

Abstract
The Ras/Erk and NF-κB pathways play critical roles in cell proliferation and are known to drive oncogenesis when over-activated. Herein we report a gatekeeper function of the two pathways by working in synergy to suppress liver tumorigenesis. Hepatocyte-specific deletion of both Shp2/Ptpn11 and Ikkβ in mice, which promote Ras/Erk and NF-κB signaling, respectively, exacerbated chemical carcinogenesis and even triggered spontaneous development of hepatocellular carcinoma (HCC). We show that the unanticipated severe tumor phenotype was contributed collectively by severe cholestasis, metabolic changes, upregulated cell cycle progression and disruption of circadian rhythm in mutant hepatocytes. Remarkably, human HCCs with dysregulated circadian gene expression displayed downregulation of Ras/Erk and NF-κB signaling and poor prognosis. Together, these data indicate that at the ground state, the two central pathways, previously known as oncogenic, cooperate to sustain tumor-suppressive physiological homeostasis and to prevent hepatic damage. Disruption of this intricate signaling network is carcinogenic in the liver.
Keywords: Hepatocellular carcinoma, NF-κB pathway, Ras/MAPK pathway, Shp2, circadian dysregulation
Introduction
Hepatocellular carcinoma (HCC) is a rising cause of cancer mortality worldwide [1]. Large scale genomic and molecular analyses of HCC specimens suggest the involvement of a heterogenous collection of tumor initiating oncogenic pathways [2]. Consistent with this heterogeneity, no effective targeted therapies have been established; the multi-kinase inhibitors that block Raf and VEGFR signaling offer only limited therapeutic benefit to HCC patients [3, 4], underscoring the complexity of liver tumorigenesis [5].
The Ras protooncogenes constitute the most frequently mutated gene family in human cancer [6–8]. RASopathies represent a group of developmental disorders driven by aberrant Ras/MAPK signaling [8] that are frequently associated with increased risk of cancer. Shp2 is a cytoplasmic tyrosine phosphatase that promotes signaling from receptor tyrosine kinases (RTKs) to the Ras/Erk pathway [9]. Dominant active mutations in PTPN11/Shp2 augment Erk signaling in patients with leukemias and solid tumors [9, 10]. Concurrent inhibition of Shp2 and other components in the Ras/Erk pathway was shown to effectively suppress tumor cell proliferation [11, 12]. The NF-κB family consists of five DNA-binding proteins in homo- or heterodimers [13]. The inactive dimers are retained in the cytoplasm by binding to the IκB inhibitor proteins. Ikkβ is a critical component of the IκB kinase (IKK) complex, which mediates phosphorylation and degradation of IκB, leading to nuclear translocation and activation of NF-κB. Overactivation of NF-κB leads to pleiotropic oncogenic processes [13], and disrupting the pathway has been shown to suppress tumor progression in a mouse HCC model [14]. Indeed, NF-κB is constitutively active in many types of cancer [13].
However, ablating either Shp2 or Ikkβ in hepatocytes ironically aggravates HCC induced by a chemical carcinogen [15, 16]. One possible explanation for the paradoxical anti-tumoral effect of the oncoproteins is forced activation of compensatory pathway(s) triggered by disruption of one critical oncogenic route [5]. Elucidating the compensatory mechanisms will hopefully guide design of more efficacious HCC therapy by simultaneously blocking parallel pathways. Toward this goal, we generated a double knockout (DKO) mouse line with both Shp2 and Ikkβ deleted in hepatocytes. Surprisingly, concurrent disruption of the Ras/MAPK and NF-κB pathways induced more severe liver tumorigenesis. The DKO mice displayed multiple liver injuries, chronic inflammation, severe cholestasis, changed metabolic pathways, deregulated cell cycle gene expression and circadian clock dysregulation. These mutually enhancing disorders and hepatic damages drove spontaneous HCC development in the liver. Thus, this study revealed a previously unrecognized tumor-inhibiting role of baseline Ras/MAPK and NF-κB signaling, associated with hepatic protection and physiological homeostasis. These two pathways are actually dispensable for hepatocyte proliferation and even malignant transformation in the liver.
Materials and Methods
Mice.
C57BL/6 mice were maintained in 12-hour light/dark cycles with standard chow and water ad libitum. Mice with hepatocyte-specific Shp2 deletion (Alb-Cre:Shp2f/f:SKO) were previously generated by G.S.F’s group. Ikkβf/f mice were generated by M.K.’s group at UCSD. These lines were crossed to generate double-knockout mice (DKO, Alb-Cre:Ikkβf/f:Shp2f/f). Mice lacking the Alb-Cre transgene were used as wild-type controls (WT). All mouse husbandry and procedures were performed in compliance with protocols approved by the UCSD Institutional Animal Care and Use Committee (protocol S09108).
Genotyping was performed by PCR analysis of genomic DNA extracted from tail snips using primers listed in Supplemental File 1.
Mouse livers and other tissue samples were collected during the same four-hour period each day (ZT 7-11, with vivarium lights-on time designated ZT 0). For tumor initiation, mice were intraperitoneally injected with 25 mg/kg diethylnitrosamine (DEN; N0258, Sigma-Aldrich) at postnatal day 15.
Histology and immunostaining.
Liver samples harvested from mice were fixed in Z-Fix (175, Anatech) and paraffin-embedded. Hematoxylin and Eosin (H&E) staining was performed by the UCSD Histology Core. Hepatocyte proliferation was assessed by costaining for Ki67 (eBioscience, 47569880; RRID:AB_2688065) and HNF4α (Santa Cruz Biotechnologies, 8987; RRID:AB_2116913). Detection of macrophages was performed by staining paraffin sections for F4/80 (eBioscience, 14-4801-81; RRID:AB_467557). Picro-Sirius Red staining for detection of fibrosis was performed according to kit instructions (American MasterTech, STPSR).
Primary hepatocyte isolation and culture.
Hepatocytes were isolated by the two-step perfusion method. Briefly, mice were humanely euthanized and livers were immediately perfused with perfusion buffer (HBSS without calcium and magnesium, HEPES, EGTA), then with digestion buffer (HBSS with calcium and magnesium, HEPES, collagenase H (11074059001, Sigma)). Digested livers were suspended in cold PBS, then centrifuged three times at 50g for three minutes to obtain a pure hepatocyte fraction. For RNA-Seq analysis, isolated hepatocytes were immediately flash-frozen. For growth factor stimulation experiments and in vitro circadian experiments, freshly isolated hepatocytes were cultured as follows.
Immediately after isolation, hepatocytes were plated on collagen-coated (C8919, Sigma) plates in Williams’ Media E (W1878, Sigma) supplemented with 10% FBS and Pen/Strep (15140, Gibco). For signaling experiments, hepatocytes were serum-starved, then stimulated with mouse recombinant IL-6 (BioLegend, 575702), TNFα (BioLegend, 575202), or mouse recombinant HGF (Sigma, SRP3300) before harvesting in RIPA. For circadian time course experiments, primary hepatocytes were allowed to adhere and recover overnight in complete media. In order to synchronize the cultures, they were serum-starved for 12 hours, then serum shocked with 50% FBS for two hours. Synchronized hepatocytes were harvested in Trizol reagent at four-hour intervals over a course of 24 hours. Time points were designated by the Zeitgeiber Time (ZT) relative to the start of serum shock (ZT0).
Immune cell isolation and flow cytometric analysis.
Spleens and liver-draining lymph nodes were collected from humanely euthanized mice between 8–12 weeks of age and homogenized through a 70 μm cell strainer, then further suspended using a 27-gauge needle. Liver non-parenchymal cells were isolated by two-step perfusion as described in “primary hepatocyte isolation and culture”. Hepatocytes were removed from the cell suspension by sequential centrifugation at 50xg; the supernatants containing NPCs were centrifuged at 300xg for 5 minutes to pellet the cells. Red blood cells were removed from NPCs, splenocytes, and lymph node suspensions by incubating in ACK lysis buffer for 5 minutes at room temperature. Cleared cells were pelleted by centrifugation at 500 g for 5 minutes, then washed twice in phosphate-buffered saline (PBS).
Surface marker staining was performed as follows: First, viability staining was performed for 20 minutes on ice in PBS with Tonbo Ghost Dye UV450, with all samples protected from light through this and subsequent steps. Conjugated antibodies were added and cells were incubated 15–20 minutes on ice. See Supplemental File 1 for antibody panels used. Cells were washed 3 times in staining buffer (PBS with 5% FBS and 0.1% sodium azide).
Intracellular staining was performed using the Foxp3 Fix/Perm solution (BioLegend) according to manufacturer’s instructions.
Immunoblotting.
Lysates from whole liver or isolated hepatocytes were prepared in RIPA (10mM Tris-HCl, 1mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 140 mM NaCl) with protease inhibitors and phosphatase inhibitors. SDS-PAGE and immunoblotting were performed according to standard procedures using the antibodies listed in Supplemental File 1.
Serum Biochemistry.
Serum was collected from 2-month-old male mice sacrificed between ZT7-11 and immediately flash-frozen. Serum AST and ALT were measured using commercially available kits (Sekisui).
Total bile acid analysis.
Serum bile acids were measured using a commercially available kit (Abcam, ab239702) according to kit instructions. Liver total bile acids were extracted from 50 mg liver and homogenized in 1 ml of 70% ethanol at room temperature. The suspension was incubated for 3 hours at 50 °C, then clarified by centrifugation at 10,000 rpm for 10 minutes at room temperature. The supernatant was used for analysis of bile acid levels.
RNA Sequencing.
