

Abstract
Dopamine transporter mediates the neurotransmitter dopamine homeostasis in a sodium-dependent manner. The transport process involves an alternating access of a substrate to the extracellular and intracellular spaces, which is associated with different conformational states of the transporter. However, the underlying mechanism of modulation of the state transition remains elusive. Here we present a computational simulation study of human dopamine transporter to explore its two end states (outward-facing open and inward-facing open) that have not been determined experimentally. We show that the full-length transporter may tend to adopt the inward-facing open state in its free state. The binding of an amphetamine may not trap the transporter in the outward-facing open state with increasing length of the N-terminal. Furthermore, we identify distinct patterns in the interaction networks between the N-terminal and the intracellular region that could stabilize the state of the transporter, independent of substrate binding and phosphorylation. Our results reveal the essential role of the N-terminal dynamics in modulating the functional states of the dopamine transporter, providing molecular insights into the coupling of conformational transition and substrate passage in neurotransmitter transporters.
Keywords: N-terminal, human dopamine transporter, conformational states, molecular dynamics simulations, N-terminal phosphorylation
1. Introduction
The human dopamine (DA) transporter (hDAT), serotonin transporter (hSERT), and leucine transporter (LeuT) are membranes of the family of neurotransmitter:sodium symporters (NSSs), which utilize the transmembrane sodium gradient as the driving force to reuptake the corresponding neurotransmitter (DA, serotonin, or leucine) across the cell membrane [1-3]. The malfunction of NSSs is associated with various neurological diseases including depression, anxiety, attention deficit hyperactivity disorder (ADHD), and Parkinson’s disease [4]. Moreover, the NSSs are also targets for psychostimulant drugs such as cocaine (COC) and amphetamine (AMPH) [5]. The competitive binding of COC and AMPH to DAT can inhibit the uptake of the neuronal DA, increase extracellular DA level, and consequently lead to the rewarding and addictive effects [6, 7].
Although no crystal structures of hDAT are available to date, the structure of hDAT is believed to adopt a similar topology with those crystal structures of Drosophila melanogaster DAT (dDAT), bacterial homolog LeuT, and hSERT, which contain 12 transmembrane helices and intracellular N- and C-termini [2, 8]. The NSS transporters have been suggested to work through an alternating access mechanism [9, 10], that is, they undergo conformational transitions between an outward-open facing (or outward-open) and an inward-open facing (or inward-open) state alternatively to enable the substrate to access the extracellular and intracellular side of the membrane. The crystal structures of dDAT in complex with various substrates have been solved in the outward-open conformation [8]. In addition to the outward-open state, the inward-open conformation of bacterial homolog LeuT and hSERT have also been reported [11, 12]. However, the conformational dynamics, especially the inward-open conformation of hDAT in substrate-free state remain largely elusive.
Sequence alignment shows that hDAT shares ~52%, ~48%, and ~20% sequence identity with dDAT, hSERT, and LeuT, respectively, indicating that the structure of dDAT could be the best template to model the structure of hDAT (Fig. S1). One major difference between the sequences of hDAT and dDAT is that hDAT contains a longer N-terminal region. In the crystal structure of dDAT in complex with AMPH (PDB ID: 4XP9) [8], the N-terminal region containing residues 1–24 was not determined, which corresponds to the residues 1–57 in the N-terminal of hDAT. Although the mobile N-terminus in the crystal structures of dDAT is absent, it is relevant to the function of DAT, especially the N-terminal phosphorylated DAT [13]. In contrast to the normal DA uptake, the phosphorylation of five N-terminal serine residues (2, 4, 7, 12, and 13) have been demonstrated to facilitate DAT-mediated DA efflux without affecting other aspects of DAT physiology [14, 15]. One of underlying mechanisms that regulate AMPH-induced DA efflux implied that N-terminal phosphorylation increases the availability of inward-open states of DAT [14]. Interactions of N-terminus of hDAT with phosphatidylinositol 4,5-biphosphate (PIP2) lipids have also been indicated to trigger the conformational transition from the outward-open to the inward-open state [16, 17]. In addition to hDAT, previous studies suggested that the N-terminal of LeuT could not enter the intracellular vestibule but may affect the intracellular sodium affinity to the inward-open states [18]. However, it remains largely unknown how the dynamics of intracellular N-terminus could impact the conformational states of hDAT defined by the transmembrane domains.
To elucidate the role of the N-terminal in the conformational transition of hDAT and regulation of AMPH-induced DA efflux, we investigated how the dynamics of the N-terminal and the binding of AMPH modulate the conformational states of hDAT in different forms by performing extensive molecular dynamics (MD) simulations. Our results indicate that in the absence of a substrate, hDAT favors an inward-open conformation. The outward-open state could be stabilized by AMPH binding, but more dependent on the N-terminal dynamics. The interactions between the N-terminal and intracellular region modulate the conformational transitions of hDAT. Moreover, N-terminal phosphorylation promotes the inward-open states of hDAT, and may thus mediate the AMPH-induced DA efflux. Our work demonstrates the feasibility of exploring functionally relevant states of hDAT by engineering its N-terminal.
2. Methods
2.1. Modeling of hDAT and hDAT in complex with AMPH
The X-ray crystal structure of dDAT in complex with AMPH (PDB ID: 4XP9) was used to construct the wild-type structure of dDAT [8]. The detailed protocol can be found in our previous studies of dDAT in complex with DA, COC, and METH [19, 20]. Briefly, the flexible loop region containing residues Ser162–Val202, which is missing in the crystal structure, was modelled and optimized using MODELLER program (version 9.23) [21]. The mutated residues in the crystal structure were reverted to the wild-type amino acids. The two sodium ions (Na+), one chloride (Cl−) ion, and water molecules found in the crystal structure were kept in the final modeled structure. This wild-type dDAT structure containing residues (25–599) was then used as the template to construct the homology models of the hDAT structure containing residues (1–604). In particular, the N-terminal was modeled using the automatic loop refinement approach implemented in MODELLER. In an early computational study, the N-terminal was found to be able to adopt specific secondary structures although it is highly dynamic. However, it should be noted that the N-terminal was simulated in the absence of the other part of DAT, and the effect of the transmembrane domain on the N-terminal remains unclear [22]. Therefore, in this work, the N-terminal was initially modeled as a loop and our results show that it could display secondary structures including α-helix and β–strand during current simulations (see Results). In the modeled hDAT structure, one Na+ ion (Na1) is coordinated by residues Ala77, Asn82, S321, and Asn353; and the other Na+ ion (Na2) is coordinated by residues Gly75, Val78, Leu418, Asp421, and Ser422. In addition, the Cl− ion is coordinated by residues Tyr102, Gln317, Ser321, and Ser357. These ions are positioned equivalently to those in the crystal structure of dDAT structure (Fig. S2). Moreover, a disulfide bond between residues Cys180 and Cys189 was introduced in the structure of hDAT, corresponding to the disulfide bond between residues Cys148 and Cys157 in the crystal structure of dDAT (PDB ID: 4XP9) [8].
Because the initial position of the long N-terminal relative to the transmembrane region may establish different interactions, to enhance sampling, two structural models of hDAT with different N-terminal positions were constructed (Fig. 1). In both models, the N-terminal region remains in the cytosol. However, in Model1, the N-terminal region spans the intracellular side of the transmembrane core. In contrast, in Model2, the N-terminal region was initially positioned on the right side of the transmembrane helices. To construct the phosphorylated hDAT (p-hDAT), five serine residues (2, 4, 7, 12, and 13) in the N-terminal were phosphorylated simultaneously. For comparison, two N-terminal truncated hDAT forms, hDAT(25-604) and hDAT(59-604) with the first 24 and 58 residues being deleted, were also constructed and simulated.
Fig. 1.

(A and B) Initial conformations of hDAT(1–604) in Model1 and Model2. (C and E) Representative conformations of hDAT(1–604) over 1-μs MD simulations from side and intracellular views for Model1. (D and F) Representative conformations of hDAT(1–604) over 1-μs MD simulations from side and intracellular views for Model2. The 12 TM helices, five serine residues, and two truncation points (V24 and Q58) are labeled. The N-terminal region is colored in red and the TM1a and TM6a are colored in blue. EC and IC denote extracellular and intracellular, respectively. Note that Ser4 is masked in (F).
