

Abstract
The extracellular matrix (ECM) of the brain is comprised of unique glycan “sulfation codes” that influence neurological function. Perineuronal nets (PNNs) are chondroitin sulfate-glycosaminoglycan (CS-GAG) containing matrices that enmesh neural networks involved in memory and cognition, and loss of PNN matrices are reported in patients with neurocognitive and neuropsychiatric disorders including Alzheimer’s disease (AD). Using liquid hromatography tandem mass spectrometry (LC-MS/MS), we show that patients with a clinical diagnosis of AD-related dementia undergo a re-coding of their PNN-associated CS-GAGs that correlates to Braak stage progression, hyperphosphorylated tau (P-tau) accumulation, and cognitive impairment. As these CS-GAG sulfation changes are detectable prior to the regional onset of classical AD pathology, they may contribute to the initiation and/or progression of the underlying degenerative processes and implicate the brain matrix sulfation code as a key player in the development of AD clinicopathology.
Keywords: Extracellular matrix, glycosaminoglycans, Alzheimer’s disease, chondroitin sulfates, brain, tau, perineuronal nets, cognition, mass spectrometry
1 |. NARRATIVE
Alzheimer’s Disease (AD) is the leading cause of progressive dementia worldwide and accounts for an estimated 60-80% of all dementia cases1. The progressive decline in cognition, learning, and memory correlated with this disease is due to neurocircuit dysfunction characterized by neuronal hyperactivation2,3, which occurs in the early stages of prodromal AD4,5. While extracellular β-amyloid (Aβ) deposition and intracellular accumulation of hyperphosphorylated tau (P-tau) and associated neurofibrillary tangles (NFTs) are the primary neuropathologic criteria for AD diagnosis6, therapeutic targeting of these classical pathologies have continuously failed to provide clinical benefit7 and may reflect in part a lack of consideration of non-neuronal factors that underlie neurocircuit integrity. Among these non-neuronal factors includes the loss of perineuronal nets (PNNs), specialized extracellular matrix (ECM) structures that enmesh and regulate the activity of key neurons in a circuit, which can in turn predispose to network hyperactivity characteristic of early AD8. In the current work, we showed by mass spectrometry that changes in brain chondroitin sulfate-glycosaminoglycan (GAG) sulfation patterns, which can destabilize PNNs and predispose to their loss over time9, was an early-stage pathology observed in demented AD patients compared to non-demented controls. Following a brief overview of known associations of PNN matrix dysfunction with AD and other neurocognitive disorders, we then detail differences in the brain matrix CS-GAG sulfation patterns between AD and non-demented postmortem brain tissue. Changes in CS-GAG composition began prior to the onset of AD clinicopathology and correlated with increasing Braak stage, P-tau accumulation, and with reduced cognitive function. Alteration of the brain’s CS-GAG ‘sulfation code’ therefore represents an early-stage pathology that may predispose to PNN dysfunction and defective neurocircuitry characteristic of AD. Based on these insights, we discuss potential mechanisms directly linking changes in CS-GAG sulfation patterns with the development of AD clinicopathology.
1.1 |. PNNs and the CS-GAG ‘sulfation code’
PNNs are synapse-stabilizing ECM structures comprised of CS-GAG chains attached to CS proteoglycans (CSPG) and assembled on hyaluronan (HA) glycosaminoglycan backbones10. PNN function and stability are influenced by the composition of these sulfated CS-GAGs, which consist of repeating disaccharide units of sulfated glucuronic acid (GlcA) and N-acetylgalactosamine (GalNac)10. Five differentially-sulfated CS isomer units exist in the human brain (CS-A, -C, -D, -E and -O), with their relative abundance comprising a matrix ‘sulfation code’ that controls key neurological functions11. These include neuroplasticity and synaptic stability (e.g., CS-A, CS-C9), protein-glycan binding interactions (e.g., CS-O, CS-D, CS-E12,13), and neuroinflammation and regeneration (e.g., CS-C, CS-E14). Changes in PNN integrity and/or its CS-GAG sulfation code have been implemented in long-term neurocircuit synaptic dysfunction, and as such, decoding changes in these CS-GAG sulfation patterns so as to better understand the role of matrix remodeling in neurocognitive disorders is now of intense interest to the biomedical community.
1.2 |. Reorganization of PNNs during aging, AD, and other neurocognitive disorders
Histochemical identification of brain PNN structures is accomplished by antibody detection of the underlying CSPG lattice, formed primarily by the CSPG aggrecan, and Wisteria floribunda agglutinin (WFA) lectin labeling of the incorporated CS-GAGs15–17. In humans and rodents, stable PNN structures are absent early in development, presumably allowing for enhanced neurocircuit plasticity and experience-dependent circuit organization17,18. The lack of mature PNNs in the juvenile brain also associates with a developmental CS-GAG sulfation code high in CS-C isomer abundance, which prevents premature formation of PNNs during this timeframe9,15,19. Meanwhile, the establishment of stable PNNs coincides with closure of so-called ‘critical periods’ of brain development and is directed by the adult CS-GAG sulfation code that is highly abundant in the CS-A isomer9,15, which stabilizes PNNs and limits the plasticity of the enmeshed neuron. Recent evidence in mice suggests that overabundance of CS-A incorporation into PNNs during advanced aging prevents plasticity of experience-driven circuitry, thus restricting new memory formation and contributing to age-related cognitive decline20.
In contrast, patients with AD exhibit a decrease in cortical PNNs, as measured by the loss of lectin labeling of the PNN associated CS-GAGs21,22. Paradoxically, abundance of the PNN-associated CSPG, aggrecan, is reportedly unchanged in AD23,24. Notably, neurons ensheathed by either WFA+ (CS-GAGs) or aggrecan+ (CSPG) PNNs remain devoid of intracellular P-tau accumulation and are believed to be ‘protected’ from neurofibrillary tangle formation, whereas extracellular Aβ deposits show significant overlap with PNN structures but exhibit no congruence with PNN loss in AD23,24. Decreased cortical PNNs have also been described in other neurocognitive disorders including schizophrenia25–27 and bipolar disorder28, and similar to AD, the magnitude of the reduction in WFA+ PNNs was reported to be much higher than aggrecan+ PNNs25. Since the negative sulfation charges on the WFA+ PNN CS-GAGs are predicted to assist with rapid ion buffering intrinsic to the fast-spiking neurons enmeshed within these matrices29, the disproportionate loss of PNN CS-GAGs relative to the underlying CSPG protein lattice may disrupt cation buffering and thus predispose to neurocircuit hyperactivity common to AD and other neurocognitive disorders2–5,30.
Inconsistent with histochemical evidence of reduced PNN CS-GAG labeling, Huynh and colleagues recently reported that the total abundance of hippocampal CS-GAGs remains unchanged in AD31, but that AD sulfated GAGs display altered binding capacities to extracellular factors compared to non-demented controls. Specifically, whereas AD GAGs exhibit decreased binding to endogenous growth factors (e.g., FGF2 and VEGF), binding to AD-associated neuropeptides (e.g., Tau) was increased31. Together, these findings raise three key possibilities: 1) that changes in the PNN CS-GAG sulfation code, rather than total abundance, underlie preferential shifts in sulfated GAG binding activity in the AD brain, 2) since WFA lectin labeling of PNNs depends upon the CS-GAG sequence9,12,32, the loss of WFA+ PNNs in AD involves changes in PNN CS-GAG sulfation patterns independent to the loss of PNN matrices per se, and 3) that changes in PNN sulfation, rather than abundance, promote AD pathology. Consistent with these possibilities is the persistence of aggrecan+ PNNs despite loss of WFA+ PNNs in patients with AD23,24, which offers a cautionary note for studies that rely on histological labeling to quantify PNNs without addressing potential changes in CS-GAG sulfation patterns.
1.3 |. Perineuronal net CS-GAG sulfation patterns are altered in AD
We previously validated the use of liquid chromatography–tandem mass spectrometry with multiple reaction monitoring (LC-MS/MS + MRM) as a sensitive and accurate method for quantifying the relative abundance of CS isomers extracted from mouse, rat, and human brain tissue15,16. To determine if the PNN CS-GAG sulfation code is altered in AD, we employed LC-MS/MS + MRM to quantify the relative abundance of each CS isomer (ΔA, ΔC, ΔD, ΔE, ΔO) from the prefrontal neocortex (middle frontal gyrus – MFG) of human subjects with and without AD neuropathologic change and associated dementia. Compared to non-demented controls, our analysis revealed that CS-GAGs from AD brain are consistently hypersulfated compared to non-demented controls, as evidenced by a decrease in the nonsulfated ΔO content and corresponding increases in the sulfated ΔC and ΔE. Notably, the total amount of CS-GAGs isolated during the glycan extraction did not differ between AD and non-demented controls, consistent with previous reports that despite evidence of PNN loss21,22, total CS-GAG abundance is unaffected in AD brain31.
