

Abstract
MALT1 is the effector protein of the CARMA/Bcl10/MALT1 (CBM) signalosome, a multi-protein complex that drives pro-inflammatory signaling pathways downstream of a diverse set of receptors. While CBM activity is best known for its role in immune cells, emerging evidence suggests that it plays a key role in the pathogenesis of solid tumors, where it can be activated by selected G protein-coupled receptors (GPCRs). Here, we demonstrated that overexpression of GPCRs implicated in breast cancer pathogenesis, specifically the receptors for Angiotensin II and thrombin (AT1R and PAR1), drove a strong epithelial-to-mesenchymal transition (EMT) program in breast cancer cells that is characteristic of claudin-low, triple-negative breast cancer (TNBC). In concert, MALT1 was activated in these cells and contributed to the dramatic EMT phenotypic changes through regulation of master EMT transcription factors including Snail and ZEB1. Importantly, blocking MALT1 signaling, through either siRNA-mediated depletion of MALT1 protein or pharmacologic inhibition of its activity, was effective at partially reversing the molecular and phenotypic indicators of EMT. Treatment of mice with mepazine, a pharmacologic MALT1 inhibitor, reduced growth of PAR1+, MDA-MB-231 xenografts and had an even more dramatic effect in reducing the burden of metastatic disease. These findings highlight MALT1 as an attractive therapeutic target for claudin-low TNBCs harboring overexpression of one or more selected GPCRs.
Keywords: Angiotensin, thrombin, AT1R, AGTR1, PAR1, F2R, GPCR, EMT, breast cancer, MALT1, invasion, migration, metastasis
INTRODUCTION
Inappropriate activation of G protein-coupled receptors (GPCRs) underlies the pathogenesis of several malignant tumors and is increasingly implicated in breast cancer (1–7). While the consequences of pathogenic GPCR signaling are diverse and depend upon the specific subtype of GPCR, our group has shown that overexpression of two Gαq/11-coupled GPCRs, either the type I Angiotensin II receptor (AT1R; product of the AGTR1 gene) or the protease-activated receptor-1 (PAR1; product of the F2R gene), is sufficient to activate the NF-κB pathway in breast cancer cells through a signaling complex composed of the proteins CARMA3, Bcl10, and MALT1 (CBM signalosome) (8, 9). Activation of NF-κB through this mechanism is linked to cell proliferation and survival, as well as the elaboration of secreted factors that support tumor angiogenesis and other changes in the tumor microenvironment (1, 8, 9). MALT1 is the effector protein of the CBM signalosome and drives the induction of the NF-κB pathway in part through its activity as a protease, selectively cleaving substrates that include CYLD, A20, RelB, Roquin-1/2, and Regnase-1, all of which are important regulators of the NF-κB signaling program (10, 11).
In the current study, we demonstrate that overexpression of either AT1R (AGTR1) or PAR1 (F2R) drives pronounced molecular and phenotypic alterations characteristic of epithelial-to-mesenchymal transition (EMT) in breast cancer models, and that MALT1 is crucially important in mediating these alterations. Further, we demonstrate that several recently identified chemical inhibitors of MALT1 protease activity are effective at reversing some of the molecular signatures of EMT and at inhibiting cancer cell migration, invasion, and in vivo metastatic spread, phenotypes that are linked to EMT. These results highlight the pleotropic actions of GPCR/MALT1 signaling in breast cancer and nominate MALT1 as an attractive therapeutic target to prevent metastatic dissemination.
Triple-negative breast cancers (TNBCs) comprise ~15% of all invasive breast cancer cases and are defined by the lack of currently targetable molecular drivers (12, 13). Specifically, these tumors are negative for human epidermal growth factor receptor-2 (HER2), estrogen receptor (ER), and progesterone receptor (PR). As such, the mainstays of neoadjuvant and adjuvant medical treatment are non-specific chemotherapeutics, and clinical outcomes are comparatively poor in part due to the lack of applicable targeted therapies (13, 14). Intense effort is currently being directed towards identifying novel molecular drivers of TNBC and developing increasingly specific therapies for patients with this aggressive subtype of breast cancer. TNBCs of the claudin-low type are particularly associated with EMT (15, 16), and in the current study, we find evidence that the GPCR/MALT1 signaling axis plays a prominent role in promoting the EMT program in these tumors. This finding raises the possibility that MALT1 inhibitors could represent a novel class of precision therapeutic for claudin-low TNBC, a category of breast cancer for which there is a desperate need for new and effective management strategies.
Intriguingly, MALT1 inhibitors also directly affect lymphocytes and other immune cells, and have recently been shown to tip the balance of the immune system to promote anti-tumor immunity (11, 17–22). Thus, for TNBC tumors that rely on intrinsic GPCR/MALT1 signaling, MALT1 inhibitors may be particularly efficacious due to their potential to act both on tumor cells and on the host immune system, possibly priming tumors for response to immunotherapy. Overall, our work nominates Gαq/11-coupled GPCRs as a class of receptor important for the pathogenesis of claudin-low (or mesenchymal-type) TNBC and identifies MALT1 as a downstream therapeutic target in this difficult-to-treat breast cancer subtype.
MATERIALS AND METHODS
Antibodies and other reagents
A detailed description of reagents and their sources can be found in the Supplementary Methods.
Cell lines and cell culture
BT549, ZR75–1, MDA-MB-231, Hs578T, and Hs606T cells were obtained directly from ATCC, with cell lines authenticated by short tandem repeat (STR) profiling by the source. Luciferase/GFP dual-labeled MDA-MB-231 cell-line (Cat No: SL018), authenticated and tested to be free of mycoplasma, was purchased from Genecoepia (Rockville, MD). Frozen aliquots of cells were prepared upon receipt and cell lines were passaged for less than 6 months. The stable ZR75-Neo and ZR75-AT1R (previously designated as ZR75-AGTR1) cell lines were established as described (8). MCF7 and MCF7-PAR1 (previously designated as MCF7-N55) cell lines were generated in the L. Covic laboratory as described (6). BT549, ZR75–1, ZR75-AT1R, MCF7, and MCF7-PAR1 cells were grown in phenol red-free RPMI1640 media (catalog no: 11835030, Gibco), while MDA-MB-231 cells were grown in DMEM-Glutamax media (catalog no: 10566016, Gibco). Both media formulations were supplemented with 10% FBS, 1% penicillin/streptomycin (Gibco), and MycoZap Prophylactic (catalog no: VZA-2032, Lonza). Lenti-Pac 293Ta cells (catalog no: CLv-PK-01) were purchased from GeneCopoeia for lentiviral packaging and were grown in DMEM-Glutamax media. All cells were grown at 37°C in a 5% CO2 incubator. Cell lines were regularly monitored for mycoplasma contamination using the mycoplasma MycoAlert Detection Kit (catalog no: LT07–318, Lonza). All cell lines were periodically reauthenticated by STR profiling utilizing one of two services (ATCC or UAGC).
Transient siRNA transfections and lentiviral shRNA transductions
ON-TARGET plus SMARTpool siRNAs targeting CARMA3 (catalog no: L-004395-00-0020), Bcl10 (catalog no: L-004381-00-0020), MALT1 (catalog no: L-005936-00-0020), AGTR1 (catalog no: L-005428-00-0020), IKKα (catalog no: L-003473-00-0020), and IKKβ (catalog no: L-003503-00-0020) were obtained from GE Dharmacon. Nontargeting siRNA pools (catalog no: D-001810-10-50) were used as controls. PAR1 siRNAs (GGCUACUAUGCCUACUACUdTdT, AGAUUAGUCUCCAUCAAUAdTdT) were synthesized by Sigma-Aldrich. Lipofectamine RNAiMAX (catalog no: 13778150, Thermo Fisher Scientific) was utilized to reverse transfect SMARTpool siRNAs (20 nmol/L) into cells following the manufacturer’s protocol. Knockdown efficiencies for the intended targets were determined by immunoblot assays after 48–72 hours. In the case of AGTR1, knockdown efficiencies were determined by TaqMan RT-PCR assays (Thermo Fisher) as described (8).
TRIPZ Inducible Lentiviral Human MALT1 shRNAs were purchased from GE Dharmacon (Clone IDs: V2THS_84224, V2THS_84226, V3THS_378343). TRIPZ inducible lentiviral MALT1 shRNAs were transfected separately into Lenti-Pac 293Ta cells along with second generation lentiviral packaging plasmids (Addgene) using Lipofectamine 3000 transfection reagent (catalog no: L3000015, Thermo Fisher Scientific). Lentiviral particles were harvested 72 hours after transfection, concentrated with Lenti-Pac Lentivirus Concentration Solution (catalog no: LT007, GeneCopoeia), and then used to transduce BT549 and MDA-MB-231 cells for 24 hours. Selection was accomplished by culturing the transduced cells with puromycin. Cells containing inducible MALT1 shRNAs were treated with 2 μg/ml doxycycline (Dox) for 5 days before harvesting for immunoblot analysis.
SDS-PAGE, immunoblotting, and quantitative RT-PCR
Cell lysates were prepared with RIPA buffer (Cat No: 89901, Thermo Fisher) containing HALT Protease and Phosphatase Inhibitor cocktail (Cat No: 78440, Thermo Fisher), loaded onto 4–15% Mini-PROTEAN® TGX™ Precast Protein Gels (catalog no: 4561084, BioRad), and transferred to 0.2 μm nitrocellulose membranes (catalog no: 1620112, BioRad). Blots were probed with indicated primary antibodies (listed in Supplementary Methods) and developed with Pierce™ ECL Western Blotting Substrate (catalog no: 32106, Thermo Fisher). Total RNA was isolated from cell cultures and evaluated by RT-PCR using TaqMan gene expression assays (Thermo Fisher) as described (8) and as detailed in the Supplementary Methods.
Immunofluorescence and confocal microscopy
ZR75–1, ZR75-Neo, and ZR75-AT1R cells were plated on glass-bottom 35 mm dishes (D35-20-0-N, Cellvis) at 1 × 105 cells/dish. Cells were fixed with 2% paraformaldehyde and permeabilized with 0.1% Triton X-100 in PBS. Cells were then blocked for 60 minutes and incubated overnight with mouse anti-E-cadherin or mouse anti-N-cadherin primary antibodies (1:400), followed by goat anti-rabbit (Alexa Fluor 488) or goat anti-mouse (Alexa Fluor 568) secondary antibodies for 1 hour. Confocal microscopy was performed using a Zeiss LSM 710 with a 63× oil objective. Images were collected and processed using Zen software (Carl Zeiss, Inc.).
Cell migration and invasion assays
2D cell migration assays were performed following the IncuCyte ZOOM 96-well Scratch Wound Cell Migration assay protocol (Sartorius). Invasion assays were performed using a modified Boyden chamber assay protocol. A detailed description of both methods is provided in Supplementary Methods.