Whole-liver samples or freshly isolated hepatocytes were collected during the same time period each day (ZT 7-11), and immediately frozen. RNA was extracted from samples of whole liver using RNeasy Microarray Tissue Mini Kit (73304, Qiagen) or from isolated hepatocytes using RNeasy Plus Micro Kit (74034, Qiagen). Libraries were prepared using Illumina TruSeq v2 kit according to manufacturer’s instructions. Sequencing was performed at UCSD Genomics Core on the HiSeq4000 platform. Raw data quality control was performed using fastqc. Sequenced reads were mapped to the mm9 reference genome using STAR, and gene differential expression analysis was performed using Cuffdiff with fragments per kilobase million (FPKM) values. To compare different expression levels of genes between KO and WT, we used the average FPKM in each group (n=3). Significantly differentially expressed genes were determined with |log2FC| > 0.5 and q-value < 0.05. Changes unique to DKO were found by overlapping the significantly differentially expressed genes in DKO vs. WT, DKO vs. IKO, and DKO vs. SKO comparisons; for a gene to be considered differentially expressed in DKO, it must have |log2FC| > 0.5 and q-value < 0.05 compared to each other genotype.
For gene expression heatmaps, we first compared gene expression among the 4 groups (WT, SKO, IKO, DKO) with function scale () in Rstudio, where we scaled the genes in range (−1 to 1), as indicated in each color key. Each column in the heatmap represents average expression level of the three biological replicates within each genotype. Heatmaps were made with R package heatmap.2. For pathway analysis, we first separated up- and down-regulated genes, then analyzed each list using the GSEA software’s pre-ranked tool. The top dysregulated gene sets for each genotype comparison, or for the DKO-unique dysregulated genes, were ranked in bar plots in RStudio, with package ggplot. The abbreviated pathway names are listed in Supplemental File 1 with respective original names. Enrichment plots for immune- and metabolism-related pathways were selected from the top 20 positively and negatively enriched pathways in the DKO vs. WT comparison, respectively. See Supplemental File 2 for the full pathway enrichment results for all whole-liver and hepatocyte comparisons between genotypes.
TCGA analysis.
The raw counts of TCGA hepatocellular carcinoma (LIHC) patient transcriptomic dataset were downloaded from Genomic Data Commons Data Portal (https://portal.gdc.cancer.gov/). This dataset includes 371 HCC patient samples and 50 adjacent normal liver tissues. The R package DESeq2 was used to normalize raw counts across all samples. Next, we divided all HCC samples into three groups: (1) circadian clock-normal, (2) circadian clock-mid, (3) circadian clock-dysregulated, based on whether circadian clock genes were dysregulated or not in these patient samples compared to non-tumor tissues. In detail, we checked expression of 7 core clock genes, including CLOCK, ARNTL, PER1, PER2, PER3, CRY1, and CRY2. For each gene, the mean and standard deviation (sd) of normalized values in non-tumor tissues were calculated. We set (mean-sd) as lower bound and (mean+sd) as upper bound, then we defined a HCC patient with this circadian clock gene considered dysregulated if its normalized value was higher than upper bound or lower than lower bound. We repeated this process for all 7 circadian clock genes. Next, we assigned patients with at most 2 dysregulated circadian clock genes into “normal” group, patients with at least 5 dysregulated circadian clock genes into “dysregulated” group, and others into “mid” group. With sub-clustering of all HCC patients, we first performed differential expression analysis between “normal” and “dysregulated” groups using DESeq2. The genes with adjusted p value < 0.05 were defined as differentially expressed. We specifically checked components from Ras/MAPK signaling pathway and NF-κB pathway, then made volcano plots with differentially expressed genes in pathways above. Next, in order to further verify whether activities of these two pathways were inhibited in dysregulated group, we collected downstream targets of Ras/MAPK pathway as well as NF-κB pathway from Msigdb gene sets HALLMARK_TNFA_SIGNALING_VIA_NFKB and BILD_HRAS_ONCOGENIC_SIGNATURE, respectively. The pathway activity was defined as the sum of rank-transformed normalized values of all genes in corresponding gene set. The boxplot was made using Wilcoxon rank sum test.
Survival analysis was performed using OncoLnc (http://www.oncolnc.org/), with “high” and “low” expression defined as the upper and lower quartiles of expression for each gene. For expression levels of individual clock genes linked to altered survival, the raw counts in tumor and nontumor liver samples from the TCGA cohort were normalized using DESeq2, as described above. To assess association of individual clock gene disruption with broader circadian deregulation within tumors, patients with upper- or lower- quartile expression of select individual clock genes were divided into “normal”, “mid” and “dysregulated” groups, as previously defined.
qRT-PCR.
RNA from whole liver or from freshly isolated hepatocytes was isolated using a standard phenol-chloroform extraction with Trizol reagent according to manufacturer’s protocol. cDNA was prepared using High-Capacity cDNA Reverse Transcription Kit (Life Technologies, 4368813). Quantitative reverse transcription was performed using PowerUP SYBR Green PCR Master Mix (Invitrogen, A25743) and the primers listed below on the Stratagene MX3005P. Ct values for each transcript were normalized to β-actin and fold change was determined by the ΔΔCt method compared to control. Primer sequences are listed in Supplemental File 1.
Statistical Analysis.
Multiple comparisons were performed using one-way ANOVA with Tukey post-hoc analysis in GraphPad Prism. P<.05 was considered statistically significant. (*P<0.05; **P<0.01; ***P<0.001). Data are presented as box plots with center line representing mean and whiskers indicating range.
Data and materials availability:
The transcriptomic data generated in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE158381 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE158381).
An original mouse line (Alb-Cre;Ikkβf/f;Shp2f/f) was generated in this study; please contact the corresponding author for resource sharing.
Results
Shp2 and Ikkβ cooperatively suppress liver tumorigenesis
To interrogate pathway crosstalk, we crossed Shp2f/f:Albumin-Cre mice [17] with Ikkβf/f mice [15], to generate a DKO mouse line lacking both Shp2 and Ikkβ in hepatocytes (Supp. Fig. S1A,B). The DKO mice were born at the expected Mendelian frequency with no obvious health problems at young ages, similar to single Shp2 (SKO) or Ikkβ (IKO) hepatocyte-specific knockouts. We first examined the susceptibility of all three mutant mouse lines to diethylnitrosamine (DEN)-induced carcinogenesis, which entails multiple mutations frequently detected in human HCCs [18]. Since both the Ras/MAPK and NF-κB pathways constitute major signaling nodes controlling cellular growth and survival, we speculated that dual inhibition of these pathways might counteract the compensatory proliferation and/or malignant transformation previously reported in either single-knockout, as is the case for combined deletion of Shp2 and Stat3 [16]. We compared liver tumor loads in WT, SKO, IKO and DKO mice at 5 and 8 months post DEN injection (Fig. 1A, B; Supp. Fig. S1C,D). The DKO mice unexpectedly exhibited much higher tumor burdens than IKO and SKO, and showed tumors at the liver surface as early as 5 months after DEN injection, when only a few small macroscopic nodules and microscopic lesions were detected in some IKO and SKO mice (Fig. 1A–C; Supp. Fig. S1D). Enhanced HCC development in DKO mice became even more striking at 8 months post DEN (Fig. 1A–B; Supp. Fig. S1C). Histopathological examination of liver sections identified the tumors as HCC (Fig. 1C; Supp. Fig. S1E). These results indicate that, contrary to our predictions, simultaneous disruption of Ras/MAPK and NF-κB signaling further aggravated chemical carcinogenesis.
Fig. 1. Shp2 and Ikkβ cooperate to suppress carcinogen-induced and spontaneous liver tumorigenesis.