To model the structure of hDAT in complex with AMPH, AutoDock Vina program [23] was applied to dock one AMPH molecule into the central binding site of hDAT. The hDAT structure was represented with the CHARMM36m force field parameters [24], and the AMPH structure was represented with the CHARMM General Force Field (CGenFF) [25] along with the RESP partial charges [26] (Fig. S3). Note that the structure of AMPH is positively charged in the quaternary ammonium motif. In the docking process, the protein structure was modeled as rigid while the AMPH structure was modeled as flexible. The optimal binding pose with the lowest binding energy was obtained and compared with the interacting residues in the binding site of the crystal structure. AMPH was found to interact with hDAT in a similar manner as to bind to the central binding site of dDAT (Fig. S4).
2.2. MD simulations of hDAT, p-hDAT, AMPH-bound hDAT, and AMPH-bound p-hDAT
The hDAT structure was embedded into a POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) lipid bilayer, and the orientation of the hDAT structure relative to the membrane was defined by aligning to the dDAT structure in the orientation of protein in membrane database (PDB ID: 4M48) [27]. The complex was then solvated by adding a 25 Å thick water layer (TIP3P water molecules) below and above the lipid bilayer. The salt concentration (NaCl) in each system was 0.15 M. The protein and lipid structures were represented with the CHARMM36m and CHARMM36 force field parameters, respectively [24, 28]. The CGenFF was used to generate the topology and force field parameters for the structure of AMPH [25], with the partial charges replaced with the calculated RESP charges [26] (Fig. S3). The CHARMM-GUI web server was used to generate the starting structures and configuration files for MD simulations [29, 30]. The resulting system contains ~240 POPC lipids and a total of ~135,000–140,000 atoms.
MD simulations were carried out using GROMACS simulation package (version 2019.4) [31-33]. Each system was first energy minimized for 10,000 steps, followed by six stages of equilibration with the harmonic constraints exerted on lipid, hDAT, and AMPH heavy atoms. The force constants for lipid head group was decreased from 1000 kJ/(mol·nm2) to 400 kJ/(mol·nm2), 200 kJ/(mol·nm2), 40 kJ/(mol·nm2), and 0 kJ/(mol·nm2), whereas the force constants for protein backbone and sidechain (denoted as backbone/sidechain) were gradually decreased from 4000/2000 kJ/(mol·nm2) to 2000/1000 kJ/(mol·nm2), 1000/500 kJ/(mol·nm2), 500/200 kJ/(mol·nm2), 200/50 kJ/(mol·nm2), and 50/0 kJ/(mol·nm2) during the equilibration procedures. The temperature was kept constant at 310 K using the Berendsen thermostat with a coupling parameter of 0.1 ps [34], and the pressure was controlled at 1.0 bar using the Berendsen barostat with a time constant 5.0 ps for the last four stages of equilibration [34]. The semiisotropic pressure coupling was applied for membrane simulations. A cutoff of 12 Å was applied for the van der Waals interactions and the long-range electrostatic interactions were treated using the particle mesh Ewald method [35]. The integration time step was 2 fs and trajectory was saved every 10 ps. The production dynamics were performed at the constant temperature (310 K) controlled by the Nosé-Hoover thermostat [36, 37] and constant pressure (1.0 bar) controlled by the Parrinello-Rahman [38, 39] barostat without any restraints. The production simulations were carried out for at least 1 microsecond (μs) for each system. The simulated systems were listed in Table 1. Note that the initial conformations of the transmembrane bundles are the same in all systems and in the same outward-open state.
Table 1.
Summary of the simulated systems and main results.
| System | Distance (Å) | State | Na+ | Supporting Information |
|
|---|---|---|---|---|---|
| F76--S422 | F76--S429 | ||||
| Free state | |||||
| hDAT(59–604) | 10.7±0.5 | 16.1±0.5 | IF | None | Fig. S5 |
| hDAT(25–604) (Model1) | 8.4±0.5 | 12.3±0.9 | IF | None | Fig. S5 |
| hDAT(25–604) (Model2) | 9.8±0.5 | 14.7±0.6 | IF | None | Fig. S5 |
| hDAT(1–604) (Model1) | 9.5±0.6 | 14.0±0.5 | IF | None | Fig. 3; Fig. S6 |
| hDAT(1–604) (Model2) | 4.3±0.3 | 11.4±0.3 | OF | Na2 | Fig.3 |
| p-hDAT((1–604)) (Model1) | 9.0±0.7 | 13.9±0.6 | IF | Na1 | Figs. S7-S9 |
| p-hDAT((1–604)) (Model2) | 8.9±0.4 | 14.2±0.5 | IF | Na2 | Figs. S7-S9 |
| In complex with AMPH | |||||
| hDAT(59–604) | 4.4±0.2 | 11.7±0.3 | OF | Na1; Na2 | Fig. S10; Fig. S11 |
| hDAT(25–604) (Model1) | 4.7±0.3 | 12.3±0.4 | OF | Na1; Na2 | Fig. S10 |
| hDAT(25–604) (Model2) | 8.4±0.5 | 13.4±0.6 | IF | Na2 | Fig. S10 |
| hDAT(1–604) (Model1) | 8.6±0.4 | 14.0±0.7 | IF | Na2 | Fig. S12; Fig. S14 |
| hDAT(1–604) (Model2) | 7.7±0.4 | 12.8±0.6 | IF | Na2 | Fig. S12; Fig. S14 |
| p-hDAT(1–604) (Model1) | 4.4±0.3 | 11.0±0.5 | OF | Na2 | Fig. S13; Fig. S14 |
| p-hDAT(1–604) (Model2) | 8.4±0.4 | 13.9±0.5 | IF | Na1; Na2 | Fig. S13; Fig. S14 |
| With C-terminal/Full-length | |||||
| hDAT(59–620) (free state) | 4.2±0.3 | 11.4±0.4 | OF | None | Fig. S15 |
| hDAT(1–620) (Model2, free state) | 10.1±0.4 | 15.2±0.5 | IF | Na1; Na2 | Fig. S15 |
| p-hDAT(1–620) + AMPH (Model1) | 4.4±0.3 | 11.0±0.6 | OF | Na2 | Fig. S15 |
| hDAT(1–620) (designed) | 4.7±0.4 | 111.3±0.6 | OF | Na2 | Fig. S16 |
Notes:
The distances of F76--S422 and F76--S429 are averaged over the last 500-ns trajectory of each simulation
Model1 and Mode2 correspond to the initial conformation of hDAT(1–604) shown in Fig. 1.
Na+ denotes the sodium ions remained in the initial binding sites. The coordination environments of Na1 and Na2 are shown in Fig. S28 and Fig. S2. None indicates the release of both Na1 and Na2.
IF and OF refer to the inward-facing open (or inward-open for simplicity) and outward-facing open (or outward-open for simplicity) conformation (or state), respectively.
For the artificially designed full-length hDAT, the C-terminal tail was initially placed inside the membrane.
2.3. Analysis methods
The convergence of simulation was assessed by the fraction of native contacts Q, which was calculated using the formula [40]:
where heavy atoms i and j in residues θi and θj (∣θi −θj∣>3) are in contact if the distance between them is less than 5.0 Å. rij (X) is the distance between i and j in conformation X; r0ij (X) is the distance in the native state (starting conformation). β is a smoothing parameter taken to be 5 Å−1 and the factor λ accounts for fluctuations when the contact is formed, taken to be 1.8 for the all-atom model.
The number of conformational states of hDAT was characterized by normal-mode frequencies, a method proposed by Benjamin and co-authors [41]. The distribution of the eigenvalues for the first eigenvector was obtained by applying a Gaussian network model (GNM) on the last 500-ns trajectory generated from MD simulations. The MDAnalysis package was used to perform the calculations [42, 43].
3. Results
3.1. Characteristic structural features for the outward-open and inward-open states of hDAT
The crystal structures of LeuT and hSERT have been captured in both outward-open and inward-open states [11, 12]. To distinguish the two states, two residue-based distances were first defined, i.e., the distance between residues Phe76 and Ser422 and the distance between Phe76 and Ser429. The residue numbering is based on the sequence of hDAT, and for the equivalent residues in other transporters, see Fig. 2 and Fig. S1. Residue Phe76 corresponds to Phe43 in the TM1a of dDAT, which is a key interacting residue in the central binding site of dDAT. Residue Ser422 is equivalent to Ser421 in the TM8 of dDAT, which interacts with AMPH (Fig. S4) and also coordinates with Na2 (Fig. S2) in the crystal structure of dDAT in complex with AMPH. No interactions between Ala428 (Ser429 in hDAT) and AMPH were observed from the crystal structures of dDAT in the outward-open conformations. As shown in Fig. 2, the outward-open conformations of transporters display two characteristic distances, the distance between Phe76 and Ser422, which varies from 3.9 to 4.4 Å, and the distance between Phe76 and Ser429, which is between 10.9 and 12.7 Å. In contrast, in the inward-open conformations of LeuT and hSERT, these two distances increase to 9.0–11.0 Å and 14.6–20.0 Å, respectively. The above results demonstrate that we may be able to distinguish the conformations of hDAT by defining the distances between those three residues. However, the distances in the outward-open occluded and inward-open occluded states closely resemble the outward-open conformations, indicating that they are less sensitive to those intermediate states. Thus, we focus on the two end states, i.e., the outward-open and inward-open states (for other states, see Discussion). In addition to the distance criteria, we also performed molecular docking to check the difference in the outward-open and inward-open conformations. We found that substrate (DA or AMPH) occupied distinct binding sites in the outward-open and inward-open conformations, a similar result was also found for the binding of ibogaine to hSERT (see Results and Discussion below, e.g. Fig. S29).
