta signaling. Genes Dev 19, 965–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talevich E, Shain AH, Botton T, and Bastian BC (2016). CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput Biol 12, e1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treiber T, Mandel EM, Pott S, Gyory I, Firner S, Liu ET, and Grosschedl R (2010). Early B cell factor 1 regulates B cell gene networks by activation, repression, and transcription-independent poising of chromatin. Immunity 32, 714–725. [DOI] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. (2013). From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 43, 11 10 11–11 10 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Zang C, Taing L, Arnett KL, Wong YJ, Pear WS, Blacklow SC, Liu XS, and Aster JC (2014). NOTCH1-RBPJ complexes drive target gene expression through dynamic interactions with superenhancers. Proc Natl Acad Sci U S A 111, 705–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub AS, Li CH, Zamudio AV, Sigova AA, Hannett NM, Day DS, Abraham BJ, Cohen MA, Nabet B, Buckley DL, et al. (2017). YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell 171, 1573–1588 e1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng AP, Ferrando AA, Lee W, Morris J.P.t., Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, and Aster JC (2004). Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306, 269–271. [DOI] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H (2009). Ggplot2 : elegant graphics for data analysis (New York: Springer; ). [Google Scholar]
- Yashiro-Ohtani Y, Wang H, Zang C, Arnett KL, Bailis W, Ho Y, Knoechel B, Lanauze C, Louis L, Forsyth KS, et al. (2014). Long-range enhancer activity determines Myc sensitivity to Notch inhibitors in T cell leukemia. Proc Natl Acad Sci U S A 111, E4946–4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, and Ren B (2017). The Three-Dimensional Organization of Mammalian Genomes. Annu Rev Cell Dev Biol 33, 265–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yui MA, and Rothenberg EV (2014). Developmental gene networks: a triathlon on the course to T cell identity. Nat Rev Immunol 14, 529–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, et al. (2012). The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481, 157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Ultra-deep genomic sequencing of parental and GSI-resistant DND41 cells. Related to Figure 1.
Table S2: Differential compartments in GSI-resistant and parental cells. Related to Figure 1.
Table S3: Differential TAD boundaries in GSI-resistant and parental cells. Related to Figure 1.
Table S4: Annotation of loci connected by statistically significant long-range interactions in DND41 and MB157. Related to Figures 2 and 5.
Table S5: Differential chromatin accessibility in T-ALL and TNBC cell lines. Related to Figures 4, 5, and 6.
Table S6: Differential gene expression in T-ALL and TNBC cell lines. Related to Figures 2, 4, 5, 6, and 7.
Table S7: Pathways and ontologies associate with GSI-resistance, as well as TCF1- and EBF1-dependent genes in DND41 and MB157 cells. Related to Figures 2, 4, and 5.
Data Availability Statement
Next-generation sequencing data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Original western blot images have been deposited at Mendeley and are publicly available as of the date of publication. Microscopy data reported in this paper will be shared by the lead contact upon request.