1.4 |. Alterations in CS-GAG sulfation patterns occur prior to the development of AD clinicopathology
To further explore the relationship between changes in brain PNN CS-GAG sulfation code and the development of AD clinicopathology, we correlated changes in each CS isomer to an established, three-categorical Braak stage patient criteria classification scheme: Braak 0-I/II (non-demented), Braak III/IV (non-demented), and Braak V/VI (demented)33. Surprisingly, changes in matrix CS-GAG sulfation were detectable in non-demented patients prior to the development of AD clinicopathology, and they became more pronounced as the Braak stage increased. Thus, although changes in CS-GAG sulfation patterns were detected before the appearance of AD clinicopathology, the magnitude of these changes correlated with the severity of the AD pathogenesis. These findings are in agreement with a recent paper that identified genes involved in chondroitin sulfate glycan metabolism (including CS-C biosynthesis) as a key biochemical pathway underlying vulnerability to P-tau accumulation and neurodegeneration in AD34.
1.5 |. Accumulation of P-tau exacerbates changes in CS-GAG sulfation patterns
Previous work demonstrates that P-tau accumulation occurs exclusively in neurons devoid of PNN structures21,22 and that genes involved in CS-C biosynthesis associates with brain regions vulnerable to P-tau accumulation34. Combined with our finding that the AD neocortex associated with a significant increase in the CS-C isomer, we next considered the possibility that changes in the PNN CS-GAG sulfation code may be intensified with the neuropathologic accumulation of P-tau in AD vulnerable brain regions. By comparing changes in each CS isomer against the number of P-tau+ inclusions, we found that in the MFG, P-tau accumulation correlated negatively with ΔO and positively with ΔC, ΔE, ΔD and with overall hypersulfation of CS-GAGs. To determine whether these changes in the MFG CS-GAG sulfation code was a localized pathological event, or instead represented a global feature of the AD brain, we performed the same CS-GAG analysis on the cerebellar cortex (CER), a brain region largely devoid of neurofibrillary tangles in AD35, from the same AD and non-demented control patients. As no significant differences were observed in the relative abundance of any CS isomer within the CER, we infer that altered CS-GAG sulfation is a feature of brain regions vulnerable to AD neuropathology.
1.6 |. Changes in CS-GAG sulfation patterns correlate with impaired cognitive function
In rodents, alterations in PNN CS-GAG sulfation patterns attenuates mature PNN formation and alters neurocircuit activity of the enmeshed neurons9. Highlighting this relationship is a study by Miyata, et al, showing that overexpression of the CS-C isomer in mouse brain caused a decrease in the relative abundance of non-sulfated CS-O isomer in association with increased CS-C and hypersulfation of CS-GAGs. Of note, this effect decreased PNN labeling by WFA, enhanced experience-driven plasticity, and induced aberrant firing rates in inhibitory parvalbumin neurons9. If changes in the PNN CS-GAG sulfation code in patients with AD (e.g., decreased ΔO; increased ΔC and ΔE) result in similar PNN matrix reorganization and impaired synaptic processing, they can be predicted to correlate with the degree of cognitive impairment. To investigate this hypothesis, we evaluated the relationship between changes in each CS isomer within the MFG and cognitive function assessed by the Mini-Mental State Examination (MMSE). Here, we found intact cognitive function was found to correlate positively with ΔO, and negatively with CS-E and hypersulfation of CS-GAGs .
1.7 |. Limitations
Among several limitations of this study is that our findings do not account for pre-mortem drug treatments that may have influenced brain PNN CS-GAG sulfation patterns independent of clinical diagnosis. Second, we cannot account for brain matrix glycan changes that occur between MMSE testing and time of death. Third, our identification of P-tau accumulation within this study did not differentiate between glial and neuronal P-tau inclusions. Finally, we acknowledge that our CS-GAG analyses may also be detecting changes in CS sulfation patterns of non-PNN matrices in the brain including the interstitial matrix and so-called ‘glial-scarring’.
1.8 |. Conclusions and directions for future research.
Reports of reduced histochemical labeling of PNNs by WFA in AD brain21,22 can potentially be explained either by reduced CS-GAG abundance or by altered CS-GAG composition (or both). Thus, our finding that the AD brain is characterized by CS-GAG hypersulfation (decreased non-sulfated CS-O and increased sulfated CS-C and CS-E) with no difference in total CS-GAG abundance implicates changes in CS-GAG composition as a main contributor to reduced histochemical detection of PNNs in AD. This conclusion is consistent with evidence that show 1) CS-GAG abundance is unaltered in AD brain31, 2) WFA preferentially binds PNN-associated CS-GAGs through the CS-O unit12, and 3) overexpression of the CS-C unit reduces both abundance of the CS-O isomer and WFA labeling of PNNs9. Further studies are warranted to quantify the extent to which reduced WFA histochemical labeling of PNNs in AD brain is driven by changes in the CS-GAG sulfation code vs. reduced PNN matrix abundance itself.
Our findings also provide important implications for previous work showing preferential loss of PNN labeling around P-tau+ neurons in AD21,22 by linking this phenomenon to changes in the CS-GAG sulfation code, rather than (or in addition to) PNN loss per se. What is less clear is the causal relationship between these findings: does P-tau accumulation alters CS-GAG sulfation patterns, or alternatively, does altered CS-GAG sulfation predisposes to P-tau pathology? Although both scenarios are not mutually exclusive, given the evidence that neurocircuit hyperexcitability induces neuronal P-tau accumulation36,37, our findings support a model in which PNN matrix dysfunction arising from changes in the CS-GAG sulfation patterns favor neurocircuit hyperexcitability that in turn then predisposes to P-tau accumulation. This model is consistent with both our finding that changes in CS-GAG sulfation patterns occur prior to the appearance of P-tau inclusions in non-demented Braak III/IV individuals, and with published evidence that sulfated glycans isolated from AD tissue exhibit increased binding capacity to tau31. Thus, changes in the CS-GAG sulfation code and/or hypersulfation of CS-GAGs in AD brain may render PNNs ineffective in their ability to control neuronal excitability and exacerbate P-tau accumulation, potentially through altered protein-glycan binding interactions. Importantly, this model places changes in CS-GAG sulfation patterns upstream of P-tau accumulation and/or ‘seeding’ events, and therefore provides a novel therapeutic target for early-stage interventions with the potential to reduce and/or prevent the formation of neurofibrillary tangles.
The surprising reduction of MMSE scores in association with increased ΔO and ΔE, but not ΔC, raises the possibility that each CS isomer may play a differential role in influencing cognitive function in AD. Further supporting this possibility is that the CS-C and CS-E isomers exert opposing effects within the brain; whereas CS-E unit inhibits neurite extension and axonal recovery38, the CS-C unit enhances glia-associated repair and neuronal differentiation14. As CS-GAGs are present in both PNN and interstitial glial-scar forming matrices10, we cannot exclude the possibility that changes in the CS-GAG sulfation patterns are reflective in part of changes in non-PNN matrices that are also present in the tissue samples. For example, CS-C and CS-E units are components of the ‘glial scar’38–41 that function to seal off sites of CNS injury, which can be both beneficial and detrimental for regeneration42. Whereas these densely-packed CS-GAGs of the glial scar may separate toxic AD neuropathology from the healthy neuropil, they also inhibit CNS regeneration, and as such, glia-associated changes in the CS-GAG sulfation code through chronic glial scaring may exacerbate the AD pathogenesis43. Studies that clarify the relationship between changes in PNN CS-GAG sulfation, neuroinflammation, and gliosis are therefore warranted.
In rodents, healthy aging associates with a progressive decrease in the CS-C isomer throughout the brain20, but in contrast, we found increased abundance of the CS-C isomer in the prefrontal cortex (MFG) of AD patients compared to non-demented controls. Moreover, this increase was associated with degree of dementia, Braak stage, and P-tau inclusions. As such, the paradoxical increase in the CS-C isomer in AD suggests that this dementing disease does not simply represent an accelerated aging phenotype but instead showcases a maladaptive matrix response unique to the AD pathogenesis. In addition to CS-GAGs, hyaluronan (HA) is another PNN associated glycosaminoglycan that is altered in the AD brain; whereas CS-GAG abundance remains unchanged, HA abundance is increased in the parietal44 and temporal45 cortices of patients with AD. Since changes in HA molecular weight is an underlying contributor to PNN destabilization32, alterations in both HA molecular weight and CS-GAG sulfation patterns may be closely linked to PNN dysfunction in the demented brain.
