

Abstract
Background:
Genetic mutations in triggering receptor expressed on myeloid cells-2 (TREM2) have been strongly associated with increased risk of developing Alzheimer’s disease (AD) and other progressive dementias. In the brain, TREM2 protein is specifically expressed on microglia suggesting their active involvement in driving disease pathology. Using various transgenic AD models to interfere with microglial function through TREM2, several recent studies provided important data indicating a causal link between TREM2 and underlying amyloid-β (Aβ) and tau pathology. However, mechanisms by which TREM2 contributes to increased predisposition to clinical AD and influences its progression still remain largely unknown.
Methods:
Spontaneous and brainstem nucleus pontis oralis stimulation-induced hippocampal oscillation paradigm was used to investigate the impact of TREM2 haploinsufficiency TREM2(Het) or total deficiency TREM2(Hom) on hippocampal network function in wild-type and amyloid-β overproducing Tg2576 mice under urethane anesthesia.
Results:
Partial (TREM2(Het)) or total (TREM2(Hom)) deletion of TREM2, led to increased incidence of spontaneous epileptiform seizures in both wild-type and Tg2576 mice. Importantly, deficiency of TREM2 in Tg2576 mice significantly diminished power of theta oscillation in the hippocampus elicited by brainstem-stimulation compared to wild-type mice. However, it did not affect hippocampal theta-phase gamma-amplitude coupling significantly, since over a 60% reduction was found in coupling in Tg2576 mice regardless of TREM2 function.
Conclusion:
Our findings indicate a role for TREM2-dependent microglial function in the hippocampal neuronal excitability in both wild type and Aβ overproducing mice, whereas deficiency in TREM2 function exacerbates disruptive effects of Aβ on hippocampal network oscillations.
Keywords: TREM2, microglia, hippocampus, seizure, theta oscillation, Alzheimer’s disease
Introduction
The prevalence of Alzheimer’s disease (AD), a devastating neurodegenerative disorder, is increasing worldwide and there is not yet any approved disease-modifying therapeutic that would affect disease progression [1]. Amyloid pathology has long been the major focus of drug development, since the spread and accumulation of amyloid-β (Aβ) in soluble misfolded oligomeric forms and in in extracellular deposits are thought to be the upstream initiating factor of the disease. However, clinical trials targeting Aβ have yielded disappointing results leading to increased efforts to identify new disease drivers and their possible interactions with the canonical AD hallmarks Aβ and hyperphosphorylated tau [2].
Evidence of the critical contribution of microglial activation in neuronal dysfunctions and cell death has fundamentally shifted our understanding of the pathogenesis of several neurodegenerative diseases, including AD [3, 4]. Microglia, the resident innate immune cells of the brain, have a pivotal role in providing homeostatic control by clearance of apoptotic debris and misfolded proteins, secretion of neurotrophic factors, inflammatory response to injury, and remodeling neuronal connections [5]. Recent experimental studies also demonstrated that microglia are involved in regulation of higher brain functions and behavior through modulation of neuronal activity [6, 7]. Unfortunately, the most of these important microglia functions can significantly deteriorate as a result of genetic mutations or ageing process [8–10]. Genome-wide association studies have linked coding variants in triggering receptor expressed on myeloid cells-2 (TREM2), which is exclusively found on microglia in the brain, with increased risk for AD, frontotemporal dementia and Nasu-Hakola disease characterized with progressive presenile dementia and bone cysts [11–13]. In particular, naturally occurring missense variants p.R47H and p.R62H that confer a partial loss of function of TREM2 has been implicated in developing late onset AD [14]. Interestingly, carriers of these TREM2 variants also showed faster rate of cognitive decline compared with non-carriers [15]. In addition, patients with a rare homozygous mutations p.Thr66Met in TREM2 had early onset frontotemporal-like dementia, with hippocampal deficits and presence of epileptic seizures [16].
To investigate genotype-phenotype correlation between TREM2 and AD, transgenic mouse lines modeling AD pathology with partial or total TREM2 deficiency were generated. In these mice, it has been observed that TREM2 haploinsufficiency caused a strong reduction in the number of microglia surrounding plaques [17], with decreased plaque compaction and severe axonal dystrophy, similarly to results from postmortem brain samples of AD patients with TREM2 mutation [18]. Interestingly, total deficiency of TREM2 signaling was shown to affect amyloid pathology in transgenic APP/PS1 mice differently depending on the stage of disease progression, as it reduced amyloid accumulation at early stage and intensified amyloid accumulation at late stage of disease [19]. In the context of tau pathology, findings on TREM2 deficiency are also discordant. Decreased TREM2 function exacerbates tau hyperphosphorylation [20] and facilitates seeding and spreading of tau aggregates around Aβ plaques in the early phase of AD [21, 22], but during the later symptomatic phase appears to slow down tau-mediated neurodegeneration, as seen in TREM2 p.R47H variant carriers [23]. Although these studies have implicated distinctive TREM2-dependent microglial role in AD at the cellular level, data on its functional relevance are largely missing.
In the present neurophysiological study, we assessed hippocampal network oscillations in amyloid-β overproducing Tg2576 transgenic mice with normal and deficient TREM2 function with aim to elucidate potential contribution of this immunoreceptor on specific oscillatory dynamic changes associated with AD pathophysiology.
Methods
Mice
Transgenic Tg2576 (B6;SJL-Tg(APPSWE)2576Kha) hemizygous mice and their wild-type controls were obtained by Biogen through a Research Crossbreeding agreement with Taconic Biosciences Inc. (Germantown, NY). The Tg2576 mice model AD pathology by overexpressing a human amyloid precursor protein (isoform 695) with the Swedish mutation (KM670/671NL), which resulted in high levels of Aβ, extensive development of amyloid plaques, and cognitive deficits [24]. These mice were crossed with TREM2 deficient mice to yield Tg2576;TREM2 genotypes. Tg2576(Hemi);TREM2(WT/Het/Hom) experimental cohorts were generated by breeding Tg2576(Hemi);TREM2(Het) mice with Tg2576 (WT);TREM2(Het) mice and were maintained on a mixed B6/SJL background. TREM2 deficient mice were generated from Trem2tm1(KOMP)Vlcg founders obtained from UC Davis KOMP Repository and maintained on a C57BL/6 mice background. In these mice, a LacZ reporter cassette replaces the entire coding sequence of the endogenous Trem2 locus, resulting in a loss of TREM2 function [25]. Breeding was carried out at Biogen and The Jackson Laboratory (Bar Harbor, ME). All animals were littermates to ensure that genetic background and environment were homogeneous. For experiments, mice cohorts were transferred to Yale University where they were housed in Yale Animal Resources Center managed facility, in a temperature and humidity-controlled room with 12:12-h light-dark cycle and free access to food and water at all times.
A total of 65 male mice were used in the study (Tg2576(WT)TREM2(WT) = 13; Tg2576(WT)TREM2(Het) = 9; Tg2576(WT)TREM2(Hom) = 12; Tg2576(Hemi)TREM2(WT) = 13; Tg2576(Hemi)TREM2(Het) = 9; Tg2576(Hemi)TREM2(Hom) = 9). All animals were at the age between 9 and 12 months. Neurophysiological recordings were done after acclimation period of at least 30 days, with all measures taken to minimize pain or discomfort of animals. All procedures were performed in accordance with National Institutes of Health guidelines (NIH publications No. 86–23, revised 1996) and were approved by the Institutional Animal Care and Use Committees of each respective institution.