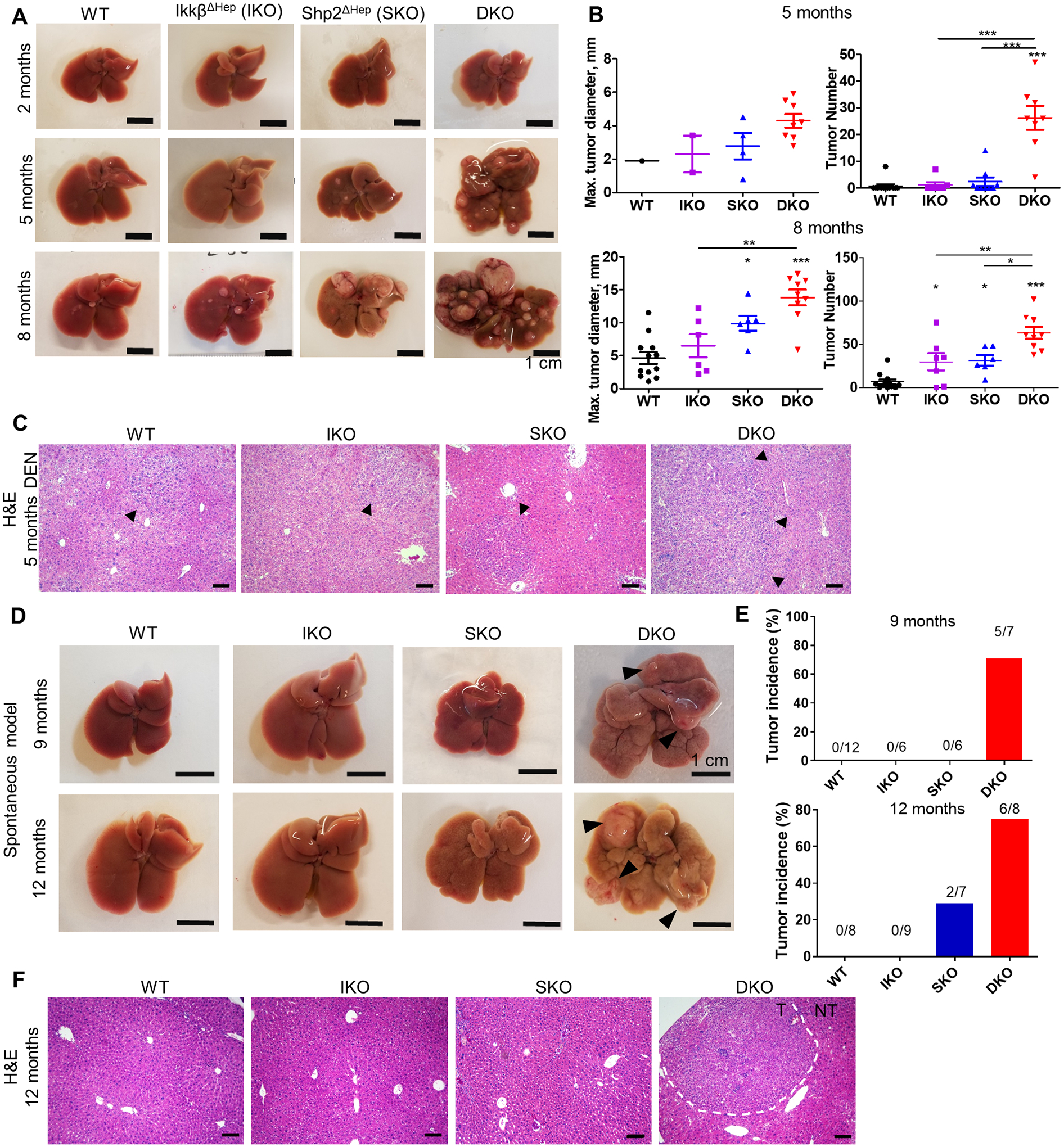
(A) Representative images of livers from DEN-treated male mice at 2, 5, and 8 months of age. Scale bar= 1cm. (B) Quantification of maximal tumor diameters (left) and tumor numbers (right) per liver in DEN-treated mice at 5 and 8 months of age. (C) Representative H&E staining of liver sections from 8-month-old DEN-treated mice. Arrows indicate microscopic nodules or, in DKO, macroscopically visible tumor margins. Scale bar=100 μm. (D) Representative images of livers from untreated male mice at 9 and 12 months of age. Arrowheads highlight macroscopic tumors in the DKO liver. (E) Tumor incidences, as percentages of total mice, in 9- or 12-month-old untreated male mice, with the number of tumor-bearing mice over the total number of mice indicated above the bars for each genotype. (F) H&E staining of liver sections from 12-month-old untreated male mice. Dotted lines indicate boundaries between tumor (T) and non-tumor (NT) areas. Scale bar=100 μm.
Even more surprising was the detection of spontaneous HCC development in DKO mice, without any exogenous tumor-promoting treatment, which was not observed in either IKO or SKO livers (Fig. 1D–E; Supp. Fig. S1F,G). When examining the mouse colonies without DEN injection, we observed spontaneous liver tumors in DKO mice at 9 months of age, a time at which no tumor nodules were detected in WT, IKO or SKO mice (Fig. 1D–E; Supp. Fig. S1G). At 12 months, 75% of DKO mice showed large liver tumors, whereas only 29% of SKO and no IKO mice showed small benign hepatocellular adenomas (HCA) (Fig. 1D–E; Supp. Fig. S1H). The DKO livers exhibited substantial cytological atypia, and the spontaneously developed tumors in DKO liver exhibited typical HCC morphology (Fig. 1F; Supp. Fig. S1G). These results indicate ironically that concurrent inactivation of two pro-oncogenic pathways, Ras/MAPK and NF-κB, drives carcinogenesis in the liver.
Dual Shp2 and Ikkβ ablation triggers more severe hepatic damages and a tumor-permissive microenvironment
To dissect the factors underlying spontaneous liver tumorigenesis, we examined the liver phenotypes of young, non-DEN-treated mice of the four genotypes prior to tumor emergence. At 2 months of age, the DKO livers already displayed irregular lobe edges and visible crevices (Fig. 2A). The IKO livers had no gross structural or developmental defects and the SKO livers exhibited mild fibrosis around the portal triads at the early stage; these fibrotic and periportal changes were dramatically enhanced in DKO mice (Fig. 2B–C). Consistent with the anatomic lesions, qRT-PCR detected progressively increased levels of fibrosis-related Col1α1 and Tgfβ mRNAs in IKO, SKO and DKO livers at 2 months (Fig. 2D); however, additional molecular and pathological analyses are required to characterize the fibrosis phenotype in DKO livers. Serum AST and ALT levels were slightly elevated in single KO and significantly higher in double KO mice (Fig. 2E). SKO mice displayed modestly increased serum and liver bile acids (BA) (Fig. 2F–G), reinforcing a role for Shp2 in control of BA biosynthesis [19]. Although deleting Ikkβ alone did not alter BA levels, both the intrahepatic and serum BA amounts were markedly elevated in DKO mice (Fig. 2F–G), indicative of severe cholestasis induced by synergistic effect of dual gene deletion.
Fig. 2. Shp2 and Ikkβ cooperate to suppress liver damage.
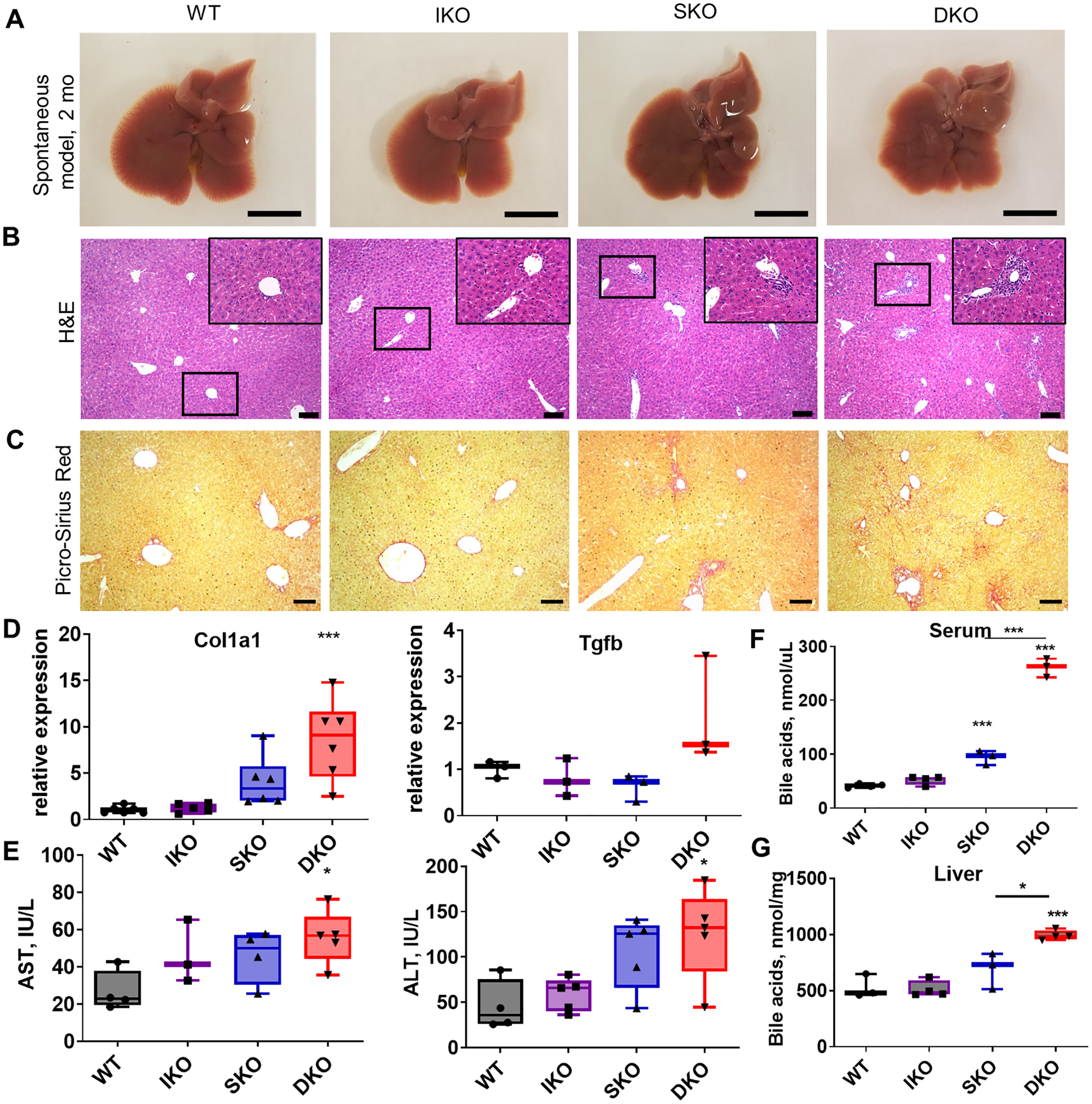
(A) Representative images of livers from 2-month-old untreated male mice. Scale bar=1 cm. (B) H&E staining of sections from untreated livers at 2 months. Insets represent further 4x magnification of indicated regions. Scale bar=100 μm. (C) Picro-Sirius Red staining for fibrosis in livers from 2-month-old mice. Scale bar=100 μm. (D) mRNA levels of fibrosis-associated genes in whole-liver lysates from 2-month-old mice normalized to β-actin (n=3–6/genotype). (E) Serum ALT and AST (n=3–5/genotype). (F) Total serum bile acids for 2-month-old male mice (n=3–5/genotype). (G) Total liver bile acids for 2-month-old male mice (n=3–4/genotype).