Consistent with this notion is growing evidence linking changes of PNN CS-GAG composition and/or abundance to CNS disease states. For example, Brewton, et al linked PNN loss in the auditory cortex to sensory processing deficits and age-related hearing loss46. Similarly, we recently reported that in a rodent model of type 2 diabetes, glucoregulatory circuit dysfunction associates with PNN loss and changes in the CS-GAG sulfation code in the mediobasal hypothalamus16. These findings support the possibility that specific regional changes in PNN CS-GAGs may manifestation into specific disease outcomes depending on the inflicted brain region. Taken together, our findings highlight the importance of future studies that 1) identify additional brain disorders associated with regional changes in PNN CS-GAG composition, 2) determine whether these ECM changes are a cause or a consequence of the underlying disease process, and 3) determine if clinical outcomes in conditions such as AD can be improved by therapeutic strategies that restore proper PNN composition and function. One such treatment may include central administration of fibroblast growth factor-1 (FGF-1), which induces robust formation of PNN matrices and normalizes brain CS-GAG sulfation in rodents16. The translation of FGF-1 and other compounds that modify brain matrix sulfation are of critical therapeutic interest for individuals with AD and other neurocognitive disorders.
2. CONSOLIDATED RESULTS AND STUDY DESIGN
2.1 |. Changes in the PNN CS-GAG sulfation code: a novel AD neuropathology
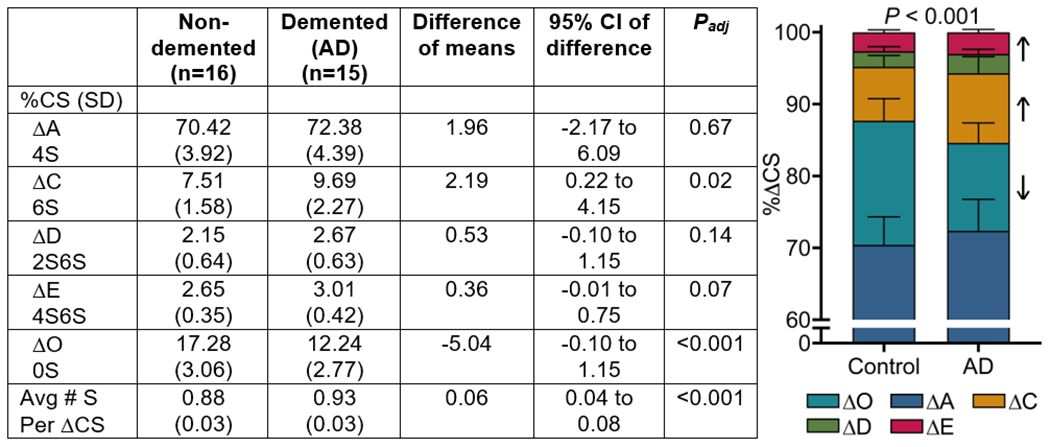
Perineuronal nets (PNNs) are unique ECM structures that stabilize circuit interactions involved in memory and cognition (Figure 1A)10. They are comprised of chondroitin sulfate proteoglycans (CSPGs) that are decorated with attached CS-glycosaminoglycans (CS-GAGs) and attached to hyaluronan (HA) backbones (Figure 1B). The diverse biological functions carried out by PNNs and other CS-GAG containing matrices are determined by the incorporation of five differentially-sulfated CS isomer units (CS-A, -C, -D, -E and -O) (Figure 1C)10, with their relative abundance to one another comprising a matrix ‘sulfation code’ that controls key neurological functions11. LC-MS/MS + MRM analysis of CS-GAG sulfation patterns in the MFG of patients with AD (Figure S1) showed hypersulfation of PNN CS-GAGs (P < 0.001) compared to non-demented controls, evident by the decrease in nonsulfated ΔO (P < 0.001) and corresponding increases in monosulfated ΔC and disulfated ΔE (P = 0.02 and P = 0.07, respectively) (Tables 1,2 and Table S1). AD associated changes in CS-GAG sulfation patterns did not correspond with a decrease in total CS-GAG abundance (Figure S2), consistent with previous reports that total CS-GAG abundance remains unaltered in the AD brain31.
Fig 1.
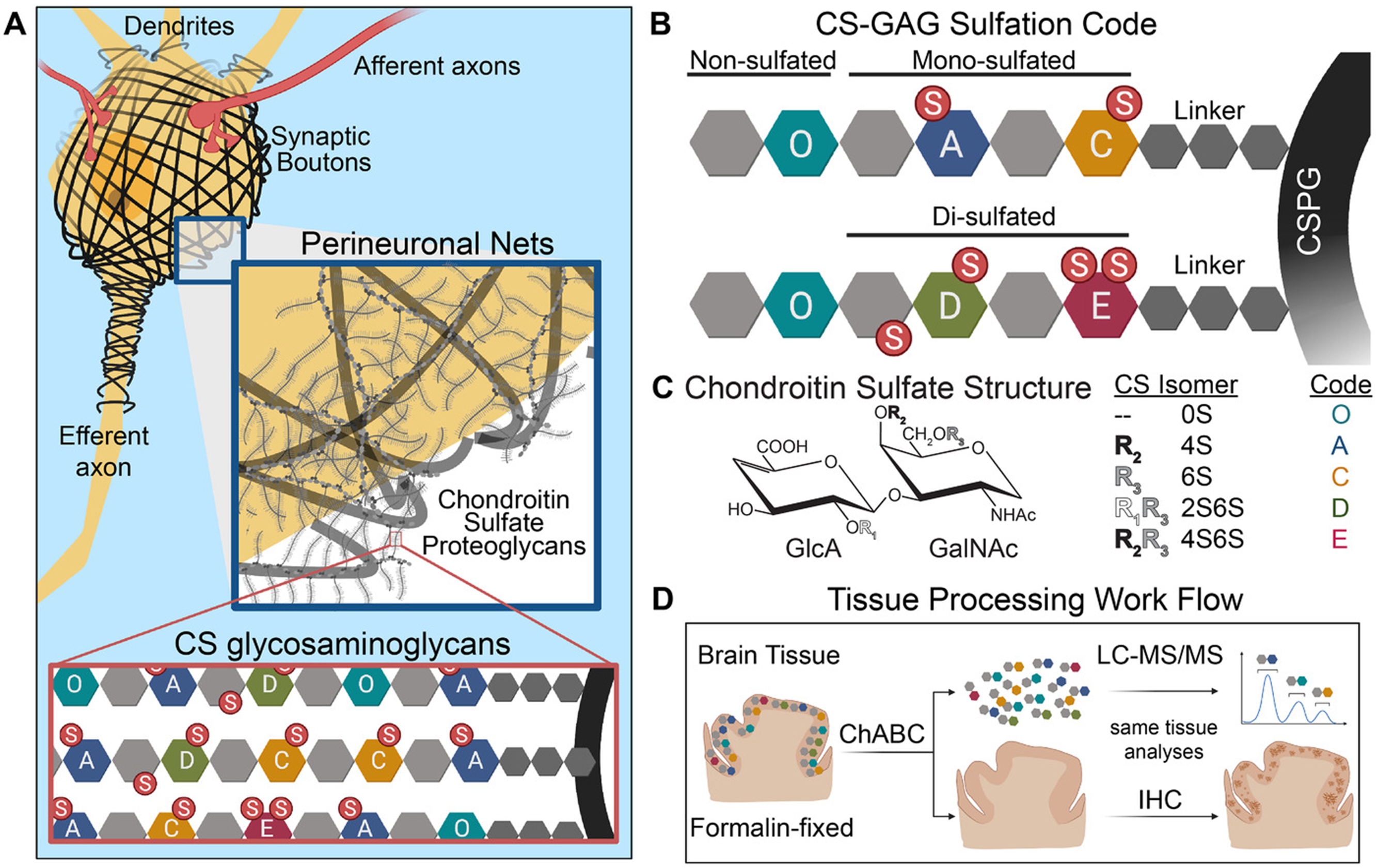
Schematic of perineuronal net chondroitin sulfate-glycosaminoglycan (CS-GAG) sulfation patterns in the brain. A, Depiction of a neuron enmeshed by sulfated CS-GAG containing perineuronal nets. B-C, The brain CS-GAG sulfation code is comprised of five CS isomers (CS-O, -A, -C-, -D, -E) distinguished by their unique sulfation pattern (non-, mono-, and di-sulfated), the relative abundance of which can influence perineuronal net stability and biological function. D, Experimental workflow for the release, isolation, and purification of CS disaccharides from formalin-fixed human brain tissue using chondroitinaseABC (ChABC). Isolated CS isomers are quantitatively analyzed by liquid chromatography tandem mass spectrometry (LC-MS/MS) and the remaining brain tissue is sequentially labeled for protein expression using immunohistochemistry
Table 1.