Orthotopic xenografts
All animal procedures were performed in accordance with the NIH and institutional guidelines, and were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Pittsburgh. 4–6-week-old female NOD Scid Gamma (NSG) mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) were utilized for xenografts and purchased from Jackson laboratories. Early passage MDA-MB-231-Luc cells (1 × 106) were collected using Accutase (Sigma) and resuspended in 50 μl of DMEM serum free media and mixed on ice with 50 μl of growth factor-reduced Cultrex BME (Cat No: 3433-010-01, Trevigen). The cell-BME mixture (100 μl) was injected into the fourth mammary gland of NSG mice (left side, 1 injection per mouse). Tumor growth was monitored bi-weekly by digital caliper measurement, V= (width2 × length) / 2. Once tumors reached 40–50 mm3, mice were randomized to receive daily IP injections of either vehicle control (5% DMSO) or mepazine (Cat no: 5005000001, Sigma; 16 mg/kg body weight prepared in 5% DMSO). Animals were sacrificed once tumors reached the maximal acceptable size allowed by IACUC guidelines (~6–7 weeks). Excised tumors were photographed and weighed. Portions of tumor were fixed in 10% buffered formalin or flash-frozen in liquid nitrogen for protein extraction. Evaluation of ZEB1 and snail expression in xenografts is described in Supplementary Methods. Liver and lungs were also excised, fixed, and sectioned to generate full cross-sections at three different tissue levels to evaluate for micrometastatic disease. As described in the Supplementary Methods, sections were immunostained with a vimentin/Ki-67 antibody cocktail, and micrometastatic burden was quantified from the resultant stains using QuPath.
IVIS Imaging
Metastatic disease burden was monitored weekly by bioluminescent imaging of control or mepazine treated mice, using an IVIS Lumina S5 system (Perkin Elmer). D-Luciferin (Cat No: LUCK-1G; Gold Biotechnology) was resuspended in sterile PBS (Ca2+ or Mg2+ free) and injected IP at a dose of 150 mg/kg body weight. 10–15 min after luciferin injection, mice were anesthetized with isoflurane/oxygen and positioned on a warmed stage in the IVIS imaging chamber to expose the mammary fat pad and ventral body surface. Imaging was conducted using consistent image settings for all mice (height, binning, FStop) and exposure time (0.25–60s). Utilizing a primary tumor mask, regions of interest representing metastatic disease were identified and quantified as total photons/sec, using the Living Image software (Perkin Elmer).
NanoString gene expression analysis, IPA, and GSEA
RNA was extracted from BT549 cells 72 hours after MALT1 siRNA transfection with RNeasy Plus Kit (Qiagen) and quantitated by NanoDrop. The quality of RNA was analyzed with RNA Nanochips (catalog no: 5067–1511) on an Agilent 2100 Bioanalyzer. RNA samples were then assessed using the nCounter Pan-Cancer Progression Panel (NanoString) according to the manufacturer’s directions. In short, 100 ng of total RNA was hybridized overnight at 65°C, then run on a NanoString Prep Station at maximum sensitivity. Cartridges were scanned on a NanoString Digital Analyzer at 555 fields of view. Raw count data was normalized using the nSolver analysis software version 3.0, which normalizes samples according to positive and negative control probes and the geometric mean of six housekeeping probes. Genes with normalized counts less than 20 were considered as background and were not included in the analysis. Heatmaps were generated by using the UCSC Xena Browser and Morpheus (https://software.broadinstitute.org/Morpheus).
The Core Analysis function within the IPA software (version 31823283; http://www.ingenuity.com) (Qiagen Bioinformatics) was used to perform pathway analyses with the NanoString data. In parallel, GSEA analyses were performed using the GSEA software package (GSEA v2.2.3) and molecular signatures available from the Broad Institute.
Bioinformatic analyses of public databases
Publicly available gene expression data were obtained from cited studies via cBioportal (www.cbioportal.org), the USCS Xena Browser (http://xena.ucsc.edu), and the NCBI Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo). Heatmaps were generated using the Xena Browser and Morpheus.
TNBCs (n=195) were identified in the TCGA breast cancer dataset using the approach described by Bareche et al (23). GSEA and IPA were applied to this subset of breast cancer cases, querying relationships to AGTR1, F2R, and MALT1. EMT scoring analysis was performed using a previously developed pan-cancer EMT signature (24, 25). The EMT score was calculated as the mean expression of the mesenchymal-related genes subtracted by the mean expression of epithelial-related genes; scores greater than 0 indicate a more mesenchymal phenotype. Correlations between individual genes were generated using data downloaded from cBioportal, with linear regressions generated in GraphPad Prism (v8).
Statistical Analysis
Statistical analyses were performed with GraphPad Prism software (v8). P values were calculated using the two-tailed Student t test with or without Welch’s correction, or two-way ANOVA with Sidak’s correction as appropriate. Statistical analyses applied to bioinformatic gene expression datasets include the Mann-Whitney U test / Wilcoxon rank-sum test with adjusted P value using Benjamin and Hochberg correction or Bonferroni correction. Significance was determined at P < 0.05.
RESULTS
AT1R and PAR1 drive EMT in breast cancer cells
We and others previously identified a subset of GPCRs that can signal via the CBM complex in a range of epithelial and mesenchymal cells (1, 26–30). These include the chief receptor for the peptide hormone angiotensin-II (AT1R) and a protease-activated receptor that responds to thrombin (PAR1). In breast cancer models, we found that activation of these GPCRs induces assembly of the CBM signalosome which in turn activates NF-κB to promote cell proliferation and survival (8, 9). During the course of our work, we noticed that exogenous expression of these GPCRs was associated with a change in morphology suggestive of EMT. To study this in more detail, we individually expressed each receptor in breast cancer lines (ZR75–1 and MCF-7) well-known for their prominent epithelioid characteristics including rounded morphology, high E-cadherin expression, and undetectable expression of mesenchymal markers such as vimentin and N-cadherin. Strikingly, we found that enforced AT1R expression in the ZR75–1 line leads cells to gradually assume a spindled appearance that is characteristic of the EMT phenotype (Fig. 1A). In concert, cells undergo a complete cadherin switch, with loss of E-cadherin from the cell surface and upregulation of N-cadherin, along with induction of Vimentin and the master EMT transcription factors, Snail and ZEB1 (Fig. 1B and C). In similar fashion, enforced PAR1 expression in MCF-7 cells leads to cell spindling, loss of E-cadherin, and induction of Vimentin, Snail, and ZEB1 (Fig. 1D and E). N-cadherin is not induced in this model, demonstrating that there are likely to be context-dependent factors that influence how these GPCRs regulate the overall EMT program.
Figure 1. Upregulation of multiple GPCRs promotes breast cancer EMT.
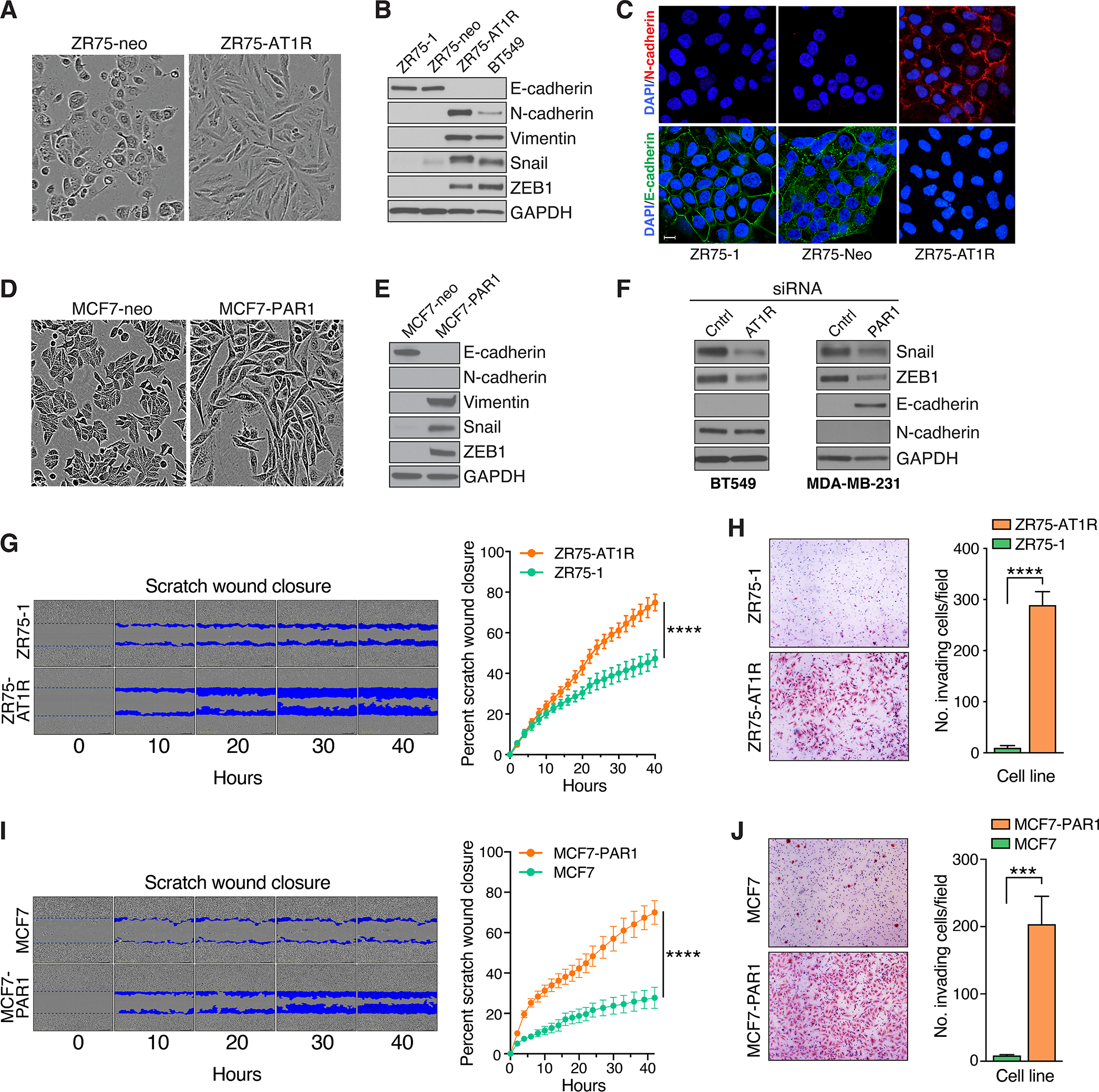
A-C, Effect of stable AT1R expression in ZR75–1 cells on cell morphology (A), expression of EMT markers by immunoblot analysis (B), and plasma membrane expression of both E- and N-cadherin species by immunofluorescence staining and confocal microscopy (C). Scale bar, 5 μm. D and E, Effect of stable PAR1 expression in MCF7 cells on cell morphology (D) and expression of EMT markers (E). F, Impact of siRNA-mediated AT1R or PAR1 knockdown in BT549 and MDA-MB-231 cells, respectively, on EMT markers. G, Effect of stable AT1R expression in ZR75–1 cells on cell migration, as measured continuously over time. A representative time-course is shown at left, with blue pseudocolor mask highlighting the progressive migration of cells into scratch wounds placed at time 0 hrs. Quantification of scratch would closure is shown at right, plotted as a continuous function of time (mean ± SD, n=10), ****, P < 0.0001, two-way ANOVA. H, Effect of stable AT1R expression in ZR75–1 cells on invasiveness, as measured using matrigel-coated Boyden chambers. Representative images of invaded cells are shown at left. Quantification of cell invasion is shown at right (mean ± SEM, n=13), ****, P < 0.0001, two-tailed t test with Welch’s correction. I, Effect of stable PAR1 expression in MCF7 cells on cell migration, using the same scratch wound assay described in panel G (mean ± SD, n=12), ****, P < 0.0001, two-way ANOVA. J, Effect of stable PAR1 expression in MCF7 cells on invasiveness, using the same matrigel invasion assay described in panel H (mean ± SEM, n=15), ***, P < 0.001, two-tailed t test with Welch’s correction.