Neurophysiological recordings
Hippocampal neuronal network integrity was tested using brainstem stimulation-induced theta oscillation paradigm, which was employed in previous neurophysiological studies of transgenic rodent models of AD pathology [26–29].
Briefly, mice were anesthetized with urethane (1.5 g/kg) given intraperitoneally and placed in a Kopf stereotaxic frame (Tujunga, CA) on a temperature-regulated heating pad set to maintain body temperature at 37°C (Physitemp Instruments Inc., Clifton, NJ). After achieving surgical plane of anesthesia which was controlled by the absence of the tail pinch reaction, left parietal craniotomies were performed over the hippocampus and rostral brainstem for insertion of the two concentric stainless-steel bipolar electrodes (NE-100X, Rhodes Medical Instruments, Woodland Hills, CA). For recording of local field potentials (LFPs), one electrode was placed in the dorsal hippocampal CA1 region, while another was lowered in the nucleus pontis oralis (nPO) for electrical stimulation. Coordinates for both target areas were determined relative to bregma and brain surface and were as follows: CA1, anteroposterior −2.0 mm, lateral 1.5 mm, and dorsoventral 1.5 mm; nPO, anteroposterior −4.0 mm, lateral 1.2 mm, and dorsoventral 3.3 mm [30]. Following placements of the electrodes, animals remained in the stereotaxic frame for LFPs recording which began after stabilization period of at least 30 minutes. Each recording lasted for approximately 60 minutes, during which time initial dose of urethane was sufficient to provide stable plane of anesthesia [31]. At the end of each recording, mice were sacrificed, brain tissue collected and histologically processed for verification of electrodes positions. The nPO stimulation protocol consisted of a train of 0.3 ms square pulses delivered over a period of 6 seconds at a rate of 250 Hz, which was provided by Isoflex stimulus isolator unit (A.M.P.I. Instruments, Jerusalem, Israel) and repeated every 100 seconds. The stimulation-response relationship was determined in each animal using 3 cycles of increasing current in steps of 20 μA, with power and frequencies averaged for each stimulation intensity. For quantitative analyses, LFPs were amplified by an A-M System (Carlsborg, WA) with filters set between 1 Hz and 500 Hz, digitized at a rate of 2 kHz, and collected on a computer via a CED Micro1401-3 interface and Spike2 software (Cambridge Electronic Design, Cambridge, UK).
Stimulation-induced hippocampal oscillation episodes were analyzed for total theta power and peak theta frequency using a fast Fourier transform in Spike2 at a spectral resolution of 0.24 Hz. Total theta power was calculated by summing power values between 3 Hz and 9 Hz, and peak frequency was identified by determining where the peak power occurred. Estimating phase-amplitude coupling between theta and gamma oscillations, a neural signature of coordinated memory formation and storage, was performed using custom scripts written in Matlab (Mathworks, Natick, MA). All offline filtering was done using eegfilt.m from EEGLAB toolbox [32]. To avoid possible stimulation artefacts, the first second of each 6-second-long stimulation episode was omitted in all analyses. Seizures were identified visually by sudden large positive and/or negative deflections of hippocampal oscillations by neurophysiologist during EEG recordings, and confirmed during off-line analysis by a computational neuroscientist as instantaneous increase in spontaneous LFPs wideband power (4 z-score difference for 2 s or longer). The strength of theta-gamma coupling was assessed using modulation index (MI) as described previously [28, 33, 34]. Specifically, using the Hilbert transformation we extracted the instantaneous phase in LFP signal containing oscillations from 3 to 10 Hz (phase frequencies), and the amplitude envelope in signal containing oscillations from 20 to 100 Hz (amplitude frequencies). For each animal an MI value was estimated for each frequency pair composed by one slow and one fast oscillation of the LFPs recorded during stimulation with current intensity inducing 50% of the maximal theta oscillation power in the stimulation-response curves. An important characteristic of this metric is that it is not influenced by the absolute amplitude of theta or gamma oscillations, but depends on the relative changes of the gamma oscillation envelope as a function of the theta oscillation phase [33]. To generate the heat-maps, the MI values over 3 stimulations from each animal in a group were averaged and expressed using a color-coded scale.
