

Abstract
Novel SARS coronavirus or SARS-CoV-2 is a novel coronavirus that was identified and spread from Wuhan in 2019. On January 30th, the World Health Organization declared the coronavirus outbreak as a Global Public Health Emergency. Although Remdesivir and Molnupiravir are FDA-approved drugs for COVID-19, finding new efficient and low-cost antiviral drugs against COVID-19 for applying in more countries can still be helpful. One of the potential sources for finding new and low-cost drugs is the herbal compounds in addition to repurposing FDA-approved drugs. So, in this study, we focused on finding effective drug candidates against COVID-19 based on the computational approaches. As ACE2 serves as a critical receptor for cell entry of this virus. Inhibiting the binding site of SARS-CoV-2 on human ACE2 provides a promising therapeutic approach for developing drugs against SARS-CoV-2. Herein, we applied a bioinformatics approach to identify possible potential inhibitors of SARS-CoV-2. A library of FDA-approved compounds and five natural compounds was screened using Smina docking. Top-docking compounds are then applied in Molecular Dynamics (MD) simulation to assess the stability of ACE2-inhibitor complexes. Results indicate that Luteolin and Chrysin represent high conformation stability with ACE2 during 120 ns of Molecular Dynamics simulation. The binding free energies of Luteolin and Chrysin were calculated by the Molecular Mechanics/Poisson–Boltzmann Surface Area method (MM/PBSA) which confirmed the relative binding free energy of these drugs to ACE2 in favor of the effective binding. So, Luteolin and Chrysin could sufficiently interact with ACE2 and block the Spike binding pocket of ACE2 and can be a potential inhibitor against the binding of SARS-CoV-2 to ACE2 receptor which is an early stage of infection. Luteolin and Chrysin could be suggestive as beneficial compounds for preventing or reducing SARS-CoV-2 transmission and infection which need experimental work to prove.
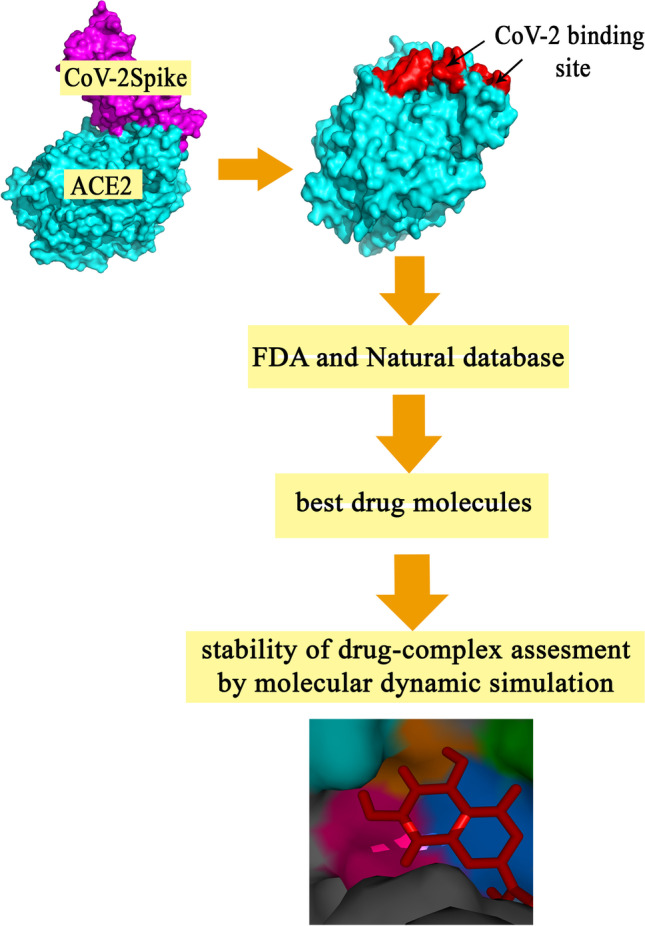
Graphical abstract
Supplementary Information
The online version contains supplementary material available at 10.1007/s00894-022-05059-1.
Keywords: SARS-CoV-2, Spike protein, Molecular Dynamics simulation, Molecular Docking Simulation, Luteolin, Chrysin, Angiotensin-converting enzyme 2
Introduction
SARS-CoV-2 is a coronavirus belonging to the β-genus of the Coronaviridea family [1]. This virus is a single-stranded enveloped RNA virus that shows high identity to SARS-CoV [2]. It affects the human respiratory system causing mild to severe acute respiratory syndrome in addition to other symptoms and signs in other organs [3]. Transmission mode of this virus is mostly by respiratory droplets from animal or human to human [1, 4]. The mortality rate of COVID-19 is said to be 2–5% which could increase to 8% in people aged 70 to 79 years and reach 14.8% in people over 80 years old though unidentified carriers who show no symptom can massively overestimate the mortality rate. These non-diagnosed patients can cause disease in susceptible persons by the transmission of the virus to them. The mortality of this disease has surpassed 5,500,000 which urged scientists to find a suitable modality for treatment or inhibition of this disease [5]. Currently, the only FDA-approved drugs against COVID-19 are Remdesivir and Molnupiravir though some studies indicate that Remdesivir caused no significant change in the reduction of the mortality rate in patients [6–8]. In this way, using a drug discovery policy based on repurposing of the existing drug compounds by computational methods can be an effective track to shorten the time and budget needed for the drug discovery. This drug designing approach has been utilized for many years for different diseases. For example, by repurposing strategy, Favipiravir and Sofosbuvir as influenza and hepatitis C drugs were found to be potentially inhibitory against Ebola and Zika viruses [9].
The structure of coronavirus is composed of two groups of proteins. One group is structural proteins composed of Spike (S), Nucleocapsid (N), Matrix (M), and Envelope (E). Another group is non-structural proteins such as RNA-dependent RNA (RdRp) (nsp12) [10]. Among these proteins, Spike protein is responsible for the specificity of coronavirus facilitating the entrance of this virus to cells [11]. Also, this protein of SARS-CoV-2 represents 10- to 20-fold higher binding affinity to angiotensin-converting enzyme 2 (ACE2) receptor in comparison with SARS-CoV [12]. Spike protein contains two subunits namely S1 and S2. S1 subunit is less conserved than S2 subunit [13] sharing 76% identity to SARS-CoV [3]. This subunit is responsible for the attachment of SARS-CoV-2 to human ACE2 receptor [2] entailing the receptor-binding domain used for detection of SARS-CoV-2 by quantitative RT-PCR [14]. S2 subunit is responsible for the fusion to the cellular membrane, passing the virus to the cells. S1 subunit is composed of two domains, namely SA and SB. Various coronaviruses use distinct domains of the S1 subunit to enter the host cells. SARS-CoVs and several SARS-related coronaviruses (SARSr-CoV) bind to the human ACE2 using the SB domain of S1. Firstly, distal S1 subunit binds to human ACE2 then furin site which is located between S1 and S2 subunit will be cleaved. This interaction triggers changes in the conformation of the S2 subunit activating the fusion of the virus to the cellular membrane. Moreover, S proteins of all coronaviruses have an additional cleavage site called Ś located upstream of the furin site. This site has been presumed to be cleaved by the host receptor changing the conformation of the S2 subunit irreversibly. The probable host protease for this action is Cathepsin L and TMPRSS2 prime [13]. ACE2 receptor exists on ciliated bronchial epithelial cells [4], the heart, kidney, blood vessels, and intestinal cells in the human body [15]. For this reason, failure of these organs has been observed in patients affected with COVID-19 [16]. Moreover, SARS-CoV-2 Spike protein in comparison with the Spike protein of SARS-CoV demonstrates a higher affinity for ACE2 [15] which leads to significantly higher infectivity and transmissibility [17]. Likewise, Zhou and co-workers found that cells that do not express the ACE2 receptor are protected from SARS-CoV-2. The only exception is seen in mice [[14. Sui et al. designed a monoclonal IgG1 antibody against the core region of S protein of SARS-CoVs which could reduce the viral replication up to four times. Data analysis of this antibody demonstrated that this antibody by interacting with S protein fragment (amino acids 324-503) which is responsible for the attachment to the ACE2 receptor-binding domain (residues 318-510) could reduce the viral replication [18]. Although Monteil et al.’s research found out that ACE2-Fc could not completely neutralize coronavirus and its effectiveness is dose-dependent [16], Biagioli et al. found that decreasing the expression of ACE2 by Pelargonidin, a natural flavonoid can act as an agonist to decrease the expression of ACE2 leading to attenuation of SARS-CoV-2 infection [19]. An alternative strategy for controlling this infection is blocking or reducing the accessibility of the ACE2 receptor by targeting the ACE2 binding site for the Spike protein and inhibiting the interaction of ACE2/SARS-CoV-2 by the small molecules [20]. As amino acids of Spike SARS-CoV-2 which play critical roles in binding to human ACE2 were dispersed throughout the Spike surface, finding a specific region for designing small molecules is highly challenging. We focused on targeting the amino acids on the human ACE2 receptor which play critical roles in binding to SARS-CoV-2. This strategy was planned to inhibit the interaction of Spike protein and the ACE2 receptor. This study aims to repurpose FDA-approved and natural compounds against the S1 binding site on the human ACE2 that is responsible for SARS-CoV-2 entrance at the very first step of infection. The whole strategy of this work is depicted in Fig. 1.