The EMT alterations induced by either AT1R or PAR1 recapitulate what is naturally seen in BT549 and other claudin-low TNBC cell lines (Fig. 1B). Yet we observed these alterations when expressing the GPCRs in ZR75–1 and MCF-7 cells, both of which represent ER+, Luminal A lines. We therefore asked if GPCR expression might induce TNBC subtype switching, in concert with driving an EMT program. Indeed, we found that stable expression of either AT1R or PAR1, in ZR75–1 and MCF-7 cells respectively, completely abolished ER expression at both the mRNA and protein levels after serial passage (Supplementary Fig. S1A). Thus, the GPCR-induced EMT alterations may be part of a broader program related to intrinsic subtype switching and the emergence of claudin-low TNBC cells. Additional studies are underway to identify the mechanisms by which these GPCRs drive such profound ER downregulation.
Our previous work demonstrated that AT1R levels are particularly high in BT549 cells while PAR1 levels are high in MDA-MB-231 cells, another claudin-low TNBC line with prominent EMT features (8, 9). We therefore asked if siRNA-mediated knockdown of endogenous AT1R or PAR1 in these respective lines would be sufficient to reverse their EMT phenotype. Transient siRNA transfection was effective at knocking down both the AT1R and PAR1 targets in the respective lines (Supplementary Fig. S1B). Interestingly, transient AT1R knockdown was also effective at partially reversing the EMT molecular signature in BT549 cells, as evidenced by a reduction in Snail and ZEB1 levels, but was not sufficient to reverse the cadherin switch and initiate E-cadherin expression (Fig. 1F, left). This suggests that AT1R signaling has a persistent effect on the cadherin genes, keeping them in a “locked-down” state even after AT1R levels are diminished. Further work will be required to determine if this is mediated by epigenetic remodeling of these genes and what other manipulations might be required to achieve reversal of the cadherin switch once AT1R signaling is suppressed. Transient PAR1 knockdown similarly reduced Snail and ZEB1 expression in MDA-MB-231 cells, and in this case did lead to pronounced restoration of E-cadherin levels (Fig. 1F, right). Interestingly, N-cadherin is undetectable in MDA-MB-231 cells at baseline, underscoring the complexity of EMT, so that PAR1 knockdown could not be expected to further reduce the levels of N-cadherin.
To further study the functional roles of AT1R and PAR1 in the EMT phenotype, we asked if expression of these receptors could promote phenotypic behaviors associated with EMT. Indeed, we found that overexpression of either AT1R or PAR1 in the ZR75–1 and MCF-7 models, respectively, is sufficient to promote both cell migration and invasion through matrigel, phenotypic features of advanced EMT (Fig. 1G–J). Taken together, the results of overexpression and knockdown experiments support the notion that AT1R and PAR1 are both strong drivers of EMT in breast cancer and are likely to play a significant role in claudin-low TNBC.
To query the relationship between these GPCRs and EMT in primary human tumor specimens, we performed gene set enrichment analysis (GSEA) with the 195 TNBC samples in the TCGA collection and observed an exceptionally strong link between either AGTR1 or F2R gene expression and EMT signatures (Supplementary Fig. S2A–D). In this regard, the association between either AGTR1 or F2R and ZEB1 expression was particularly notable (Supplementary Fig. 2E). Perhaps most remarkable, we found that all six cases in the TCGA collection diagnosed as “spindled cell/sarcomatoid metaplastic carcinoma”, the rare breast cancer subtype with the most dramatic features of EMT, are clustered among cases with the highest combined AGTR1/F2R expression levels (Supplementary Fig. S2F). Taken together, this analysis of clinical samples provides strong correlative support for the notion that expression of one or both of these GPCRs provides signals to drive the EMT process in the setting of TNBC.
MALT1 regulates molecular markers of EMT in GPCR-positive, claudin-low TNBC
Since multiple EMT transcription factors are regulated by NF-κB (31–36), we reasoned that the GPCR/CBM signaling axis, which we previously demonstrated controls NF-κB activation in breast cancer models, could be responsible for driving at least some aspects of the GPCR-induced EMT program. Indeed, we found that inhibition of NF-κB through the use of either a chemical inhibitor of IKKβ or through siRNA-mediated knockdown of IKK complex subunits (IKKα or IKKβ) is sufficient to inhibit Snail expression in the AGTR1+, claudin-low BT549 cell line (Supplementary Fig. S3A and B).
We then individually knocked down the CBM signalosome components (CARMA3, Bcl10, and MALT1) in BT549 cells, and found that in each case this strongly abrogates both Snail and ZEB1 expression (Fig. 2A, left). We then focused on MALT1, the effector protein of the CBM signalosome, and found that MALT1 knock-down also reduces N-cadherin expression (Fig. 2A, right). Interestingly, Vimentin and E-cadherin protein levels are unaffected, indicating that blockade of CBM signalosome activity alone does not mediate a complete reversal of the EMT program within the time frame of the transient siRNA experiment (Fig. 2A). To extend these observations even more broadly, we evaluated two additional AGTR1+, claudin-low TNBC lines, Hs578T and Hs606T, which we previously showed exhibit active AT1R signaling (8), and found that knocking down MALT1 in these lines similarly abrogates both ZEB1 and N-cadherin expression (Supplementary Fig. S3C).
Figure 2. MALT1 expression is linked to EMT in GPCR+ breast cancer.
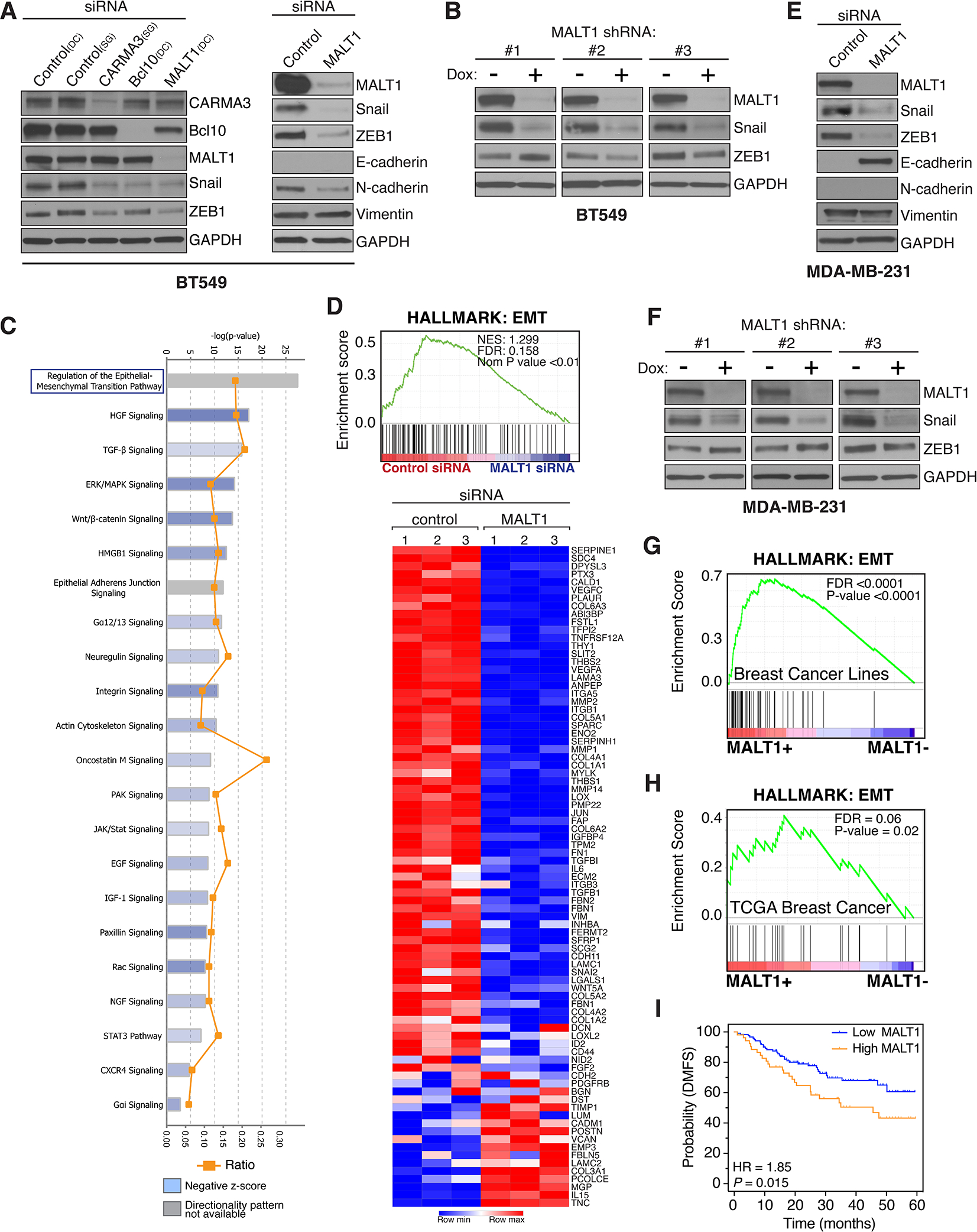
A and B, Effect of CARMA3, Bcl10, or MALT1 knockdown, in AT1R+ BT549 cells, on markers of EMT. Knockdown was accomplished by either transient siRNA transfection (A) or by stably-integrated doxycycline-inducible shRNAs (B). In the case of shRNA-mediated knockdown, cells were treated for 5 days with 2 μg/ml doxycycline to induce shRNA expression prior to harvesting and immunoblot analysis. DC, Dharmacon siRNA; SG, Sigma siRNA. C and D, IPA and GSEA linking MALT1 levels with EMT in BT549 cells. Cells were subjected to MALT1 knockdown using transient siRNA transfection, in biologic triplicate, and gene expression profiles were subsequently analyzed using the NanoString Pan-Cancer Progression Panel. Gene expression data was analyzed by IPA to identify pathways impacted by the loss of MALT1, revealing EMT as the top-most significantly affected pathway (C). The same data was evaluated using GSEA and the Hallmark EMT signature (D). The heatmap shows individual genes included in the Hallmark EMT gene set and their specific alterations in response to MALT1 knockdown. E and F, Effect of transient (E) or stable (F) MALT1 knockdown, in PAR1+ MDA-MB-231 cells, on markers of EMT. Analyses were carried out using an approach identical to that used for BT549 cells (panels A and B). G, GSEA demonstrates an association between MALT1 expression and the EMT Hallmark signature in breast cancer cell lines included in the Hoeflich dataset (37). H, GSEA demonstrating an association between MALT1 expression and the EMT Hallmark signature in TCGA breast cancer cases. I, High MALT1 expression is associated with worse DMFS in Grade-3 TNBC with 5 years of follow-up. Dataset from Gyorffy et al (73) and analyzed using KM-plotter (www.kmplot.com).