Statistical analysis
All data were initially determined to be suitable for parametric analysis according to normality and homoscedasticity using the Shapiro-Wilk and Brown–Forsythe tests, respectively. Statistical analyses were performed by 1-way ANOVA with Tukey’s post hoc test for assessing stimulus-response relationship in elicited theta power or peak frequency, or with Dunnett’s post hoc test for comparisons of average MI values between the groups (GraphPad Prism Software, Inc, La Jolla, CA). Data are expressed as the mean ± standard error of the mean (SEM). Differences were considered significant when P < 0.05.
Results
Behavioral observations
Inspecting the animals before neurophysiological experiments revealed a striking pathologic self-grooming in individually housed TREM2 homozygous and heterozygous mutant mice. Due to excessive grooming behavior these mice displayed large hairless patches on their lateral body surface (Fig. 1A). Although the extent of severe fur loss varied between these animals, close physical examination of the affected area by researcher and attending veterinarian from animal facility did not find any significant skin ulceration or inflammation that would have required particular treatment. The similar signs of compulsory over-grooming behavior were not detected in corresponding wild-type or transgenic Tg2576 mice with intact TREM2 function.
Fig.1.
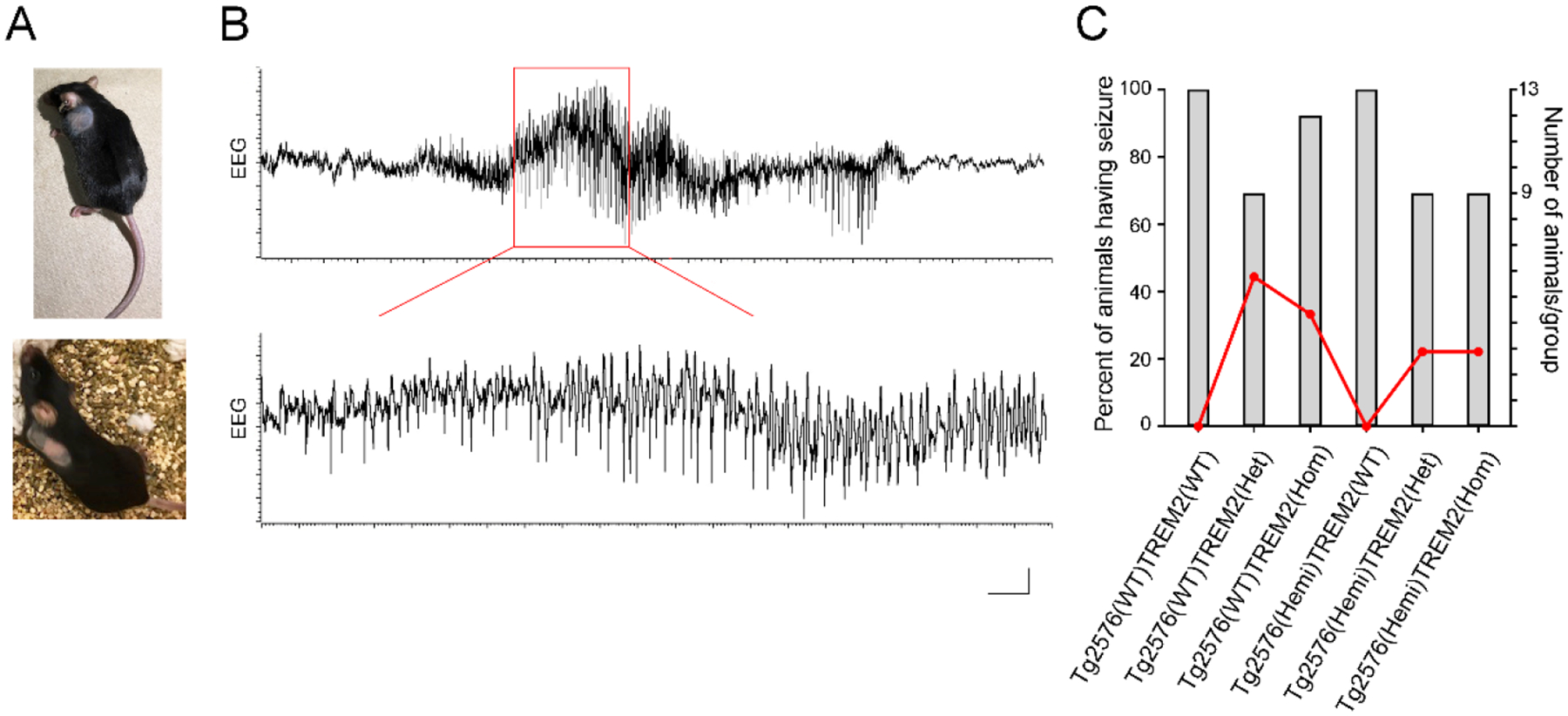
Pathological behavior and increased seizure susceptibility in control and Tg2576 mice with partial or complete TREM2 loss of function mutations. A) Photographs showing large patches of missing hair caused by excessive self-grooming of individually housed TREM2 homozygous mutants crossed with transgenic AD mice (Tg2576(Hemi); upper panel) and their littermate wild-type controls (Tg2576(WT); lower panel). The same compulsive hair removal feature was found in TREM2 heterozygous mice of either genotype. B) Typical EEG recording of spontaneous epileptiform activity in the hippocampal CA1 region of TREM2 deficient mice under anesthesia. Trace shows aberrant large amplitude spiking discharges from Tg2576(WT)TREM2(Het) mouse lasting over 15 seconds. Enlarged segment of the trace (marked in red) is at the bottom. C) Distribution of susceptibility to epileptic seizures between different genotype groups of mice. In wild-type controls, seizures were detected in 44% of TREM2 heterozygous and 33% of TREM2 homozygous groups. In transgenic Tg2576 mice, they were present in 22% of mice from TREM2 heterozygous and homozygous groups. Scale bar: 0.02 mV, 1 s (upper trace); 0.02 mV, 0.2 s (lower trace).
Epileptiform brain activity
TREM2 homozygous and heterozygous mutant mice had high susceptibility to spontaneous epileptic seizure activity. During hippocampal EEG recordings we observed in some of these mice suddenly occurring episodes of synchronous large amplitude spike discharges lasting several seconds (average duration 15.61 ± 1.56 s) and followed by a depression of neuronal field potentials (Fig. 1B). These epileptiform seizures were present in 44% of Tg2576(WT)TREM2(Het) (4 out of 9 mice), 33% of Tg2576(WT)TREM2(Hom) (4 out of 12 mice), and 22% of either Tg2576(Hemi)TREM2(Het) or Tg2576(Hemi)TREM2(Hom) (2 out of 9 mice, in each) groups (Fig. 1C). In contrast, no spontaneous paroxysmal spiking discharges were seen in the hippocampus of mice with intact TREM2 function, regardless of the presence of Tg2576(Hemi) and Tg2576(WT) genotypes. These results indicate increased neuronal excitability only in mice with impaired TREM2 function.
Changes in hippocampal oscillations