This paper does not report any original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-MYC [Y69] | Abcam | Cat# ab32072, RRID:AB_731658 |
| Anti-EBF-1 Antibody | Millipore | Cat# AB10523, RRID:AB_2636856 |
| Anti-Early B-cell factor (EBF1) Antibody | Sigma-Aldrich | Cat# ABE1294, RRID:AB_2893472 |
| Rabbit anti-EBF1 | (Boller et al., 2016) | N/A |
| TCF1 (C46C7) Rabbit mAb antibody | CST | Cat# 2206, RRID:AB_2199300 |
| LEF1 (D6J2W) Rabbit mAb | CST | Cat# 76010, RRID:AB_2799877 |
| Maml1 (D3K7B) Rabbit mAb | CST | Cat# 12166, RRID:AB_2797838 |
| Cleaved Notch1 (Val1744) (D3B8) Rabbit mAb | CST | Cat# 4147, RRID:AB_2153348 |
| Caspase-3 Antibody | CST | Cat# 9662, RRID:AB_331439 |
| Annexin V Recom Horizon V450 50 test antibody | BD | Cat# 560506, RRID:AB_2869356 |
| Monoclonal Anti-β-Actin antibody, clone AC-74 | Sigma-Aldrich | Cat# A5316, RRID:AB_476743 |
| Calnexin (C5C9) Rabbit mAb antibody | CST | Cat# 2679, RRID:AB_2228381 |
| GAPDH (D16H11) XP Rabbit mAb antibody | CST | Cat# 5174, RRID:AB_10622025 |
| Rabbit polyclonal anti-H3 acetyl-K27 | Active Motif | Cat# 39133, RRID:AB_2561016 |
| Tri-Methyl-Histone H3 (Lys27) (C36B11) Rabbit mAb | CST | Cat# 9733, RRID:AB_2616029 |
| Rabbit polyclonal anti-H3 mono methyl-K4 | Abcam | Cat# ab8895, RRID:AB_306847 |
| Rabbit anti-SMC1 Antibody, Affinity Purified | Bethyl | Cat# A300–055A, RRID:AB_2192467 |
| Rabbit polyclonal anti-CTCF | Millipore | Cat# 07–729, RRID:AB_441965 |
| YY1 antibody (pAb) | Active Motif | Cat# 61779, RRID:AB_2793763 |
| Cas9 (7A9-3A3) antibody | CST | Cat# 14697, RRID:AB_2750916 |
| Rabbit Anti-Lamin B1 Polyclonal Antibody | Abcam | Cat# ab16048, RRID:AB_443298 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 | Thermo | Cat# A10042, RRID:AB_2534017 |
| Goat Anti-Rabbit IgG, H & L Chain Specific Peroxidase Conjugate antibody | Millipore | Cat# 401315, RRID:AB_437787 |
| Goat Anti-Mouse IgG, H & L Chain Antibody, Peroxidase Conjugated | Millipore | Cat# 401215, RRID:AB_10682749 |
| Bacterial and virus strains | ||
| One Shot Stbl3 Chemically Competent E.coli | Thermo | Cat# C737303 |
| Chemicals, peptides, and recombinant proteins | ||
| Trypsin (2.5%) | Gibco | Cat# 15090-046 |
| gamma-Secretase Inhibitor XXI (compound E) | Calbiochem | Cat# 565790 |
| FuGene HD | Promega | Cat# E2311 |
| Penicillin-Streptomycin | Corning | Cat# 30-002-CI |
| MEM Non-Essential Amino Acids | Gibco | Cat# 11140-050 |
| Sodium Pyruvate | Gibco | Cat# 11360-070 |
| 2-mercaptoethanol | Sigma | Cat# M3148 |
| RPMI 1640 | Corning | Cat# 10-040-CM |
| DMEM | Corning | Cat# 10-013-CV |
| Dulbecco’s Phosphate-Buffered Salt Solution 1X | Corning | Cat# 21031CV |
| HyClone Fetal bovine serum | Thermo Fisher Scientific | Cat# SH30070.03 |
| L-glutamine | Corning | Cat# 25-005-CI |
| Polybrene | Sigma-Aldrich | Cat# H9268 |
| Gibco Puromycin Dihydrochloride (10 mg/mL) | Thermo | Cat# A1113803 |
