

Abstract
Purpose:
Neuroendocrine prostate cancer (NEPC) is a resistance phenotype that emerges in men with metastatic castration-resistant prostate adenocarcinoma (CR-PRAD) and has important clinical implications, but is challenging to detect in practice. Herein, we report a novel tissue-informed epigenetic approach to non-invasively detect NEPC.
Experimental Design:
We first performed methylated immunoprecipitation and high-throughput sequencing (MeDIP-seq) on a training set of tumors, identified differentially methylated regions between NEPC and CR-PRAD, and built a model to predict the presence of NEPC (termed NEPC Risk Score). We then performed MeDIP-seq on cell-free DNA (cfDNA) from two independent cohorts of men with NEPC or CR-PRAD and assessed the accuracy of the model to predict the presence NEPC.
Results:
The test cohort comprised cfDNA samples from 48 men, 9 with NEPC and 39 with CR-PRAD. NEPC Risk Scores were significantly higher in men with NEPC than CR-PRAD (P=4.3x10−7) and discriminated between NEPC and CR-PRAD with high accuracy (AUROC 0.96). The optimal NEPC Risk Score cut-off demonstrated 100% sensitivity and 90% specificity for detecting NEPC. The independent, multi-institutional validation cohort included cfDNA from 53 men, including 12 with NEPC and 41 with CR-PRAD. NEPC Risk Scores were significantly higher in men with NEPC than CR-PRAD (P=7.5x10−12) and perfectly discriminated NEPC from CR-PRAD (AUROC 1.0). Applying the pre-defined NEPC Risk Score cut-off to the validation cohort resulted in 100% sensitivity and 95% specificity for detecting NEPC.
Conclusions:
Tissue-informed cfDNA methylation analysis is a promising approach for non-invasive detection of NEPC in men with advanced prostate cancer.
Keywords: Prostate cancer, Metastatic Castration-Resistant Prostate Cancer, Neuroendocrine Prostate Cancer, Biomarkers, DNA Methylation, Epigenetics
INTRODUCTION
Neuroendocrine prostate cancer (NEPC) can arise as a resistance mechanism to androgen deprivation therapy (ADT) and androgen receptor signaling inhibitors (ARSIs) in men with metastatic castration-resistant prostate cancer (mCRPC).(1,2) Present in up to 17% of men with mCRPC, NEPC is associated with poor response to ARSIs and shorter overall survival (OS).(1,3) However, NEPC tumors are more likely to respond to platinum-based chemotherapy and several novel NEPC-directed therapies are in clinical development.(4)
The current approach to diagnosing NEPC – performing tissue biopsy for pathologic tumor analysis – has significant shortcomings. There is a lack of consensus pathological criteria for defining NEPC and, due to intra-patient tumor heterogeneity, biopsy samples may not represent a patient’s overall disease burden.(5–7) Consequently, NEPC diagnosis is often delayed or missed and reported rates likely underestimate the prevalence of this aggressive disease variant. The lack of a biomarker for early and accurate detection is a significant barrier to improving outcomes for men who develop NEPC.
Liquid biopsies are well-suited to address this unmet need. Most clinical cell-free DNA (cfDNA) tests detect somatically acquired tumor mutations or copy number alterations. However, the defining genetic hallmark of NEPC, deleterious alterations in RB1 and/or TP53, are present in more than one-third of castration-resistant prostate adenocarcinoma (CR-PRAD) tumors and thus cannot unambiguously discriminate between the tumor subtypes.(1) In contrast, vast DNA methylation differences exist between NEPC and CR-PRAD.(5) Cell-free methylated DNA immunoprecipitation and high-throughput sequencing (cfMeDIP-seq), a highly sensitive method for genome-wide cfDNA methylation profiling, capable of non-invasive cancer detection and discriminating between tumor types, is well-suited to non-invasively detect NEPC.(8–10)
In this manuscript, we evaluate the ability of cfMeDIP-seq to detect NEPC in men with mCRPC. We first perform methylation profiling on a training set of NEPC and PRAD tumors to identify methylation sites enriched in each tumor type. We then establish the ability to implement tissue-informed analysis of cfMeDIP-seq data to detect NEPC in cfDNA from men with NEPC or CR-PRAD. Finally, in an independent cfDNA cohort from men with NEPC or CR-PRAD, we confirm the analytical and clinical validity of this approach for accurate, non-invasive detection of NEPC.
MATERIALS AND METHODS
Subjects and samples
Plasma samples were collected from men with mCRPC diagnosed and treated at the Dana-Farber Cancer Institute (DFCI), Brigham and Women’s Hospital, or Weill Cornell Medicine (WCM) between April 2003 and August 2021. Two genitourinary pathologists (H.K.T. and M.S.H.) confirmed the presence of high-grade neuroendocrine carcinoma of prostate origin according to modern conventions based on histologic review of available material, re-interpretation of original reports, and integration of available molecular results.(11) CR-PRAD patients had castration-resistant prostate adenocarcinoma with no pathologic evidence of neuroendocrine differentiation throughout their disease course. All patients provided written informed consent. The use of samples was approved by the DFCI (01-045 and 09-171) and WCM (1305013903). Studies were conducted in accordance with recognized ethical guidelines. The previously-described LuCaP PDXs were derived from resected metastatic prostate cancer with informed consent of patient donors under a protocol approved by the University of Washington Human Subjects Division IRB.(12)
Sample processing
cfDNA samples were processed by the following method. Peripheral blood was collected in EDTA Vacutainer tubes (BD), and processed within 3 hours of collection. Plasma was separated by centrifugation at 2,500 g for 10 minutes, transferred to microcentrifuge tubes, and centrifuged at 2,500 g at room temperature for 10 minutes to remove cellular debris. The supernatant was aliquoted into 1-2 mL aliquots and stored at −80°C until the time of DNA extraction. cfDNA was isolated from 1 mL of plasma, using the Qiagen Circulating Nucleic Acids Kit (Qiagen), eluted in AE buffer, and stored at −80°C. DNA from the LuCaP PDXs was extracted using the DNeasy Blood and Tissue Kit (Qiagen). Genomic DNA was sheared using a Covaris Sonicator E220 and AMPure XP beads (Beckman Coulter) were used to size select 150-250 bp DNA fragments.
cfDNA tumor content calculation
Low-pass whole genome sequencing (LPWGS) was performed on all cfDNA samples. The ichorCNA R package was used to infer copy number profiles and cfDNA tumor content from read abundance across bins spanning the genome using default parameters.(13)
cfMeDIP-seq protocol
cfMeDIP-seq was performed using previously published methods.(10) cfDNA library preparation was performed using KAPA HyperPrep Kit (KAPA Biosystems) according to the manufacturer’s protocol. We then performed end-repair, A-tailing, and ligation of NEBNext adaptors (NEBNext Multiplex Oligos for Illumina kit, New England BioLabs). Libraries were digested using the USER enzyme (New England BioLabs). λ DNA, consisting of unmethylated and in vitro methylated DNA, was added to prepared libraries to achieve a total amount of 100 ng DNA. Methylated and unmethylated Arabidopsis thaliana DNA (Diagenode) was added for quality control. MeDIP was performed using the MagMeDIP kit (Diagenode) following the manufacturer’s protocol. Samples were purified using the iPure Kit v2 (Diagenode). Success of the immunoprecipitation was confirmed using qPCR to detect recovery of the spiked-in Arabidopsis thaliana methylated and unmethylated DNA.