Evaluation of elicited hippocampal theta oscillation and theta-phase gamma-amplitude coupling in rodents under anesthesia was shown to provide valuable insights into hippocampus network functioning related to memory and cognitive processes in behavior-independent manner [35, 36]. Therefore, similarly to our previous studies, we applied high-frequency electrical stimulation of the brainstem nPO to induce regular theta oscillation (3–9 Hz) in the hippocampal CA1 region with a current-dependent increase of peak frequency in urethane anesthetized mice (Fig. 2A). In these experiments analyses were performed only in animals in which hippocampal epileptiform activity did not develop during baseline recording or at any stage of brainstem stimulation thus potentially resulting in instability of neurophysiological activity. Quantitative input-output analyses of elicited hippocampal oscillation showed a significant difference in absolute theta power (F(5, 48) = 16.55; P < 0.001), but not peak theta frequency (F(5,48) = 0.51; P = 0.769) in response to varying stimulus intensities between the groups (Fig. 2B,D). Further AUC analysis with post hoc comparisons for theta power changes (stimulation intensities 0.02 – 0.12 mA) showed a marked reduction in theta power in Tg2576(Hemi)TREM2(Het) and even more significant decrease in Tg2576(Hemi)TREM2(Hom) mice when compared with control mice (Tg2576(WT)TREM2(WT) vs. Tg2576(Hemi)TREM2(Het), P = 0.047; Tg2576(WT)TREM2(WT) vs. Tg2576(Hemi)TREM2(Hom), P = 0.003, Tukey’s post hoc test), indicating that deficiency of TREM2, and particularly its elimination leads to disruption of theta oscillation in the hippocampus of transgenic Aβ overproducing mice (Fig. 2C).
Fig.2.
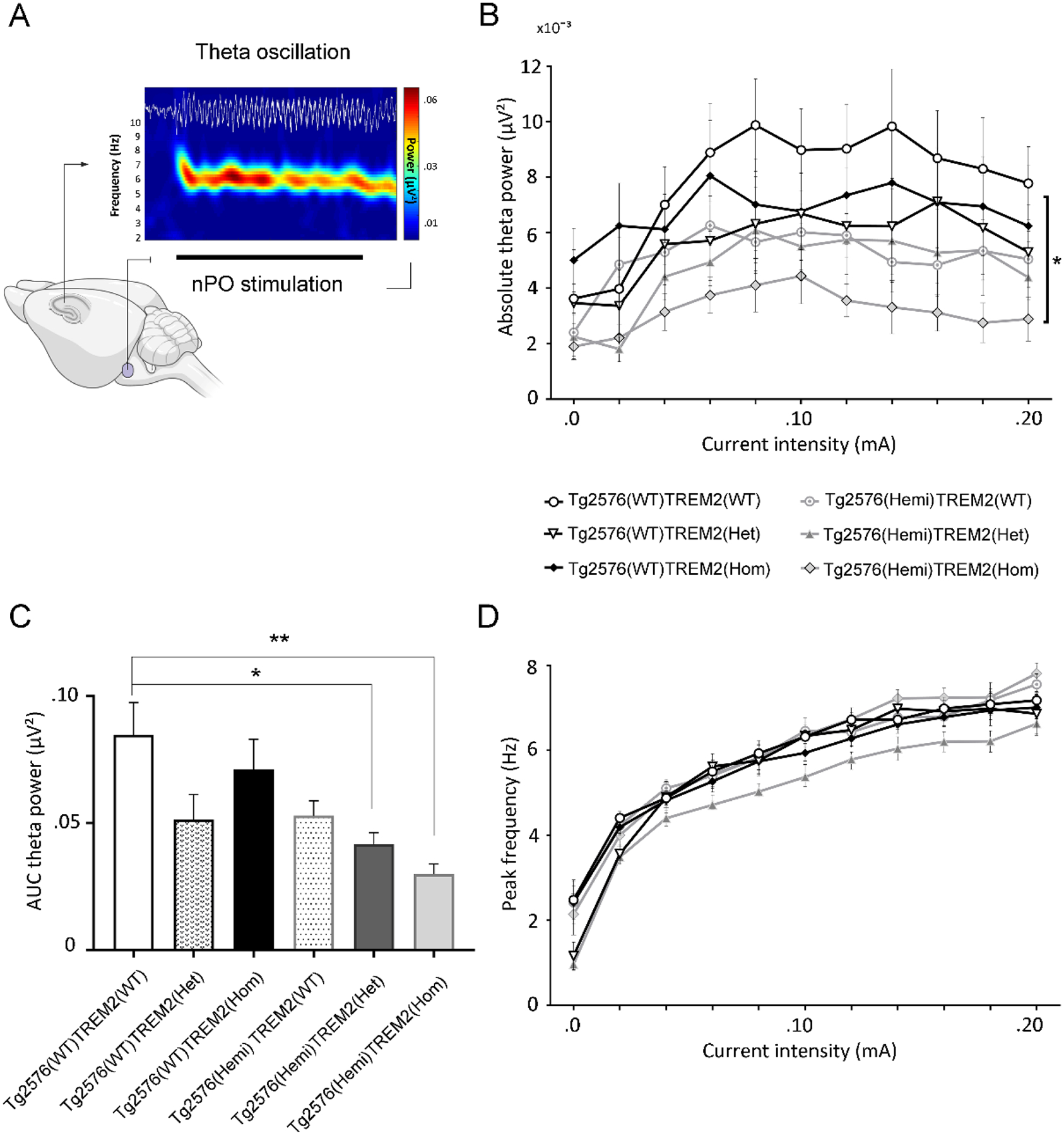
Disruption of elicited hippocampal oscillations in amyloid-β overproducing Tg2576 mice with partial or complete TREM2 loss of function mutations. A) High-frequency electrical stimulation of brainstem nucleus pontis oralis (nPO) induces highly regular theta oscillations in the hippocampal CA1 region of anesthetized mice, characterized by relatively constant frequency and spectral content as shown in the time-frequency decomposition map (horizontal black bar under the map marks the period of stimulation; the color-coded scale denotes EEG power). B-D) Stimulus-response relationship for absolute theta power and peak frequency over increasing stimulus intensities, and area under the curve (AUC) analyses showed significant decline in elicited theta power (B,C), but not peak theta frequency (D) in transgenic Tg2576 mice with both TREM2 heterozygous and homozygous mutations compared to respective wild-type controls. (*P < 0.05, **P < 0.01, 1-way ANOVA with Tukey’s post hoc test). Scale bar: 0.2 mV, 1 s.
In further assessment of oscillatory dynamics in the hippocampus during stimulation, theta-phase gamma amplitude coupling was measured in subsets of wild-type and transgenic Tg2576 mice with intact and total TREM2 deletion. In line with our previous studies, a strong coupling between phase of theta and amplitude of low gamma (30–60 Hz) was observed in Tg2576(WT)TREM2(WT) mice, whereas in other groups of mice this coupling was attenuated (Fig. 3A). Comparing the degree of theta-phase gamma-amplitude coupling calculated as modulation index (MI) significant difference was found among groups (F(3,26) = 3.65, P = 0.026). Further post hoc analysis using Dunnett’s test showed significantly lower coupling in Tg2576(Hemi)TREM2(WT) mice (P = 0.021) and Tg2576(Hemi)TREM2(Hom) mice (P = 0.048) comparing to wild-type controls (Tg2576(WT)TREM2(WT), (Fig. 3B). TREM2 deficiency in Tg2576(WT)TREM2(Hom) mice indicated a trend to reduced coupling, but it did not reach a significant level.
Fig.3.
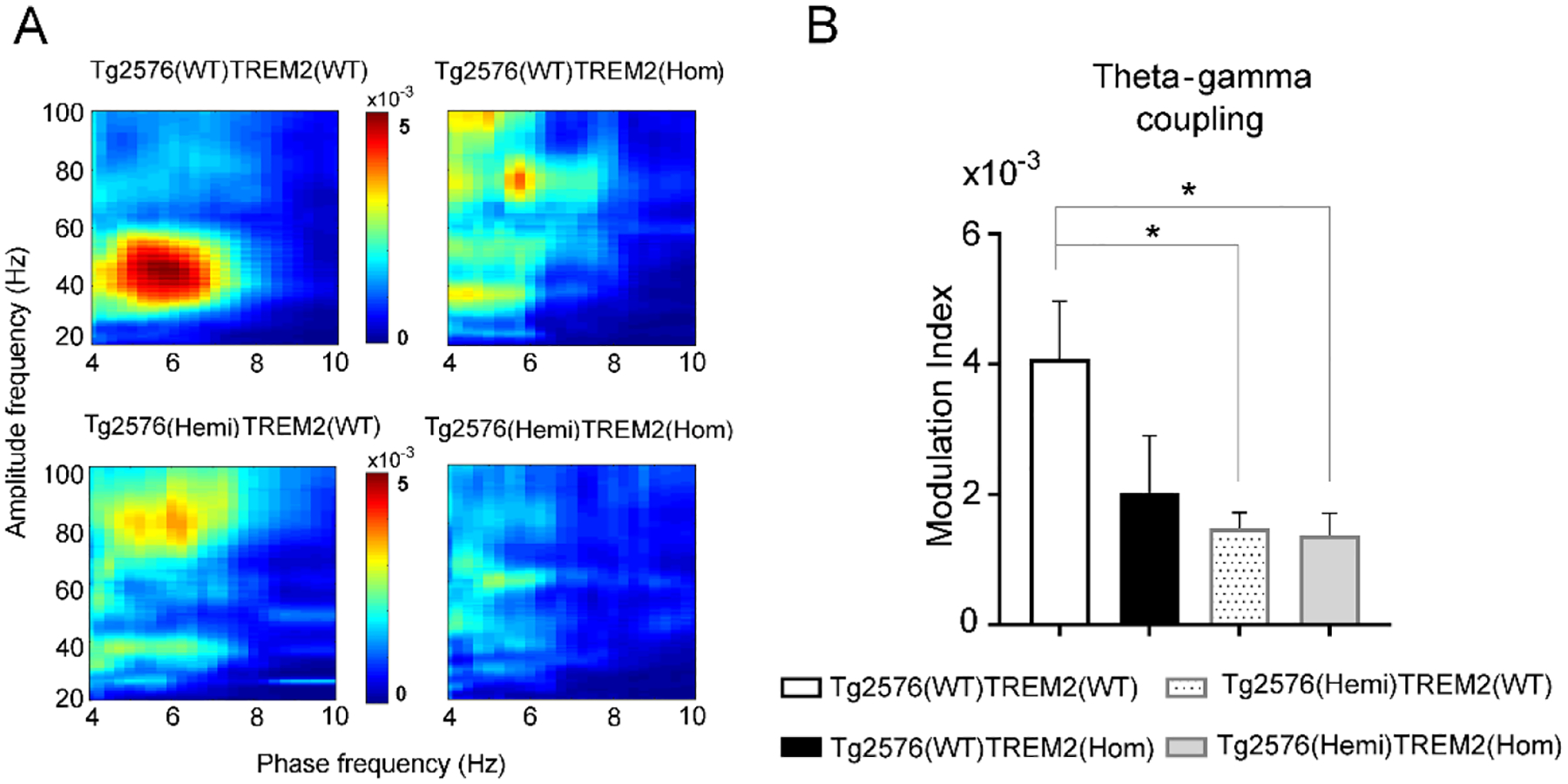
Reduction of phase-amplitude coupling between hippocampal theta and gamma oscillations in amyloid-β overproducing Tg2576 mice with both intact and deficient TREM2 function. Theta phase-gamma amplitude coupling during nPO-stimulation induced oscillations in hippocampus of anesthetized mice is calculated as Modulation Index (MI). A) Heat-maps with averaged theta-gamma coupling over all animals in each group indicate the strength of theta phase-nested distribution of gamma power (30–60 Hz). B) Comparison of average MI between groups revealed significantly decreased coupling in transgenic Tg2576 mice, regardless whether TREM2 was present or knock-out, compared to controls. (*P < 0.05, 1-way ANOVA with Dunnett’s post hoc test).