To further test the role of MALT1, we used a lentiviral approach to establish BT549 cells with stably-integrated, doxycycline (Dox)-inducible, MALT1 shRNAs. Three different shRNA sequences were used, each of which mediated a marked reduction in MALT1 levels in BT549 cells following 5 days of Dox treatment (Fig. 2B). As seen with transient siRNA-mediated MALT1 knockdown, Snail expression was also reduced as a consequence of shRNA-mediated MALT1 loss. However, there was no consistent effect on ZEB1 expression levels with shRNA-mediated MALT1 knockdown, unlike what we had observed with the siRNA approach (Fig. 2B). The explanation for this difference is unclear but could relate to the engagement of feedback loops to restore ZEB1 levels in the setting of prolonged MALT1 knockdown with shRNAs. To test for a broader role of MALT1 in driving EMT, beyond selected EMT transcription factors such as Snail and ZEB1, we transiently knocked down MALT1 in BT549 cells and assayed alterations in the transcriptomic landscape using the NanoString PanCancer Progression Panel, which covers 770 genes related to tumor progression. Results demonstrated that MALT1 levels in BT549 cells are strongly associated with an overall EMT signature, as determined independently by both Ingenuity Pathway and Gene Set Enrichment analyses (IPA and GSEA) (Fig. 2C and D).
We next tested if the contribution of MALT1 to EMT is also evident in the PAR1+, MDA-MB-231 cell model of claudin-low TNBC. Similar to what we observed with AT1R+ cells, we found that siRNA-mediated MALT1 knockdown in MDA-MB-231 cells results in a dramatic reduction in both Snail and ZEB1 (Fig. 2E). Remarkably, E-cadherin expression emerges in this context (Fig. 2E), an effect not observed after MALT1 knockdown in AGTR1+, BT549 cells (Fig. 2A). We also established MDA-MB-231 cells with three different stably-integrated, Dox-inducible, MALT1 shRNAs and demonstrated that in each case, Dox-induced MALT1 knockdown results in a marked reduction in Snail, but not ZEB1, recapitulating what is seen with BT549 cells (Fig. 2F).
To test for a relationship between MALT1 levels and EMT across an even broader range of cell line models, we interrogated the dataset of Hoeflich et al, which includes gene expression profiles for 51 breast cancer lines (37). Interestingly, cell lines with high MALT1 expression were significantly enriched for the TNBC subtype, as compared to those with low MALT1 which were enriched for Luminal (hormone receptor-positive) and HER2-amplified subtypes (Supplementary Fig. S4A and B). Next, we compared the gene expression profiles of high MALT1-expressing lines to those with low MALT1 and identified 1010 significantly differentially expressed genes (Supplementary Fig. S4C and Supplementary Table 1A and B). We then performed GSEA and identified EMT as the top enriched pathway in the high MALT1 group (Fig. 2G). We also tested for an association between MALT1 and EMT in breast cancer specimens in the TCGA collection and similarly found that EMT is the top enriched Hallmark gene signature associated with MALT1 (Fig. 2H). Likewise, the Charafe breast cancer mesenchymal UP signature (38) is one of the top curated gene signatures associated with MALT1 (Supplementary Fig. S4D). Finally, because EMT is associated with metastatic dissemination, we tested for a relationship between MALT1 levels and distant metastasis free survival (DMFS). Results showed that for high-grade TNBC, high MALT1 expression is indeed associated with a statistically significant worse DMFS (Fig. 2I).
Pharmacologic inhibition of MALT1 protease activity abrogates expression of Snail and ZEB1
As the effector protein of the CBM complex, MALT1 directs downstream NF-κB signaling through two essential activities (10, 22). First, MALT1 acts as a scaffold to recruit additional molecules that directly activate the IKK complex (Fig. 3A). Second, MALT1 acts as a caspase-like protease to cleave and neutralize a limited number of substrates, including CYLD, A20, and RelB, several of which are negative regulators of the NF-κB pathway (Fig. 3A). In this way, the scaffolding and enzymatic activities of MALT1 work in concert for optimal NF-κB activation. Having established a critical role for MALT1 in maintaining features of EMT in both AT1R+ and PAR1+ breast cancer cell lines, and a correlative link between MALT1 levels and EMT in human specimens, we next asked if pharmacologic inhibition of MALT1 activity is effective at abrogating EMT.
Figure 3. Inhibiting MALT1 protease activity abrogates Snail and ZEB1 expression in GPCR+ breast cancer cells.
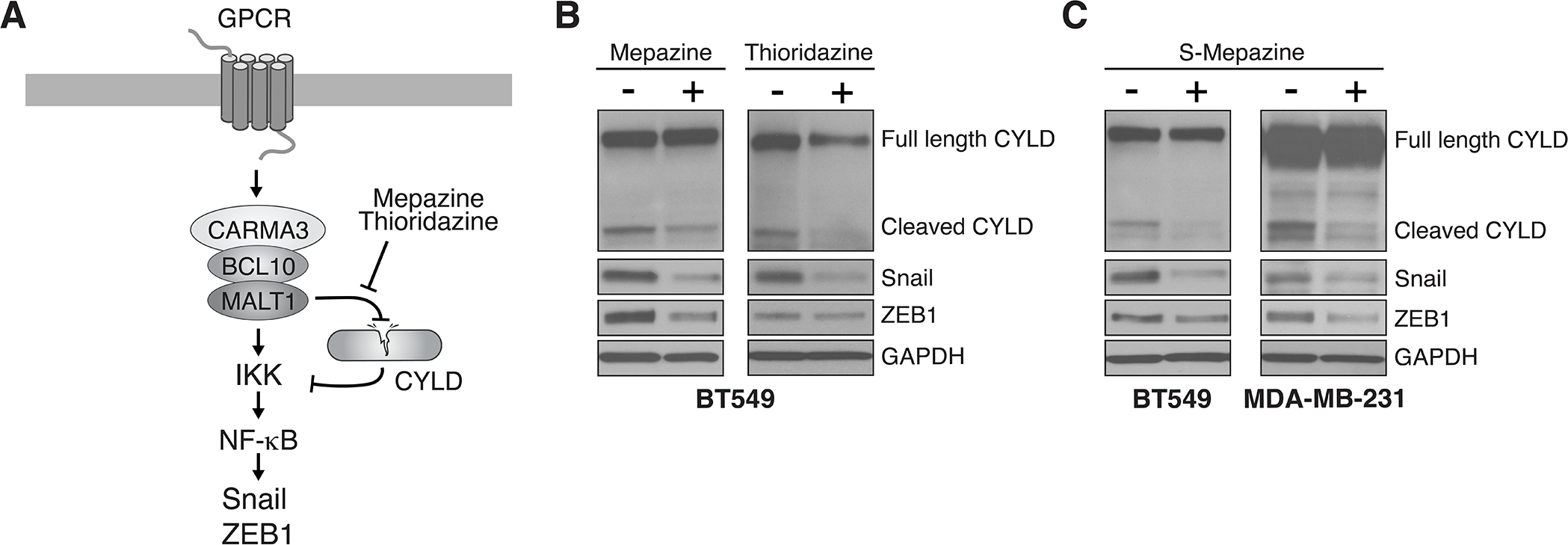
A, Schematic of CBM signaling downstream of GPCRs, highlighting the dual role of MALT1 as a protease and as a scaffolding protein, both of which are activities that contribute to NF-κB activation. B, Effect of pharmacologic MALT1 protease inhibition on expression of Snail and ZEB1. BT549 cells were treated ± 10 μM mepazine or 5 μM thioridazine for 2 days before harvesting and immunoblot analysis. C, Effect of pharmacologic MALT1 protease inhibition with the s-enantiomer of mepazine. BT549 or MDA-MB-231 cells were treated ± 10 μM s-mepazine for 2 days before harvesting and immunoblot analysis.
Currently, no small-molecule inhibitors of MALT1 scaffolding activity have been developed. However, several drugs and drug-like molecules have been identified that act as specific inhibitors of MALT1 protease activity (11). To test if inhibiting only the protease arm of MALT1 is sufficient to block certain aspects of the EMT program, we treated BT549 cells with mepazine or thioridazine, two phenothiazines that act as potent MALT1 protease inhibitors (39). While untreated cells showed evidence of constitutive CYLD cleavage, consistent with AT1R-driven MALT1 activation, treatment with either of these phenothiazines inhibited the cleavage, as evidenced by a reduction in levels of the major CYLD cleavage fragment (Fig. 3B). Coincident with MALT1 protease blockade, we also observed a reduction in the levels of both Snail and ZEB1. These findings were recapitulated with S-mepazine, an enantiomer of mepazine with significantly higher MALT1 binding affinity and inhibitory potential (40) (Fig. 3C). In the PAR1+, MDA-MB-231 cell model, S-mepazine was similarly effective at both blocking CYLD cleavage and suppressing Snail and ZEB1 levels (Fig. 3C). Finally, we analyzed two additional AT1R+ claudin-low breast cancer lines, Hs578T and Hs606T, and again observed constitutive CYLD cleavage that is abrogated by treatment with either mepazine or thioridazine; ZEB1 levels showed a corresponding decrease with these treatments (Supplementary Fig. S5). Taken together, our analyses of cell line models and human specimens reveal that MALT1 levels and constitutive protease activity contribute to several aspects of the EMT program in GPCR-positive, claudin-low TNBC.
MALT1 is required for EMT-associated cell migration, invasion, and metastasis in AT1R+ and PAR1+ breast cancer models
Cells that have undergone EMT characteristically take on enhanced migratory and matrix invasive properties that are ultimately associated with metastasis to distant sites (41). To test for a role of MALT1 in driving these phenotypic properties in AT1R+ and PAR1+ breast cancer cell models, we knocked down MALT1 in BT549 and MDA-MB-231 cells, respectively. Results demonstrated that MALT1 knockdown clearly reduced migration and invasion in both cell types (Fig. 4A–C). Inhibition of MALT1 protease activity via treatment with mepazine had a similar effect (Fig. 4D–F). To determine how pharmacologic MALT1 protease inhibition would impact tumor progression in vivo, we established orthotopic xenografts of luciferase-expressing MDA-MB-231 cells in NSG mice. Once tumors reached a size of ~50 mm3, mice were randomized into two groups and received daily IP injections of either mepazine or vehicle control, up to day 40. Mepazine treatment caused a small but statistically significant reduction in primary tumor growth over time (Fig. 5A and B). Strikingly, nuclear ZEB1 expression was reduced in tumor cells in the mepazine treated xenografts, as determined by immunohistochemical staining and quantitative image analysis (Fig. 5C). Snail expression could not be reliably quantified via immunohistochemistry due to non-specific tissue immunoreactivity of commercially available antibodies. However, western blot analyses of tumor lysates demonstrated a similar significant reduction in Snail levels across the mepazine treated cohort (Fig. 5D).