Next-generation sequencing library construction
KAPA HiFi Hotstart ReadyMix (KAPA Biosystems) and NEBNext Multiplex Oligos for Illumina (New England Biolabs) were added to a final concentration of 0.3 uM and libraries were amplified as follows: activation at 95°C for 3 minutes, amplification cycles of 98°C for 20 seconds, 65°C for 15 seconds, 72°C for 30 seconds, and a final extension of 72°C for 1 minute. Samples were pooled and sequenced (Novogene Corporation, CA) on Illumina HiSeq 4000 to generate 150 bp paired-end reads.
Quality control and processing of sequencing reads
After sequencing, the quality and quantity of the raw reads were examined using FastQC version 0.11.5 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) and MultiQC version 1.7.(14) Raw reads were quality and adapter trimmed using Trim Galore! version 0.6.0 (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) using default settings in paired-end mode. The trimmed reads then were aligned to hg19 using Bowtie2 version 2.3.5.1 in paired-end mode and all other settings default.(15) The SAMtools version 1.10 software suite was used to convert SAM alignment files to BAM format, sort and index reads, and remove duplicates.(16) The R package RSamtools version 2.2.1 was used to calculate the number of unique mapped reads. Saturation analyses to evaluate reproducibility of each library were carried out using the R Bioconductor package MEDIPS version 1.38.0.(17)
Tissue-informed approach to NEPC detection
To identify DMRs between NEPC and PRAD tumors, we first binned the genome into 300 base-pair windows and tested each window for differential methylation between NEPC and PRAD samples using limma-voom (using R package limma version 3.42.0) on TMM-normalized counts (using R package edgeR version 3.28.0).(18,19) Only bins with a total count above a fixed threshold were tested for differential methylation, where the threshold was set at 20% of the total number of samples across both groups. We restricted our search to bins within annotated CpG islands and FANTOM5 enhancers and excluded regions of high signal or poor mappability.(20,21) We selected DMRs with read enrichment in NEPC compared to PRAD PDXs at FDR-adjusted P<1.0x10−6 and log2 fold-change >3. We removed windows with peaks in MeDIP-seq data from white blood cells (as determined by MACS2, version 2.1.2) to minimize signal from blood cell-derived cfDNA.(22) Using the MeDIPs R package, we calculated a CpG-normalized relative methylation scores across 300 bp windows for each cfDNA sample.(17,23) We then summed relative methylation scores in cfDNA at NEPC-enriched PDX DMRs for each sample and normalized this value to the sum of rms values across all 300bp windows. This value was termed “NEPC Methylation Value.” The same process was performed for PRAD-enriched PDX DMRs to derive a “PRAD Methylation Value.” We then calculated the log2 ratio of the NEPC Methylation Value to the PRAD Methylation Value and normalized these values to the median score in cfDNA from eight healthy cancer-free controls. This value was termed the “NEPC Risk Score.” This approach is summarized in Fig. 1A.
Figure 1.
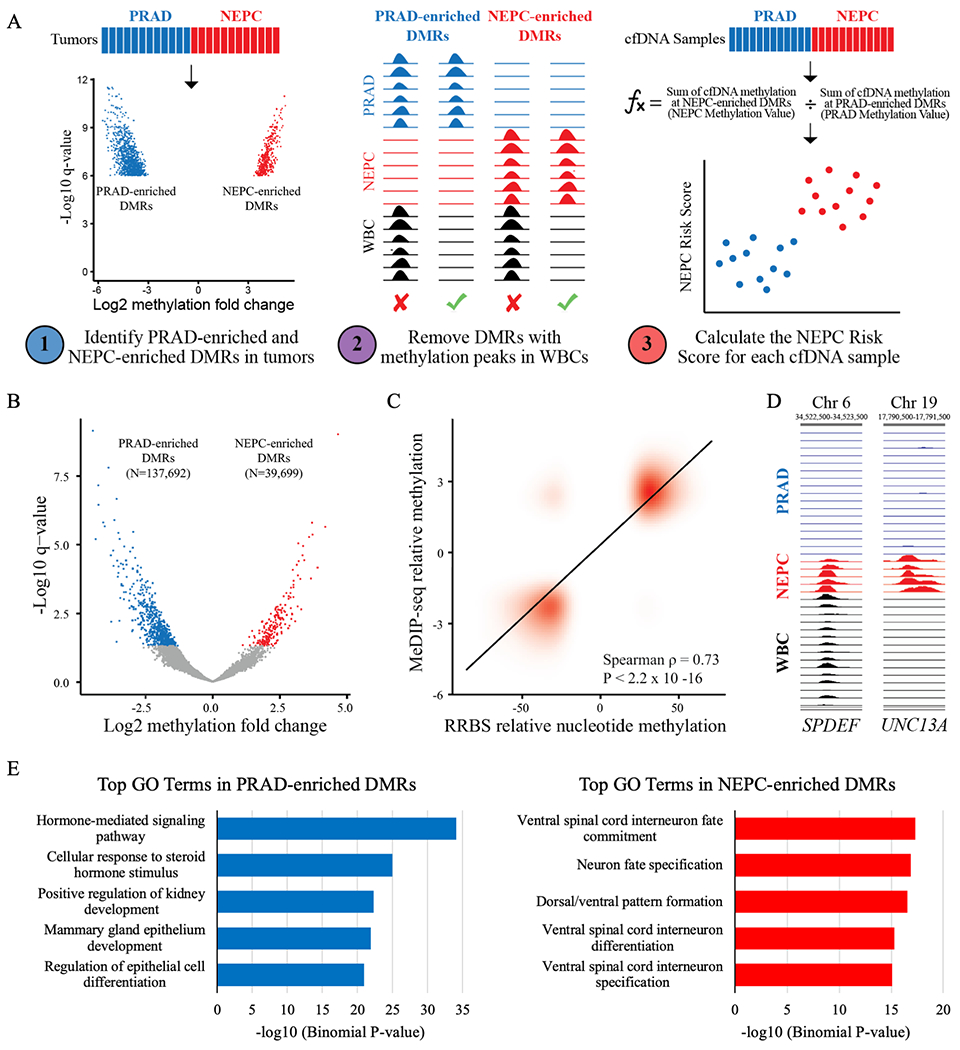
Identification of tumor-derived PRAD-enriched and NEPC-enriched DMRs. A) Overview of the methods used to detect the presence of NEPC based on tissue-informed cfDNA analysis. B) Volcano plot showing differentially methylated regions (DMRs) between PRAD (N=24) and NEPC (N=5) patient-derived xenografts. Red and blue dots represent NEPC-enriched PRAD-enriched (N=137,692) and NEPC-enriched (N=39,699) DMRs, respectively, with FDR-adjusted P<0.05. C) Correlation between tumor-derived DMRs with differentially methylated nucleotides in reduced representation bisulfite sequencing (RRBS) data from CR-PRAD and NEPC tumors.(5) D) Tracks depict methylation at the SPDEF gene and UNC13A gene determined by MeDIP-seq in PRAD tumors, NEPC tumors, and white blood cells (WBCs). E) The top 5 gene ontology (GO) enrichment terms for PRAD-enriched and NEPC-enriched DMRs after removing sites with DNA methylation in WBCs.