Discussion
Microglial TREM2-mediated signaling impairment has been proposed as a risk factor for the development and progression of AD, yet its contribution to different aspects of disease-related neuronal dysfunctions remains ambiguous. Here we provide in vivo evidence that TREM2 deficiency increases susceptibility for spontaneous epileptiform seizures in the hippocampus of both control and Aβ overproducing Tg2576 mice. TREM2 deficiency also significantly aggravates compromised hippocampal theta oscillation activity in these transgenic AD mice but without further influencing their already markedly diminished theta-phase gamma-amplitude coupling as compared to control counterparts.
The epileptiform activity has been recognized as an important clinical entity of AD, since several retrospective, prospective and cross-sectional studies identified a significantly higher incidence of unprovoked seizures in AD patients than in the age-matched control subjects [reviewed in 37, 38]. Furthermore, these seizures were found to be predominantly non-convulsive with focal temporolimbic localization, occurring even at an early, prodromal stage of AD [39, 40], and more frequently in male patients [41]. Importantly, evidence suggests that increased subclinical epileptiform activity in AD patients, which is indicative of neuronal hyperexcitability, might contribute to their accelerated cognitive decline [38]. Similar network hyperexcitability and spontaneous non-convulsive seizures were also detected in several transgenic models of AD with overt Aβ expression in the brain [29, 42, 43] after chronic cortical and hippocampal EEG monitoring. Strikingly, in this study, we found a higher propensity to epileptiform activity in both partial and total TREM2 deficient mice regardless of Aβ pathology. Intact TREM2 function is required for microglia-mediated remodeling of synaptic connections by phagocytic elimination of supernumerary dendritic spines, which is an important process for the maturation of neural networks and maintenance of delicate balance between excitation and inhibition during development and in adulthood [5]. Accordingly, it has been shown that mice with ablated TREM2 on microglia have a higher density of dendritic spines due to suppressed phagocytic capacity and consequently defective synapse pruning [44]. Since spine synapses closely correspond to the excitatory synapses [45], an increase in their number can induce an excitatory gain offsetting the balance between excitation and inhibition in the complex neuronal networks, thus modulating neurotransmission and behavioral responses. Recent studies revealed a spectrum of abnormalities in TREM2 deficient mice including enhanced excitatory neurotransmission and reduced synaptic plasticity in the hippocampus, impaired long-range functional connectivity between cortical and hippocampal regions, and behavioral alterations such as excessive self-grooming, a trait typically associated with animals’ increased anxiety and perseveration [44, 46, 47]. This TREM2-dependent amplification of excitatory input in the network due to microglial failure to prune synapses may also lower the threshold for seizure activity facilitating spontaneous epileptogenesis as observed here. Indeed, genetically determined defects in microglial elimination of excessive excitatory synapses in the neocortex and hippocampus during development [48, 49], or brain-wide pharmacological depletion of microglial phagocytosis in adult mice [6] both resulted in increased susceptibility to behavioral seizures. Our finding of a high rate of seizures among TREM2 haploinsufficient mice suggests that even a partial deficit in microglial TREM2 function is enough to induce significant perturbation in this mechanism of controlling synaptic balance.
Over the course of AD development, aberrant network excitability has been linked to Aβ pathology [50]. Recent data suggest that this dysfunction is likely initiated by preexisting neuronal activity, stemming from impaired baseline excitatory transmission, which triggers a vicious cycle where the soluble Aβ species induce hyperexcitability before the emergence of plaques [51]. This hyperexcitability not only increases the potential for epileptic seizures but might also exacerbate AD pathology by increasing diffusion and deposition of Aβ [52], and impairing the function of microglia [53]. It has been demonstrated that neuronal hyperactivity-evoked widespread release of adenosine triphosphate (ATP) may damage microglial phagocytosis and apoptotic debris clearance, and enhance proinflammatory cytokines secretions [53]. Interestingly, new evidence showed that microglial sensing of elevated extracellular ATP can in turn activate negative feedback mechanism to suppress neuronal activity via adenosine [6], further remodeling circuit function. In this view, an increase in TREM2 that parallels with the rise of Aβ levels seen in transgenic mice [12], and at early AD stage in patients [54] might have the compensatory function to prevent or mitigate alterations due to this initial detrimental events. However, during disease progression microglia in response to oligomeric and plaque Aβ challenge tend to undergo substantial ultrastructural changes leading to aberrant activation involved in Aβ-mediated neuronal dysfunctions and pathologic loss of synapses in cognitive-related brain regions [55, 56]. This aligns with the notion of the negative impact of impaired TREM2 signaling on Aβ pathology during the late stage of AD [14, 19].
Neurophysiological studies revealed a close association between profound alterations in EEG signals and cognitive dysfunctions in AD patients and transgenic animals carrying pathological genes for Aβ overproduction and tau hyperphosphorylation [57]. Specifically, generalized EEG slowing with spectral profile shift to the low frequencies has been linked to cognitive deterioration in patients [58, 59], whereas in transgenic models with demonstrated learning and memory deficits, multiple oscillatory alterations in cortical and hippocampal networks have been identified [29, 42, 43]. Previously, we reported several distinctive hippocampal oscillatory abnormalities that clearly reflected AD-related impairment of functional network integrity, such as the reduction in theta band power and weakening of theta-phase gamma-amplitude coupling in transgenic Aβ overproducing APP/PS1 and 5xFAD mice [26, 28] and TgF344-AD rats [29], as well as tau Tg4510 mice [27] recorded under urethane anesthesia. We also observed that these oscillatory dysfunctions are age-dependent and directly correlate with plaque load in the hippocampus [26, 29]. Hippocampal theta oscillation has been linked to cognitive processes, and the correlation between hippocampal theta activity and learning and memory has been indicated in both experimental animals and humans [60–63]. Hence, it has been proposed that theta oscillation could serve as a surrogate measure to quantify the cognitive abilities that depend on hippocampal function, but also to predict cognitive effects of drugs [35, 63]. Accordingly, pharmaco-EEG experimental studies showed that drugs currently in use for symptomatic AD treatment, donepezil and memantine, both increase the power of elicited hippocampal theta oscillation [64, 65] in anesthetized rodents. Further, the coupling between the phase of theta and amplitude of gamma oscillations in the hippocampus has been consistently shown to implicate effective memory encoding and consolidation in humans [66] and experimental animals [33, 67, 68]. In line with these findings, multiple studies found a reduction in hippocampal theta-gamma coupling in relation to AD pathology [28, 29, 69, 70]. Consistent with these observations, here we find that Tg2576 mice also showed compromised elicited theta oscillation in response to brainstem stimulation, and importantly that this deficit is significantly aggravated in animals with deficient TREM2 function. Moreover, we observed that transgenic Tg2576 mice displayed weakened hippocampal theta-phase gamma amplitude coupling as compared to their littermate controls, but this impairment contrary to theta power was not dependent on TREM2 function. Therefore, current findings implicate microglial TREM2 important role in slowing down certain robust aspects of AD-related oscillatory impairments such as hippocampal theta power decline, but not in mitigating other more subtle oscillatory abnormalities such as a reduction in theta-gamma coupling that depends on fine-tuned interaction between multiple neurotransmitter systems and cell types, and thereby is more vulnerable to disease pathology.