Fig. 1.

Schematic view of the whole strategy in this study
Materials and methods
Protein preparation
The structure of human ACE2 was retrieved from protein data bank (www.rcsb.org) with PDB ID: 6VW1 which is a newly released crystal structure of human ACE2 that is complexed with the 2019-nCoV chimeric receptor-binding domain. Protein was prepared for docking using AutoDock Tools 4.2. The ligands, ions, and all the water molecules were removed from the PDB file. Charges and missing hydrogen atoms were added to the human ACE2 receptor in the AutoDock Tools environment and were saved in pdbqt format to be used in the following steps.
Ligand preparation
In total, 1615 FDA-approved drugs were retrieved from PubChem, and then unsuitable, organic polymers, and inorganic compounds were manually excluded. Previous studies demonstrated the antiviral activity of Luteolin, Chrysin, emodin, rhein, and quarcitin against severe acute respiratory syndrome coronavirus (SARS-CoV) by affecting the virus entry to the cells [21, 22]. Therefore, these compounds were added to FDA-approved drug library. Ligand structures were prepared by the AutoDock Tools 4.2. Then, OpenBabel (version 2.4.1) was used to convert SDF to PDB format. Nonpolar hydrogen bonds were merged, Gasteiger-Marsili charges were assigned, atoms were adjusted to the AutoDock atom types, and the rotatable bonds were assigned and saved in pdbqt format. Finally, compounds were converted to Structure Data Format (SDF) using an Open Babel software for further docking.
Binding pocket selection and generation of grid box for docking studies
The binding site of the human ACE2 receptor which is interacting with the 2019-nCoV chimeric receptor-binding domain was analyzed by LigPlot software. Moreover, the hot spot amino acids of this region were illustrated by the Robetta alanine scanning server (http://old.robetta.org/). The Robetta alanine scanning server scans at the first finding interface residues on both proteins and starts to substitute each of the residues with alanine and then measures the free binding energy of the complex compared to the native complex. If the difference between free binding energy was more than 2 kcal mol−1 that amino acid position was identified as the hot spot residue. Summing up these results was used to locate the binding pockets for virtual screening. After marking the critical amino acids, a 20 × 18 × 22 Å (x, y, and z) grid box with 1-Å grid spacing was selected by using AutoDock Tools 4.2.
Virtual screening and Molecular Docking studies
A structure-based virtual screening approach was applied to find potential compounds for inhibiting angiotensin-converting enzyme 2 (ACE2). FDA-approved libraries were docked in the selected binding pocket using Smina software (http://smina.sf.net) [23]. Smina is a fork of AutoDock Vina focused on the improvement of scoring and energy minimization. The best Smina docking result was ranked based on the binding affinity which is computed according to Koes et al. [23].
Molecular Dynamics simulation (MD)
Molecular Dynamics (MD) technique was performed using GROMACS version 5 [24]. As GROMACS software could not prepare the topology file and its parameters for ligands compounds, the PRODRG server was used to retrieve these files for each ligand [25]. The topology parameters for the protein structure were prepared by the Gromacs program. Thereafter, protein-ligand complexes were immersed in a simulation box filled with SPC (simple point charge) water molecules [26]. To give neutral conditions in terms of electrical charge, a suitable number of ions were added to the simulation box. The total charge of the system was neutralized by adding an appropriate number of Na and Cl ions instead of solvent molecules in the simulation box. Three-dimensional periodic boundary conditions were applied to the system. For all MD simulations, the energy minimization process was comprised of two parts: firstly, systems were balanced at 300 K for 100 ps using NVT (constant number of particles, volume, and temperature) and secondly, 1000 ps NPT (constant number of particles, pressure, and temperature) equilibration of the system was performed using Parrinello–Rahman barostat to obtain constant temperature and pressure in 300 K and 1.0 bar. The long-range electrostatics was treated with particle mesh Ewald (PME) [27] algorithm by 10 Å cutoff distance. Van der Waals (VDW) interactions were computed with a 1-nm cutoff. The LINear Constraint Solver (LINCS) algorithm was used to constrain the length of the covalent bonds [28]. Finally, for all MD simulations, the first 10 ns was taken as equilibrium time leading to 120 ns with 2-fs time steps.
Calculating the binding free energy
The relative binding free energy in the protein-ligand complex was calculated using the Molecular Mechanics/Poisson–Boltzmann Surface Area (MM/PBSA) method developed by Rashmi Kumara [29]. MM/PBASA analyzes different interaction energies such as electrostatic interactions, van der Waals interactions, polar solvation energy, and nonpolar solvation. For each protein-ligand complex, the binding free energy (ΔG) was calculated from the last 20 ns of simulation trajectories. The MM-PBSA for contributing residues provides the critical amino acid which can help to design the inhibitors.
Results
The human ACE2 binding site region of SARS-CoV-2 is similar to SARS-CoV
To determine the key residues involved in the binding regions, the crystal structure of the 2019-nCoV chimeric receptor binding domain with human ACE2 complex (PDB ID: 6VW1) was analyzed with LigPlot and Robetta webservers. The result demonstrated that Ser19, Gln24, Thr27, Phe28, His34, Glu35, Asp38, Tyr41, Gln42, Leu45, Tyr83, Asn330, Lys353, Gly354, Asp355, and Arg357 were key residues having a critical role in the binding regions. These residues were used for Molecular Docking. Also, comparing the PDB structures of SARS-CoV with SARS-CoV-2 during the interaction with human ACE2 indicated that SARS-CoV-2 had relatively similar interaction binding sites to SARS-CoV-2 except for Ser19, Leu45, Leu79, Gln325, Gly326, and Glu329 which are not involved in the interaction in SARS-CoV-2. Interestingly, according to these outcomes, we found that SARS-CoV-2 displayed a higher affinity for interaction with Lys353, Gly354, Asp355, and Arg357 which are less favored in SARS-CoV. Data is shown in Fig. 2. In addition, the key residues of the RBD domain of Spike protein of SARS-CoV-2 are given from PDB code 6VW1 too. Data showed that Gln498, Tyr505, Gly502, Asn501, Gly496, Ala475, Phe456, Thr500, Leu455, Tyr489, Tyr453, Asn487, Phe486, Tyr449, and Gln493 play a critical role in interaction with human ACE2. So, these amino acids were chosen for Molecular Docking. Moreover, the interacting residues of ACE2-SARS-CoV-2 variant omicron (PDB ID:7t9k) revealed that amino acids Phe456, Tyr449, Ser496, Arg498, Tyr453, Ser494, The500, Arg493, Gly476, Tyr489, Ala475, and Phe486 of the Spike protein are in interaction with the amino acids Thr27, Asp38, Gln42, Asn330, His34, Arg357, Tyr41, Gln35, Gly354, Lys353, Gln24, Lys31, Tyr83, Met82, Phe28, Asp355, and Leu79 in ACE2 which except Ser19 and Leu45, all amino acids are in common with the Wuhan SARS-CoV-2 while the interacting amino acids of Spike changed between these two variants (Table S1).
Fig. 2.

Human ACE2 sequence. Amino acids of ACE2 which interact with the Spike protein of SARS-CoV and SARS-CoV-2 are shown with asterisks. Amino acids which play a role in interaction with ACE2 in SARS-CoVs are Ser19(S), Gln24(Q), Thr27(T), Phe28(F), Asp30(D), Lys31(K), His34(H), Glu37(E), Asp38(D), Tyr 41(Y), Gln42(Q), Leu45(L), Leu79(L), Met82(M), Tyr83(Y), Gln325(Q), Gly326(G), Glu329(E), Asn330(N), Lys353(K), Gly354(G), Asp355(D), and Arg357(R)
The best drug candidates were selected according to their binding affinity and molecular interaction with ACE2
The top 20 drug candidates were selected based on the lowest docking score which indicated higher binding affinity and targeting more residues on the human ACE2 capable of interaction with SARS-CoV-2 (Table 1). Minimized affinity illustrated that Ursodeoxycholic acid showed the best binding affinity of −8.7 and Rhein had the worst binding affinity of −6.8 among these drug candidates. The schematic docking results are given in Fig. 3.