Statistical analysis
Comparisons between two groups were calculated using a Wilcoxon rank-sum test. To determine the accuracy of the NEPC Risk Score for discriminating between cfDNA samples from men with NEPC versus CR-PRAD, the AUROC was calculated using JMP version 16. The optimal cut-off for classifying NEPC versus CR-PRAD samples based on NEPC Risk Scores in the cfDNA test cohort was calculated using Youden’s index (J = sensitivity + specificity – 1). The optimal cut-off was determined as the point with the maximum index value. OS was defined as time from radiographic evidence of metastatic disease to death. Living patients were censored at the last evaluation. OS was estimated using the Kaplan-Meier method. P-values were calculated using log-rank test. All P-values were two-sided.
Data and materials availability
The cfMeDIP-seq NGS data for patient samples that support the findings of this study are available upon request from the corresponding author (M.L.F.) to comply with the DFCI ethics regulations to protect patient privacy. All requests for raw and analyzed data will be promptly reviewed by the Belfer Office for Dana-Farber Innovations to verify if the request is subject to any intellectual property or confidentiality obligations. Any data and materials that can be shared will be released via a Data Transfer Agreement. All code used to process the data and carry out the analyses described in the methods is in a publicly available GitHub repository at: https://github.com/scbaca/cfmedip.
RESULTS
Identification of NEPC- and PRAD-enriched DMRs in a tumor training set
Prior applications of cfMeDIP-seq for non-invasive cancer detection identified DMRs directly in cfDNA between highly disparate patient groups, such as cancer versus no cancer.(8–10) However, as men with mCRPC who develop NEPC often have concurrent PRAD, this limits the ability to identify NEPC-specific DMRs directly in cfDNA. To address this unique challenge, we developed a novel tissue-informed strategy for analyzing cfMeDIP-seq data (Fig. 1A). We first performed MeDIP-seq on 29 LuCaP PDXs, including 5 NEPC and 24 PRAD tumors (Supplementary Table S1).(12) We chose to analyze PDXs based on recent single-cell analyses of mCRPC clinical biopsy specimens, which revealed significant intra-tumoral heterogeneity, including admixed NEPC and PRAD cell populations.(24,25) In contrast, the LuCaP PDXs, which have undergone comprehensive pathologic and molecular characterization, provide a more pure source of NEPC and PRAD tumor cells.(12)
Differential methylation analysis of the LuCaP PDXs identified 39,699 NEPC-enriched and 137,692 PRAD-enriched DMRs (FDR-adjusted P<0.05) (Fig. 1B).(12) To ensure that the PDX methylation data is representative of clinical biopsy specimens, we compared the LuCaP-derived NEPC- and PRAD-enriched DMRs to DNA methylation data generated from an independent set of castration-resistant NEPC and PRAD tumors using reduced-representation bisulfite sequencing.(5) We observed a high correlation between NEPC- and PRAD-enriched DMRs from the LuCaP PDXs and the clinical biopsy specimens (ρ=0.73; P<2.2x10−16) (Fig. 1C).
We then identified a subset of NEPC- and PRAD-enriched DMRs that could be used to non-invasively detect NEPC. Using a stringent cut-off of FDR-adjusted P<1.0x10−6 and log2 fold-change >3, we identified 432 NEPC-enriched and 1,086 PRAD-enriched DMRs. As the majority of cfDNA is derived from leukocytes, we removed sites that were methylated in WBCs from age-matched male controls (N=1,165), resulting in a final set of 76 NEPC-enriched and 277 PRAD-enriched DMRs. The SPDEF gene highlights the importance of this step. While SPDEF was methylated in NEPC and unmethylated in PRAD tumors, it is also methylated in WBCs (Fig. 1D). The inability to determine whether a methylated cfDNA fragment at this locus originated from NEPC or WBCs renders it uninformative for detecting NEPC and could contribute to misclassification. As exemplified in UNC13A, a gene associated with neural signaling, the final set of DMRs are methylated in one tumor type and unmethylated in the opposite tumor type and WBCs. Consequently, cfDNA fragments at these loci indicate the presence of NEPC or PRAD.
To ensure that the final set of tumor-derived DMRs retained biological relevance, we assessed nearby genes for Gene Ontology (GO) term enrichment.(26) The top GO terms in NEPC-enriched DMRs pertained to neural development and differentiation, whereas PRAD-enriched DMRs related to hormone signaling and epithelial cell differentiation, suggesting that the final set of tumor-derived DMRs reflect the divergent gene regulatory programs of NEPC and PRAD (Fig. 1E).
Classification of NEPC and CR-PRAD samples in a cfDNA test cohort
To evaluate the ability to accurately detect NEPC using the novel tissue-informed approach, we analyzed a test cohort of plasma cfDNA samples from 56 men with mCRPC, including 11 with NEPC and 45 with CR-PRAD. We first performed LPWGS on all cfDNA samples and calculated tumor content using ichorCNA, an analytical tool that estimates cfDNA tumor fraction based on somatic copy number alterations.(13) Based on the ichorCNA lower limit of detection (3% tumor fraction), 48 (86%) of the 57 cfDNA samples had detectable tumor DNA including 9 (82%) and 39 (87%) of NEPC and CR-PRAD patients, respectively.(13) cfDNA samples with less than 3% tumor content were excluded from methylation analysis (Supplementary Fig. S1). These results compare favorably to a published cfDNA analysis of 269 samples from men with metastatic prostate cancer that detected tumor DNA in 83% of samples using LPWGS and hybrid-capture targeted sequencing.(27)
Characteristics of men in the cfDNA test cohort at the time of plasma collection are listed in Table 1. Consistent with known decoupling of prostate specific antigen (PSA) from its typical association with disease burden in NEPC, the median PSA was 0.37 for NEPC patients (range 0.03-3.7) and 140 for CR-PRAD patients (range 0.79-4305).(11) Median cfDNA tumor content was 15% for men with NEPC (range 5.1-75%) and 21% for those with CR-PRAD (range 3.3-80%) (P=0.89) (Table 1; Supplementary Fig. S2A).
Table 1.