Fig. 2.

(A) Characteristic structural features used to define the outward-facing open (or outward-open for simplicity) and inward-facing open (or inward-open) states of hDAT. The equivalent residues for Phe76, Ser422, and S429 of hDAT in dDAT, LeuT, and hSERT are shown in the insect. The distances between Cα atoms of Phe76 and Ser422 and between Phe76 and Ser429 are used to denote the corresponding distances in different transporters. (B) The relative positions of Phe76, Ser422, and Ser429 in the outward-open (blue) and inward-open (red) states of hDAT(1–604). (C) The relative positions of Phe76, Ser422, and Ser429 in the outward-facing (blue) and inward-facing (red) states of AMPH-bound p-hDAT(1–604). OF, IF, OFo, and, IFo denote outward-facing open, inward-facing open, outward-facing occluded, and inward-facing occluded, respectively.
3.2. N-terminal could shift the conformational states of free hDAT
Fig. 3 shows results obtained from simulations of hDAT(1–604) that started from the outward-open conformation. The fraction of native contacts Q shown in Figs. 3A and 3B suggest that each hDAT model reached equilibrium as no significant change in intramolecular contacts were observed after 200-ns simulations. Figs. 3C and 3D show results of the distribution of the eigenvalues for the first eigenvector. A single peak centered at 0.05 and 0.06 appear for the two hDAT models with different initial N-terminal positions, respectively, indicating that there is only one state for both models of hDAT. Figs. 3E-H shows the time change of the distance between Phe76 and S422 (Figs. 3E and 3F) and between Phe76 and Ser429 (Figs. 3G and 3H). For Model1 of hDAT, the distribution of the distance of Phe76-Ser422 has a peak centered at 9 Å and the distance of Phe76-Ser429 has a peak centered at 14 Å, implying the inward-open state. In contrast, for Model2 of hDAT, the distances of Phe76-Ser422 and Phe76-Ser429 have peaks at 4 Å and 12 Å, respectively, suggesting that this model remained in the initial outward-open state. Thus, in the presence of N-terminal, our simulations could capture both outward- and inward-open states of free hDAT(1–604).
Fig. 3.
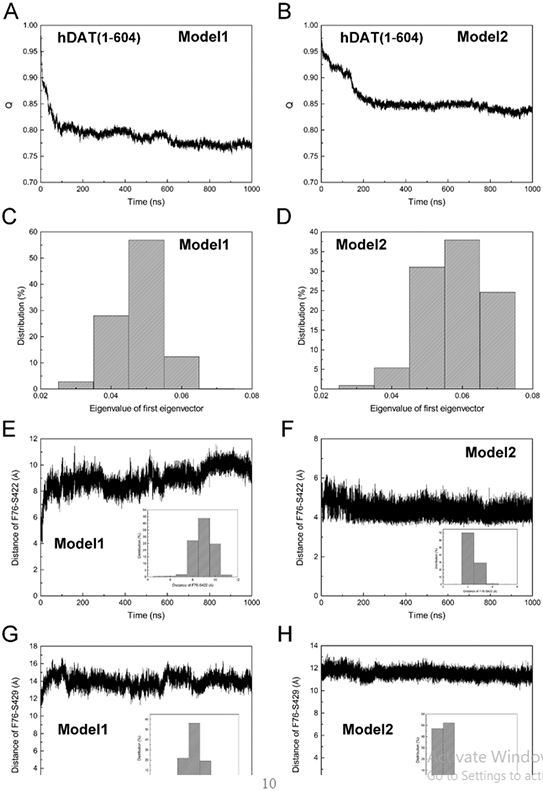
Comparison of conformational states of hDAT(1–604) in the inward-open (A, C, E, and G) and outward-open states (B, D, F, and H). (A and B) The fraction of native contacts Q. (C and D) The distribution of the eigenvalue of the first eigenvector. (E and F) The time change of the distance between Cα atoms of Phe76 and Ser422 over 1-μs MD simulations. (G and H) The time change of the distance between Cα atoms of Phe76 and Ser429 over 1-μs MD simulations. The distribution of the distance is shown in the inset of each figure. Model1 and Model2 correspond to the structural models shown in Fig. 1.
To further examine the effects of N-terminal dynamics on the conformational transition of hDAT, different truncated forms of hDAT(1–604) were simulated and results were summarized in Table 1. Note that all simulations yielded single state according to the GNM method (more discussion on this method can be found in Fig. S9). Elimination of the N-terminal residues 1–58 of hDAT, i.e., hDAT(59–604), resulted in the inward-open state. Similarly, the inward-open states were also observed for simulations of two models of hDAT(25–604) in which the first 24 residues were removed. Moreover, upon phosphorylation of the N-terminal serine residues (2, 4, 7, 12, and 13), the Model1 of p-hDAT(1–604) shifted to the inward-open state as observed in its wild-type. The finding that the Model2 of p-hDAT(1–604) also shifted to the inward-open state, in contrast to its wild-type that remained in the outward-open state, suggested that the N-terminal phosphorylation could promote the conformational transition. Taken together, the above results imply that the N-terminal, especially residues 1–24, could either maintain the initial outward-open state of hDAT(1–604) or shift it to the inward-open state, and the N-terminal phosphorylated hDAT(1–604) tends to populate the inward-open conformations.
3.3. N-terminal could alter the conformational states of AMPH-bound hDAT
It is well recognized that the conformational dynamics of neurotransmitter transporters are coupled with the binding of substrates [10]. To characterize the substrate-dependent conformational states of hDAT, we simulated various forms of hDAT(1–604) in complex with AMPH, and observed different scenarios compared to free states of hDAT(1–604) (Table 1). Specifically, the structural model AMPH-bound hDAT(59–604) remained in the outward-open state, consistent with previous simulations of AMPH-bound hDAT(58–598) in a POPC membrane [44]. Such results seemed also consistent with the outward-open state observed in the crystal structure of dDAT(25–602) in complex with AMPH [8]. However, the binding of AMPH may not always trap hDAT in the outward-open state. For AMPH-bound hDAT(25–604), the Model1 was maintained in the outward-open state whereas the Model2 shifted to the inward-open state, indicating that increasing the length of the N-terminal (excluding residues 1–24) could alter the conformational states of AMPH-bound hDAT(25–604). Further inclusion of residues 1–24 resulted in the inward-open states of AMPH-bound hDAT(1–604) in both models, highlighting the critical regulation role of N-terminal region. Of interest, similar to AMPH-bound hDAT(25–604), both outward-open (Model1) and inward-open (Model2) states were observed in AMPH-bound p-hDAT(1–604). Note that the N-terminal contains 11 charged residues including 5 lysines (3, 5, 19, 27, and 35), 1 arginine (Arg51), and 5 glutamic acids (20, 28, 30, 35, and 56). Phosphorylation of the 5 serine residues (2, 4, 7, 12, and 13) resulted in a highly negatively charged N-terminal, which was found to promote the inward-open states of free p-hDAT(1–604), but lead to different states of AMPH-bound p-hDAT(1–604). The above results intrigued us to examine if the regulation mechanism of the N-terminal is dependent on the binding of AMPH or phosphorylation or both.
3.4. N-terminal alters the conformational states of hDAT by forming distinct interaction networks
When the N-terminal was truncated, hDAT(59–604) and AMPH-bound hDAT(59–604) were observed in the inward-open and outward-open conformation, respectively. The latter is associated with the reduced conformational flexibility of hDAT due to the binding of AMPH. The conformational transition involved movements of transmembrane helices (TMs) 1 and 6 in which there are hinge regions between TM1a and TM1b and between TM6a and TM6b. To identify the difference in the movement of TM1a and TM6a, the angles between TM1a/TM6a with respect to z-axis were calculated and summarized in Table S1. Indeed, the binding of AMPH induced TM1a to move closer to the TM scaffold as the angle of TM1a relative to z-axis decreased by 10°. Meanwhile, a tilting of 3° away from the central binding site was observed for TM6a compared to the free hDAT(59–604). However, with the increasing length in the N-terminal, no direct link between the TM angles and specific conformational states could be established due to the fluctuations of these angles induced by either the dynamics of the N-terminal or the binding of AMPH or both. Figs. 2B and 2C show the difference in the TM angels between the two states in hDAT(1–604) and AMPH-bound p-hDAT(1–604) after superposition of the TM core (TM3, 4, 8, and 9). Compared to the outward-open conformation, both TM1a and 6a shifted away from the central site in the inward-open conformations, which could affect the binding affinity of APMH and consequently enable AMPH to alternatively access to the intracellular side.