Table 1.
The top 20 drug candidates from Smina docking output against human ACE2. Drugs were ranked from the lowest to the highest docking scores. The highest and lowest docking scores of human ACE2 belong to Ursodeoxycholic acid and Rhein drugs, respectively. The docking result of these compounds against the Spike protein of SARS-CoV-2 indicates a moderate binding affinity of them
| Number | Name | Docking score (kcal mol−1) | |
|---|---|---|---|
| ACE2 | Spike | ||
| 1 | Ursodeoxycholic acid | −8.74 | −6.83 |
| 2 | Pimozide | −8.52 | −6.58 |
| 3 | Indacaterol | −8.14 | −7.46 |
| 4 | Rofecoxib | −8.05 | −6.52 |
| 5 | Olsalazine | −8.05 | −6.77 |
| 6 | Meclizine | −7.92 | −6.89 |
| 7 | Methylergometrine | −7.85 | −6.6 |
| 8 | Labetalol | −7.77 | −6.85 |
| 9 | Lumacaftor | −7.74 | −8.76 |
| 10 | Luteolin | −7.77 | −6.99 |
| 11 | Granisetron | −7.69 | −6.36 |
| 13 | Levothyroxine | 7.57 | −6.01 |
| 14 | Ketoprofen | 7.57 | −7.09 |
| 15 | Cilostazol | −7.52 | −6.27 |
| 16 | Folic acid | −7.18 | −6.80 |
| 17 | Chrysin | −7.12 | −6.48 |
| 18 | Triazolam | −7.08 | −6.99 |
| 19 | Triamterene | −7.07 | −6.57 |
| 20 | Rhein | −6.88 | −6.25 |
Fig. 3.

Docking result of the candidate drugs against the SAR-CoV-2 binding site on ACE2. Results indicate that all candidate drugs could interact effectively with the ACE2. The minimized affinity of Smina docking results in Luteolin, Chrysin, Pimozide, and Ursodeoxycholic acids is −7.7, −7.1, −8.5, and −8.7, respectively
Also, this library was docked against the binding site of the RBD domain of the Spike protein from SARS-CoV-2. Top twenty compounds which showed the highest binding affinities are mentioned in Table S2. The highest binding affinity belonged to Porfimer and the lowest binding affinity belonged to Imatinib. Moreover, results indicated that the top 20 compounds which had the highest binding affinity to the human ACE2 protein have moderate binding affinity to the Spike protein of SARS-CoV-2 (Table 1). The tertiary structure of the docking result between the RBD domain of the Spike protein and the selected compounds is given in Fig. S1. These compounds can potentially interact with the ACE2 too.
Drug candidates which made stable complex with human ACE2 during simulation were selected
To study the stability of 20 drug candidates during interaction with ACE2, 120 ns of Molecular Dynamics simulation was applied. Luteolin-ACE2 and Chrysin-ACE2 are indicated as the best drug candidates which still effectively interact with ACE2 and reach steady-state in 120 ns (Fig. 4). On the other hand, Pimozide-ACE2 and Ursodeoxycholic acid-ACE2 in complex with ACE2 were just stable until 100 ns of simulation.
Fig. 4.

Molecular Dynamics simulation analysis of Luteolin and Chrysin throughout the interaction with ACE2 receptor during 120 ns of the simulation. A RMSD plots represent that Luteolin-ACE2 and Chrysin-ACE2 reach a steady-state during 120 ns of simulation with the RMSD around 0.7 nm. Free ACE2 is defined as the receptor in the RMSD plot. B RMSD plots of Luteolin and Chrysin during 120 ns of simulation. C Rg plot indicates that Luteolin-ACE2 and Chrysin-ACE2 have the Rg values of around 2.1 which is less than the Rg value of 2.4 for the free form of ACE2 (defined as the receptor in the plot). D SASA plot presents that Luteolin-ACE2 and Chrysin-ACE2 have similar SASA values which are less than the free form of the ACE2. E Hydrogen bond plot figures out that Luteolin-ACE2 has a higher number of hydrogen bonds than Chrysin. F RMSF plot demonstrating that Luteolin has higher RMSF fluctuation than Chrysin and the free form of ACE2 (defined as the receptor in the plot)
Root mean square deviation (RMSD) plot of Luteolin-ACE2 and Chrysin-ACE2 complexes showed that they reach steady-state around 80 ns of simulation (Fig. 4A). The RMSD plot of Luteolin and Chrysin during 120 ns of simulation represented that their RMSD values are 0.7 nm which is below 1nm implying low fluctuation during the simulation. The result demonstrated that the RMSD values of Luteolin-ACE2 and Chrysin-ACE2 show lower fluctuation compared to the free ACE2 which is depicted as the receptor in Fig. 4. This means that none of these drugs caused any drastic changes in the ACE2 structure through their attachment. The RMSD plot of Luteolin and Chrysin during 120 ns of the simulation represented that Chrysin showed stable fluctuation during 120 ns of simulation. Although Luteolin showed higher fluctuation during simulation, the rate of its fluctuation was very low (< 0.15) which was indicative of no drastic change in its structure during simulation (Fig. 4B). Rg value represents the compactness and relaxedness of the complex structure. Output revealed that Luteolin and Chrysin after attachment to the ACE2 reduced the Rg value of ACE2 from 2.4 to 2.1 nm. Therefore, it seems that Luteolin and Chrysin induce the relaxedness and compactness of the ACE2 structure (Fig. 4C). Both Luteolin-ACE2 and Chrysin-ACE2 complexes had similar SASA values which were lower than free ACE2. This demonstrated that these two drugs caused positional changes in the ACE2 surface residues which could result in lower availability of the critical amino acids on the ACE2 receptor for the attachment to SARS-CoV-2 Spike protein (Fig. 4D). These results are compatible with the secondary structure analysis of free ACE2 compared to Luteolin and Chrysin during 120 ns of simulation which showed a higher number of alpha-helix structures in ACE2 complex during interaction with Luteolin and Chrysin than the free form of ACE2. These alpha helixes were especially in residues number of 300–350 (Fig. 5). This result is compatible with the structural alignment of the free ACE2 and ACE2 during interaction with Luteolin and Chrysin after 120 ns of a simulation showing lower Rg values for ACE2 due to interacting with Luteolin and Chrysin. No severe change in ACE2 was observed after interaction with Luteolin and Chrysin (Fig. S2). Also, analysis of the Rg plot of RBD binding interface of ACE2 during interaction with Luteolin and Chrysin with the PDB files of 1R4L and 1R42 as the close and open forms represented that the Rg value of ACE2 during interaction with Luteolin and Chrysin was lower than 1R4L and 1R42 though Rg value of 1R42 was much lower and near to the Rg value of Luteolin-ACE2 and Chrysin-ACE2 at the end of the simulation (Fig. S3). This result showed that the interaction of Luteolin and Chrysin does not modify the RBD binding site region of the ACE2 which is responsible for the stability of ACE2 structure and causes side effects.
Fig. 5.

Secondary structure analysis plot during 120 ns of the simulation. A The free form of the ACE2. B Luteolin-ACE2 complex. C Chrysin-ACE2 complex. D Pimozide-ACE2 complex. E Ursodeoxycholic acid-ACE2 complex. Data showed all compounds caused no changes in the ACE2 structure during the simulation and no drastic change in the secondary structure was observed during the simulation except for Pimozide which increases the alpha-helix structure around 200-250 region compared to free ACE2. Luteolin and Chrysin caused more stable structures
The hydrogen bond plot demonstrates that Luteolin-ACE2 has more hydrogen bonds compared to the Chrysin-ACE2 (Fig. 4E). Also, both drugs showed higher hydrogen bond numbers at the end of the simulation. To study fluctuations of each ACE2 residue during simulation, the RMSF (root mean square fluctuation) plot was analyzed. Results demonstrated that Luteolin-ACE2 showed higher RMSF fluctuations than Chrysin-ACE2 but its RMSF value was below 1 nm which indicates that this drug did not cause huge structural changes in the ACE2 (Fig. 4F).