Another notable phenotype in the DKO mice was enlargement of spleen and liver-draining lymph nodes as early as 2 months of age (Supp. Fig. S2A–C). We performed flow cytometric analysis of the immune cell sub-populations in the liver, spleen and lymph nodes (Supp. Fig. S2D–F). We detected lowered CD4/CD8 cell ratios and NKT percentages in SKO and DKO livers, relative to WT and IKO livers. Treg cells were elevated in SKO but not in DKO liver (Supp. Fig. S2F). However, we did not observe significant or biased changes in the spleen of DKO mice, with only lower dendritic cells (DCs) in IKO and monocytes in SKO mice (Supp. Fig. S2D). Similar trends were detected in the lymph nodes, with slightly higher ratios of B and NKT cells in DKO mice (Supp. Fig. S2E). Thus, the enlarged spleen was likely related to portal hypertension due to liver injuries rather than a specific immune response.
To decipher the molecular mechanisms underlying the histopathological changes, we performed RNA-seq analysis of the whole liver from 2-month-old mice without DEN treatment. We compared the WT liver transcriptome with those of IKO, SKO or DKO livers, and performed gene set enrichment analysis (GSEA) for differentially expressed genes (Fig. 3A–B; Supp. Fig. 3A–C). With the complete results presented in Supplemental File 2, the analysis of canonical pathways (CP) showed relatively fewer significant transcriptomic changes in the IKO/WT than the SKO/WT comparison (Supp. Fig. S3B), consistent with the severity of respective liver damages and tumor loads. Shared changes in both IKO and SKO livers included cytokine and interferon signaling and innate immune systems, indicating hepatic injuries and inflammatory responses (Supp. Fig. S3B,C). The DKO liver had most of the changes detected in IKO and SKO; however, the DKO/WT comparison also revealed more prominent changes in cell cycle and metabolic pathways (Fig. 3B; Supp. Fig. S3C). Indeed, the DKO liver was particularly distinguished by upregulation in cell cycle, cell division and chromosome segregation pathways. Homeostatic processes and cytokine responses were altered in DKO compared to WT, consistent with the trends seen in the single knockouts (Fig. 3B; Supp. Fig. S3C).
Fig. 3. Inflammatory and metabolic perturbations in mutant livers at early stage.
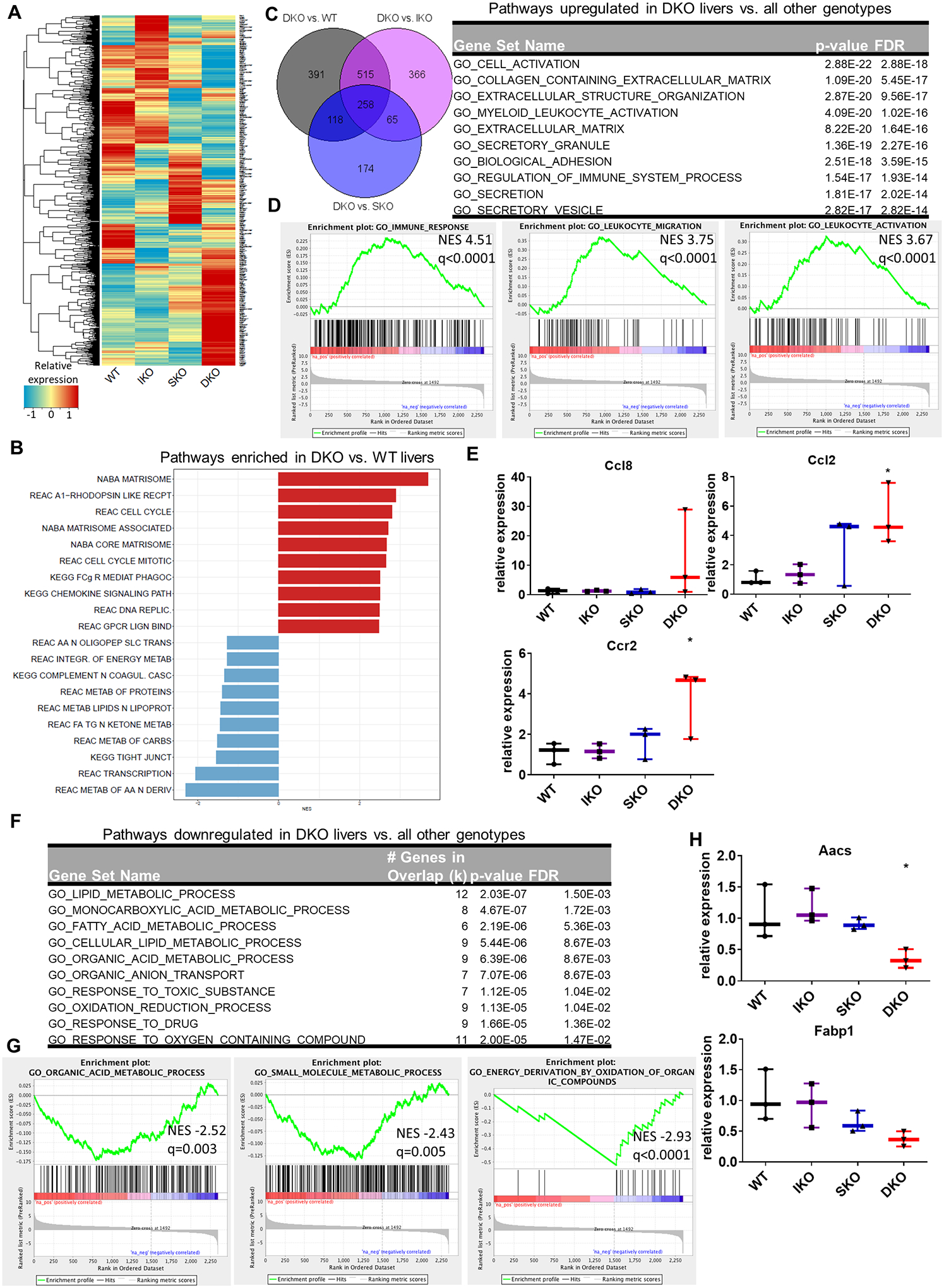
(A) Heatmap of gene expression changes in IKO, SKO, and DKO livers from untreated male mice compared to WT, generated by RNA-Seq of whole-liver transcripts. Color key indicates the relative expression of each gene. (B) Top ten positive and negative gene set enrichment pathways in DKO liver compared to WT using GSEA canonical pathways (CP). (C) Left, overlap of genes upregulated in DKO vs. indicated genotype from 2-month-old whole liver samples. Right, gene set enrichment of the 258 genes upregulated only in DKO livers compared to all other genotypes. (D) Enrichment plots of immune-related gene sets for DKO vs WT livers, all located among the top 20 positively enriched gene sets for this genotype comparison. NES; normalized enrichment score. q indicates FDR q value for each enrichment plot. (E) qRT-PCR analysis of mRNAs associated with macrophage recruitment in 2-month-old livers. (F) Gene set enrichment of the 83 genes uniquely downregulated in DKO livers compared to all other genotypes. (G) Enrichment plots of metabolic gene sets in DKO liver compared to WT, all located within the top 20 negatively enriched genesets for this genotype comparison. (H) qPCR validation of mRNA levels of lipid metabolic enzymes in whole-liver lysates from 2-month-old male mice (n=3/genotype).
We then focused on changes unique to the DKO liver transcriptome only, by identifying genes that were differentially expressed in the DKO livers compared to all other genotypes, and then by determining to which functional gene sets the 258 DKO-unique upregulated genes belonged (Fig. 3C). These upregulated genes were involved in extracellular matrix deposition, and various immune-related pathways such as myeloid and leukocyte activation (Fig. 3C–D). In particular, mRNAs for the macrophage-recruiting cytokine Ccl2 and its cognate receptor Ccr2 were significantly upregulated in the DKO liver tissue only, as detected by qRT-PCR (Fig. 3E). Additionally, expression of the monocyte chemoattractant Ccl8 showed a trend towards upregulation in DKO (Fig. 3E). Immunostaining for the macrophage marker F4/80 also demonstrated increased macrophage presence in DKO livers (Supp. Fig. S4A).
Also noted in the DKO liver were altered metabolic pathways (Fig. 3B,F). By probing the gene-set segregation of DKO-specific downregulated transcripts, we found that perturbed metabolic gene sets included metabolism of lipids and amino acids, transport of metabolic components, and oxidative processes (Fig. 3F–G; Supp. Fig. S4B). Heatmaps of select metabolic pathways revealed modestly altered patterns of gene expression in each single knockout, with the DKO most profoundly perturbed compared to WT (Supp. Fig. S4C). qRT-PCR validated expression changes in select metabolic genes identified by the RNA-Seq data, especially the downregulated expression of the fatty acid metabolic genes Aacs and Fabp in DKO liver (Fig. 3H). Together, these results suggest that removal of both Ikkβ and Shp2 caused more severe hepatic damages, metabolic changes, chronic inflammation and immune suppression, showing additive or synergistic effects of dual gene deletion. These changes collectively contribute to an apparently tumor-promoting niche in DKO livers.