| Complete Protease Inhibitor Cocktail Tablets | Roche | Cat# 11697498001 |
| Complete Mini EDTA-free Protease Inhibitor Cocktail | Roche | Cat# 11836170001 |
| Recombinant Protein G Agarose | Invitrogen | Cat# 15920-010 |
| Lithium Chloride, 8M | Sigma-Aldrich | Cat# 7447-41-8 |
| Sodium bicarbonate | Sigma-Aldrich | Cat#144-55-8 |
| RNase A | Roche | Cat# 10109169001 |
| Proteinase K | Invitrogen | Cat# 25530-049 |
| Pierce™ 16% Formaldehyde | Thermo | Cat# 28908 |
| EGS | Thermo | Cat# 21565 |
| Glycine | Invitrogen | Cat# 15527-013 |
| MboI | NEB | Cat# R0147 |
| SalI-HF | NEB | Cat# R3138S |
| AscI | NEB | Cat# R0558S |
| NEBNext End Repair Module | NEB | Cat# E6050 |
| Klenow Fragment (3’ to 5’ exo-) | NEB | Cat# M0212 |
| CIP | NEB | Cat# M0290 |
| TO-PRO3 Ready Flow Reagent | Thermo | Cat# R37170 |
| Hygromycin B Gold 1 g (10 × 1 ml) | Invivogen | Cat# ant-hg-1 |
| Biotin-14-dATP 200 μl (1 mM) | Jena Bioscience | Cat# NU-835-BIO14-S |
| Maxima H Minus Reverse Transcriptase (200 U/μL) | Thermo | Cat# EP0752 |
| T4 DNA Ligase | NEB | Cat# M0202L |
| DNA Polymerase I, Large (Klenow) Fragment | NEB | Cat# M0210S |
| Triton X-100 | Roche | Cat# 10789704001 |
| BSA, 50mg/ml | Invitrogen | Cat# AM2616 |
| SurePAGE, Bis-Tris, 10×8, 4–12%, 15 wells | GenScript | Cat# M00654 |
| MES SDS Running Buffer Powder | GenScript | Cat# M00677 |
| PageRuler Prestained Protein Ladder, 10 to 180 kDa | Thermo | Cat# 26616 |
| PageRuler Plus Prestained Protein Ladder, 10 to 250 kDa | Thermo | Cat# PI26619 |
| Tween 20 | BIO-RAD | Cat# 170-6531 |
| Protein A Magnetic Beads | Pierce | Cat# 88846 |
| Dynabeads MyOne Streptavidin C1 | Invitrogen | Cat# 65001 |
| Dynabeads Protein G for Immunoprecipitation | Invitrogen | Cat# 10003D |
| Phenol-chloroform-isoamyl (PCI) | Invitrogen | Cat# 15593049 |
| Dimethylformamide | Sigma-Aldrich | Cat# D4551 |
| Power SYBR™ Green PCR Master Mix | Applied Biosystems | Cat# 4367659 |
| Tris-HCI, pH 7.5, 1M | Invitrogen | Cat# 15567-027 |
| Tris-HCI, pH 8.0, 1M | Invitrogen | Cat# 15568-025 |
| EDTA, pH 8.0, 0.5M | Invitrogen | Cat# 15575-020 |
| Sodium Chloride | Fisher BioReagents | Cat# 7647-14-5 |
| MgCl2, 1M | Thermo | Cat# AM9530G |
| NP-40 (Igepal CA-630) | Sigma | Cat# I8896 |
| Sodium deoxycholate | Sigma-Aldrich | Cat# D6750 |
| SDS solution, 10% | Invitrogen | Cat# 15553-027 |
| Geneticin Selective Antibiotic (G418) (50 mg/mL) | Thermo | Cat# 10131035 |
| Doxycycline | Sigma | Cat# D9891 |
| Gibco Blasticidin S HCl (10 mg/mL) | Thermo | Cat# A1113903 |
| Phusion PCR Master Mix | NEB | Cat# M0531 |
| Phusion Flash High-Fidelity PCR Master Mix | Thermo | Cat# F548S |
| NEBNext High-Fidelity 2X PCR Master Mix | NEB | Cat# M0541 |
| AMPure XP | Beckman Coulter | Cat# A63881 |
| SuperScript III Reverse Transcriptase | Invitrogen | Cat# 18080093 |
| Gateway™ LR Clonase™ II Enzyme mix | Invitrogen | Cat# 11791020 |