Also, the Molecular Dynamics simulation of the Luteolin and Chrysin complex with the Spike protein of SARS-CoV-2 during 120 ns of the simulation indicated that these drugs could make stable interaction with this protein during 120 ns of the simulation (Fig. 6). The RMSD plot represented that both small molecules reach steady-state around 80–90 ns of the simulation with the RMSD value less than 0.5 nm which indicated stable interaction of these molecules with the Spike protein. Luteolin has less RMSD value (0.25 nm) than Chrysin (0.4 nm) which means higher stability of Luteolin in the interaction with the Spike protein in comparison with Chrysin (Fig. 6A). The RMSD plot of Luteolin and Chrysin indicated that Chrysin had less fluctuation during simulation than Luteolin (Fig. 6B). Rg plot represented both drug candidates have less Rg value than the free Spike protein. Also, Chrysin was more stable and showed a lower Rg value than Luteolin during simulation (Fig. 6C). This result showed that the Spike protein had a more relaxed and compact structure in the interaction with Luteolin-Spike and Chrysin-Spike than its free form. SASA plot revealed that both drug candidates had less SASA value than the free form of the Spike protein which demonstrated that due to the interaction of the drugs candidates with the Spike protein, surface residues of the Spike protein were not accessible to solvent (Fig. 6D). The hydrogen bond plot revealed that Luteolin had more hydrogen bonds than Chrysin which means it made a more stable interaction with the Spike protein (Fig. 6E). RMSF plot analysis disclosed that both drug candidates had low fluctuation values (less than 0.5 nm) while Chrysin showed higher fluctuation in 440-440 and 495-505 residues in comparison with Luteolin (Fig. 6F).
Fig. 6.

Molecular Dynamics simulation analysis of Luteolin and Chrysin throughout the interaction with the Spike protein during 120 ns of the simulation. A RMSD plots represent that Luteolin-Spike reaches a steady-state during 90 ns with RMSD value around 0.2 nm, Chrysin-Spike reaches a steady-state during 80 ns of simulation with RMSD around 0.4 nm. B RMSD plots of Luteolin and Chrysin during 120 ns of simulation. C Rg plot indicates that Luteolin-Spike and Chrysin-Spike have an Rg value of around 1.8 which is less than the Rg value of Luteolin-Spike and free Spike. D SASA plot presents that Luteolin-Spike and Chrysin-Spike have less SASA values than the free Spike. E Hydrogen bond plots figure out that Luteolin-Spike has a higher number of hydrogen bonds than Chrysin. F RMSF plot demonstrating that Chrysin has higher RMSF fluctuation than Luteolin and free Spike, especially in regions 440-440 and 495-505
The result of the Molecular Dynamics simulation for Pimozide-ACE2 and Ursodeoxycholic acid-ACE2 represented that both drugs were stable around 120 ns of simulation. Although a slight fluctuation was observed for both drugs (Fig. 7A), the RMSD values were 0.7 nm which was similar to the RMSD value of the free form of the ACE2. Also, the RMSD plot of just Pimozide and Ursodeoxycholic acid figured out that both small molecules were steady-state during simulation (Fig. 7) though Pimozide showed a more stable structure than Ursodeoxycholic acid. The Rg plot analysis represents that both drugs had lower Rg values than the free form of the ACE2 which means that both drugs after binding to the ACE2 made this structure compact and relaxed. Although free ACE2 showed higher fluctuation in the Rg plot, secondary structure analysis of the free and the complex form of ACE2 revealed that no structure change occurred during simulation (Fig. 5). Moreover, Ursodeoxycholic acid-ACE2 showed higher Rg fluctuation than Pimozide-ACE2 which indicates that this drug caused more structural changes in ACE2 than Pimozide (Fig. 7C). The Rg plot analysis of the RBD binding site interface of ACE2 illustrated that due to the binding of Ursodeoxycholic acid and Pimozide to ACE2, these regions became more stable than 1R4L as an open structure of the free form of ACE2 (Fig. 3S). This means that like Rg plot and secondary structure prediction, attachment of these compounds does not make the binding site structure of ACE2 unstable which might trigger side effects. SASA value indicates that both drugs have less SASA value than the free form of ACE2 (Fig. 7D). This could be due to the occupation of the surface area of ACE2 by the small molecules. Hydrogen bonds of both drugs illustrate that both drugs have the same numbers of hydrogen bonds (Fig. 7E). RMSF plot represents that Ursodeoxycholic acid-ACE2 shows higher fluctuation than the free form of ACE2. In contrast, Pimozide-ACE2 has similar fluctuation to the free form of ACE2. This output means that Pimozide makes fewer structural changes in the ACE2 than Ursodeoxycholic acid (Fig. 7F).
Fig. 7.

Molecular dynamics simulation of Pimozide and Ursodeoxycholic acid throughout the interaction with the ACE2 receptor during 120 ns of the simulation. A RMSD plot of Pimozide and Ursodeoxycholic acid shows that both drugs have the same RMSD value to the free form of ACE2. Besides, both drugs represent a slight fluctuation around 100 ns of the simulation. B RMSD plot of Pimozide and Ursodeoxycholic acid during simulation. Data shows that Ursodeoxycholic acid has less RMSD value than Pimozide. C Rg plot of Pimozide and Ursodeoxycholic acid showing lower Rg value than the free form of ACE2 which indicates both drugs caused a more relax and compact structure compared to the free form of ACE2. D SASA plot of Pimozide and Ursodeoxycholic acid illustrates that both drugs make surface structure changes in ACE2 compared to the free form of ACE2. E Hydrogen bond plot of Pimozide and Ursodeoxycholic acid shows that both drugs have the same hydrogen bond. F RMSF plot of Pimozide and Ursodeoxycholic acid indicates that Ursodeoxycholic acid has more fluctuation than Pimozide and the free form of ACE2. Also, Pimozide shows the same fluctuation compared to the free form of ACE2
Molecular Dynamics simulation of Ursodeoxycholic acid and Pimozide in the interaction with the Spike protein during 120 ns is represented in Fig. 8. RMSD plot indicated that the free Spike protein has less fluctuation than the Pimozide-Spike and the Ursodeoxycholic acid-Spike complexes while Ursodeoxycholic acid-Spike showed more stability than the Pimozide-Spike complex (Fig. 8A). The RMSD plot of drugs indicated that Ursodeoxycholic acid-Spike protein was stable showing less RMSD value (0.1 nm) than Pimozide (Fig. 8B). The Rg plot indicated that both Ursodeoxycholic acid-Spike and Pimozide-Spike had lower Rg values (around 1.75 to 1.8) but this value was not significantly different from the Rg value of the free form of the Spike protein (1.85). Also, free Spike protein showed less fluctuation during the simulation in comparison with the Ursodeoxycholic acid-Spike and Pimozide-Spike complexes (Fig. 8C). SASA plot represented that both Ursodeoxycholic acid-Spike and Pimozide-Spike had less fluctuation than the free form of the Spike protein (Fig. 8D). The hydrogen bond plot showed that Ursodeoxycholic acid made more hydrogen bonds with the Spike protein than Pimozide (Fig. 8E). RMSF plot analysis revealed that Ursodeoxycholic acid-Spike and Pimozide-Spike had higher fluctuation than the free form of the Spike protein especially in 365-375 regions of the Spike while in 495-505 and 515-530 regions of the Spike, Ursodeoxycholic acid showed higher fluctuation (Fig. 8F).
Fig. 8.

Molecular Dynamics simulation analysis of Pimozide and Ursodeoxycholic acid throughout the interaction with the Spike protein during 120 ns of simulation. A RMSD plots represent that the Pimozide-Spike and Ursodeoxycholic acid-Spike complexes have higher RMSD values than the free form of the Spike. B RMSD plots of Pimozide and Ursodeoxycholic acid during 120 ns of the simulation. Data show that Ursodeoxycholic acid has lower fluctuation with an RMSD value of 0.1 nm than Pimozide. C Rg plot indicates that Pimozide-Spike and Ursodeoxycholic acid-Spike have an Rg value less than the free form of the Spike. D SASA plot presents that Ursodeoxycholic acid-Spike and Pimozide-Spike have lower SASA values than the free form of the spike. E Hydrogen bond plot figures out that Ursodeoxycholic acid has a higher number of hydrogen bonds than Pimozide. F RMSF plot demonstrates that Ursodeoxycholic acid and Pimozide have higher RMSF fluctuation than the free form of the Spike
After simulation, drug candidates can tightly interact with ACE2 through the critical amino acids
The PDB files of each small molecule and ACE2 complex were extracted from the steady-state simulation and analysis in-depth with the LigPlot software [30]. The drugs’ binding affinity for the ACE2 was computed by the Prodigy web server [31]. Results are presented in Table 2 while the tertiary structures of drugs-ACE2 complexes are shown in Fig. 9. It became clear that Luteolin and Chrysin showed approximately similar binding affinity forming tight interaction with the human ACE2 (Fig. 9B, C). The LigPlot analysis and binding affinity assessment of these drugs are given in Table S3 and their tertiary structures are represented in Fig. 9C and D.