Cell cycle related genes are particularly upregulated in DKO hepatocytes
The above-described hepatic disorders vividly illustrate the drastically augmented susceptibility to chemical carcinogenesis in DKO liver. However, these pathogenic alterations are apparently not sufficient to drive spontaneous HCC development. To identify hepatocyte-intrinsic oncogenic signal(s), we isolated fresh hepatocytes from untreated 2-month-old mice of each genotype and performed RNA-Seq analysis. This approach allowed us to parse out hepatocyte-autonomous changes from microenvironmental factors; the results of these comparative pathway analyses are shown in Supplemental File 2. Comparison of the different transcriptomes (Supp. Fig. S5A–B) revealed higher ROS levels, disordered metabolic pathways and reduced cell proliferation in IKO hepatocytes, while SKO hepatocytes displayed changes in innate immunity and interferon response, as well as epigenetic regulators. Notably, comparing DKO hepatocytes with WT controls revealed markedly increased expression of genes involved in cell mitosis, chromosome segregation and antigen processing (Fig. 4A).
Fig. 4. Shp2- and Ikkβ-deficient hepatocytes exhibit proliferative advantage in vivo.
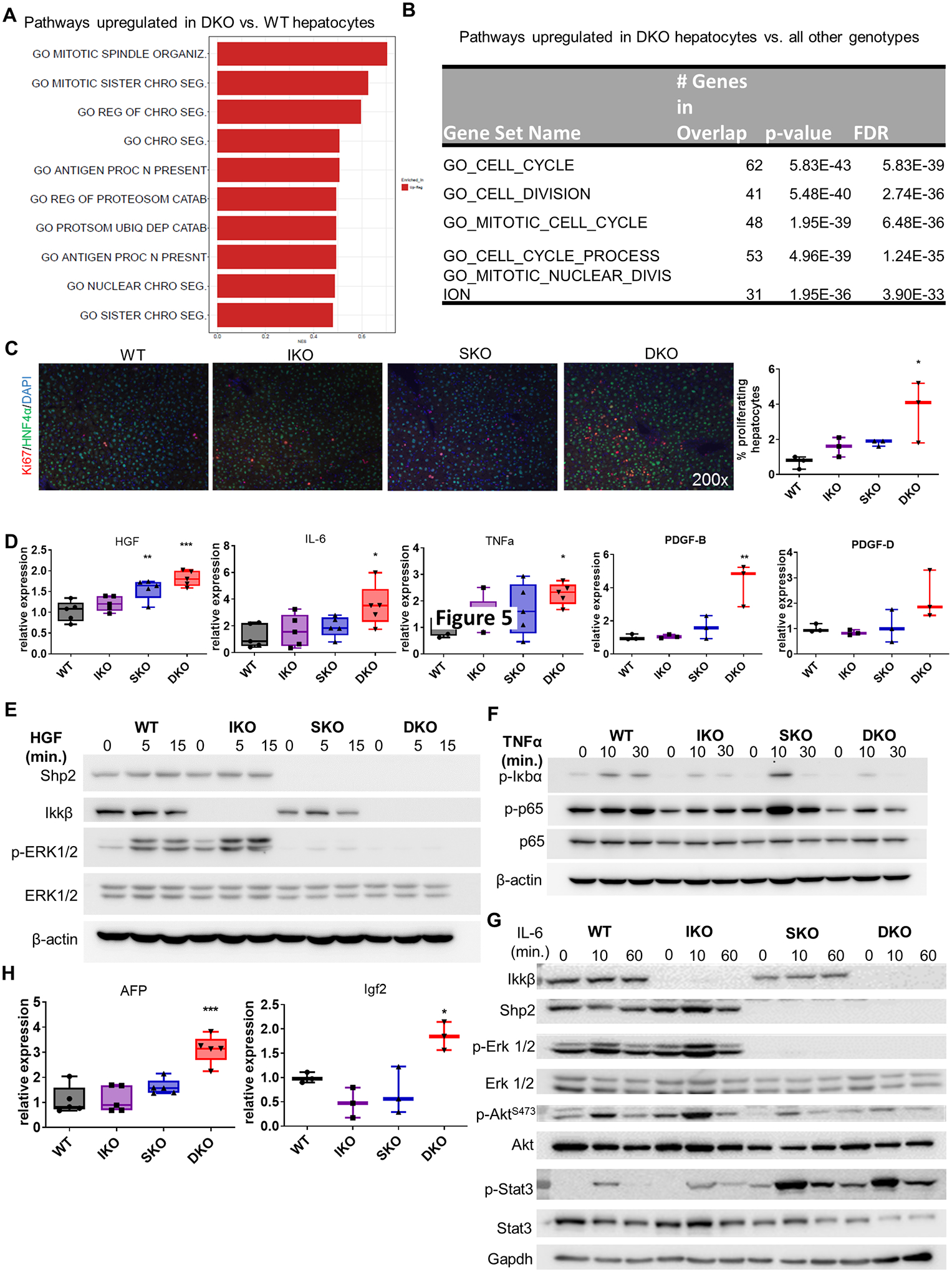
(A) Gene set enrichment analysis of upregulated genes (DEGs) in hepatocytes freshly isolated from DKO untreated male mice compared to WT. (B) Gene set enrichment analysis of genes uniquely upregulated in DKO hepatocytes compared to all other genotypes. (C) Left, co-immunofluorescence of the proliferation marker Ki67 and the hepatocyte marker HNF4α in sections of untreated livers from 2-month-old male mice. Right, quantification of proliferating hepatocytes (Ki67+ HNF4α+ cells). Each point represents the average quantification of 4–5 fields of view for a single mouse. (D) mRNA expression of growth-stimulating factors in 2-month-old livers (n=3–5/genotype). (E) Immunoblot of ERK activation in primary hepatocytes from indicated genotypes treated with 20 ng/ml mouse recombinant HGF. (F) Immunoblot of NF-κB pathway constituents in primary hepatocytes of the indicated genotype treated with 10 ng/ml mouse recombinant TNFα. (G) Immunoblot of various growth-promoting pathway members in isolated hepatocytes of the indicated genotype treated with 20 ng/ml mouse recombinant IL-6. (H) Expression of onco-fetal mRNAs in 2-month-old livers (n=3–5/genotype).
We then compared the transcriptome of DKO hepatocytes against WT, IKO, and SKO groups to identify changes induced specifically by dual Ikkβ and Shp2 removal (Fig. 4B; Supp. Fig. S5C, Supplemental File 3). This analysis revealed unique upregulation of 148 genes only in DKO hepatocytes, a large proportion of which were integral pro-mitotic genes or part of cell cycle-related pathways (Fig. 4B, Supp. Fig. S5C; Supplemental File 3). These data suggest that a distinguishing feature of DKO hepatocytes is, surprisingly, enhanced cell cycle progression and cell division, despite concurrent disruption of two proliferative pathways. Consistent with the RNA-Seq data, immunostaining of liver sections from 2-month-old mice detected significantly increased hepatocyte proliferation in DKO livers using Ki67 and HNF4α antibodies (Fig. 4C). qRT-PCR analysis showed marked upregulation of transcripts for multiple growth factors, including Hgf, IL-6, TNFα, PDGF-B, and PDGF-D, which are known to spur compensatory hepatocyte proliferation in damaged livers (Fig. 4D). As many of these factors mediate liver regeneration via pathways expected to be impaired in the DKO model, we isolated hepatocytes from WT, IKO, SKO and DKO livers and examined the activation status of Ras/MAPK and NF-κB pathway components in response to several growth factors. Removal of Shp2 indeed blunted Erk activation by HGF, and loss of Ikkβ suppressed IκBα and p65/RelA phosphorylation following TNF stimulation (Fig. 4E–F). Following IL-6 stimulation, p-Erk and p-Akt levels were severely reduced in SKO and DKO hepatocytes but Stat3 phosphorylation increased in SKO and DKO (Fig. 4G). Therefore, both the Ras/MAPK and NF-κB pathways were indeed disrupted in hepatocytes deficient for Shp2 and Ikkβ. Furthermore, the expression of onco-fetal genes Afp and Igf2 were upregulated in DKO livers in young mice at the pre-neoplastic stage, before detection of macroscopic tumors (Fig. 4H). These results argue that the Ras/MAPK and NF-κB pathways are less critical for driving cell division than previously thought, and are actually dispensable for hepatocyte proliferation and transformation in the liver.