| Gateway™ BP Clonase™ II Enzyme mix | Invitrogen | Cat# 11789020 |
| poly-L-lysine-treated glass slides | Thermo | Cat# P4981-001 |
| Polyvinylsulfonic acid (PVSA) | Sigma Aldrich | Cat# 278424 |
| 20x SSC Buffer | Corning | Cat# 46-020-CM |
| Slowfade | Invitrogen | Cat# 336936 |
| Critical commercial assays | ||
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat# G7571 |
| DC Protein Assay Reagents Package | Bio-rad | Cat# 5000116 |
| QIAquick PCR Purification Kit | QIAGEN | Cat# 28106 |
| MinElute Reaction Cleanup Kit | QIAGEN | Cat# 28204 |
| DNeasy Blood & Tissue Kit | QIAGEN | Cat# 69504 |
| RNeasy Plus Mini Kit | QIAGEN | Cat# 74136 |
| RNeasy Plus Micro Kit | QIAGEN | Cat# 74034 |
| SeqCap Adapter Kit A | Roche | Cat# 7141530001 |
| KAPA Library Quant Kit | Roche | Cat# KK4824 |
| ZymoPURE Plasmid Miniprep Kit 100 preps | Zymo | Cat# D4210 |
| Zymoclean Gel DNA Recovery Kit | Zymo | Cat# D4007 |
| NEBuilder HiFi DNA Assembly Cloning Kit | NEB | Cat# E5520S |
| NEBNext Ultra II DNA Library Prep Kit | NEB | Cat# E7645S |
| HiScribe T7 High Yield RNA Synthesis Kit | NEB | Cat# E2040S |
| SMARTer Stranded Total RNA Sample Prep Kit - HI Mammalian | Takara | Cat# 634873 |
| NucleoBond Xtra Maxi EF | Takara | Cat# 740424 |
| ECL Prime Western Blotting Detection Reagents | Cytiva | Cat# RPN2232 |
| D1000 ScreenTape | Agilent | Cat# 5067-5582 |
| D1000 Reagents | Agilent | Cat# 5067-5583 |
| High Sensitivity D1000 ScreenTape | Agilent | Cat# 5067-5584 |
| High Sensitivity D1000 Reagents | Agilent | Cat# 5067-5585 |
| Genomic DNA ScreenTape | Agilent | Cat# 5067-5365 |
| Genomic DNA Reagents | Agilent | Cat# 5067-5366 |
| RNA ScreenTape | Agilent | Cat# 5067-5576 |
| RNA ScreenTape Ladder | Agilent | Cat# 5067-5578 |
| RNA ScreenTape Sample Buffer | Agilent | Cat# 5067-5577 |
| Qubit dsDNA HS Assay Kit | Invitrogen | Cat# Q32851 |
| Qubit ssDNA Assay Kit | Invitrogen | Cat# Q10212 |
| Arima-HiC Kit (8 Reactions) | Arima | Cat# A510008 |
| Accel-NGS 2S Plus DNA Library Kit (24 rxns) | Swift | Cat# 21024 |
| 2S SET A INDEXING KIT (12 INDICES, 48 RXNS) | Swift | Cat# 26148 |
| NEBNext Multiplex Oligos for Illumina (Dual Index Primers Set 1) | NEB | Cat# E7600S |
| NEBNext Multiplex Oligos for Illumina (Dual Index Primers Set 2) | NEB | Cat# E7780S |
| Nextera XT Index Kit v2 Set A (96 indexes, 384 samples) | Illumina | Cat# FC-131-2001 |
| Illumina Tagment DNA Enzyme and Buffer Kit | Illumina | Cat# 20034197 |
| Nextseq 500/550 high output kit v2.5 (75 cycles) | Illumina | Cat# 20024906 |
| NovaSeq 6000 S1 Reagent Kit v1.5 (100 cycles) | Illumina | Cat# 20028319 |
| Deposited data | ||
| Raw and analyzed data | This study | GSE173872 |
| MB157 HiChIP | (Petrovic et al., 2019) | GSE116869 |
| Experimental models: Cell lines | ||
| HPB-ALL | DSMZ | Cat# ACC 483 |
| KOPTK1 | (Wang et al., 2014) | N/A |
| Granta-519 | (Ryan et al., 2017) | N/A |
| DND41 | DSMZ | Cat# ACC 525 |