Table 2.
Amino acid analysis of the candidate compounds against SARS-CoV-2-binding site on the human ACE2. The bold amino acids are critical amino acids on ACE2 binding according to the crystal structure analysis of the SARS-CoV-2 complex with the ACE2
| Small molecule drug name | Binding residues on human ACE2 | KD (∆Gnonelec(kcal mol−1) | |
|---|---|---|---|
| Non-hydrogen bond | Hydrogen bond | ||
| Luteolin | Phe40, Trp349, Asp350, Gly352, Lys353, Phe389, Phe390, Arg393 | Glu37, Gly354, Asp382, Leu391 | −8.8 |
| Chrysin | Phe40, Ala99, Trp69, Leu100, Asn103, Phe390, | Asp350, Gly352, Arg393 | −8.4 |
Fig. 9.

Tertiary structure analysis of drug candidates—ACE2 after MD simulation. A Tertiary structure of the free form of the ACE2. The binding site of ACE2 is depicted within the circle. B The tertiary structure of the Luteolin in complex with the ACE2. C Tertiary structure of the Chrysin in the complex with ACE2. D The tertiary structure of Ursodeoxycholic acid in complex with ACE2. E The tertiary structure of Pimozide in complex with ACE2. As shown, Luteolin and Chrysin drug candidates interact with the ACE2 binding pocket. Also, Ursodeoxycholic acid and Pimozide drug candidates still interact with the ACE2 binding pocket. The structures were created by PyMoL software
The Luteolin and Chrysin interact with ACE2 by van der Waals and electrostatic interaction
MM/PBSA methodology was incorporated with MD simulation to compute the binding free energy of protein-ligand complexes. The calculated DG binding energies for Luteolin and Chrysin with the ACE2 protein were found to be −83.410 and −118.524, respectively. It was found that Chrysin bound more tightly to the ACE2 compared to Luteolin. Total free energy can be decomposed to van der Waals (DGvdw), electrostatic (DGele), polar solvation energy (DGpol), and nonpolar interactions (DGnonpol). Electrostatic (DGele), nonpolar interactions (DGnonpol), and van der Waals interactions (DGvdw) contributed positively to the binding process (Table 3). The polar component of solvation (DG polar) contributed negatively to the binding process. Particularly, van der Waals interactions play an important role in the attachment to the ACE2 receptor. Due to the exploration of the key residues involved in protein-ligand interaction, the individual residue energy contribution was calculated by the MM/PBSA method. Luteolin showed the highest contribution with Asp38, Phe40, Trp349, Asp350, Leu351, Gly352, Asp355, Tyr385, Ala386, Gln388, Pro389, Phe390, and Leu391 with the binding energies ranging from −1.0923 to −9.4637 kcal mol−1. In Chrysin, Glu37, Asp38, Phe40, Trp69, Ala99, Leu100, Asn103, Asp355, Asp382, Ala386, Phe390, and Leu391 residues contributed mostly in the protein-ligand interaction with the binding energies ranging from −1.0669 to −12.1387 kcal mol−1. This MM/PBSA analysis confirmed Luteolin and Chrysin can affect the critical amino acids in the ACE2. A graphical representation of each residue in the binding energy is shown in Fig. 10A. Other drug candidates have low binding energies which are mentioned in Table S4. Also, the role of each amino acid residue in the binding energy of Pimozide and Ursodeoxycholic acid is represented in Fig. 10B.
Table 3.
Contribution of each energy component in the binding of Luteolin and Chrysin to the ACE2 in a simulated system
| Compound | ∆Evdw | ∆Eele | ∆Gpol | ∆Gnonpo | ∆Gbind |
|---|---|---|---|---|---|
| Luteolin | −114.89 ± 16.850 | −1.15 ± 3.979 | 40.52 ± 41.893 | −7.89 ± 8.483 | −83.41 ± 16.085 |
| Chrysin | −159.14 ± 14.302 | −40.23 ± 25.900 | 94.40 ± 25.894 | −13.54 ± 0.788 | −118.52 ± 18.415 |
Fig. 10.

Binding free energy of critical amino acids in ACE2 binding site computed with MMPBSA. A Luteolin and Chrysin. B Pimozide and Ursodeoxycholic acid; result indicates that Ursodeoxycholic acid has near-zero binding free energy value compared to Pimozide
MM/PBSA analysis of Luteolin and Chrysin to the Spike protein showed that the binding affinities of these compounds to the Spike were lower than ACE2 (Table S5). MM/PBSA analysis of Pimozide and Ursodeoxycholic acid represented that Pimozide had a higher number of van der Waals, electrostatic, and nonpolar interactions than Ursodeoxycholic acid but the binding energy of Ursodeoxycholic acid was more than Pimozide (Table S6).
Discussion
ACE2 is a critical receptor for SARS-CoVs interacting with the Spike protein of these viruses. The attachment of the Spike protein to ACE2 plays an important role in the infection and replication of the virus. The higher affinity of the Spike protein for the human ACE2 correlates with the severity and contagiousness of the disease. Although SARS-CoV-2 is similar to the SARS-CoV, it interacts with the ACE2 more efficiently. So higher affinity of SARS-CoV-2 Spike protein for the ACE2 could be responsible for a higher human-human transmission of this virus. On the other hand, amino acid analysis between Wuhan SARS-CoV-2 and SARS-CoV-2 variant omicron revealed that despite differences in the RBD amino acids between these two variants, they both interact with the same amino acids on the ACE2 receptor. Because of that, inhibiting the attachment of SARS-CoV-2 to the ACE2 receptor could effectively prevent the spreading of current and possibly future variants of this virus.
According to Li et al.’s findings, fourteen key residues of SARS-CoV are responsible for the binding to the human ACE2. Sequence alignment of SARS-CoV and SARS-CoV-2 Spike proteins by the Clustal omega software revealed that eight of these amino acids are identical between SARS-CoV and SARS-CoV-2 while six of them are semi identical. These semi-conserved residues are related to the higher transmissibility of SARS-CoV-2. Among these six amino acids, Asn479 and Thr487 are critical for the binding of SARS-CoV to human ACE2 affecting the binding affinity. These two amino acids are replaced with Gln493 and Asn501 in SARS-CoV-2. So, presumably, they could provoke higher human-human transmissibility [32]. These data are presented in Table 4 too. Analysis of the crystal structure of SARS-CoV demonstrates that Asn479 interacts with His34 while Thr487 interacts with Tyr41, Lys353, and Asp355 on the ACE2 receptor. Data analysis of the crystal structure of SARS-CoV-2 elucidated that Gln493 interacts with Glu35 and Lys31 on the human ACE2. Also, Asn501 of the SARS-CoV-2 interacts with Tyr41 of the human ACE2.
Table 4.
Fourteen key amino acids of SARS-CoV and SARS-CoV-2 playing roles in the interaction with the human ACE2. Amino acids in italics are identical between SARS-CoV and SARS-CoV-2. Asn479 and Thr487 amino acids which are marked with an asterisk are critical in the interaction of SARS-CoV and ACE2 which are replaced by Gln493 and Asn501 in SARS-Co-V-2, respectively
| Type of virus | Amino acid number | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SARS-CoV | T402 | R426 | Y436 | Y440 | Y442 | L472 | N473 | Y475 | N479* | Y484 | T486 | T487* | G488 | Y491 |
| SARS-CoV-2 | T415 | N439 | Y451 | Y453 | L455 | F486 | N487 | Y489 | Q493 | Q498 | T500 | N501 | G502 | Y505 |
Data analysis of the crystal structures of SARS-CoV and SARS-CoV-2 Spike proteins presented that both viruses have a similar binding site which interacts with Ser19, Gln 24, Thr27, Phe28, Asp30, Lys31, Hi34, Glu37, Asp38, Tyr41, Gln42, Leu45, Leu79, Met82, Tyr83, Gln325, Gly326, Glu329, Asn330, Lys353, Gly354, Asp355, and Arg357 on the human ACE2.