Circadian genes are uniquely deregulated in DKO hepatocytes
Besides the upregulated transcripts, we also examined the functions of genes downregulated only in DKO hepatocytes. In contrast to the enhanced proliferation of DKO hepatocytes, IKO and SKO hepatocytes shared downregulation of cell division processes (Supp. Fig. S5B). In comparison to WT controls, DKO hepatocytes demonstrated the metabolic changes already detected in whole liver (Fig. 5A). Intriguingly, despite being freshly isolated during the same time interval as all other genotypes (Zeitgeiber Time 7–11), DKO hepatocytes exhibited changes to circadian pathways (Fig. 5A). To confirm that these processes were unique to the DKO hepatocytes, we interrogated the 90 genes that were downregulated only in DKO hepatocytes, as compared to WT, IKO and SKO (Fig. 5B; Supplemental File 3). Interestingly, the most significant gene set changes were those involved in control of circadian clock (Fig. 5B). Specifically, we observed dramatically reduced expression of 13 circadian genes in DKO hepatocytes, including inhibitory components Bhlhe40 (Dec1) and Bhlhe41 (Dec2); transcription factor Klf10; core clock inhibitors Cry2, Per2, and Per3; and the clock-implicated nuclear receptor Rorc (Fig. 5C). Among them, the majority (10/13) were transcription factors that are negative or positive clock regulators. In agreement with the RNA-Seq data, qRT-PCR analysis detected very low levels of mRNAs for Per2, Per3, Cry2 and Dbp in primary hepatocytes isolated from 2-month-old DKO mice, as well as altered levels of the clock effectors Bmal1 and Clock (Fig. 5D). Of note, the transcripts encoding negative regulatory genes were expressed at similar or even higher levels in IKO and SKO hepatocytes than the WT (Fig. 5C), suggesting that their drastic inhibition in DKO hepatocytes was unlikely due to a simple additive effect of the two gene deletions. Rather, it appears to be a specific defect in hepatocytes induced only by combined disruption of the two pathways. To further examine the effect of dual pathway blockade on the expression patterns of circadian genes, we monitored clock gene expression over a 24-hour period in serum-shock-synchronized primary hepatocytes isolated from WT and DKO mice. The expression of the negative clock regulatory genes Per2, Cry2, and Dec1 were consistently lower in DKO than WT hepatocytes (Fig. 5E). While Bmal expression was only slightly altered in DKO, Clock mRNA levels were markedly higher at most time points in DKO hepatocytes (Fig. 5E). Modest induction of circadian transcripts was observed in DKO hepatocytes in vitro, after serum synchronization, which displayed similar kinetics to WT. However, the amplitude of induction was generally lower in DKO hepatocytes. These results suggest that circadian deregulation is a cell-autonomous effect of Ikkβ and Shp2 deletion. Given that disruptions of circadian rhythm or deletion of clock regulators in murine liver were sufficient to induce HCC [20], clock dysfunction may be a key contributor to spontaneous HCC development in DKO mice (Fig. 5F).
Fig. 5. The circadian clock is dysregulated in DKO hepatocytes.
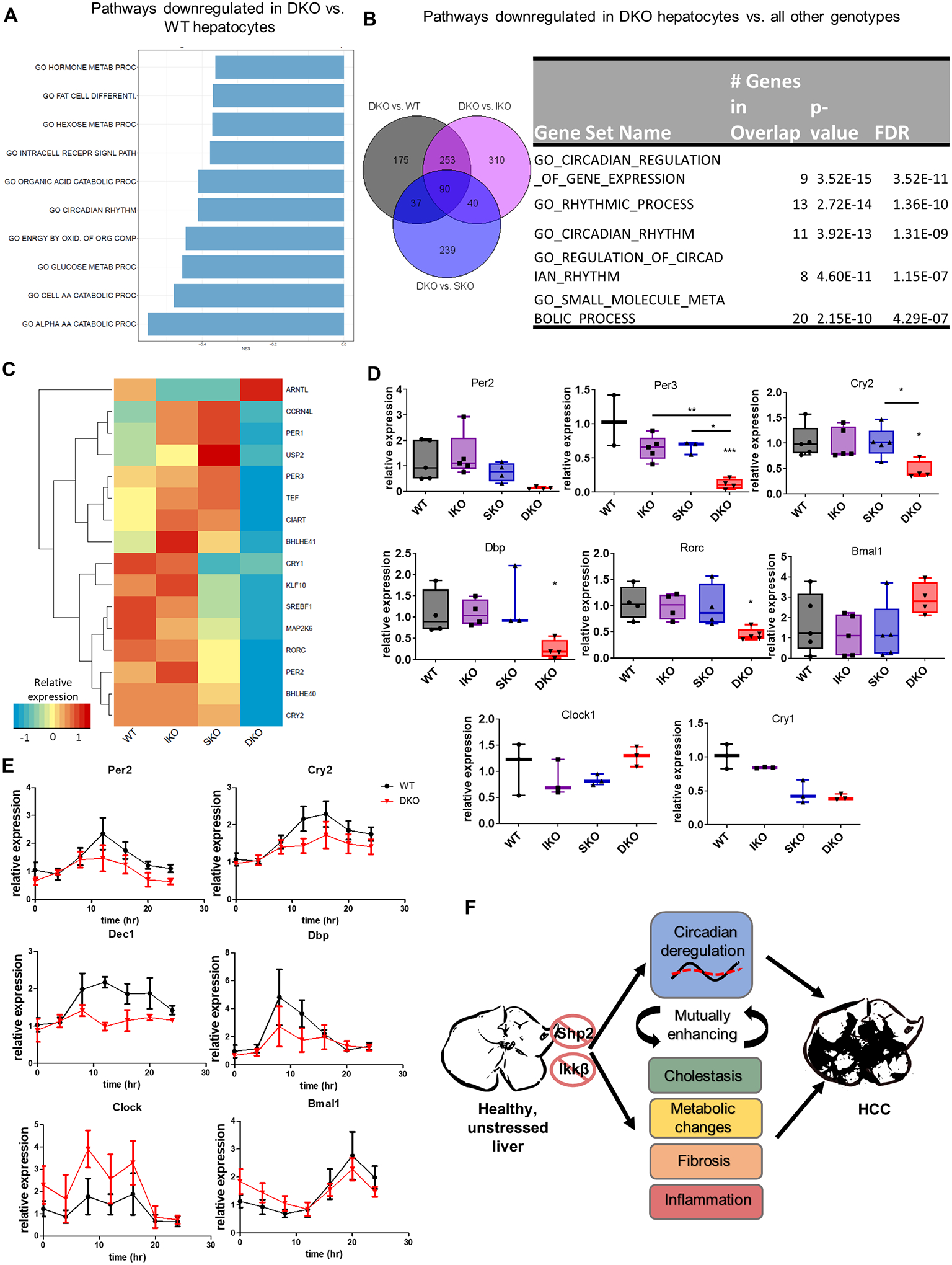
(A) Gene set enrichment analysis of downregulated genes in DKO vs. WT hepatocytes using GO-BP terms. (B) Left, overlap of genes downregulated in DKO vs. indicated genotype from RNA-Seq analysis of 2-month-old hepatocytes. Right, gene set enrichment of the 90 genes uniquely downregulated in DKO hepatocytes. (C) Heatmap showing relative expression levels of 16 differentially-expressed circadian genes identified from RNA-Seq analysis (each column represents the average expression of n=3/genotype). (D) qPCR validation of circadian clock gene expression in isolated hepatocytes (n=3–5/genotype). (E) Time course of circadian clock gene expression in synchronized primary hepatocytes collected from WT and DKO mice. Relative mRNA abundance of each gene was determined by qPCR. (F) A model to illustrate spontaneous HCC development in the DKO mice. Dual Shp2 and Ikkβ deletion triggers circadian dysregulation as well as cholestasis, metabolic changes, fibrosis and inflammation, which may be mutually enhancing and eventually lead to HCC.
Disordered expression of circadian clock genes in human HCC
To explore the relevance of the mouse data to human HCC, we analyzed 371 HCC patient samples and 50 adjacent normal liver tissues in the TCGA dataset. Based on expression levels of circadian clock genes in tumor relative to non-tumor tissues, we divided HCC samples into three groups: (1) circadian clock-normal, (2) circadian clock-mid, (3) circadian clock-dysregulated. Specifically, we checked expression of 7 core clock genes, including Clock, Arntl, Per1, Per2, Per3, Cry1, and Cry2. Patients with at most 2 dysregulated circadian genes were assigned into “normal” group, patients with at least 5 dysregulated clock genes into “dysregulated” group, and the others into “mid” group. With sub-clustering of all HCC patients, we performed differential expression analysis between “normal” and “dysregulated” groups using DESeq2. The genes with adjusted p value < 0.05 were defined as differentially expressed. We specifically checked components in the Ras/MAPK or NF-κB pathways, and generated volcano plots with differentially expressed genes in the two pathways (Fig. 6A–B). Multiple constituents from both pathways exhibited altered expression in clock-dysregulated HCC patients (Fig. 6A–B). Next, to verify whether activities of these two pathways were inhibited in the “clock-dysregulated” group, we collected downstream targets of Ras/MAPK and NF-κB pathways from Msigdb gene sets, respectively. The pathway activity was defined as the sum of rank-transformed normalized values of all genes in a corresponding gene set. These analyses revealed a correlation between dysregulated circadian genes with downregulation of both Ras/MAPK and NF-κB pathways in HCC patients (Fig. 6C–D).
Figure 6. Circadian deregulation in human HCCs.