Patient characteristics at the time of cfDNA collection in the test and validation cohorts of men with mCRPC.
| Test Cohort | Validation Cohort | |||
|---|---|---|---|---|
| NEPC N = 9 | PRAD N = 39 | NEPC N = 12 | PRAD N = 41 | |
| Median cfDNA Tumor Content (Range) | 15% (5.1-75%) | 21% (3.3-80%) | 23% (3.4-43%) | 16% (3.8-49%) |
| Median Age (Range) | 72 (60-84) | 71 (61-92) | 71 (54-91) | 70 (49-86) |
| Median PSA (Range) | 0.37 (0.03-3.7) | 140 (0.79-4305) | 0.33 (0.01-6.23) | 112 (4.5-1821) |
| De Novo NEPC | 3 (33%) | N/A | 2 (17%) | N/A |
| Prior Local Therapy | 5 (56%) | 27 (69%) | 5 (42%) | 26 (63%) |
| Prior ADT | 4 (44%) | 39 (100%) | 8 (67%) | 41 (100%) |
| Prior Abiraterone or Enzalutamide | 0 (0%) | 36 (92%) | 4 (33%) | 39 (95%) |
| Prior Docetaxel | 2 (22%) | 25 (64%) | 2 (17%) | 35 (85%) |
| Prior EP Chemotherapy | 7 (78%) | 0 (0%) | 8 (67%) | 0 (0%) |
| Liver Metastases | 3 (33%) | 15 (38%) | 8 (67%) | 13 (32%) |
Abbreviations: mCRPC, metastatic castration-resistant prostate cancer; cfDNA, cell-free DNA; NEPC, neuroendocrine prostate cancer; PRAD, prostate adenocarcinoma; PSA, prostate-specific antigen; N/A, not applicable; ADT, androgen deprivation therapy; ARSI, androgen receptor signaling inhibitor; EP, etoposide plus platinum.
To evaluate to the ability to detect NEPC in cfDNA from men with mCRPC, we first performed cfMeDIP-seq on the test cohort samples. We then derived an NEPC Methylation Value and PRAD Methylation Value for each sample by summing the methylated cfDNA fragments at tissue-derived NEPC-enriched and PRAD-enriched DMRs, respectively (Fig. 1A). An NEPC Risk Score was calculated for each sample as the normalized ratio of the NEPC Methylation Value versus the PRAD Methylation Value.
We observed significantly higher NEPC Methylation Values in men with NEPC than CR-PRAD (median 8.1x10−6 versus 6.3x10−6; P=0.0025) (Fig. 2A). In contrast, PRAD Methylation Values were significantly higher in men with CR-PRAD than NEPC (median 5.4x10−5 versus 4.1x10−5; P=4.3x10−6) (Fig. 2B). NEPC Risk Scores were significantly higher in men with NEPC than those with CR-PRAD (median 0.35 versus −0.14; P=4.3x10−7) (Fig. 2C). The AUROC for accurate classification of men with NEPC versus CR-PRAD based on NEPC Risk Score was 0.96. The optimal NEPC Risk Score cut-off (high >0.15 versus low ≤0.15) demonstrated 100% sensitivity and 90% specificity for detecting NEPC. Further, high versus low NEPC Risk Score was associated with significantly shorter OS from the time of metastatic prostate cancer (hazard ratio [HR]=2.5; 95% confidence interval [95%CI]=1.2-4.8; P=0.017) (Fig. 2D). Median OS was 32 months shorter for men with high (14 months) versus low (46 months) NEPC Risk Scores.
Figure 2.
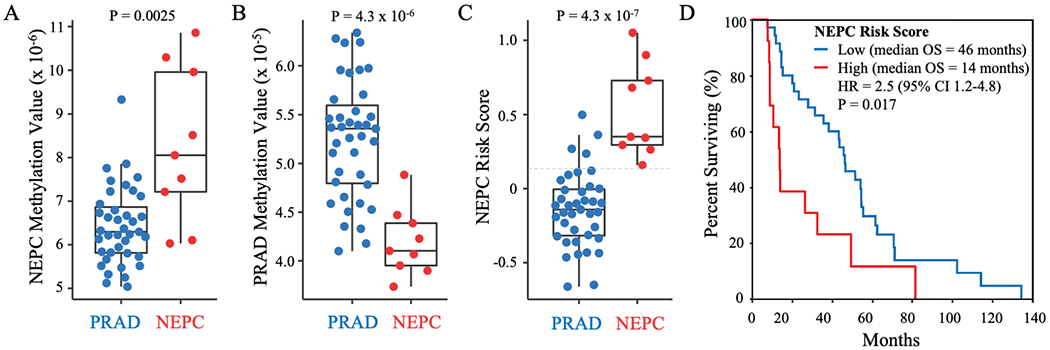
Classification of NEPC and PRAD samples in the cfDNA test cohort. NEPC Methylation Values (A), PRAD Methylation Values (B), and NEPC Risk Scores (C) in cfDNA samples from men with PRAD or NEPC in the test cohort. P-Values calculated using a two-sided Wilcoxon rank-sum test. Optimal cut-off (indicated by dotted gray line) was determined in this cohort using Youden’s J statistic. D) Kaplan-Meier curve for overall survival (OS) from the time of metastatic disease for men with high (>0.15) versus low (≤0.15) NEPC Risk Score relative to the cut-off.
Classification of NEPC and CR-PRAD samples in an independent cfDNA validation cohort
To assess the reproducibility of this approach and the NEPC Risk Score cut-off, we identified an independent multi-institutional validation cohort of plasma samples from 73 men with mCRPC at DFCI and WCM, including 16 men with NEPC and 57 with CR-PRAD. cfDNA LPWGS identified tumor DNA in 53 (73%) of samples including 12 (75%) and 48 (72%) of NEPC and CR-PRAD patients, respectively. cfDNA samples with less than 3% tumor content were excluded from methylation analysis (Supplementary Fig. S1). Median cfDNA tumor content was 23% for NEPC patients (range 3.4-43%) and 16% for CR-PRAD patients (range 3.8-39%) in the test cohort (P=0.49) (Supplementary Fig. S2B; Table 1). Median PSA was 0.33 (range 0.01-6.23) in men with NEPC versus 112 (range 4.5-1821) in those with CR-PRAD. Differences between men with NEPC and CR-PRAD in the cfDNA validation cohort were similar to those observed in the cfDNA test cohort (Table 1).
As we observed in the test cohort, NEPC samples in the cfDNA validation cohort exhibited significantly higher NEPC Methylation Values (median 9.6x10−6 versus 6.4x10−6; P=1.5 x 10−4), lower PRAD Methylation Values (median 4.5x10−5 versus 5.5x10−5; P=0.0013), and higher NEPC Risk Scores (median 0.69 versus −0.19; P=7.5x10−12) than those with CR-PRAD (Figs. 3A–C). The AUROC for accurate classification of men with NEPC versus CR-PRAD based on NEPC Risk Score was 1.0. Applying the NEPC Risk Score cut-off derived in the test cohort (high >0.15 versus low ≤0.15) to the cfDNA validation cohort resulted in 100% sensitivity and 95% specificity for detecting NEPC. High versus low NEPC Risk Score was associated with significantly shorter OS from the time of metastatic prostate cancer (HR=4.3, 95%CI=2.9-8.9; P=3.2x10−4) (Fig. 3D). Median OS was 36 months shorter for men with high (20 months) versus low (56 months) NEPC Risk Scores. Notably, there was no association of cfDNA tumor content with OS across the two cfDNA cohorts (Supplementary Fig. S3), suggesting that the negative correlation between NEPC Risk Score and OS is driven by different tumor biology and not higher disease burden.