| MB157 | ATCC | Cat# CRL-7721 |
| HEK293T | ATCC | Cat# CRL-3216 |
| Oligonucleotides | ||
| Primer: EEF1A1 qPCR forward: TTGTCGTCATTGGACACGTAG | (Petrovic et al., 2019) | N/A |
| Primer: EEF1A1 qPCR reverse: TGCCACCGCATTTATAGATCAG | (Petrovic et al., 2019) | N/A |
| Primer: MYC qPCR forward: ACCCTCTCAACGACAGCAGC | (Petrovic et al., 2019) | N/A |
| Primer: MYC qPCR reverse: ACTCCGTCGAGGAGAGCAGA | (Petrovic et al., 2019) | N/A |
| Primer: CD300A qPCR forward: GCGGCCAAGACCTCAACAA | This study | N/A |
| Primer: CD300A qPCR reverse: TGAGTTCACCACCTCCTCAGT | This study | N/A |
| Primer: DTX1 qPCR forward: ATGGAGCGACTGGTCACAGC | This study | N/A |
| Primer: DTX1 qPCR reverse: CCGTAGATGGCCTTGCAGGT | This study | N/A |
| Primer: CR2 qPCR forward: CACACACCGGGAGTTCCTTA | This study | N/A |
| Primer: CR2 qPCR reverse: ACAACGGATGGTGCTCTCTC | This study | N/A |
| Primer: LEF1 qPCR forward: TAATGCACGTGAAGCCTCAGCA | This study | N/A |
| Primer: LEF1 qPCR reverse: CACTCAGCAACGACATTCGCTC | This study | N/A |
| Primer: TCF7 qPCR forward: CCAACCATCAAGAAGCCCCT | This study | N/A |
| Primer: TCF7 qPCR reverse: TGTGCACTCTGCAATGACCT | This study | N/A |
| Primer: CD79A qPCR forward: GCACCAAGAACCGAATCATC | This study | N/A |
| Primer: CD79A qPCR reverse: CTTCTCGTTCTGCCATCGTT | This study | N/A |
| Primer: pDONR221-LEF1 PCR forward: GGGGACAAGTTTGTACAAAAAAGCAGGCTTCATGCCCCAACTCTCCGGAGGA | This study | N/A |
| Primer: pDONR221-LEF1 PCR reverse: GGGGACAAGTTTGTACAAAAAAGCAGGCTTCATGCCCCAACTCTCCGGAGGA | This study | N/A |
| Primer: pDONR221-TCF7 PCR forward: GGGGACAAGTTTGTACAAAAAAGCAGGCTTCAGATCGGCCACCATGTATCC | This study | N/A |
| Primer: pDONR221-TCF7 PCR reverse: GGGGACCACTTTGTACAAGAAAGCTGGGTCGttatctagagcactgtcatcgg | This study | N/A |
| Primer: TRE3G-EBF1-puro PCR forward: TACCCTCGTAAAACCGGTCGATGTTTGGGATTCAGGAAAG | This study | N/A |
| Primer: TRE3G-EBF1-puro PCR reverse: GTACAAGAAAGCTGGGTCGGTCACATAGGAGGAACAATC | This study | N/A |
| sgRNA: MYC targeting sequence E3g5: GCGTGACCAGATCCCGGAGT | This study | N/A |
| sgRNA: LEF1 targeting sequence g3: CCCTCCTACTCGAGTTATTC | This study | N/A |
| sgRNA: TCF7 targeting sequence g5: TGTTCGTAGAGAGAGAGTTG | This study | N/A |
| sgRNA: EBF1 targeting sequence g7: TCTCATGTGTGAGCAAGACT | This study | N/A |
| Secondary Oligopaint probe: /5Alex488N/CACACGCTCTTCCGTTCTATGCGACGTCGGTG/3AlexF488N/ | (Beliveau et al., 2018) | N/A |
| Secondary Oligopaint probe: /5ATTO565N/ACACCCTTGCACGTCGTGGACCTCCTGCGCTA/3ATTO565N/ | (Beliveau et al., 2018) | N/A |
| Secondary Oligopaint probe: /5Alex647N/TGATCGACCACGGCCAAGACGGAGAGCGTGTG/3AlexF647N/ | (Beliveau et al., 2018) | N/A |
| Recombinant DNA | ||
| LentiV_Cas9_puro | (Grevet et al., 2018) | Addgene Cat# 108100 |
| LRG2.1 | (Grevet et al., 2018) | Addgene Cat# 108098 |
| LRCherry2.1 | (Grevet et al., 2018) | Addgene Cat# 108099 |
| Gateway pDONR221 | Invitrogen | Cat# 12536017 |