The conformational transition from the initial outward-open to the inward-open state was observed in the free hDAT(1–604), AMPH-bound hDAT(25–604), and AMPH-bound p-hDAT(1–604) (Table 1). To gain insight into the molecular determinant for specific conformational states, the contact maps, especially the electrostatic interaction networks, between the N-terminal and the intracellular region were calculated. Since the N-terminal in the cytosol, only interactions with those residues in the intracellular end were established. Fig. 4 shows the contact maps calculated for hDAT(1–604) and AMPH-bound p-hDAT(1–604) in either an outward-open or an inward-open state. For other systems, results were shown in Figs. S21-S26, and Tables S2-S9. Fig. 5 shows the interaction networks calculated for hDAT(1–604) and AMPH-bound p-hDAT(1–604) in the same states as shown in Fig. 4. It was found that when there are weak or no interactions between the N-terminal and the intracellular portion and C-terminal region, hDAT(1–604) would populate the inward-open conformation. Otherwise, hDAT would sample the outward-open conformation. Figs. 4D and 5D reveal that only one N-terminal residue Glu30 in AMPH-bound p-hDAT(1–604) (Model2) interacted with Arg60, and the inward-open state was identified. Of importance, despite the significant differences between hDAT(1–604) (Model2) and AMPH-bound p-hDAT(1–604) (Model1), both systems displayed similar interaction patterns (Figs. 4B and 4C; and Figs. 5B and 5C), i.e., the N-terminal interacted with TM1a, the C-terminal, and most intracellular loops (ILs) simultaneously. Specifically, the N-terminal of hDAT(1–604) (Model2) interacted with TM1a, ILs 1–4, and C-terminal; and the N-terminal of AMPH-bound p-hDAT(1–604) (Model1) interacted with TM1a, IL1, ILs 3–5, and the C-terminal. In contrast, the lack of interactions between the N-terminal residues 1–10 and ILs led to the inward-open conformation of hDAT(1–604) (Model1) (Figs. 4A and 5A). Consistent results were obtained when applying this criterion to other systems. For instance, the system AMPH-bound hDAT(25–604) (Model1) adopted the outward-open state (Table 1), which could be attributed to the interactions between its N-terminal and ILs (1–5), TM5, TM8, and C-terminal (Fig. S20). Similarly, for hDAT(25–604) (Model2) and AMPH-bound hDAT(25–604) (Model2), the inward-open conformation would be expected because of the collapse in their N-termini (Fig. S22). The formation of relatively stable β-strands in the N-terminal of both p- hDAT(1–604) models also contributed to the weak association between the N-terminal and the intracellular portion (Figs. S8 and S27). The intracellular side views of hDAT(1–604) in the inward-open (Fig. 1E) and outward-open conformations (Fig. 1F) demonstrated such difference, indicating that the N-terminal could stabilize the outward-open state by attaching to the intracellular end and forming a closed loop.
Fig. 4.

Contact maps calculated for hDAT(1–604) (A and B) and AMPH-bound p-hDAT(1–604) (C and D) in the outward-facing open (OF) and inward-facing open (IF) states. N-term, 1a, 3, 5, 8, C-term denote N-terminal, TM1a, TM3, TM5, TM8, and C-terminal, respectively. IL denotes intracellular loop. A contact occurs if any atom in the N-terminal region is within 3 Å of any atom in the TM domains, and only a contact frequency >0.5 (50%) is shown. Contact frequency was calculated using the last 500-ns trajectory of each simulation. Model1 and Model2 correspond to the two models shown in Fig. 1.
Fig. 5.

Interaction networks calculated for hDAT(1–604) (A and B) and AMPH-bound p-hDAT(1–604) (C and D) in the outward-facing and inward-facing states. Model1 and Model2 correspond to the two models shown in Fig. 1. The N-terminal tail (1–58) is shown in red. The 12 transmembrane helices (TM), intracellular loops (IL), AMPH, and phosphorylated serines (p-S2, p-S4, p-S7, p-S12, and p-S13) are labelled.
As mentioned above, the N-terminus includes 11 charged residues (5 lysines, 1 arginine, and 5 glutamic acids), and its electrostatic interactions with specific charged resides in the intracellular region may be the driving force for the association of N-terminal with the intracellular region. The details in the contact frequency > 50% were summarized in Table S2-S9. For hDAT(1–604) in the inward-open state (Fig. S25), Glu36, Asp436, and Glu437 interact with Lys35, Arg51, and Lys133. Also, Glu56 interacts with Lys66 and Lys337. These electrostatic interactions appear to form the core of the interaction network, facilitating the formation of other types of interactions involving hydrogen bonding and salt-bridges. Fig. S25 also shows the interaction network for hDAT(1–604) in the outward-open state. In the electrostatic interaction core, Arg51 interacts with Asp507, and Glu56 interacts with Arg125 and Lys337. Although the involving residues are less than those in the inward-open conformation as shown in Fig. S25, their contact frequencies are > 85%, higher than those observed in the inward-open conformation (54%–84%) (Table S3). Such a stable interaction center could promote the N-terminal to form an interaction network that spans a wider area of the intracellular end, prevent water penetration, and consequently, stabilize hDAT(1–604) in the outward-open conformation. Similar findings were also found in AMPH-bound p-hDAT(1–604) in which those phosphorylated serines were also involved in establishing the electrostatic interaction center (Fig. S26). In contrast, if the N-terminal collapsed in the intracellular solution, AMPH-bound p-hDAT(1–604) was found to adopt the inward-open state (Fig. 5D). Together, these results suggest that the N-terminal could trap hDAT(1–604) in an outward-open conformation through interactions with more residues located in the intracellular portion, especially with residues in those intracellular loops and C-terminal. On the other hand, an inward-open conformation would be expected when the N-terminal collapses or less interactions with those intracellular loops and C-terminal are established.
3.5. C-terminal may shift the conformational state of hDAT by altering interactions of N-terminal with intracellular side
Since we focus on the role of N-terminal dynamics, the C-terminal residues 605–620, which was modeled as disordered using the crystal structure of dDAT, was not included in the models of hDAT(1–604). To examine the influence of this C-terminal, we first constructed the structural model hDAT(59–620) which displayed the same initial outward-open conformation as hDAT(59–604) (Table 1). However, different from hDAT(59–604), hDAT(59–620) was observed to maintain the outward-open state, indicating that the flexible C-terminal tail may stabilize the outward-open conformation of N-terminal truncated hDAT. This intrigues us to further investigate the role of the C-terminal in the conformational states of hDAT with N-terminal. In the second model including C-terminal, the last snapshot of the simulations of hDAT(1–604) in the outward-open state (Model2) was used to construct the full-length hDAT structure containing residues 1–620. Results from our MD simulations of this model showed a conformational transition from the initial outward-open state to the inward-open state. We speculated that the presence of the C-terminal tail may disrupt the interaction network required to maintain the outward-open conformation. The calculated contact maps shown in Fig. 6 confirmed this assumption. Compared to hDAT(1–604) (Fig. 4B), in the full-length hDAT (Fig. 6A), the lack of interactions between the N-terminal residues 50–60 and ILs 3 and 5 suggests a weak association between the N-terminal and the intracellular portion (Fig. 6C). These weakening effects could be ascribed to the transient interactions (occupancy <50%, Fig. 6B) between the C-terminal and the N-terminal, which may impact the association of the N-terminal with the intracellular region. In this respect, the free state of full-length hDAT tends to populate the inward-open conformation. A previous experimental data has suggested that DAT C-terminal alone could not affect the substrate (DA or AMPH) binding affinity, but influence the binding affinity with N-terminal synergistically [45]. Based on our finding, this may be attributed to the inward-open conformation induced by the C-terminal, which assists in the substrate transport from the central binding site to the intracellular space. Consequently, the substrate binding affinity reduces according to our simulations resultsin.
Fig. 6.