Patient data by study group.
| Non-demented | Demented (AD) | |
|---|---|---|
| Demographic Features | ||
| Number | 16 | 15 |
| Age (years) | ||
| Mean (SD) | 85.3 (6.0) | 85.3 (6.0) |
| Median (IQR) | 86.5 (81.8-89.8) | 86.0 (81.0-90.0) |
| Sex (Male/Female) | (8/8) | (7/8) |
| Post-mortem Interval (hours) | ||
| Mean (SD) | 4.6 (1.4) | 7.4 (11.1) |
| Median (IQR) | 4.5 (3.5-5.5) | 4.3 (3.3-5.8) |
| Alzheimer’s Pathology | ||
| Number | 16 | 15 |
| Braak Stage | ||
| B1: Braak 0-II | 9 | 0 |
| B2: Braak III/IV | 7 | 0 |
| B3: Braak V/VI | 0 | 15 |
| Mean (SD) | 2 (1) | 6 (1) |
| Median (IQR) | 2 (2-3) | 6 (5-6) |
| CERAD Score | ||
| 0 | 16 | 0 |
| 3 | 0 | 15 |
| Mean (SD) | 0 (0) | 3 (0) |
| Median (IQR) | 0 (0-0) | 3 (3-3) |
| Age of tissue (years) | ||
| Mean (SD) | 10.3 (4.3) | 10.1 (4.0) |
| Median (IQR) | 9.0 (7.0-14.0) | 10.0 (7.0-13.0) |
| Cognitive Features | ||
| MMSE Score (points) | ||
| Number | 16 | 13 |
| Mean (SD) | 27.6 (2.2) | 18.8 (7.4) |
| Median (IQR) | 28.0 (26.3-29.8) | 22.0 (14.0-24.0) |
Abbreviations: AD, Alzheimer’s disease; CERAD, Consortium to Establish a Registry for Alzheimer’s Disease; MMSE, Mini-Mental State Examination; IQR, interquartile range; SD, standard deviation.
Table 2.
Relative percentages of CS isomers in the middle frontal gyrus.
|
Abbreviations: AD, Alzheimer’s disease; SD, standard deviation; CS, chondroitin sulfate.
Stats: mixed-effects, repeated measures two-way ANOVA with matched CS isomers and Šidák’s multiple comparisons. Stacked bars are represented with SD.
We next determined if changes in MFG CS-GAG sulfation patterns associate with Braak stage in AD by correlating changes in CS isomers between B0/1: Braak 0-I/II, B1: Braak III/IV, and B3: Braak V/VI stage classifications33. The progression of Braak stage associated with CS-GAG hypersulfation (P < 0.001) driven by the decrease in nonsulfated ΔO (P < 0.001) and increases in sulfated ΔC (P = 0.01), ΔE (P < 0.05), and ΔD (P = 0.09) (Figure 2A,B). Closer examination of the non-sulfated ΔO showed a Braak stage-dependent decrease between the non-demented Braak 0-II to non-demented Braak III/IV populations (P = 0.04), which is further exacerbated between non-demented Braak III/IV and demented Braak V/VI populations (P < 0.05) (Figure 2A). Complementing this effect was a Braak stage-dependent increase in CS-GAG hypersulfation between the non-demented Braak 0-II to non-demented Braak III/IV populations (P = 0.03) that also further increased between non-demented Braak III/IV and demented Braak V/VI populations (P = 0.009) (Figure 2B).
Fig 2.
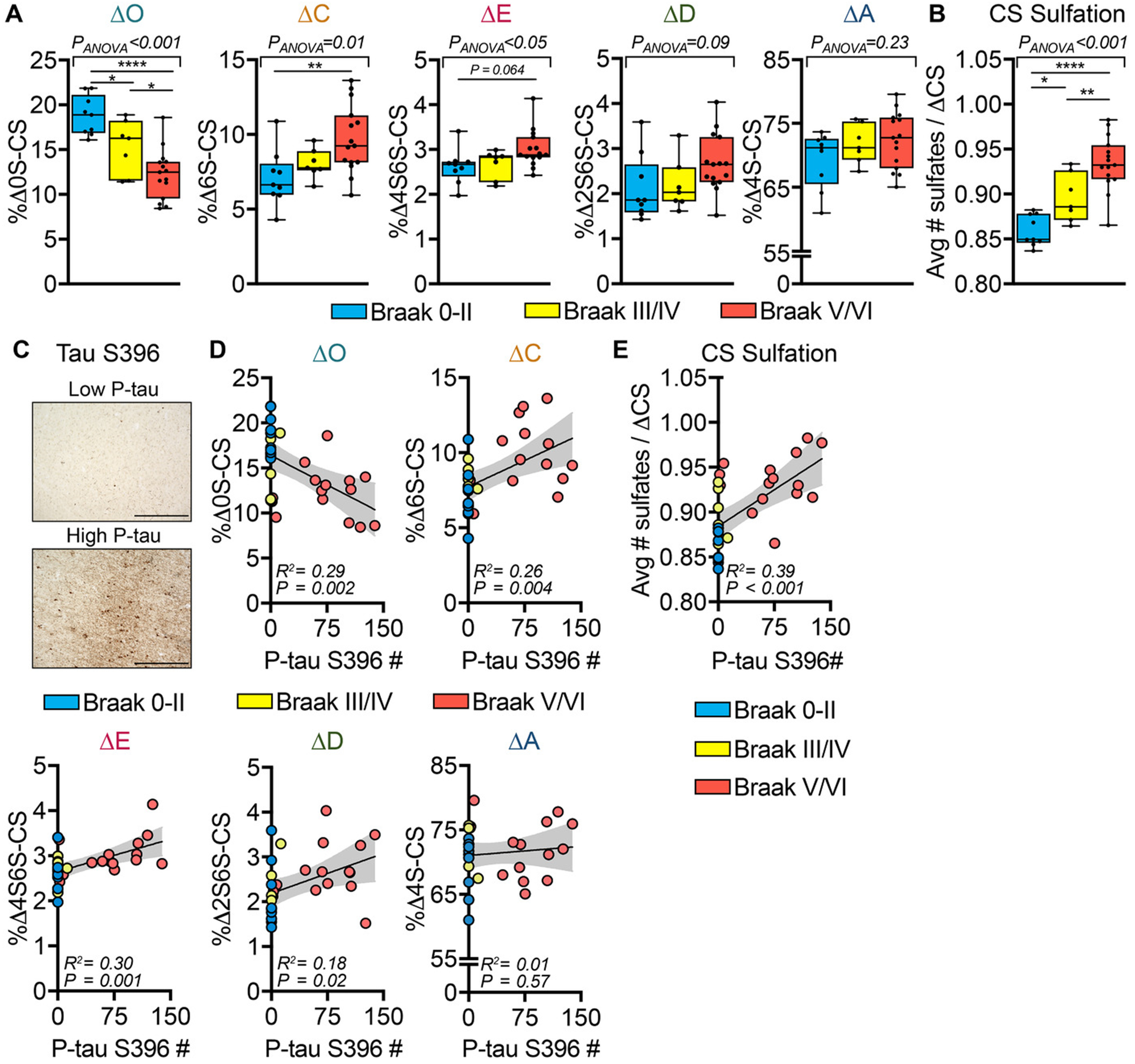
Changes in brain matrix chondroitin sulfate-glycosaminoglycan (CS-GAG) sulfation patterns correlate to Braak stage and phosphorylated tau (p-tau) accumulation. Demented (Alzheimer’s disease) and non-demented controls were stratified by pathology according to Braak stage: B0/1, Braak 0-II (blue); B2, Braak III/IV (yellow); and B3, Braak V/VI (red). A, The relative abundance of the five perineuronal net CS isomers (ΔO, ΔA, ΔC, ΔD, and ΔE) isolated from the middle frontal gyrus (MFG) and (B) CS-GAG hypersulfation were correlated to Braak stage. C, After CS-GAG extraction, the MFG tissue sections were histochemically labeled for p-tau S396 (see Figure 1D). MFG p-tau+ inclusion number was correlated against changes in (D), each ΔCS and (E), CS-GAG hypersulfation from CS-GAGs isolated from the same tissue sections. A-B, n = 7–15 subjects per stage; mean ± standard deviation (SD). Data were analyzed using a mixed-effects, one-way analysis of variance model with Tukey’s multiple comparison (* P < .05, ** P < .01, **** P < .0001). C, Scale bar: 500 μm. MFG p-tau+ inclusions were correlated against the relative abundance of each ΔCS isomers isolated from the same tissue sections. D, n = 31 subjects; mean ± SD. Data were analyzed using linear regression with the 95% confidence bands shown in gray
Based on the observation that P-tau accumulation occurs exclusively in neurons devoid of PNN structures21,22, we next correlated changes in CS-GAG sulfation patterns with P-tau S396 labeling of the same tissue used in the CS isomer extraction (Figure 1D), which showed robust accumulation of P-tau+ inclusions within layer V of the MFG of AD patients (Figure 2C). P-tau inclusion number correlated negatively with the ΔO (P = 0.002) and positively with ΔC (P = 0.004), ΔE (P = 0.001), ΔD (P = 0.02) (Figure 2D) as well as CS-GAG hypersulfation (P < 0.001) (Figure 2E). In contrast to the MFG, CS-GAG sulfation patterns in the cerebellar cortex (CER), a brain region largely devoid of neurofibrillary tangles in AD35, from the same AD and non-demented control patients showed no significant changes in the relative abundance of any CS isomer (Table S3), suggesting that altered CS-GAG sulfation is a regionally-distinct occurrence within AD brain.