Figure 3.
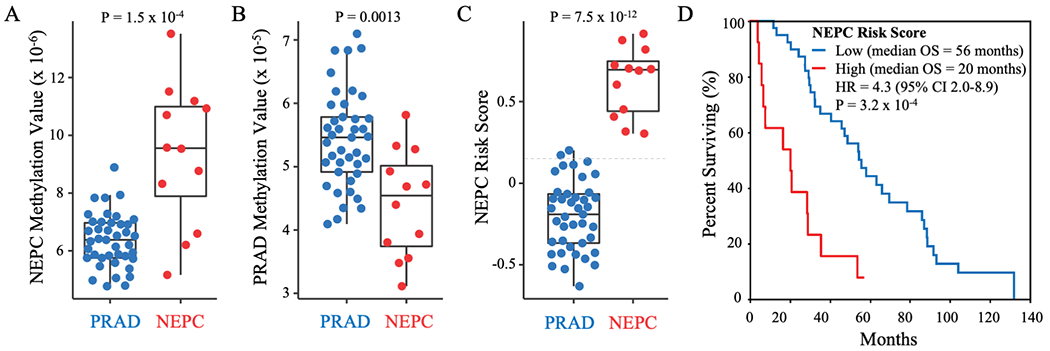
Classification of NEPC and PRAD samples in the cfDNA validation cohort. NEPC Methylation Values (A), PRAD Methylation Values (B), and NEPC Risk Scores (C) in cfDNA samples from men with NEPC or PRAD in the validation cohort. P-Values calculated using a two-sided Wilcoxon rank-sum test. The optimal NEPC Risk Score cut-off determined in the independent cfDNA test cohort is indicated by dotted gray line. D) Kaplan-Meier curve for overall survival (OS) from the time of metastatic disease for men with high (>0.15) versus low (≤0.15) NEPC Risk Score relative to the cut-off determined in the independent cfDNA test cohort.
Patient vignettes highlight NEPC risk factors in misclassified CR-PRAD samples
To understand potential factors driving misclassification, we reviewed medical histories for the six patients with CR-PRAD with NEPC Risk Scores >0.15 across the two cfDNA cohorts. Five of these patients had clinical, radiographic, and genomic features associated with NEPC (Fig. 4). The two patients with the highest NEPC Risk Score (0.50 and 0.36) both previously received abiraterone, docetaxel, cabazitaxel, and were on enzalutamide at the time of cfDNA collection. The first patient’s CT scan six days after cfDNA collection showed marked increase in metastatic tumor burden, including new liver metastases. He subsequently experienced clinical deterioration and died five weeks later. The second patient previously underwent somatic tumor profiling revealing two-copy RB1 deletion. He experienced clinical deterioration and died 8 weeks after cfDNA collection. The next three patients had all received prior abiraterone and/or enzalutamide. The first (NEPC Risk Score of 0.27) was progressing on abiraterone and underwent tumor biopsy at the time of cfDNA collection showing poorly differentiated carcinoma harboring single-copy RB1 loss and two deleterious TP53 alterations. The second patient (NEPC Risk Score of 0.24) previously received abiraterone and was progressing on enzalutamide at the time of cfDNA collection. Genomic profiling two months earlier showed that the patient’s tumor harbored biallelic loss of RB1 and TP53. The third patient (NEPC Risk Score of 0.20) previously received abiraterone and at the time of cfDNA collection was progressing on docetaxel with CT scan showing new liver metastases. He experienced clinical deterioration and died two months later. These hypothesis-generating vignettes suggest the possibility that the cfDNA NEPC Risk Score may identify occult NEPC not detected through routine clinical care.
Figure 4.

cfDNA from men with CR-PRAD with high NEPC Risk Scores display clinical and genomic features of NEPC.
Association of the plasma cfDNA methylome with NEPC Risk Score and tumor content
Prior data suggest that the plasma cfDNA methylome strongly correlates with tumor content in men with metastatic prostate cancer.(28) As such, we sought to understand the association of the cfDNA methylome in this cohort with tumor content. We first performed principal component analysis (PCA) of the genome-wide methylome data (Fig. 5A) and the methylation data at the NEPC- and PRAD-enriched DMRs included in the NEPC Risk Score (Fig. 5B) for the 101 cfDNA samples included in the NEPC Risk Score analyses. In the genome-wide data, the first principal component (PC1) is driven by an outlier sample with high CpG enrichment relative to the others. There was otherwise no separation of NEPC and CR-PRAD samples in PC1 and PC2 in the genome-wide methylome data (Fig. 5A). However, at the DMR sites, PC1 and PC2 clearly separated NEPC and CR-PRAD samples (Fig. 5B).
Figure 5.
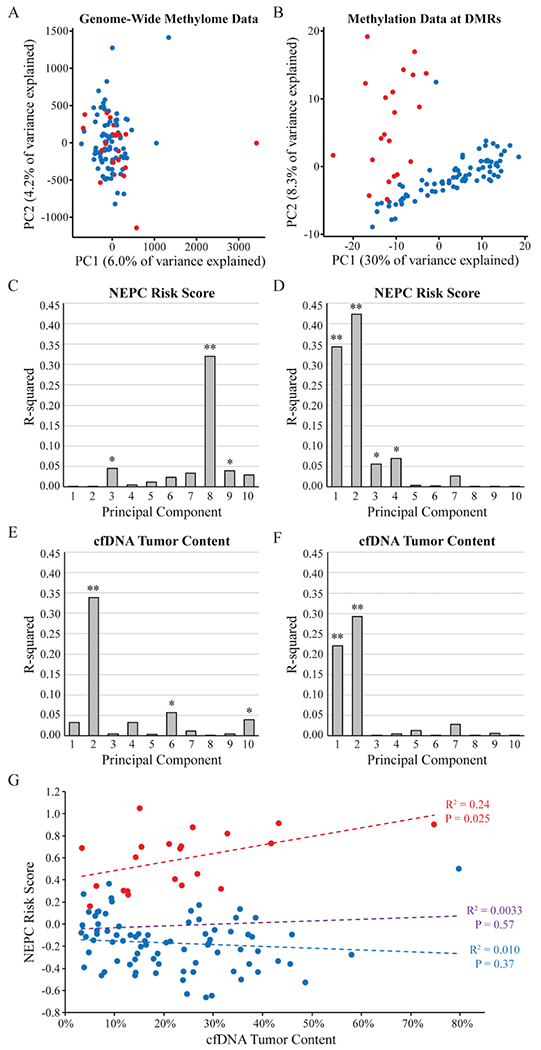
Association of the plasma cfDNA methylome with NEPC Risk Score and tumor content. A) Principal component analysis (PCA) of the genome-wide methylome for 101 plasma cfDNA samples from men with CR-PRAD or NEPC. B) PCA of the 101 plasma cfDNA samples limiting to the NEPC- and PRAD-enriched DMRs included in the NEPC Risk Score. Correlation between NEPC Risk Score with the top 10 principal components (PCs) for the cfDNA genome-wide methylome data (C) and restricted to the DMR sites (D). Correlation between cfDNA tumor content with the top 10 PCs for the cfDNA genome-wide methylome data (E) and restricted to the DMR sites (F). Correlation between NEPC Risk Score and each PC was measured using the coefficient of determination (R2). * P<0.05; ** P<1x10−6. G) Correlation between NEPC Risk Score and tumor content for the 101 cfDNA samples from men with NEPC and CR-PRAD. Dotted lines show the linear regression for the NEPC samples (red), CR-PRAD samples (blue), and all samples (purple).