Contact map and interaction network calculated for the full-length hDAT (A–C), AMPH-bound p-hDAT (D–F), and the artificially designed full-length hDAT (G–I) where the C-terminal tail was tapped in membrane. The label coloring schemes are the same as used in Figs. 4 and 5. For the details of interacting residues, see Table S7. The N-terminal tail (1–58) is shown in red, and the C-terminal tail (605–620) is shown in yellow. The 12 transmembrane helices (TM), intracellular loops (IL), AMPH, and phosphorylated serines (p-S2, p-S4, p-S7, p-S12, and p-S13) are labelled. For the details of interacting residues, especially the electrostatic interactions, see Tables S7-S9.
To further assess the effect of the C-terminal residues 605–620 on the outward-open state of p-hDAT, in the third structural model, the last snapshot taken from the simulations of AMPH-bound p-hDAT(1–604) (Model1) was used to construct the full-length p-hDAT in complex with AMPH by adding the C-terminal residues 605–620. Our simulation results demonstrated that this full-length p-hDAT remained in the outward-open state (Table 1). Compared to p-hDAT(1–604) (Fig. 4C), a similar interaction network between the N-terminal and the intracellular portion was observed (Fig. 6D). The interactions of the N-terminal residues 16–22 with ILs 2–4 implied more interactions involved, as demonstrated in Fig. 6F. In addition, transient interactions between the N- and C- termini were also identified in p-hDAT (Fig. 6E), but such interactions appeared to have little effect on the interaction network because of the strong electrostatic interactions (including hydrogen bonding) established between those phosphorylated serines and the C-terminal (Fig. 6F).
However, what would happen when there are little interactions between the C- and N- terminal regions? Could the full-length hDAT behave similarly to the C-terminal truncated hDAT? To answer these questions, we took the last snapshot from the simulations of Model 2 of hDAT(1–604) in the outward-open state, and added residues 605–620. Different from the above full-length hDAT, we purposely restrained the position of the C-terminal by placing residues Arg615 and Lys619 inside the lipids (Fig. S16). Due to the electrostatic interactions between the two residues and lipid head groups, interactions between the C- and N- termini were largely blocked over simulations. Fig. 6G shows the calculated interaction map between the N-terminal and the intracellular region, which is quite similar to that of C-terminal truncated hDAT(1–604) shown in Fig. 4B. As a result, the outward-open state was observed (Table 1). Figs. 6H and 6I confirm that the C-terminal interacted weakly with IL4, as well as the N-terminal. Although artifact was introduced through the insertion of C-terminal tail into membrane, the present results highlighted the critical role of N-terminal in determining the conformational states of hDAT. Unlike the N-terminal, the C-terminal is much shorter, and could not form stable interactions with the intracellular region as the N-terminal does. Taken together, the above results indicated that the full-length hDAT might populate the inward-open conformation, and the C-terminal alone may not be able to modulate the state transitions.
3.6. Mapping the substrate release path in hDAT
In addition to the residue distances used to define the conformational states of hDAT, we further characterized the interactions between AMPH and hDAT in different conformations, and results are shown in Fig. S28. Compared to the initial outward-open state, a similar binding site was found in AMPH-bound hDAT(59–604) and hDAT(25–604) in the outward-open conformations. In the outward-open state of p-hDAT, AMPH was found to move deeper towards the intracellular side and therefore interact with Phe320, Ser321, and Gly323 less frequently, but interact with Gly327 and Val328 more frequently. However, for hDAT in the inward-open states, no interaction with Gly327 was found, and AMPH moved further towards the intracellular end and interacted with Ser429 located in TM8. Thus, this interacting residue Ser429 can be used as an indicator to distinguish hDAT is either in the outward-open or in the inward-open state, which supports our use of the distance between Phe76 (always an interacting residue) and Ser429 to define the conformational states of hDAT (Fig. 2).
Although AMPH was observed to move deeper into the binding site of hDAT in the inward-open conformations, further investigations are needed to identify the release path of AMPH from the extracellular into the intracellular side. One outward-open conformation was selected from the simulations of hDAT(1–604) (Model2), with a typical distance of 4 Å between Phe76 and Ser422, and 12 Å between Phe76 and Ser429. And one inward-open conformation was selected from the simulations of p-hDAT(1–604) (Model2), with a typical distance of 9 Å between Phe76 and Ser422, and 14 Å between Phe76 and Ser429. Molecular docking was performed with AutoDock Vina [23]. The same search space centered on the central binding site was used and a total of 20 binding poses of AMPH was obtained for each docking study. Fig. 7A shows the result of docking AMPH to hDAT(1–604) in the outward-open conformation. The primary binding site for AMPH is located in the central binding site. Fig. 7B shows the result of docking AMPH to p-hDAT(1–604) in the inward-open conformation. No AMPH binds to the central binding site and the primary binding site involves TM1a, TM6b, TM5, and TM8. Taken these results together, Fig. 7C illustrates the whole path that connects the central binding site to the intracellular side. A similar substrate path could also be mapped for hSERT by docking ibogaine to both outward-open and inward-open conformations of hSERT (Fig. S29). Such results suggest that different neurotransmitter transporters may share the same substrate release path that involves TM1a, TM6b, TM5, and TM8.
Fig. 7.

(A) Docking of AMPH to hDAT(1–604) in the outward-open conformation. (B) Docking of AMPH to p-hDAT(1–604) in the inward-open conformation. (C) Illustration of the path for AMPH release from the central binding site. (D) Trajectory of the center of mass of DA in p-hDAT(1–604) over 1-μs MD simulations. The initial DA binding pose and residues Phe76, Ser422, and Ser429 are shown. (E) The minimal distance between DA and Ser429, and between extracellular (Tyr156 and Phe320) and intracellular (Arg85 and Asp476) gating residues over the 1-μs MD simulations of DA-bound p-hDAT(1–604) in the inward-open state. For comparison, the minimal distance between AMPH and Ser429 was also calculated based on the simulations of AMPH-bound p-hDAT(1–604) in the inward-open state (Model2). In (A) and (B), a total of 20 binding poses of AMPH was shown. TM12 was not labelled as it was covered by other TMs. For clarity, in (D) and (E), the trajectory was saved every 1 ns.
The binding of AMPH to hDAT competitively inhibit DA reuptake. AMPH could also induce reverse transport of intracellular DA to the extracellular side (DA efflux), and the N-terminal phosphorylation of hDAT has been reported to play an essential role [14]. To investigate the relevance of such conformations to DA efflux, MD simulations of DA-bound p-hDAT(1–604) were also performed. The same inward-open conformation of p-hDAT(1–604) that was used for docking AMPH was taken as the initial conformation for the simulations. The force field parameters for DA were taken from our previous study [19]. The initial binding pose of DA was obtained by molecular docking with AutoDock Vina [23]. Fig. 7D shows the trajectory of DA in p-hDAT(1–604); and Fig. 7E shows the changes in the distance between DA and Ser429 over simulations. Compared to AMPH, the fluctuation of the distance between DA and Ser429 indicated a weaker binding to p-hDAT. The distance between the extracellular (Tyr156 and Phe320) gating residues suggested that DA may access to the binding site of p-hDAT from the extracellular side, corresponding to the normal inward transport of DA. However, the distance between the intracellular (Arg85 and Asp476) gating residues also suggested that DA may access to the binding site from the intracellular side, but because its binding was weaker than AMPH, the presence of AMPH may facilitate the release of DA by competitively binding to p-hDAT, corresponding to the DAT-mediated AMPH-induced DA efflux [14]. Direct observation of DA transport from simulations may require computational time scales far beyond current capabilities. Nonetheless, our results imply that AMPH and DA display distinct dynamics in the binding pocket of p-hDAT(1–604) captured in the inward-open state, which may have implications for the mechanism of AMPH-induced DA efflux.
3.7. Binding of AMPH stabilizes the bound sodium ion
hDAT is one of sodium-dependent neurotransmitter transporters [46]. The dynamics of the initially bound sodium ions Na1 and Na2, especially Na2, are of particular importance (Fig. S28). For example, the Na2 was shown to mediate Li− permeation and intracellular gating opening in a Cl--dependent manner in mouse DAT [47]. Also, the association of substrate transport and Na2 release in a transition state of LeuT has been shown experimentally [48, 49]. In addition, in hSERT, distinct roles of Na1 and Na2 in transport process has been reported [50]. As shown in Table 1, in the absence of AMPH, the release of both Na1 and Na2 were found in the N-terminal truncated forms of hDAT, as well as in the inward-open state of hDAT(1–604). While in the outward-open state, the Na2 stayed in its initial position over simulations. N-terminal phosphorylation resulted in the release of Na1 or Na2. The full-length hDAT seems an exception in which both Na1 and Na2 were stable. Further investigations are required to establish the connection between the conformational state of the substrate-free hDAT and the binding of Na ions, especially Na2. The binding of AMPH stabilized the occupation of Na2 in all forms and states of hDAT, and even Na1 when hDAT was in the outward-open states but not phosphorylated. Together, the above results indicated that the binding of AMPH increases the stability of Na2 in general.