Braak stage and P-tau accumulation closely parallel the progressive decline of cognitive function in AD47, which can be assessed using the Mini-Mental State Examination (MMSE). Consistent with the expected inverse relationship between P-tau accumulation in the MFG and cognitive function48, we found P-tau inclusion number within the MFG correlated negatively with MMSE scoring (P < 0.001) (Figure 3A). MMSE scoring also correlated positively with ΔO (P < 0.001) and negatively with CS-E (P = 0.006) (Figure 3B) and CS-GAG hypersulfation (P < 0.001) (Figure 3C), while no significant association was observed between MMSE score and ΔA, ΔC, or ΔD units (Figure S3). Taken together, our findings revealed significant shifts in the PNN CS-GAG sulfation code both prior to and after the development of AD clinicopathology (Figure 3D).
Fig 3.
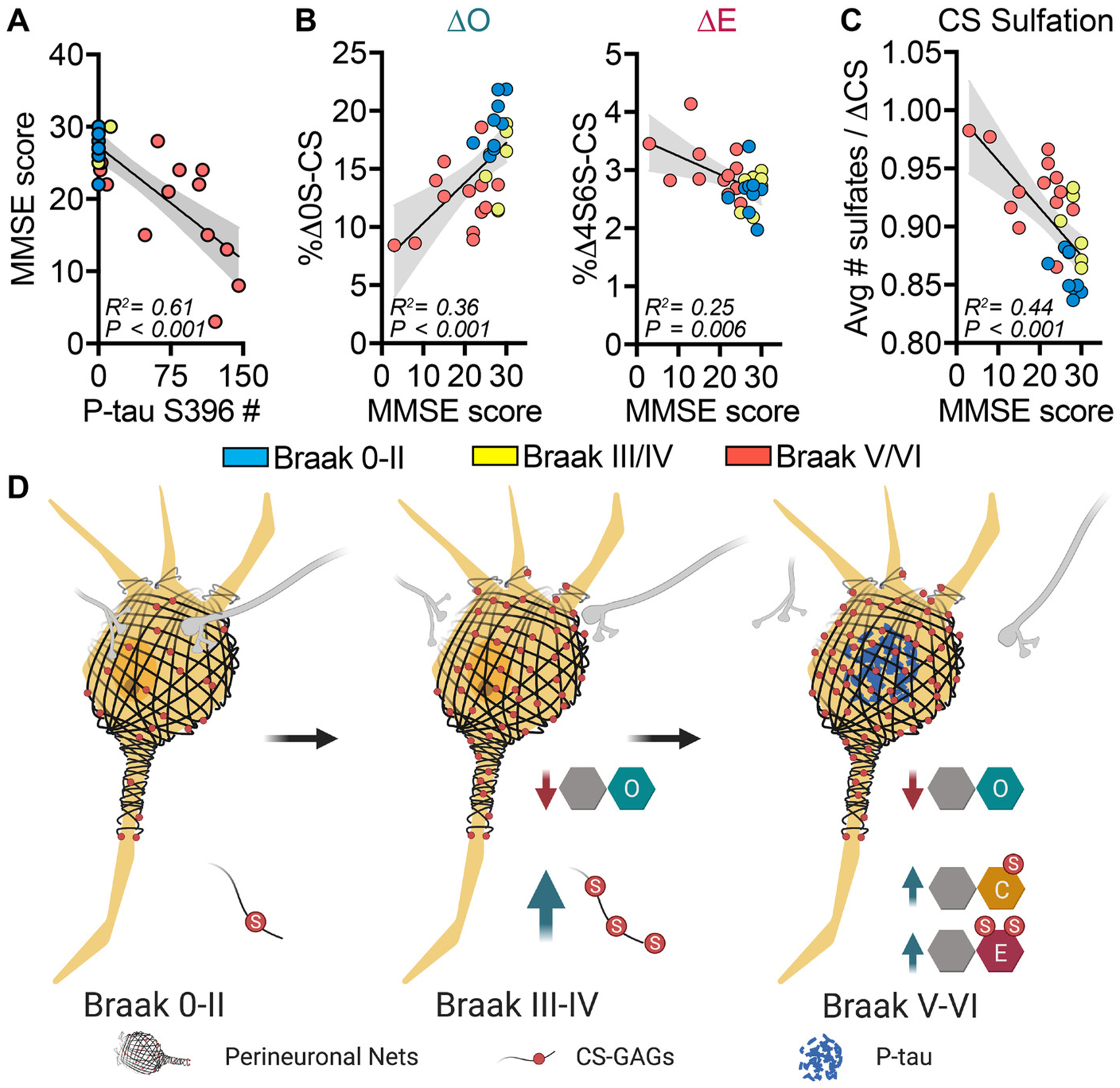
Changes in brain chondroitin (CS)-O, CS-E, and CS-sulfate-glycosaminoglycan (CS-GAG) hypersulfation correspond to altered cognitive function. Demented (Alzheimer’s disease) and non-demented individuals were stratified by pathology according to Braak stage: B0/1, Braak 0-II (blue); B2, Braak III/IV (yellow); and B3, Braak V/VI (red). A, Mini-Mental State Examination (MMSE) scores negatively associate with phosphorylated tau (p-tau) inclusion number in the middle frontal gyrus (MFG). B, MMSE scores positively associate with CS-O and negatively associate with CS-E and (C) negatively associate with CS-GAG hypersulfation in the MFG. D, Model depicting changes in perineuronal net CS-GAG sulfation code in relation to AD clinicopathology. A-C, n = 29 subjects; mean ± standard deviation. Data were analyzed using linear regression with the 95% confidence bands shown in gray
2.2. |. Human Participants
This postmortem cohort study utilized human brain tissue provided by the University of Washington (UW) BioRepository and Integrated Neuropathology (BRaIN) laboratory and derived from research donors from the UW Alzheimer’s Disease Research Center (ADRC) clinical core, the Adult Changes in Thought (ACT) Study, and the Seattle Longitudinal Study (SLS) collected from years 2002 to 2018. The current study utilized both prefrontal cortex (middle frontal gyrus – MFG) and cerebellar cortex (CER) sections dissected from 16 subjects with dementia and neuropathologically confirmed high AD neuropathologic change (CERAD frequent plaques (3) and Braak stage V-VI) and from 16 control (non-demented) subjects with no or low AD neuropathologic change (CERAD none/sparse and Braak stage 0-III). A predetermined exclusion criterion of postmortem interval (PMI) >48 hours resulted in the exclusion of 1 demented individual prior to data collection. The characteristics of the AD and non-demented individuals including age, sex, PMI, age of tissue, Braak stage, CERAD score, MMSE scoring, and study origins are outlined in Table 1 and Table S1.
2.3 |. CS disaccharide isolation and quantification
CS disaccharide from formalin-fixed MFG and CER samples (30 μm) were isolated and quantified according to our previously published methods15,16. ChondroitinaseABC (ChABC) enzyme, a combinatorial lyase/enolase that selectively degrades all CS-GAG chains into their individual disaccharide units49–51, was used to release CS isomers from the fixed tissue for sequential LC-MS/MS + MRM analysis (Figure 1D). Quantification of the total amount of glycans released after ChABC digestion was performed using cetylpyridinium chloride (CPC) turbidimetry analysis52 and normalized to total tissue area. The isolation and quantification of CS isomers were completed blinded and twice from the same cohort, and the quantifications for each analysis then averaged.