Figure 4. Inhibiting MALT1 effectively blocks migration and invasion of GPCR+ breast cancer cells.
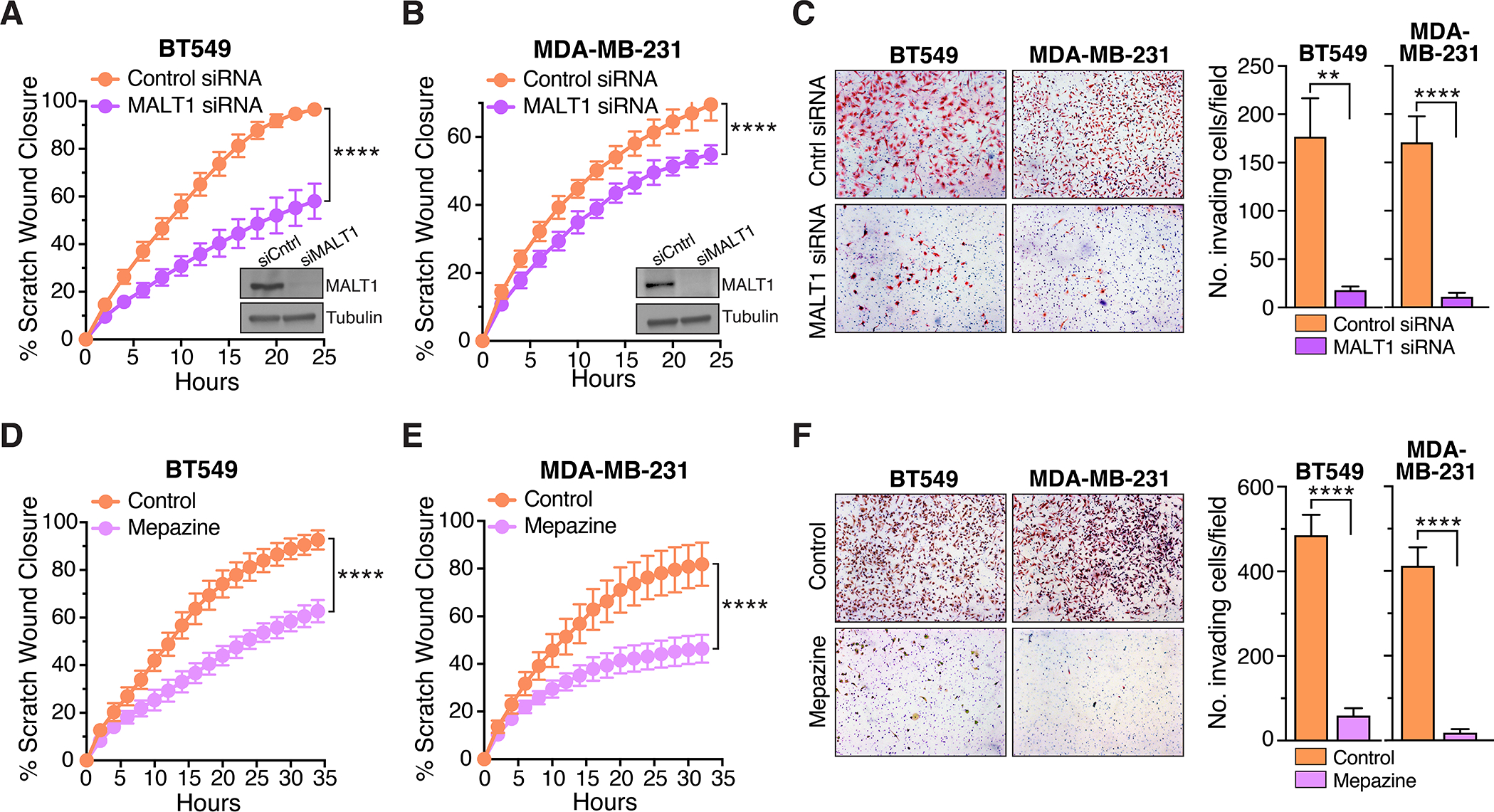
A and B, Effect of MALT1 knockdown on migration of BT549 (A) and MDA-MB-231 (B) cells in the scratch wound assay. Quantification of scratch would closure is plotted as a continuous function of time (mean ± SD, n=8–10), ****, P < 0.0001, two-way ANOVA. The degree of MALT1 knockdown is shown in representative immunoblot inserts. C, Effect of MALT1 knockdown on BT549 and MDA-MB-231 cell invasiveness, as measured using matrigel-coated Boyden chambers. Representative images of invaded cells are shown at left. Quantification of cell invasion is shown at right (mean ± SEM, n=12), **, P < 0.01; ****, P < 0.0001, two-tailed t test with Welch’s correction. D and E, Effect of mepazine (10 μM) on migration of BT549 (D) and MDA-MB-231 (E) cells in the scratch wound assay. Quantification of scratch would closure is plotted as a continuous function of time (mean ± SD, n=8–12), ****, P < 0.0001, two-way ANOVA. F, Effect of mepazine (10 μM) on BT549 and MDA-MB-231 cell invasiveness, as measured using matrigel-coated Boyden chambers. Representative images of invaded cells are shown at left. Quantification of cell invasion is shown at right (mean ± SEM, n=12–13), ****, P < 0.0001, two-tailed t test with Welch’s correction.
Figure 5. MALT1 inhibition abrogates metastasis of GPCR+ breast cancer xenografts.
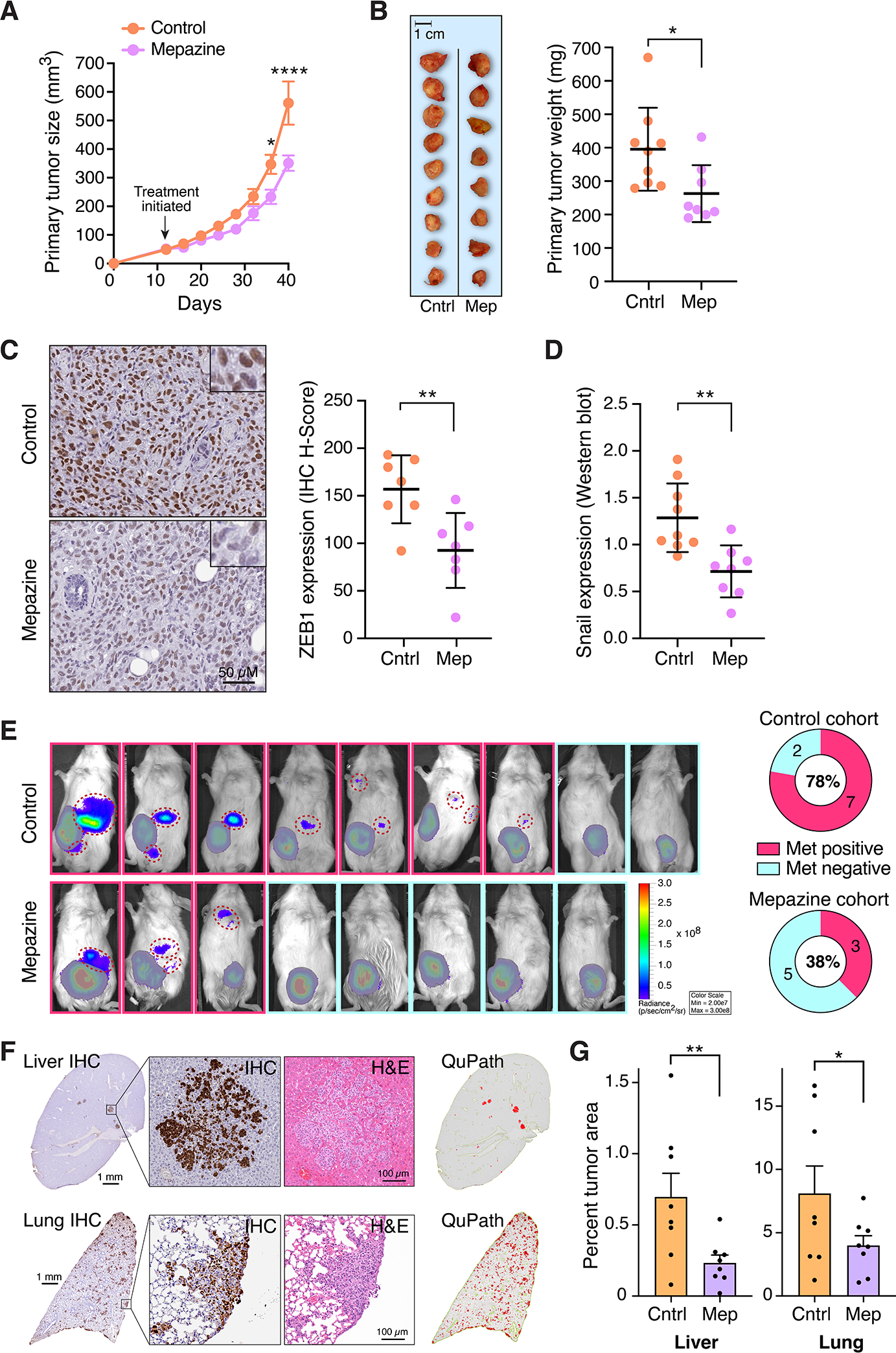
A and B, Effect of mepazine on growth of MDA-MB-231 orthotopic xenografts in NSG mice. Daily IP injections of 5% DMSO vehicle or mepazine (16 mg/kg) were initiated when tumors reached ~50 mm3. Tumor size was measured over time with calipers (mean ± SEM, n=8–9), *, P < 0.05; ****, P < 0.0001, two-way ANOVA with Sidak’s correction for multiple comparisons (A). Excised tumors were weighed at 40 days (mean ± SD, n=8–9), *, P < 0.05, two-tailed t test with Welch’s correction (B). C, Effect of mepazine on nuclear ZEB1 expression in xenografts, as measured by immunohistochemical staining and quantitative image analysis. Representative images are shown at left; ZEB1 quantification is shown at right and expressed as H-score (mean ± SD, n=7), **, P <0.01, two-tailed t test. D, Effect of mepazine on Snail expression in xenograft lysates, as measured by western blot analysis. Snail band intensity for each sample was normalized to β-actin (mean ± SD, n=8–9), **, P < 0.01, two-tailed t test. E, Representative IVIS images of the same MDA-MB-231 xenografts described in panels A and B, immediately prior to tumor harvesting. Signals from primary tumors are covered by a grayed out pseudo-mask to highlight the presence of tumor cell signals emanating from lesions that represent either distant metastases or the spread of tumors outside the confines of the mammary fat pad (individual metastatic lesions are identified by hashed red circles). Quantification of the proportion of mice with metastatic spread is shown at right. F, Representative photomicrographs of liver and lung sections from a control-treated NSG mouse, 40 days after orthotopic implantation with MDA-MB-231 cells in the mammary fat pad. Immunohistochemical (IHC) stains using a vimentin/Ki-67 cocktail are shown alongside corresponding H&E stains. QuPath image conversions of the IHC stains, generated to annotate and quantify micrometastatic lesions, are shown at right. G, QuPath quantification of micrometastatic tumor burden in liver and lung, for all mice included in the study (mean ± SEM, n=8, *, P<0.05; **, P <0.05, one-tailed t test. Cntrl, control-treated; Mep, Mepazine-treated.