4. Discussion
Based on available crystal structures of dDAT, LeuT, and hSERT, as well as substrate-protein interactions, we first applied two distances between Phe76 and Ser422 and between Phe76 and Ser429 to determine the conformations of these transporters in either the outward-open or the inward-open state (Fig. 2 and Table 1). For hSERT and LeuT, the distances between extracellular and intracellular gating residues are often used to define the two states of transporters. For example, the distances between intracellular (Cα atoms of Trp82 and Tyr350, hSERT numbering) gating residues in the crystal structures of hSERT are 5.9 Å (PDB ID: 6DZY, outward-open state), and 28.1 Å (PDB ID: 6DZZ, inward-open state) [12], respectively. The large difference is because TM1a undergoes a notable movement in the inward-open structure. The corresponding gating residue Trp63 of hDAT is also located in TM1a but no significant movement of TM1a was observed in all inward-open conformations due to the presence of the N-terminal in our simulations (Table S1). In fact, TM1a in the outward-open state of hDAT(1–604) displayed an average angle of 19° with respect to z-axis, whereas it showed an angle of 13° in the inward-open state of p-hDAT(1–604) (Table S1), suggesting that the distance criterion for gating residues may not be applicable to discriminate the conformational states of hDAT. The conformational states defined by the distance criterion were further confirmed by molecular docking studies. AMPH was predicted to bind to the central binding region of hDAT in the outward-open state; whereas in the inward-open state of p-hDAT, AMPH prefers to bind to a region below the central site (Fig. S28). In addition, the indicative role of Ser429 was also highlighted in the analyses of the interacting residues since AMPH was only found to interact with Ser429 in the inward-open states of hDAT (Fig. 6). Further structural comparison between the representative conformations of hDAT and crystal structures of dDAT and hSERT were provided in Figs. S30 and S31.
Between the outward-open and inward-open states, there may exist two intermediate states, outward-occluded and inward-occluded states. The outward-occluded state has been captured in the crystal structures of hSERT in complex with ibogaine (PDB ID: 6DZV) [12] and substrate-bound LeuT as well as substrate-free LeuT (PDB ID: 2A65, [51] and 5JAE [52]). The inward-occlude state has been reported in the crystal structure of LeuT in complex with L-phenylalanine (PDB ID: 6XWM) [53]. As shown in Fig. 2, the distance criterion seems not feasible to distinguish these states. Compared to hSERT in the outward-open state (PDB ID: 6DZY) where the distances of Phe76-Ser422 and Phe76-Ser429 are 4.9 Å and 11.7 Å. respectively, the corresponding distances are 4.2 Å and 10.9 Å in hSERT in the outward-occluded conformation (PDB ID: 6DZV). Also, it seems not desirable to apply the distances between gating residues to distinguish the above two states because the difference in the extracellular gating residues is < 0.2 Å and in the intracellular is 1.1 Å. Thus, other structural features are required because these states may involve various moments in TMs and ILs. For example, compared to the outward-open conformation, changes in the positions of TM1a, TM6a, and ILs were identified in the outward-occluded hSERT [12]. In the inward-occluded conformation of LeuT, a notable tilting of TM5 was found [53]. Different from the end states where the distances between residues Phe76, Ser422, and S429 are well preserved, these intermediate states were captured with various rearrangement in TMs and ILs, reflecting conformational polymorphism. Fig. 3 shows that in addition to the predominant conformations of hDAT, other less populated states may also exist. Considering the sequence difference between hDAT and other transporters and the lack of experimental structure of DAT (either dDAT or hDAT) in these intermediate states, it remains challenging to characterize their structural properties from current MD simulations.
By performing extensive unbiased all-atom MD simulations in POPC membrane, we investigated the conformational states of hDAT under different conditions (Table 1). Starting from the same outward-open conformation, we observed that in the absence of both N- and C-termini, free hDAT(59–604) (Table 1) changed its conformation and populated the inward-open state. However, in the presence of C-terminal (hDAT(59–620) in Table 1) or AMPH (hDAT(59–604) + AMPH in Table 1), these N-terminal truncated forms of hDAT were found to be able to maintain the outward-open states, which could be attributed to different reasons. The effect of C-terminal on the conformational stability resembles a previous finding that the existence of a fluctuating tail helps to stabilize a vulnerable β-hairpin structure by transferring entropy to the surrounding solution [54]. However, the binding of AMPH blocks the movements of hinges located in TM1 and TM6, and retains hDAT in the outward-open state. Such a result is in agreement with previous studies using this truncated form of hDAT to investigate protein-ligand interactions in various environments [44, 55-58]. Yet unbiased MD simulations may not reach the time scale required to observe the conformational transition along substrate transport. One enhanced-sampling method, accelerated molecular dynamics, with a biased potential energy added to the system, has been successfully applied to explore the conformational states associated with DA translocation through hDAT [59].
The effects of N-terminal dynamics on the conformational transition were first demonstrated in the truncated form of hDAT (hDAT(25–604) in Table 1). Of interest, two independent simulations with different initial N-terminal positions in intracellular side showed that hDAT(25–604) preferred the inward-open states. The binding of APMH could retain hDAT(25–604) in the outward-open state in one simulations but not in the other one, raising the question of whether the conformational transition of hDAT is dominated by AMPH binding or the N-terminal dynamics or both. By adding residues 1–24, results from two independent simulations show that the free state hDAT(1–604) could populate either the outward-open or the inward-open state, demonstrating the regulation role of N-terminal residues 1–24. In contrast, AMPH-bound hDAT(1–604) underwent conformational transitions in both models, suggesting that the binding of AMPH failed to stabilize the outward-open conformation with increasing length of the N-terminal. Moreover, N-terminal phosphorylation shifted hDAT(1–604) from the initial outward-open state to the inward-open states in two independent simulations, but still stayed in the outward-open state in one of simulations of AMPH-bound p-hDAT(1–604). Together, these results demonstrate the regulation role of N-terminal in the conformational transition of hDAT(1–604) from the same outward-open state to the inward-open states, which could be further altered by phosphorylation.
By examination of interaction networks between the N-terminal and the intracellular portion of hDAT (Figs. 4 and 5), a common mechanism of retaining the outward-open state emerges from our results. When the N-terminal spanned the intracellular end and interacted with several TMs (1a, 5, and 8), most ILs, and C-terminal simultaneously, hDAT was observed to populate the outward-open state, and the electrostatic interactions between charged residues formed the core of the network. Because similar networks were identified in hDAT(1–604), AMPH-bound hDAT(25–604), and AMPH-bound p-hDAT(1–604), this modulation mechanism seems independent of AMPH binding and phosphorylation. In contrast, in the case where interactions network collapses (e.g., N-terminal dissociates from the intracellular side) or shrinks (e.g., N-terminal encompasses less area of the intracellular side), the inward-open state of hDAT would be expected. This conclusion was further supported by MD simulations of the full-length hDAT and AMPH-bound p-hDAT. Results from the simulations of the full-length hDAT revealed that the C-terminal weakened the electrostatic core and thus altered the interaction network between the N-terminal and the intracellular portion. Consequently, this full-length hDAT changes its states from the outward-open to the inward-open state. In contrast, the full-length p-hDAT in complex with AMPH exhibited a similar interaction network as found in those outward-open states. Thus, it retained in the outward-open state. An early experimental study of hDAT suggested that a cation-π interaction between Arg60 and Tyr335 could stabilize a salt bridge between Arg60 and Asp436 in the intracellular interaction network, which played an import role in regulating substrate transport [60]. However, the underlying molecular mechanism remained to be elucidated. So far, few MD simulations were performed to investigate the conformational states of hDAT including the N-terminal. In the simulation study of the full-length DA-bound hDAT in a mixed membrane containing POPC, POPE(1-palmatoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine), POPS(1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine), PIP2, and cholesterol. The regulation role of N-terminal on the conformational transition of hDAT was addressed for the first time, which involves interactions between the N-terminal and IL4 that are regulated by the highly charged PIP2 [17]. Because the N-terminal interacted primarily with IL4, a shrinking interaction network was anticipated, and as a result, the inward-open conformation was observed. This finding is in agreement with the present results, providing additional support for the regulation mechanism proposed here. Of interest, compared to POPC, POPE lipid increases bilayer’s rigidity and compactness [61], and thus may prevent conformational transition of DAT which involves movements of TMs. Also, cholesterol has been suggested to be able to maintain the outward-open conformation of DAT [58, 62, 63]. In this regard, the observation of the inward-open conformations of hDAT in the present and previous studies imply the determinant role of interactions between the N-terminal and the intracellular portion in state-to-state transition of hDAT.