| pInducer20 | Addgene | Cat# 44012 |
| pINDUCER21 (ORF-EG) | Addgene | Cat# 46948 |
| pENTR221-TCF7 | Addgene | Cat# 79498 |
| pDONR221-EBF1 | DNASU | Cat# HsCD00296820 |
| pDONR221-LEF1 | This study | N/A |
| pDONR221-TCF7 | This study | N/A |
| pInducer20-LEF1 | This study | N/A |
| pInducer20-TCF7 | This study | N/A |
| pInducer20-EBF1 | This study | N/A |
| pInducer21-EBF1 | This study | N/A |
| TRE3G-TRF1-puro | (Dilley et al., 2016) | N/A |
| CMV-rtTA3G-blast | (Dilley et al., 2016) | N/A |
| TRE3G-EBF1-puro | This study | N/A |
| Software and algorithms | ||
| Benchling | N/A | https://www.benchling.com |
| BEDTools v2.25.0 | (Quinlan and Hall, 2010) |
https://bedtools.readthedocs.io/en/stable; RRID:SCR_006646 |
| biomaRt v2.28.0 | (Durinck et al., 2005) | https://www.rdocumentation.org/packages/biomaRt/versions/2.28.0; RRID:SCR_006442 |
| BWA v0.7.13 | (Li and Durbin, 2009) | http://bio-bwa.sourceforge.net; RRID:SCR_010910 |
| DESeq2 v1.10.1 | (Love et al., 2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html; RRID:SCR_015687 |
| FlowJo v9.7.7 | FlowJo LLC | https://www.flowjo.com; RRID:SCR_008520 |
| ggplot2 v2.2.1 | (Wickham, 2009) | https://ggplot2.tidyverse.org; RRID:SCR_014601 |
| HiC-Pro v2.5.0 | (Servant et al., 2015) | https://github.com/nservant/HiC-Pro |
| HOMER v4.8 | (Heinz et al., 2010) | http://homer.ucsd.edu/homer; RRID:SCR_010881 |
| JASPAR | (Sandelin et al., 2004) | http://jaspar.genereg.net; RRID:SCR_003030 |
| Juicer tools v1.7.6 | (Durand et al., 2016b) | https://github.com/aidenlab/juicer |
| Juicebox v1.0 | (Durand et al., 2016a) | https://www.aidenlab.org/juicebox |
| MACS v2.0.9 | (Zhang et al., 2008) | https://github.com/taoliu/MACS; RRID:SCR_013291 |
| Mango v1.2.0 | (Phanstiel et al., 2015) | https://github.com/dphansti/mango |
| MEME v4.11.1 | (Bailey et al., 2015) | http://meme-suite.org/index.html; RRID:SCR_001783 |
| MSigDB v6.1 | (Liberzon et al., 2015) | http://software.broadinstitute.org/gsea/msigdb/index.jsp; RRID:SCR_003199 |
| normR v1.6.0 | (Helmuth and Chung, 2018) | https://bioconductor.org/packages/release/bioc/html/normr.html; RRID:SCR_006442 |
| Picard v2.1.0 | Broad Institute | https://broadinstitute.github.io/picard; RRID:SCR_006525 |
| pheatmap v 1.0.10 | R. Kodle | https://github.com/raivokolde/pheatmap; RRID:SCR_016418 |
| RepeatMasker | AFA Smit, R Hubley, P. Green | http://www.repeatmasker.org; RRID:SCR_012954 |
| SAMTools v1.3 | (Li et al., 2009) | http://samtools.sourceforge.net; RRID:SCR_002105 |
| STAR v2.5 | (Dobin et al., 2013) | https://github.com/alexdobin/STAR; RRID:SCR_015899 |
| Sushi v1.18.0 | (Phanstiel et al., 2014) | https://bioconductor.org/packages/release/bioc/html/Sushi.html; RRID:SCR_006442 |
| Trim Galore v0.4.1 | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore; RRID:SCR_011847 |
| UCSC tools v329 | (Kent et al., 2010) | https://github.com/ucscGenomeBrowser/kent |