We next quantified the correlation between each of the first 10 PCs with NEPC Risk Score and cfDNA tumor content. For the genome-wide data, not until PC8, which explained 2.2% of variance, was there a robust correlation with NEPC Risk Score (R2=0.32; P=7.3x10−10) (Fig. 5C; Supplementary Fig. S4A). In contrast, when limiting to the DMRs included in the NEPC Risk Score, PC1 (R2=0.34; P=1.2x10−10), which explained 30% of variance, and PC2 (R2=0.42; P=2.0x10−13), which explained 8.3% of variance, both demonstrated robust correlation with NEPC Risk Score (Fig. 5D; Supplementary Fig. S4B). We then quantified the correlation between the top PCs and cfDNA tumor content. In the genome-wide methylome data, PC2, which explained 4.2% of variance, correlated with cfDNA tumor content (R2=0.34; P=1.7x10−10) (Fig. 5E; Supplementary Fig. S4A). This result affirms the prior finding that the prostate cancer plasma cfDNA methylome correlates with cfDNA tumor content.(28) When limiting to the DMRs included in the NEPC Risk Score, PC1 (R2=0.22; P=7.2x10−7) and PC2 (R2=0.29; P=5.1x10−9) correlated with cfDNA tumor content, though not to the same extent they correlated with NEPC Risk Score (Fig. 5F; Supplementary Fig. S4B).
Finally, we assessed the correlation between NEPC Risk Score and cfDNA tumor content (Fig. 5G). Across all NEPC and CR-PRAD cfDNA samples, there was no correlation between NEPC Risk Score and tumor content (R2=0.0033; P=0.57); there was also no correlation in the PRAD samples (R2=0.010; P=0.37). NEPC Risk Score and tumor content did significantly correlate in the cfDNA samples from men with NEPC (R2=0.24; P=0.025). Given this association, suggesting lower NEPC Risk Scores in men with lower cfDNA tumor content, we evaluated the diagnostic performance of the NEPC Risk Score in the NEPC and CR-PRAD samples across the two cohorts with cfDNA tumor content less than 10%. The NEPC Risk Score in these patients resulted in an AUROC of 0.93; applying the NEPC Risk Score cut-off of 0.15 resulted in 100% sensitivity and 82% specificity for detecting NEPC (data not shown).
DISCUSSION
NEPC is an aggressive, clinically actionable resistance phenotype that can emerge in men with mCRPC. The challenge of identifying NEPC in routine clinical practice leads to delays in diagnosis and underdiagnosis. In the largest study to date of cfDNA from men with NEPC, we present a novel approach for tissue-informed analysis of cfMeDIP-seq data, leading to highly accurate non-invasive detection of NEPC. We first identified methylation sites enriched in a training set of NEPC and PRAD tumors. Tissue-informed methylation analysis of two independent cohorts of cfDNA from men with pathologically confirmed NEPC or CR-PRAD, demonstrated high accuracy (AUROC of 0.96 and 1.0). Importantly, applying a diagnostic cut-off derived in the cfDNA test cohort to the independent validation cohort resulted in 100% sensitivity and 95% specificity for detecting NEPC. Finally, in both cfDNA cohorts, a high NEPC Risk Score was associated with significantly shorter OS (HR of 2.5 and 4.3). This work strongly supports the analytical and clinical utility of tissue-informed analysis of cfMeDIP-seq data to non-invasively detect NEPC in men with mCRPC.
The current approach for diagnosing NEPC has significant shortcomings that could be overcome with a liquid biopsy. The lack of consensus pathological criteria highlights the advantage of an objective biomarker. Herein, we demonstrated that tissue-informed analysis of cfMeDIP-seq data provides a quantitative readout (NEPC Risk Score) that discriminated between men with NEPC or CR-PRAD with high accuracy in two independent cfDNA cohorts. The ability to apply a diagnostic cut-off from one cfDNA cohort to an independent cohort while maintaining excellent diagnostic accuracy suggests this quantitative approach is robust and reproducible. An additional benefit of a liquid biopsy to detect NEPC is that cfDNA may be more representative of a patient’s overall tumor burden than tissue biopsy of a single metastatic focus.(29) Intra-patient tumor heterogeneity is well-established in mCRPC.(6) This is highly relevant in this clinical context, as NEPC often emerges as a treatment-resistant subclone and can be missed due to sampling error with a single tissue biopsy. Future studies analyzing cfDNA from patients undergoing autopsy or correlating cfDNA results with molecular imaging could provide further insight into how representative NEPC Risk Scores are of intra-patient NEPC versus PRAD tumor burden. However, we establish is this report that the NEPC Risk Score is a highly accurate quantitative non-invasive biomarker that is highly predictive of the presence or absence of NEPC in men with mCRPC.
Detecting NEPC has important prognostic implications. Compared to men with CR-PRAD, NEPC is associated with shorter OS.(1) Consistent with this known negative prognostic association, we observed that high NEPC Risk Score (relative to the diagnostic cut-off) was associated with significantly shorter OS in two independent cfDNA cohorts. Aggarwal et al reported a HR of 1.8 (95%CI=1.0-3.2) for OS in men with versus without treatment-emergent small-cell neuroendocrine prostate cancer.(1) The HRs for OS in our cohorts were 2.5 (95%CI=1.2-4.8) and 4.3 (95%CI=2.0-8.9). The worse prognosis of men with high NEPC Risk Score in our study may reflect differences in patient characteristics. We included only men with morphologic high-grade neuroendocrine carcinoma and not PRAD with expression of neuroendocrine markers by immunohistochemistry – the latter is not as clearly associated with a virulent clinical course as the former.(11)
Early and accurate detection of NEPC also has important therapeutic implications. While men with NEPC are characteristically unresponsive to ARSIs, they are more likely to respond to platinum-based chemotherapy.(4) Consequently, NCCN guidelines recommend treatment with platinum chemotherapy combined with etoposide or a taxane.(30) Histology-specific treatment recommendations highlight the immediate clinical actionability of detecting NEPC. With several ongoing clinical trials testing novel therapeutic approaches in men with NEPC, the implications of detecting this disease variant will likely increase in the coming years.(31) While the association of the NEPC Risk Score with response to prostate cancer therapy remains to be established, we encourage incorporating cfDNA collection into prospective clinical trials to facilitate future studies to develop and validate non-invasive biomarkers to identify patients likely to benefit from NEPC-directed therapy.