It is important to note that all neurophysiological recordings in this study were performed under urethane anesthesia, which may have confounding influences on reported findings. Even though urethane does not markedly affect neurotransmission inducing spontaneous brain state alterations that resemble physiological sleep [31], it has been shown that subthreshold excitability in the hippocampus can be compromised given its suppressive effects on neuronal spike activity and spike synchrony therein [71, 72]. This might be the reason why we haven’t observed EEG epileptiform activity in Tg2576 mice with intact TREM2 function unlike previous studies when this transgenic model was analyzed in non-anesthetized condition [42, 73]. Nevertheless, our results of high seizure susceptibility in hippocampal EEG of Tg2576 TREM2 deficient mice, even during urethane anesthesia, indicate that diminished TREM2 function leads to enhanced neuronal excitability and aggravated Aβ-induced hippocampal oscillatory deficit. Studies on mice with TREM2 deficiency could contribute to our understanding the pathological role of microglia function in heightened neuronal excitability in its contribution to AD disease progression, and potentially identify novel therapeutic targets.
Acknowledgements:
This study was supported by Biogen, Inc., and NIH grants AG067329, AG052986 and AG051459 (TLH).
Footnotes
Conflict of interests: Dr. Mihály Hajós is an employee of and owns stocks in Cognito Therapeutics, shareholder of Biogen and Pfizer. The other authors have no conflicts of interest.
References
- [1].Alzheimer’s Association (2020) 2020 Alzheimer’s disease facts and figures. Alzheimers Dement 16, 391–460. [DOI] [PubMed] [Google Scholar]
- [2].Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K (2020) Alzheimer’s disease drug development pipeline: 2020. Alzheimers Dement (N Y) 6, e12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bartels T, De Schepper S, Hong S (2020) Microglia modulate neurodegeneration in Alzheimer’s and Parkinson’s diseases. Science 370, 66–69. [DOI] [PubMed] [Google Scholar]
- [4].Tejera D, Heneka MT (2019) Microglia in Neurodegenerative Disorders. Methods Mol Biol 2034, 57–67. [DOI] [PubMed] [Google Scholar]
- [5].Li Q, Barres BA (2018) Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol 18, 225–242. [DOI] [PubMed] [Google Scholar]
- [6].Badimon A, Strasburger HJ, Ayata P, Chen X, Nair A, Ikegami A, Hwang P, Chan AT, Graves SM, Uweru JO, Ledderose C, Kutlu MG, Wheeler MA, Kahan A, Ishikawa M, Wang YC, Loh YE, Jiang JX, Surmeier DJ, Robson SC, Junger WG, Sebra R, Calipari ES, Kenny PJ, Eyo UB, Colonna M, Quintana FJ, Wake H, Gradinaru V, Schaefer A (2020) Negative feedback control of neuronal activity by microglia. Nature 586, 417–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Yasumoto Y, Stoiljkovic M, Kim JD, Sestan-Pesa M, Gao XB, Diano S, Horvath TL (2020) Ucp2-dependent microglia-neuronal coupling controls ventral hippocampal circuit function and anxiety-like behavior. BioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Mosher KI, Wyss-Coray T (2014) Microglial dysfunction in brain aging and Alzheimer’s disease. Biochem Pharmacol 88, 594–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Olah M, Patrick E, Villani AC, Xu J, White CC, Ryan KJ, Piehowski P, Kapasi A, Nejad P, Cimpean M, Connor S, Yung CJ, Frangieh M, McHenry A, Elyaman W, Petyuk V, Schneider JA, Bennett DA, De Jager PL, Bradshaw EM (2018) A transcriptomic atlas of aged human microglia. Nat Commun 9, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Streit WJ, Xue QS (2012) Alzheimer’s disease, neuroprotection, and CNS immunosenescence. Front Pharmacol 3, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Dardiotis E, Siokas V, Pantazi E, Dardioti M, Rikos D, Xiromerisiou G, Markou A, Papadimitriou D, Speletas M, Hadjigeorgiou GM (2017) A novel mutation in TREM2 gene causing Nasu-Hakola disease and review of the literature. Neurobiol Aging 53, 194 e113–194 e122. [DOI] [PubMed] [Google Scholar]
- [12].Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate A, Rademakers R, Morgan K, Powell J, St George-Hyslop P, Singleton A, Hardy J, Alzheimer Genetic Analysis G (2013) TREM2 variants in Alzheimer’s disease. N Engl J Med 368, 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Jin SC, Benitez BA, Karch CM, Cooper B, Skorupa T, Carrell D, Norton JB, Hsu S, Harari O, Cai Y, Bertelsen S, Goate AM, Cruchaga C (2014) Coding variants in TREM2 increase risk for Alzheimer’s disease. Hum Mol Genet 23, 5838–5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Carmona S, Zahs K, Wu E, Dakin K, Bras J, Guerreiro R (2018) The role of TREM2 in Alzheimer’s disease and other neurodegenerative disorders. Lancet Neurol 17, 721–730. [DOI] [PubMed] [Google Scholar]
- [15].Del-Aguila JL, Fernandez MV, Schindler S, Ibanez L, Deming Y, Ma S, Saef B, Black K, Budde J, Norton J, Chasse R, Alzheimer’s Disease Neuroimaging I, Harari O, Goate A, Xiong C, Morris JC, Cruchaga C (2018) Assessment of the Genetic Architecture of Alzheimer’s Disease Risk in Rate of Memory Decline. J Alzheimers Dis 62, 745–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Le Ber I, De Septenville A, Guerreiro R, Bras J, Camuzat A, Caroppo P, Lattante S, Couarch P, Kabashi E, Bouya-Ahmed K, Dubois B, Brice A (2014) Homozygous TREM2 mutation in a family with atypical frontotemporal dementia. Neurobiol Aging 35, 2419 e2423–2419 e2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ulrich JD, Finn MB, Wang Y, Shen A, Mahan TE, Jiang H, Stewart FR, Piccio L, Colonna M, Holtzman DM (2014) Altered microglial response to Abeta plaques in APPPS1–21 mice heterozygous for TREM2. Mol Neurodegener 9, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, Luo W, Colonna M, Baddeley D, Grutzendler J (2016) TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 90, 724–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jay TR, Hirsch AM, Broihier ML, Miller CM, Neilson LE, Ransohoff RM, Lamb BT, Landreth GE (2017) Disease Progression-Dependent Effects of TREM2 Deficiency in a Mouse Model of Alzheimer’s Disease. J Neurosci 37, 637–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bemiller SM, McCray TJ, Allan K, Formica SV, Xu G, Wilson G, Kokiko-Cochran ON, Crish SD, Lasagna-Reeves CA, Ransohoff RM, Landreth GE, Lamb BT (2017) TREM2 deficiency exacerbates tau pathology through dysregulated kinase signaling in a mouse model of tauopathy. Mol Neurodegener 12, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Leyns CEG, Gratuze M, Narasimhan S, Jain N, Koscal LJ, Jiang H, Manis M, Colonna M, Lee VMY, Ulrich JD, Holtzman DM (2019) TREM2 function impedes tau seeding in neuritic plaques. Nat Neurosci 22, 1217–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, Poliani PL, Cominelli M, Grover S, Gilfillan S, Cella M, Ulland TK, Zaitsev K, Miyashita A, Ikeuchi T, Sainouchi M, Kakita A, Bennett DA, Schneider JA, Nichols MR, Beausoleil SA, Ulrich JD, Holtzman DM, Artyomov MN, Colonna M (2020) Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med 26, 131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gratuze M, Leyns CE, Sauerbeck AD, St-Pierre MK, Xiong M, Kim N, Serrano JR, Tremblay ME, Kummer TT, Colonna M, Ulrich JD, Holtzman DM (2020) Impact of TREM2R47H variant on tau pathology-induced gliosis and neurodegeneration. J Clin Invest 130, 4954–4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274, 99–102. [DOI] [PubMed] [Google Scholar]