Moreover, we show that the inward-open conformations of hDAT defined in this work may be functionally relevant. When AMPH bound to hDAT in the inward-open conformation, it moved deeper towards the intracellular end (Fig. S28). Molecular docking studies also confirmed the difference in the conformational states between the outward-open and inward-open states, showing that AMPH occupied two distinct sites (Fig. 7). The substrate release path in hDAT was then identified, involving TMs 1a, 6b, 5, and 8 (Fig. 7). Since no other favorable binding regions in the intracellular side of p-hDAT were found from our docking studies, we speculate that reverse transport of DA (i.e., DA efflux) may share the same path. Compared to AMPH, DA seemed unable to bind to the inward-open conformation of p-hDAT stably (Fig. 7E), and thus likely to be blocked by AMPH after DA enters the central site from the intracellular side, which is in line with the exchange mechanism of p-hDAT-mediated AMPH-induced DA efflux [15, 64]. In addition, we showed that the bound Na2 could remain in its binding site in both states of hDAT, a result that is different from previous computational study showing that the transition of DA-bound hDAT to the inward-open state was accompanied by the release of Na2 [17, 65]. The difference in the substrates DA and AMPH may contribute to the distinct results, as AMPH is a potent inhibitor of hDAT and could induce DA efflux [66]. Compared to the dynamics of DA in the inward-open state of p-hDAT (Fig. 7E), the stable binding of AMPH and Na2 indicates a significantly slow AMPH transport and associated release of Na2.
5. Conclusions
In this study, we presented comprehensive investigation on the mechanism that modulates the conformational states of hDAT, which involves the interactions between the N-terminal and the intracellular region of hDAT. A general mechanism emerges from our results, which suggests that when the N-terminal is open to encompass a large area of the intracellular portion mainly by interactions with the intracellular loops and C-terminal, hDAT populates the outward-open conformation. Otherwise, when the N-terminal collapses in cytosol or its position is restrained, the inward-open conformation of hDAT is expected. Our results also provide novel insights into how the N-terminal phosphorylation facilitates DAT-mediated AMPH-induced DA efflux. Our results suggest that it seems plausible to capture hDAT in a specific conformational state by alteration of its N-terminal or interactions between the N-terminal and intracellular region via protein engineering.
Supplementary Material
Acknowledgements
The authors acknowledge support from the NIH (Grant #GM121275), and the Texas Advanced Computing Center (TACC) at The University of Texas at Austin for providing HPC resources that have contributed to the research results reported within this paper. URL: http://www.tacc.utexas.edu.
Footnotes
Declaration of Competing Interest
The authors declare no competing financial interest.
References
- [1].Cheng MH, Bahar I, Monoamine transporters: structure, intrinsic dynamics and allosteric regulation, Nat Struct Mol Biol, 26 (2019) 545–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Joseph D, Pidathala S, Mallela AK, Penmatsa A, Structure and Gating Dynamics of Na+/Cl–Coupled Neurotransmitter Transporters, Front Mol Biosci, 6 (2019) 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Leviel V, Dopamine release mediated by the dopamine transporter, facts and consequences, J Neurochem, 118 (2011) 475–489. [DOI] [PubMed] [Google Scholar]
- [4].Vaughan RA, Foster JD, Mechanisms of dopamine transporter regulation in normal and disease states, Trends Pharmacol Sci, 34 (2013) 489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sitte HH, Freissmuth M, Amphetamines, new psychoactive drugs and the monoamine transporter cycle, Trends Pharmacol Sci, 36 (2015) 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zhu J, Reith ME, Role of the dopamine transporter in the action of psychostimulants, nicotine, and other drugs of abuse, CNS Neurol Disord Drug Targets, 7 (2008) 393–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Verma V, Classic Studies on the Interaction of Cocaine and the Dopamine Transporter, Clin Psychopharmacol Neurosci, 13 (2015) 227–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wang KH, Penmatsa A, Gouaux E, Neurotransmitter and psychostimulant recognition by the dopamine transporter, Nature, 521 (2015) 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Forrest LR, Zhang YW, Jacobs MT, Gesmonde J, Xie L, Honig BH, Rudnick G, Mechanism for alternating access in neurotransmitter transporters, Proc Natl Acad Sci U S A, 105 (2008) 10338–10343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nielsen AK, Möller IR, Wang Y, Rasmussen SGF, Lindorff-Larsen K, Rand KD, Loland CJ, Substrate-induced conformational dynamics of the dopamine transporter, Nat Commun, 10 (2019) 2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Krishnamurthy H, Gouaux E, X-ray structures of LeuT in substrate-free outward-open and apo inward-open states, Nature, 481 (2012) 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Coleman JA, Yang D, Zhao Z, Wen P-C, Yoshioka C, Tajkhorshid E, Gouaux E, Serotonin transporter–ibogaine complexes illuminate mechanisms of inhibition and transport, Nature, 569 (2019) 141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Foster JD, Pananusorn B, Vaughan RA, Dopamine Transporters Are Phosphorylated on N-terminal Serines in Rat Striatum, J Biol Chem, 277 (2002) 25178–25186. [DOI] [PubMed] [Google Scholar]
- [14].Eric N, Khoshbouei H, Sen N, Guptaroy B, Johnson LA, Lund D, Gnegy ME, Galli A, Javitch JA, N-Terminal Phosphorylation of the Dopamine Transporter Is Required for Amphetamine-Induced Efflux, PLoS Biology, 2 (2004) e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Robertson SD, Matthies HJG, Galli A, A Closer Look at Amphetamine-Induced Reverse Transport and Trafficking of the Dopamine and Norepinephrine Transporters, Mol Neurobiol, 39 (2009) 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hamilton PJ, Belovich AN, Khelashvili G, Saunders C, Erreger K, Javitch JA, Sitte HH, Weinstein H, Matthies HJG, Galli A, PIP2 regulates psychostimulant behaviors through its interaction with a membrane protein, Nat Chem Biol, 10 (2014) 582–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Khelashvili G, Stanley N, Sahai MA, Medina J, LeVine MV, Shi L, De Fabritiis G, Weinstein H, Spontaneous Inward Opening of the Dopamine Transporter Is Triggered by PIP2-Regulated Dynamics of the N-Terminus, ACS Chem Neurosci, 6 (2015) 1825–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Khan JA, Sohail A, Jayaraman K, Szöllősi D, Sandtner W, Sitte HH, Stockner T, The Amino Terminus of LeuT Changes Conformation in an Environment Sensitive Manner, Neurochem Res, 45 (2019) 1387–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Xu L, Chen LY, Identification of A New Allosteric Binding Site for Cocaine in Dopamine Transporter, J Chem Inf Model, 60 (2020) 3958–3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Xu L, Chen LY, Association of Sigma-1 Receptor with Dopamine Transporter Attenuates the Binding of Methamphetamine via Distinct Helix-Helix Interactions, Chem Biol Drug Des, 97 (2021) 1194–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Webb B, Sali A, Comparative Protein Structure Modeling Using MODELLER, Curr Protoc Bioinformatics, 54 (2016) 5.6.1–5.6.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Khelashvili G, Doktorova M, Sahai MA, Johner N, Shi L, Weinstein H, Computational modeling of the N-terminus of the human dopamine transporter and its interaction with PIP2 -containing membranes, Proteins, 83 (2015) 952–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Trott O, Olson AJ, AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J Comput Chem, 31 (2010) 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, de Groot BL, Grubmüller H, MacKerell AD, CHARMM36m: an improved force field for folded and intrinsically disordered proteins, Nat Methods, 14 (2016) 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, Mackerell AD, CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields, J Comput Chem, 31 (2010) 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang J, Cieplak P, Kollman PA, How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules?, J Comput Chem, 21 (2000) 1049–1074. [Google Scholar]