Successful cfDNA-based biomarkers must be accurate, cost-effective, and practical to implement in clinical practice. Beltran et al previously reported the feasibility of non-invasively detecting NEPC-specific DNA methylation in cfDNA using whole-genome bisulfite sequencing (WGBS).(7) However, test characteristics for distinguishing men with NEPC versus CR-PRAD using this approach were not reported. Compared to WGBS, cfMeDIP-seq has several advantages. The high cost of whole-genome sequencing currently limits the ability to implement WGBS in clinical practice. In contrast, by only sequencing methylated cfDNA, cfMeDIP-seq provides genome-wide methylation data at a fraction of the cost of WGBS. Further, cfMeDIP-seq only requires 5-10 ng of cfDNA, which can be obtained from 1 ml of plasma. While direct comparison of the performance of these approaches will be informative, the low cost, small sample requirement, and high sensitivity highlight advantages of cfMeDIP-seq as the basis for a clinical biomarker.
The methods presented in this manuscript represent an important advance for developing cfDNA methylation-based clinical biomarkers. Several recent publications highlight advantages of epigenetic compared to genetic liquid biopsy approaches.(32–34) Whereas previous studies performed unsupervised analysis of cfMeDIP-seq data, we developed a novel tissue-informed method, which benefits from the strength of its biological basis. Tissue-informed analysis ensures that the model is built upon molecular features known to be present in and specific to the tumor of interest. This contrasts with the tumor-naïve approach of developing a methylation signature directly in cfDNA (e.g., from individuals with cancer versus without cancer), which risks overfitting due to signal unrelated to cancer (e.g., sex, age, smoking status, comorbid disease, etc). Principal component analysis emphasized the value of this tissue-informed approach. In the genome-wide cfDNA methylome data, not until PC8, which explained only 2.2% of variance, did we observe a correlation with the methylation signal that distinguished NEPC from CR-PRAD samples. However, the methylation data at the tissue-derived NEPC- and PRAD-enriched DMRs revealed a strong correlation between NEPC Risk Score and PCs 1 and 2, which explained nearly 40% of variance. We believe that the methods presented herein will help facilitate detection of clinically relevant cancer phenotypes. Aberrant DNA methylation is present across tumor types with several clinically actionable subtypes harboring distinct methylation profiles, such as IDH-mutant gliomas and microsatellite instability (MSI) high uterine and colon cancers.(35–38) As our understanding of clinically-actionable epigenetically-distinct cancer phenotypes evolves, the methods presented in this manuscript will facilitate non-invasive detection of these tumor subtypes.
While our results strongly support the feasibility of using cfMeDIP-seq to non-invasively detect NEPC in men with mCRPC, it is appropriate to recognize several potential limitations. First, is the number of patients with NEPC included in the study. The cfDNA test and validation cohorts contained 21 men with NEPC. While this number is small in absolute terms, it represents the largest analysis to date of cfDNA from men with pathologically-confirmed NEPC. While similarly high accuracy was observed across two independent cfDNA cohorts, the reproducibility and clinical validity will benefit from further prospective validation. It is also important to acknowledge that this was a retrospective and heterogeneous cohort, which introduces potential confounding factors, mainly patient selection. This limits our ability to conclusively establish that NEPC Risk Score is associated with inferior clinical outcomes. However, that the NEPC Risk Score performs so well despite the heterogeneity in the cohort speaks to the robustness of the methylation signal and the ability of the assay and methods to detect it. Another limitation is that NEPC patients in this study were limited to men with high-grade neuroendocrine carcinoma and did not include those with PRAD with neuroendocrine differentiation.(11) Thus, we cannot comment on whether the NEPC Risk Score discriminates between these two variants. Likewise, all men in the NEPC group had biopsy-proven high-grade neuroendocrine carcinoma at the time of plasma collection. We did not evaluate plasma samples in men prior to NEPC diagnosis when the relative abundance of NEPC-derived cfDNA may be lower and thus harder to detect. As such, we were not able to establish how early in the disease course this biomarker can detect treatment-emergent NEPC. Finally, as we limited the methylation analysis to men with detectable circulating tumor DNA by ichorCNA, we did not assess how the assay performs in men with very low cfDNA tumor content. More work is needed to fully establish the relationship between NEPC Risk Score with cfDNA tumor content. Several additional questions remain: What is the analytical limit of detection of this cfDNA methylation-based approach for detecting NEPC? What is the optimal timing to evaluate cfDNA in men with mCRPC to identify NEPC? How long before clinical diagnosis can NEPC be detected in cfDNA? Will early detection and initiation of platinum-based chemotherapy improve clinical outcomes for men diagnosed with NEPC? We hope to address these questions in future studies.
In summary, we demonstrated the analytical and clinical utility of tissue-informed cfDNA methylation analysis to non-invasively detect NEPC in men with mCRPC. These findings support further studies to establish the prognostic and predictive value of the cfDNA NEPC Risk Score in men with mCRPC. Finally, the novel methods reported in this manuscript are an important step towards broadening the clinical applicability of blood-based epigenomic assays by providing a framework for non-invasive detection of tumor subtypes with distinct DNA methylation profiles.
Supplementary Material
STATEMENT OF TRANSLATIONAL RELEVANCE.
Early detection of neuroendocrine prostate (NEPC) is challenging in clinical practice, but has important prognostic and therapeutic implications for men with metastatic castration-resistant prostate cancer (mCRPC). In the largest study to date of cell-free DNA (cfDNA) from men with NEPC, we developed and validated a non-invasive NEPC Risk Score using tissue-informed cfDNA methylation analysis. Applying the NEPC Risk Score to cfDNA from two independent cohorts of men with mCRPC resulted in highly accurate discrimination between men with versus without NEPC. In both cohorts, high NEPC Risk Score was associated with significantly worse overall survival. These data strongly support the clinical utility of this cfDNA methylation-based NEPC Risk Score in men with mCRPC to non-invasively identify those who should be considered for platinum-based chemotherapy or clinical trials of novel NEPC-directed therapies.