2.4 |. Immunohistochemistry
After CS-GAG extraction, the same tissue sections were utilized for immunohistochemistry analysis for P-tau S396 (Figure 1D). Brightfield microscopy was performed using a Keyence BZ-X800 microscope. A compete tile-image of the entire tissue section was taken at 2 x magnification followed by six random images at 10 x magnification within cortical layer V of the tissue, and these images consisted of three superficial and three deep layer images each as previously described53. P-tau positive cell number was quantified by unbiased counting and averaging of positive inclusions from all six 10 x images using Fiji open-source imaging software54 specific for DAB-tau imaging analysis55 and the Analyze Particles function (Size inclusion criteria: 0.00003-Infinity mm2; Circularity inclusion criteria: 0.02-1.00). Quantitative histological analyses were performed blinded by two independent investigators and results were then averaged.
3 |. DETAILED METHODS AND RESULTS
3.1 |. Patient tissue sampling
Sample size for this study was determined based on the differences of the means generated from a pilot cohort of 4 non-demented and 5 demented (AD) MFG samples provided by the OHSU Layton Aging and Alzheimer’s Disease Center and Oregon Brain Bank and analyzed for the CS-O isomer. Statistically significant differences at alpha 0.05 (p=0.05) specified n=14/group at 95% power, n=11/group at 90% power, n=8/group at 80% power and n=4/group at 50% power. The three-categorical Braak stage criteria was created using recommendation in the National Institute of Aging Alzheimer’s Association (NIA-AA) guidelines for the pathologic assessment of AD neuropathologic change: B0/1 (Braak 0-I/II); B2 (Braak III/IV); B3 (Braak V/VI)33.
To determine whether sampling location within the MFG influences CS-GAG sulfation, two samples analyzed from within the same MFG region (i.e., A01 and A14) of a single donor male, 73 years of age, PMI 4.5 h, with AD diagnosis (Table S3). CS-GAG sulfation patterns were found to be reproducible across multiple different areas of the MFG collected from the same patient and thus, differences in CS-GAG sulfation patterns in AD cannot be attributed to variation in tissue sampling within the MFG.
3.2 |. Brain tissue processing
Brain tissue was fixed in 10% neutral buffered formalin (NBF) for 3 weeks, coronally sectioned into whole-mount blocks, and stored in NFB until further processing. Human brain sections were cryopreserved in 30% sucrose, frozen in O.C.T. compound, and acclimated at −20°C overnight. Tissues were then cut with a Leica CM1950 cryostat at 30 μm-thick serial sections and stored in 0.1 M phosphate buffered saline (PBS) + 0.02% sodium azide at 4°C as free-floating sections before processing. Sections were washed 3 x in Optima LC/MS-grade water and 1 x in 50 mM ammonium bicarbonate (pH 7.6) at room temperature. ChABC (Sigma, C3667) was reconstituted (500 mU/mL) in 50 mM ammonium bicarbonate (pH 7.6) and incubated with free-floating brain tissue at 37°C in a Thermo Scientific MaxQ4000 orbital shaker for 24 h. Supernatants were collected and filtered through sterile 100k MWCO centrifugal filters to remove any tissue debris. The supernatant was dehydrated using a SpeedVac Concentrator and the lyophilized product was reconstituted in 30 μL of LC/MS-grade water for LC-MS/MS analysis.
Brain samples for intraregional analyses was provided by the Oregon Health & Science University (OSHU) Layton Aging and Alzheimer’s Disease Center and Oregon Brain Bank. Here, brain samples were frozen as fresh whole-mount blocks and then postfixed in 4% paraformaldehyde (PFA) for 5 days before processing. All UW and OHSU donor brain tissues were collected with informed consent and in ethical compliance with NIH guidelines for the use of human subjects.
3.3 |. LC-MS/MS + MRM quantification of isolated CS disaccharides
The CS isomers isolated from the MFG and CER were analyzed using a triple quadrupole mass spectrometer equipped with an electrospray ion source (Waters Xevo TQ-S) operated in negative mode ionization. Liquid chromatography tandem mass spectrometry (LC-MS/MS) was performed using a Waters Acquity I-class ultra-performance liquid chromatographic system (UPLC) coupled to the same Waters Xevo TQ-S system. CS disaccharides were resolved by porous graphitic chromatography (Hypercarb column; 2.1 x 50 mm, 3 μm; ThermoFisher) as described previously15 and assigned the following multiple reaction monitoring (MRM) channels: ΔA (4S), m/z 458 > 300; ΔC (6S), m/z 458 > 282; ΔD (2S6S) and ΔE (4S6S), m/z 268 > 282; ΔO (0S), m/z 378 > 175. MassLynx software version 4.1 (Waters) was used to acquire and quantify all data. Under the conditions described above, the ratios between peak areas produced from equimolar CS standard mixes were normalized to the highest peak intensity and relative quantification of each CS isomer within a sample was achieved using a modified peak area normalization function as previously described15,56,57. Each CS isomer was expressed as a percentage of the relative abundance of each CS isomer within a brain sample. CS sulfation was computed using the weighed mean formula for the average number of sulfates per ΔCS:
3.4 |. Immunohistochemical Labeling
Initial immunohistochemical labeling of phosphorylated tau was completed by the UW ADRC to determine Braak score and a second immunohistochemical labeling was performed during this study after the glycan extraction (Figure 1D) such that a direct comparison of CS-GAG sulfation patterns and P-tau+ inclusions from the same tissue could be compared. 30 μm-thick sections of MFG underwent antigen retrieval in 10 mM trisodium citrate (pH 8.5) at 92°C for 20 m and then washed in 0.1M PBS (PBS) for 10 m to remove citrate. Tissues were permeabilized for 60 m at room temperature in PBS + 0.2% Triton X-100 (PBS-T). After another PBS wash, the tissue slices were then incubated in Vector® BLOXALL® (SP-6000-100; Vector Laboratories, Burlingame, CA) in PBS for 20 m and then washed in PBS. Tissue slices were blocked in 1.5% goat serum in PBS for 2 h at 37°C and incubated in primary antibody (1:1,000 dilution, rabbit P-tau S396; Anaspec AS-54977) in 2.5% goat serum in PBS overnight at 4°C. In the morning, tissue slices were then washed with PBS, incubated in 0.05% biotinylated goat anti-rabbit IgG secondary antibody in 1% goat serum in PBS for 0.5 h, incubated in Vector® VECTASTAIN® Elite ABC Reagent (PK-6101; Vector Laboratories, Burlingame, CA) for 0.5 h, washed in PBS, and then incubated with 3,3’-diaminobenzidine (DAB) peroxidase substrate. After washing 2 x in tap water, tissue slices were washed again in 0.1M PBS and cover slipped using Fluoromount-G® (ThermoFisher, 4958-02).
3.5 |. Statistical analyses
Non-demented and demented (AD) individuals were matched by age, sex, PMI, and age of the tissue (Table 1 and Table S1). A mixed-effects, repeated measures two-way ANOVA with matched CS isomers and Geisser-Greenhouse correction was used to fit a full model (isomer effect, dementia effect, and isomer/dementia interaction effect) for the primary analyses comparing CS isomer changes between non-demented and demented groups, and then secondary analysis corrected using Šidák’s multiple comparisons computed the mean percent difference and 95% CI for each of the CS isomers changed in AD diagnosis. The comparison of the three-group Braak stage CS isomer comparison utilized a mixed-effects, one-way ANOVA for the primary analysis to compare changes in CS isomers between Braak grouping, and then secondary analysis corrected using Tukey’s multiple comparisons and a single pooled variance computed the mean percent difference and 95% CI for isomer changes between each of the three Braak grouping. Linear regression with the 95% CI bands of the best-fit line was used to compare CS isomers to P-tau, P-tau to MMSE score, and CS isomers to MMSE score. Statistical analyses were completed using GraphPad Prism® 9.0 (Graph Pad Software, Inc., La Jolla, CA). Error bars represent the standard deviation (SD) of the mean. Investigators were blinded to group conditions during the quantitative mass spectrometry and histochemical analyses.
3.6 |. Data Availability.