- [25].Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, Xu G, Margevicius D, Karlo JC, Sousa GL, Cotleur AC, Butovsky O, Bekris L, Staugaitis SM, Leverenz JB, Pimplikar SW, Landreth GE, Howell GR, Ransohoff RM, Lamb BT (2015) TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J Exp Med 212, 287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Scott L, Feng J, Kiss T, Needle E, Atchison K, Kawabe TT, Milici AJ, Hajos-Korcsok E, Riddell D, Hajos M (2012) Age-dependent disruption in hippocampal theta oscillation in amyloid-beta overproducing transgenic mice. Neurobiol Aging 33, 1481 e1413–1423. [DOI] [PubMed] [Google Scholar]
- [27].Scott L, Kiss T, Kawabe TT, Hajos M (2016) Neuronal network activity in the hippocampus of tau transgenic (Tg4510) mice. Neurobiol Aging 37, 66–73. [DOI] [PubMed] [Google Scholar]
- [28].Stoiljkovic M, Kelley C, Hajos GP, Nagy D, Koenig G, Leventhal L, Hajos M (2016) Hippocampal network dynamics in response to alpha7 nACh receptors activation in amyloid-beta overproducing transgenic mice. Neurobiol Aging 45, 161–168. [DOI] [PubMed] [Google Scholar]
- [29].Stoiljkovic M, Kelley C, Stutz B, Horvath TL, Hajos M (2019) Altered Cortical and Hippocampal Excitability in TgF344-AD Rats Modeling Alzheimer’s Disease Pathology. Cereb Cortex 29, 2716–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Paxinos GE, Franklin K (2012) Paxinos and Franklin’s The mouse brain in stereotaxic coordinates., Academic Press. [Google Scholar]
- [31].Maggi CA, Meli A (1986) Suitability of urethane anesthesia for physiopharmacological investigations in various systems. Part 1: General considerations. Experientia 42, 109–114. [DOI] [PubMed] [Google Scholar]
- [32].Delorme A, Makeig S (2004) EEGLAB: an open source toolbox for analysis of single-trial EEG dynamics including independent component analysis. J Neurosci Methods 134, 9–21. [DOI] [PubMed] [Google Scholar]
- [33].Tort AB, Komorowski RW, Manns JR, Kopell NJ, Eichenbaum H (2009) Theta-gamma coupling increases during the learning of item-context associations. Proc Natl Acad Sci U S A 106, 20942–20947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Onslow AC, Bogacz R, Jones MW (2011) Quantifying phase-amplitude coupling in neuronal network oscillations. Prog Biophys Mol Biol 105, 49–57. [DOI] [PubMed] [Google Scholar]
- [35].McNaughton N, Kocsis B, Hajos M (2007) Elicited hippocampal theta rhythm: a screen for anxiolytic and procognitive drugs through changes in hippocampal function? Behav Pharmacol 18, 329–346. [DOI] [PubMed] [Google Scholar]
- [36].Stoiljkovic M, Kelley C, Nagy D, Hajos M (2015) Modulation of hippocampal neuronal network oscillations by alpha7 nACh receptors. Biochem Pharmacol 97, 445–453. [DOI] [PubMed] [Google Scholar]
- [37].Giorgi FS, Saccaro LF, Busceti CL, Biagioni F, Fornai F (2020) Epilepsy and Alzheimer’s Disease: Potential mechanisms for an association. Brain Res Bull 160, 107–120. [DOI] [PubMed] [Google Scholar]
- [38].Vossel KA, Tartaglia MC, Nygaard HB, Zeman AZ, Miller BL (2017) Epileptic activity in Alzheimer’s disease: causes and clinical relevance. Lancet Neurol 16, 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lam AD, Deck G, Goldman A, Eskandar EN, Noebels J, Cole AJ (2017) Silent hippocampal seizures and spikes identified by foramen ovale electrodes in Alzheimer’s disease. Nat Med 23, 678–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Vossel KA, Beagle AJ, Rabinovici GD, Shu H, Lee SE, Naasan G, Hegde M, Cornes SB, Henry ML, Nelson AB, Seeley WW, Geschwind MD, Gorno-Tempini ML, Shih T, Kirsch HE, Garcia PA, Miller BL, Mucke L (2013) Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol 70, 1158–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lyou HJ, Seo KD, Lee JE, Pak HY, Lee JH (2018) Association of Alzheimer’s Disease with the Risk of Developing Epilepsy: a 10-Year Nationwide Cohort Study. Dement Neurocogn Disord 17, 156–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kam K, Duffy AM, Moretto J, LaFrancois JJ, Scharfman HE (2016) Interictal spikes during sleep are an early defect in the Tg2576 mouse model of beta-amyloid neuropathology. Sci Rep 6, 20119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L (2007) Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 55, 697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Filipello F, Morini R, Corradini I, Zerbi V, Canzi A, Michalski B, Erreni M, Markicevic M, Starvaggi-Cucuzza C, Otero K, Piccio L, Cignarella F, Perrucci F, Tamborini M, Genua M, Rajendran L, Menna E, Vetrano S, Fahnestock M, Paolicelli RC, Matteoli M (2018) The Microglial Innate Immune Receptor TREM2 Is Required for Synapse Elimination and Normal Brain Connectivity. Immunity 48, 979–991 e978. [DOI] [PubMed] [Google Scholar]
- [45].Berry KP, Nedivi E (2017) Spine Dynamics: Are They All the Same? Neuron 96, 43–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Jadhav VS, Lin PBC, Pennington T, Di Prisco GV, Jannu AJ, Xu G, Moutinho M, Zhang J, Atwood BK, Puntambekar SS, Bissel SJ, Oblak AL, Landreth GE, Lamb BT (2020) Trem2 Y38C mutation and loss of Trem2 impairs neuronal synapses in adult mice. Mol Neurodegener 15, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kalueff AV, Stewart AM, Song C, Berridge KC, Graybiel AM, Fentress JC (2016) Neurobiology of rodent self-grooming and its value for translational neuroscience. Nat Rev Neurosci 17, 45–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Chu Y, Jin X, Parada I, Pesic A, Stevens B, Barres B, Prince DA (2010) Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc Natl Acad Sci U S A 107, 7975–7980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zhou YD, Lee S, Jin Z, Wright M, Smith SE, Anderson MP (2009) Arrested maturation of excitatory synapses in autosomal dominant lateral temporal lobe epilepsy. Nat Med 15, 1208–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, Garaschuk O (2008) Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 321, 1686–1689. [DOI] [PubMed] [Google Scholar]