The mepazine-induced suppression of EMT transcription factors was associated with a reduction in metastatic spread that was even more notable than the reduction in primary tumor growth. Specifically, 78% of mice in the control group showed evidence of gross metastatic spread by IVIS imaging, versus only 38% in the mepazine treated group (Fig. 5E). Additionally, histologic evaluation of liver and lungs revealed a substantial number of micrometastases in control-treated mice that were not readily detected by IVIS (Fig. 5F). In comparison, mepazine-treated mice showed a lower overall micrometastatic burden, as demonstrated by QuPath-assisted quantitative image analysis of liver and lung tissue sections (Fig. 5G). These results suggest that while MALT1 does play a role in primary tumor growth of the PAR1+ MDA-MB-231 xenografts, its impact may be even more closely tied to EMT-associated phenotypic changes and metastatic dissemination.
Finally, because mepazine has been demonstrated to have cellular effects that cannot be attributed solely to MALT1 inhibition (42–44), we sought to evaluate the impact of a newer and even more selective small molecule MALT1 protease inhibitor, MLT-748 (45), on the behavior of GPCR+ breast cancer cells. To this end, we treated both the AT1R+, BT549 and the PAR1+, MDA-MB-231 cell lines with MLT-748 and observed clear inhibition of both cell migration and invasion (Fig. 6A–C), recapitulating what we had seen with mepazine. To confirm that MLT-748 is active at inhibiting MALT1 in these cell lines, we probed lysates for CYLD and found that MLT-748 treatment completely eliminates the CYLD cleavage fragment that is produced as a consequence of MALT1 proteolytic activity (Fig. 6D). MLT-748 is amongst the most potent and selective of the currently available MALT1 protease inhibitors and the CYLD cleavage assay showed that MLT-748 very effective at inhibiting the MALT1 target in these cancer cells. Nevertheless, we noted that MLT-748 was somewhat less effective than mepazine at blocking breast cancer cell migration and invasion. There could be multiple explanations for this observation, including differential stability or metabolism of the two compounds within breast cancer cells, the potential for these inhibitors to differentially impair MALT1 activity against selected substrates, and the possibility that mepazine could be acting through multiple targets beyond MALT1, some of which may also be important for EMT. Currently, work is underway to expand these findings and optimize the in vivo pharmacokinetic properties of highly selective MALT1 protease inhibitors related to MLT-748 in order to thoroughly evaluate their long-term activities against breast cancer xenograft models in mice.
Figure 6. A highly specific, next-generation small molecule inhibitor of MALT1 is effective at abrogating migration and invasion of GPCR+ breast cancer cells.
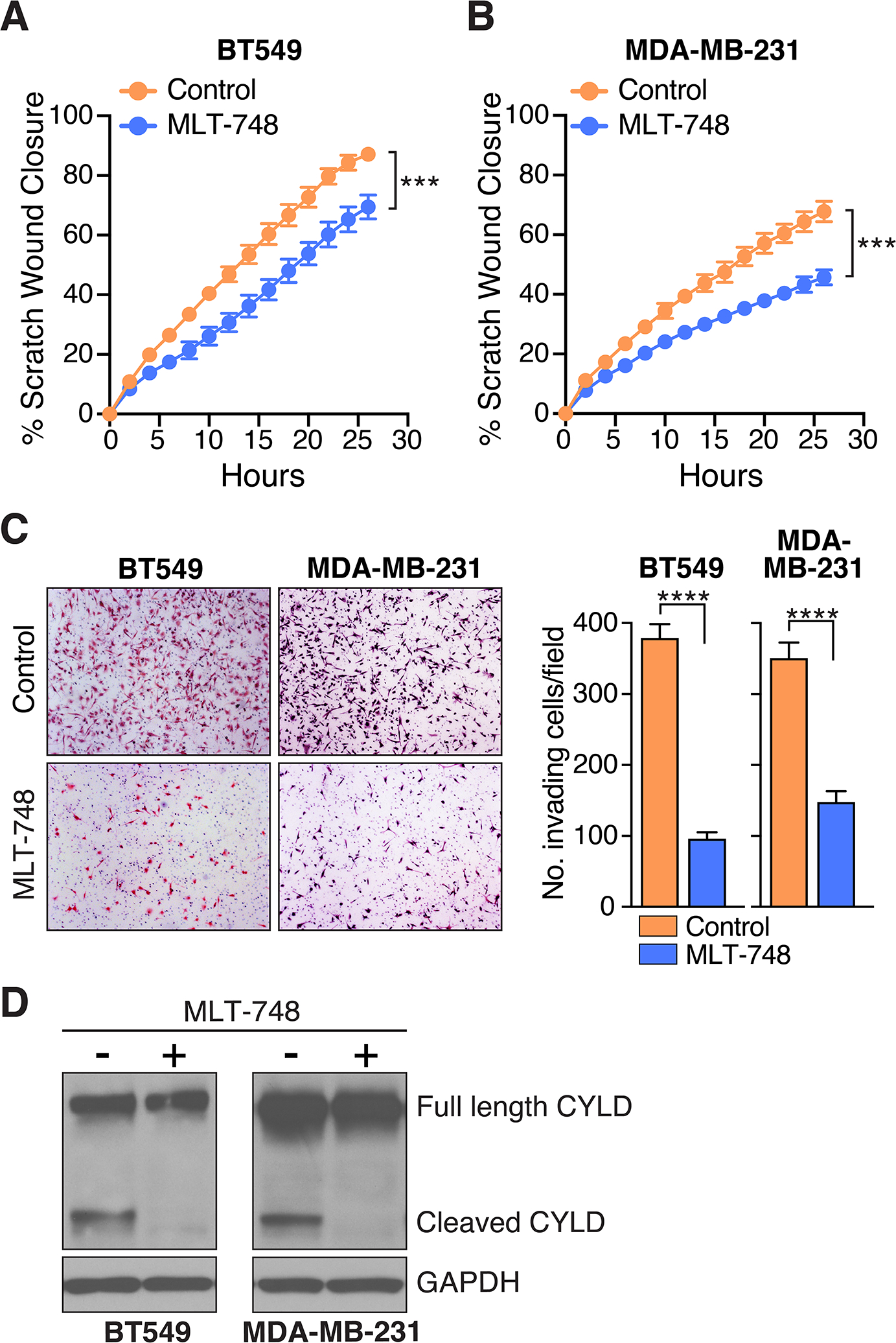
A and B, Effect of MLT-748 on migration of BT549 (A) and MDA-MB-231 (B) cells in the scratch wound assay. Quantification of scratch would closure is plotted as a continuous function of time (mean ± SD, n=8–10), ***, P < 0.001, two-way ANOVA. C, Effect of MLT-748 on BT549 and MDA-MB-231 cell invasiveness, as measured using matrigel-coated Boyden chambers. Representative images of invaded cells are shown at left. Quantification of cell invasion is shown at right (mean ± SEM, n=12), ****, P < 0.0001, two-tailed t test with Welch’s correction. D, Effect of MLT-748 on MALT1-dependent CYLD cleavage in BT549 and MDA-MB-231 cells. Cells were treated ± a single dose of 20 μM MLT-748 for 5 days before harvesting and immunoblot analysis.
DISCUSSION
While significant progress has been made in developing molecularly-informed, targeted therapies for the treatment of breast cancer, the TNBC subtype of breast cancer has remained a major challenge. Defined by the absence of known drivers, TNBCs have thus far not presented tractable therapeutic targets, leading the field to continue searching for key oncogenic drivers or signaling pathways that might reveal an “Achilles’ heel” for these aggressive cancers. We report here that two Gαq/11-coupled GPCRs, AT1R and PAR1, function similarly to one another to drive a strong EMT program in TNBC linked to metastatic dissemination. These findings support those of other researchers, who have also provided evidence implicating GPCR-driven EMT in breast cancer and in other solid tumors (6, 46–53). Particularly intriguing is the recent finding that Twist, a master EMT transcription factor, directly induces PAR1 gene expression (54), raising the possibility that in some cases a vicious positive feedback loop could be established whereby PAR1-induced EMT promotes further PAR1 expression.
Most importantly, we demonstrate that the GPCR-directed EMT program involves signaling through the CARMA3/Bcl10/MALT1 (CBM) complex, whereby MALT1 functions as the key effector molecule. Since MALT1 is a targetable enzyme, with several novel small molecules now identified as potent inhibitors of its protease activity (11), this discovery nominates MALT1 as a potentially attractive therapeutic target. Moreover, because previous work has demonstrated that several GPCRs, beyond AT1R and PAR1, can similarly engage the CBM complex (1), we speculate that MALT1 functions as a major signaling node to integrate signals emanating from a spectrum of cell surface GPCRs. For example, the lysophosphatidic acid receptors (LPARs), the receptor for CXCL12 (CXCR4), and the receptor for CXCL8/IL-8 (CXCR2) are all known to signal through the CBM signalosome [see comprehensive review; (1)]. These receptors are also recognized pathogenic drivers of breast cancer, particularly for the TNBC subtype. In the current manuscript, we provide evidence that simultaneous, coordinated expression of both AT1R and PAR1 is associated with spindle-cell metaplastic breast cancer, a TNBC subtype that shows the most dramatic features of EMT. Thus, it is tempting to speculate that targeting MALT1, as a node or hub that integrates upstream signals from multiple GPCRs, may be an effective approach to treating such tumors, as opposed to individually targeting distinct GPCRs that may be acting in a redundant fashion.
Signaling through the MALT1-dependent NF-κB pathway represents only one mechanism by which this family of GPCRs could impact cancer cell phenotype, and EMT in particular. Numerous other mechanisms have been identified for regulating master EMT transcription factors. Key mediators include the WNT ligands, TGFβ, mitogenic growth factors, hypoxia/HIF1α, Notch signaling, and the Hippo-YAP/TAZ pathways, among others (55, 56). Indeed, recent work has highlighted the role of TAZ signaling downstream of PAR1 in breast cancer (54, 57). The diversity of mechanisms controlling EMT likely explain our observation that MALT1 depletion or pharmacologic inhibition does not completely reverse EMT at the level of molecular markers or phenotype. Further, the individual EMT transcription factors, cadherins, and mesenchymal proteins most impacted by MALT1 signaling appear to differ somewhat, depending on cellular context and the major driving GPCRs present within a particular cancer line or tumor. Thus, effective therapeutic strategies for abrogating EMT may require combining MALT1 inhibitors with selected inhibitors of complementary pathways. Additional work is ongoing to identify the complementary pathways that are most critical in the context of GPCR/MALT1 signaling and empirically test a range of therapeutic combinations.
It is important to note that a recognized role for MALT1 in solid tumors is rapidly expanding and is not limited to TNBC, or even to breast cancer. Recently, multiple groups have highlighted a critical role for MALT1 in a diverse range of solid tumors that include glioblastoma, pancreatic adenocarcinoma, ovarian adenocarcinoma, melanoma, oral cancer, and non-small cell lung cancer (58–65). A key challenge for the field will be to devise a precision medicine approach to identify which tumors harness MALT1 signaling most robustly, and through which receptors, in order to identify those tumors that may be particularly sensitive to MALT1 inhibitor treatment.