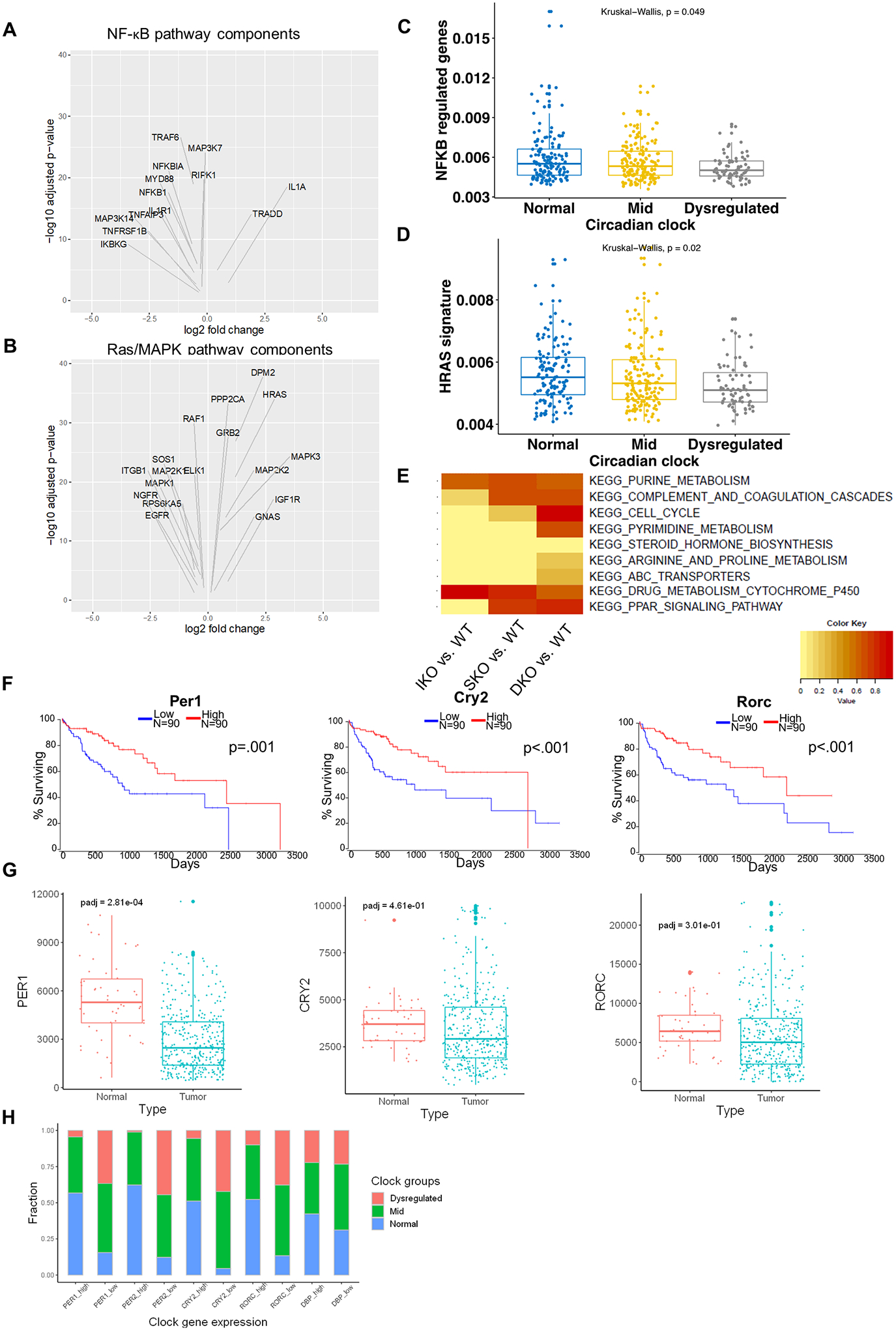
(A) Volcano plots of differentially expressed NF-κB pathway components in clock-dysregulated human HCCs (adjusted p<0.05). (B) Volcano plots of differentially expressed Ras/MAPK pathway components in clock-dysregulated human HCCs (adjusted p<0.05). (C) NF-κB target gene expression profiles in human HCCs with Normal, Mid (<5 clock genes disrupted), or Dysregulated (>5 clock genes disrupted) circadian status. (D) HRas target gene expression profiles in human HCCs as stratified in C. (E) Enrichment for KEGG pathways previously identified to correlate with disrupted circadian processes in human HCC. DEGs from hepatocytes isolated from 2-month-old livers were analyzed to determine similarity to this “circadian-disrupted” signature. (F) Association between clock gene expression and overall survival in HCC patients. TCGA-LIHC cohort data were analyzed and Kaplan-Meier plots were generated using OncoLnc; upper and lower quartiles of gene expression were used to determine “high” and “low” expression, respectively. (G) Normalized reads of the indicated circadian transcripts in normal (non-tumor liver) and tumor (HCC) tissues in patients from the TCGA-LIHC cohort. Box-and-whisker plots indicate quartiles of expression for each gene. (H) Fractions of patients with Normal, Mid, or Dysregulated circadian status in patients with upper-quartile (High) or lower-quartile (Low) expression of the indicated clock gene.
To determine whether this DKO tumor model resembled clock-disrupted human HCC, we further assessed enrichment for gene sets that were previously reported to associate with low expression of a signature of clock genes in TCGA HCC data (Fig. 6E) [21]. The majority of these gene sets were also altered in DKO hepatocytes, with lower enrichment seen in IKO or SKO hepatocytes. We then determined whether individual circadian genes downregulated in the DKO mouse model were associated with HCC prognosis. Indeed, lower quartile expression of Per1, Rorc, and Cry2 was significantly associated with poorer overall survival (Fig. 6F). We also examined the expression levels of these genes in the TCGA cohort and confirmed that expression of these clock genes was significantly lower in tumors than in non-tumor liver tissues (Fig. 6G). Finally, we asked whether these individual circadian gene disruptions were associated with broader circadian deregulation in HCC. To address this, we compared the proportion of patients with normal or dysregulated clock gene sets, as defined for Fig. 6C–D, between the upper- and lower-quartile expression levels of individual clock genes. The patients with low expression of these genes in tumors tended to have disruptions to other clock genes, as indicated by a higher proportion of patients falling within the mid- or dysregulated groups (Fig. 6H). Thus, deregulated expression of clock genes was linked to disease progression and prognosis in HCC patients. Together, these results suggest that this novel mechanism of HCC development unveiled in our mouse model is very likely implicated in pathogenesis of human liver cancer.
Discussion
A large body of literature has documented critical roles of the Ras/MAPK and NF-κB pathways in driving carcinogenesis in various cell types and organs [6–8, 13]. Accordingly, these two pathways are currently hot drug targets pursued for oncological treatment worldwide. However, this study showed that a compound mutant mouse line with both pathways disrupted in hepatocytes was hypersensitive to DEN, which was known to induce oncogenic mutations frequently detected in HCC patients [18]. Consistent with a previous prediction [5], the augmented susceptibility to DEN was associated with more severe cholestasis, apparent fibrosis and chronic inflammation in the young DKO liver. Transcriptomic analysis revealed broad hepatic metabolic disorders, including changes in metabolism of fatty acids, amino acids, and bile acids. While some changes were shared with single knockout livers, the DKO mice exhibited the most severe perturbations, due to additive or synergistic effects of dual gene deletion.
The most unanticipated result in this study is the spontaneous hepatocarcinogenesis in DKO mice without any exogenous challenge. This finding is surprising because the severe liver disorders that aggravated DEN-induced tumors were likely not sufficient to be carcinogenic drivers. Transcriptomic analysis of isolated hepatocytes identified severe disruption of core circadian control gene expression as the most prominent feature in DKO hepatocytes at 2 months, prior to tumor appearance but concurrent with accelerated proliferation and expression of HCC marker genes. Of note, previous studies demonstrated that chronic jetlag in mice was sufficient to drive HCC development, and targeted deletion of clock regulators or effectors was also found to drive spontaneous HCC development in mice [20]. Therefore, we propose here that clock disruption contributes to the spontaneous HCC development in DKO mice. Consistent with the mouse results, TCGA data analysis revealed a potentially similar mechanism of tumorigenesis involved in human HCC. In human liver cancer, as in our DKO mouse model, circadian dysfunction was coupled to enhanced proliferation and disruption of numerous metabolic processes [21]. Epidemiological links of chronic circadian disruption, such as shift work, to increased rates of human cancer has prompted the IARC to list circadian disruption as a probable carcinogen. Lower expression of several clock genes deregulated in the DKO model was associated with worse prognosis in human HCC. Together, these observations suggest a functional link between the circadian disruption, broad liver pathologies, and spontaneous tumor phenotype in our DKO model.
This study unexpectedly disclosed a physiological function of the Ras/MAPK and NF-κB pathways working in concert to maintain circadian homeostasis at the ground state. A variety of negative regulatory clock TFs were consistently downregulated, which could contribute to the observed increased expression of Bmal1. As downregulation of multiple clock genes occurred only in the DKO hepatocytes, it is unlikely that the circadian deregulation was secondary to liver damages. Given that it was not observed in either IKO or SKO hepatocytes, the dramatic dysregulation in DKO hepatocytes was evidently not an additive effect of dual gene deletion; rather, it appears to be a systemic disorder that stems from disruption of an intricate signaling network constituted by two classical signaling pathways. This finding is consistent with the highly heterogenous nature of HCC-associated oncogenic mutations [22], which frequently does not follow a clearly defined mutational signature.
Contrary to the previously defined pro-proliferative effects of MAPK and NF-κB pathways, the DKO hepatocytes displayed dramatically increased proliferation and upregulated expression of cell cycle genes. Tumor nodules were detected in DKO livers in 9 months, displaying a phenotype much faster than that produced by deletion of clock genes [20]. We propose that this accelerated tumor phenotype was driven by the mutually promoting effects of circadian dysregulation and multiple liver disorders, especially the strikingly elevated BA levels in the DKO liver (Fig. 2G). Circadian dysfunction has been shown to disrupt BA homeostasis [23], and aberrant BA production influenced hepatic expression of clock genes [24]. Therefore, the circadian disruption and cholestasis-associated liver damages may constitute a destructive feedback loop spurring excessive compensatory hepatocyte proliferation and eventually driving tumorigenesis (Fig. 5F). The initial trigger leading to this pathogenic downward spiral was ironically the combined suppression of two well characterized oncogenic pathways. Although circadian deregulation is apparently a contributor to the pathologic phenotype, we cannot exclude contributions from as-yet-unidentified pathways. For example, while canonical NF-κB signaling is suppressed in this model, we cannot exclude a possibility that Ikkβ deletion might influence non-canonical NF-κB signaling [25, 26], which remains to be explored.