- [51].Zott B, Simon MM, Hong W, Unger F, Chen-Engerer HJ, Frosch MP, Sakmann B, Walsh DM, Konnerth A (2019) A vicious cycle of beta amyloid-dependent neuronal hyperactivation. Science 365, 559–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Yamamoto K, Tanei ZI, Hashimoto T, Wakabayashi T, Okuno H, Naka Y, Yizhar O, Fenno LE, Fukayama M, Bito H, Cirrito JR, Holtzman DM, Deisseroth K, Iwatsubo T (2015) Chronic optogenetic activation augments abeta pathology in a mouse model of Alzheimer disease. Cell Rep 11, 859–865. [DOI] [PubMed] [Google Scholar]
- [53].Abiega O, Beccari S, Diaz-Aparicio I, Nadjar A, Laye S, Leyrolle Q, Gomez-Nicola D, Domercq M, Perez-Samartin A, Sanchez-Zafra V, Paris I, Valero J, Savage JC, Hui CW, Tremblay ME, Deudero JJ, Brewster AL, Anderson AE, Zaldumbide L, Galbarriatu L, Marinas A, Vivanco M, Matute C, Maletic-Savatic M, Encinas JM, Sierra A (2016) Neuronal Hyperactivity Disturbs ATP Microgradients, Impairs Microglial Motility, and Reduces Phagocytic Receptor Expression Triggering Apoptosis/Microglial Phagocytosis Uncoupling. PLoS Biol 14, e1002466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Suarez-Calvet M, Kleinberger G, Araque Caballero MA, Brendel M, Rominger A, Alcolea D, Fortea J, Lleo A, Blesa R, Gispert JD, Sanchez-Valle R, Antonell A, Rami L, Molinuevo JL, Brosseron F, Traschutz A, Heneka MT, Struyfs H, Engelborghs S, Sleegers K, Van Broeckhoven C, Zetterberg H, Nellgard B, Blennow K, Crispin A, Ewers M, Haass C (2016) sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO Mol Med 8, 466–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].El Hajj H, Savage JC, Bisht K, Parent M, Vallieres L, Rivest S, Tremblay ME (2019) Ultrastructural evidence of microglial heterogeneity in Alzheimer’s disease amyloid pathology. J Neuroinflammation 16, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B (2016) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Babiloni C, Blinowska K, Bonanni L, Cichocki A, De Haan W, Del Percio C, Dubois B, Escudero J, Fernandez A, Frisoni G, Guntekin B, Hajos M, Hampel H, Ifeachor E, Kilborn K, Kumar S, Johnsen K, Johannsson M, Jeong J, LeBeau F, Lizio R, Lopes da Silva F, Maestu F, McGeown WJ, McKeith I, Moretti DV, Nobili F, Olichney J, Onofrj M, Palop JJ, Rowan M, Stocchi F, Struzik ZM, Tanila H, Teipel S, Taylor JP, Weiergraber M, Yener G, Young-Pearse T, Drinkenburg WH, Randall F (2020) What electrophysiology tells us about Alzheimer’s disease: a window into the synchronization and connectivity of brain neurons. Neurobiol Aging 85, 58–73. [DOI] [PubMed] [Google Scholar]
- [58].Babiloni C, Del Percio C, Lizio R, Noce G, Cordone S, Lopez S, Soricelli A, Ferri R, Pascarelli MT, Nobili F, Arnaldi D, Fama F, Aarsland D, Orzi F, Buttinelli C, Giubilei F, Onofrj M, Stocchi F, Stirpe P, Fuhr P, Gschwandtner U, Ransmayr G, Caravias G, Garn H, Sorpresi F, Pievani M, D’Antonio F, De Lena C, Guntekin B, Hanoglu L, Basar E, Yener G, Emek-Savas DD, Triggiani AI, Franciotti R, Frisoni GB, Bonanni L, De Pandis MF (2017) Abnormalities of Cortical Neural Synchronization Mechanisms in Subjects with Mild Cognitive Impairment due to Alzheimer’s and Parkinson’s Diseases: An EEG Study. J Alzheimers Dis 59, 339–358. [DOI] [PubMed] [Google Scholar]
- [59].Rodriguez G, Copello F, Vitali P, Perego G, Nobili F (1999) EEG spectral profile to stage Alzheimer’s disease. Clin Neurophysiol 110, 1831–1837. [DOI] [PubMed] [Google Scholar]
- [60].Buzsaki G (2002) Theta oscillations in the hippocampus. Neuron 33, 325–340. [DOI] [PubMed] [Google Scholar]
- [61].Cornwell BR, Arkin N, Overstreet C, Carver FW, Grillon C (2012) Distinct contributions of human hippocampal theta to spatial cognition and anxiety. Hippocampus 22, 1848–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Lega BC, Jacobs J, Kahana M (2012) Human hippocampal theta oscillations and the formation of episodic memories. Hippocampus 22, 748–761. [DOI] [PubMed] [Google Scholar]
- [63].Vertes RP (2005) Hippocampal theta rhythm: a tag for short-term memory. Hippocampus 15, 923–935. [DOI] [PubMed] [Google Scholar]
- [64].Guadagna S, Bundgaard C, Hovelso N, Volbracht C, Francis PT, Egebjerg J, Sotty F (2012) Memantine potentiates hippocampal theta oscillations at a therapeutic dose in anesthetized mice: a mechanistic link to its cognitive-enhancing properties. Neuropharmacology 62, 2208–2218. [DOI] [PubMed] [Google Scholar]
- [65].Kinney GG, Patino P, Mermet-Bouvier Y, Starrett JE Jr., Gribkoff VK (1999) Cognition-enhancing drugs increase stimulated hippocampal theta rhythm amplitude in the urethane-anesthetized rat. J Pharmacol Exp Ther 291, 99–106. [PubMed] [Google Scholar]
- [66].Axmacher N, Henseler MM, Jensen O, Weinreich I, Elger CE, Fell J (2010) Cross-frequency coupling supports multi-item working memory in the human hippocampus. Proc Natl Acad Sci U S A 107, 3228–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Canolty RT, Knight RT (2010) The functional role of cross-frequency coupling. Trends Cogn Sci 14, 506–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Trimper JB, Stefanescu RA, Manns JR (2014) Recognition memory and theta-gamma interactions in the hippocampus. Hippocampus 24, 341–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Goutagny R, Gu N, Cavanagh C, Jackson J, Chabot JG, Quirion R, Krantic S, Williams S (2013) Alterations in hippocampal network oscillations and theta-gamma coupling arise before Abeta overproduction in a mouse model of Alzheimer’s disease. Eur J Neurosci 37, 1896–1902. [DOI] [PubMed] [Google Scholar]
- [70].Gurevicius K, Lipponen A, Tanila H (2013) Increased cortical and thalamic excitability in freely moving APPswe/PS1dE9 mice modeling epileptic activity associated with Alzheimer’s disease. Cereb Cortex 23, 1148–1158. [DOI] [PubMed] [Google Scholar]
- [71].Suzuki SS, Smith GK (1988) Spontaneous EEG spikes in the normal hippocampus. V. Effects of ether, urethane, pentobarbital, atropine, diazepam and bicuculline. Electroencephalogr Clin Neurophysiol 70, 84–95. [DOI] [PubMed] [Google Scholar]
- [72].Yagishita H, Nishimura Y, Noguchi A, Shikano Y, Ikegaya Y, Sasaki T (2020) Urethane anesthesia suppresses hippocampal subthreshold activity and neuronal synchronization. Brain Res 1749, 147137. [DOI] [PubMed] [Google Scholar]
- [73].Chan J, Jones NC, Bush AI, O’Brien TJ, Kwan P (2015) A mouse model of Alzheimer’s disease displays increased susceptibility to kindling and seizure-associated death. Epilepsia 56, e73–77. [DOI] [PubMed] [Google Scholar]