The EMT phenotype has been linked to other pathogenic properties in cancer, including stemness and resistance to chemotherapy (66). We therefore speculate that MALT1 inhibition may have effects that go beyond inhibiting phenotypic behaviors directly attributable to EMT, namely cell migration and invasion. Indeed, we have previously shown that blocking CBM signaling activity has pleiotropic effects on intrinsic properties of breast cancer cells and on the breast cancer microenvironment (8, 9). It will therefore be important to examine the role of MALT1 in maintaining stemness and in conferring chemoresistance, particularly in the setting of TNBC, since this could have important implications for the role of MALT1 in dormancy and recurrence following initially effective neoadjuvant and/or adjuvant therapy.
One of the most important processes linked to EMT is the escape from anti-tumor immune surveillance (67, 68). We previously showed that MALT1 signaling downstream of PAR1 is essential for expression of cytokines that are known to negatively impact immune cell functionality, including IL-1β and IL-8 (9). Motivated by these findings, work is underway to thoroughly understand the impact of inhibiting MALT1 in tumor cells on the surrounding tumor immune contexture, via disruptions in paracrine signaling from cancer cell to immune cell. At the same time, it is intriguing to note that the CBM complex was originally described as a key signaling module in lymphocytes, whereby it controls lymphocyte activation downstream of the antigen receptor (69). Interestingly, the overall role of the CBM complex is different for each distinct lymphocyte subset. Most notably, MALT1 proteolytic activity appears to be especially important for Treg function. Inhibition of the protease arm of MALT1, but not the scaffolding arm, via the use of MALT1 chemical inhibitors or genetic means, has been shown to alter the balance of the immune system by disproportionately suppressing Foxp3+ Treg activity and thereby enhancing overall effector T cell activity (18, 19, 21, 70–72). As such, MALT1 protease inhibitors have the potential to enhance anti-tumor immunity through their direct actions on Treg cells, and recent work by multiple groups nicely demonstrates that one of these MALT1 inhibitors, mepazine, does indeed enhance anti-tumor immunity in both melanoma and colorectal cancer models (19, 21). Taken together, our findings and those of groups working on the immunoregulatory impact of MALT1 inhibitors, suggest that mepazine or other similar MALT1 protease inhibitors might have the greatest potential for clinical impact if utilized to treat tumors that show intrinsic, cancer cell dependence on MALT, such as the GPCR+ TNBC subtype studied here. In such tumors, these inhibitors would be expected to act at two levels – first as inhibitors of cancer cell proliferation, migration, invasion, and EMT, and second, as modulators of anti-tumor immunity through direct, coordinated actions on T cells in the microenvironment. It is in this context, where MALT1 protease inhibitors would be expected to simultaneously work on two cell populations (cancer cells and immune cells), that synergistic therapeutic benefit might be realized. To heighten responses even further, combining a MALT1 inhibitor with a checkpoint inhibitor could be a logical choice.
It is important to note that the xenograft studies carried out here were performed in immunocompromised mice. As a result, the ability of mepazine to inhibit MDA-MB-231 tumor growth and dissemination likely reflects the direct actions of MALT1 protease inhibition in tumor cells. Further work will be required to evaluate the impact of pharmacologic MALT1 protease inhibitors on MALT1-dependent human tumors in mouse hosts with a humanized immune system, in order to unmask the potential synergistic value of targeting both tumor cells and immune cells. Alternatively, mouse tumor lines can be screened for MALT1 dependence to identify appropriate models for evaluation in immunocompetent, syngeneic hosts.
In summary, the current work identifies MALT1 as a novel signaling molecule in a subset of GPCR+ TNBCs, with a major role in promoting EMT, cell migration, invasion and metastasis. Because MALT1 is targetable through a rapidly growing suite of compounds including the phenothiazines, which have a history of clinical use in psychiatry, the opportunity exists to apply or repurpose these compounds as cancer therapeutics, if appropriate cases can be selected through precision medicine efforts. Especially exciting is the idea of simultaneously leveraging MALT1 inhibition in tumor cells and immune cells in these selected tumors, for maximum efficacy.
Supplementary Material
Implications:
This study nominates a GPCR/MALT1 signaling axis as a pathway that can be pharmaceutically targeted to abrogate EMT and metastatic progression in TNBC, an aggressive form of breast cancer that currently lacks targeted therapies.
Acknowledgments:
The authors thank all members of the Lucas and McAllister labs for advice and support, and Lambertus (Bert) Klei for advice on statistical analyses. This work was supported by funding from the NIH (R01 HL082914 to P.C. Lucas and L.M. McAllister-Lucas) and by a UPMC Hillman Development Fund awarded via P30CA047904 from the NCI (to P.C. Lucas). J.-Y. Lee was supported by a RAC Fellowship Award from UPMC Children’s Hospital of Pittsburgh. D. Hu was supported by a gift from the NSABP Foundation. Z. Cai was supported by a China Scholarship Council award through the Tsinghua School of Medicine (Beijing, China). D. Krappmann was supported by the Deutsche Forschungsgemeinschaft (SFB1335 P07-ID360372040). This project used the Animal Imaging and Histology core facilities of the University of Pittsburgh Department of Pediatrics, Rangos Research Center.
Footnotes
Authors’ Disclosures
P.C. Lucas is Director of Pathology at the NSABP Foundation. P.C. Lucas and L.M. McAllister-Lucas are shareholders of Amgen. L.M. McAllister-Lucas has served as a compensated presenter to the Board of Directors of Schrödinger, Inc. F. Bornancin is an employee and shareholder of Novartis Pharma AG. No disclosures were reported by the other authors.
REFERENCES
- 1.McAuley JR, Freeman TJ, Ekambaram P, Lucas PC, McAllister-Lucas LM. CARMA3 Is a Critical Mediator of G Protein-Coupled Receptor and Receptor Tyrosine Kinase-Driven Solid Tumor Pathogenesis. Front Immunol 2018;9:1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lappano R, Jacquot Y, Maggiolini M. GPCR Modulation in Breast Cancer. Int J Mol Sci 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dorsam RT, Gutkind JS. G-protein-coupled receptors and cancer. Nat Rev Cancer 2007;7:79–94. [DOI] [PubMed] [Google Scholar]
- 4.Rhodes DR, Ateeq B, Cao Q, Tomlins SA, Mehra R, Laxman B, et al. AGTR1 overexpression defines a subset of breast cancer and confers sensitivity to losartan, an AGTR1 antagonist. Proc Natl Acad Sci U S A 2009;106:10284–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arakaki AKS, Pan WA, Trejo J. GPCRs in Cancer: Protease-Activated Receptors, Endocytic Adaptors and Signaling. Int J Mol Sci 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boire A, Covic L, Agarwal A, Jacques S, Sherifi S, Kuliopulos A. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 2005;120:303–13. [DOI] [PubMed] [Google Scholar]
- 7.Yin YJ, Salah Z, Grisaru-Granovsky S, Cohen I, Even-Ram SC, Maoz M, et al. Human protease-activated receptor 1 expression in malignant epithelia: a role in invasiveness. Arterioscler Thromb Vasc Biol 2003;23:940–4. [DOI] [PubMed] [Google Scholar]
- 8.Ekambaram P, Lee JL, Hubel NE, Hu D, Yerneni S, Campbell PG, et al. The CARMA3-Bcl10-MALT1 Signalosome Drives NFkappaB Activation and Promotes Aggressiveness in Angiotensin II Receptor-Positive Breast Cancer. Cancer Res 2018;78:1225–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McAuley JR, Bailey KM, Ekambaram P, Klei LR, Kang H, Hu D, et al. MALT1 is a critical mediator of PAR1-driven NF-kappaB activation and metastasis in multiple tumor types. Oncogene 2019;38:7384–98. [DOI] [PubMed] [Google Scholar]
- 10.Juilland M, Thome M. Holding All the CARDs: How MALT1 Controls CARMA/CARD-Dependent Signaling. Front Immunol 2018;9:1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jaworski M, Thome M. The paracaspase MALT1: biological function and potential for therapeutic inhibition. Cell Mol Life Sci 2016;73:459–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harbeck N, Penault-Llorca F, Cortes J, Gnant M, Houssami N, Poortmans P, et al. Breast cancer. Nat Rev Dis Primers 2019;5:66. [DOI] [PubMed] [Google Scholar]
- 13.Garrido-Castro AC, Lin NU, Polyak K. Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov 2019;9:176–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCann KE, Hurvitz SA, McAndrew N. Advances in Targeted Therapies for Triple-Negative Breast Cancer. Drugs 2019;79:1217–30. [DOI] [PubMed] [Google Scholar]
- 15.Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res 2010;12:R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fougner C, Bergholtz H, Norum JH, Sorlie T. Re-definition of claudin-low as a breast cancer phenotype. Nat Commun 2020;11:1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bertossi A, Krappmann D. MALT1 protease: equilibrating immunity versus tolerance. EMBO J 2014;33:2740–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng L, Deng N, Yang N, Zhao X, Lin X. Malt1 Protease Is Critical in Maintaining Function of Regulatory T Cells and May Be a Therapeutic Target for Antitumor Immunity. J Immunol 2019;202:3008–19. [DOI] [PubMed] [Google Scholar]
- 19.Di Pilato M, Kim EY, Cadilha BL, Prussmann JN, Nasrallah MN, Seruggia D, et al. Targeting the CBM complex causes Treg cells to prime tumours for immune checkpoint therapy. Nature 2019;570:112–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martin K, Touil R, Kolb Y, Cvijetic G, Murakami K, Israel L, et al. Malt1 Protease Deficiency in Mice Disrupts Immune Homeostasis at Environmental Barriers and Drives Systemic T Cell-Mediated Autoimmunity. J Immunol 2019;203:2791–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosenbaum M, Gewies A, Pechloff K, Heuser C, Engleitner T, Gehring T, et al. Bcl10-controlled Malt1 paracaspase activity is key for the immune suppressive function of regulatory T cells. Nat Commun 2019;10:2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruland J, Hartjes L. CARD-BCL-10-MALT1 signalling in protective and pathological immunity. Nat Rev Immunol 2019;19:118–34. [DOI] [PubMed] [Google Scholar]