In addition to the crystal structure analysis, according to Li et al.’s study, Lys31, Tyr41, Met82, Tyr83, Pro84, Lys353, Asp355, and Arg357 on the human ACE2 are critical for the attachment of the S1 domain of Spike protein on SARS-CoV. Moreover, replacing the amino acids 30-40 and 90-93 of human ACE2 with the corresponding amino acids in civet ACE2 increases the binding affinity of SARS-CoV to ACE2 indicating that these regions influence the binding affinity of the Spike protein to the human ACE2 [33]. Also, Qiu et al. found that Thr20, Lys31, Tyr41, Lys68, Tyr83, Lys353, Asp355, Arg357, and Met383 in the ACE2 are critical for the interaction of SARS-CoV-2 with the ACE2 [34]. Likewise, Hussain and co-workers found that each amino acid substitution at positions Thr27, Glu35, Glu37, Met82, Glu329, and Met383 could disturb the interaction of Gln42 in the ACE2 with Tyr449 in SARS-CoV-2. Also, Lys31, Lys353, and Pro389 in the ACE2 play a critical role in SARS-CoV-2 interaction [35].
In this work, we attempted to find FDA-approved drug or natural compounds against the ACE2 binding site for inhibiting the interaction between the SARS-CoV-2 Spike protein and the human ACE2 receptor. Based on this approach, FDA-approved and natural drug databases were used for virtual screening against the ACE binding site. It was revealed that two candidates from the FDA-approved database (Pimozide and Ursodeoxycholic acid) and two natural compounds from natural resources (Luteolin and Chrysin) could effectively bind to the human ACE2 binding site, but just Luteolin and Chrysin showed stability during binding to the ACE2 throughout 120 ns of the Molecular Dynamics simulation. In addition, docking the current libraries against the Spike protein showed that all these drug candidates could interact with the Spike protein. This data is compatible with Guler et al.’s work which found Chrysin could interact with both human ACE2 and RBD region of SARS-CoV-2 [36]. Also, according to this study, Chrysin has a higher docking score to ACE2 (−8.48 kcal mol−1) than the RBD region of SARS-CoV-2 (−7.48 kcal mol−1) which is compatible with our data that found Chrysin has a better docking score to ACE2 (−7.12 kcal mol−1) than the RBD region of SARS-CoV-2 (−6.48 kcal mol−1). Moreover, Carino et al. found that though Ursodeoxycholic acid could interact with the RBD region of SARS-CoV-2, this interaction was not stable [37] which is in accordance with our study which found that Ursodeoxycholic acid could not make stable interaction with the Spike protein of SARS-CoV-2.
Luteolin (3,4,5,7-tetrahydroxy flavone) is a naturally common flavone; dietary sources such as carrot, cabbage, artichoke, tea, celery, and apple have a great amount of Luteolin. Luteolin possesses antioxidant, anticancer, anti-apoptotic, anti-inflammatory, neuroprotective, and cardioprotective effects [38]. Luteolin has also been extensively investigated due to its antiviral effect against Japanese encephalitis virus (JEV) [39], coxsackie virus B3 (CVB3) [32], Epstein–Barr virus (EBV) [32], and HIV-1 [32]. Luteolin has antiviral activity against influenza type A by affecting the endocytic pathway [40]. Also, Luteolin could act as a general antiviral compound by inhibiting alpha glycosidase activity which is essential for the viral envelope glycoproteins [41]. In influenza type A similar to SARS-CoV-2, ACE2 plays an important role in severe acute lung injury [42]. Docking of Luteolin illustrates that this drug can interact with Glu37, Lys353, Ala386, Met383, and Phe356 on the ACE2 receptor while the critical SAR-CoV-2-ACE2 interaction by hydrogen bonds is formed by three of them (Glu37, Lys353, and Met383). Molecular Dynamics simulation illustrates that the Luteolin-ACE2 complex is stable during simulation forming a compact structure with the ACE2. Moreover, Luteolin does not induce a conformational change in the ACE2 receptor. This result is compatible with Li et al.’s findings which observed that no large conformational change happened after interaction of SAR-CoV binding region domain and ACE2 [33]. Also, this drug creates the highest number of hydrogen bonds (4.3 hydrogen bonds) with the ACE2 receptor which would be increased at the end of the simulation. The LigPlot analysis of the Luteolin-ACE2 complex after simulation reveals that this drug interacts with Glu37, Asp382, Leu391, and Gly354 residues on the ACE2 receptor by the hydrogen bonds which are the most favorable interaction bond. Moreover, Luteolin could interact with ACE2 by Phe40, Trp349, Asp350, Gly352, Lys353, Phe389, Phe390, Leu391, and Arg393 residues with the hydrophobic bonds. Based on the crystal structure analysis of ACE2 in the interaction with SARS-CoVs, Glu37, Lys353, and Gly354 are critical residues on the ACE2 for binding to SARS-CoVs. Hence, interaction with these amino acids especially Gly37 and Lys353 that interact with ACE2 by the hydrogen bonds can effectively block ACE2. This data is in accordance with Choudhary et al.’s results which found that eptifibatide acetate, TNP, GNF5, GR 127935, hydrochloride hydrate, and RS504393 drugs could bind to Glu37, Lys353, Asp382, Phe390, Leu391, Gly354, Trp349, ASP350, and Gly352 on the ACE2 [43] but in contrast with data of Cheng et al. which reported the docking result of flavonoid in citrus fruits against ACE2. These compounds can bind to Tyr515, Glu402, Glu398, Asn394, Pro146, Leu143, Lys131, Asn277, Arg273, His505, Lys562, Glu564, Gly205, Trp349, Ala348, and Trp69 on the ACE2 which are different from the Luteolin binding site on the ACE2 except for Leu351 and Asp350 [44] and Joshi et al. virtual screening of phytochemical compounds against ACE2. These compounds can interact with Tyr515, Arg273, Thr347, Phe504, His505, His345, Glu375, Tyr510, Glu402, His378, Pro346, His374, Asn149, Glu145, Lys353, Ser409, Phe274, Glu406, Thr276, Thr371, Asp367, Thr445, Arg518, Tyr515, Leu370, Cys344, and Asp368 [45].
Chrysin (5,7-dihydroxy-2-phenyl-4H-Chromen-4-one) is a natural flavonoid. Chrysin is abundantly found in honey and many plants such as propolis, blue passionflower (Passiflora caerulea), Radix Scutellariae, Cytisus multiflorus, Scutellaria immaculata [46], Desmos cochinchinensis [47], Pelargonium crispum [48], Oroxylum indicum [49], and even mushrooms [50]. Chrysin is an aromatase inhibitor showing antioxidant, anti-inflammatory, anticancer, and antiviral characteristics [51, 52]. Docking output analysis of Chrysin revealed that this drug could interact with the ACE2 through Ala348 and Arg393 by hydrogen and hydrophobic bonds, respectively. Although this drug makes a hydrophobic bond with the critical amino acid of Arg93 on the ACE2 receptor, Molecular Dynamics simulation indicates that this drug can interact with the ACE2 in a compact and stable mode. Simulation of the interaction shows that this drug attaches to Asp350, Gly352, and Arg393 by hydrogen bonds and interacts with Phe40, Ala99, Trp69, Leu100, Asn103, and Phe390 by hydrophobic bonds. While Lys353, Gly354, Asp355, and Arg357 are the most potent binding sites on the ACE2, the interaction of Chrysin with Phe40, Asp350, and Gly352 on the ACE2 can interfere with the binding of SARS-CoV-2. This data is in agreement with data from Choudhary et al. who found that eptifibatide acetate, TNP, GNF5, GR 127935, hydrochloride hydrate, and RS504393 drugs could bind to Phe40, Phe390, Gly352, Tyr69, Asp350, and Arg393 residues and inhibit SARS-CoV-2 attachment to the ACE2 [43]. Also, it is against Cheng et al. and Jochi et al.’s studies showing that their suggested compounds interact with the ACE2 by different binding sites except for Trp69 and Asp350 in Chen’s study [44].