The data that support the findings of this study are available from the corresponding author upon request.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to the subjects who donated their brains for this research and their loved ones, for the expert technical assistance of Lisa Keene, Kim Howard, Katelyn Kern, and Amanda Keen, and administrative support of Aimee Schantz (University of Washington, BioRepository and Integrated Neuropathology (BRaIN) Laboratory), biostatistical support by Sarah E. Holte was provided by the University of Washington Nutrition and Obesity Research Center (P30 DK035816) from the National Institute of Diabetes and Digestive and Kidney Disease (NIH-NIDDK). Histochemical work was supported by the University of Washington Medicine Diabetes Institute (UWMDI) and mass spectrometry work was supported by University of Washington School of Pharmacy Mass Spectrometry Center. This work was also supported by NIH-NIDDK grants DK122662 (KMA), DK101997 and DK-720269 (MWS), DK114474 (JMS), DK007742 (KLF), and DK007247 (NER and CLF); and the National Institute of Aging (NIH-NIA) grants AG066509 (KMA), AG065426 (CSL), P30 AG066509 (UW Alzheimer’s Disease Research Center), U01 AG006781 (Adult Changes in Thought study), and P50 AG066518 (Oregon Alzheimer’s Disease Research Center). Additional funding provided by the Nancy and Buster Alvord Endowment (CDK), and the NIH-NIDDK Diseases–funded Nutrition Obesity Research Center (DK035816). Additional funding to support these studies was provided to JMS by the UW Royalty Research Fund (RRF A139339), ITHS Young Investigator Catalyst Award to KMA (BR 013035) and funding to KMA and JMS by the UW Diabetes Research Center (DRC) NIH-NIDDK sponsored grant DK017047. Some figures were created using BioRender.com.
DISCLOSURES:
KMA reports NIH support and a travel award from the University of Washington; AFL reports NIH support, a travel award from Brain & Brain PET, a provisional patent for CTE treatment, and is a Board Member for CTEC, LLC (Prophos Neurosciences); NER reports NIH funding, UW-Madison Student Research Grant, a travel award to Central Society for Clinical and Translational Research, a travel award for American Aging Association, and holds a provisional patent for Compositions and Methods for Restoring Metabolic Health; CLF reports NIH support; WAB reports NIH support, consulting fees for the Federal Trade Commission, and a leadership role for Siber (nonprofit); CDK reports NIH and DoD support, a travel award for a NIH grant review session; CSL reports NIH support and a relationship with the National Academy of Neuropsychology; MWS reports NIH support, a honoraria for the Albert Einstein School of Medicine, a honoraria for the University of Toronto, a honoraria for the University of Colorado, and a Research Agreement with Novo Nordisk; JSA reports NIH support, a UW Royalty Research Funds award, a travel award to Helmholtz Diabetes Conference, a travel award to Symposium on Hormones and Cell Regulation, a travel award to Japan Diabetes Society, and a travel award to Japan Insulin Study Group; RLW reports NIH support; KLF reports NIH support ; BAP, VN, NS, and SJH report no disclosures.
ABBREVIATIONS:
- PNN
perineuronal net
- CS
chondroitin sulfate
- GAG
glycosaminoglycan
- CSPG
chondroitin sulfate proteoglycan
- HA
hyaluronan
- ECM
extracellular matrix
- LC-MS/MS
liquid chromatography tandem mass spectrometry
- MRM
multiple reaction monitoring
References
- 1.2020 Alzheimer’s disease facts and figures, <https://www.alz.org/alzheimers-dementia/facts-figures> (2020). [DOI] [PubMed]
- 2.Terry RD, Masliah E, Salmon DP et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30, 572–580, doi: 10.1002/ana.410300410 (1991). [DOI] [PubMed] [Google Scholar]
- 3.Busche MA & Konnerth A Neuronal hyperactivity--A key defect in Alzheimer’s disease? Bioessays 37, 624–632, doi: 10.1002/bies.201500004 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Dickerson BC, Salat DH, Greve DN et al. Increased hippocampal activation in mild cognitive impairment compared to normal aging and AD. Neurology 65, 404–411, doi: 10.1212/01.wnl.0000171450.97464.49 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haberman RP, Branch A & Gallagher M Targeting Neural Hyperactivity as a Treatment to Stem Progression of Late-Onset Alzheimer’s Disease. Neurotherapeutics 14, 662–676, doi: 10.1007/s13311-017-0541-z (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Braak H & Braak E Neuropathological stageing of Alzheimer-related changes. Acta neuropathologica 82, 239–259, doi: 10.1007/bf00308809 (1991). [DOI] [PubMed] [Google Scholar]
- 7.Mehta D, Jackson R, Paul G et al. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010-2015. Expert Opin Investig Drugs 26, 735–739, doi: 10.1080/13543784.2017.1323868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reichelt AC Is loss of perineuronal nets a critical pathological event in Alzheimer’s disease? EBioMedicine 59, doi: 10.1016/j.ebiom.2020.102946 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miyata S, Komatsu Y, Yoshimura Y et al. Persistent cortical plasticity by upregulation of chondroitin 6-sulfation. Nature neuroscience 15, 414–422, s411-412, doi: 10.1038/nn.3023 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Testa D, Prochiantz A & Di Nardo AA Perineuronal nets in brain physiology and disease. Seminars in cell & developmental biology 89, 125–135, doi: 10.1016/j.semcdb.2018.09.011 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Smith PD, Coulson-Thomas VJ, Foscarin S et al. “GAG-ing with the neuron”: The role of glycosaminoglycan patterning in the central nervous system. Exp Neurol 274, 100–114, doi: 10.1016/j.expneurol.2015.08.004 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Nadanaka S, Miyata S, Yaqiang B et al. Reconsideration of the Semaphorin-3A Binding Motif Found in Chondroitin Sulfate Using Galnac4s-6st-Knockout Mice. Biomolecules 10, doi: 10.3390/biom10111499 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Djerbal L, Lortat-Jacob H & Kwok J Chondroitin sulfates and their binding molecules in the central nervous system. Glycoconjugate journal 34, 363–376, doi: 10.1007/s10719-017-9761-z (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Woerly S & Marchand R Collagen-cliondroitin-6-sulfate hydrogel implants in CNS lesion cavities favor glial repair, the differentiation of co-implanted neurons and the growth of axons. Restor Neurol Neurosci 3, 95–99, doi: 10.3233/rnn-1991-3206 (1991). [DOI] [PubMed] [Google Scholar]
- 15.Alonge KM, Logsdon AF, Murphree TA et al. Quantitative analysis of chondroitin sulfate disaccharides from human and rodent fixed brain tissue by electrospray ionization-tandem mass spectrometry. Glycobiology, doi: 10.1093/glycob/cwz060 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alonge KM, Mirzadeh Z, Scarlett JM et al. Hypothalamic perineuronal net assembly is required for sustained diabetes remission induced by fibroblast growth factor 1 in rats. Nature Metabolism, doi:Accepted for Publication. PMC Journal— In Process (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lensjo KK, Christensen AC, Tennoe S et al. Differential expression and cell-type specificity of perineuronal nets in hippocampus, medial entorhinal cortex, and visual cortex examined in the rat and mouse. eNeuro 4, doi: 10.1523/eneuro.0379-16.2017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rogers SL, Rankin-Gee E, Risbud RM et al. Normal Development of the Perineuronal Net in Humans; In Patients with and without Epilepsy. Neuroscience 384, 350–360, doi: 10.1016/j.neuroscience.2018.05.039 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miyata S & Kitagawa H Chondroitin 6-Sulfation Regulates Perineuronal Net Formation by Controlling the Stability of Aggrecan. Neural Plast 2016, 1305801, doi: 10.1155/2016/1305801 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foscarin S, Raha-Chowdhury R, Fawcett JW et al. Brain ageing changes proteoglycan sulfation, rendering perineuronal nets more inhibitory. Aging (Albany NY) 9, 1607–1622, doi: 10.18632/aging.101256 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baig S, Wilcock GK & Love S Loss of perineuronal net N-acetylgalactosamine in Alzheimer’s disease. Acta neuropathologica 110, 393–401, doi: 10.1007/s00401-005-1060-2 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Kobayashi K, Emson PC & Mountjoy CQ Vicia villosa lectin-positive neurones in human cerebral cortex. Loss in Alzheimer-type dementia. Brain research 498, 170–174 (1989). [DOI] [PubMed] [Google Scholar]
- 23.Brückner G, Hausen D, Härtig W et al. Cortical areas abundant in extracellular matrix chondroitin sulphate proteoglycans are less affected by cytoskeletal changes in Alzheimer’s disease. Neuroscience 92, 791–805, doi: 10.1016/s0306-4522(99)00071-8 (1999). [DOI] [PubMed] [Google Scholar]