- 23.Bareche Y, Venet D, Ignatiadis M, Aftimos P, Piccart M, Rothe F, et al. Unravelling triple-negative breast cancer molecular heterogeneity using an integrative multiomic analysis. Ann Oncol 2018;29:895–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mak MP, Tong P, Diao L, Cardnell RJ, Gibbons DL, William WN, et al. A Patient-Derived, Pan-Cancer EMT Signature Identifies Global Molecular Alterations and Immune Target Enrichment Following Epithelial-to-Mesenchymal Transition. Clin Cancer Res 2016;22:609–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res 2013;19:279–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grabiner BC, Blonska M, Lin PC, You Y, Wang D, Sun J, et al. CARMA3 deficiency abrogates G protein-coupled receptor-induced NF-kappaB activation. Genes Dev 2007;21:984–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klemm S, Zimmermann S, Peschel C, Mak TW, Ruland J. Bcl10 and Malt1 control lysophosphatidic acid-induced NF-kappaB activation and cytokine production. Proc Natl Acad Sci U S A 2007;104:134–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McAllister-Lucas LM, Ruland J, Siu K, Jin X, Gu S, Kim DS, et al. CARMA3/Bcl10/MALT1-dependent NF-kappaB activation mediates angiotensin II-responsive inflammatory signaling in nonimmune cells. Proc Natl Acad Sci U S A 2007;104:139–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang D, You Y, Lin PC, Xue L, Morris SW, Zeng H, et al. Bcl10 plays a critical role in NF-kappaB activation induced by G protein-coupled receptors. Proc Natl Acad Sci U S A 2007;104:145–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wegener E, Krappmann D. CARD-Bcl10-Malt1 signalosomes: missing link to NF-kappaB. Sci STKE 2007;2007:pe21. [DOI] [PubMed] [Google Scholar]
- 31.Chua HL, Bhat-Nakshatri P, Clare SE, Morimiya A, Badve S, Nakshatri H. NF-kappaB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: potential involvement of ZEB-1 and ZEB-2. Oncogene 2007;26:711–24. [DOI] [PubMed] [Google Scholar]
- 32.Huber MA, Azoitei N, Baumann B, Grunert S, Sommer A, Pehamberger H, et al. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest 2004;114:569–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Julien S, Puig I, Caretti E, Bonaventure J, Nelles L, van Roy F, et al. Activation of NF-kappaB by Akt upregulates Snail expression and induces epithelium mesenchyme transition. Oncogene 2007;26:7445–56. [DOI] [PubMed] [Google Scholar]
- 34.Li CW, Xia W, Huo L, Lim SO, Wu Y, Hsu JL, et al. Epithelial-mesenchymal transition induced by TNF-alpha requires NF-kappaB-mediated transcriptional upregulation of Twist1. Cancer Res 2012;72:1290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Min C, Eddy SF, Sherr DH, Sonenshein GE. NF-kappaB and epithelial to mesenchymal transition of cancer. J Cell Biochem 2008;104:733–44. [DOI] [PubMed] [Google Scholar]
- 36.Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP. Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell 2009;15:416–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoeflich KP, O’Brien C, Boyd Z, Cavet G, Guerrero S, Jung K, et al. In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin Cancer Res 2009;15:4649–64. [DOI] [PubMed] [Google Scholar]
- 38.Charafe-Jauffret E, Ginestier C, Monville F, Finetti P, Adelaide J, Cervera N, et al. Gene expression profiling of breast cell lines identifies potential new basal markers. Oncogene 2006;25:2273–84. [DOI] [PubMed] [Google Scholar]
- 39.Nagel D, Spranger S, Vincendeau M, Grau M, Raffegerst S, Kloo B, et al. Pharmacologic inhibition of MALT1 protease by phenothiazines as a therapeutic approach for the treatment of aggressive ABC-DLBCL. Cancer Cell 2012;22:825–37. [DOI] [PubMed] [Google Scholar]
- 40.Schlauderer F, Lammens K, Nagel D, Vincendeau M, Eitelhuber AC, Verhelst SH, et al. Structural analysis of phenothiazine derivatives as allosteric inhibitors of the MALT1 paracaspase. Angew Chem Int Ed Engl 2013;52:10384–7. [DOI] [PubMed] [Google Scholar]
- 41.Yang J, Antin P, Berx G, Blanpain C, Brabletz T, Bronner M, et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bardet M, Unterreiner A, Malinverni C, Lafossas F, Vedrine C, Boesch D, et al. The T-cell fingerprint of MALT1 paracaspase revealed by selective inhibition. Immunol Cell Biol 2018;96:81–99. [DOI] [PubMed] [Google Scholar]
- 43.Meloni L, Verstrepen L, Kreike M, Staal J, Driege Y, Afonina IS, et al. Mepazine Inhibits RANK-Induced Osteoclastogenesis Independent of Its MALT1 Inhibitory Function. Molecules 2018;23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Unterreiner A, Stoehr N, Huppertz C, Calzascia T, Farady CJ, Bornancin F. Selective MALT1 paracaspase inhibition does not block TNF-alpha production downstream of TLR4 in myeloid cells. Immunol Lett 2017;192:48–51. [DOI] [PubMed] [Google Scholar]
- 45.Quancard J, Klein T, Fung SY, Renatus M, Hughes N, Israel L, et al. An allosteric MALT1 inhibitor is a molecular corrector rescuing function in an immunodeficient patient. Nat Chem Biol 2019;15:304–13. [DOI] [PubMed] [Google Scholar]
- 46.Oh E, Kim JY, Cho Y, An H, Lee N, Jo H, et al. Overexpression of angiotensin II type 1 receptor in breast cancer cells induces epithelial-mesenchymal transition and promotes tumor growth and angiogenesis. Biochim Biophys Acta 2016;1863:1071–81. [DOI] [PubMed] [Google Scholar]
- 47.Ma Y, Xia Z, Ye C, Lu C, Zhou S, Pan J, et al. AGTR1 promotes lymph node metastasis in breast cancer by upregulating CXCR4/SDF-1alpha and inducing cell migration and invasion. Aging (Albany NY) 2019;11:3969–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tekin C, Shi K, Daalhuisen JB, Ten Brink MS, Bijlsma MF, Spek CA. PAR1 signaling on tumor cells limits tumor growth by maintaining a mesenchymal phenotype in pancreatic cancer. Oncotarget 2018;9:32010–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhong W, Chen S, Qin Y, Zhang H, Wang H, Meng J, et al. Doxycycline inhibits breast cancer EMT and metastasis through PAR-1/NF-kappaB/miR-17/E-cadherin pathway. Oncotarget 2017;8:104855–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Y, Liu J, Ying X, Lin PC, Zhou BP. Twist-mediated Epithelial-mesenchymal Transition Promotes Breast Tumor Cell Invasion via Inhibition of Hippo Pathway. Sci Rep 2016;6:24606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang E, Cisowski J, Nguyen N, O’Callaghan K, Xu J, Agarwal A, et al. Dysregulated protease activated receptor 1 (PAR1) promotes metastatic phenotype in breast cancer through HMGA2. Oncogene 2016;35:1529–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wen J, Zhao Z, Huang L, Wang L, Miao Y, Wu J. IL-8 promotes cell migration through regulating EMT by activating the Wnt/beta-catenin pathway in ovarian cancer. J Cell Mol Med 2020;24:1588–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sobolik T, Su YJ, Wells S, Ayers GD, Cook RS, Richmond A. CXCR4 drives the metastatic phenotype in breast cancer through induction of CXCR2 and activation of MEK and PI3K pathways. Mol Biol Cell 2014;25:566–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Y, Liao R, Chen X, Ying X, Chen G, Li M, et al. Twist-mediated PAR1 induction is required for breast cancer progression and metastasis by inhibiting Hippo pathway. Cell Death Dis 2020;11:520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sabbah M, Emami S, Redeuilh G, Julien S, Prevost G, Zimber A, et al. Molecular signature and therapeutic perspective of the epithelial-to-mesenchymal transitions in epithelial cancers. Drug Resist Updat 2008;11:123–51. [DOI] [PubMed] [Google Scholar]
- 56.Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol 2019;20:69–84. [DOI] [PubMed] [Google Scholar]
- 57.Arakaki AKS, Pan WA, Wedegaertner H, Roca-Mercado I, Chinn L, Gujral TS, et al. alpha-arrestin ARRDC3 tumor suppressor function is linked to GPCR-induced TAZ activation and breast cancer metastasis. J Cell Sci 2021:[Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jacobs KA, Andre-Gregoire G, Maghe C, Thys A, Li Y, Harford-Wright E, et al. Paracaspase MALT1 regulates glioma cell survival by controlling endo-lysosome homeostasis. EMBO J 2020;39:e102030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Konczalla L, Perez DR, Wenzel N, Wolters-Eisfeld G, Klemp C, Luddeke J, et al. Biperiden and mepazine effectively inhibit MALT1 activity and tumor growth in pancreatic cancer. Int J Cancer 2020;146:1618–30. [DOI] [PubMed] [Google Scholar]
- 60.Yang F, Liu X, Liu Y, Liu Y, Zhang C, Wang Z, et al. miR-181d/MALT1 regulatory axis attenuates mesenchymal phenotype through NF-kappaB pathways in glioblastoma. Cancer Lett 2017;396:1–9. [DOI] [PubMed] [Google Scholar]
- 61.Wang Y, Zhang G, Jin J, Degan S, Tameze Y, Zhang JY. MALT1 promotes melanoma progression through JNK/c-Jun signaling. Oncogenesis 2017;6:e365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pan D, Jiang C, Ma Z, Blonska M, You MJ, Lin X. MALT1 is required for EGFR-induced NF-kappaB activation and contributes to EGFR-driven lung cancer progression. Oncogene 2016;35:919–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pan D, Zhu Y, Zhou Z, Wang T, You H, Jiang C, et al. The CBM Complex Underwrites NF-kappaB Activation to Promote HER2-Associated Tumor Malignancy. Mol Cancer Res 2016;14:93–102. [DOI] [PubMed] [Google Scholar]
- 64.Mahanivong C, Chen HM, Yee SW, Pan ZK, Dong Z, Huang S. Protein kinase C alpha-CARMA3 signaling axis links Ras to NF-kappa B for lysophosphatidic acid-induced urokinase plasminogen activator expression in ovarian cancer cells. Oncogene 2008;27:1273–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rehman AO, Wang CY. CXCL12/SDF-1 alpha activates NF-kappaB and promotes oral cancer invasion through the Carma3/Bcl10/Malt1 complex. Int J Oral Sci 2009;1:105–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol 2017;14:611–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kudo-Saito C, Shirako H, Takeuchi T, Kawakami Y. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell 2009;15:195–206. [DOI] [PubMed] [Google Scholar]
- 68.Dongre A, Rashidian M, Reinhardt F, Bagnato A, Keckesova Z, Ploegh HL, et al. Epithelial-to-Mesenchymal Transition Contributes to Immunosuppression in Breast Carcinomas. Cancer Res 2017;77:3982–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lucas PC, McAllister-Lucas LM, Nunez G. NF-kappaB signaling in lymphocytes: a new cast of characters. J Cell Sci 2004;117:31–9. [DOI] [PubMed] [Google Scholar]
- 70.Demeyer A, Staal J, Beyaert R. Targeting MALT1 Proteolytic Activity in Immunity, Inflammation and Disease: Good or Bad? Trends Mol Med 2016;22:135–50. [DOI] [PubMed] [Google Scholar]
- 71.Demeyer A, Skordos I, Driege Y, Kreike M, Hochepied T, Baens M, et al. MALT1 Proteolytic Activity Suppresses Autoimmunity in a T Cell Intrinsic Manner. Front Immunol 2019;10:1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Martin K, Junker U, Tritto E, Sutter E, Rubic-Schneider T, Morgan H, et al. Pharmacological Inhibition of MALT1 Protease Leads to a Progressive IPEX-Like Pathology. Front Immunol 2020;11:745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat 2010;123:725–31. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.