Binding affinities for both drug candidates in either the Prodigy webserver or Smina docking are consistent with each other. Comparing the binding affinities of Azithromycin and Hydroxychloroquine with the binding affinity of the studied drug candidates demonstrates that Azithromycin and Hydroxychloroquine have binding affinities of −5.2 and −3.7 kcal mol−1 against the ACE2, respectively [53] while the binding affinities of Luteolin and Chrysin to the ACE2 are −8.8 and −8.4, respectively. This data illustrates that the introduced drug candidates can attach to the ACE much stronger than Azithromycin and Hydroxychloroquine. Also, comparing the binding affinities of Luteolin and Chrysin with the binding affinities of eptifibatide acetate, TNP, GNF5, GR 127935, hydrochloride hydrate, and RS504393 drugs according to Choudhary et al. study [43] demonstrates that except GR hydrochloride which has the binding affinity of −11.23 kcal mol−1 against ACE2, other drugs’ binding affinities to the ACE2 are similar to Luteolin and Chrysin. Moreover, according to Chen et al.’s finding, Naringin which is the best citrus flavonoid compound against ACE2 has a binding affinity of −6.85 kcal mol−1 which is weaker than the binding affinities of Luteolin and Chrysin against ACE2 [44].
Docking score and prodigy webserver results indicate that Luteolin has a higher binding affinity to ACE2 than Chrysin. Additionally, MM/PBSA demonstrates that Chrysin binds more tightly to the ACE2 and the binding free energy of Chrysin is better than Luteolin [54, 55].
This study indicated that consuming honey, carrot, cabbage, artichoke, tea, celery, apple propolis, blue passionflower (Passiflora caerulea), Radix Scutellariae, Cytisus multiflorus, Scutellaria immaculata, Desmos cochinchinensis, Pelargonium crispum, Oroxylum indicum, and even mushrooms could have a beneficial effect for preventing or reducing the transmission and infectivity of SARS-CoV-2.
Conclusions
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is spreading rapidly all over the world. The bioinformatics approach could be a very useful tool to identify possible inhibitors of 2019-nCoV. In this study, we used virtual screening-based Molecular Docking and Molecular Dynamics simulation to identify the possible inhibitors against ACE2. Candidates with better docking scores were selected for Molecular Dynamics simulation. Our study disclosed that Luteolin and Chrysin showed a strong affinity for the active site of the ACE2. MD and MM/PBSA analysis showed that ACE2-inhibitor complexes displayed structural stability with suitable binding energies compared to other FDA-approved drugs and natural drug databases. Screening the compounds by the above-mentioned computational methods showed that these compounds could be used as possible lead compounds against SARS-CoV-2 nevertheless experimental assays to finalize and validate our findings will be essential to support the clinical validation of these natural compounds. After their approval in the clinics, new natural-based drug compounds against the SARS-CoV-2 can be developed and used in the future.
Supplementary Information
Below is the link to the electronic supplementary material.
Abbreviations
- ACE2
Angiotensin-converting enzyme 2
- kcal mol−1
Kilocalorie per mole
- MD
Molecular Dynamics simulation
- MMPBSA
Molecular Mechanics/Poisson–Boltzmann Surface Area
- PDB
Protein Data Bank
- RMSD
Root Mean Square Deviation
- SASA
Surface accessible area
- RMSF
Root Mean Square Fluctuation
Author contribution
BS contributed in the investigation, data analysis, and writing the draft of the manuscript. LM contributed in the investigation and data analysis. LT-T contributed in the conception, resources, and editing the manuscript.
Funding
This study was supported by the National Institute for Medical Research Development (NIMAD) (Islamic Republic of Iran, Grant Number: 994135).
Data availability
All data will be available upon request.
Code availability
N/A
Declarations
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Shanker A, Bhanu D, Alluri A, Gupta S Whole genome sequence analysis and homology modelling of a 3C like peptidase and a non-structural protein 3 of the SARS-CoV-2 shows protein ligand interaction with an Aza-peptide and a noncovalent lead inhibitor with possible antiviral properties. Preprint. 2020
- 2.Zhu Y, Yu D, Yan H, Chong H, He Y (2020) Design of potent membrane fusion inhibitors against SARS-CoV-2, an emerging coronavirus with high fusogenic activity. Journal of Virology [DOI] [PMC free article] [PubMed]
- 3.Zhou Y, Hou Y, Shen J, Huang Y, Martin W. Cheng F Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell discovery. 2020;6(1):1–18. doi: 10.1038/s41421-020-0153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rodriguez-Morales AJ, Bonilla-Aldana DK, Tiwari R, Sah R, Rabaan AA. Dhama K COVID-19, an emerging coronavirus infection: current scenario and recent developments-an overview. J Pure Appl Microbiol. 2020;14:6150. doi: 10.22207/JPAM.14.1.02. [DOI] [Google Scholar]
- 5.Gautret P, Lagier J-C, Parola P, Meddeb L, Mailhe M, Doudier B et al (2020) Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. International journal of antimicrobial agents 105949 [DOI] [PMC free article] [PubMed]
- 6.Russo A, Binetti E, Borrazzo C, GentiliniCacciola E, Battistini L, Ceccarelli G, et al. Efficacy of remdesivir-containing therapy in hospitalized COVID-19 patients: a prospective clinical experience. J Clin Med. 2021;10(17):3784. doi: 10.3390/jcm10173784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsu J (2020) Covid-19: what now for remdesivir? bmj. 371 [DOI] [PubMed]
- 8.Whitley R. Molnupiravir - a step toward orally bioavailable therapies for Covid-19. N Engl J Med. 2021 doi: 10.1056/NEJMe2117814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muralidharan N, Sakthivel R, Velmurugan D, Gromiha MM (2020) Computational studies of drug repurposing and synergism of lopinavir, oseltamivir and ritonavir binding with SARS-CoV-2 protease against COVID-19. Journal of Biomolecular Structure and Dynamics. 1-6 [DOI] [PubMed]
- 10.Elfiky AA (2020) Anti-HCV, nucleotide inhibitors, repurposing against COVID-19. Life sciences. 117477 [DOI] [PMC free article] [PubMed]
- 11.Zhang C, Zheng W, Huang X, Bell EW, Zhou X. Zhang Y Protein structure and sequence reanalysis of 2019-nCoV genome refutes snakes as its intermediate host and the unique similarity between its spike protein insertions and HIV-1. J Proteome Res. 2020;19(4):1351–1360. doi: 10.1021/acs.jproteome.0c00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sinha SK, Shakya A, Prasad SK, Singh S, Gurav NS, Prasad RS et al (2020) An in-silico evaluation of different Saikosaponins for their potency against SARS-CoV-2 using NSP15 and fusion spike glycoprotein as targets. Journal of Biomolecular Structure and Dynamics. 1-12 [DOI] [PMC free article] [PubMed]
- 13.Walls AC, Park Y-J, Tortorici MA, Wall A, McGuire AT, Veesler D (2020) Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell [DOI] [PMC free article] [PubMed]
- 14.Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hasan A, Paray BA, Hussain A, Qadir FA, Attar F, Aziz FM et al (2020) A review on the cleavage priming of the spike protein on coronavirus by angiotensin-converting enzyme-2 and furin. Journal of Biomolecular Structure and Dynamics. 1-9 [DOI] [PMC free article] [PubMed]
- 16.Monteil V, Kwon H, Prado P, Hagelkrüys A, Wimmer RA, Stahl M et al (2020) Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell [DOI] [PMC free article] [PubMed]