- 24.Morawski M, Bruckner G, Jager C et al. Involvement of perineuronal and perisynaptic extracellular matrix in Alzheimer’s disease neuropathology. Brain pathology (Zurich, Switzerland) 22, 547–561, doi: 10.1111/j.1750-3639.2011.00557.x (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Enwright JF, Sanapala S, Foglio A et al. Reduced Labeling of Parvalbumin Neurons and Perineuronal Nets in the Dorsolateral Prefrontal Cortex of Subjects with Schizophrenia. Neuropsychopharmacology 41, 2206–2214, doi: 10.1038/npp.2016.24 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mauney SA, Athanas KM, Pantazopoulos H et al. Developmental pattern of perineuronal nets in the human prefrontal cortex and their deficit in schizophrenia. Biol Psychiatry 74, 427–435, doi: 10.1016/j.biopsych.2013.05.007 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berretta S, Pantazopoulos H, Markota M et al. Losing the sugar coating: Potential impact of perineuronal net abnormalities on interneurons in schizophrenia. Schizophrenia Research 167, 18–27, doi: 10.1016/j.schres.2014.12.040 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alcaide J, Guirado R, Crespo C et al. Alterations of perineuronal nets in the dorsolateral prefrontal cortex of neuropsychiatric patients. International Journal of Bipolar Disorders 7, 24, doi: 10.1186/s40345-019-0161-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Härtig W, Derouiche A, Welt K et al. Cortical neurons immunoreactive for the potassium channel Kv3.1b subunit are predominantly surrounded by perineuronal nets presumed as a buffering system for cations. Brain research 842, 15–29, doi: 10.1016/s0006-8993(99)01784-9 (1999). [DOI] [PubMed] [Google Scholar]
- 30.Whitfield-Gabrieli S, Thermenos HW, Milanovic S et al. Hyperactivity and hyperconnectivity of the default network in schizophrenia and in first-degree relatives of persons with schizophrenia. Proceedings of the National Academy of Sciences of the United States of America 106, 1279–1284, doi: 10.1073/pnas.0809141106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huynh MB, Ouidja MO, Chantepie S et al. Glycosaminoglycans from Alzheimer’s disease hippocampus have altered capacities to bind and regulate growth factors activities and to bind tau. PLoS One 14, e0209573, doi: 10.1371/journal.pone.0209573 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sugitani K, Egorova D, Mizumoto S et al. Hyaluronan degradation and release of a hyaluronan-aggrecan complex from perineuronal nets in the aged mouse brain. Biochimica et Biophysica Acta (BBA) - General Subjects 1865, 129804, doi: 10.1016/j.bbagen.2020.129804 (2021). [DOI] [PubMed] [Google Scholar]
- 33.Montine TJ, Phelps CH, Beach TG et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta neuropathologica 123, 1–11, doi: 10.1007/s00401-011-0910-3 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grothe MJ, Sepulcre J, Gonzalez-Escamilla G et al. Molecular properties underlying regional vulnerability to Alzheimer’s disease pathology. Brain 141, 2755–2771, doi: 10.1093/brain/awy189 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jacobs HIL, Hopkins DA, Mayrhofer HC et al. The cerebellum in Alzheimer’s disease: evaluating its role in cognitive decline. Brain 141, 37–47, doi: 10.1093/brain/awx194 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Yanamandra K, Kfoury N, Jiang H et al. Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron 80, 402–414, doi: 10.1016/j.neuron.2013.07.046 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu JW, Hussaini SA, Bastille IM et al. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci 19, 1085–1092, doi: 10.1038/nn.4328 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown JM, Xia J, Zhuang B et al. A sulfated carbohydrate epitope inhibits axon regeneration after injury. Proc Natl Acad Sci U S A 109, 4768–4773, doi: 10.1073/pnas.1121318109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Properzi F, Carulli D, Asher RA et al. Chondroitin 6-sulphate synthesis is up-regulated in injured CNS, induced by injury-related cytokines and enhanced in axon-growth inhibitory glia. The European journal of neuroscience 21, 378–390, doi: 10.1111/j.1460-9568.2005.03876.x (2005). [DOI] [PubMed] [Google Scholar]
- 40.Gilbert RJ, McKeon RJ, Darr A et al. CS-4,6 is differentially upregulated in glial scar and is a potent inhibitor of neurite extension. Mol Cell Neurosci 29, 545–558, doi: 10.1016/j.mcn.2005.04.006 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Moeendarbary E, Weber IP, Sheridan GK et al. The soft mechanical signature of glial scars in the central nervous system. Nat Commun 8, 14787, doi: 10.1038/ncomms14787 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang T, Dai Y, Chen G et al. Dissecting the Dual Role of the Glial Scar and Scar-Forming Astrocytes in Spinal Cord Injury. Front Cell Neurosci 14, 78–78, doi: 10.3389/fncel.2020.00078 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Henstridge CM, Hyman BT & Spires-Jones TL Beyond the neuron–cellular interactions early in Alzheimer disease pathogenesis. Nature Reviews Neuroscience 20, 94–108, doi: 10.1038/s41583-018-0113-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reed MJ, Damodarasamy M, Pathan JL et al. Increased Hyaluronan and TSG-6 in Association with Neuropathologic Changes of Alzheimer’s Disease. J Alzheimers Dis 67, 91–102, doi: 10.3233/jad-180797 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jenkins HG & Bachelard HS Glycosaminoglycans in Cortical Autopsy Samples from Alzheimer Brain. Journal of neurochemistry 51, 1641–1645, doi: 10.1111/j.1471-4159.1988.tb01135.x (1988). [DOI] [PubMed] [Google Scholar]
- 46.Brewton DH, Kokash J, Jimenez O et al. Age-Related Deterioration of Perineuronal Nets in the Primary Auditory Cortex of Mice. Frontiers in aging neuroscience 8, doi: 10.3389/fnagi.2016.00270 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Malpas CB, Sharmin S & Kalincik T The histopathological staging of tau, but not amyloid, corresponds to antemortem cognitive status, dementia stage, functional abilities and neuropsychiatric symptoms. International Journal of Neuroscience, 1–10, doi: 10.1080/00207454.2020.1758087 (2020). [DOI] [PubMed] [Google Scholar]
- 48.Sabbagh MN, Cooper K, DeLange J et al. Functional, global and cognitive decline correlates to accumulation of Alzheimer’s pathology in MCI and AD. Curr Alzheimer Res 7, 280–286, doi: 10.2174/156720510791162340 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hamai A, Hashimoto N, Mochizuki H et al. Two distinct chondroitin sulfate ABC lyases. An endoeliminase yielding tetrasaccharides and an exoeliminase preferentially acting on oligosaccharides. J Biol Chem 272, 9123–9130, doi: 10.1074/jbc.272.14.9123 (1997). [DOI] [PubMed] [Google Scholar]
- 50.Prabhakar V, Capila I, Soundararajan V et al. Recombinant expression, purification, and biochemical characterization of chondroitinase ABC II from Proteus vulgaris. J Biol Chem 284, 974–982, doi: 10.1074/jbc.M806630200 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamagata T, Saito H, Habuchi O et al. Purification and properties of bacterial chondroitinases and chondrosulfatases. J Biol Chem 243, 1523–1535 (1968). [PubMed] [Google Scholar]
- 52.Kwok JCF, Foscarin S & Fawcett JW in Extracellular Matrix (eds Jennie B Leach & Elizabeth M Powell) 23–32 (Springer; New York, 2015). [Google Scholar]
- 53.Cummings BJ, Mason AJ, Kim RC et al. Optimization of techniques for the maximal detection and quantification of Alzheimer’s-related neuropathology with digital imaging. Neurobiol Aging 23, 161–170, doi: 10.1016/s0197-4580(01)00316-5 (2002). [DOI] [PubMed] [Google Scholar]
- 54.Schindelin J, Arganda-Carreras I, Frise E et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682, doi: 10.1038/nmeth.2019 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Crowe AR & Yue W Semi-quantitative Determination of Protein Expression using Immunohistochemistry Staining and Analysis: An Integrated Protocol. Bio Protoc 9, e3465, doi: 10.21769/BioProtoc.3465 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu S, Dong J, Zhou W et al. A rapid and precise method for quantification of fatty acids in human serum cholesteryl esters by liquid chromatography and tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 960, 222–229, doi: 10.1016/j.jchromb.2014.04.040 (2014). [DOI] [PubMed] [Google Scholar]
- 57.Han CY, Kang I, Harten IA et al. Adipocyte-Derived Versican and Macrophage-Derived Biglycan Control Adipose Tissue Inflammation in Obesity. Cell Reports 31, 107818, doi: 10.1016/j.celrep.2020.107818 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request.