Although each signaling pathway has received extensive analysis individually, how they intertwine is complex and unpredictable. While deleting both Shp2 and Pten in blood cells showed mutually neutralizing effects in myeloproliferation and leukemogenesis [27], simultaneous removal of Shp2 and Pten in hepatocytes accelerated NASH-HCC development [28]. Although loss of either Shp2 or β-catenin promoted DEN-induced HCC [5], deleting both suppressed liver tumorigenesis (Chen et al., unpublished data). This study has unveiled previously unrecognized roles of two central signaling pathways in sustaining circadian rhythm and physiological homeostasis, providing a fresh view on their baseline gatekeeper function. Due to the limited efficacy of monotherapy that targets a single oncogenic pathway, one prevailing view in cancer therapy is to simultaneously block multiple pathways to optimize the anti-tumor effect. However, the data presented here raise a cautionary note on the concept of combinatorial targeting.
Supplementary Material
Implications.
We demonstrate here that basal levels of the Ras/MAPK and NF-κB pathways, while promoting tumorigenesis if overactivated, are required to maintain physiological homeostasis and regulate circadian rhythm in the liver, which are anti-tumorigenic.
Acknowledgments:
RNA Sequencing was performed at the UCSD Genomics Core. Histological consultation was provided by Dr. N. Varki from the UCSD Mouse Phenotyping Core. Research reported in this publication was supported by NIH award F31CA228420 to KLH and NCI grants R01CA239629 and R01CA236074 to G.S.F.
Footnotes
Competing interests: Authors declare no competing interests.
References
- [1].Stewart BW, Wild CP, IARC World Cancer Report 2014, 2014, pp. 19; 403–412.
- [2].Zucman-Rossi J, Villanueva A, Nault JC, Llovet JM, Genetic Landscape and Biomarkers of Hepatocellular Carcinoma, Gastroenterology, 149 (2015) 1226–1239.e1224. [DOI] [PubMed] [Google Scholar]
- [3].Llovet JM, Villanueva A, Lachenmayer A, Finn RS, Advances in targeted therapies for hepatocellular carcinoma in the genomic era, Nature reviews. Clinical oncology, 12 (2015) 408–424. [DOI] [PubMed] [Google Scholar]
- [4].Bruix J, Qin S, Merle P, Granito A, Huang YH, Bodoky G, Pracht M, Yokosuka O, Rosmorduc O, Breder V, Gerolami R, Masi G, Ross PJ, Song T, Bronowicki JP, Ollivier-Hourmand I, Kudo M, Cheng AL, Llovet JM, Finn RS, LeBerre MA, Baumhauer A, Meinhardt G, Han G, Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial, The Lancet, 389 (2017) 56–66. [DOI] [PubMed] [Google Scholar]
- [5].Feng GS, Conflicting Roles of Molecules in Hepatocarcinogenesis: Paradigm or Paradox, Cancer Cell, 21 (2012) 150–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Karnoub AE, Weinberg RA, Ras oncogenes: Split personalities, Nature Reviews Molecular Cell Biology, 9 (2008) 517–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Papke B, Der CJ, Drugging RAS: Know the enemy, Science, 355 (2017) 1158–1163. [DOI] [PubMed] [Google Scholar]
- [8].Simanshu DK, Nissley DV, McCormick F, RAS Proteins and Their Regulators in Human Disease, Cell, 170 (2017) 17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chan RJ, Feng G-S, PTPN11 is the first identified proto-oncogene that encodes a tyrosine phosphatase, Blood, 109 (2007) 862–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, Van der Burgt I, Crosby AH, Ion A, Jeffery S, Kalidas K, Patton MA, Kucherlapati RS, Gelb BD, Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome, Nature Genetics, 29 (2001) 465–468. [DOI] [PubMed] [Google Scholar]
- [11].Lamarche MJ, Chan HM, Fekkes P, Garcia-fortanet J, Acker MG, Antonakos B, Chen C.H.-t., Chen Z, Cooke VG, Dobson JR, Deng Z, Fei F, Firestone B, Fodor M, Fridrich C, Gao H, Grunenfelder D, Hao H.-x., Jacob J, Ho S, Hsiao K, Kang ZB, Karki R, Kato M, Larrow J, Bonte LRL, Lenoir F, Liu G, Liu S, Majumdar D, Meyer MJ, Palermo M, Perez L, Pu M, Price E, Quinn C, Shakya S, Shultz MD, Slisz J, Venkatesan K, Wang P, Warmuth M, Williams S, Yang G, Yuan J, Zhang J.-h., Zhu P, Ramsey T, Keen NJ, Sellers WR, Stams T, Fortin PD, Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases, Nature, 535 (2016) 148–152. [DOI] [PubMed] [Google Scholar]
- [12].Ahmed TA, Adamopoulos C, Karoulia Z, Wu X, Sachidanandam R, Aaronson SA, Poulikakos PI, SHP2 Drives Adaptive Resistance to ERK Signaling Inhibition in Molecularly Defined Subsets of ERK-Dependent Tumors, Cell Reports, 26 (2019) 65–78.e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Taniguchi K, Karin M, NF-B, inflammation, immunity and cancer: Coming of age, Nature Reviews Immunology, 18 (2018) 309–324. [DOI] [PubMed] [Google Scholar]
- [14].Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, Ben-Neriah Y, NF-kappaB functions as a tumour promoter in inflammation-associated cancer, Nature, 431 (2004) 461–466. [DOI] [PubMed] [Google Scholar]
- [15].Maeda S, Kamata H, Luo JL, Leffert H, Karin M, IKKβ couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis, Cell, 121 (2005) 977–990. [DOI] [PubMed] [Google Scholar]
- [16].Bard-Chapeau EA, Li S, Ding J, Zhang SS, Zhu HH, Princen F, Fang DD, Han T, Bailly-Maitre B, Poli V, Varki NM, Wang H, Feng GS, Ptpn11/Shp2 Acts as a Tumor Suppressor in Hepatocellular Carcinogenesis, Cancer Cell, 19 (2011) 629–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bard-Chapeau EA, Hevener AL, Long S, Zhang EE, Olefsky JM, Feng GS, Deletion of Gab1 in the liver leads to enhanced glucose tolerance and improved hepatic insulin action, Nat Med, 11 (2005) 567–571. [DOI] [PubMed] [Google Scholar]
- [18].Connor F, Rayner TF, Aitken SJ, Feig C, Lukk M, Santoyo-Lopez J, Odom DT, Mutational landscape of a chemically-induced mouse model of liver cancer, Journal of Hepatology, 69 (2018) 840–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Li S, Hsu DDF, Li B, Luo X, Alderson N, Qiao L, Ma L, Zhu HH, He Z, Suino-Powell K, Ji K, Li J, Shao J, Xu HE, Li T, Feng GS, Cytoplasmic tyrosine phosphatase shp2 coordinates hepatic regulation of bile acid and FGF15/19 signaling to repress bile acid synthesis, Cell Metabolism, 20 (2014) 320–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kettner NM, Voicu H, Finegold MJ, Coarfa C, Sreekumar A, Putluri N, Katchy CA, Lee C, Moore DD, Fu L, Circadian Homeostasis of Liver Metabolism Suppresses Hepatocarcinogenesis, Cancer Cell, 30 (2016) 909–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Liu Z, Yu K, Zheng J, Lin H, Zhao Q, Zhang X, Feng W, Wang L, Xu J, Xie D, Zuo ZX, Liu ZX, Zheng Q, Dysregulation, functional implications, and prognostic ability of the circadian clock across cancers, Cancer Medicine, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Nault JC, Paradis V, Cherqui D, Vilgrain V, Zucman-Rossi J, Molecular classification of hepatocellular adenoma in clinical practice, Journal of Hepatology, 67 (2017) 1074–1083. [DOI] [PubMed] [Google Scholar]
- [23].Ma K, Xiao R, Tseng HT, Shan L, Fu L, Moore DD, Circadian dysregulation disrupts bile acid homeostasis, PLoS ONE, 4 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Govindarajan K, MacSharry J, Casey PG, Shanahan F, Joyce SA, Gahan CGM, Unconjugated bile acids influence expression of circadian genes: A potential mechanism for microbe-host crosstalk, PLoS ONE, 11 (2016) 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kucharzewska P, Maracle CX, Jeucken KCM, van Hamburg JP, Israelsson E, Furber M, Tas SW, Olsson HK, NIK-IKK complex interaction controls NF-kappaB-dependent inflammatory activation of endothelium in response to LTbetaR ligation, J Cell Sci, 132 (2019). [DOI] [PubMed] [Google Scholar]
- [26].Kuo SH, Tsai HJ, Lin CW, Yeh KH, Lee HW, Wei MF, Shun CT, Wu MS, Hsu PN, Chen LT, Cheng AL, The B-cell-activating factor signalling pathway is associated with Helicobacter pylori independence in gastric mucosa-associated lymphoid tissue lymphoma without t(11;18)(q21;q21), J Pathol, 241 (2017) 420–433. [DOI] [PubMed] [Google Scholar]
- [27].Zhu HH, Luo X, Zhang K, Cui J, Zhao H, Ji Z, Zhou Z, Yao J, Zeng L, Ji K, Gao W-Q, Zhang Z-Y, Feng G-S, Shp2 and Pten have antagonistic roles in myeloproliferation but cooperate to promote erythropoiesis in mammals, Proceedings of the National Academy of Sciences of the United States of America, 112 (2015) 13342–13347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Luo X, Liao R, Hanley KL, Zhu HH, Malo KN, Hernandez C, Wei X, Varki NM, Alderson N, Chu C, Li S, Fan J, Loomba R, Qiu SJ, Feng GS, Dual Shp2 and Pten Deficiencies Promote Non-alcoholic Steatohepatitis and Genesis of Liver Tumor-Initiating Cells, Cell Reports, 17 (2016) 2979–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.