- [27].Lomize MA, Pogozheva ID, Joo H, Mosberg HI, Lomize AL, OPM database and PPM web server: resources for positioning of proteins in membranes, Nucleic Acids Res, 40 (2012) D370–D376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Pastor RW, Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types, The Journal of Physical Chemistry B, 114 (2010) 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Jo S, Kim T, Iyer VG, Im W, CHARMM-GUI: A web-based graphical user interface for CHARMM, J Comput Chem, 29 (2008) 1859–1865. [DOI] [PubMed] [Google Scholar]
- [30].Lee J, Cheng X, Swails JM, Yeom MS, Eastman PK, Lemkul JA, Wei S, Buckner J, Jeong JC, Qi Y, Jo S, Pande VS, Case DA, Brooks CL, MacKerell AD, Klauda JB, Im W, CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field, J Chem Theory Comput, 12 (2015) 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Berendsen HJC, van der Spoel D, van Drunen R, GROMACS: A message-passing parallel molecular dynamics implementation, Comput Phys Commun, 91 (1995) 43–56. [Google Scholar]
- [32].Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E, GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX, 1–2 (2015) 19–25. [Google Scholar]
- [33].Kutzner C, Páll S, Fechner M, Esztermann A, Groot BL, Grubmüller H, More bang for your buck: Improved use of GPU nodes for GROMACS 2018, J Comput Chem, 40 (2019) 2418–2431. [DOI] [PubMed] [Google Scholar]
- [34].Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR, Molecular dynamics with coupling to an external bath, J Chem Phys, 81 (1984) 3684–3690. [Google Scholar]
- [35].Darden T, York D, Pedersen L, Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems, J Chem Phys, 98 (1993) 10089–10092. [Google Scholar]
- [36].Nosé S, A unified formulation of the constant temperature molecular dynamics methods, J Chem Phys, 81 (1984) 511–519. [Google Scholar]
- [37].Hoover WG, Canonical dynamics: Equilibrium phase-space distributions, Phys Rev A, 31 (1985) 1695–1697. [DOI] [PubMed] [Google Scholar]
- [38].Parrinello M, Rahman A, Crystal Structure and Pair Potentials: A Molecular-Dynamics Study, Phys Rev Lett, 45 (1980) 1196–1199. [Google Scholar]
- [39].Parrinello M, Rahman A, Polymorphic transitions in single crystals: A new molecular dynamics method, J App Phys, 52 (1981) 7182–7190. [Google Scholar]
- [40].Best RB, Hummer G, Eaton WA, Native contacts determine protein folding mechanisms in atomistic simulations, Proc Natl Acad Sci U S A, 110 (2013) 17874–17879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hall BA, Kaye SL, Pang A, Perera R, Biggin PC, Characterization of Protein Conformational States by Normal-Mode Frequencies, J Am Chem Soc, 129 (2007) 11394–11401. [DOI] [PubMed] [Google Scholar]
- [42].Michaud-Agrawal N, Denning EJ, Woolf TB, Beckstein O, MDAnalysis: A toolkit for the analysis of molecular dynamics simulations, J Comput Chem, 32 (2011) 2319–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Gowers R, Linke M, Barnoud J, Reddy T, Melo M, Seyler S, Domański J, Dotson D, Buchoux S, Kenney I, Beckstein O, MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations, Proceedings of the 15th Python in Science Conference, (2016) 98–105. [Google Scholar]
- [44].Cheng MH, Block E, Hu F, Cobanoglu MC, Sorkin A, Bahar I, Insights into the Modulation of Dopamine Transporter Function by Amphetamine, Orphenadrine, and Cocaine Binding, Front Neurol, 6 (2015) 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Sweeney CG, Tremblay BP, Stockner T, Sitte HH, Melikian HE, Dopamine Transporter Amino and Carboxyl Termini Synergistically Contribute to Substrate and Inhibitor Affinities, J Biol Chem, 292 (2017) 1302–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kanner BI, Zomot E, Sodium-Coupled Neurotransmitter Transporters, Chem Rev, 108 (2008) 1654–1668. [DOI] [PubMed] [Google Scholar]
- [47].Borre L, Andreassen TF, Shi L, Weinstein H, Gether U, The Second Sodium Site in the Dopamine Transporter Controls Cation Permeation and Is Regulated by Chloride, J Biol Chem, 289 (2014) 25764–25773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Terry DS, Kolster RA, Quick M, LeVine MV, Khelashvili G, Zhou Z, Weinstein H, Javitch JA, Blanchard SC, A partially-open inward-facing intermediate conformation of LeuT is associated with Na+ release and substrate transport, Nat Commun, 9 (2018) 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].LeVine MV, Terry DS, Khelashvili G, Siegel ZS, Quick M, Javitch JA, Blanchard SC, Weinstein H, The allosteric mechanism of substrate-specific transport in SLC6 is mediated by a volumetric sensor, Proc Natl Acad Sci U S A, 116 (2019) 15947–15956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Felts B, Pramod AB, Sandtner W, Burbach N, Bulling S, Sitte HH, Henry LK, The Two Na+ Sites in the Human Serotonin Transporter Play Distinct Roles in the Ion Coupling and Electrogenicity of Transport, J Biol Chem, 289 (2014) 1825–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E, Crystal structure of a bacterial homologue of Na+/Cl--dependent neurotransmitter transporters, Nature, 437 (2005) 215–223. [DOI] [PubMed] [Google Scholar]
- [52].Malinauskaite L, Said S, Sahin C, Grouleff J, Shahsavar A, Bjerregaard H, Noer P, Severinsen K, Boesen T, Schiøtt B, Sinning S, Nissen P, A conserved leucine occupies the empty substrate site of LeuT in the Na+-free return state, Nat Commun, 7 (2016) 11673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Gotfryd K, Boesen T, Mortensen JS, Khelashvili G, Quick M, Terry DS, Missel JW, LeVine MV, Gourdon P, Blanchard SC, Javitch JA, Weinstein H, Loland CJ, Nissen P, Gether U, X-ray structure of LeuT in an inward-facing occluded conformation reveals mechanism of substrate release, Nat Commun, 11 (2020) 1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Xu L, Nussinov R, Ma B, Allosteric stabilization of the amyloid-β peptide hairpin by the fluctuating N-terminal, Chem Commun, 52 (2016) 1733–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Wang P, Zhang X, Fu T, Li S, Li B, Xue W, Yao X, Chen Y, Zhu F, Differentiating Physicochemical Properties between Addictive and Nonaddictive ADHD Drugs Revealed by Molecular Dynamics Simulation Studies, ACS Chem Neurosci, 8 (2017) 1416–1428. [DOI] [PubMed] [Google Scholar]
- [56].Xue W, Wang P, Tu G, Yang F, Zheng G, Li X, Li X, Chen Y, Yao X, Zhu F, Computational identification of the binding mechanism of a triple reuptake inhibitor amitifadine for the treatment of major depressive disorder, Phys Chem Chem Phys, 20 (2018) 6606–6616. [DOI] [PubMed] [Google Scholar]
- [57].Wang P, Fu T, Zhang X, Yang F, Zheng G, Xue W, Chen Y, Yao X, Zhu F, Differentiating physicochemical properties between NDRIs and sNRIs clinically important for the treatment of ADHD, Biochim Biophys Acta Gen Sub, 1861 (2017) 2766–2777. [DOI] [PubMed] [Google Scholar]
- [58].MacKerell A, Zeppelin T, Ladefoged LK, Sinning S, Periole X, Schiøtt B, A direct interaction of cholesterol with the dopamine transporter prevents its out-to-inward transition, PLOS Computat Biol, 14 (2018) e1005907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Cheng Mary H., Bahar I, Molecular Mechanism of Dopamine Transport by Human Dopamine Transporter, Structure, 23 (2015) 2171–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Kniazeff J, Shi L, Loland CJ, Javitch JA, Weinstein H, Gether U, An Intracellular Interaction Network Regulates Conformational Transitions in the Dopamine Transporter, J Biol Chem, 283 (2008) 17691–17701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Venable RM, Brown FLH, Pastor RW, Mechanical properties of lipid bilayers from molecular dynamics simulation, Chem Phys Lipids, 192 (2015) 60–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Hong WC, Amara SG, Membrane Cholesterol Modulates the Outward Facing Conformation of the Dopamine Transporter and Alters Cocaine Binding, J Biol Chem, 285 (2010) 32616–32626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Jones KT, Zhen J, Reith MEA, Importance of cholesterol in dopamine transporter function, J Neurochem, 123 (2012) 700–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Kahlig KM, Galli A, Regulation of dopamine transporter function and plasma membrane expression by dopamine, amphetamine, and cocaine, Eur J Pharmacol, 479 (2003) 153–158. [DOI] [PubMed] [Google Scholar]
- [65].Razavi AM, Khelashvili G, Weinstein H, A Markov State-based Quantitative Kinetic Model of Sodium Release from the Dopamine Transporter, Sci Rep, 7 (2017) 40076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Sulzer D, Sonders MS, Poulsen NW, Galli A, Mechanisms of neurotransmitter release by amphetamines: A review, Prog Neurobiol, 75 (2005) 406–433. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.