ACKNOWLEDGMENTS:
J. Berchuck is supported by the Department of Defense (W81XWH-20-1-0118). S. Baca is supported by a fellowship from the PhRMA Foundation and the Kure It Cancer Research Foundation. K. Korthauer is supported by the Natural Sciences and Engineering Research Council of Canada. Establishment and characterization of the LuCaP PDX models has been supported by the Pacific Northwest Prostate Cancer SPORE (P50CA97186), the Department of Defense Prostate Cancer Biorepository Network (W81XWH-14-2-0183), the National Cancer Institute P01 CA163227, the Prostate Cancer Foundation, the Institute for Prostate Cancer Research, and the Richard M. Lucas Foundation. M. Pomerantz, H. Beltran, and M. Freedman are supported by a DFCI Medical Oncology grant award. H. Beltran is supported by the National Cancer Institute (5R37CA241486). M. Freedman is supported by the Claudia Adams Barr Program for Innovative Cancer Research, the H.L. Snyder Medical Research Foundation, and the Cutler Family Fund for Prevention and Early Detection.
Footnotes
Conflict of Interest Statement: JEB, SCB, and MLF are listed as inventors on patents filed that pertain to the data presented in this manuscript. All other author disclosures are outside the scope of the current manuscript.
REFERENCES
- 1.Aggarwal R, Huang J, Alumkal JJ, Zhang L, Feng FY, Thomas GV, et al. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. J Clin Oncol Off J Am Soc Clin Oncol. 2018;36:2492–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci U S A. 2019;116:11428–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci. National Academy of Sciences; 2019;116:11428–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Humeniuk MS, Gupta RT, Healy P, McNamara M, Ramalingam S, Harrison M, et al. Platinum sensitivity in metastatic prostate cancer: does histology matter? Prostate Cancer Prostatic Dis. 2018;21:92–9. [DOI] [PubMed] [Google Scholar]
- 5.Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016;22:298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JMC, Papaemmanuil E, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015;520:353–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beltran H, Romanel A, Conteduca V, Casiraghi N, Sigouros M, Franceschini GM, et al. Circulating tumor DNA profile recognizes transformation to castration-resistant neuroendocrine prostate cancer. J Clin Invest. 2020;130:1653–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shen SY, Singhania R, Fehringer G, Chakravarthy A, Roehrl MHA, Chadwick D, et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature. 2018;563:579–83. [DOI] [PubMed] [Google Scholar]
- 9.Shen SY, Burgener JM, Bratman SV, De Carvalho DD. Preparation of cfMeDIP-seq libraries for methylome profiling of plasma cell-free DNA. Nat Protoc. 2019;14:2749–80. [DOI] [PubMed] [Google Scholar]
- 10.Nuzzo PV, Berchuck JE, Korthauer K, Spisak S, Nassar AH, Abou Alaiwi S, et al. Detection of renal cell carcinoma using plasma and urine cell-free DNA methylomes. Nat Med. 2020;26:1041–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Epstein JI, Amin MB, Beltran H, Lotan TL, Mosquera J-M, Reuter VE, et al. Proposed morphologic classification of prostate cancer with neuroendocrine differentiation. Am J Surg Pathol. 2014;38:756–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nguyen HM, Vessella RL, Morrissey C, Brown LG, Coleman IM, Higano CS, et al. LuCaP Prostate Cancer Patient-Derived Xenografts Reflect the Molecular Heterogeneity of Advanced Disease an--d Serve as Models for Evaluating Cancer Therapeutics. The Prostate. 2017;77:654–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adalsteinsson VA, Ha G, Freeman SS, Choudhury AD, Stover DG, Parsons HA, et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat Commun. 2017;8:1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinforma Oxf Engl. 2016;32:3047–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinforma Oxf Engl. 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lienhard M, Grimm C, Morkel M, Herwig R, Chavez L. MEDIPS: genome-wide differential coverage analysis of sequencing data derived from DNA enrichment experiments. Bioinforma Oxf Engl. 2014;30:284–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cavalcante RG, Sartor MA. annotatr: genomic regions in context. Bioinformatics. Oxford Academic; 2017;33:2381–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amemiya HM, Kundaje A, Boyle AP. The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci Rep. 2019;9:9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pelizzola M, Koga Y, Urban AE, Krauthammer M, Weissman S, Halaban R, et al. MEDME: an experimental and analytical methodology for the estimation of DNA methylation levels based on microarray derived MeDIP-enrichment. Genome Res. 2008;18:1652–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cejas P, Xie Y, Font-Tello A, Lim K, Syamala S, Qiu X, et al. Subtype heterogeneity and epigenetic convergence in neuroendocrine prostate cancer. Nat Commun. 2021;12:5775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dong B, Miao J, Wang Y, Luo W, Ji Z, Lai H, et al. Single-cell analysis supports a luminal-neuroendocrine transdifferentiation in human prostate cancer. Commun Biol. 2020;3:778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mayrhofer M, De Laere B, Whitington T, Van Oyen P, Ghysel C, Ampe J, et al. Cell-free DNA profiling of metastatic prostate cancer reveals microsatellite instability, structural rearrangements and clonal hematopoiesis. Genome Med. 2018;10:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu A, Cremaschi P, Wetterskog D, Conteduca V, Franceschini GM, Kleftogiannis D, et al. Genome-wide plasma DNA methylation features of metastatic prostate cancer. J Clin Invest. American Society for Clinical Investigation; 2020;130:1991–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wyatt AW, Annala M, Aggarwal R, Beja K, Feng F, Youngren J, et al. Concordance of Circulating Tumor DNA and Matched Metastatic Tissue Biopsy in Prostate Cancer. JNCI J Natl Cancer Inst [Internet]. 2017. [cited 2019 Aug 11];109. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6440274/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.NCCN Clinical Practice Guidelines in Oncology: Prostate Cancer (Version 1.2022) [Internet]. 2021. Available from: https://www.nccn.org/professionals/physician_gls/pdf/prostate.pdf
- 31.Berchuck JE, Viscuse PV, Beltran H, Aparicio A. Clinical considerations for the management of androgen indifferent prostate cancer. Prostate Cancer Prostatic Dis. 2021; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lasseter K, Nassar AH, Hamieh L, Berchuck JE, Nuzzo PV, Korthauer K, et al. Plasma cell-free DNA variant analysis compared with methylated DNA analysis in renal cell carcinoma. Genet Med Off J Am Coll Med Genet. 2020;22:1366–73. [DOI] [PubMed] [Google Scholar]
- 33.Parikh AR, Seventer EEV, Siravegna G, Hartwig AV, Jaimovich A, He Y, et al. Minimal Residual Disease Detection using a Plasma-Only Circulating Tumor DNA Assay in Colorectal Cancer Patients. Clin Cancer Res [Internet]. American Association for Cancer Research; 2021. [cited 2021 May 5]; Available from: https://clincancerres.aacrjournals.org/content/early/2021/04/28/1078-0432.CCR-21-0410 [Google Scholar]
- 34.Liu MC, Oxnard GR, Klein EA, Swanton C, Seiden MV, Liu MC, et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann Oncol. Elsevier; 2020;31:745–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saghafinia S, Mina M, Riggi N, Hanahan D, Ciriello G. Pan-Cancer Landscape of Aberrant DNA Methylation across Human Tumors. Cell Rep. 2018;25:1066–1080.e8. [DOI] [PubMed] [Google Scholar]
- 36.Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cancer Genome Atlas Research Network, Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.