- 17.Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh C-L, Abiona O, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sui J, Li W, Roberts A, Matthews LJ, Murakami A, Vogel L, et al. Evaluation of human monoclonal antibody 80R for immunoprophylaxis of severe acute respiratory syndrome by an animal study, epitope mapping, and analysis of spike variants. J Virol. 2005;79(10):5900–5906. doi: 10.1128/JVI.79.10.5900-5906.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Biagioli M, Marchianò S, Roselli R, Di Giorgio C, Bellini R, Bordoni M, et al. Discovery of a AHR pelargonidin agonist that counter-regulates Ace2 expression and attenuates ACE2-SARS-CoV-2 interaction. Biochemical Pharmacology. 2021;188:114564. doi: 10.1016/j.bcp.2021.114564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jia H, Neptune E. Cui H Targeting ACE2 for COVID-19 therapy: opportunities and challenges. Am J Respir Cell Mol Biol. 2021;64(4):416–425. doi: 10.1165/rcmb.2020-0322PS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ho T-Y, Wu S-L, Chen J-C, Li C-C. Hsiang C-Y Emodin blocks the SARS coronavirus spike protein and angiotensin-converting enzyme 2 interaction. Antiviral Res. 2007;74(2):92–101. doi: 10.1016/j.antiviral.2006.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yi L, Li Z, Yuan K, Qu X, Chen J, Wang G, et al. Small molecules blocking the entry of severe acute respiratory syndrome coronavirus into host cells. J Virol. 2004;78(20):11334–11339. doi: 10.1128/JVI.78.20.11334-11339.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koes DR, Baumgartner MP. Camacho CJ Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J Chem Inf Model. 2013;53(8):1893–1904. doi: 10.1021/ci300604z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1:19–25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- 25.Schüttelkopf AW. Van Aalten DM PRODRG: a tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60(8):1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 26.Wu Y, Tepper HL. Voth GA Flexible simple point-charge water model with improved liquid-state properties. The Journal of chemical physics. 2006;124(2):024503. doi: 10.1063/1.2136877. [DOI] [PubMed] [Google Scholar]
- 27.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H. Pedersen LG A smooth particle mesh Ewald method. J Chem Phys. 1995;103(19):8577–8593. doi: 10.1063/1.470117. [DOI] [Google Scholar]
- 28.Hess B, Bekker H, Berendsen HJ. Fraaije JG LINCS: a linear constraint solver for molecular simulations. J Comput Chem. 1997;18(12):1463–1472. doi: 10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H. [DOI] [Google Scholar]
- 29.Kumari R, Kumar R, Consortium OSDD Lynn A g_mmpbsa—a GROMACS tool for high-throughput MM-PBSA calculations. J Chem Inform Model. 2014;54(7):1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- 30.Wallace AC, Laskowski RA. Thornton JM LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng Des Sel. 1995;8(2):127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 31.Xue LC, Rodrigues JP, Kastritis PL, Bonvin AM. Vangone A PRODIGY: a web server for predicting the binding affinity of protein–protein complexes. Bioinformatics. 2016;32(23):3676–3678. doi: 10.1093/bioinformatics/btw514. [DOI] [PubMed] [Google Scholar]
- 32.Li F, Li W, Farzan M. Harrison SC Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309(5742):1864–1868. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- 33.Li W, Zhang C, Sui J, Kuhn JH, Moore MJ, Luo S, et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005;24(8):1634–1643. doi: 10.1038/sj.emboj.7600640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qiu Y, Zhao Y-B, Wang Q, Li J-Y, Zhou Z-J, Liao C-H et al (2020) Predicting the angiotensin converting enzyme 2 (ACE2) utilizing capability as the receptor of SARS-CoV-2. Microbes and infection [DOI] [PMC free article] [PubMed]
- 35.Hussain M, Jabeen N, Raza F, Shabbir S, Baig AA, Amanullah A et al (2020) Structural variations in human ACE2 may influence its binding with SARS‐CoV‐2 spike protein. Journal of medical virology [DOI] [PMC free article] [PubMed]
- 36.Guler HI, Fulya A, Zehra C, Yakup K, BELDUZ AO, CANAKCI S et al (2021) Targeting CoV-2 spike RBD and ACE-2 interaction with flavonoids of Anatolian propolis by in silico and in vitro studies in terms of possible COVID-19 therapeutics. bioRxiv [DOI] [PMC free article] [PubMed]
- 37.Carino A, Moraca F, Fiorillo B, Marchianò S, Sepe V, Biagioli M, et al. Hijacking SARS-CoV-2/ACE2 receptor interaction by natural and semi-synthetic steroidal agents acting on functional pockets on the receptor binding domain. Front Chem. 2020;8:846. doi: 10.3389/fchem.2020.572885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luo Y, Shang P. Li D Luteolin: a flavonoid that has multiple cardio-protective effects and its molecular mechanisms. Front Pharmacol. 2017;8:692. doi: 10.3389/fphar.2017.00692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fan W, Qian S, Qian P. Li X Antiviral activity of luteolin against Japanese encephalitis virus. Virus Res. 2016;220:112–116. doi: 10.1016/j.virusres.2016.04.021. [DOI] [PubMed] [Google Scholar]
- 40.Yan H, Ma L, Wang H, Wu S, Huang H, Gu Z, et al. Luteolin decreases the yield of influenza A virus in vitro by interfering with the coat protein I complex expression. J Nat Med. 2019;73(3):487–496. doi: 10.1007/s11418-019-01287-7. [DOI] [PubMed] [Google Scholar]
- 41.Godinho PI, Soengas RG. Silva VL Therapeutic potential of glycosyl flavonoids as anti-coronaviral agents. Pharmaceuticals. 2021;14(6):546. doi: 10.3390/ph14060546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang P, Gu H, Zhao Z, Wang W, Cao B, Lai C, et al. Angiotensin-converting enzyme 2 (ACE2) mediates influenza H7N9 virus-induced acute lung injury. Sci Rep. 2014;4:7027. doi: 10.1038/srep07027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Choudhary S, Malik YS, Tomar S, Tomar S (2020) Identification of SARS-CoV-2 cell entry inhibitors by drug repurposing using in silico structure-based virtual screening approach. Chemrxiv [DOI] [PMC free article] [PubMed]
- 44.Cheng L, Zheng W, Li M, Huang J, Bao S, Xu Q et al (2020) Citrus fruits are rich in flavonoids for immunoregulation and potential targeting ACE2 [DOI] [PMC free article] [PubMed]
- 45.Joshi T, Joshi T, Sharma P, Mathpal S, Pundir H, Bhatt V, et al. In silico screening of natural compounds against COVID-19 by targeting Mpro and ACE2 using molecular docking. Eur Rev Med Pharmacol Sci. 2020;24:4529–4536. doi: 10.26355/eurrev_202004_21036. [DOI] [PubMed] [Google Scholar]
- 46.Siddiqui A, Badruddeen AJ, Uddin MSS, Khan MI, Khalid M, et al. A naturally occurring flavone (chrysin): chemistry, occurrence, pharmacokinetic, toxicity, molecular targets and medicinal properties. J BiolActive Prod Nature. 2018;8(4):208–227. [Google Scholar]
- 47.Bajgai SP, Prachyawarakorn V, Mahidol C, Ruchirawat S. Kittakoop P Hybrid flavan-chalcones, aromatase and lipoxygenase inhibitors, from Desmos cochinchinensis. Phytochemistry. 2011;72(16):2062–2067. doi: 10.1016/j.phytochem.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 48.Williams CA, Harborne JB, Newman M, Greenham J. Eagles J Chrysin and other leaf exudate flavonoids in the genus Pelargonium. Phytochemistry. 1997;46(8):1349–1353. doi: 10.1016/S0031-9422(97)00514-1. [DOI] [PubMed] [Google Scholar]
- 49.Zaveri M, Khandhar A. Jain S Quantification of baicalein, chrysin, biochanin-A and ellagic acid in root bark of Oroxylum indicum by RP-HPLC with UV detection. Eurasian J Anal Chem. 2008;3(2):245–257. [Google Scholar]
- 50.Anandhi R, Annadurai T, Anitha TS, Muralidharan AR, Najmunnisha K, Nachiappan V, et al. Antihypercholesterolemic and antioxidative effects of an extract of the oyster mushroom, Pleurotus ostreatus, and its major constituent, chrysin, in Triton WR-1339-induced hypercholesterolemic rats. J Physiol Biochem. 2013;69(2):313–323. doi: 10.1007/s13105-012-0215-6. [DOI] [PubMed] [Google Scholar]
- 51.Balam FH, Ahmadi ZS. Ghorbani A Inhibitory effect of chrysin on estrogen biosynthesis by suppression of enzyme aromatase (CYP19): a systematic review. Heliyon. 2020;6(3):e03557. doi: 10.1016/j.heliyon.2020.e03557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Salimi A, Pourahmad J (2018) Role of natural compounds in prevention and treatment of chronic lymphocytic leukemia. In: Polyphenols: prevention and treatment of human disease. Elsevier, pp 195-203
- 53.Sandeep S, McGregor K (2020) Energetics based modeling of hydroxychloroquine and azithromycin binding to the SARS-CoV-2 spike (S) Protein-ACE2 Complex
- 54.Wang E, Sun H, Wang J, Wang Z, Liu H, Zhang JZ, et al. End-point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design. Chem Rev. 2019;119(16):9478–9508. doi: 10.1021/acs.chemrev.9b00055. [DOI] [PubMed] [Google Scholar]
- 55.Zhang X, Perez-Sanchez H. C Lightstone F A comprehensive docking and MM/GBSA rescoring study of ligand recognition upon binding antithrombin. Curr Top Med Chem. 2017;17(14):1631–1639. doi: 10.2174/1568026616666161117112604. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data will be available upon request.
N/A